

Abstract
Aggregates of Aβ peptide and the microtubule-associated protein tau are key molecular hallmarks of Alzheimer’s disease (AD). However, the interaction between these two pathologies and the mechanisms underlying disease progression have remained unclear. Numerous failed clinical trials suggest the necessity for greater mechanistic understanding in order to refine strategies for therapeutic discovery and development. To this end, we have generated a transgenic Caenorhabditis elegans model expressing both human Aβ1-42 peptide and human tau protein pan-neuronally. We observed exacerbated behavioral dysfunction and age-dependent neurodegenerative changes in the Aβ;tau transgenic animals. Further, these changes occurred in the Aβ;tau transgenic animals at greater levels than worms harboring either the Aβ1-42 or tau transgene alone and interestingly without changes to the levels of tau expression, phosphorylation or aggregation. Functional changes were partially rescued with the introduction of a genetic suppressor of tau pathology. Taken together, the data herein support a synergistic role for both Aβ and tau in driving neuronal dysfunction seen in AD. Additionally, we believe that the utilization of the genetically tractable C. elegans model will provide a key resource for dissecting mechanisms driving AD molecular pathology.
Introduction
Alzheimer’s disease (AD) is the most common form of dementia afflicting a growing elderly population. With the population of affected individuals expected to rise to 131 million by 2050 placing an enormous burden on caretakers and healthcare systems, there is an urgent need for effective disease modifying treatments (1). While rare genetic mutations in the gene encoding amyloid precursor protein (APP) and the presenilin genes PSEN1 and PSEN2 have been found to cause early onset familial AD, these represent only a small fraction of AD cases whereas the majority of cases are late onset occurring sporadically (1). The lack of causal genetic variants and poor understanding of the biology of late onset disease have posed challenges to developing a mechanistic understanding of AD pathogenesis, progression and drug discovery (2, 3). Despite many compounds showing promise in preclinical animal models, a therapy to modify disease onset or progression has yet to succeed in human clinical trials. This points to a need for a more thorough mechanistic understanding of disease onset and progression.
Pathologically, AD is characterized by the accumulation of extracellular Aβ plaques composed of aggregated Aβ peptides originating from processed APP as well as intraneuronal neurofibrillary tangles (NFTs) composed of the microtubule-associated protein tau (4). The relationship between these two protein pathologies and mechanistic disease progression is not yet well understood. However, it has long been hypothesized that the deposition of these protein aggregates acts synergistically to drive the pathological progression of AD and neuron death. The amyloid cascade hypothesis has been favored by many in the field for well over a decade and provides a hypothetical explanation linking both Aβ and tau pathologies. This hypothesis describes increased abundance of toxic Aβ peptide, potentially oligomeric Aβ, triggering a series of downstream events ultimately leading to tau hyperphosphorylation and the formation of NFTs (5–7). The direct evidence supporting this pathological relationship remains circumstantial and the interaction between these two pathologies remains unclear.
Much effort has been given to the development of therapies targeting Aβ under the assumption that Aβ pathology develops prior to and triggers the formation of tau pathology downstream. Numerous clinical trials have tested such proposed treatments, yielding no treatment that successfully slows or halts progression of AD. Some candidate therapies have successfully decreased the burden of Aβ, though they fail to impact cognitive impairment (8–10). The repeated failures to find effective disease modifying therapies have elucidated the need for a greater understanding of the mechanistic pathology and progression (11, 12) and perhaps support the need for simultaneous targeting of amyloid beta and tau molecular pathologies.
While Aβ has received much of the field’s attention for the past two decades, mounting evidence supports the idea that the microtubule-associated protein tau encoded by the MAPT gene plays a critical role in the AD pathogenesis. Mutations in the MAPT gene cause inherited frontotemporal lobar degeneration in which neuropathology includes neuronal and glial lesions positive for tau protein in the absence of Aβ pathology accompanied by obvious brain atrophy and loss of neurons (13–15). Additionally, traumatic brain injury, aging and neuronal stress can lead to the deposition of pathological tau, neurodegeneration and dementia, further suggesting that tau may play a key role in disease progression outside of an Aβ-induced pathway (16–18). We hypothesize that both pathologies are important in the development and progression of AD and therefore seek to further understand their interaction in the context of driving pathological protein accumulation, neuronal dysfunction and neurodegeneration.
The nematode, Caenorhabditis elegans, provides a number of experimental advantages including a developed and fully mapped nervous system, tractable genetics and short lifespan (19). These features allow for relatively rapid and broad genetic investigation of age-related neurodegeneration. Transgenic C. elegans strains expressing toxic Aβ1-42 or tau have been developed independently. Each strain on its own models respective molecular and cellular aspects of Aβ or tau protein accumulation seen in AD. Strains expressing Aβ1-42 accumulate oligomer forming peptides and exhibit impaired chemotaxis ability and increased sensitivity to serotonin resulting in paralysis indicative of a compromised nervous system (20). In previous work, we generated a variety of transgenic strains expressing wildtype and mutant human tau isoforms with varying levels of tau protein expression driving a range of severity in the tau-related phenotypes including accumulation of pathological tau species, behavioral dysfunction, neurodegeneration and shortened life span (21–23).
In order to investigate possible synergism between Aβ and tau pathologies in AD, we have generated a bigenic C. elegans model that pan-neuronally expresses both the toxic Aβ1-42 peptide and the wildtype 4R1N isoform of human tau from independent transgenes. Here we have utilized this model to examine the effects of pathological protein expression on pathological protein accumulation, behavior and neuronal integrity. The evidence presented suggests that Aβ and tau may interact synergistically leading to neuronal dysfunction.
Results
Co-expression of Aβ1-42 peptide and tau in neurons causes behavioral abnormalities
To explore the consequences of combined amyloid and tau expression in C. elegans neurons, we crossed pre-existing amyloid and tau transgenic strains to generate a bigenic amyloid/tau transgenic line. We examined the neuronal function in this line by assessing the swimming behavior exhibited by worms placed in liquid. Staged day one adult worms were placed in M9 buffer to instigate swimming behavior over the duration of 1 min captured by video. Videos were then analyzed using automated video analysis algorithms within WormTracker software, to count the number of mid-point body bends per minute. Use of this software allowed for user-independent analysis of worm behavior. Our analysis revealed that worms expressing only Aβ1-42 (Aβ1-42-Tg) exhibited similar swimming ability compared to non-transgenic (non-Tg, N2) worms as measured by the number of body bends per minute when maintained at 15°C, but worms subjected to temperature upshift for 24 h at either 20 or 25°C to increase levels of Aβ1-42 peptide expression exhibited observable, though not statistically significant, mild motor deficits seen in decreased body bends as compared to non-Tg worms (Fig. 1A and B). Tau expression alone (tau-Tg) consistently exhibited significant motor deficits across all three temperatures as compared to non-Tg worms at the same temperatures. We consistently observed the most drastic motor deficits in worms expressing both Aβ1-42 and tau (Aβ1-42;tau-Tg). These worms exhibited mean body bend per minute counts which were significantly different from non-Tg, Aβ1-42-Tg and tau-Tg worms at each temperature.
