

Abstract
Objective
Serum uric acid is the end-product of purine metabolism and at high levels is a risk factor for several human diseases including gout and cardiovascular disease. Heritability estimates range from 0.32 to 0.63. Genome-wide association studies (GWAS) provide an unbiased approach to identify loci influencing serum uric acid. Here, we performed the first GWAS for serum uric acid in continental Africans, with replication in African Americans.
Methods
Africans (n = 4126) and African Americans (n = 5007) were genotyped on high-density GWAS arrays. Efficient mixed model association, a variance component approach, was used to perform association testing for a total of ~ 18 million autosomal genotyped and imputed variants. CAVIARBF was used to fine map significant regions.
Results
We identified two genome-wide significant loci: 4p16.1 (SLC2A9) and 11q13.1 (SLC22A12). At SLC2A9, the most strongly associated SNP was rs7683856 (P = 1.60 × 10−44). Conditional analysis revealed a second signal indexed by rs6838021 (P = 5.75 × 10−17). Gene expression and regulatory motif data prioritized a single-candidate causal variant for each signal. At SLC22A12, the most strongly associated SNP was rs147647315 (P = 6.65 × 10−25). Conditional analysis and functional annotation prioritized the missense variant rs147647315 (R (Arg) > H (His)) as the sole causal variant. Functional annotation of these three signals implicated processes in skeletal muscle, subcutaneous adipose tissue and the kidneys, respectively.
Conclusions
This first GWAS of serum uric acid in continental Africans identified three associations at two loci, SLC2A9 and SLC22A12. The combination of weak linkage disequilibrium in Africans and functional annotation led to the identification of candidate causal SNPs for all three signals. Each candidate causal variant implicated a different cell type. Collectively, the three associations accounted for 4.3% of the variance of serum uric acid.
Introduction
The most abundant antioxidant in human plasma is uric acid (1). Endogenous and exogenous sources account for approximately two-thirds and one-third, respectively, of the total uric acid load (2). The primary endogenous source is purine degradation in the liver, such as that following extrusion of nuclei from erythroblasts (3,4). Exogenous sources include purine-rich foods such as organ meats and seafood (5). Approximately two-thirds of elimination of uric acid occurs via the kidneys while the remainder occurs via the intestines (6).
High serum uric acid is a risk factor for several diseases including gout, hypertension, diabetes, kidney disease and cardiovascular disease (7–9) and is moderately to strongly heritable, with estimates in adults ranging from 0.32 to 0.63 (10–22). In individuals without gout, serum uric acid ranges from 2.6 to 6.0 mg/dl in premenopausal women and from 3.5 to 7.2 mg/dl in men and postmenopausal women (23). Meta-analysis of GWAS in European-ancestry individuals identified 26 loci, collectively accounting for 7.0% of the variance in serum uric acid levels (24). We previously reported the first GWAS of serum uric acid in African Americans (25). The most strongly associated locus in African Americans, at or near the gene SLC2A9 (25), was the most strongly associated locus in the European meta-analysis. The second most strongly associated locus in African Americans, at or near the gene SLC22A12 (26), was the fifth most strongly associated locus in the European meta-analysis. It is not known if these results reflect European ancestry in admixed African Americans or if these associations are also present in the background of African ancestry. To address this question, we describe the first GWAS of serum uric acid in continental Africans.
Results
As expected, males had higher serum uric acid levels than females in both the discovery and replication samples (Table 1). After adjusting for the effects of sex, age, age2, BMI, hypertension, T2D, eGFR and PCs, the heritability for serum uric acid in AADM was estimated as 0.600 (SD 0.048). Genomic inflation factors were approximately 1.01, suggesting no residual population stratification in either the discovery or replication analyses (Supplementary Material, Fig. S1). Two regions, 4p16.1 and 11q13.1, were identified as genome-wide significant in the discovery analysis of continental Africans and subsequently replicated in African Americans (Fig. 1). All genome-wide significant SNPs (P < 5 × 10−8) in the meta-analysis of discovery and replication samples are listed in Supplementary Material, Table S1.
Table 1.
Study characteristics for the discovery and replication samples
| Discovery (Africans) | Replication (African Americans) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Overall | Female | Male | P value | Overall | Female | Male | P value | |||
| N | Mean (SD) | Mean (SD) | Mean (SD) | N | Mean (SD) | Mean (SD) | Mean (SD) | |||
| Age (years) | 4126 | 51.01 (12.78) | 50.44 (12.25) | 51.85 (13.49) | 0.0006 | 5113 | 47.93 (13.16) | 48.04 (12.98) | 47.74 (13.46) | 0.4254 |
| BMI (kg/m2) | 4126 | 26.62 (5.66) | 27.94 (6.06) | 24.67 (4.32) | <0.0001 | 5098 | 29.77 (7.15) | 30.90 (7.55) | 27.91 (6.00) | <0.0001 |
| SBP (mm Hg) | 4117 | 136.50 (23.86) | 135.77 (24.39) | 137.57 (23.02) | 0.0161 | 5109 | 126.82 (20.93) | 125.91 (20.97) | 128.30 (20.78) | <0.0001 |
| DBP (mm Hg) | 4116 | 81.79 (13.49) | 81.82 (13.48) | 81.76 (13.52) | 0.8962 | 5109 | 79.28 (12.44) | 77.95 (11.69) | 81.47 (13.28) | <0.0001 |
| Uric acid (mg/dl) | 4126 | 5.11 (1.66) | 4.71 (1.49) | 5.70 (1.73) | <0.0001 | 5007 | 5.87 (1.71) | 5.42 (1.59) | 6.61 (1.63) | <0.0001 |
| eGFR(ml/min/1.73 m2) | 4125 | 98.74 (35.87) | 98.71 (35.27) | 98.77 (36.75) | 0.9538 | 5012 | 87.75 (31.31) | 85.96 (30.62) | 90.68 (32.21) | <0.0001 |
| N (%) | N (%) | N (%) | N (%) | N (%) | N (%) | |||||
| Type 2 diabetes | 4126 | 2068 (50.12) | 1253 (50.85) | 815 (49.04) | 0.2528 | 5028 | 823 (16.4) | 544 (17.5) | 279 (14.6) | 0.008 |
| Hypertension | 4117 | 2253 (54.70) | 1367 (55.57) | 886 (53.41) | 0.1713 | 5096 | 2429 (47.7) | 1537 (48.6) | 892 (46.1) | 0.0897 |
Figure 1.
