

Abstract
Schistosoma spindale and Schistosoma indicum are ruminant-infecting trematodes of the Schistosoma indicum group that are widespread across Southeast Asia. Though neglected, these parasites can cause major pathology and mortality to livestock leading to significant welfare and socio-economic issues, predominantly amongst poor subsistence farmers and their families. Here we used mitogenomic analysis to determine the relationships between these two sympatric species of schistosome and to characterise S. spindale diversity in order to identify possible cryptic speciation. The mitochondrial genomes of S. spindale and S. indicum were assembled and genetic analyses revealed high levels of diversity within the S. indicum group. Evidence of functional changes in mitochondrial genes indicated adaptation to environmental change associated with speciation events in S. spindale around 2.5 million years ago. We discuss our results in terms of their theoretical and applied implications.
Subject terms: Molecular ecology, Phylogenetics, Parasite evolution
Introduction
Schistosoma blood flukes infect over 240 million people and at the very least 165 million cattle worldwide1,2. In the mammalian host the parasite causes chronic disease that leads to wasting, anaemia, fibrosis and inflammatory enlargement of internal organs and even cancer2. Yet, despite their socio-economic importance, species within the Schistosoma indicum group from Southeast Asia are amongst the most neglected of all the schistosomes3. The S. indicum group contains three species: Schistosoma spindale, Schistosoma indicum and Schistosoma nasale, all of which utilise the snail Indoplanorbis exustus as an intermediate host and are widespread across Southeast Asia from India through to the Malay Archipelago4. These schistosome species primarily infect bovids, including domestic cattle and water buffalo, and in endemic areas prevalence can be as high as 100%4.
Schistosoma spindale and S. indicum are considered to be sympatric, often occurring in mixed infections and both causing hepato-intestinal schistosomiasis resulting in reduced milk yield, wasting, as well as liver fibrosis due to the occurrence of granulomas around trapped parasites eggs4. Across the Indian subcontinent these parasites can cause severe mortality, with outbreaks leading to high death rates in herds4. Also, species within the S. indicum group are a major cause of human cercarial dermatitis, as a result of the cercaria penetrating the skin, which has become a significant public health problem for people living in endemic regions5. Furthermore, experimental evidence has shown that schistosomula can migrate to the lungs or central nervous system in ‘incompatible’ mammalian hosts causing severe pathologies beyond cercarial dermatitis6.
Molecular studies have been of great importance in the study of schistosomes, providing vital new information on species identification, population movement, introgression and monitoring of the impact of mass drug administration. Such studies have aided in the focusing of limited resources available for control7–9. However, few molecular studies have been undertaken on members of the S. indicum group, with the majority focused primarily on species identification and phylogenetics using mitochondrial markers, particularly the species barcoding gene the cytochrome c oxidase subunit 1 (cox1), the 16 s and 12 s ribosomal subunit RNA3,10–12. To date, Attwood et al.10 and Devkota et al.11 have provided the most detailed account of the evolution of S. indicum group using phylogenetic comparisons to illustrate the complexity of the radiation of the group across Southeast Asia and noting the high levels of diversity of S. spindale. Devkota et al.11 also utilised cox1, 16 s and 12 s to identify schistosome species parasitizing I. excutus from Nepal, demonstrating a division between S. spindale populations and a potentially undescribed species which was referred to as Schistosoma cf. indicum. The study highlighted the need for continued molecular epidemiological surveys of the S. indicum group but also showed that the group is more diverse than originally thought which could account for heterogeneity in infection and pathology11.
Over the past decade, full mitochondrial genomes have been used for comparative analysis within and between species, predominantly in the Schistosoma mansoni, Schistosoma haematobium and Schistosoma japonicum groups13–15. However, only a single complete mitochondrial genome has been published for S. spindale, using specimens from Sri Lanka which resolved the relationship between the S. spindale and S. haematobium groups15. In this current study we aimed to assess the differentiation between the sympatric parasites S. indicum and S. spindale by sequencing and comparing complete mitochondrial genomes of isolates from Bangladesh with that of the published mitochondrial genome of a S. spindale isolate from Sri Lanka. Furthermore, we assessed the divergence between isolates of S. spindale to clarify the level of differentiation between different populations and identify the suspected occurrence of cryptic species11. Complete mitochondrial genomes of S. spindale from Bangladesh and Sri Lanka were compared to provide genomic insights into the divergence between the north and the south of the Indian subcontinent. We predicted that, overall, the two new mitochondrial genomes of S. spindale and S. indicum would show a great deal of similarity to each other in terms of genome size and structure, owing to the close relationship between these species. We hypothesised that there would be high levels of diversity between the new S. spindale data from Bangladesh and the reference mitochondrial genome from Sri Lanka, with the new sequences being more similar to samples from Nepal in comparative genes, which could be evidence of cryptic speciation in S. spindale.
Results
Divergence across the mitochondrial genomes of S. spindale and S. indicum
Using the published Sri Lankan S. spindale mitochondrial genome (accession: DQ157223) as reference, complete mitochondrial genomes of S. spindale and S. indicum (accession: MN637820 and MN637821 respectively) were assembled and annotated, all showing the derived gene order shared with the African S. mansoni and S. haematobium groups (Fig. 1)13. No major differences in the position or size of the genes were seen. Likewise, although no differences were identified between the Sri Lankan and Bangladeshi S. spindale, there were, however, eight tRNAs with distinct structures observed in S. indicum, which included those carrying the anticodon for cysteine (C), histidine (H), isoleucine (I), asparagine (N), glutamine (Q), arginine (R), tryptophan (W) and tyrosine (Y) (Supplementary Fig. S1).
Figure 1.

Circularised mitochondrial genomes of Schistosoma indicum and Schistosoma spindale from Bangladesh. Graphical representations generated by Genome VX.
Divergence between mitochondrial genomes was assessed by calculating pairwise averaged nucleotide divergence (Dxy) for each of the individual protein coding and ribosomal coding genes. Comparisons were performed between both S. spindale isolates with S. indicum, alongside direct comparisons between the Bangladeshi and Sri Lankan S. spindale. The cytochrome b (cytb) gene had the highest levels of Dxy at 0.17461, with the ATP synthase F0 subunit 6 (atp6) gene showing the lowest level of Dxy at 0.09162 when comparing S. spindale to S. indicum (Fig. 2a). Notably, the cytochrome oxidase subunit 1 (cox1) gene appeared to have the second highest Dxy at 0.15652, all other genes had a Dxy within a narrow divergence range of 0.10752–0.12445. Unlike the 12 s ribosomal gene, which displayed low Dxy at 0.09211, the 16 s gene had a Dxy of 0.12081 comparable to that of the other protein coding genes such as the NADH dehydrogenases (nad1-6) (Fig. 2a). Although, the inter S. spindale comparisons showed lower levels of Dxy, the distribution of divergence across the mitochondrial genome was different with nad5 showing the highest Dxy at 0.13863. Unlike the interspecies comparisons, cox1 also showed low levels of Dxy at 0.09127. However, the ribosomal genes had the lowest levels of Dxy with 16 s at 0.0625 and 12 s at 0.03289 in the inter S. spindale comparison (Fig. 2a). In order to account for the impact of such divergence on genes and their resultant proteins the total number of amino acid replacements per gene was estimated for comparisons of both S. spindale and S. indicum as well as between S. spindale from Sri Lanka and Bangladesh. Although the inter-species comparisons showed a generally higher rate of proportional amino acid replacements of 0.106–0.195 than the inter S. spindale comparison at 0.056–0.161, both comparisons showed nad4L to have the highest proportion of amino acid replacement (Fig. 2b).
Figure 2.