Figure 1.
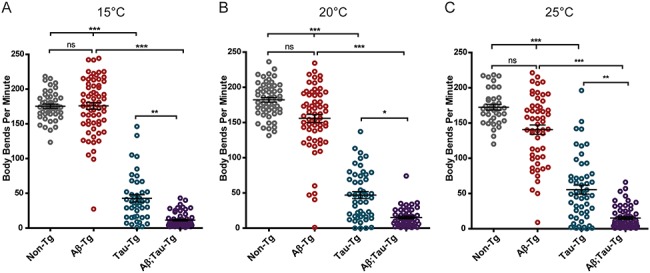
Aβ1-42 and tau neuronal co-expression exacerbates motility defects in C. elegans model as compared to tau expression alone. Staged L4 worms were either kept at 15°C (A) or temperature upshifted for 24 h at 20°C (B) or 25°C (C) to upregulate Aβ1-42 and then subjected to a swimming assay video captured for 1 min swimming duration. Videos were analyzed by automated WormTracker software to count the number of body bends per minute. Aβ1-42-Tg worms showed a similar body-bend mean (175.9 ± 5.0 SEM) to the mean of non-Tg worms (175.4 ± 3.0 SEM) at 15°C, but trended toward reduced body-bend means at 20°C (Aβ1-42-Tg: 155.8 ± 5.8 SEM; non-Tg: 182.2 ± 3.2 SEM) and 25°C (Aβ1-42-Tg: 140.6 ± 6.6 SEM; non-Tg 172.5 ± 4.0 SEM) as compared to non-Tg worms. tau-Tg worms had consistently lower body-bend means at all three temperatures (42.7 ± 5.3 SEM, 46.9 ± 4.9 SEM and 55.46 ± 6.4 SEM at 15, 20 and 25°C, respectively) compared to non-Tg controls. Aβ1-42;tau-Tg demonstrated the lowest body-bend means at all three temperatures (11.5 ± 1.3 SEM, 15.2 ± 1.9 SEM, and 15.2 ± 2.0 SEM at 15, 20 and 25°C, respectively). (N = 2, n ≥ 38, Kruskal–Wallis ANOVA, post hoc Dunn’s comparison. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001).
The presence of Aβ1-42 peptide does not alter tau expression or tau phosphorylation
In order to gain a greater understanding of the underlying mechanism of the observed behavioral and neuroanatomical phenotypes, we examined tau protein expression with and without Aβ1-42 transgene expression. Whole worm lysates from staged day one adult worms were subjected to SDS-PAGE and western blots probed with a pan-tau antibody to assess total tau expression. We did not observe a significant change to the levels of expression of tau in the presence of Aβ1-42 (Fig. 2A), confirmed by quantitation of western blots (Fig. 2B).
Figure 2.
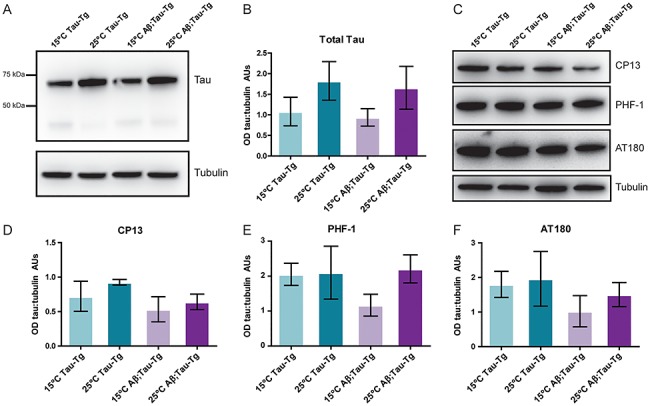
Expression of Aβ1-42 does not alter levels of tau expression or tau phosphorylation in the C. elegans model. Worms grown to L4 stage were either maintained at 15°C or upshifted to 25°C for 24 h until day one adult stage when populations were harvested. Pellets were approximately 50–100 mg of adult worms (~50 000 individual animals). To assess changes to overall tau expression, blots were probed with a pan-tau antibody (A, representative blot) and quantitated in (B). Aβ1-42;tau-Tg worms were compared to tau-Tg worms; however, no statistically significant change in the level of tau expression was observed. To assess tau phosphorylation, blots were probed with CP-13(ptau Ser202), PHF-1 (ptau Ser396/Ser404) and AT180 (ptau Thr 231) (C, representative blots) and quantitated in (D–F). Aβ1-42;tau-Tg worms were compared to tau-Tg worms; however, no statistically significant change in the level of tau expression was observed. Student’s t-test. Error bars represent SEM. N = 3 populations.
Tau contains numerous phosphorylation sites and becomes hyperphosphorylated in AD (4). Additionally, human tau expressed in worms is phosphorylated by endogenous kinases (23). We further hypothesized that while the presence Aβ1-42 peptide may not alter the levels of total tau expression in worm neurons, it may affect tau phosphorylation. Again, we used whole worm lysates of staged day one adult worms which we subjected to SDS-PAGE and western blots that were then probed with phospho-tau antibodies PHF-1, CP-13 and AT180 which detect phosphorylation at specific phospho-sites (p-Ser396/Ser404, p-Ser202 and p-Thr 231 respectively) of tau commonly associated with tauopathy (Table 1). Though levels of phosphorylation were somewhat variable, we observed no clear trend in phosphorylation at any of the probed phosphorylation sites, suggesting that expression of Aβ1-42 peptide in worm neurons does not significantly alter levels of tau phosphorylation (Fig. 2C–F).
Table 1.