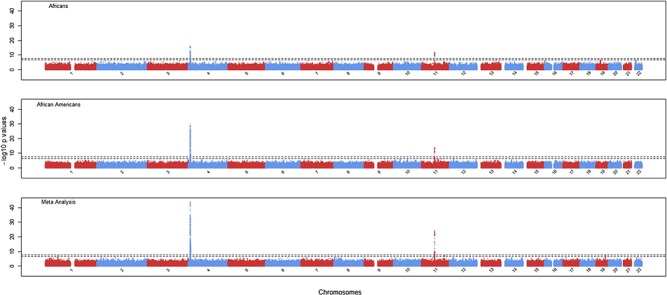
GWAS Manhattan plots for Africans (n = 4126, top panel), African Americans (n = 5007, middle panel) and meta-analysis (n = 9133, bottom panel). The two dotted lines represent genome-wide significance at −log10 (5 × 10−8) and genome-wide suggested significance at −log10 (5 × 10−7), respectively.
At 4p16.1, 503 SNPs from 9819 to 10 382 kb in or near the gene solute carrier family 2 member 9 (SLC2A9, GeneID: 56606, also known as GLUT9) and the neighboring gene WD repeat domain 1 (WDR1, GeneID: 9948) reached genome-wide significance with directional consistency (Fig. 2 and Supplementary Material, Table S1). The lead SNP was rs7683856 (P = 1.60 × 10−44); association at this SNP explained 2.2% of the phenotypic variance (Table 2). Meta-analysis conditional on rs7683856 revealed a secondary signal indexed by rs6838021 (P = 5.75 × 10−17) that explained 0.9% of the phenotypic variance (Table 2 and Supplementary Material, Fig. S2). Meta-analysis conditional on rs7683856 and rs6838021 revealed no additional signal at this locus (Supplementary Material, Fig. S2).
Figure 2.
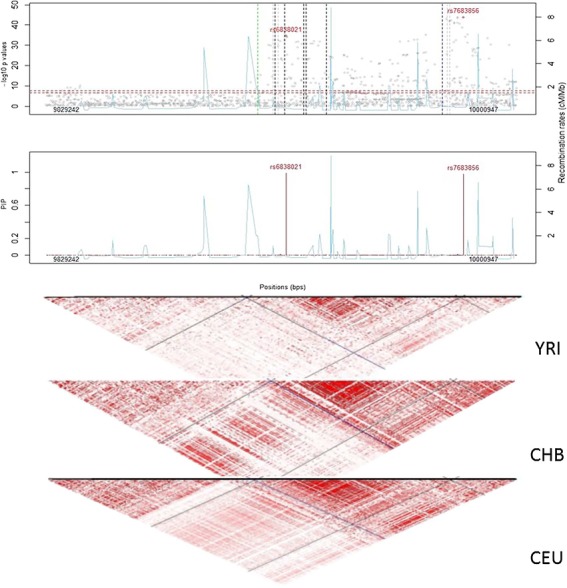
Regional association plots at SLC2A9. (Top) Regional Manhattan plot. Vertical dotted lines represent the positions for reported genetic variants associated with serum urate (blue, black, green and gray dotted lines represent African, European, Chinese and other ancestral backgrounds, respectively). Sky blue lines depict recombination rates. (Middle) Posterior inclusion probability (PIP) plots from fine-mapping. (Bottom) LD plots for Africans (YRI), Europeans (CEU) and Chinese (CHB). The diagonal lines represent pairwise LD for rs6838021 (blue lines) and rs7683856 (black lines).
Table 2.
GWAS and conditional single SNP association results in top regions
| Conditional on | Chr:Pos (bp) | Gene (functional class) | SNP | Ref/Alt | Africans (AADM) | African Americans (ARIC + HUFS) | Meta-analysis | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | AAF | Beta | SE | P value | r 2 | N | AAF | Beta | SE | P value | r 2 | AAF | Beta | SE | P value | r 2 | |||||
| NO | 4:10000947 | SLC2A9 (intron) | rs7683856 | A/G | 4126 | 0.784 | 0.216 | 0.027 | 6.94E−16 | 0.016 | 5007 | 0.775 | 0.281 | 0.024 | 1.62E−30 | 0.028 | 0.779 | 0.251 | 0.018 | 1.60E−44 | 0.022 |
| 11:64367854 | SLC22A12 (nonsynonymous) | rs147647315 | G/A | 4126 | 0.019 | −0.579 | 0.082 | 1.82E−12 | 0.012 | 5007 | 0.012 | −0.688 | 0.091 | 5.14E−14 | 0.011 | 0.015 | −0.628 | 0.061 | 6.65E−25 | 0.012 | |
| rs7683856 | 11:64367854 | SLC22A12 (nonsynonymous) | rs147647315 | G/A | 4126 | 0.019 | −0.597 | 0.083 | 6.66E−13 | 0.013 | 5007 | 0.012 | −0.711 | 0.090 | 2.53E−15 | 0.012 | 0.015 | −0.649 | 0.061 | 1.24E−26 | 0.013 |
| 4:9927620 | SLC2A9 (intron) | rs6838021 | T/C | 4126 | 0.449 | 0.113 | 0.024 | 1.90E−06 | 0.006 | 5007 | 0.505 | 0.153 | 0.022 | 3.12E−12 | 0.012 | 0.480 | 0.135 | 0.016 | 5.75E−17 | 0.009 | |
| rs7683856, rs6838021 | 11:64367854 | SLC22A12 (nonsynonymous) | rs147647315 | G/A | 4126 | 0.019 | −0.587 | 0.083 | 1.33E−12 | 0.013 | 5007 | 0.012 | −0.709 | 0.089 | 2.07E−15 | 0.012 | 0.015 | −0.644 | 0.061 | 2.17E−26 | 0.012 |
| rs7683856, rs6838021, rs147647315 | 11:64369267 | SLC22A12 (insertion) | rs150284736 | −/CCCTG | 4126 | 0.216 | 0.070 | 0.026 | 0.008 | 0.002 | 5007 | 0.206 | 0.123 | 0.025 | 7.32E−07 | 0.005 | 0.211 | 0.098 | 0.018 | 5.37E−08 | 0.003 |
From 1395 SNPs with rsIDs mapping to SLC2A9, rs7683856 and rs6838021 had marginal posterior inclusion probabilities of 0.989 and 0.976, respectively (Fig. 2). The two SNPs rs7683856 and rs6838021 are weakly correlated in African-ancestry individuals (r2 = 0.07 in Africans (AADM or YRI) and 0.12 in African Americans (ARIC and HUFS) in contrast to 0.37 in CHB and 0.60 in CEU). These results are consistent with the results from the conditional analysis of two signals at this locus.
