

Abstract
Advances in molecular imaging modalities have accelerated the diagnosis and treatment of human disease. However, tumors less than 1-cm in size still remain difficult to localize by conventional means because of the difficulty in specific targeting/delivery to the tumor site. Furthermore, high nonspecific uptake in the major organs and persistent background retention result in low tumor-to-background ratio. In this study, we demonstrate targeting and therapy of gastrointestinal stromal tumors (GIST) using nonsticky and renal clearable theranostic nanoparticles (a.k.a. H-Dots). H-Dots are not only able to target GIST for image-guided surgery but also to tailor fate and deliver imatinib (IM) anticancer drug resulting in efficient treatment of unresectable GIST. In addition, H-Dots enable monitoring targetability, pharmacokinetics, and drug delivery, and show therapeutic efficacy in GIST-bearing xenograft mice followed by surgical resection. More importantly, IM loaded H-Dots exhibit lower uptake into the immune system, while free IM accumulated in spleen/liver, improved tumor selectivity, and increased tumor suppression compared to free IM. Precisely designed H-Dots can be used as a promising theranostic nanoplatform that can potentially reduce the side effects of conventional chemotherapies.
Keywords: Renal clearance, theranostics, optical imaging, nanoparticles, drug delivery
Graphical Abstract
H-Dot is a nonsticky and renal clearable theranostic nanoplatform designed to target gastrointestinal stromal tumors (GIST). H-Dots not only target GIST for image-guided surgery but also tailor the fate of anticancer drugs to the tumor site precisely, resulting in the treatment of unresectable GIST without nonspecific uptake of off-target H-Dots.
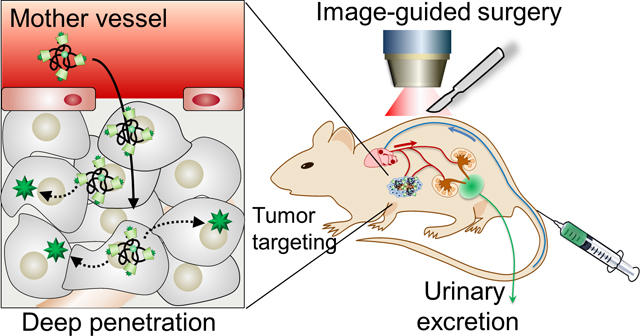
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal malignancies of the gastrointestinal tract. Surgical resection with negative margins is the standard for GIST therapy, which allows most patients with operable GIST to be cured by primary surgery.[1] However, a critical clinical challenge with GIST surgery lies in its localization of small tumors.[2] Despite advances in imaging techniques and the development of new localization procedures, tumors less than 1 cm in size remain difficult to localize by conventional means[3] because of the difficulty in specific delivery of contrast agents to the tumor site and a low tumor-to-background ratio (TBR) resulting from high nonspecific uptake and background retention. Chemotherapeutic agents such as imatinib (IM) can be used to treat GIST with locally advanced, recurrent, primary unresectable, and metastasized tumors.[4] IM is the first approved selective tyrosine kinase inhibitor for KIT and platelet derived growth factor receptor alpha in GIST.[5] Importantly, IM continuation is crucial for long-term survival of advanced GIST patients since IM typically suppresses GIST growth but does not eradicate the tumors. IM can occasionally cause serious adverse effects including myelosuppression, tumor bleeding, gastrointestinal perforation, interstitial pneumonia, and severe skin symptoms.[6]
Recently, drug delivery technology has advanced, making it possible for small molecules to be delivered to target tumors with the use of nanoparticles (NPs).[7] This development has had significant clinical implications such as enhancement of the anti-tumor effect, reduction of adverse effects by suppressing nonspecific uptake by other organs, and decreased dosage by increasing intratumoral drug accumulation. Very recently, ultra-small silica NPs (<10 nm in diameter) were reported by Bradbury and coworkers and were applied as a drug delivery vehicle for inducing ferroptosis and regression of tumor.[8] In addition to silica NPs, Zheng group has intensively studied renal clearable ultra-small gold NPs (<2 nm core diameter) to target tumor and deliver the anticancer drug, doxorubicin.[9] Previously, we also reported that an ultra-small (< 5.5 nm) zwitterionic organic nanocarrier, termed Harvard-Dot or H-Dot, systemically travels the whole body through the bloodstream without nonspecific tissue uptake, then eventually clears out to the urine exclusively (>80% at 4 h post-injection).[10] However, image-guided surgical intervention and therapeutic efficacy of H-Dots have not yet been verified. Here, we demonstrate that the H-Dot, as a theranostic nanoplatform, facilitates determination of surgical margin by NIR image-guidance, and anticancer drug delivery to target tissues. The H-Dots include a near-infrared (NIR; 650–900 nm) fluorescent moiety which enables us to monitor targeting, pharmacokinetics, drug delivery, and therapeutic efficacy in vivo following surgical resection. When delivered by H-Dots, IM shows lower uptake into the immune system, improved tumor selectivity, and increased tumor suppression compared to free IM. H-Dots are a promising theranostic nanoplatform for both image-guidance and drug delivery with the ability to reduce the adverse effects associated with previous theranostic systems.
