

Graphical abstract
Abbreviations: QuEChERS, Quick, Easy, Cheap, Effective, Rugged, Safe; MP, microplastics; μFTIR, micro Fourier transform infrared; ATR, attenuated total reflection; MPF, microplastic free; SPT, sodium polytungstate; QA, quality assurance; QC, quality control; PTFE, polytetrafluoroethylene; UR, user requirements; SR, system requirements; TS, technical solution; SI, supplementary information; MQ (water), MilliQ (water)/ ultrapure water; SOPs, standard operation procedures
Method name: A QuEChERS approach to extract microplastics from environmental samples
Keywords: Sequential protocol, Purification technique, Chemical digestion, Handbook for laboratory work, Manual for good practices on microplastic extraction, Contamination prevention, Recovery rates, Wastewater, Particle-rich
Abstract
The identification of microplastics (MP), especially small (<500 μm) MP, using automated surface-chemistry approaches requires the best possible reduction of natural particles whilst preserving the integrity of the targeted synthetic polymers particles. In general, both natural and synthetic particles can be highly diverse physically and chemically and MP extraction, particularly from complex matrices such as sediments, sludge and soils, requires efficient method pipelines. Our paper presents a universal framework of modular protocols (presented in a decision tree) that fulfil predefined user requirements (QuEChERS: Quick, Easy, Cheap, Effective, Rugged, Safe) as well as providing best practises for reasonable MP working conditions within a standard laboratory. New procedures and technical innovations for density separation of particle-rich matrices are presented, such as a spiral conveyor developed and validated for MP recovery. In sharing such best-practice protocols, we aim to help in the push towards MP quantification method standardisation.
-
•
Publication of protocols of an entire MP extraction (10 μm – 5 mm) pipeline for particle-based analysis of various environmental matrices
-
•
Modularity: Optimised quantitative sample preparation adapted to particle sizes and sample matrices
-
•
New protocols and technical innovations (e.g. spiral conveyor) optimise MP extraction
Specification Table
| Subject Area: | Environmental Science |
| More specific subject area: | Microplastic pollution |
| Method name: | A QuEChERS approach to extract microplastics from environmental samples |
| Name and reference of original method: |
|
| Resource availability: |
Solutions/liquids/reagents
Devices
|
Method details
Protocols background
Microplastics (MP) are particulate synthetic polymerised hydrocarbons which are foreign to natural cycles and act as persistent pollutants if occurrent in the environment. It is to be expected that globally rising production volumes induce rinsing levels of this environmental pollutant if the development of waste management systems do not keep pace and non-circular economic practices persist. Commonly reported sizes of MP range between 1 μm to 5 mm. In contrast to many other environmental pollutants (e.g. heavy metals, pesticides), MP act as dispersed or suspended solid particles and their number and size determine distribution patterns and possible encounter and interaction rates with biota. Therefore, reliable techniques determining numbers and sizes of MP in various environmental matrices are greatly needed.
MP research has intensified considerably since being first scientifically described. Sampling, extraction and analysis methods have been developed and improved continuously to reach higher accuracies. However, the extraction of especially small MP (<500 μm) from various environmental matrices remains very time consuming and a general lack of standards persists. Especially particle-rich matrices such as sediments, sludge and soils require more efficient MP extraction pipelines. Ongoing developmental processes in this research field (see recent review by Zarfl [1]) produce a large variety of protocols which usually depend on the conditions and resources of the laboratories, as well as the sample type and research questions involved. There are numerous scientific publications on MP extraction of a specific sample matrix [[2], [3], [4], [5]], which can be disadvantageous, both in terms of additional costs and required expertise when diversifying the MP extraction work between several sample types.
Objectives
We present a comprehensive compilation of both modified and new methods for MP purification, organised into individual methodological modules designed to be adaptable to the sample type proposed for analysis, and flexible enough to be implemented in a general-purpose research laboratory. Although an optimal working condition for MP sample treatment would require a purpose-built clean lab facility (see SI.1 for further information), the de facto condition for many researchers is to work in a standard laboratory, often used by several other research disciplines simultaneously. Yet some adaptations to a general lab are highly encouraged, the most important of which is the installation of a laminar flow bench to be used exclusively as an MPF zone for all steps involving work on open MP samples. Within the proposed protocol modules are also novel technical steps to further improve both accuracy and efficiency of MP purification. This is especially important given the increasing shift from visual analysis of large MP only towards a focus on the full spectrum of MP size and type using highly-precise, automated, particle-based analysis approaches. Therefore, the overarching aim of this paper is to enable and encourage an adoption of MP-suitable working conditions. To pursue this goal, the following objectives are satisfied:
-
•
a set of coordinated protocols and devices are presented
-
•
a technical solution to density separations is demonstrated that addresses shortcomings of existing methods
-
•
advise is given to the reader on how to construct a full workflow from the provided protocols, which is adapted to the specific sample properties
Analytical requirements: when every particle counts
The endpoint of automated particle-based analyses, i.e. via micro Fourier transform infrared (μFTIR) or Raman spectroscopy, is the quantification of MP particles based on their chemical compositions or fingerprints without prior visual inspection. Independent of the sample type, the analysis time is mainly dependent on the final number of particles that has to be measured. Thus, when extracting MP from environmental samples, the overarching goal is the best possible reduction of natural particles while preserving the targeted synthetic polymers in numbers, sizes, shapes as well as their chemical composition. The final particle number also greatly depends on the particle size limit. While a common definition of the lower MP size is not yet established, the presented protocols are designed and applied for MP of 10 μm to 5 mm. It should be noted, that MP cover a size range differing up to 500-fold. An analysis attempt in one go would lead to restricted efficiency and quality in the analysis, i.e. by larger particles eclipsing smaller ones. We suggest several options to size-fractionate large, manually handleable particles from smaller MP and prepare them for single particle analysis, e.g. attenuated total reflection (ATR) FTIR (personal communication with Fischer) [6].