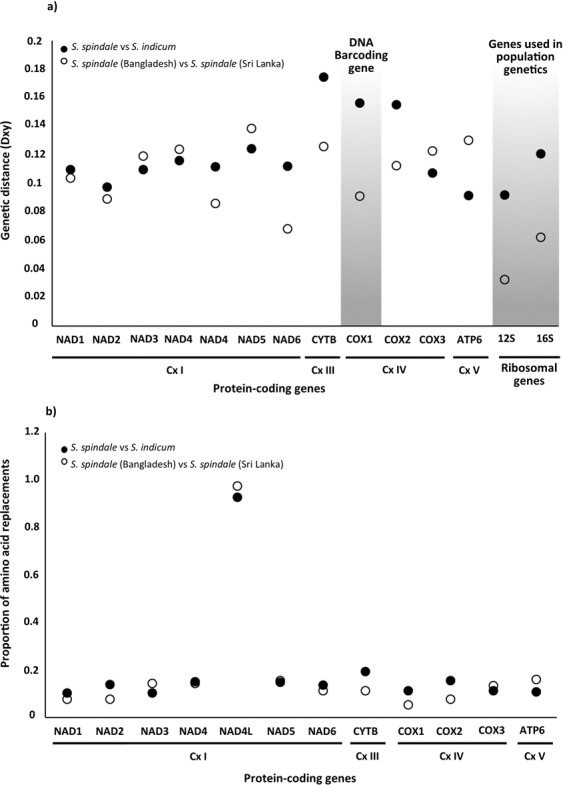
Nucleotide divergence and amino acid replacements in mitochondrial genes between S. indicum and S. spindale, and isolates of S. spindale. Where (a) is nucleotide divergence based on pairwise genetic distance calculated as the average number of nucleotide substitutions per site (Dxy) with blocked areas highlighting the species identification cox1 gene and the ribosomal genes used in population genetic studies. (b) is the proportion of amino acid differences calculated as the total number of replacements within each protein sequence divided by the protein length. The oxidative phosphorylation system complex (Cx) is highlighted.
Owing to the small number mitochondrial genomes being compared in this study it was not possible to use standard population genetic methods to detect the occurrences of positive selection on each of the genes within and between species. Nevertheless, to account for the levels of amino acid replacements seen across the mitochondrial genomes indirect assessment of selection were performed by assessing the potential impact of amino acid changes and identifying the regions within the proteins that these mutations occur. Provean15, protein variation effect analyses, identified a total of 33 functionally altering amino acid replacements across nine proteins when the Bangladeshi S. spindale was compared to S. indicum, and 29 in Sri Lankan S. spindale and S. indicum comparison (Supplementary Table S1). However, only nine functionally altering amino acid replacements were found across five proteins when the Bangladeshi and Sri Lankan S. spindale were compared (Supplementary Table S1). Overall, when the proportion of functionally altering amino acid changes were calculated per gene, cytb showed the highest proportion of functional mutations followed by nad4L in comparisons between S. spindale and S. indicum and between the S. spindale isolates (Fig. 3a). Also, the NADH dehydrogenases appeared to have the highest proportion of function altering mutations relative to other genes excluding those discussed above. Interestingly, the cox1 gene showed the lowest level frequency of function altering amino acid replacements between S. indicum and S. spindale, and none were detected in the intra-species comparisons (Fig. 3a). The TMHMM16 analyses indicated that the majority of the functional altering amino acid replacements occurred within the transmembrane domains of the proteins and not within the functional domains exposed on either the outer or inner surface of the mitochondria (Fig. 3b). However, in the comparisons of S. spindale and S. indicum there were two amino acid replacements in the region of the protein exposed to the mitochondrial matrix inside the mitochondria in cytochrome oxidase subunit 2 protein (cox2), three found in cytb, two in nad1, one in nad5 and a further two in nad1. Such replacements were not as extensive in the inter S. spindale comparisons with only four alterations being found in the domains exposed to the mitochondrial matrix, two of them in cox2 and the others in cytb (Fig. 3a,b; Supplementary Table S1). Fewer alterations were found within domains of the protein exposed to the surface of mitochondria. When S. spindale and S. indicum were compared, only four such alterations were identified, a single alteration in cox1, cytb, nad4 and nad5. Notably, in the inter S. spindale comparisons a single alteration was found in cytb (Supplementary Table S1).
Figure 3.
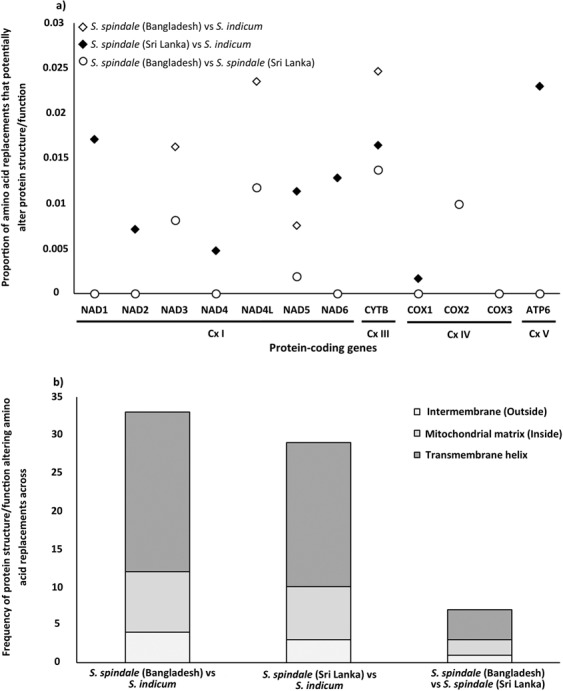
Proportion of amino acid changes with functional effects on mitochondrial proteins and the frequency and locality of replacements across the protein in comparisons between S. indicum and S. spindale, and isolates of S. pindale. Where (a) is the proportion of amino acid replacements identified to have a potential functional change to the resultant protein as predicted by Provean26. (b) is the frequency of potential functional changing replacements found within the different domains of the mitochondrial proteins as identified by TMHMM16 searches.
Phylogenetic reconstruction of the S. indicum group
Owing to the paucity of comparable mitochondrial genomic data for the S. indicum group, cox1, 16 s and 12 s gene markers were employed to provide a phylogenetic perspective of the group, consistent with that employed to date across the majority of published studies and hence the largest comparable data sets available (Supplementary Tables S2–S4). Initial calculations of phylogenetically informative sites showed the cox1 to have 299 out of 985, the 16 s 237 out of 632, and the 12 s 76 out of 333 and subsequent maximum likelihood (ML) phylogenetic reconstruction (Fig. 4ai–ci) showed similar overall topologies with well supported clades with nodal bootstrap values > 50 for each of the gene markers. Each phylogeny showed S. nasale to be the most basal within the group and S. spindale the most derived taxa. In all phylogenies there was a clear separation between S. cf. indicum and S. indicum. Topologies of the cox1 and 16 s phylogenies appeared identical with S. cf. indicum resolving as an intermediate between S. spindale and S. indicum (Fig. 4ai–bi). However, in the 12 s analyses S. cf. indicum appeared as a paraphyletic sister taxon to an S. indicum and S. spindale clade. Clear distinction was identified between the S. spindale populations which split into two well supported clades with each tree producing a distinct Nepal/Bangladesh and a Sri Lanka/Thailand/Malaysia cluster, with the exception of the 16 s phylogeny which showed a single sequence from Bangladesh within the Sri Lanka/Thailand/Malaysia lineage. Although phylogenetic trees constructed using Bayesian inferences (BI) produced well supported phylogenies with high posterior nodal support values and broadly illustrated the same over relationships as the ML analyses, only the 12 s phylogenetic topologies were identical between analyses (Fig. 4ci,cii). There were notable differences in the relationships between S. indicum and S. cf. indicum, and unlike the cox1 and 16 s ML phylogenies, the BI analyses resolved S. indicum in different phylogenetic positions, showing S. indicum to be a sister taxa to S. cf. indicum in the cox1 phylogeny, with the 16 s showing S. indicum to share a distinct clade with S. nasale (Fig. 4ai and ii,bi and ii). Similarly, the 16 s BI analyses also showed the S. cf. indicum to be a sister taxa to S. spindale. The BI phylogenies showed the same relationships between S. spindale isolates as the ML analyses across all three markers with the prominent two major clusters occurring as described above (Fig. 4a–c).
Figure 4.

Maximum likelihood and Bayesian inference phylogenies of mitochondrial genes from the Schistosoma indicum species group. Schistosoma incognitum was employed as the outgroup. Were (a) are phylogenies constructed using the cox1 gene using the HKY + G + I model for both ML (ai) and bi (aii) analyses; (b) are phylogenies constructed using the 16 s ribosomal sequence using the GTR + G model for both ML (bi) and bi (ii) analyses; (c) are phylogenies constructed using the 12 s ribosomal sequence HKY + G for both ML (ci) and bi (cii) analyses. Nodal support values are provided by 1000 bootstrap replicates in the ML analyses and posterior nodal values for the bi analyses. The position of Schistosoma indicum is highlighted in blue, with the Schistosoma spindale cluster from Sri Lanka/Thailand/Malaysia being highlighted in pink, and the S. spindale cluster from Nepal/Bangladesh highlighted in green.