Nonsticky and renal-clearable properties of H-Dots given by the zwitterionic surface and the size smaller than glomerular filtration threshold show wide distribution to the central (Vc) and peripheral compartments (Vp), including tumor volume (Vt), as illustrated in Figure 1a. After the distribution phase, renal clearable NPs are rapidly cleared from the major organs through urinary excretion resulting in less background retention in organs and tissues.[10–11] Due to very low background tissue retention, renal clearable NPs can exhibit high tumor-to-background signal ratio for tumor targeting via the enhanced permeability and retention (EPR) effect, despite the lack of active targeting moieties.[9a,c] On the other hand, nonrenal clearable and nonspecific NPs, which are mostly larger in size and/or lipophilic surface, are retained in the central and peripheral compartments including immune-related organs such as liver and spleen.[7a,12] This can cause slow elimination, unwanted release, and potential toxicities due to the anti-cancer drug/carrier itself.[7b,13] We have previously demonstrated the fate control of inorganic or organic NPs in the body by modulating physicochemical properties such as size, shape, surface charges, and hydrophilicity/lipophilicity.[10,13–14] Based on our previous studies, H-Dots have been designed with zwitterionic surface on ε-polylysine backbone to achieve nonsticky property and with cyclodextrin for hydrophobic drug delivery (Figure 1b). It can make an inclusion complex with the anti-cancer drug, IM (imatinib mesylate), and deliver and release IM to a GIST tumor. The detailed synthetic scheme for H-Dots is shown in Figure S1.
Figure 1. Concept of nonsticky and renal clearable theranostic nanoparticle (H-Dots).

A) Schematic diagram for comparison of pharmacokinetics/dynamics, distribution, and clearance between renal clearable NPs and nonspecific NPs. B) Chemical structures of H-Dot and imatinib anticancer drug for image guided GIST therapy. H-Dot, composed of NIR-fluorescent cyclodextrin-grafted polylysine (ZW800-CDPL±), has balanced charges and its overall size is less than 5.5 nm in HD. H-Dots can make an inclusion complex with the anticancer drug, imatinib. The targeted H-Dots enable performance of image-guided therapy including surgical intervention and drug delivery.
First, we verified that H-Dots can function as a nanocarrier to deliver the anticancer drug, IM, and change its biodistribution. Prior to this, the terminus of IM was modified with an amino group by following a previously reported synthetic route,[15] and ZW800–1 NIR fluorescent dye was conjugated to it (Figure S2a,b). The final conjugate (IM-ZW) was confirmed by HPLC-MS (Figure S2c). In the previous literature, conjugate with the terminal position of IM did not interfere the affinity of the IM to KIT on GIST cells. In addition, cyclic RGD and antibody conjugated with ZW800 was able to specifically bind target tumor with very high tumor-to-background ratio without any accumulation in liver or spleen.[16] We summarized physicochemical properties of IM-ZW and confirmed in vitro cell binding of IM-ZW against GIST-T1 cells (Figure S3). IM-ZW or IM-ZW/H-Dot inclusion complex (in this case, H-Dots were not conjugated with any NIR fluorophores) were injected into tumor bearing mice, and tumor targeting and biodistribution were assessed. It should be noted that IM-ZW alone showed NIR fluorescence signals in liver and spleen, but did not localize to the tumor site until 24 h post-injection, (Upper panel in Figure 2a, and Figure S4 for up to 72 h; SBR in the tumor was less than 2 in Figure 2b). In contrast, IM-ZW/H-Dot showed good tumor accumulation (SBR~4) and the remainder of the IM-ZW/H-Dot was excreted into the bladder resulting in kidney signals at 24 post-injection (Figure 2a,b). Since H-Dots are cleared by renal filtration, it is important to confirm that H-Dots do not cause renal dysfunction and that they are not retained in the kidneys after injection. To confirm that these two things are true, H-Dots were injected intravenously to CD-1 mice and sacrificed after 5 days. Another CD-1 mouse was injected with H-Dots 1 day prior to imaging for signal comparison. As shown in the NIR fluorescence image (Figure S5), kidney signals at 5-day post-injection were faint compared with those at 1-day post-injection, indicating active filtration of H-Dots without accumulation or blockage in the kidney over time.
Figure 2. Biodistribution, tumor targetability and pharmacokinetics of IM-ZW and IM-ZW/H-Dot.