All the methods proposed here have been developed with the “QuEChERS” (quick, easy, cheap, effective, rugged and safe) approach in mind [7,8], and are presented in a way reminiscent of systems engineering [9,10]. Fig. 1 visually demonstrates how these approaches have been applied to the presented MP methodology; giving users the methods to achieve their desired goals whilst meeting QuECheERS standards.
Fig. 1.

The scheme detailing the three main components of a system engineering approach is meant to be read from the kernel (the “black box”) of a yet to be analysed MP sample. Under the overall goal of enabling a sample to be submitted for efficient microspectroscopic particle identification, the main objectives are the near-complete reduction of natural substances and the preservation of the contained MP. Users requirements (UR, placed around the kernel) formulate typical considerations of MP researchers working under these objectives. They are summarised here by the targets of a QuEChERS approach. One or more UR define a system requirement (SR, in rounded boxes) that specifies what a solution must provide in order to achieve the UR targets. The coloured circles indicate which SR are derived from which UR. Finally, the approach culminates in technical specifications (TS) that satisfy the previously defined requirements. Part of the solutions are the equipment (chemicals, laboratory set up, devices) and procedures used, presented here together as protocols or manuals ready for MP laboratory work (m0–m6).
In order to analyse a variety of sample matrices, which can differ markedly in composition (i.e. water vs soil), different methodological steps may be needed. By segmenting the methods into modules, certain procedures can be included or omitted as required by the sample type in question, greatly increasing the flexibility of the proposed methods. In order to ascertain which modules should be combined and in what order for a given sample matrix, a decision tree is an ideal way to guide a user to what specific methods they require.
The protocol tree: a decision tree for the protocol modules
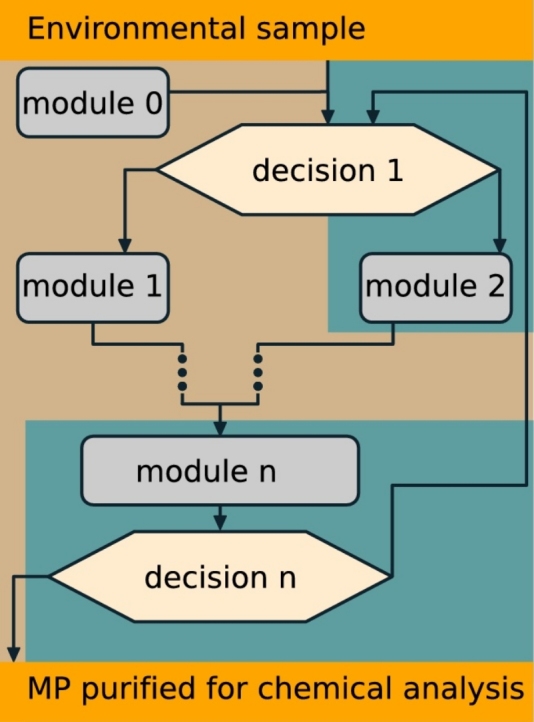
We used a decision tree to visualise the complete workflow of connectible protocol modules (see section Protocol modules m0–m6), referred to as the protocol tree (Fig. 2).
Fig. 2.

The decision tree comprises the procedural steps involved in the preprocessing of environmental samples for later particle-based MP chemical analysis. The decision tree starts after accomplished sampling with the sample being transferred from the sampling device, if applicable. It consists of a number of protocol modules equipped with indices (m0–m6) each of them referring to the detailed protocols. The digestion module m4 is further subdivided by specific treatments in a scheme below. These protocol modules are linked through various decisional paths that have to be taken for a complete pretreatment. The background colours indicate whether the sample is in dry (brown) or wet (blue) condition. Vacuum-filtration often finalises wet modules and is included in the respective protocol modules. As filtration is an important intermediate step, it is indicated by a respective symbol (close up of a filter unit, see legend).