Antibodies used
| Antigen | Clone/product name | Dilution | Host species | Source | Catalog # |
|---|---|---|---|---|---|
| β-Tubulin | E7 mAb | 1:5000 | Mouse | Developmental Studies Hybridoma Bank (Iowa City, IA, USA) |
N/A |
| Pan-tau | SP70 pAb | 1:1000 | Rabbit | Rockland Immunochemicals Inc. (Limerick, PA, USA) |
200-C01-B33 |
| ptau Ser202 | CP13 mAb | 1:1000 | Mouse | Peter Davies (Litwin-Zucker Research Center for the Study of Alzheimer’s Disease, The Feinstein Institute of Medical Research, Northwell Health, Manhasset, NY, USA) |
N/A |
| ptau Ser396/Ser404 | PHF-1 mAb | 1:2000 | Mouse | ||
| ptau Thr 231 | AT180 | 1:1000 | Mouse | ThermoFisher, Waltham, MA | MN040 |
| tau (total) | K9JA (DAKO) pAb | 1:1000 | Rabbit | Agilent Technologies, Inc. (Santa Clara, CA, USA) | A002401-2 |
| Aβ | H31L21 | Rabbit | ThermoFisher, Waltham, MA | 700 254 | |
| Aβ(1-16) | 6E10 | 1:1000 | Mouse | Biolegend, San Diego, CA, USA | 803 001 |
| Aβ(17-24) | 4G8 | 1:2000 | Mouse | Biolegend, San Diego, CA, USA | 800 701 |
| 20 Ab Mouse | Horseradish Peroxidase α-Ms IgG (H + L) | 1:5000 | Goat | Jackson Immunoresearch (West Grove, PA, USA) | 115-035-146 |
| 20 Ab Rabbit | Horseradish Peroxidase α-Rb IgG (H + L) | 1:5000 | Mouse | Jackson Immunoresearch (West Grove, PA, USA) | 211-032-171 |
| 20 Ab Mouse | F(ab’) Ms IGG (H + L), Alexa647 | 1:2000 | Goat | ThermoFisher, Waltham, MA | A-21237 |
| 20 Ab Rabbit | F(ab’) Rb IGG (H + L), Alexa594 | 1:2000 | Goat | ThermoFisher, Waltham, MA | A-11072 |
The presence of Aβ1-42 peptide does not alter tau aggregation, but tau does increase Aβ1-42 accumulation
The primary component of NFTs observed in AD is aggregated tau protein (4). While C. elegans models of tauopathy do not exhibit mature NFTs composed of paired helical filaments, they can exhibit amorphous pre-tangle aggregated forms of tau. We hypothesized that expression of tau and Aβ1-42 peptide together may increase tau aggregation or amyloid accumulation. To test this, we subjected staged day one adult worm pellets to sequential tau fractionation enabling the separation of soluble tau from less soluble aggregate forms. We observed no significant changes in levels of tau in any of the fractions, RAB-soluble, RIPA-soluble or FA fractions as compared to tau-only worms, suggesting that tau aggregation remains unaltered by the presence of Aβ1-42 peptide in this worm model (Fig. 3A and B). However, using a Luminex-based immunodetection assay, we do observe a significant increase in total detectable amyloid when co-expressed with tau (Aβ1-42;tau-Tg) (Fig. 3C).
Figure 3.
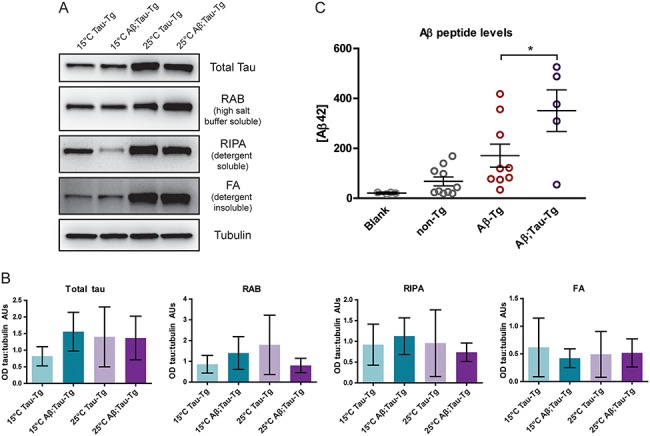
Expression of Aβ1-42 does not alter levels of tau aggregation in the C. elegans model. Worms grown to L4 stage were either maintained at 15°C or upshifted to 25°C for 24 h until day one adult stage when they were harvested. Pellets (~100 mg each) were subjected to sequential fractionation using buffers of iteratively increasing solubilizing strength to separate soluble tau from less soluble forms. Total tau (both soluble and insoluble tau), soluble tau (RAB fraction), detergent soluble (RIPA fraction) and detergent insoluble, formic acid soluble tau (FA fraction). (A) Blots of fractions were probed with a pan-tau antibody and quantitated in (B). Aβ1-42;tau-Tg worms were compared to tau-Tg worms; however, no statistically significant change in the level of tau expression was observed. Quantitation of western blots comparing fractions of sequentially extracted soluble and insoluble tau or tubulin control. (N = 3 populations, Kruskal–Wallis ANOVA, Dunn’s post hoc comparison. Error bars represent SEM.) (C) Pellets were subjected to amyloid peptide extraction and measured by Luminex assay to quantitate Aβ1-42 peptide levels. Aβ1-42-Tg worms were compared to Aβ1-42;tau-Tg worms. A significant increase in Aβ1-42 levels was induced by tau expression (N = 2, n ≥ 5, ANOVA, post hoc Tukey’s comparison, P < 0.05 Error bars represent SEM).
Aβ1-42 peptide and tau co-expression leads to aberrant neuronal morphology
Eight chemosensory neurons, six amphid neurons in the head and two phasmid neurons in the tail, are exposed to the environment via cilia which enable the uptake of lipophilic dyes, such as DiO, into the cell bodies of these neurons upon environmental exposure. Mutations in genes necessary for the development or maintenance of these neurons lead to altered dye-filling phenotypes; therefore, dye-filling of amphid and phasmid neurons, or lack thereof, can be used to score alterations in neuron anatomy and morphology (24–26). Here we exposed staged day one adult worms that had been subjected to 24-h incubation at 25°C (from L4 to day one adult stage) to DiO dye and used fluorescent microscopy to score for the number of dye-filled amphid neurons. Both non-Tg and tau-Tg worms exhibit dye-filling of essentially all amphid neurons, suggesting that moderate levels of the wild type tau expression do not alter the anatomy of chemosensory amphid neurons (Fig. 4A). Worms expressing Aβ1-42 alone (Aβ1-42-Tg) showed a modestly reduced dye-filling phenotype, whereas worms neuronally co-expressing Aβ1-42 and tau (Aβ1-42;tau-Tg) showed a drastic reduction in the number of dye-filled neurons suggesting that co-expression of both transgenes alters the anatomy of amphid neurons or their processes preventing normal dye-filling (Fig. 4A. Representative images shown in Fig. 4D). To assess the effects of aging on neuronal morphology, we also assessed the dye-filling phenotype at day 2 and day 4 of adulthood (Fig. 4A–C). By the second day of adulthood, Aβ1-42-Tg and Aβ1-42;tau-Tg worms showed fewer dye-filled neurons as compared to non-Tg or tau-Tg worms, which were not significantly different. Whereas Aβ1-42-Tg exhibited decreasing numbers of dye-filled neurons with age, we do see a floor effect at day 4 where both Aβ1-42-Tg and Aβ1-42;tau-Tg animals exhibit similarly few dye-filled amphids (with means less than 1, Fig. 4C). Even at day 4 of adulthood, there was no significant change in the number of dye-filled neurons in tau-Tg worms when compared with non-Tg.