A) Comparisons of biodistribution and tumor targetability of IM-ZW and IM-ZW/H-Dot. 10 nmol of IM-ZW and IM-ZW/H-Dot in 5% BSA saline were injected separately into GIST bearing mice, and NIR imaging was carried out at 24 h post-injection. The left side and abdominal region without surgical opening; abdominal region with surgical opening; and resected organs. Abbreviations used are: Tu, Tumor, Bl, bladder; Du, duodenum; He, heart; In, intestine; Ki, kidneys; Li, liver; Lu, lungs; Mu, muscle; Pa, pancreas; Sp, spleen. B) Signal to background ratio (SBR) of each resected organ from mice injected with IM-ZW and IM-ZW/H-Dot, respectively. C) Blood concentration (%ID g−1) decay curve, and Half-life of IM-ZW and IM-ZW/H-Dot. Blood samples from the IM-ZW or IM-ZW/H-dot injected mouse were collected from the tail vein at time points: 1, 3, 5, 10, 30, 60, 120, 180, and 240 min. D) Pharmacokinetic parameters of IM-ZW and IM-ZW/H-dot. A urine sample was collected at 4 h post-injection. (n = 3, mean ± s.e.m., *P <0.05)
Additionally, we investigated pharmacokinetic parameters of IM-ZW and IM-ZW/H-Dot after a single intravenous injection (Figure 2c). Blood half-life was calculated by analyzing the NIR fluorescent signal of mouse serum at each time point. The blood concentration curves demonstrated that both IM-ZW and IM-ZW/H-Dot exhibit two-compartment model profiles; rapid initial decays indicate efficient initial distribution into major organs. Interestingly, the blood curve profile of IM/H-Dot showed ideal PK pattern for tumor targeting: after being intravenously injected, it rapidly distributed into first compartment, circulated in the body with the reasonable area under the curve (AUC) to meet the target, and rest of off-targeting H-Dots can be eventually excreted through urine within 4 h (Figure 2d). In addition, volume of distribution (Vd) of IM-ZW/H-Dot (245 mL kg−1) was 24.3% of the body volume which is very close to the volume of the extracellular fluids (~20%). This indicates that H-Dot can distribute whole body while it does not stick to the peripheral compartment as explained in Figure 1. Although, the half-life of IM-ZW/H-Dot (t½β = 47.32 min) is shorter than that of IM-ZW in the elimination phase, the AUC values of IM-ZW/H-Dot (1647 %ID·min g−1) and IM-ZW (1635 %ID·min g−1) were similar. In contrast, IM-ZW showed approximately 2-fold longer half-life (t½β = 91.12 min) and high Vd (608 mL kg−1) value than those of H-Dot. These half-life and AUC expand are because of nonspecific uptake of IM in mostly liver and spleen as off target organs. These results suggest that IM-ZW/H-Dots distributed into the interstitial space with evasion of nonspecific interactions associated with plasma proteins and central compartments, resulting in better tumor targetability. Overall, even after a payload containing an anticancer drug, H-Dots were able to systemically circulate and distribute to the whole body, target tumor tissue, and then clear efficiently from the body.
Prior to carrying out in vivo therapeutic efficacy studies, the IM/H-Dot inclusion complex was tested for pH-induced drug release (Figure 3a). Cy3 conjugated IM was used to confirm the release by measuring the absorbance value of Cy3. IM-Cy3/H-Dot inclusion complex was incubated in 30% FBS/PBS for 24 h and then IM-Cy3 release was observed at each time point over 24 h. Since the hydrophobic interaction between IM and the nonpolar cavity of β-CD was reduced in the acidic environment due to the monoprotonation of the cyclic tertiary amine of piperazine (Figure 1b), 55% of the IM was released at pH 6.0 after incubation for 12 h.[17] In contrast, IM/H-Dot was relatively stable at pH 7.4. These results suggest that the inclusion complex is stable in neutral physiological environments, but loses stability and efficiently releases the drug in acidic tumor microenvironments (pH ~6.0). Next, in vitro therapeutic efficacy and kinetic studies were conducted (all the concentrations of IM/H-Dot are based on H-Dot). KIT receptor-positive GIST-T1 cells were incubated in a 96 well plate and were treated with IM alone, H-dot alone, and IM/H-dot complex over the range of 10 μM to 0.1 nM in a dose-dependent manner. The number of IM in H-Dot was calculated by measuring the UV absorption value of IM, which was ~4.3 (Figure S6). After incubation for 24 h, the cell viability was assessed by colorimetric assay with cell counting kit-8. The results showed no cytotoxicity was observed in the H-Dot alone treatment group, even at the highest dose administered, 10 μM (Figure 3b). In addition, GIST-T1 cell morphology showed a normal elongated spindle-shape (Figure 3c), which indicates H-Dot nanocarriers are safe. In contrast, IM alone and IM/H-Dot treated groups showed significant and dose-dependent cytotoxicity effects (Figure 3b,c). The half maximal effective concentration (EC50) for IM/H-Dot was calculated to be 4.21 nM against GIST-T1 cells which is similar with IM alone (7.82 nM). These in vitro results suggest that IM/H-Dots are able to efficiently deliver IM to tumor cells with better performance characteristics than free IM.
Figure 3. In vitro therapeutic efficacy test.

A) pH dependent drug release profile of the IM/H-Dot complex incubating in 30% FBS for 24 h (*P <0.05). B) GIST-T1 cell growth inhibitory effect with different concentrations of IM alone, H-Dots alone, and IM/H-Dot complex. (n = 5, mean ± s.e.m.). C) The morphology of GIST-T1 before and after the treatment of IM/H-Dot complex was compared using microscopic images (×20). Scale bar, 100 μm. Cytotoxicity of IM/H-Dot complex was evidenced by altered morphology of GIST-T1 cells after treatment.
Figure 4 and Figure S7 demonstrate how image-guided surgery using H-Dots can be used to completely remove tumors with narrow margins. Cancerous tissue is specifically targeted by fluorescent H-Dots, which shows high signal to background ratios, making complete tumor resection possible. This technique is also able to distinguish tumors less than 1 cm from the surrounding tissue, ensuring that after resection, no tumor remains. In Figure 4, the tumor is clearly outlined and differentiated from the surrounding tissue. After resection of this large tumor, a concealed, nonvisible tumor less than 1 cm is removed, made possible by our image guidance system (K-FLARE).