How to use the protocol tree and modules?
The sample matrix and the numbers and the sizes of targeted MP determine the route through the tree. They define the optimal protocol pipeline along the tree structure adapted for a given individual case. Essential advancements of the presented methods originate from newly developed equipment applied in the protocols and are described in the respective section (New Equipment). All other materials needed are listed in the beginning of the paper. General MP laboratory procedures, including the cleaning of equipment and laboratory, MPF-filtration of solutions, etc. are described in the general module (m0). Further general measures of quality assurance and control (QA/QC) are described in the respective SI section (SI.2). To cover the diversity of samples in volume and composition some protocol modules contain different optional paths to follow, indicated by an arrow (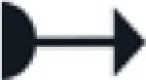 ).
).
Modularity and variations due to sample composition
The two widely used techniques to extract MP for a particle-based analysis are a separation by particle density (MP typically range from 900 to 1600 kg m−3) which primarily eliminates the mineral fraction (e.g. quartz, ρ = 2650 kg m−3), and chemical destruction i.e. digestion of particulate organic matter (see e.g. review by [11]). Covering the entire range of possible sample matrices (conceivably substances like honey, household dust, food waste, construction rubble, etc.) would make the decision tree increasingly complex. The focus of the presented protocol workflows is on environmental samples, such as water, soils, sludge, sediments and biota. Biota, for instance, comprise a large range of different biogenic material compositions (chitin, keratin, lipids, etc.) ranging from small zooplankton to stomach contents of birds, some of which may require a repetition or skipping of modules. In most of these cases a freeze drying step may be omitted if the dry weight is not required. Besides, freeze drying facilitates the following digestion step by breaking up cell walls (i.e. in samples rich in plant-based material). In case of a sample being largely made of animal tissue, this time-consuming step may be left out and still yields readily digestible samples. Also, a density separation module might not be necessary, if only negligible amounts of dense non-MP particles remain after digestion. Generally, fewer treatment modules, hence less sample manipulation, is preferential when dealing with potentially small numbers of MP. The required minimum particle size determines parts of the procedural details (e.g. m3 or m6) to assure that also the smallest targeted MP can be reliably quantified.
Digestion modules (m4.1–m4.3) more often necessitate a case-by-case adaptation and are therefore a good example of methodological variation based on sample type. While a purely oxidative treatment with hydrogen peroxide can successfully be applied for purification of water or sediment samples (m4.1), it often fails to degrade animal tissues in reasonable time [2]. A combined alkaline-oxidative treatment based on diluted potassium hydroxide and sodium hypochlorite [12,13] serves as a more appropriate choice for many biota samples (m4.2). The high fat, oil or wax content that can be present in biota samples can in principal be broken up by simple alkaline hydrolysis. However, this process may require longer reaction times or higher temperatures than what is desirable for MP purifications. Therefore, remaining lipid constituents are recommended here to be removed by an additional n-pentane based washing module instead (m4.3). In contrast to the other digestion and density separation modules, the application of a n-pentane based treatment has not yet been described for MP sample purification. The effectiveness of the proposed method in terms of achieving better spectral qualities in Raman or μFTIR analysis is promising for sample matrices where lipids could not be sufficiently removed by the other protocols. Apart from biota samples, the application of the n-pentane treatment was preliminarily tested for primary sludge samples from waste water treatments plants that contain potentially high levels of fats and oils, partly due to artificial substitution. This is especially challenging during MP extraction as fats and oils enclose sample material and this way reduce the effectiveness of the other treatments. A proposed digestion pipeline for these specific sample matrices is a treatment with n-pentane enclosed by repeated hydrogen peroxide digestions. However, the n-pentane module is presented here as a preliminary method, as its inertness to targeted polymers is currently being evaluated (R. Lenz, unpublished data). The general principle is based on classic organic chemistry procedures to dissolve or extract emulsified or deposited lipids from a sample and was applied before in a similar manner e.g. in infant milk fat extraction protocols [14,15]. Fischer and Scholz-Böttcher, 2017 [16] used petroleum ether to prepare greasy fish stomach samples for a mass-based MP analysis (Pyrolysis-Gas Chromatography - Mass-Spectrometry). Compared to n-pentane, use of petroleum can reduce operating costs but has higher health risk due to its hexane contents. The lipid removal potential and resistance of polymers can be expected to be comparable between the two chemicals. The following section presents all protocol modules that build the protocol tree.