Functional annotation revealed four SNPs in strong linkage disequilibrium with rs7683856 spread over 4.7 kb. All five SNPs were cis-eQTLs for SLC2A9 in skeletal muscle, with all alternate (Alt) alleles associated with higher expression. Of these five SNPs, rs7678287 showed the strongest association with gene expression (P = 0.000030). Furthermore, rs7678287 is located within two regulatory motifs. At the SIX5 motif, the LOD scores for the Ref and Alt alleles were both 11.9, indicating no difference in binding potential and hence no expectation of differential expression. At the RREB1 motif, the LOD scores for the Ref and Alt alleles were 14.6 and 2.9, respectively, indicating that RREB1 is predicted to bind better to the sequence containing the Ref allele. RREB1 is a zinc finger transcription factor that binds to RAS-responsive elements of gene promoters and is expressed in skeletal muscle. The Alt allele is associated with higher gene expression and higher serum uric acid, consistent with RREB1 acting as a repressor and with transport from muscle to blood.
Functional annotation revealed 12 SNPs in strong linkage disequilibrium with rs6838021. Nine SNPs were cis-eQTLs for SLC2A9, all in subcutaneous adipose tissue. The two SNPs rs7670751 and rs5028843 showed the strongest association with gene expression (P = 0.000027 for both SNPs). The SNP rs7670751 is located within regulatory motifs for MAFF and PTF1A. MAFF is expressed in subcutaneous adipose tissue. PTF1A is pancreas-specific transcription factor 1a and is not expressed in subcutaneous adipose tissue. The SNP rs5028843 is located within a regulatory motif for FXR. FXR (also known as NR1H4) is not expressed in subcutaneous adipose tissue. At the MAFF motif, the LOD scores for the Ref and Alt alleles were 7.7 and 11.5, respectively. The Alt allele is associated with lower gene expression and lower serum uric acid. MAFF lacks a transactivation domain; homodimers may therefore act as repressors. Taken together, these results support rs7670751 as the causal variant, with better binding of MAFF associated with lower gene expression.
At 11q13.1, 61 SNPs across 506 kb showed genome-wide significance and directional consistency (Fig. 3 and Supplementary Material, Table S1). The strongest association occurred at rs147647315 (P = 6.65 × 10−25); association at this SNP explained 1.2% of the phenotypic variance (Table 2). Meta-analysis conditional on rs147647315 revealed no secondary signal (Supplementary Material, Fig. S2). rs147647315 had a marginal posterior inclusion probability of 0.996 (Fig. 3). Posterior inclusion probabilities supported only one signal, consistent with the conditional analysis. The derived A allele at rs147647315 has a minor allele frequency of 1.6% in the 1000 Genomes AFR superpopulation and is monomorphic in the EUR superpopulation. rs147647315 is not an eQTL for SLC22A12 and does not overlap any known regulatory motifs. The G>A mutation at rs147647315 is missense, causing an arginine to histidine substitution in all five transcripts of SLC22A12. SIFT predicts that the change is deleterious in all five transcripts of SLC22A12. PolyPhen predicts that the change is benign in transcripts ENST00000336464, ENST00000377574 (the principal isoform) and ENST00000473690, but probably damaging in transcripts ENST00000377567 (a transcript not supported by either an EST or an mRNA) and ENST00000377572. The PHRED-scaled CADD score was 23.3, placing this SNP in the top 0.5% of the most deleterious substitutions genome-wide.
Figure 3.
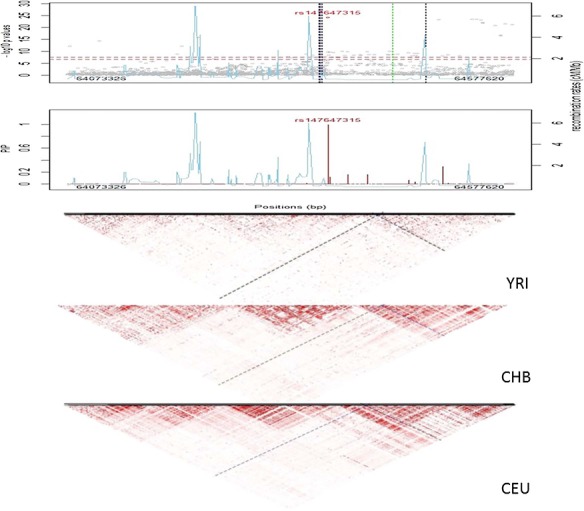
Regional association plots at SLC22A12. (Top) Regional Manhattan plot. Vertical dotted lines represent the positions for reported genetic variants associated with serum urate (blue, black, green and gray dotted lines represent African, European, Chinese and other ancestral backgrounds, respectively). Sky blue lines depict recombination rates. (Middle) Posterior inclusion probability (PIP) plots from fine-mapping. (Bottom) LD plots for Africans (YRI), Europeans (CEU) and Chinese (CHB). The diagonal lines represent pairwise LD for rs147647315 (blue lines).