Due to the low levels of divergence identified within Nepal/Bangladesh and Sri Lanka/Thailand/Malaysia clades, single representation of cox1 sequences were used from the complete mitochondrial genomes of S. spindale from Sri Lanka and Bangladesh, along with those from S. indicum, in order to estimate divergence times within the S. indicum group using BEAST17,18. Owing to the lack of sequence data for the S. indicum group, a range of schistosome species were used to aid the estimation of divergence times across Schistosoma available cox1 data (Supplementary Table S5), as well as sequences for Bivetellobilharzia nairi, Schistosomatium douthitti and Trichobilharzia regenti, the latter of which were used as the outgroup. The cox1 marker has been historically used to calculate divergence time between schistosome species and populations, and the use of other Schistosoma species allowed the calibration of the clock analyses using predicted divergence times between S. mansoni and S. japonicum, S. incognitum and S. mansoni, and S. mansoni and S. haematobium as highlighted by Lawton et al.19. This approach allowed time to the most recent common ancestor (TMRCA) for each node to be calculated across the schistosome phylogeny. Effective sample size (ESS) was in excess of the 200 thresholds, providing reliable posterior probabilities and a greater level of statistical certainty of estimated divergence times17,18. The Bayesian timetree produced by BEAST provided the accepted standard phylogenetic topology of the schistosomes as highlighted Lawton et al.19. However, in the time tree S. nasale appears as a sister taxon to the S. haematobium group rather than clustering with the rest of the S. indicum group as also seen in the cox1 phylogeny produced by Devkota et al.11. Thus, the clock is considered only to be a conservative estimate of divergence time for the group and predicts the emergence of the ancestor of the S. indicum group approximately 9.35 million years ago (MYA). The timetree resolved S. spindale and S. indicum as sister taxa and estimated their divergence at approximately 6.75 MYA with S. indicum from Bangladesh and S. cf. indium from Nepal predicted to have diverged from each other approximately 5.74 MYA. However, the divergence of the two S. spindale lineages was estimated to have occurred much more recently around 2.51 MYA (Fig. 5).
Figure 5.

Schistosoma Bayesian timetree based on molecular clock analyses. Molecular clock created in BEAST showing the mean time of most recent common ancestor (MRCA) for all available schistosome species (blue) and the posterior probabilities (red). The white bars at each node show the highest 95% posterior density (HPD) interval for the main nodes an indicator of the estimated range of time of divergence. Molecular clock predicts divergence of the S. indicum group at 9.35 MYA. This molecular clock predicts the S. indicum group to be paraphyletic with S. nasale as the most derived of the group and separate from the other species. The MRCA between the two populations of S. spindale is predicted to be approximately 2.51 MYA.
Divergence in schistosoma spindale
Haplotype network analyses of S. spindale mirrored the relationships identified in the phylogenetic analyses, where the cox1 and 16 s showed two distinct clusters, the first comprising of haplotypes from Nepal and Bangladesh, with haplotypes being shared between both localities (Figs. 4a,b and 6a,b). The other cluster contained sequences from Sri Lanka and Thailand or Malaysia, but within the 16 s haplotype network a single sample from Bangladesh clustered with the Sri Lankan and Thai samples. Despite the occurrence of fewer mutations between sequences, the network produced for 12 s showed a similar clustering of the haplotypes but separated Thailand haplotypes from those of Sri Lanka more readily, which was also reflected in the occurrence of separate Sri Lankan, Thai and Nepal/Bangladesh clusters in the ML phylogenetic reconstruction (Figs. 4c and 6c).
Figure 6.
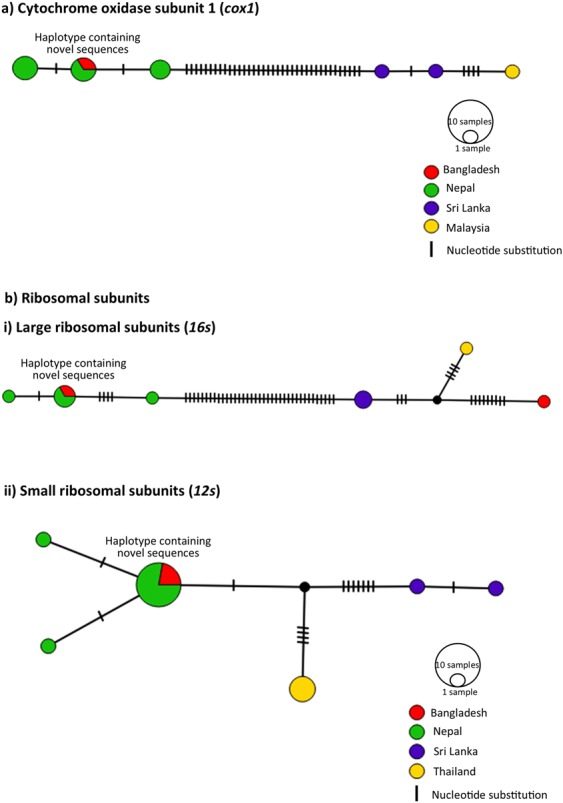
Population structuring of S. spindale. Haplotype networks created in PopART using TCS network method, with country of origins represented as colours and size of circle indicating number of sequences in single haplotype. Dashes represent single mutations.
Both the cox1 and 16 s networks revealed substantial divergence between the Nepal/Bangladesh and a Sri Lanka/Thailand/Malaysia clusters. In order to account for the occurrence of any potential speciation events, uncorrected p-distances between sequences were calculated as a measure of divergence. When all S. spindale sequences were assessed, there were elevated levels of divergence based on average uncorrected p-distance with cox1 showing 4.25% and 16 s showing 4.27% divergence (Table 1). Despite the occurrence of three separate linages in 12 s, the average divergence was relatively low compared to the other markers (1.24%). However, when divergence was compared between lineages, the Nepal/Bangladesh and Sri Lanka/Thailand/Malaysia clusters the cox1 possessed the highest level of divergence (8.9%) followed by the 16 s with a divergence of 7.3% (Table 1).
Table 1.
Uncorrected P distance calculation within and between Schistosoma spindale populations.
| Comparisons | Genes | |||
|---|---|---|---|---|
| Cox1 | 16s | 12s | ||
| Within clades | All | 0.0425 | 0.0427 | 0.0123 |
| Bangladesh/Nepal clade | 0.0058 | 0.0056 | 0.0027 | |
| Sri Lanka/Malaysia clade | 0.0092 | na | na | |
| Sri Lanka/Thailand/Bangladesh clade | na | 0.0169 | na | |
| Sri Lanka clade | na | na | 0.003 | |
| Thailand clade | na | na | 0 | |
| Between clades | Bangladesh/Nepal clade vs Sri Lanka/Malaysia clade | 0.089 | na | na |
| Bangladesh/Nepal clade vs Sri Lanka/Thailand/Bangladesh clade | na | 0.073 | na | |
| Sri Lanka clade vs Thailand clade | na | na | 0.032 | |
| Sri Lanka clade vs Bangladesh/Nepal clade | na | na | 0.028 | |
| Thailand vs Bangladesh/Nepal clade | na | na | 0.016 | |
The data indicates that the S. spindale population from Nepal and Bangladesh may be a separate species to the population from the rest of Southeast Asia as there is a high level of distance seen within the species as a whole yet when each population is analysed separately this level drops dramatically and the distance between the populations is comparatively high.
The same pattern was reflected in the automatic barcode gap discovery (ABGD)20 analyses used to determine molecular operational taxonomic units as potential species. When assessing the complete data for the S. indicum group, five distinct groups were identified within cox1 and 16 s data sets at the species cut-off value of 0.0359 (>3% divergence based on uncorrected p-distance) indicating five separate species within the S. indicum group: S. nasale, S. indicum, S. cf. indicum, with S. spindale split into two distinct species groups (Sri Lanka/Malaysia and Bangladesh/Nepal). Conversely, although the 12 s resolved differences between S. nasale, S. indicum, and S. cf. indicum it did not separate S. spindale into two distinct taxonomic units. In order to validate the findings described above the Birky21 4X ratio was applied to provide a lineage specific perspective of species delimitation rather than depending on average divergence estimations based on gross pairwise differences between DNA sequences. In contrast to the previous analyses, the 4X ratio calculations failed to differentiate S. spindale in to distinct species and resolved all sequences from across Southeast Asia as a single species, indicated by all values of K/θ = <4 (cox1- 2.45, 16s- 2.78 and 12s- from 0.87–1.63) (Table 2).