Protocol modules
m0 – General module
 Irrespective of the protocol pathway selected it is recommended to read the general module as a prerequisite for many other modules as it covers overarching procedural steps when working with MP. It only contains procedures that are specific for the here applied protocols and materials and/or are not yet exhaustively covered in the MP literature and/or are not standard laboratory routines.1
Irrespective of the protocol pathway selected it is recommended to read the general module as a prerequisite for many other modules as it covers overarching procedural steps when working with MP. It only contains procedures that are specific for the here applied protocols and materials and/or are not yet exhaustively covered in the MP literature and/or are not standard laboratory routines.1
Clean bench SOPs: Ensure to keep all surfaces in the laminar flow bench MPF and ready to use. Solely use tissues made of non-disintegrating natural fibres. All water used to clean surfaces should be MPF water. Before use, make sure to rinse all materials with MPF water or muffle at 500 °C. Before muffling, cover with aluminium foil and open again only inside the clean bench to avoid contamination. All sample containers should be rinsed with MPF water when entering the clean bench coming from non-MPF conditions (such as field sampling or cold storage). Additionally, vessels like beakers or Erlenmeyer flasks which hold samples inside the bench are to be thoroughly rinsed with MPF water or MPF MQ before being poured into a filtration or another container. This is important, as a complete separation of droplets from the inner and outer walls cannot be guaranteed during pouring and rinsing procedures. Contamination should also be prevented by entering the bench with arms and hands when working in it. Always wear a 100% cotton lab coat and nitrile gloves kept over the lab coat sleeves to protect the skin and avoid contamination by synthetic clothes. Before entering always use abundant MPF water to wash your hands/gloves.
Cleaning fine stainless steel filters (e.g. 10 μm): After usage, rinse all stainless steel filters with MPF water from the tap to remove coarse dirt. Put the filters in a glass beaker, fill with MPF water and cover with aluminium foil (at all times). Apply repetitive ultrasonic bath sessions with changing power values (50–100%).2 Recommended are 10 min at 35 kHz and 225 W (for a volume of the ultrasonic bath of 18.7 l). In the initial session, diluted H2O2 can be added if the filters contain organic matter. Alternatively few drops of MPF Tween80 can be added. Rinse the beaker and filters and renew the MPF water in between subsequent baths to make sure that all particles are removed from the both the filters and the residual water. It is advisable to apply a minimum of 3 repetitions. However, if the water in the beaker remains cloudy or hazy after sonification the procedure should be repeated. After the final treatment, discard the water from the beaker, rinse and fill up with fresh MPF water. At this point, the MPF filters are ready to be used. Keep them inside the bench in a beaker. Just before use, rinse again with MPF water using tweezers and a syringe.
MPF solutions: It is essential when working with MP to ensure that all solutions which get in contact with the sample and/or are used in the clean bench are MPF. Use a vacuum filtration system to filter all solutions (apart from tap water, see tap water filter in the SI.3). Use a filter below your minimum size threshold to ensure a complete removal, e.g. the presented protocols predominantly use 10 μm as the minimum size threshold, a 1.5 μm glass fibre filter (cleaned beforehand3) is used for the last filtration. Make sure that the solutions are filtered in pre-cleaned MPF glass bottles and are covered with aluminium foil inside the bench. Appropriate labelling of samples and solutions is part of standard laboratory routines. During MP-extraction it can help to consistently label all the MPF solutions accordingly with “MPF”. Inside the clean bench, keep one glass syringe for each specific solution in use and avoid cross-over.
SPTrecovery: The SPT solution can be used for many density separation cycles either by recycling at the manufacturer (if available) or by vacuum filtration in the laboratory. After usage of the SPT solution leave it to settle in a collection flask (>24 h). Pour the supernatant into a separate collection flask. As soon as stirred up particles appear, leave the remaining solution to completely settle again for later recovery. Check pH of recovered SPT before use and adjust to pH 3 if necessary by adding droplets of HCl (10%). Check the density by weighing a defined volume (e.g. 100 ml) and adjust to 1800 kg m−3 by dissolving the required amount of crystalline SPT salt in the recovered solution. Vacuum-filter the SPT solution onto a MPF glass fibre filter as explained in the MPF solution section above. In case of high particle content, a pre-filtration on a filter with larger pore sizes (e.g. on 10 μm stainless steel filters) may be applied beforehand to expedite the process. Reconfirm pH and density again after filtration.
Explanatory remarks
Starting condition: Any sampling equipment or specific pretreatment step, such as the removal of sample material from a given sampling device, must be completed beforehand. The assumed sample condition for the following protocols begins with the sample material in a dedicated vessel (e.g. glass beaker).
Finishing condition: The aspiration of a complete pipeline is to bring an environmental MP sample from any possible compartment to an acceptable condition for particle-based chemical analysis. The presented protocols assume that there is a temporal or spatial discontinuity between the sample purification and the analysis, which is why treatments end with readily purified MP particles suspended in MPF MQ water, stored in a sealed MPF glass container (e.g. Erlenmeyer flasks with ground glass joint). This specific procedure is described in detail below (Final Filtration) and is to be conducted at the end of a protocol pipeline. As it would be repetitive to include them in every respective section they are stated here as a general instruction. In case the sample will be submitted to chemical analysis instantly within the same lab, the finalising filtration may be conducted directly onto the desired analysis substrate.