Figure 4.
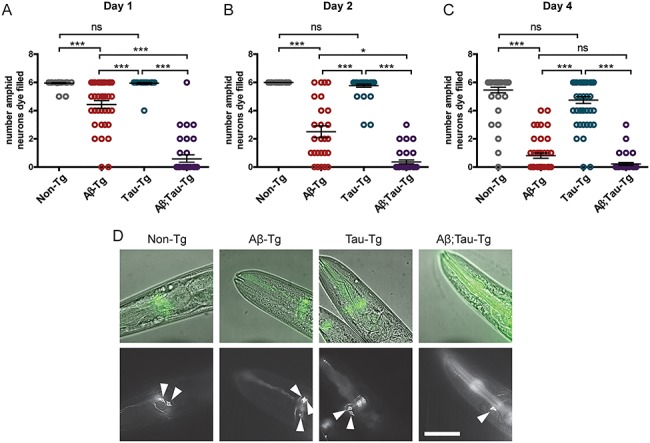
Aβ1-42 and tau co-expression drastically alters dye-filling phenotype of chemosensory amphid neurons in C. elegans model. Worms were exposed to fluorescent lipophilic dye which is taken up by six environmentally exposed amphid neurons of normal morphology. Staged L4 worms were temperature upshifted at 25°C for 24 h until day-one adult stage. After exposure to dye, worms were scored for the number of fluorescent amphid neurons. (A) Dye-filling phenotype of day 1 adult worms. Non-Tg worms and tau-Tg worms demonstrated wildtype dye-filling phenotypes with respective means of 5.95 ± 0.03 SEM and 5.94 ± 0.06 SEM dye-filled neurons. Aβ1-42-Tg worms averaged 4.43 ± 0.27 SEM dye-filled amphid neurons, whereas Aβ1-42;tau-Tg worms averaged 0.77 ± 0.26 SEM dye-filled neurons. (B) Dye-filling phenotype of day 2 adult worms. Tau-Tg worms maintained wildtype dye-filling phenotype averaging 5.77 ± 0.11 SEM dye-filled neurons as compared to non-Tg worms that averaged 6.00 ± 0.00 SEM filled neurons. Aβ1-42-Tg and Aβ1-42;tau-Tg worms demonstrated significantly fewer filled neurons with means of 2.50 ± 0.42 SEM and 0.36 ± 0.14 SEM neurons, respectively. (C) Dye-filling phenotype of day 4 adult worms. Aβ1-42-Tg and Aβ1-42;tau-Tg have fewer dye-filled neurons as compared to wildtype, exhibiting means of 0.81 ± 0.19 SEM and 0.22 ± 0.10 SEM, respectively, as compared to non-Tg worms with a mean of 5.46 ± 0.20 SEM filled neurons, whereas tau-Tg worms maintain no significant difference as compared to non-Tg worms (4.75 ± 0.24 mean dye-filled neurons). (N = 3, n ≥ 26, Kruskal–Wallis ANOVA, post hoc Dunn’s comparison. Error bars represent SEM. *P < 0.05, ***P < 0.001). (D) Representative images of selected focal planes of day 1 adult worms. Aβ1-42;tau-Tg worm images shown with two neurons. While the mean was approximately one, given that few worms observed with only one neuron, zero and two or more neurons were generally observed. Scale bar 50 μm.
Suppression of tau by sut-2, a strong suppressor of tau pathology, ameliorates motor deficits in Aβ1-42;tau transgenic animals
Loss-of-function mutations in C. elegans sut-2, a homolog to mammalian SUT-2 (MSUT2), confer strong suppression of tau toxicity and motor deficits in worm tauopathy models (27, 28). To explore the contribution of Aβ1-42 and tau to the cumulative toxicity, we crossed the strain CK181, containing a sut-2 loss of function (lof) mutation into the Aβ1-42;tau-Tg line obtaining a strain homozygous for both Aβ1-42 and tau transgenes and the sut-2 mutation. Due to low brood size as a result of the sut-2 mutation, these worms and the control strains were maintained at 20°C to generate large enough populations to subject to the automated swimming assay described earlier. Swimming videos of staged day-one adult worms were collected and analyzed by the WormTracker software and again body bends were counted over minute duration after acclimation. tau-Tg;sut-2 lof worms demonstrated greater swimming ability as seen by the increased number of body bends as compared to tau-Tg worms without the sut-2 mutation. The data suggests that sut-2 loss of function suppressed tau motility deficits seen in the tau-Tg worms (Fig. 5A). Importantly, reduction in motility defects as seen by the increased number of body bends in Aβ1-42;tau-Tg;sut-2 lof worms as compared Aβ1-42;tau-Tg without the sut-2 mutation indicates that sut-2 loss of function provided a partial rescue of tau-induced motility defects in Aβ1-42;tau-Tg worms as well (Fig. 5B).
Figure 5.
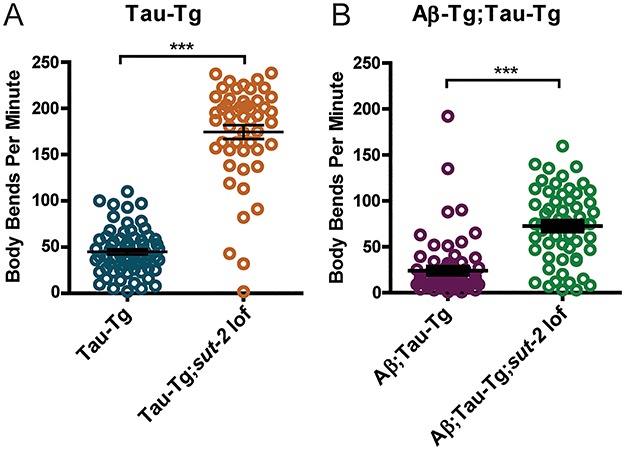
Loss of function mutant of sut-2 suppresses induced motility defects in both tau-Tg and Aβ1-42;tau-Tg worms. After 24-h temperature upshift at 25°C, day-one adult worms were subjected to a swimming assay to assess motility. One-minute videos were captured and subsequently analyzed by automated WormTracker software counting the number of body bends per minute. (A) Suppression of tau phenotype by a sut-2 loss of function mutation was observed in tau-Tg background, where tau-Tg worms showed a mean of 44.9 ± 3.0 SEM body bends per minute versus tau-Tg;sut-2 lof worms that showed a mean of 174.5 ± 7.4 SEM body bends per minute. (B) Notably, Aβ1-42;tau-Tg worms showed a mean of 24.09 ± 3.5 SEM while Aβ1-42;tau-Tg;sut-2 lof showed a partial rescue of tau-induced motility defects with a mean of 72.6 ± 5.0 SEM body bends per minute. For reference, non-transgenic worms typically have a mean of approximately 150-200 body bends per minute. (N = 2, n ≥ 50, unpaired t-test. Error bars represent SEM. ***P < 0.0001).