Table 2.
4X rule calculation for species delimitation.
| Gene | Cox1 | 16s | 12s | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Clade | Nepal/Bangladesh | Sri Lanka/Malaysia | Nepal/Bangladesh | Sri Lanka/Malaysia | Nepal/Bangladesh | Thailand | Nepal/Bangladesh | Sri Lanka | Thailand | Sri Lanka |
| n | 8 | 3 | 5 | 4 | 11 | 3 | 11 | 2 | 3 | 2 |
| K | 0.105 | 0.065 | 0.016 | 0.024 | 0.03 | |||||
| d | 0.003 | 0.009 | 0.003 | 0.015 | 0.001 | 0 | 0.001 | 0.003 | 0 | 0.003 |
| π = d*(n/n − 1) | 0.0034 | 0.0135 | 0.0038 | 0.0227 | 0.0011 | 0.006 | 0.0011 | 0.006 | 0 | 0.006 |
| θ = π/(1 − 4π/3) | 0.0104 | 0.0428 | 0.0037 | 0.0233 | 0.0033 | 0.0184 | 0.0033 | 0.0184 | 0 | 0.0184 |
| K/θ | 2.451 | 2.7809 | 0.8676 | 1.3013 | 1.6267 | |||||
Calculations used to delimit species under the 4X rule (Birky, 2013) for cox1, 16s and 12s show that under a conservative model the two S. spindale population represent a single species as all results for K/θ are below 4, with the exception of the 12s populations from Thailand and Sri Lanka. Where n = numbers of sequences; K = total pairwise differences between clades; d = pairwise differences within clades.
In order to account for the cause of the divergence between the S. spindale isolates, alterations in physiochemical properties of the cox1 sequences were identified as an indicator of the impact of codon specific selection across the S. spindale phylogeny using TreeSAAP22. By utilising a sliding window approach, TreeSAAP22 compared the total number of observed amino acid replacements relative to those evolving under neutrality across the S. spindale cox1 phylogeny. The analyses detects the occurrence of 20 physiochemical properties across eight categories, where 1–3 are considered to have little impact on the resulting protein, 4–5 having medium effect, and 6–8 having a substantial effect, altering the biochemistry of the protein and considered to be evidence of codon specific positive selection. Only seven physiochemical alterations appeared to have occurred within the cox1 protein, with four possessing z-scores > ±3.09 with P < 0.001. The properties which had the most profound effect were in category level 7 and were alterations in isoelectric point (z = 4.248) and short medium range non-bond energy (z = 3.995) (Table 3). Category level 6 alterations were also shown for partial specific volume (z = 4.543) and helical contact area (z = 3.85). This is illustrative of the phenotypic impacts of the mutations between the S. spindale isolates and relates the divergence between isolates to potential subtle local adaptive differences. When the same analyses were performed for S. indicum and S. cf. indicum cox1 no significant differences were identified (data not shown).
Table 3.
Property showing signs of adaptive changes in Schistosoma spindale cox1.
| Property | Categories (Z-scores) | ||
|---|---|---|---|
| 6 | 7 | 8 | |
| Equilibrium constant (ionization of COOH) | 1.792* | ||
| Isoelectric point | 4.248*** | ||
| Short and medium range non-bonded energy | 3.995*** | ||
| Solvent accessible reduction ratio | 2.168* | ||
| Power to be at the C-terminal | −1.78* | ||
| Partial specific volume | 4.543*** | ||
| Helical contact area | 4.385*** | ||
TreeSAAP results for cox1 analysis of the two S. spindale samples reveal that isoelectric point and short and medium range non-bonded energy show the most prominent signs adaptation. Only changes with P < 0.001 (***) are regarded as highly significant and useful in terms of adaptation. These adaptive changes in isoelectric point have been previously linked which adaptive changes to abiotic pressures.
Discussion
Comparative analyses of the complete mitochondrial genomes of S. spindale and S. indicum revealed that, although variation and diversity exist, nucleotide divergence (Dxy) is reasonably low between species (Dxy = 0.09–0.17). Interestingly, with the exception of cox1, nad6, atp6, 16 s and 12 s the intra-species comparison of the Bangladeshi and Sri Lankan S. spindale broadly showed the same regions of the mitochondrial genome to display the same patterns of divergence as that seen between S. spindale and S. indicum. Such patterns have been suggested to reflect the major processes driving species level divergence as illustrated with the S. japonicum group14 and in comparisons of S. japonicum populations between mainland Asia and surrounding Islands14,23. However, owing to the sympatric nature of host-sharing in S. spindale and S. indicum, divergence in such regions of the mitochondrial genomes could be indicative of the establishment of speciation barriers, giving rise to alterations in the function of genes and ribosomal structures especially tRNAs24,25. Although it was not possible to assess the occurrence of positive selection across the mitochondrial genomes using standard population genetic approaches in this study due to low sample size, amino acid replacement analyses did illustrate high rates of substitution. Likewise, the Provean26 and TMHMM26 analyses highlighted a number of potentially function altering amino acid changes in regions of the peptides directly involved in the electron transport machinery of the mitochondria. Although most of the genes required for mitochondrial function are encoded by the nucleus, genes for five enzymatic complexes have been retained in the mitochondria in Schistosoma13. These complexes include those composed of the genes which regulate oxidative phosphorylation (OXPHOS) processes including complex 1 (Cx I) containing nad1-6, complex 3 (Cx III) containing cytb, complex 4 (Cx IV) containing cox1-3, and complex 5 (Cx V) comprising of the single atp6 gene responsible for ATP synthesis27. In our study, Cx I and Cx III were shown to contain the majority of amino acid replacements potentially affecting protein function, illustrating the signature of positive selection. Although the majority of mutations were found in transmembrane regions of proteins, which may result in subtle alterations in protein shape, the inter-species comparison of cytb, nad1 and nad4L showed the highest proportion of function altering changes, with cytb and nad1 possessing mutations in the protein region directly within the mitochondrial matrix, potentially affecting the efficacy of the electron transport chain. Several studies have shown similar patterns of mutation in Cx I and Cx III invertebrates that have been linked to not only local environmental adaptation within species but also the establishment of cyto-nuclear incompatibilities between closely related sympatric species and ultimately causing hybrid break down24,25.
Ribosomal genes have also been implicated in translational deficiency in hybrids, as nuclear ribosomal subunits required for their translation are highly divergent between species. This results in the inability of accurate transcription and translation of the mitochondrial ribosomal subunits and tRNAs, with the subsequent failure of mitochondrial function in hybrid offspring25. Such a process could account for the alternative tRNA structures observed between S. spindale and S. indicum; tRNA function is structure-dependent and single nucleotide mutations in the anticodon loops of mitochondrial tRNAs can reduce OXPHOS capacity owing to reduced translational efficacy in hybrids due to species specific mutations28,29. Hybridisation events among schistosomes are becoming well documented, however, there are no detailed studies of the phenomenon within the S. indicum species group30. Despite their sympatric nature, inter-species mating between S. spindale and S. indicum and the production of viable hybrid eggs have been recorded only under laboratory conditions31. Therefore, alterations within the structure of the tRNAs alongside the divergence within the OXPHOS Cx I and Cx III, may be indicative of the development of initial cyto-nuclear speciation barriers between S. indicum and S. spindale. However, it is important to note that in order to truly disentangle the mechanism of speciation processes in the S. indicum group, increased sampling and genome sequencing of S. indicum and S. spindale from a range of geographical locations across Asia is essential.