Figure 4. In vivo image-guided surgical intervention.

A) Tumors were resected under NIR fluorescence imaging guidance. White arrows indicate small size tumors (less than 1cm) which remained after first resection and were then removed with image guidance. Scale bar, 1 cm. B) Distributions of fluorescence intensities in the tumor area (white dotted line #1 and #2) and background tissue (white dotted line #3). The fluorescence intensities were measured using ImageJ software.
Therapeutic efficacy of IM/H-Dot was investigated on xenograft GIST mice and compared to those of free IM, H-Dot, and saline (Figure 5). All the treatment doses were injected intravenously (daily injection on weekdays) into GIST bearing mice for 2 weeks. Accumulation in tumors, biodistribution and tumor growth suppression were monitored by NIR imaging. As shown in Figure 5a, IM/H-Dot accumulated in tumors and untargeted IM/H-Dot was excreted to urinary bladder after injection. As a result, very low nonspecific uptake and background signal were observed. Drug delivery was monitored by signal intensity changes and correlated well with times of injection (Figure 5b). Relative tumor volume v/v0 (v is the volume after growth, v0 is the initial tumor volume) was successfully suppressed overtime in the IM/H-Dot treatment group (Figure 5c) which was 2.6 times smaller than the saline control group. In contrast, tumor size with the treatment of IM alone was 1.8 times bigger than IM/H-Dot complex group. This indicates that IM/H-Dot treatment significantly enhanced anticancer efficacy with respect to that of free IM due to IM/H-Dot inclusion complex is able to evade nonspecific uptake by off target organs and tissues, and delivers the IM more efficiently to the tumor. This evading capability of H-Dot is because of its zwitterionic surface that renders it “nonsticky.” In H-Dot alone injection, signal also continuously increased in tumor site by observing NIR imaging while no tumor inhibition effect was observed, which is similar with saline treatment as a control group. Furthermore, almost no change in mouse body weight was observed when the mice were treated with IM/H-dot (Figure 5d). Therefore, our H-Dot based theranostic nanoplatform is very promising and successful to for both image-guided resection and therapy.
Figure 5. In vivo tumor therapeutic efficacy of IM/H-Dot complex.
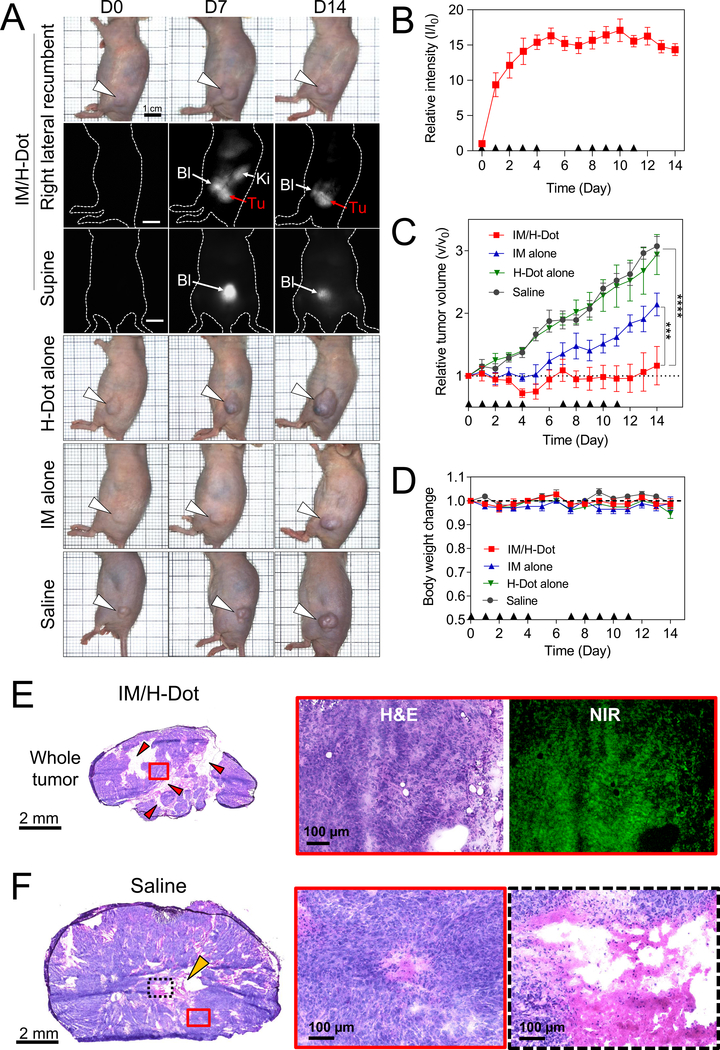
A) Representative images of each mouse treated with IM/H-Dot, IM, H-Dot, and saline. Each dose was intravenously injected for 14 days (daily injection on weekdays). See detailed dose information in Table S2. B) Relative intensity at tumor site from mice treated with IM/H-Dots C) Relative tumor volume up to 14 days post-injection. The tumor volume of IM/H-Dot complex injected mice was significantly lower than that of IM, saline, and H-Dot injected mice (n = 5, mean ± s.e.m., ***P <0.001, ****P <0.0001). D) Normalized body weight of mice in each group for 14 days. E) H&E staining and NIR fluorescence images of a resected tumor after treatment with IM/H-Dots. Red arrow heads indicate apoptosis sites. F) H&E staining images of saline control group. Yellow arrow head indicates necrosis site.