Ten variants in the SLC2A9 locus have been catalogued as being significantly associated with serum uric acid (Supplementary Material, Table S2). These variants clustered into two regions, one around rs76836856 and the other around rs6838021 (Fig. 2). These two signals are in different LD blocks in the 1000 Genomes YRI sample in contrast to being in one large block in both CEU and CHB (Fig. 2).
Thirty SNPs associated with serum uric acid were reported in > 140 000 European-ancestry individuals (24). We attempted to replicate these findings using exact (i.e. querying the reported SNP) and local replication (i.e. querying SNPs in LD (r2 ≥ 0.3) with the reported SNPs within a defined window of 250 kb). Of 26 loci that we could assess, 16 loci in or near SLC2A9, ABCG2, REFB1, SLC17A1, SLC16A9, SLC22A11, NRXN2, INHBE, ORC4L, TMEM171, PRKAG2, STC1, HNF4G, A1CF, OVOL1 and MAF replicated in our set of African-ancestry individuals (Supplementary Material, Table S3).
Discussion
GWAS have been useful in detecting common genetic variants influencing serum uric acid levels. These loci implicate genes involved in uric acid secretion (NTP1, ABCG2, MRP4, OAT1 and OAT3) and reabsorption (OAT4, OAT10, SLC22A12 and SLC2A9). Genetic variation in SLC2A9 accounts for 1.7–5.3% of the variance in serum uric acid (24,27–29). In the Framingham Heart Study (28), rs16890979 (a missense variant in SLC2A9) was the most significant SNP (P = 7.0 × 10−168), with association at this SNP explaining 5.3% of the phenotypic variance. In our study of African-ancestry individuals, rs16890979 was significantly associated with serum uric acid (P = 5.79 × 10−24). However, we found that rs16890979 was in strong linkage disequilibrium with the more strongly associated rs6838021 (r2 = 0.72 in YRI and 0.97 in CEU).
Our results indicated two distinct signals within the SLC2A9 locus. The stronger effect was indexed by rs7683856 and accounted for 2.2% of the phenotypic variance. The weaker effect was indexed by rs6838021 and accounted for 0.9% of the phenotypic variance. Our results indicated one signal within the SLC22A12 locus, indexed by rs147647315 and accounting for 1.2% of the phenotypic variance. Taken together, these three signals explain 4.3% of the total variance of serum uric acid or approximately 7.2% of the heritable component of serum uric acid.
For the primary association indexed by rs76838056, our analyses support rs7678287 as the candidate causal variant. The combination of association testing and functional annotation provides evidence that (i) RREB1 binds to rs7678287 in skeletal muscle, (ii) RREB1 binds worse to the Alt allele and (iii) the Alt allele is associated with higher gene expression of SLC2A9 and higher serum uric acid. Furthermore, the gene RREB1 is a known GWAS locus for serum uric acid (8). For the secondary association indexed by rs6838021, our analyses support rs7670751 as the candidate causal variant. The combination of association testing and functional annotation provides evidence that (i) MAFF binds to rs7670751 in subcutaneous adipose tissue, (ii) MAFF binds better to the Alt allele and (ii) the Alt allele is associated with lower gene expression of SLC2A9 and lower serum uric acid.
We attempted to replicate 30 SNPs reported in meta-analysis of European-ancestry individuals. Associations in genes implicated in reabsorption such as SLC2A9, SLC22A12, SLC22A6 and SLC2A14 as well as genes implicated in secretion such as ABCG2 were replicated in our study of individuals with African ancestry. African American populations contribute to the refinement of GWAS signals because of weaker LD than in populations of Asian or European ancestry. In contrast to African Americans, continental African populations, in particular sub-Saharan African populations, can offer even better resolution because of the absence of recent admixture with European ancestry. The local replication approach has been successful in African-ancestry individuals for T2D, fasting insulin, insulin resistance and serum bilirubin (30–32). For loci discovered in Asian- and European-ancestry populations, fine-mapping in populations with African ancestry is useful for localization of association signals and discovery of causal variants.
In summary, using a high coverage SNP array in conjunction with an improved imputation reference panel, we identified association with serum uric acid in SLC2A9 and SLC22A12 in a large (n~9000) meta-analysis of African-ancestry individuals. Based on genome-wide meta-analysis, conditional analysis, fine-mapping and functional annotation, we inferred the presence of two causal variants at SLC2A9 and one causal variant at SLC22A12. These three associations may influence serum uric acids levels through effects in skeletal muscle, subcutaneous adipose tissue and the kidneys. Sixteen genes previously implicated in uric acid secretion and reabsorption, including the two highlighted in our study, were replicated. One potential issue is that the discovery study was enriched for cases of type 2 diabetes whereas the replication studies were not. Although Mendelian randomization studies have shown that serum uric acid is not a causal risk factor for type 2 diabetes (33–35), the difference in study design between discovery and replication could still matter if serum uric acid is a causal risk factor for other diseases that tend to co-occur with type 2 diabetes. A limitation of our study is that we relied on data sets of African Americans for replication because of the absence of a second continental African data set. To fully capitalize on genetic diversity in African-ancestry individuals, we encourage the expansion of studies of continental Africans.