Speciation across the Schistosoma genus is highly complex with little known about the processes and mechanisms affecting inter-species divergence or intra-species. The phylogenetic analyses here support that of Devkota et al.11 showing clear distinctions between S. indicum and S. cf. indicum, as well as between populations of S. spindale. With the exception of the BI 16 s phylogeny all other analyses showed S. nasale to be basal within the S. indicum group forming a paraphyletic sister clade to the closely related S. indicum, S. cf. indicum and their sister taxa S. spindale. Close evolutionary relationships between the S. indicum and S. haematobium groups appears to be associated with both groups having evolved to infect ungulates. The molecular clock predicted the divergence of the S. indicum group in the late Miocene, approximately 9.35 MYA, substantially earlier than previous predictions of approximately two million years ago during the major mammal migration from Africa to Asia10. This coincides with a major change in plant flora across Africa as a result of reductions in temperature, which lead to the domination and radiation of bovids around 7–15 MYA32. Interestingly, Gauffre – Autelin et al.33 indicated that Indoplanorbis is also most likely to have an African origin and diverged from Bulinus, the intermediate host for the S. haematobium group, in the early Miocene, 17–38 MYA. Indoplanorbis would have then undergone a dispersal along the middle eastern land connection before radiating across Asia from the mid to late Miocene 5–15 MYA. This Indoplanobis dispersal event coincides with the mass movement of animals back and forth between Africa and Eurasia32 providing a potential mechanism for the proto-S. indicum group to leave Africa and disperse into Asia. The molecular clock analyses predicted the divergence of S. spindale and S. indicum at approximately 6.74 MYA with S. indicum and S. cf. indicum diverging approximately 5.74 MYA The divergence event between S. spindale and S. indicum coincides with the late Miocene period of tectonic and climatic instability; during this period there would have been a rapid uplift of the Qinghai–Tibetan Plateau which would have initiated and subsequently intensified the monsoons. This event would have isolated several bovid species carrying ancestral parasite populations giving rise to S. spindale and S. indicum. Again, the timing of this radiation event coincides with a major diversification event of Indoplanorbis across Asia33. It could be suggested that proto-S. indicum would have become isolated on the Indian subcontinent and rapidly radiated across the continent giving rise to S. indicum in India, Bangladesh and Pakistan and S. cf. indicum in Nepal. This may account for the specificity of S. indicum on the Indian subcontinent and its absence elsewhere across South East Asia, but it is clear that there is a direct association between the movement of definitive bovids hosts, the radiation of Indoplanorbis as an intermediate host and the diversification of the S. indicum group.
Comparisons between S. spindale from Bangladesh and Sri Lanka here showed distinct nucleotide divergence across the mitochondrial genome with some genes showing similar levels of divergence as seen between S. spindale and S. indicum. Similarly, both phylogenetic and haplotype network reconstruction highlighted clear separation between S. spindale populations producing Nepal/Bangladesh and a Sri Lanka/Thailand/Malaysia clusters. When several methods of species delimitation were applied to the data mixed results were obtained. The ABGD analyses indicated the presence of two species and p-distance calculations favoured patterns that indicate speciation in S. spindale with intraspecific divergence below 1% and interspecific divergence around 10X that of intraspecific34. Conversely the K/θ ratio method failed to predict separate species21. Such divergence between populations has been shown to occur in other animal groups and is considered to represent evidence of a speciation event in action35–38. These patterns are the result of vicariance where populations have become isolated as a result of a geographical barrier, leading to rapid speciation, which can be followed by secondary contact of populations before full speciation barriers have arisen35–38. The molecular clock estimated divergence between the two clusters of S. spindale in the late Pliocene-early Pleistocene, around 2.5 MYA, a time when sea levels were lower and most of the Southeast Asian islands were part of the continental landmass39. This may account for the close relationship between parasites from the Sri Lanka, Malaysia and Thailand, as the ancestral population would have moved freely across the region with their hosts40. The timing of this divergence also coincides with the last major uplift of the Himalayas, of between 2200–3400 metres since the Pliocene, providing the vicariance event required to separate the S. spindale populations41. However, it is important to consider that the relationships between S. spindale populations within the Sri Lanka/Thailand/Malaysia cluster may in fact result from recent invasion events due to the movement of cattle between these regions as a result of human migration and trade in livestock10. This could account for the 16 s Bangladeshi haplotype which fell within the Sri Lanka/Thailand/Malaysia cluster and the general lack of diversity.
Although substantial amino acid replacements were identified across the peptides produced by mitochondrial genes when the S. spindale isolates were compared, only eight replacements across four genes appeared to have any potential functional effect based on the Provean and TMHMM analyses. As with the inter-species comparison, genes within Cx I and Cx III appeared to have the highest number of functional replacements with cytb having the highest proportion of such mutations overall. Replacements were found in both regions of the cytb proteins that directly interact with the internal matrix and those that are exposed to the intermembrane on the surface of the mitochondria. Again, owing to the high level of conservation in the cytb protein due to its functional role in creation of proton gradient and electron transfer to Cx IV mutation in this protein would have profound effect of the efficiency of the respiration processes42. Interestingly, the cox 2 gene of Cx IV also showed functional amino acid changes in its peptide in the regions also exposed to the internal mitochondrial matrix. Such alterations have been shown to reduce coupling efficiency of the OXPHOS across the electron transport chain in other animals and have been linked to adaptations to colder climates42,43.
Although not detected by the Provean26 analyses conducted on the genes from the complete mitochondrial genomes, when cox1 was compared between the Nepal/Bangladesh and Sri Lanka/Thailand/Malaysia clusters of S. spindale substantial nucleotide differences were identified, several of which were non-synonymous. TreeSAAP22 revealed a number of profound changes in physiochemical properties including isoelectric point, which has been linked with adaptation to low temperature or high-altitude environments44–49. Alterations in isoelectric point can cause changes in the folding of the cytochrome oxidase subunit proteins, resulting in proton leakage making the OXPHOS pathway less efficient50. Both analyses from the complete mitochondrial genomes of S. spindale and the inter population variation analyses of the Nepal/Bangladesh and Sri Lanka/Thailand/Malaysia clusters are indicative of adaptation to colder climates or hypoxic environments, as a less efficient OXPHOS pathway results in increased metabolic heat47, and increases expression of hypoxia specific genes49,51. Such adaptations have been noted in a number of organisms from similar environments to that of the Nepalese/Bangladeshi S. spindale, particularly from the Tibetan plateau, such as the plateau pika and Tibetan horse and antelope45,46,52. Evidence of similar adaptation has also been seen in Schistosoma turkestanicum inhabiting colder temperate climates53. This provides further evidence of the occurrence of a vicariance event separating the S. spindale linages. Thus, the data shown here supports a speciation event captured in process with the Nepal/Bangladesh cluster initially isolated by the up lift of the Nepalese plateau, and a Sri Lanka/Thailand/Malaysia cluster which most likely represents the S. spindale that radiated across South East Asia. However, further sampling, detailed assessment of host use and deeper genetic analyses is required in order to resolve the Nepal/Bangladesh and a Sri Lanka/Thailand/Malaysia clusters as distinct species.
Diversity and speciation events in Schistosoma lack detailed understanding, and this is especially true for the S. indicum group and its members. Thus, the prevalence of each species, information which is vital in planning effective treatment regimes, and the development and monitoring of control programmes, remains uncertain. Mitochondrial molecular markers have been shown to be vital in the identification of different schistosome species and for providing initial insights into the evolution and epidemiology of parasite populations. Although it is preferable to use complete mitochondrial genomes to address evolutionary issues within and between members of the S. indicum group, a paucity in published comparative molecular data exists. The cox1, 16 s and 12 s can provide a reliable species identification and initial insights in population biology of the S. indicum group that could inform control procedures and aid in assessing the impact of these parasites on food security and socioeconomic status of communities that depend on farmed animals, as well as the public health implications the S. indicum group poses to the people within these communities54.
Materials and Methods
Parasite material and DNA extraction
Adult S. spindale and S. indicum samples were collected from cattle and goat carcasses from local meat markets in Bangladesh as part of an ongoing study by the University of Agricultural and Veterinary Sciences, Bangladesh. Worms were identified morphologically based on the number of testes in males and ovaries in females and ten paired worms of each species were sent to Kingston University for genome analyses. Owing to low DNA concentrations from extractions of individual worms, pools of five males were used for DNA extraction for each species. Sample DNA was extracted using the Qiagen DNeasy blood and tissue extraction kit (Qiagen Inc.).