Discussion
The presence of both Aβ plaques and NFTs composed of aggregated tau protein distinguishes AD from most other single proteinopathy-driven dementia disorders (4). Early onset familial AD is caused by mutations in APP, Presenilin and genes whose protein products are responsible for amyloid processing leading to an over-abundance of Aβ peptide in the brain (1). While amyloid deposition has been hypothesized to trigger NFT formation and neuron death, reduction of Aβ load has not been shown to ameliorate cognitive deficits suggesting that the relationship between these two proteins and the progression of AD pathology remains more complex than the unidirectional linear relationship proposed by the amyloid cascade hypothesis. As there have been no causative mutations for late onset sporadic disease identified to date, the mechanistic underpinnings of age-dependent disease onset and progression have remained elusive.
We sought to develop a more robust model of AD in C. elegans by expressing both Aβ and tau peptides in a developed yet simple nervous system which would allow investigation into the interaction between the two molecular pathologies of AD that underlies neuronal dysfunction. To this end, we created a double transgenic worm line that pan-neuronally expresses Aβ1-42 paired with pan-neuronal expression of wildtype human 4R1N tau protein. Independently, each transgene results in relatively modest behavioral phenotypes, whereas their co-expression results in significantly greater deficits, suggesting that the combination of both Aβ1-42 and tau together is important for the onset of neuronal dysfunction and degeneration.
To address synergistic amyloid/tau toxicity, we examined neuronal structural integrity using a simple dye-filling assay. C. elegans has eight chemosensory neurons exposed to the environment that take up lipophilic dye upon environmental exposure (29). Using this approach, one can rapidly assess neuronal anatomy without the use of transgenic reporters which can compromise neuron function. We found that both the tau transgenic parental strains exhibited no significant dye filling or structural defects while the amyloid strain exhibited very modest dye-filling defects at day 1 but progressed to moderate loss of dye filling amphids on day 2 and severe loss of amphids dye filling by day 4. In contrast, the bigenic amyloid/tau strain showed consistent and significant severe defects in both dye-filling and neuronal structural abnormalities by day 1 and did not progress beyond that indicative of a neurodegenerative floor effect. Taken together, these data indicate synergism between the two molecular pathologies of AD driving neurodegenerative changes in a simple transgenic model.
Several bigenic mouse models have been generated to understand AD progression and the relationship of Aβ and tau pathologies. These models vary greatly in their recapitulation of disease features including impaired cognition, neuronal dysfunction, Aβ deposition and plaques, presence of pathological tau, and neurodegeneration. Popular mouse models such as 3x-Tg, APP/PS1/rTg21221, TauPS2APP and Tg2576/tauP301L/APPswe have utilized overexpression and/or familial disease-causing mutations to drive both Aβ accumulation and plaque formation, pathological tau deposition and tangle formation in mouse brains (30–34). However, the vast majority of AD cases possess no causal mutations, and no mutations in MAPT have been associated with AD. In order to better replicate the protein interactions in most disease cases, the C. elegans model reported here utilizes the wild type human Aβ peptide and wildtype human tau coding sequences but did not yield an observable increase to aggregated or phosphorylated tau. In contrast, a separate, recently reported C. elegans model of AD demonstrated accumulation of Aβ and tau aggregates by expressing the same Aβ transgene seen here in combination with the aggregate-prone ΔK280 mutant tau (35). Cognitive deficits as indicated through behavioral assays appear in several mouse models (i.e. 3x-Tg, APPNL-G-F/MAPT, PLB-triple and TauPS2APP) prior to or without plaque or tangle formation and are seldom accompanied by neuron loss (30, 31, 33, 36, 37). Similarly, locomotor deficits indicating neuronal dysfunction in our C. elegans model are present without plaques or tangles. The model described in Wang et al. demonstrated several phenotypes indicating neuronal dysfunction driving impaired egg laying, deficits in chemotaxis, associative learning and normal slowing of locomotion in response to finding a food source (35). Dystrophic neurites have been observed in bigenic mouse models with and without tau tangle pathology (32, 33, 36). The dye-filling assays completed on the Aβ1-42;tau-Tg AD worm model presented in this work indicated aberrant neuronal anatomy of sensory neurons suggestive of neurodegeneration. Likewise, the worm model described in Wang et al. demonstrated age-related degeneration of mechanosensory neurons (35). Both transgenic C. elegans models recapitulate behavioral abnormalities indicative of neuronal dysfunction and neuronal loss, characteristic features of human AD. However, the model we present here expresses wildtype human tau, providing greater similarity to sporadic human AD where only wild type tau and Aβ are present.
We have previously shown and replicated here that sut-2 loss of function mutations fully suppress tau toxicity in worms harboring the tau transgene alone. However, loss of sut-2 only partially rescues the motility phenotype in the amyloid/tau bigenic animals. The Aβ1-42;tau-doubleTg demonstrates profound behavioral deficits without altering tau expression, phosphorylation or aggregation. However, suppression of tauopathy phenotypes using sut-2 loss of function does not fully rescue behavioral deficits in the presence of amyloid. This suggests that tau/amyloid synergism drives toxicity through mechanisms independent of either tau or amyloid alone. This is particularly surprising in light of the fact that the tau transgene alone drives much more severe behavioral dysfunction than the amyloid transgene alone. Combined with the neuronal morphology data shown in Figure 4, the sut-2 suppression findings support a novel mechanism of amyloid/tau synergistic toxicity.