Materials and Methods
Study design
Individuals included in the GWAS discovery samples were drawn from the Africa America Diabetes Mellitus (AADM) study, a large, ongoing genetic epidemiology study of type 2 diabetes (T2D) and related traits in Africans (30,36,37). Although AADM originated with a sib-pair design, subsequent enrollment included both unrelated individuals and extended families. Demographic information was collected using standardized questionnaires across the AADM study centers in Nigeria (Ibadan, Lagos and Enugu), Ghana (Accra and Kumasi) and Kenya (Eldoret). Anthropometric, medical history and clinical examination parameters were obtained by trained study staff during a clinic visit. Weight was measured in light clothes on an electronic scale to the nearest 0.1 kg, and height was measured with a stadiometer to the nearest 0.1 cm. Body mass index (BMI) was computed as weight (kg) divided by the square of height in meters (m2). Blood samples were drawn after an overnight fast of at least 8 h. The definition of T2D was based on the American Diabetes Association (ADA) criteria. Controls were required to have fasting plasma glucose < 110 mg/dl or 2 h post load of < 140 mg/dl and no clinical features suggestive of diabetes (the classical symptoms being polyuria, polydipsia and unexplained weight loss).
Biochemistry
Fasting serum samples were assayed for uric acid using a COBAS® Analyzer Series (Roche Diagnostics, Indianapolis, Indiana). Serum creatinine levels were estimated on fasting samples using the modified Jaffe method. The estimated glomerular filtration rate (eGFR) was calculated using the simplified Modification of Diet in Renal Disease Study equation (38).
Genotyping and imputation
A total of 5231 individuals from the AADM study were genotyped on high-density GWAS arrays: 1808 samples were genotyped using the Affymetrix® Axiom® Genome-Wide PanAFR Array Set, and 3423 samples were genotyped using the Illumina Multi-Ethnic Genotyping Array. After technical quality control and appropriate sample- and SNP-level exclusionary filtering (individual call rate ≤ 95%, SNP call rates ≤ 95%, Hardy–Weinberg equilibrium (HWE) P value < 1 × 10−6 and minor allele frequency (MAF) < 0.01) (37), imputation was performed using the African Genome Resources Panel available from the Sanger Imputation Service (https://imputation.sanger.ac.uk/) (39). The imputation reference panel comprised 4956 individuals, including all 2504 from the 1000 Genomes Project Phase 3, ~ 2000 individuals from Uganda (Baganda, Banyarwanda, Barundi and others) and ~ 100 individuals from each of a set of populations from Ethiopia (Gumuz, Wolayta, Amhara, Oromo and Somali), Egypt, Namibia (Nama/Khoe-San) and South Africa (Zulu), yielding 9912 haplotypes for 93 421 145 SNPs. Pre-phasing was performed with EAGLE version 2.0.5 (40), and imputation was performed using PBWT (41). Imputed variants with MAF ≥ 0.01 and imputation info ≥0.30 were retained, leaving 18 219 730 variants. Coordinates are given based on the hg19 genome build. The samples (by ethnic groups) clustered as expected (Supplementary Material, Fig. S3) based on principal components (PCs) of the genotypes computed using SNPRelate (42).
Association analysis
Serum uric acid levels were first log-transformed and then regressed on age, age2 and sex. The resulting residuals were skewed and therefore were ranked and inverse-normalized. Association analysis was performed using the EPACTS (Efficient and Parallelizable Association Container Toolbox) pipeline (http://genome.sph.umich.edu/wiki/EPACTS) (43), using imputed dosages and adjusting for genetic relatedness, BMI, hypertension, T2D, eGFR and the first three principal components. Principal components were obtained from the R package SNPRelate (42) using genotyped but not imputed SNPs. Within EPACTS, we performed single variant EMMAX association analysis. The genome-wide significance level α was declared to be 5 × 10−8.
Replication study
Replication was assessed in 5113 African Americans obtained from the Atherosclerosis Rick in Communities study (ARIC, n = 3137) (44) and the Howard University Family Study (HUFS, n = 1976) (45). ARIC is a prospective study of atherosclerosis in healthy middle-aged adults and HUFS is a population-based study. As with the discovery study, serum uric acid levels were first log-transformed and then regressed on age, age2 and sex. The resulting residuals were ranked and inverse-normalized. Genotyping in ARIC and HUFS was performed using the Affymetrix® Genome-Wide SNP Array 6.0. Imputation and association analysis were performed as described previously. We adjusted for genetic relatedness, BMI, T2D, eGFR, hypertension and the first two principal components. METAL (46) was used to perform inverse variance-weighted fixed-effect meta-analysis of the discovery and replication samples.
Functional annotation
Using HaploReg v4.1 (47), we annotated SNPs with respect to sequence conservation across mammals, promoter histone marks, enhancer histone marks, chromatin state, protein binding and regulatory motifs. We queried the index SNP plus all SNPs in strong pairwise linkage disequilibrium (r2 ≥ 0.8) in the 1000 Genomes AFR superpopulation. The Genotype-Tissue Expression Project was interrogated regarding cis-eQTLs and tissue-specific gene expression (https://gtexportal.org/home/). PHRED-scaled Combined Annotation Dependent Depletion (CADD, version 1.4) scores for the predicted deleteriousness of variants were retrieved from https://cadd.gs.washington.edu/snv. The top (i.e. most deleterious across the entire genome) 10% of raw scores have a scaled score of 10, the top 1% of raw scores have a scaled score of 20, the top 0.1% of raw scores have a scaled score of 30, etc.