Mitochondrial genome sequencing, assembly and annotation
The DNA samples were submitted to the Natural History Museum (London) sequencing facility to be sequenced using Illumina MiSeq next generation sequencing (NGS) (Illumina, Inc). Each DNA extract was indexed, and libraries prepared for NGS sequencing using the TrueSeq Nano DNA Sample Preparation Kit (Illumina, Inc). Both the S. indicum and S. spindale libraries were then run independently on a MiSeq Illumina sequencer (pipeline 1.9) yielding approximately 300 bp long paired-end reads. The resultant DNA sequence reads were then analysed using CLC genomics workbench 7.7.5 (https://www.qiagenbioinformatics.com/). As with other helminths such as Paragonimus westermani55, Fasciolopsis buski56, Dirofilaria species57 and Trichuris species58 reads were trimmed and adaptors removed using CLC, based on quality reports of length distribution, GC content and nucleotide contribution. The refined sequence reads were then used to assemble the mitochondrial genomes of S. spindale and S. indicum by mapping sequences to the published Sri Lankan S. spindale mitochondrial genome using parameters specified (Supplementary Fig. S2). There were no allelic forms identified in the mapped reads for either S. indicum or S. spindale respectively, and once assembled each novel genome sequence was locally realigned and the consensus was extracted for further analysis. Sequences were annotated based on homology with the Sri Lankan reference sequence, allowing the identification of individual genes and coding regions in the newly assembled genomes. MITOS was used to confirm genome annotation and produce secondary tRNA structures by uploading mitochondrial genomes to the server (http://mitos.bioinf.uni-leipzig.de/index.py). GenomeVX was used to create a circularized graphical representation of the mitochondrial genome (http://wolfe.ucd.ie/GenomeVx/) with positions of all genes specified based on the annotations from the genome assembly.
Measuring divergence and estimating phylogenetic relationships of the S. indicum group
To assess the level of divergence between S. indicum and S. spindale the mitochondrial genomes of S. spindale and S. indicum from Bangladesh Sri Lankan S. spindale were aligned against each other using MUSCLE (https://www.ebi.ac.uk/Tools/msa/muscle/). Sequences for individual genes within the OXPHOS complexes were extracted from the alignments and pairwise average nucleotide divergence (Dxy) was calculated for both inter and intra species comparisons using DNASP v5.1059. As only a few complete mitochondrial genomes are available for the S. indicum group, it was not possible to accurately measure the occurrence of positive selection across the mitochondrial genome based on the ratio of synonymous and non-synonymous mutations to provide initial insight into the divergence between parasite species and isolates. Instead indirect measures were performed which would allow the comparison of differences between resultant protein peptide sequences. The proportion of amino acid replacements per gene was estimated for comparisons between S. spindale and S. indicum, as well as between both isolates of S. spindale. In both cases total number of amino acid differences between each protein was calculated in MEGA version X60, and then divided by the total number of amino acids within the protein produced by each gene. To assess the functional effect of each amino acid change Provean26, the protein variation effect analyser was used to assess differences both within and between species. By evaluating each protein sequence variation in an evolutionary context Provean26 predicts the likelihood of an amino acid substitution having a functional effect on the protein. The default confidence threshold of −2.5 was used as recommended by Choi and colleagues15 to determine if an amino acid replacement was likely to have a functional effect on the resultant protein. In order to identify the region of the protein that these amino acid replacements occurred within domains were predicted for each protein using TMHMM26 webserver (http://www.cbs.dtu.dk/services/TMHMM/) which employs a hidden Markov model to predicted transmembrane regions within a peptide sequence. Mitochondrial proteins are considered to have three separate domains: (i) those exposed to the inside of the mitochondria and interact directly with the mitochondrial matrix; (ii) those within the mitochondrial membrane providing structural support for the protein; (iii) those that are on the surface of the mitochondria and between the inner and outer membrane. Owing to their functional roles in regulating the electron transport chain it is considered that the OXPHOS complexes are under strong purifying selection, thus any amino acid replacements within the domains of such proteins could indicate alterations in function that has occurred as a result of positive selection. Subsequently, the proportion of putative function altering amino acid replacements were was also calculated between S. spindale and S. indicum as well as between the Bangladeshi and Sri Lankan S. spindale isolates.
Owing to the lack of complete mitochondrial genomes of other isolates of S. spindale and S. indicum, to resolve the phylogeny of the S. indicum group and disentangle inter and intra species relationship the cox1, 12 s and 16 s sequences were retrieved from the mitochondrial genomes and compared to published data representing species and isolates from across Asia (Supplementary Tables S2–4. Again, alignments were performed using MUSCLE and ML phylogenies were constructed for each gene using MEGA version X60. For each alignment nucleotide substitution models were also calculated using MEGA with the HKY + G + I indicated for the cox1 alignment, the GTR + G for 16 s and the HKY + G for the 12 s. All maximum likelihood phylogenetic analyses used 1000 bootstrap replicates to provide nodal support and owing to its historical taxonomic association with the S. indicum group and utility in other studies10,11 Schistosoma incognitum was used as an outgroup to provide a root for each of the S. indicum group phylogenies. For comparison, phylogenetic relationships were also reconstructed using Bayesian inferences (BI) implemented through BEAST v.2.4.6 for each of the gene markers. The same substitution models were used as described above and for each of the genes an MCMC chain length of 107 and a pre-burn-in of 106 was used to construct the trees.
In order to estimate times of divergence within and between members of the S. indicum group Bayesian molecular clock analyses were also performed using BEAST with known cox1 schistosome and trematode evolutionary rates of 0.035 ± 0.0071 substitutions per million61,62. In order to provide the most detail account of the divergence of the S. indicum group a full phylogenetic analyses of all Schistosoma species was performed with the the HKY + G + I substitution model again with an MCMC chain length of 107 and a pre-burn-in of 106. A strict clock approach was taken using conditions of the Yule model for speciation; an approach used in previous analysis of schistosome divergence62 and was calibrated based on published divergence time estimates between S. mansoni and S. japonicum, S. mansoni and S. incognitum, S. mansoni and S. haematobium19. Tracer 1.3 was used to check the effective sample size (ESS) was above the 200-threshold value as an indication of exploration of parameter space63. In order to summarise the posterior distribution of trees output by BEAST, the maximum clade credibility (MCC) tree was obtained using TreeAnnotator (part of the BEAST suite of programs) with a burn-in percentage of 10% and no posterior probability limit. The MCC tree was manipulated in FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/) where node ages and posterior probabilities could be assigned, and the visual appearance of the tree could be edited.
Evolutionary analyses of Schistosoma spindale
Owing to the high levels of divergence identified between isolates of S. spindale in previous studies and across the mitochondrial genome in this current study, detailed analyses between S. spindale isolates were performed based on reanalyses of the few published DNA sequences from across Asia, whilst incorporating the sequence data generated in the present study. Initially, in order to identify the occurrence of shared mitochondrial haploytpes of cox1, 12 s and 16 s between localities, as well as to determine which haplotypes clustered together more readily, the most parsimonious haplotype networks were generated using TCS as implemented through PopART64. Although only a few sequences for each gene could be used the analyses provided initial insights into divergence of S. spindale, providing perspective of any underlying population structure.
Three methods of species delimitation were utilised to identify the number of species that were represented within the sampled sequences. Firstly, uncorrected p-distance values were obtained for both within and between species as a measure of percentage of divergence for both intra and inter species assessments. This was done for each marker sequence alignment using MEGA. This allowed the identification of pairwise comparisons between isolates which were 3% or higher, which is often used as the minimum level of divergence to differentiate between species using mitochondrial markers, especially cox1 as the recognised barcoding gene65. Secondly, each of the alignments were submitted to the ABGD server20 to independently identify the number of molecular taxonomic units based on the total number of sequence clusters which grouped together within the data set. All parameters used within the ABGD analyses were assigned default settings and a 3% cut-off value, which is typically thought to be the species threshold used to identify the number of species within each group66,67. Finally, the Birky21 4X ratio was applied to the S. spindale alignments. This final method of species delimitation compares the ratio of mean pairwise difference between two clades (K) and the mean pairwise difference within each clade (θ), with K/θ ≥ 4 indicating that the clades represent separate species. As two clades are being compared then there are two values produced for θ, as per the recommendations of Birky21, the larger value is used for the final K/θ calculation as this gives more conservative results, which are less likely to return false positives.
To identify the potential cause for the level of divergence, the impact of potential positive selection was assessed within and between S. spindale isolates using TreeSAAP22 to detect evidence of physiochemical property changes to the protein sequences. Sequences for S. spindale cox1 were aligned and a Newick format ML tree was created in MEGA, from this TreeSAAP tested the sequences for alterations in physiochemical properties. The significance of such amino acid replacements was evaluated by a z-test which is also used to identify the type of selection acting upon the bases within a specific region specified within the sliding window. Only results from category 6–8 were used with high levels of significance (p < 0.001) as these categories show property changes that are having the most effect on protein function and could be related to adaptation. The same analysis was also performed on cox1 samples of S. indicum and S. cf. indicum.