 Filtration: Vacuum-filtration often finalises wet modules and is described in detail in the respective protocol modules. The symbol is to be found in the protocol tree (Fig. 2). Additional ultrasonification: Depending on filter pore size and sample composition, removing the sample particles from the filter by rinsing may be non-exhaustive4 . In such cases the filter should be additionally cleaned in an ultrasonic bath for minimum 2 minutes and residues added to the sample5. Check if additional ultasonification is needed to remove all sample particles from the filter.
Filtration: Vacuum-filtration often finalises wet modules and is described in detail in the respective protocol modules. The symbol is to be found in the protocol tree (Fig. 2). Additional ultrasonification: Depending on filter pore size and sample composition, removing the sample particles from the filter by rinsing may be non-exhaustive4 . In such cases the filter should be additionally cleaned in an ultrasonic bath for minimum 2 minutes and residues added to the sample5. Check if additional ultasonification is needed to remove all sample particles from the filter.
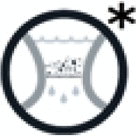 Final Filtration: The “Final Filtration” symbol is included as an optional path in the respective protocol modules where applicable. This is your last step before chemical analysis. Flush your filtering sample again using MPF MQ water to remove all solutes from the sample6 (it is marked in the protocol tree with the filtration symbol and an asterisk). At the end of a filtration the filter is removed from the apparatus using tweezers.
Final Filtration: The “Final Filtration” symbol is included as an optional path in the respective protocol modules where applicable. This is your last step before chemical analysis. Flush your filtering sample again using MPF MQ water to remove all solutes from the sample6 (it is marked in the protocol tree with the filtration symbol and an asterisk). At the end of a filtration the filter is removed from the apparatus using tweezers.
 If you see >3 MP particles too large for microspectroscopic analysis, apply the module “Size-fractionation: wet sieving & picking” (m6).
If you see >3 MP particles too large for microspectroscopic analysis, apply the module “Size-fractionation: wet sieving & picking” (m6).
Then rinse off the remaining sample particles from the filter into the same final Erlenmeyer flask using MPF MQ water. Place the filter in a clean beaker and cover it with MPF MQ water and sonicate for 2 min to remove all particles from the filter. Then rinse the filter off and add the residues into the same Erlenmeyer flask that containes the bulk of the sample. Close with glass stopper and secure with metal clip. Once the filter is cleaned without residues it can be removed. The sample should be stored in a fridge (4 °C).7
m1 – Freeze-drying
Freeze drying is usually performed for one or several of the following three reasons: a) reduction of water in the sample and breakage of particle conglomerates (resulting in a “fluffy” texture) to allow unrestricted performance of the next processing steps (e.g. digestion or density separation), b) determination of the dry weight and c) breakage of cell walls and membranes during the freezing step to increase later digestion efficiency.
1. Filter: If the sample is predominantly liquid, filter it in the vacuum filtration set up on a stainless steel filter of the lowest size threshold (e.g. 10 μm). Replace filters if clogged. Make sure to properly rinse all walls, including the sample vessel and the vacuum filtration funnel that the sample was in contact with. Depending on the sample matrix, it may be appropriate to use a diluted detergent solution (e.g. Tween80) to remove sample adherences from the glass ware. Collect all filters (containing sample material) in a glass beaker and add the sample rinsate of the filtration funnels. Cover with aluminium foil.
2. Freeze: Freeze the sample at −20 °C (∼24 h).8
3. Cover: Cover sample with an air-permeable MPF membrane with a mesh size smaller than that of the lowest detection size (dust-free tissue paper is ideal for most applications). Fix coverings firmly in place (elastic bands are appropriate). It is recommended to perform the uncovered part near an air filtration device or within an MPF clean bench to avoid contamination.
4. Freeze-dry: Freeze-dry the sample according to machine manufacturer’s guidelines until the sample is completely dry.
5. Weigh: If the sample is not further split, the dry weight is now to be determined.
m2 – Volume-fractionation (homogeneous splitting)
Depending on the initial sample volume, a volume fractionation can be necessary to reduce the amount of sample that is treated, or split into several sub-fractions (e.g., for method comparison purposes). If this step is used as sub-sampling (i.e. not all split parts are to be further processed) an effective homogenisation of the sample is most critical for a successful volume fractionation.
1. Mix: For freeze-dried samples, shake the sample with the lid closed for minimum 2 min. Then wait until all particles settled. Open the lid and mix the sample with a clean MPF long metal spoon for further minimum 2 min9 within the MPF clean bench or, if not possible, near an air filtration device. If mixing in the available sample container is restricted, transfer the sample to a MPF-cleaned bowl (i.e. ceramic or stainless steel) and proceed with the homogenisation process.