Fine-mapping and identification of candidate causal variants
Fine-mapping was performed using the R package CAVIARBF, an approximate Bayesian method that can incorporate functional annotation (48). Minimal data requirements are marginal statistical test results and linkage disequilibrium between SNPs. SNP annotations were coded for the presence (1) or absence (0) of promoter histone, enhancer histone, DNAse or bound protein as provided by HaploReg v4.1.
Transferability of previous GWAS findings
We attempted to replicate GWAS loci for uric acid and gout previously reported in meta-analysis of > 140 000 Europeans (24). Based on the association results from meta-analysis of the Africans and African Americans, we performed both exact replication (i.e. the same SNPs as previously reported) and local replication (i.e. SNPs in linkage disequilibrium with previously reported SNPs) as previously described (31). For the identification of SNPs in linkage disequilibrium (LD) with published variants, we used the EUR reference dataset (n = 503) from the 1000 Genomes Project, phase 3. To adequately account for multiple testing, we estimated the effective degrees of freedom (49) from the spectrally decomposed covariance matrix for the block of SNPs from the reference populations. Replication significance levels were either 0.05 for exact replication or 0.05 divided by the effective degrees of freedom for local replication.
We queried the GWAS Catalog (accessed March 3, 2018) for variants in the SLC2A9 region associated (P < 5 × 10−8) with urate, uric acid or serum uric acid levels (50). Genotype data from the SLC2A9 and SLC22A12 regions were extracted from three 1000 Genomes Project samples: Utah residents with Northern and Western European ancestry (CEU), Han Chinese in Beijing, China (CHB) and Yoruba in Ibadan, Nigeria (YRI). Linkage disequilibrium plots were drawn using the R package snp.plotter (51).
SNP heritability
We estimated GWAS 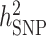 using LDAK (52,53). A total of 1 035 348 genotyped SNPs with MAF > 0.05 were used. The phenotype of serum uric acid levels and covariates sex, age, age2, BMI, T2D, hypertension, eGFR and significant PCs were used for
using LDAK (52,53). A total of 1 035 348 genotyped SNPs with MAF > 0.05 were used. The phenotype of serum uric acid levels and covariates sex, age, age2, BMI, T2D, hypertension, eGFR and significant PCs were used for 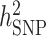 estimation. First, based on local LD, we calculated a weight for each predictor (SNP) which showed how well each SNP was tagged. Next, using these weights, we calculated a kinship matrix to improve poorly tagged predictors that had lower than average MAF. Then, we fit the linear mixed model
estimation. First, based on local LD, we calculated a weight for each predictor (SNP) which showed how well each SNP was tagged. Next, using these weights, we calculated a kinship matrix to improve poorly tagged predictors that had lower than average MAF. Then, we fit the linear mixed model 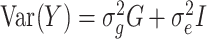 , in which Y is the vector of phenotype values, G is a kinship matrix based on weights and I is an identity matrix. Estimates of
, in which Y is the vector of phenotype values, G is a kinship matrix based on weights and I is an identity matrix. Estimates of 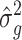 and
and 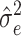 were obtained via restricted maximum likelihood (REML) (52). The proportion of phenotypic variance explained by additive genetic effects was estimated as
were obtained via restricted maximum likelihood (REML) (52). The proportion of phenotypic variance explained by additive genetic effects was estimated as 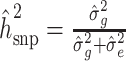 .
.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official view of the National Institutes of Health. We are thankful to the participants of the AADM project, their families and their physicians. The study was supported in part by the Intramural Research Program of the National Institutes of Health in the Center for Research on Genomics and Global Health (CRGGH). The CRGGH is supported by the National Human Genome Research Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, the Center for Information Technology and the Office of the Director at the National Institutes of Health (1ZIAHG200362). Support for participant recruitment and initial genetic studies of the parent AADM study was provided by NIH grant 3T37TW00041-03S2 from the Office of Research on Minority Health. The Atherosclerosis Risk in Communities (ARIC) Study was carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C and HHSN268201100012C. The authors thank the staff and participants of the ARIC study for their important contributions.
Conflict of Interest statement. None declared.
Funding
Funding for the ARIC Gene Environment Association Studies (dbGaP Study Accession phs000090.v2.p1, a sub-study of phs000280.v2.p1), was provided by National Human Genome Research Institute grant U01HG004402 (Eric Boerwinkle). The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
References
- 1. Ames B.N., Cathcart R., Schwiers E. and Hochstein P. (1981) Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc. Natl. Acad. Sci. USA, 78, 6858–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kang D.-H. and Johnson R.J. (2015) Uric acid metabolism and the kidney In Kimmel, P.L. and Rosenberg, M.E. (eds), Chronic Renal Disease. Academic Press, San Diego, CA, pp. 418–428. [Google Scholar]
- 3. Krafka J.J. (1929) Endogenous uric acid and hematopoiesis. J. Biol. Chem., 83, 409–414. [Google Scholar]
- 4. Keerthivasan G., Wickrema A. and Crispino J.D. (2011) Erythroblast enucleation. Stem Cells Int., 2011, 139851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi H.K., Liu S. and Curhan G. (2005) Intake of purine-rich foods, protein, and dairy products and relationship to serum levels of uric acid: the Third National Health and Nutrition Examination Survey. Arthritis Rheum., 52, 283–289. [DOI] [PubMed] [Google Scholar]