Supplementary information
Acknowledgements
The authors would like to thank Natural History Museum (London) sequencing facility for providing the Illumina MiSeq next generation sequencing (Illumina, Inc) service which was used to generate raw data for the mitochondrial genomes used in this study.
Author contributions
B.P.J., A.J.W., J.P.W., P.R.V.J.R. and S.P.L. conceived the project. B.P.J., B.F.N., H.E.B., S.W.A. and S.P.L. performed initial assembly and bioinformatic analyses of S. spindale mitochondrial genomes. B.P.J., B.F.N. and S.P.L. performed initial assembly and analyses on the S. indicum mitochondrial genome. Detailed comparative genomic analyses and phylogenetics were performed by B.P.J. and S.P.L. S.W.A. and M.M.H.M. were responsible for collecting and providing parasite material. B.P.J., A.J.W., J.P.W., P.R.V.J.R. and S.P.L. were involved in writing the final text which was subsequently checked and aprovied by all authors. This work was undertaken as part of the MSc by Research of B.P.J.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-57736-x.
References
- 1.De Bont J, Vercruysse J. The epidemiology and control of cattle schistosomiasis. Parasitology Today. 1997;13:255–262. doi: 10.1016/S0169-4758(97)01057-0. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Schistosomiasis. Available at, http://www.who.int/mediacentre/factsheets/fs115/en/ (2017) (Accessed 19 Jan. 2018).
- 3.Agatsuma T, et al. Affinities between Asian non-human Schistosoma species, the S. indicum group, and the African human schistosomes. Journal of Helminthology. 2002;76:7–19. doi: 10.1079/JOH200191. [DOI] [PubMed] [Google Scholar]
- 4.Agrawal, M. C. Schistosomes and schistosomiasis in South Asia. New Delhi: Springer (2012).
- 5.Agrawal MC, Gupta S, George J. Cercarial dermatitis in India. Bulletin of the World Health Organisation. 2000;78:278. [PMC free article] [PubMed] [Google Scholar]
- 6.Horák P, Kolářová L. Snails, waterfowl and cercarial dermatitis. Freshwater Biology. 2010;56:779–790. doi: 10.1111/j.1365-2427.2010.02545.x. [DOI] [Google Scholar]
- 7.Chevalier F, et al. Independent origins of loss-of-function mutations conferring oxamniquine resistance in a Brazilian schistosome population. International Journal for Parasitology. 2016;46:417–424. doi: 10.1016/j.ijpara.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan J, et al. Origin and diversification of the human parasite Schistosoma mansoni. Molecular Ecology. 2005;14:3889–3902. doi: 10.1111/j.1365-294X.2005.02709.x. [DOI] [PubMed] [Google Scholar]
- 9.Webster JP, Borlase AM, Rudge JW. Who acquires infection from whom and how? - disentangling multi-host and multi-mode transmission dynamics in the ‘elimination’ era. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372:20160091. doi: 10.1098/rstb.2016.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Attwood SW, et al. A DNA sequence-based study of the Schistosoma indicum (Trematoda: Digenea) group: population phylogeny, taxonomy and historical biogeography. Parasitology. 2007;134:2009–2020. doi: 10.1017/S0031182007003411. [DOI] [PubMed] [Google Scholar]
- 11.Devkota R, Brant S, Loker E. The Schistosoma indicum species group in Nepal: presence of a new lineage of schistosome and use of the Indoplanorbis exustus species complex of snail hosts. International Journal for Parasitology. 2015;45:857–870. doi: 10.1016/j.ijpara.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lockyer AE, et al. The phylogeny of the Schistosomatidae based on three genes with emphasis on the interrelationships of Schistosoma Weinland, 1858. Parasitology. 2003;126:203–224. doi: 10.1017/S0031182002002792. [DOI] [PubMed] [Google Scholar]
- 13.Zarowiecki M, Huyse T, Littlewood DJT. Making the most of mitochondrial genomes – Markers for phylogeny, molecular ecology and barcodes in Schistosoma (Platyhelminthes: Digenea) International Journal for Parasitology. 2007;37:1401–1418. doi: 10.1016/j.ijpara.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Yin M, et al. Co-dispersal of the blood fluke Schistosoma japonicum and Homo sapiens in the Neolithic. Age. Scientific Reports. 2015;5:18058. doi: 10.1038/srep18058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Littlewood DTJ, Lockyer A, Webster B, Johnston D, Le T. The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Molecular Phylogenetics and Evolution. 2006;39:452–467. doi: 10.1016/j.ympev.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 16.Krogh A, et al. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of Molecular Biology. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 17.Bouckaert R, et al. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Computational Biology. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suchard MA, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evolution. 2018;4:vey016. doi: 10.1093/ve/vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawton SP, Hirai H, Ironside JE, Johnston DA, Rollinson D. Genomes and geography: genomic insights into the evolution and phylogeography of the genus Schistosoma. Parasites & Vectors. 2011;4:131. doi: 10.1186/1756-3305-4-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puillandre N, Lambert A, Brouillet S, Achaz G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Molecular Ecology. 2011;21:1864–1877. doi: 10.1111/j.1365-294X.2011.05239.x. [DOI] [PubMed] [Google Scholar]
- 21.Birky C. Species Detection and Identification in Sexual Organisms Using Population Genetic Theory and DNA Sequences. PLoS ONE. 2013;8:e52544. doi: 10.1371/journal.pone.0052544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woolley S, Johnson J, Smith MJ, Crandall KA, McClellan DA. TreeSAAP: Selection on Amino Acid Properties using phylogenetic trees. Bioinformatics. 2003;19:671–672. doi: 10.1093/bioinformatics/btg043. [DOI] [PubMed] [Google Scholar]
- 23.Li J, et al. A specific indel marker for the Philippines Schistosoma japonicum revealed by analysis of mitochondrial genome sequences. Parasitology Research. 2015;114:2697–704. doi: 10.1007/s00436-015-4475-2. [DOI] [PubMed] [Google Scholar]
- 24.Sloan DB, Triant DA, Wu M, Taylor DR. Cytonuclear Interactions and Relaxed Selection Accelerate Sequence Evolution in Organelle Ribosomes. Molecular Biology and Evolution. 2014;31:673–682. doi: 10.1093/molbev/mst259. [DOI] [PubMed] [Google Scholar]
- 25.Burton R, Pereira R, Barreto F. Cytonuclear Genomic Interactions and Hybrid Breakdown. Annual Review of Ecology, Evolution, and Systematics. 2013;44:281–302. doi: 10.1146/annurev-ecolsys-110512-135758. [DOI] [Google Scholar]
- 26.Choi Y, et al. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Efremov. RG, Sazanov LA. Structure of membrane domain of respiratory complex I. Nature. 2011;476:414–420. doi: 10.1038/nature10330. [DOI] [PubMed] [Google Scholar]
- 28.Moreno-Loshuertos R, et al. Evolution meets disease: penetrance and functional epistasis of mitochondrial tRNA mutations. PLoS Genetics. 2011;7:e1001379. doi: 10.1371/journal.pgen.1001379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meiklejohn CD, et al. An incompatibility between amitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genetics. 2013;9:e1003238. doi: 10.1371/journal.pgen.1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leger E, Webster JP. Hybridizations within the Genus Schistosoma: implications for evolution, epidemiology and control. Parasitology. 2017;144:65–80. doi: 10.1017/S0031182016001190. [DOI] [PubMed] [Google Scholar]
- 31.Agrawal, M. C. Final report of national agricultural technology programme on diagnosis of parasitic diseases of domestic animals. Jabalpur centre, ICAR, New Delhi (2004).
- 32.Kemp, T. The origin and evolution of mammals. Oxford University Press, Oxford. (2005).