The limited diffusion into the tumor has been one of the major challenges to be addressed for drug delivery systems in the nanomedicine field. We evaluated the penetration and distribution of H-Dots by fluorescence microscopy and confirmed tumor morphological changes by H&E staining. With IM/H-Dot treatment, the boundary of the tumor was still intact, however, inside the tumor, especially near the intra-tumoral vessels, apoptosis occurred resulting in many areas with empty/low cell populations (red arrow heads in Figure 5e). In addition, strong H-Dot fluorescence signals were observed in the deep tumor tissues and the signals were evenly distributed throughout the whole tumor specimen as observed by NIR microscopy. This suggests that ultra-small-sized H-Dots can penetrate deeply due to higher permeability and delivery efficiency.[9b] In contrast, high cellularity was observed, and necrosis was identified in the center of the tumor (yellow arrow head in Figure 5f) in the saline treatment group. Furthermore, we tested single dose acute toxicity with intravenous administration (329 mg kg−1 for IM-Dot and 60 mg kg−1 for IM which are equimolar based on IM). There were no fatalities, and abnormal behavior related to toxicity was not observed. After 2 weeks post-injection, biochemical analysis and histopathological examinations were conducted. There was no evidence of tissue damage, inflammation, or morphological change in either histological or biochemical readouts in the IM/H-Dot treatment group, compared with the saline control group (Figure S8). These results demonstrate that the H-Dot based drug-carriers can significantly enhance therapeutic efficacy as well as safety because of more efficient delivery, deep tumor penetration, and fast renal excretion, limiting off-target drug effects.
Many GISTs can be treated successfully by targeting oncogenic KIT and PDGFRA with small molecule tyrosine kinase inhibitors. Several drugs have been developed that block the activation of these targets, such as imatinib mesylate (Gleevec, Novartis), which is approved by the FDA for unresectable/metastatic KIT-positive GISTs, with 80% of patients showing a clinical response in this setting. IM is a selective tyrosine kinase inhibitor that mimics ATP and binds competitively to KIT and PDGFRA, inhibiting multiple downstream signaling pathways.[18] Currently, the FDA also approved the use of IM in the adjuvant setting following gross resection of KIT-positive tumors to prevent recurrence.[19] Currently, primary GIST tumors are completely resected with an intact pseudo capsule and negative microscopic margins or by segmental resection. However, complete but conservative resection can be difficult because most GIST surgeries are performed “blindly” without efficient intraoperative image guidance to assess tumor margin. In addition, because of the high reoccurrence of GIST, it is imperative that the tumor/area around the tumor be treated with anti-cancer drug before/after surgical resection. In a clinical setting, H-Dot inclusion complexes not only localize the tumor, but prevent reoccurrence after surgical resection by delivering IM to the tumor area and surrounding tissues, exerting its anti-cancer effect on any unresectable and/or metastatic, malignant GIST tissue that may remain after surgery. In addition, we can monitor tumor growth suppression in the neoadjuvant/adjuvant treatment by NIR imaging.
Targeted drug delivery to the tumor site is another challenge. Over the past decade, various NPs, such as noble metal NPs, iron oxide NPs, carbon-based materials, polymeric NPs, and liposomes, have been developed for drug delivery. However, the lack of understanding in NPs’ fate results in less targetability as well as adverse effects in off-target organs such as liver and spleen. This becomes even more of a challenge when considering small tumors less than 1 cm. To solve these challenges, H-Dots can be employed to improve IM target specificity and pharmacodynamics. Another benefit of H-Dots-based drug delivery is that it minimizes systemic cytotoxic effects by avoiding nonspecific uptake in normal tissues. To confirm that nonspecific uptake was avoided, the biodistribution of the delivery vehicle and anticancer drugs were tracked by NIR fluorescent imaging.[20] H-Dots can control the distribution mediated pharmacokinetics by: i) protecting the drug from unwanted degradation, ii) preventing nonspecific interactions, and iii) enhancing drug absorption in target tissues, all of which are less efficient with traditional chemotherapies.[7a,13] To achieve this, rapid excretion from the body and/or efficient degradation into nontoxic products is required.[14b] Renal clearance, compared to hepatobiliary excretion, is preferred for theranostic NPs because unbound theranostic NPs predispose to rapid elimination from the body with limited cellular internalization and metabolism, thus minimizing the theranostic NP’s exposure in normal tissue.[7a,8c,13,14b]
In this study, we demonstrated nonsticky and renal clearable H-Dot GIST targeting for image-guided surgery, selective IM delivery to mutant KIT receptors, and efficient treatment of unresectable GIST. H-Dots enabled monitoring of targetability, pharmacokinetics, drug delivery, and therapeutic efficacy in GIST-bearing xenograft mice followed by surgical resection. More importantly, IM-loaded H-Dots exhibited lower uptake into the immune system, while free IM accumulated in spleen/liver, improved tumor selectivity, and increased tumor suppression compared to free IM. This demonstrates the potential for H-Dots to be used as a promising theranostic nanoplatform with the potential to reduce the side effects of conventional chemotherapies. In summary, this paper highlights how H-Dots can systemically travel through the bloodstream avoiding nonspecific tissue uptake, and then eventually be excreted in the urine. In addition, H-Dots allow optimal targeting of anticancer drugs to tumor specifically for maximizing therapeutic efficacy and minimizing drug toxicity. These studies pave the way for the development of ideal theranostic nanoparticles and the next generation of personalized medicine.