The bigenic C. elegans model presented here, as well as that described by Wang et al., clearly show that co-expression of Aβ and tau lead to distinct phenotypes when expressed independently or in combination(35). Similarly, the models discussed above each recapitulate unique combinations of AD features. Taken together, we suggest that this model captures not only the additive individual contributions of pathological tau and pathological Aβ but the combined effects unique to the Aβ/tau pathological synergistic toxicity. By this logic, therapies targeting solely Aβ or tau are bound to fail and therapeutic success requires simultaneous targeting of both pathological tau and Aβ together.
Given the combination of both Aβ and tau expression and cumulative pathology, this model provides a unique platform with which to study the mechanistic underpinnings and pathological interactions driving disease progression. Combined with the tractability of C. elegans genetics, this model offers a powerful tool enabling the search for genetic modifiers performing screens commonly utilized with nematodes. Identification of genetic modifiers of AD pathology will ultimately lead to a greater understanding of the mechanistic underpinnings of disease progression and therapeutic discovery.
Materials and Methods
C. elegans strains and maintenance
N2 Bristol (wild type)
CK1441 (wild type tau low) bkls1441[Paex-3::h4R1Ntau; Pmyo-2::dsRED]
CK144 (wild type tau high) bkIs144[Paex-3::h4R1Ntau Pmyo-2::GFP]
CK181 sut-2(bk741)
CL2355 smg-1ts dvIs50 [pCL45 (Psnb-1:: secretory signal peptide::Aβ1-42::3′ UTR(long) + Pmtl-2::GFP].
Worms were maintained at 15°C (with the exception of sut-2 strains and their controls which were maintained at 20°C due to sut-2-induced temperature sensitivity at 15°C) on NGM plates seeded with OP50 E. coli according to standards described in Brenner et al. 1974 (38). For protein extraction, worms were grown on plates containing media with five times peptone (5xPEP), nutrient rich media for robust C. elegans growth, seeded with OP50 (38).
Temperature upshift for induction of Aβ1-42
Worms were maintained at 15°C until the L4 stage when they were transferred to 20 or 25°C for appropriate induction of human amyloid beta expression (smg-1ts(snb-1/Aβ1-42) for 1, 2 or 4 days prior to experimentation. This allowed worms to be maintained under conditions of lower Aβ1-42 expression during development, but upregulate Aβ1-42 production, increasing Aβ1-42 accumulation, upon reaching the L4 stage where the nervous system is fully developed, to maintain a staged adult population for the performance of experiments. We utilized this method in order to more accurately simulate the temporal changes occurring with AD, onset of which is thought to happen in adulthood in a developed nervous system.
C. elegans motility assays and analysis
L4 stage worms were picked from mixed population plates and transferred to clean NGM growth plates for incubation at 15, 20 or 25°C for 24 h. Subsequently, worms were moved to the assay room and allowed to acclimate for 20 min. One strain at a time, worms were washed from NGM plates to food-free video plates with 2 ml of M9, allowed to acclimate to M9 buffer for 15 s prior to a 1-min video recording. Videos were collected using the WormLab platform (MBF Bioscience, VT). After videos were taken, worm movement behavior was analyzed using the WormTracker software (MBF BioScience, version 2019.1.2). Body bends from the mid-point body location of each worm tracked were counted. The total number of body bends was divided by the track length(s) to give the frequency of body bends per second. This was compared across strains within each temperature using the Kruskal–Wallis ANOVA with post hoc Dunn’s test (*P < 0.05, **P < 0.01, ***P < 0.001).
Luminex assay
Staged day one adult worms were grown on 5xPEP plates and subjected to temperature upshift at 25°C for 24 h prior to harvest. Worms were harvested and washed 3× in M9 buffer, pelleted in Eppendorf tubes and flash frozen with liquid nitrogen. Pelleted worms were resuspended in RIPA buffer (containing Complete Mini protease inhibitor cocktail) and homogenized in a bead mill (3 spins for 10 s each, samples were placed on ice for 5 min between bead-mill spins). Homogenized samples were then centrifuged for 45 min at 21 000g and 4°C. Supernatants were then removed and stored. The remaining pellets were resuspended in 5 M guanidine hydrochloride (Gu-HCl; containing Complete Mini protease inhibitor cocktail) and sonicated on ice for 10 pulses. Centrifugation for 30 min at 21 000g and 4°C produced a Gu-HCl soluble supernatant that was stored at −80°C. Total protein content was determined in Gu-HCl soluble extracts using a BCA kit (Pierce; Rockford, IL). For each sample, we loaded 20 μg of Gu-HCl soluble protein into individual wells of a 96-well plate and incubated them shaking overnight with 5000 Aβ42-antibody (clone H31L21, Life Technologies; Carlsbad, CA) conjugated magnetic beads. Samples were then washed and incubated for 3.5 h with a biotinylated Aβ42-antibody (clone 6E10, Biolegend; San Diego, CA). The samples were washed again and incubated with streptavidin-R-phycoerythrin (10 μg/ml) for 1 h. The samples were washed and resuspended in phosphate-buffered saline, and their fluorescent intensity signals were measured in a Bio-Plex 200 Luminex instrument (Bio-Rad; Hercules, CA).
Protein extraction
Protein extraction and western blotting procedures were conducted as previously described (21, 23). Briefly, to create staged populations, worms were grown at 15°C on 150 mm 5X PEP plates to generate populations for hypochlorite treatment for harvest of eggs. Harvested eggs were deposited onto 5X PEP and maintained at 15°C until the L4 stage when a portion of the plates for each strain was transferred to 25°C for induction of Aβ1-42. Worms were harvested from plates using M9 buffer. Worms were pelleted by centrifugation (3000×g 45 s) and pellets subsequently washed three times with 8 ml M9 buffer and transferred to Eppendorf tubes. Buffer was aspirated from centrifuged worms, and pellets were snap frozen with liquid nitrogen prior to storage at −80°C.
Whole worm protein lysates were created as follows. Worm pellets were thawed on ice and weighed to determine pellet mass. SDS protein sample buffer (0.046 M Tris, 0.005 M ethylenediamine tetraacetate, 0.2 M dithiothreitol, 50% sucrose, 5% sodium dodecyl sulfate, 0.05% bromophenol blue, 2× concentration) was added to the pellets at a volume (μl) four times the pellet weight (mg). Pellets were sonicated three times, 20 s each at 70% amplitude, returning to ice in between sonication sessions. Samples were boiled at 95°C for 5 min and then centrifuged at 13 000×g for 5 min. Samples were returned to ice prior to gel loading.