2. Split: Transfer the required sample (dry) weight directly into a separate glass beaker, tarred on a scale. If required, several replicate sub-fractions can be obtained in this way.
m3 – Size-fractionation (dry sieving)
To further ease processing and analysis of samples where the predominant particle size is substantially smaller than the lower MP size limit, the sample volume can be reduced by sifting out a lower particle size fraction by dry sieving. In principle, this method can also be applied to compartmentalise the sample into a variety of sample fractions of differing particle size, e.g., for differing analytical approaches (visual, surface chemistry, chromatography).10
1. Prepare: Select sieve sizes appropriate to the size-fractions desired and form a stack. Use a close-fitting stainless steel lid, alternatively thick aluminium foil, to cover the sieve(s) from the top and the bottom. Then slowly add the sample (ideally freeze-dried) to the top sieve, in an MPF clean bench or near an air filtration device. Close the sieve stack making sure there are no gaps where sample material may escape (or contaminated air may enter).
2. Sieve: Secure sieve stack in shaker. The amplitude and time can be adjusted according to how easily the sample sieves. We suggest, however, to start with lower amplitudes and shake times and document progress. Two rounds of 5−10 min at 0.75–1.5 mm amplitudes have been found to be sufficient for a variety of soil samples.
m4 – Digestion
Chemical digestion is a major step to reduce the organic matter content of a sample. A set of different digestion sub-modules are provided that are to be applied for different sample matrices (see scheme below).11

m4.1 – Hydrogen peroxide (H2O2) digestion
Hydrogen peroxide (H2O2) at 30% is a strong oxidizer. Typically, it is the primary choice for digesting water, sediment, soil, waste water and sludge samples. However, in certain cases the sample matrix might contain substances that are highly resistant to the treatment in which cases other subsequent digestion steps may be added.
1. Add: Depending on the nature of the sample, i.e. organic matter content and presence of iron ions, the reaction after the addition of H2O2 can be very energetic (bubble formation and temperature rise). Because of that, it has to be added very carefully and in small amounts. With a glass syringe, add up to 5 ml of H2O2 to the sample. For unknown sample matrix even smaller amounts (∼2 ml) should be added initially and observed.12 To anticipate heat generation or an overflow event, the beaker can be kept in a second MPF container (e.g. larger beaker or ceramic bowl) with cold MPF water to avoid temperature rise to more than 40 °C. When working in a fume hood without appropriate MPF conditions an aluminium foil lid should cover the sample at all times and only shortly opened to insert the tip of the glass syringe.
2. Proceed: Add more H2O2 with a syringe until it completely covers the whole sample. Shake the sample and rinse the walls with H2O2 to allow all particles to be in contact with the digestion solution. The beaker should be kept closed in a fume hood for minimum 24 h. Generally, additional H2O2 should be added or replaced (filter sample and add new H2O2) for as long as the digestion process continues or organic matter remains visible.13
If the sample remains with large indigestible non-MP debris manually pick each item, rinse off adherent smaller particles into the sample and discard. It is recommended to cut very large pieces (e.g. blades of grass, leaves, braches) into smaller ones to allow a more efficient disintegration of biofilms and particle conglomerates during the digestion process.
3. Filter: To endthe digestion, filter the digested sample in the vacuum filtration set up on a stainless steel filter of the lowest size threshold (e.g. 10 μm). Carefully rinse the stainless steel filter that remained in H2O2 from the previous filtration step from all sides using MPF water, hold it withtweezers and/or a gloved hand. Replace filters if clogged. Once the filter is cleaned without residues it can be removed. Make sure to properly rinse all walls, including the beaker and the vacuum filtration funnel that the sample was in contact with. Depending on the sample matrix, it may be appropriate to use a diluted detergent solution (e.g. Tween80) to remove sample adherences from the glassware.
m4.2 – Potassium hydroxide (KOH) digestion
This protocol module may be applied for samples high in protein content like animal tissue (e.g. stomach analysis). It can also be applied for environmental samples with a challenging matrix composition, that do not sufficiently digest in H2O2.
1. Prepare: A saturated KOH stock solution is prepared by dissolving 1120 g/l of dry KOH salt (pellets) in water inside a tightly-closed glass bottle. Shake well until complete dissolution. Beware of heat generation, it may be advisable to reduce the temperature with a water bath. The digestion solution is prepared according to the below recipe:
Per 1 l of digestion solution to add to a glass bottle: 700 ml of water (tap water or MilliQ)
ml NaClO (14% active chlorine)
ml KOH stock solution
After adding the active chlorine, re-seal the bottle and shake well for homogenisation. The ready digestion solution can now be MPF filtered (see General Module m0).
2. Digest: Add 5 ml of MPF-filtered digestion solution per gram wet-weight of sample14 into the sample vessel, however at least so much that the sample is completely covered with solution. Shake gently to allow the solution to reach all parts of the sample. Leave for 5 h incubation at room temperature.