Experimental
Materials:
Epsilon-Polylysine (ε-poly-l-lysine, EPL; MW ~4,000) was purchased from BOC Sciences (NY, USA). β-cyclodextrin (β-CD), Dess-Martin periodinane (DMP), sodium borohydride, succinic anhydride, bovine serum albumin (BSA), diisopropylethylamine (DIEA), dipyrrolidino(N-succinimidyloxy)carbenium hexafluorophosphate (HSPyU), ninhydrin, acetone, and ethanol were purchased from Fisher Scientific (Pittsburgh, PA), Sigma-Aldrich (Saint Louis, MO), or Acros Organics (Morris Plains, NJ). Imatinib mesylate was provided by Novartis or purchased from LC Laboratories (Woburn, MA).
Stability and pH-responsive drug release tests:
H-Dots (2 mm in water; 20 μL) were mixed with Cy3-IM solution (10 mm in DMSO/PBS, 50/50 v/v%; 40 μL) and the mixture was vortexed for 24 h. Then, Cy3-IM loaded H-Dots were purified using micro Bio-Spin P-6 gel columns (Bio-rad) to remove unbound excess Cy3-IM. To monitor the release of imatinib at different pH environments, IM/H-Dot was dispersed in 1 mL of 30% FBS, pH 6.0 and pH 7.4, respectively. 10–50 μL of the IM/H-Dot solutions were taken at each time point, and released imatinib was removed by the micro Bio-Spin P-6 gel column. The absorbance of purified IM/H-Dot solutions was then measured at 550 nm to calculate the amount of imatinib released. The percentage of the released imatinib was calculated by the following formula: (Absinitial – Abstime point)/Absinitial×100.
In vitro therapeutic efficacy test of IM/H-Dots against to GIST-T1 cells:
GIST-T1 cells, harboring a KIT activating mutation, were seeded onto sterilized 6.4 mm diameter in 96 well plates (2 × 103 cells per well) and incubated in DMEM (Mediatech, Hermdon, VA) supplemented with 10% FBS, 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin at 37°C for 24 h [21]. Then imatinib alone, H-Dots alone, and imatinib/H-Dot complex were treated to each well over the range of 0.001 to10 μm for each well. The cellular therapeutic efficacy effect of each drug dose was evaluated with cell counting kit-8 and microscopy. The mean EC50 value can be calculated as following equation:
where E is the % cell viability, Top and Bottom are plateaus in the % cell viability, x is the logarithm of the concentration, and the Hill coefficient reflects the slope of the curve.
In vivo biodistribution and pharmacokinetics of IM-ZW/H-Dot:
The procedure described in our previous report was adapted for this study and will be briefly outlined here. Animals were housed in an AAALAC-certified facility and were studied under the supervision of MGH IACUC in accordance with the approved institutional protocol (N2016000529). Six-week-old CD-1 mice (male; 25–30 g) were purchased from Charles River Laboratories (Wilmington, MA). Mice were maintained under anesthesia with isoflurane and oxygen during the preparation prior to injection. The end of the tail was cut for blood extraction. Before injection, blood was sampled in capillary tubes (Fisher Scientific, Pittsburgh, PA) as a reference. The penis was tied off using surgical sutures to prevent urination during the experiment. Mice were injected with 10 nmol of IM-ZW or with IM-ZW/H-Dot in saline. Blood was sampled in capillary tubes at the following time points (1, 3, 5, 10, 30, 60, 120, 180, and 240 min) to calculate distribution (t1/2α) and elimination (t1/2β) blood half-life values. Mice were imaged using the in-house built real-time intraoperative NIR imaging system. A 760 nm excitation laser source (4 mW cm−1) was used with white light (400–650 nm; 40,000 lux). Color and NIR fluorescence images were acquired simultaneously with customized software. After 4 h post-injection, mice were sacrificed to image organs and collected urine from the bladder. At least 3 mice were analyzed for each sample. Results were presented as a bi-exponential decay curve using Prism software version 8.2.0 (GraphPad, San Diego, CA). The volume of distribution was calculated as following equation:
where t1/2 is the blood half-life value, and AUC is the area under the curve.
In vivo therapeutic efficacy test of H-Dots:
Animals were housed in an AAALAC-certified facility and were studied under the supervision of MGH IACUC in accordance with the approved institutional protocol N2016000529. To establish tumor-xenografted nude mice, GIST cell was cultured in DMEM with 10% FBS and 100 units mL−1 of penicillin and streptomycin. NCr nu/nu mice (Taconic Farms, Germantown, NY) were inoculated via subcutaneous injection with 2×106 GIST cells suspended in 100 μL of PBS/Matrigel (50 v/v%) at the flank. Once the tumor reached a size of 0.5–0.7 cm, 15 μmol kg−1 of IM/H-Dots or intact H-Dots in saline was injected through tail vein. IM or saline were treated via intraperitoneal injection. (see Table S2 for details) Tumor mice were imaged using real-time intraoperative NIR imaging system at the following time points and then scarified for ex vivo imaging and histological evaluations. The length (L) and width (W) of the tumors were measured with a digital caliper and the volume (V) was calculated by the equation V = ½ LW2. And, the body weights were also monitored during the treatment. For histology, fluorescence microscopy was performed on a Nikon TE2000 with two custom filter sets (Chroma Technology, Brattleboro, VT, USA).