Isolation of soluble and insoluble tau
Sequential fractionation of tau protein using buffers of increasing protein solubilizing strength was conducted to separate soluble tau from insoluble tau through the collection of sequential buffer fractionation as previously described (23). Staged day one adult worm pellets were resuspended in 2 μl of RAB high salt buffer [0.1 M MES, 1 mm EGTA, 0.5 mm MgSO4, 0.75 M NaCl, 0.02 M NaF, pH 7.0, plus Complete Protease Inhibitor Cocktail (Roche Diagnostics) and 0.5 mm PMSF] per milligram of wet weight. Worms were lysed by three rounds of sonication at 70% amplitude for 8 s. An appropriate amount of worm lysate was reserved as the total fraction used to detect the relative level of total tau (soluble and insoluble combined), and the remaining RAB sample (minimum volume of 100 μl) was subjected to centrifugation at 40 000×g for 40 min. The RAB supernatants (soluble tau) were collected, and the pellets were resuspended half the volume of the starting RAB sample of RIPA buffer [150 mm NaCl/1% Nonidet P-40/0.5% deoxycholate/0.1% SDS/50 mm Tris, pH 8.0, plus Complete Protease Inhibitor Cocktail (Roche Diagnostics) and 0.5 mm PMSF], sonicated 1–2 times at 70% amplitude for 8 s and then centrifuged at 40 000×g for 20 min. The supernatants were collected as the RIPA-soluble fractions (detergent-extractable insoluble tau), and remaining pellets resuspended in a volume equivalent to the starting RAB sample of 70% formic acid (FA), sonicated 1-2 times 70% amplitude for 8 s and centrifuged under vacuum in a SpeedVac on low or medium heat until FA had evaporated. Remaining pellets (FA insoluble tau) were resuspended in a volume of sample buffer (0.046 M Tris, 0.005 M ethylenediamine tetraacetate, 0.2 M dithiothreitol, 50% sucrose, 5% sodium dodecyl sulfate, 0.05% bromophenol blue, 2x concentration) equivalent to the RAB starting sample and sonicated 1–2 times at 70% amplitude for 8 s. The RAB supernatants were boiled at 95°C for 5 min, chilled on ice for 5 min and centrifuged for 15 min at 13 000×g after which the supernatants were collected as the RAB-soluble fractions. Sample buffer was added to a final concentration of 1x from a 5× stock to total, RAB-soluble and RIPA soluble fractions. These samples were boiled for 5 min at 95°C, chilled on ice for 5 min, centrifuged at 13 000×g for 5 min and stored at −20°C in preparation for separation by SDS-PAGE.
Immunoblotting
Lysed samples were subjected to SDS-PAGE according to the manufacturer’s instructions using precast Tris-HCl gels (catalog number 3450028, Biorad, Hercules, CA). Gels were run at 200 V for 60 min and proteins transferred to PVDF membranes at 80 V for 30 min and were blocked (5% milk in PBS) directly post-transfer. Membranes were incubated with primary antibodies diluted in 5% milk-PBS block solution overnight rocking at 4°C and washed three times with PBST (10 min each wash). They were then subjected to secondary antibody incubation for 2 h rocking at room temperature, washed three times with PBST and detected using chemiluminescence kit (Biorad cat. number 1705060) and Chemidoc-it2 imaging system (UVP, CA) and VisionWorksLS software (version 8.1.2). Blots were quantitated using ImageJ (39).
Dye-filling assay
Dye-filling experiments were based on methods described in Hofler and Koelle 2011 (29). Staged adults, 24-, 48- or 96-h temperature up-shifted and control worms were incubated, rotating, at room temperature for 2 h in 50 μl of 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO’) (cat. no. D275, ThermoFisher Scientific, Waltham, MA) dye (stock 2 mg/ml in dimethyl formamide) diluted 1:200 in M9 buffer. Worms were then plated on seeded NGM plates to de-stain and recover for 2 h. To score neurodegeneration, worms were transferred to slides with 2% agar pads, immobilized with sodium azide and visualized. Microscopy and image analyses were conducted on a Delta Vision microscope (GE, Inc) using a 100x oil immersion objective, a sCMOS camera. Image analysis was performed using softWoRx 6.0 Beta software (GE, Inc). Images were subsequently adjusted for brightness and contrast using Adobe Photoshop CS5 (San Jose, CA). The six dye-filled amphid neurons present in N2 animals were counted while ancillary dye-filled cells lacking neuronal morphology were ignored. Neuron count data were analyzed with Kruskal–Wallis ANOVA with Dunn’s post hoc test within GraphPad Prism software (version 5.0) (*P < 0.05, **P < 0.01, ***P < 0.001).
Acknowledgements
We thank Jeanna Wheeler, Aristide Black, Rikki Uhrich and Ashley Yeung for essential technical assistance. We thank Chris Link and the C. elegans Genetics Center (CGC) for providing strains. We thank WormBase for essential C. elegans model organism information. We thank the Developmental Studies Hybridoma Bank (NICHD) for the β-tubulin primary antibody E7. We thank Chris Link for feedback on the manuscript.
Funding
We thank the Department of Veterans Affairs (Merit Review Grant I01BX003755 to B.C.K.) for funding and training support for SB. We also thank the National Institutes of Health R01NS064131 (B.C.K.).
Conflict of Interest statement: The authors state that they have no conflicts of interest.