- 33.Gauffre-Autelin P, von Rintelen T, Stelbrink B, Albrecht C. Recent range expansion of an intermediate host for animal schistosome parasites in the Indo-Australian Archipelago: phylogeography of the freshwater gastropod Indoplanorbis exustus in South and Southeast Asia. Parasites & Vectors. 2017;10:126. doi: 10.1186/s13071-017-2043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng Y, Li Q, Kong L, Zheng X. DNA barcoding and phylogenetic analysis of Pectinidae (Mollusca: Bivalvia) based on mitochondrial COI and 16s rRNA genes. Molecular Biology Reports. 2010;38:291–299. doi: 10.1007/s11033-010-0107-1. [DOI] [PubMed] [Google Scholar]
- 35.Bohlen J, Perdices A, Doadrio I, Economidis PS. Vicariance, colonisation, and fast local speciation in Asia Minor and the Balkans as revealed from the phylogeny of spined loaches (Osteichthyes; Cobitidae) Molecular Phylogenetics and Evolution. 2006;39:552–61. doi: 10.1016/j.ympev.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Piano F, Craddock EM, Kambysellis MP. Phylogeny of the island populations of the Hawaiian Drosophila grimshawi complex: evidence from combined data. Molecular Phylogenetics and Evolution. 1997;7:173–84. doi: 10.1006/mpev.1996.0387. [DOI] [PubMed] [Google Scholar]
- 37.Stefanni S, Knutsen H. Phylogeography and demographic history of the deep-sea fish Aphanopus carbo (Lowe, 1839) in the NE Atlantic: Vicariance followed by secondary contact or speciation? Molecular Phylogenetics and Evolution. 2006;42:38–46. doi: 10.1016/j.ympev.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 38.Sturmbauer C, Baric S, Salzburger W, Rüber L, Verheyen E. Lake level fluctuations synchronize genetic divergences of cichlid fishes in African lakes. Molecular Biology and Evolution. 2001;18:144–54. doi: 10.1093/oxfordjournals.molbev.a003788. [DOI] [PubMed] [Google Scholar]
- 39.Woodruff D, Turner L. The Indochinese-Sundaic zoogeographic transition: a description and analysis of terrestrial mammal species distributions. Journal of Biogeography. 2009;36:803–821. doi: 10.1111/j.1365-2699.2008.02071.x. [DOI] [Google Scholar]
- 40.van den Bergh G, de Vos J, Sondaar P. The Late Quaternary palaeogeography of mammal evolution in the Indonesian Archipelago. Palaeogeography, Palaeoclimatology, Palaeoecology. 2001;171:385–408. doi: 10.1016/S0031-0182(01)00255-3. [DOI] [Google Scholar]
- 41.Zhou Z, Yang Q, Xia K. Fossils of Quercus sect. Heterobalanus can help explain the uplift of the Himalayas. Chinese Science Bulletin. 2007;52:238–247. doi: 10.1007/s11434-007-0005-7. [DOI] [Google Scholar]
- 42.Morales HE, et al. Positive and Purifying selection in mitochondrial genomes of a bird with mitonuclear discordance. Molecular Ecology. 2015;24:2820–2837. doi: 10.1111/mec.13203. [DOI] [PubMed] [Google Scholar]
- 43.Gómez-Durán A, et al. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplotypes. Human Molecular Genetics. 2010;19:3343–3353. doi: 10.1093/hmg/ddq246. [DOI] [PubMed] [Google Scholar]
- 44.Xu S, et al. A Mitochondrial Genome Sequence of the Tibetan Antelope (Pantholops hodgsonii) Genomics, Proteomics and Bioinformatics. 2005;3:5–17. doi: 10.1016/S1672-0229(05)03003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu S, et al. High-Altitude Adaptation and Phylogenetic Analysis of Tibetan Horse Based on the Mitochondrial Genome. Journal of Genetics and Genomics. 2007;34:720–729. doi: 10.1016/S1673-8527(07)60081-2. [DOI] [PubMed] [Google Scholar]
- 46.da Fonseca R, Johnson W, O'Brien S, Ramos M, Antunes A. The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics. 2008;9:119. doi: 10.1186/1471-2164-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melo-Ferreira J, et al. The Elusive Nature of Adaptive Mitochondrial DNA Evolution of an Arctic Lineage Prone to Frequent Introgression. Genome Biology and Evolution. 2014;6:886–896. doi: 10.1093/gbe/evu059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gu P, et al. Evidence of adaptive evolution of alpine pheasants to high-altitude environment from mitogenomic perspective. Mitochondrial DNA. 2015;27:455–462. doi: 10.3109/19401736.2014.900667. [DOI] [PubMed] [Google Scholar]
- 49.Ma X, Kang J, Chen W, Zhou C, He S. Biogeographic history and high-elevation adaptations inferred from the mitochondrial genome of Glyptosternoid fishes (Sisoridae, Siluriformes) from the southeastern Tibetan Plateau. BMC Evolutionary Biology. 2015;15:223. doi: 10.1186/s12862-015-0508-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun J, et al. Evidence for high dispersal ability and mito-nuclear discordance in the small brown planthopper, Laodelphax striatellus. Scientific Reports. 2015;5:8045. doi: 10.1038/srep08045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castello P, David P, McClure T, Crook Z, Poyton R. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: Implications for oxygen sensing and hypoxic signalling in eukaryotes. Cell Metabolism. 2006;3:277–287. doi: 10.1016/j.cmet.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 52.Luo Y, et al. Mitochondrial genome analysis of Ochotona curzoniae and implication of cytochrome c oxidase in hypoxic adaptation. Mitochondrion. 2008;8:352–357. doi: 10.1016/j.mito.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 53.Lawton SP, Bowen L, Emery AM, Majoros G. Signatures of mito-nuclear discordance in Schistosoma turkestanicum indicate a complex evolutionary history of emergence in Europe. Parasitology. 2017;144:1752–1762. doi: 10.1017/S0031182017000920. [DOI] [PubMed] [Google Scholar]
- 54.Agrawal MC, Rao VG. Some facts on South Asian schistosomiasis and need for international collaboration. Acta Tropica. 2017;180:76–80. doi: 10.1016/j.actatropica.2017.12.022. [DOI] [PubMed] [Google Scholar]
- 55.Biswal DK, Chatterjee A, Bhattacharya A, Tandon V. The mitochondrial genome of Paragonimus westermani (Kerbert, 1878), the Indian isolate of the lung fluke representative of the family Paragonimidae (Trematoda) PeerJ. 2014;2:e484. doi: 10.7717/peerj.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biswal DK, et al. An integrated pipeline for next generation sequencing and annotation of the complete mitochondrial genome of the giant intestinal fluke, Fasciolopsis buski (Lankester, 1857) Looss, 1899. PeerJ. 2013;1:e207. doi: 10.7717/peerj.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yilmaz E, et al. The mitochondrial genomes of the zoonotic canine filarial parasites Dirofilaria (Nochtiella) repens and candidatus Dirofilaria (Nochtiella) honkongensis provide evidence for presence of cryptic species. PLoS Neglected Tropical Diseases. 2016;10:e0005028. doi: 10.1371/journal.pntd.0005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hawash MB, Andersen LO, Gasser RB, Stensvold CR, Nejsum P. Mitochondrial Genome Analyses Suggest Multiple Trichuris Species in Humans, Baboons, and Pigs from Different Geographical Regions. PLoS Neglected Tropical Diseases. 2015;9:e0004059. doi: 10.1371/journal.pntd.0004059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 60.Kumar S, et al. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dejong RJ, et al. Phylogeography of Biomphalaria glabrata and B. pfeifferi, important intermediate hosts of Schistosoma mansoni in the New and Old-World tropics. Molecular Ecology. 2003;12:3041–56. doi: 10.1046/j.1365-294X.2003.01977.x. [DOI] [PubMed] [Google Scholar]
- 62.Attwood SW, Fatih FA, Upatham ES. DNA-sequence variation among Schistosoma mekongi populations and related taxa; phylogeography and the current distribution of Asian schistosomiasis. PLoS Neglected Tropical Diseases. 2008;2:e200. doi: 10.1371/journal.pntd.0000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Systematic Biology. 2018;67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leigh JW, Bryant D. PopART: Full-feature software for haplotype network construction. Methods in Ecology and Evolution. 2015;6:1110–1116. doi: 10.1111/2041-210X.12410. [DOI] [Google Scholar]
- 65.Vilas R, Criscione C, Blouin M. A comparison between mitochondrial DNA and the ribosomal internal transcribed regions in prospecting for cryptic species of platyhelminth parasites. Parasitology. 2005;131:839. doi: 10.1017/S0031182005008437. [DOI] [PubMed] [Google Scholar]
- 66.Brant S, Loker E. Molecular Systematics of the Avian Schistosome Genus Trichobilharzia (Trematoda: Schistosomatidae) in North America. Journal of Parasitology. 2009;95:941–963. doi: 10.1645/GE-1870.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ács Z, Hayward A, Sugár L. Genetic diversity and population genetics of large lungworms (Dictyocaulus, Nematoda) in wild deer in Hungary. Parasitology Research. 2016;115:3295–3312. doi: 10.1007/s00436-016-5088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.