- 6. Xu X., Li C., Zhou P. and Jiang T. (2016) Uric acid transporters hiding in the intestine. Pharm. Biol., 54, 3151–3155. [DOI] [PubMed] [Google Scholar]
- 7. Jin M., Yang F., Yang I., Yin Y., Luo J.J., Wang H. and Yang X.F. (2012) Uric acid, hyperuricemia and vascular diseases. Front. Biosci., 17, 656–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Q.O., Kottgen A., Dehghan A., Smith A.V., Glazer N.L., Chen M.H., Chasman D.I., Aspelund T., Eiriksdottir G., Harris T.B. et al. (2010) Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ. Cardiovasc. Genet., 3, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feig D.I., Kang D.H. and Johnson R.J. (2008) Uric acid and cardiovascular risk. N. Engl. J. Med., 359, 1811–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilk J.B., Djousse L., Borecki I., Atwood L.D., Hunt S.C., Rich S.S., Eckfeldt J.H., Arnett D.K., Rao D.C. and Myers R.H. (2000) Segregation analysis of serum uric acid in the NHLBI Family Heart Study. Hum. Genet., 106, 355–359. [DOI] [PubMed] [Google Scholar]
- 11. Pilia G., Chen W.-M., Scuteri A., Orrú M., Albai G., Dei M., Lai S., Usala G., Lai M., Loi P. et al. (2006) Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLOS Genet., 2, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sulem P., Gudbjartsson D.F., Walters G.B., Helgadottir H.T., Helgason A., Gudjonsson S.A., Zanon C., Besenbacher S., Bjornsdottir G., Magnusson O.T. et al. (2011) Identification of low-frequency variants associated with gout and serum uric acid levels. Nat. Genet., 43, 1127–1130. [DOI] [PubMed] [Google Scholar]
- 13. Shriner D., Kumkhaek C., Doumatey A.P., Chen G., Bentley A.R., Charles B.A., Zhou J., Adeyemo A., Rodgers G.P. and Rotimi C.N. (2015) Evolutionary context for the association of γ-globin, serum uric acid, and hypertension in African Americans. BMC Med. Genet., 16, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang W., Zhang D., Xu C., Wu Y., Duan H., Li S. and Tan Q. (2018) Heritability and genome-wide association analyses of serum uric acid in middle and old-aged Chinese twins. Front. Endocrinol., 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang W., Miller M.B., Rich S.S., North K.E., Pankow J.S., Borecki I.B., Myers R.H., Hopkins P.N., Leppert M. and Arnett D.K. (2003) Linkage analysis of a composite factor for the multiple metabolic syndrome: the National Heart, Lung, and Blood Institute Family Heart Study. Diabetes, 52, 2840–2847. [DOI] [PubMed] [Google Scholar]
- 16. Yang Q., Guo C.-Y., Cupples L.A., Levy D., Wilson P.W.F. and Fox C.S. (2005) Genome-wide search for genes affecting serum uric acid levels: the Framingham Heart Study. Metabolism, 54, 1435–1441. [DOI] [PubMed] [Google Scholar]
- 17. Nath S.D., Voruganti V.S., Arar N.H., Thameem F., Lopez-Alvarenga J.C., Bauer R., Blangero J., MacCluer J.W., Comuzzie A.G. and Abboud H.E. (2007) Genome scan for determinants of serum uric acid variability. J. Am. Soc. Nephrol., 18, 3156–3163. [DOI] [PubMed] [Google Scholar]
- 18. Vitart V., Rudan I., Hayward C., Gray N.K., Floyd J., Palmer C.N., Knott S.A., Kolcic I., Polasek O., Graessler J. et al. (2008) SLC2A9 is a newly identified urate transporter influencing serum urate concentration. urate excretion and gout. Nat. Genet., 40, 437–442. [DOI] [PubMed] [Google Scholar]
- 19. Rule A.D., Fridley B.L., Hunt S.C., Asmann Y., Boerwinkle E., Pankow J.S., Mosley T.H. and Turner S.T. (2009) Genome-wide linkage analysis for uric acid in families enriched for hypertension. Nephrol. Dial. Transplant., 24, 2414–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Voruganti V.S., Göring H.H.H., Mottl A., Franceschini N., Haack K., Laston S., Almasy L., Fabsitz R.R., Lee E.T., Best L.G. et al. (2009) Genetic influence on variation in serum uric acid in American Indians: the strong heart family study. Hum Genet, 126, 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voruganti V.S., Nath S.D., Cole S.A., Thameem F., Jowett J.B., Bauer R., MacCluer J.W., Blangero J., Comuzzie A.G., Abboud H.E. et al. (2009) Genetics of variation in serum uric acid and cardiovascular risk factors in Mexican Americans. J. Clin. Endocrinol. Metab., 94, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. MacCluer J.W., Scavini M., Shah V.O., Cole S.A., Laston S.L., Voruganti V.S., Paine S.S., Eaton A.J., Comuzzie A.G., Tentori F. et al. (2010) Heritability of measures of kidney disease among Zuni Indians: the Zuni Kidney Project. Am. J. Kidney Dis., 56, 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Desideri G., Castaldo G., Lombardi A., Mussap M., Testa A., Pontremoli R., Punzi L. and Borghi C. (2014) Is it time to revise the normal range of serum uric acid levels? Eur. Rev. Med. Pharmacol. Sci., 18, 1295–1306. [PubMed] [Google Scholar]
- 24. Köttgen A., Albrecht E., Teumer A., Vitart V., Krumsiek J., Hundertmark C., Pistis G., Ruggiero D., O'Seaghdha C.M., Haller T. et al. (2013) Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet., 45, 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Charles B.A., Shriner D., Doumatey A., Chen G., Zhou J., Huang H., Herbert A., Gerry N.P., Christman M.F., Adeyemo A. et al. (2011) A genome-wide association study of serum uric acid in African Americans. BMC Med. Genomics, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tin A., Woodward O.M., Kao W.H.L., Liu C.