Single dose acute toxicity study:
Nine CD-1 mice (male; 25–30 g, 6-week-old) were divided into three test groups: Saline control, IM alone, and IM/H-Dot. For the single dose acute toxicity test, we determined the LD50 (IV, mouse) amount of IM to be 60 mg kg−1. This value has not been reported on, but the maximum tolerated dose for oral administration has been reported, and is set at 600 mg kg−1 (oral, rat). Because the maximum dose of parenteral administration is generally 5–10 times less than that of oral administration [22], 60 mg kg−1 has been used to be compatible with IV injection. The mice in the IM alone group were intravenously injected with 100 μL of 30.5 mM imatinib mesylate (60 mg kg−1), and the mice in the IM/H-Dot group were intravenously injected with an equimolar dose with respect to IM of IM/H-Dot (329 mg kg−1). We monitored the body weight, clinical signs (e.g. abnormal gait and posture or muscle tone), and mortality for 2 weeks. After that, all mice were euthanized, followed by collection of blood and resection of the heart, liver, spleen, and kidneys for biochemical and histological analyses.
Quantitative analysis:
The fluorescence and background intensities of a region of interest over each tissue were quantified using customized imaging software and ImageJ v1.51j8 (National Institutes of Health, Bethesda, MD). The signal-to-background ratio (SBR) was calculated as SBR = fluorescence/background, where background is the fluorescence intensity of muscle. All data depict the mean ± s.e.m. with a minimum of 3 biological replicates. A one-way ANOVA followed by Tukey’s multiple comparisons test was used to assess the statistical difference among more than two groups. P value of less than 0.05 was considered significant: *P <0.05, **P <0.01, ***P <0.001, and ****P<0.0001.
Supplementary Material
Acknowledgments:
We thank Sung Ki Kim for manuscript editing. This study was supported by NIH grants NIBIB #R01EB022230, NHLBI #R01HL143020, and NCI #R21CA223270, and the Creative Materials Discovery Program through the National Research Foundation of Korea (2019M3D1A1078938). The content expressed is solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Competing interests: The authors declare that they have no competing financial interests.
Data and materials availability: All data is available in the main text or the supplementary materials.
Contributor Information
Homan Kang, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114 (United States).
Wesley R. Stiles, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114 (United States)
Yoonji Baek, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114 (United States).
Shinsuke Nomura, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114 (United States).
Kai Bao, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114 (United States).
Shuang Hu, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114 (United States).
G. Kate Park, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114 (United States).
Min Joo Jo, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114 (United States).
I Hoseok, Department of Thoracic and Cardiovascular Surgery, Pusan National University School of Medicine and Biomedical Research Institute, Pusan National University Hospital, Busan (Republic of Korea).
Jean-Luc Coll, Cancer Targets & Experimental Therapeutics, Institute for Advanced Biosciences, University of Grenoble Alpes, INSERM-U1209, CNRS-UMR 5309- Grenoble (France).
Brian P. Rubin, Departments of Pathology and Cancer Biology, Robert J. Tomsich Pathology and Laboratory Medicine Institute, and Lerner Research Institute and Taussig Cancer Center, Cleveland Clinic, Cleveland, OH 44195 (United States)
Hak Soo Choi, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114 (United States).
REFERENCES
- [1].Joensuu H, Hohenberger P, Corless CL, The Lancet 2013, 382, 973. [DOI] [PubMed] [Google Scholar]
- [2].Chandramouly BS, Wu DW, Clinical Nuclear Medicine 1999, 24, 204. [DOI] [PubMed] [Google Scholar]
- [3].Hirshberg B, Libutti SK, Alexander HR, Bartlett DL, Cochran C, Livi A, Chang R, Shawker T, Skarulis MC, Gorden P, Journal of the American College of Surgeons 2002, 194, 761. [DOI] [PubMed] [Google Scholar]
- [4].Akahoshi K, Oya M, Koga T, Shiratsuchi Y, World J Gastroenterol 2018, 24, 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Espinosa I, Lee CH, Kim MK, Rouse BT, Subramanian S, Montgomery K, Varma S, Corless CL, Heinrich MC, Smith KS, Wang Z, Rubin B, Nielsen TO, Seitz RS, Ross DT, West RB, Cleary ML, van de Rijn M, Am J Surg Pathol 2008, 32, 210. [DOI] [PubMed] [Google Scholar]