3. Filter: To stop the digestion, filter the digested sample in the vacuum filtration set up on a stainless steel filter of the lowest size threshold (e.g. 10 μm). Carefully rinse the stainless steel filter(s) that remained in the digestive solution from the previous filtration step from all sides using MPF water, hold it by tweezers and/or gloved hand. Replace filters if clogged. Once the filter is cleaned without residues it can be removed. Make sure to properly rinse all walls, including the beaker and the vacuum filtration funnel that the sample was in contact with. Depending on the sample matrix, it may be appropriate to use a diluted detergent solution (e.g. Tween80) to remove sample adherences from the glass ware.
m4.3 – n-Pentane (C5H12) digestion
Oil and fat constituents in sample matrices can sometimes remain in the digested sample and hinder a successful spectroscopic analysis.15 Possible use-cases of n-pentane16 include digested animal tissues (e.g. in fish stomach analysis) post-KOH digestion and primary sludge samples from waste water treatment facilities post-H2O2 digestion. The procedure requires extended times with the sample exposed to air and should therefore be conducted inside the MPF clean bench. Check if your work safety regulations require wearing a gas mask (filter type “AX”).
1. Solubilise: As a precondition, the digested sample should be contained in a tightly sealed vessel (e.g. Erlenmeyer flasks with ground glass joint) and must be suspended in MPF water or MPF MQ. To better solubilise the fatty constituents, hold the sample in a 35 °C water bath during the procedure. Wait for the sample to warm up to approx. 35 °C, then add 10 ml of the acidified ethanol (pH ∼ 3.0, acidified with acidic acid) per 50 ml of sample. Close and shake the sample for 1 min. Place in the warm water bath again.
2. Wash: When the ethanol is fully mixed in the sample continue by adding 10 ml of n-pentane per 50 ml of sample. Close the lid tightly and shake for 2 min. Stop shaking and open the flask periodically to release pressure.17 Return the sample to the warm water bath and allow the phases to separate for 5 min, with a loosely-closed lid.
3. Remove: Remove the pentane phase from the top of the sample using a glass pipette.18 Stop just before the pentane layer disappears to avoid sucking the sample.19 Repeat the above steps a further two times.
4. Filter: To remove any remaining dissolved fats and pentane continue with a vacuum filtration. Filter the sample in the vacuum filtration set up onto a stainless steel filter of the lowest size threshold (e.g. 10 μm). Make sure to properly rinse all walls, including the Erlenmeyer flask and the vacuum filtration funnel that the sample was in contact with. Check if additional ultasonification is needed to remove all sample particles from the filter (see Filtration above). Once the filter is cleaned without residues it can be removed. Make sure to properly rinse all walls, including the beaker and the vacuum filtration funnel that the sample was in contact with. Depending on the sample matrix, it may be appropriate to use a diluted detergent solution (e.g. Tween80) to remove sample adherences from the glass ware.
m5 – Density separation
Density separation is a major step to reduce the inorganic matter content of a sample.
1. Transfer: Choose a separation funnel of appropriate volume.20 Transfer the sample (dry or suspended in SPT) from a beaker or filter to the separation funnel.
Size fractionation: If the sample contains particles >5 mm or larger than the outlet valve, manually pick (if just very few larger particles) or sieve the sample during transfer to prevent blockage in the outlet valve or during the stirring of the sample. Sieving: Place a glass or metal funnel21 in the top of the separation funnel with a small sieve (e.g. 4 mm pore size) inside and pass the entire sample (preferably dry) through with help of a metal spatula. Thoroughly rinse the large particles that remained on the sieve with SPT to rinse all adhering smaller particles into the sample. Optionally, retained particles can also be transferred to a beaker with a few droplets of H2O2 and SPT to ensure a complete detachment of adhering particles. The detachment process might take some minutes. The rinsate is then added to the separation funnel.
Fill approximately 10% of the separation funnel (valve closed!) with MPF SPT (1800 kg m−3).
Add a first fraction of the sample (maximum up to 5 cm high from the lower tip of the separation funnel) and shake thoroughly to ensure that all particles in the lowest tip of the separation funnel are coated by the SPT solution. Then gently swing/shake the separation funnel inclining it ∼45°.
If the sample does not contain more than approximately 5 g of solids (e.g. water samples) repeat gentle swinging of the separation funnel every 15 min during the first hour to resuspend the settled material and disintegrate agglomerates.