-T., Lu X., Nalls M.A., Shriner D., Semmo M., Akylbekova E.L., Wyatt S.B. et al. (2011) Genome-wide association study for serum urate concentrations and gout among African Americans identifies genomic risk loci and a novel URAT1 loss-of-function allele. Hum. Mol. Genet., 20, 4056–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McArdle P.F., Parsa A., Chang Y.P., Weir M.R., O'Connell J.R., Mitchell B.D. and Shuldiner A.R. (2008) Association of a common nonsynonymous variant in GLUT9 with serum uric acid levels in old order amish. Arthritis Rheum., 58, 2874–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dehghan A., Kottgen A., Yang Q., Hwang S.J., Kao W.L., Rivadeneira F., Boerwinkle E., Levy D., Hofman A., Astor B.C. et al. (2008) Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet, 372, 1953–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Denny J.C., Bastarache L., Ritchie M.D., Carroll R.J., Zink R., Mosley J.D., Field J.R., Pulley J.M., Ramirez A.H., Bowton E. et al. (2013) Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol., 31, 1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adeyemo A.A., Tekola-Ayele F., Doumatey A.P., Bentley A.R., Chen G., Huang H., Zhou J., Shriner D., Fasanmade O., Okafor G. et al. (2015) Evaluation of genome wide association study associated type 2 diabetes susceptibility loci in sub Saharan Africans. Front. Genet., 6, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen G., Bentley A., Adeyemo A., Shriner D., Zhou J., Doumatey A., Huang H., Ramos E., Erdos M., Gerry N. et al. (2012) Genome-wide association study identifies novel loci association with fasting insulin and insulin resistance in African Americans. Hum. Mol. Genet., 21, 4530–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen G., Ramos E., Adeyemo A., Shriner D., Zhou J., Doumatey A.P., Huang H.X., Erdos M.R., Gerry N.P., Herbert A. et al. (2012) UGT1A1 is a major locus influencing bilirubin levels in African Americans. Eur. J. Hum. Genet., 20, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pfister R., Barnes D., Luben R., Forouhi N.G., Bochud M., Khaw K.T., Wareham N.J. and Langenberg C. (2011) No evidence for a causal link between uric acid and type 2 diabetes: a Mendelian randomisation approach. Diabetologia, 54, 2561–2569. [DOI] [PubMed] [Google Scholar]
- 34. Sluijs I., Holmes M.V., van der Schouw Y.T., Beulens J.W.J., Asselbergs F.W., Huerta J.M., Palmer T.M., Arriola L., Balkau B., Barricarte A. et al. (2015) A Mendelian randomization study of circulating uric acid and type 2 diabetes. Diabetes, 64, 3028–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Keenan T., Zhao W., Rasheed A., Ho W.K., Malik R., Felix J.F., Young R., Shah N., Samuel M., Sheikh N. et al. (2016) Causal assessment of serum Urate levels in cardiometabolic diseases through a Mendelian randomization study. J. Am. Coll. Cardiol., 67, 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rotimi C.N., Dunston G.M., Berg K., Akinsete O., Amoah A., Owusu S., Acheampong J., Boateng K., Oli J., Okafor G. et al. (2001) In search of susceptibility genes for type 2 diabetes in West Africa: the design and results of the first phase of the AADM study. Ann. Epidemiol., 11, 51–58. [DOI] [PubMed] [Google Scholar]
- 37. Adeyemo A.A., Zaghloul N.A., Chen G., Doumatey A.P., Leitch C.C., Hostelley T.L., Nesmith J.E., Zhou J., Bentley A.R., Shriner D. et al. (2019) ZRANB3 is an African-specific type 2 diabetes locus associated with beta-cell mass and insulin response. Nat Commun, 10, 3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. National Kidney Foundation (2002) K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am. J. Kidney Dis., 39, S1–S266. [PubMed] [Google Scholar]
- 39. McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A.R., Teumer A., Kang H.M., Fuchsberger C., Danecek P., Sharp K. et al. (2016) A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet., 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loh P.R., Danecek P., Palamara P.F., Fuchsberger C., Reshef Y.A., Finucane H.K., Schoenherr S., Forer L., McCarthy S., Abecasis G.R. et al. (2016) Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet., 48, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Durbin R. (2014) Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics, 30, 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zheng X.W., Levine D., Shen J., Gogarten S.M., Laurie C. and Weir B.S. (2012) A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28, 3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kang H.M., Sul J.H., Service, S.K, Zaitlen N.A., Kong S.Y., Freimer N.B., Sabatti C. and Eskin E. (2010) Variance component model to account for sample structure in genome-wide association studies. Nat. Genet., 42, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu B., Zheng Y., Alexander D., Manolio T.A., Alonso A., Nettleton J.A. and Boerwinkle E. (2013) Genome-wide association study of a heart failure related metabolomic profile among African Americans in the Atherosclerosis Risk in Communities (ARIC) study. Genet. Epidemiol., 37, 840–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adeyemo A., Gerry N., Chen G.J., Herbert A., Doumatey A., Huang H.X., Zhou J., Lashley K., Chen Y.X., Christman M. et al. (2009) A genome-wide association study of hypertension and blood pressure in African Americans. PLOS Genet., 5, e1000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Willer C.J., Li Y. and Abecasis G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ward L.D. and Kellis M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen W., Larrabee B.R., Ovsyannikova I.G., Kennedy R.B., Haralambieva I.H., Poland G.A. and Schaid D.J. (2015) Fine mapping causal variants with an approximate Bayesian method using marginal test statistics. Genetics, 200, 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bretherton C.S., Widmann M., Dymnikov V.P., Wallace J.M. and Blade I. (1999) The effective number of spatial degrees of freedom of a time-varying field. J. Climate, 12, 1990–2009. [Google Scholar]
- 50. Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L. et al. (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res., 42, D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luna A. and Nicodemus K.K. (2007) snp.plotter: an R-based SNP/haplotype association and linkage disequilibrium plotting package. Bioinformatics, 23, 774–776. [DOI] [PubMed] [Google Scholar]
- 52. Speed D., Hemani G., Johnson M.R. and Balding D.J. (2012) Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet., 91, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Speed D. and Balding D.J. (2019) SumHer better estimates the SNP heritability of complex traits from summary statistics. Nat. Genet., 51, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.