References
- 1. Lane C.A., Hardy J. and Schott J.M. (2018) Alzheimer’s disease. Eur. J. Neurol., 25, 59–70. [DOI] [PubMed] [Google Scholar]
- 2. Anderson R.M., Hadjichrysanthou C., Evans S. and Wong M.M. (2017) Why do so many clinical trials of therapies for Alzheimer's disease fail? Lancet, 390, 2327–2329. [DOI] [PubMed] [Google Scholar]
- 3. Gauthier S., Albert M., Fox N., Goedert M., Kivipelto M., Mestre-Ferrandiz J. and Middleton L.T. (2016) Why has therapy development for dementia failed in the last two decades? Alzheimers Dement., 12, 60–64. [DOI] [PubMed] [Google Scholar]
- 4. Kumar A., Singh A. and Ekavali (2015) A review on Alzheimer's disease pathophysiology and its management: an update. Pharmacol. Rep., 67, 195–203. [DOI] [PubMed] [Google Scholar]
- 5. Selkoe D.J. (2000) Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid β-protein. Ann. N. Y. Acad. Sci., 924, 17–25. [DOI] [PubMed] [Google Scholar]
- 6. Selkoe D.J. (1991) The molecular pathology of Alzheimer's disease. Neuron, 6, 487–498. [DOI] [PubMed] [Google Scholar]
- 7. Kayed R. and Lasagna-Reeves C.A. (2013) Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis., 33(Suppl 1), S67–S78. [DOI] [PubMed] [Google Scholar]
- 8. Hawkes N. (2017) Merck ends trial of potential Alzheimer’s drug verubecestat. BMJ, 356, j845. [DOI] [PubMed] [Google Scholar]
- 9. Doody R.S., Raman R., Farlow M., Iwatsubo T., Vellas B., Joffe S., Kieburtz K., He F., Sun X., Thomas R.G. et al. (2013) A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N. Engl. J. Med., 369, 341–350. [DOI] [PubMed] [Google Scholar]
- 10. Hung S.Y. and Fu W.M. (2017) Drug candidates in clinical trials for Alzheimer's disease. J. Biomed. Sci., 24, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mehta D., Jackson R., Paul G., Shi J. and Sabbagh M. (2017) Why do trials for Alzheimer's disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs, 26, 735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mangialasche F., Solomon A., Winblad B., Mecocci P. and Kivipelto M. (2010) Alzheimer's disease: clinical trials and drug development. Lancet Neurol., 9, 702–716. [DOI] [PubMed] [Google Scholar]
- 13. Poorkaj P., Bird T.D., Wijsman E., Nemens E., Garruto R.M., Anderson L., Andreadis A., Wiederholt W.C., Raskind M. and Schellenberg G.D. (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol., 43, 815–825. [DOI] [PubMed] [Google Scholar]
- 14. Hutton M., Lendon C.L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A. et al. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature, 393, 702–705. [DOI] [PubMed] [Google Scholar]
- 15. Spillantini M.G., Murrell J.R., Goedert M., Farlow M.R., Klug A. and Ghetti B. (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U. S. A., 95, 7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hunter L.E., Branch C.A. and Lipton M.L. (2019) The neurobiological effects of repetitive head impacts in collision sports. Neurobiol. Dis., 123, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edwards G. 3rd, Zhao J., Dash P.K., Soto C. and Moreno-Gonzalez I. (2019) Traumatic brain injury induces tau aggregation and spreading. J. Neurotrauma, 37, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kovacs G.G. (2017) Tauopathies. Handb. Clin. Neurol., 145, 355–368. [DOI] [PubMed] [Google Scholar]
- 19. Corsi A.K., Wightman B., and Chalfie M.. A Transparent window into biology: A primer on Caenorhabditis elegans (June 18, 2015), WormBook, ed. The C. elegans Research Community, WormBook, doi: /10.1895/wormbook.1.177.1, “http://www.wormbook.org/” http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu Y., Wu Z., Butko P., Christen Y., Lambert M.P., Klein W.L., Link C.D. and Luo Y. (2006) Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J. Neurosci. Off. J. Soc. Neurosci., 26, 13102–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kow R.L., Sikkema C., Wheeler J.M., Wilkinson C.W. and Kraemer B.C. (2018) DOPA decarboxylase modulates tau toxicity. Biol. Psychiatry, 83, 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taylor L.M., McMillan P.J., Kraemer B.C. and Liachko N.F. (2019) Tau tubulin kinases in proteinopathy. FEBS J., 286, 2434–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kraemer B.C., Zhang B., Leverenz J.B., Thomas J.H., Trojanowski J.Q. and Schellenberg G.D. (2003) Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci., 100, 9980–9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hedgecock E.M., Culotti J.G., Thomson J.N. and Perkins L.A. (1985) Axonal guidance mutants of Caenorhabditis elegans identified by filling sensory neurons with fluorescein dyes. Dev. Biol., 111, 158–170. [DOI] [PubMed] [Google Scholar]
- 25. Perkins L.A., Hedgecock E.M., Thomson J.N. and Culotti J.G. (1986) Mutant sensory cilia in the nematode Caenorhabditis elegans. Dev. Biol., 117, 456–487. [DOI] [PubMed] [Google Scholar]
- 26. Starich T.A., Herman R.K., Kari C.K., Yeh W.H., Schackwitz W.S., Schuyler M.W., Collet J., Thomas J.H. and Riddle D.L. (1995) Mutations affecting the chemosensory neurons of Caenorhabditis elegans. Genetics, 139, 171–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guthrie C.R., Greenup L., Leverenz J.B. and Kraemer B.C. (2011) MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum. Mol. Genet., 20, 1989–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guthrie C.R., Schellenberg G.D. and Kraemer B.C. (2009) SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet., 18, 1825–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hofler C. and Koelle M.R. (2011) AGS-3 alters Caenorhabditis elegans behavior after food deprivation via RIC-8 activation of the neural G protein G alphao. J. Neurosci. Off. J. Soc. Neurosci., 31, 11553–11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Billings L.M., Oddo S., Green K.N., McGaugh J.L. and LaFerla F.M. (2005) Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron, 45, 675–688. [DOI] [PubMed] [Google Scholar]
- 31. Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed R., Metherate R., Mattson M.P., Akbari Y. and LaFerla F.M. (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron, 39, 409–421. [DOI] [PubMed] [Google Scholar]
- 32. Jackson R.J., Rudinskiy N., Herrmann A.G., Croft S., Kim J.M., Petrova V., Ramos-Rodriguez J.J., Pitstick R., Wegmann S., Garcia-Alloza M. et al. (2016) Human tau increases amyloid beta plaque size but not amyloid beta-mediated synapse loss in a novel mouse model of Alzheimer's disease. Eur. J. Neurosci., 44, 3056–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grueninger F., Bohrmann B., Czech C., Ballard T.M., Frey J.R., Weidensteiner C., von Kienlin M. and Ozmen L. (2010) Phosphorylation of tau at S422 is enhanced by Abeta in TauPS2APP triple transgenic mice. Neurobiol. Dis., 37, 294–306. [DOI] [PubMed] [Google Scholar]
- 34. Lewis J., Dickson D.W., Lin W.L., Chisholm L., Corral A., Jones G., Yen S.H., Sahara N., Skipper L., Yager D. et al. (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science, 293, 1487–1491. [DOI] [PubMed] [Google Scholar]
- 35. Wang C., Saar V., Leung K.L., Chen L. and Wong G. (2018) Human amyloid beta peptide and tau co-expression impairs behavior and causes specific gene expression changes in Caenorhabditis elegans. Neurobiol. Dis., 109, 88–101. [DOI] [PubMed] [Google Scholar]
- 36. Saito T., Mihira N., Matsuba Y., Sasaguri H., Hashimoto S., Narasimhan S., Zhang B., Murayama S., Higuchi M., Lee V.M.Y. et al. (2019) Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J. Biol. Chem., 294, 12754–12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Platt B., Drever B., Koss D., Stoppelkamp S., Jyoti A., Plano A., Utan A., Merrick G., Ryan D., Melis V. et al. (2011) Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse PLB1. PLoS One, 6, e27068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]