- [6].a) Zhang Q, Xu J, Qian Y, Chen L, Li Q, Xu K, Chen M, Sun L, He Z, Yang L, Zhang D, Wang L, Sun X, Wang Y, Xu H, Xu Z, Mol Cancer Ther 2018, 17, 2780; [DOI] [PubMed] [Google Scholar]; b) Joensuu H, Trent JC, Reichardt P, Cancer Treat Rev 2011, 37, 75. [DOI] [PubMed] [Google Scholar]
- [7].a) Kang H, Hu S, Cho MH, Hong SH, Choi Y, Choi HS, Nano Today 2018, 23, 59; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WC, Nature Reviews Materials 2016, 1, 16014. [Google Scholar]
- [8].a) Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X, Monette S, Pauliah M, Gonen M, Zanzonico P, Quinn T, Wiesner U, Bradbury MS, Overholtzer M, Nature nanotechnology 2016, 11, 977; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen F, Ma K, Madajewski B, Zhuang L, Zhang L, Rickert K, Marelli M, Yoo B, Turker MZ, Overholtzer M, Nat. Commun 2018, 9, 4141; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, Humm J, Gonen M, Kalaigian H, Schoder H, Strauss HW, Larson SM, Wiesner U, Bradbury MS, Sci. Transl. Med 2014, 6, 260ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Peng C, Xu J, Yu M, Ning X, Huang Y, Du B, Hernandez E, Kapur P, Hsieh JT, Zheng J, Angew. Chem. Int. Ed 2019, 58, 8479; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Peng C, Yu M, Hsieh JT, Kapur P, Zheng J, Angewandte Chemie 2019, 58, 12076; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Du B, Jiang X, Das A, Zhou Q, Yu M, Jin R, Zheng J, Nature nanotechnology 2017, 12, 1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kang H, Gravier J, Bao K, Wada H, Lee JH, Baek Y, El Fakhri G, Gioux S, Rubin BP, Coll JL, Choi HS, Advanced Materials 2016, 28, 8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yu M, Zheng J, ACS Nano 2015, 9, 6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Choi HS, Ipe BI, Misra P, Lee JH, Bawendi MG, Frangioni JV, Nano Lett. 2009, 9, 2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kang H, Mintri S, Menon AV, Lee HY, Choi HS, Kim J, Nanoscale 2015, 7, 18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV, Nature Biotechnology 2007, 25, 1165; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Choi HS, Frangioni JV, Molecular Imaging 2010, 9, 291; [PMC free article] [PubMed] [Google Scholar]; c) Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG, Frangioni JV, Nature Nanotechnology 2010, 5, 42; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kang H, Han M, Xue J, Baek Y, Chang J, Hu S, Nam H, Jo MJ, Fakhri GE, Hutchens MP, Choi HS, Kim J, Nat. Commun 2019, 10, 5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Wang Q, Liu F, Qi S, Qi Z, Yan XE, Wang B, Wang A, Wang W, Chen C, Liu X, Jiang Z, Hu Z, Wang L, Wang W, Ren T, Zhang S, Yun CH, Liu Q, Liu J, Eur J Med Chem 2018, 150, 366; [DOI] [PubMed] [Google Scholar]; b) Fischer JJ, Dalhoff C, Schrey AK, Graebner OY, Michaelis S, Andrich K, Glinski M, Kroll F, Sefkow M, Dreger M, Koester H, J Proteomics 2011, 75, 160. [DOI] [PubMed] [Google Scholar]
- [16].Choi HS, Gibbs SL, Lee JH, Kim SH, Ashitate Y, Liu F, Hyun H, Park G, Xie Y, Bae S, Henary M, Frangioni JV, Nat. Biotechnol 2013, 31, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Beni S, Szakacs Z, Csernak O, Barcza L, Noszal B, Eur. J. Pharm. Sci 2007, 30, 167. [DOI] [PubMed] [Google Scholar]
- [18].a) Subramanian S, West RB, Corless CL, Ou W, Rubin BP, Chu K-M, Leung SY, Yuen ST, Zhu S, Hernandez-Boussard T, Oncogene 2004, 23, 7780; [DOI] [PubMed] [Google Scholar]; b) Pelczar P, Zibat A, van Dop WA, Heijmans J, Bleckmann A, Gruber W, Nitzki F, Uhmann A, Guijarro MV, Hernando E, Gastroenterology 2013, 144, 134; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, Zhu S, Ball CA, Nielsen TO, Patel R, The American journal of pathology 2004, 165, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Demetri GD, Von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, New England Journal of Medicine 2002, 347, 472; [DOI] [PubMed] [Google Scholar]; b) Demetri GD, Benjamin RS, Blanke CD, Blay J-Y, Casali P, Choi H, Corless CL, Debiec-Rychter M, DeMatteo RP, Ettinger DS, Journal of the National Comprehensive Cancer Network: JNCCN 2007, 5, S1. [PubMed] [Google Scholar]
- [20].a) Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R, Nature Nanotechnology 2007, 2, 751; [DOI] [PubMed] [Google Scholar]; b) Li Y, Lin T.-y., Luo Y, Liu Q, Xiao W, Guo W, Lac D, Zhang H, Feng C, Wachsmann-Hogiu S, Walton JH, Cherry SR, Rowland DJ, Kukis D, Pan C, Lam KS, Nat. Commun 2014, 5, 4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Taguchi T, Sonobe H, Toyonaga S.-i, Yamasaki I, Shuin T, Takano A, Araki K, Akimaru K, Yuri K, Laboratory investigation 2002, 82, 663. [DOI] [PubMed] [Google Scholar]
- [22].Wang Y, Ning ZH, Tai HW, Long S, Qin WC, Su LM, Zhao YH, Regul. Toxicol. Pharmacol 2015, 71, 205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.