If the sample does not allow for digestion before density separation, add a few droplets of H2O2 solution during the shaking process to initiate the disintegration of particle conglomerates, wait for the reaction to proceed.22
2. Upwell: If the sample material exceeds a volume where shaking of the separation funnel becomes insufficient in freeing possible overlays of particles (as often the case for sediment and soil samples) continue with the following steps. Firstly, prefill a part of the required SPT solution into the separation funnel followed by a small amount of the sample material. Subsequently add the remaining sample material and SPT solution. Top up with a minimum of 5 cm SPT solution to form the separation phase. Thoroughly rinse all equipment that was in contact with the sample material with SPT, such as the beaker, funnel and sieve and add it to the separation funnel. Insert the spiral conveyor to the sample and mount it to the electrical overhead stirrer. Centre and place it as low as possible but make sure it does not touch the walls of the separation funnel. Run it at relatively low speed (∼35–60 rpm) for approximately 2 h. The time needed to allow sufficient reshuffling of the material depends on sample composition, amount and dimensions of the separation funnel and spiral conveyor.23 After conveying, stop the spiral conveyor and thoroughly rinse adhering material into the separation funnel with ample MPF SPT while slowly lifting it out of the separation funnel. Release the lower tip (approximately 3 cm) of the separation funnel into a beaker and re-add to the separation solution.24
3. Wait: Rinse the inner walls of the separation funnel to ensure that all particles are in the separation solution. Leave it to separate for 1.1 h per 1 cm rising distance in the fluid separation phase.25
4. Divide: Slowly (no stirring up of particles) release the sedimented fraction into a MPF beaker placed below the separation funnel and shut the valve securely before any surface floating particles could be flushed out.
5. Repeat (*XDS): Depending on the sediment composition (especially fine/organic rich samples) the density separation (DS) cycle has to be repeated (XDS times) to reach a higher purification efficiency. Therefore, refill some MPF SPT, shake and leave it again to settle for 15 h. After this, evaluate whether you need to repeat again. As a rule, apply as many separation cycles until no further material has settled.
6. Filter: When the separation is finished, rinse the supernatant upside down from the separation funnel into the vacuum filtration apparatus equipped with a stainless steel filter of the lowest size threshold (e.g. 10 μm). Replace filters if clogged. Recover the undiluted SPT solution by transferring it from the filtration apparatus to a dedicated SPT recovery container (see General module m0). Then continue to thoroughly rinse the content of the separation funnel (incl. walls and glass stopper if used) into the vacuum filtration funnel using MPF water and a syringe. Take care not to lose any droplet or particle. Rinse all walls of the top part of the vacuum filter apparatus. Once the filter is cleaned without residues it can be removed. Depending on the sample matrix, it may be appropriate to use a diluted detergent solution (e.g. Tween80) to remove sample adherences from the glass ware.
m6 – Size-fractionation (wet sieving or visual sorting)
Samples which remain relatively rich in larger particles (>500 μm) even after passing the treatment pipeline should be size-compartmentalised to ease later chemical analysis.
The sorting and picking process should always be conducted in a very conservative manner, meaning that only particles with clear indications of natural origin may be discarded and any other particle is to be picked and retained for analysis. This is important to minimise researcher bias. Whether to use 0.5 or 1 mm as a cut-off size should be decided according to the following criteria table:
| Situations suggesting a cut-off size of | ||
| 0.5 mm | 1 mm | |
| Experience level of the separator | High: picking of 500 μm particles in a conservative manner is only possible with an experienced eye and hand | Low: only for particles larger 1 mm, can an inexperienced user conduct a conservative separation after a basic introduction |
| Number of particles in 500–1000 μm and >1000 μm size range | Particles potentially disturbing the microspectroscopic analysis are numerous in the 0.5–1 mm range | Particles potentially disturbing the microspectroscopic analysis are predominantly in the >1 mm range |
| Size cut-off predefined by sampling technique | Lower than 300 μm (e.g. sediment samples, flow-through filtration water samples) | 300 μm and larger (e.g. Manta samples) |
It should be noted that a 500 μm cut-off size should preferably be used in all cases where possible and meaningful, as it will be more beneficial to ease and expedite the microspectroscopic analysis of the remaining small fraction.
Depending on the number of particles, a case by case decision needs to be taken whether wet sieving is applied before visual sorting. In general, when more then 10 particles are above the chosen upper size limit for microspectrosopic analysis, it is advisable to first wet sieve the particles according to the below steps:
Optional wet sieving prior to visual sorting and picking
1. Set up: Prepare a filtration set-up as usual (e.g. 10 or 200 μm filter depending on minimum size limit) and place an additional 1 mm26 stainless steel sieve into the vacuum filtration apparatus.
2. Rinse: Turn on the vacuum pump and transfer the sample by using ample MPF water, rinse the sample container and the filtrate thoroughly.27 Finish this step by rising the sample and all relevant walls with MPF MQ water.
3. Conserve: Stop the pump and place the 1 mm filter into a petri dish and observed it under a binocular microscope. Particles on the smaller filter (e.g. 200 μm) are filled (back) into the Erlenmeyer beakers by using MPF MQ water.
Visual sorting and picking
1. Sort on sieve: If applicable, firstly check the larger (i.e. >0.5 mm or >1 mm) fraction on the sieve (or on the original vacuum filter, if no wet sieving was applied) for prominent plastic suspects,28 separate them using fine tweezers and rinse off potentially adhering smaller particles with MPF MQ above the sample. Sandwich the separated particle between two MPF microscopy glass slides using tweezers. These “sandwiches” can then be closed with two small stripes of paper tape and labelled with a running number (e.g.: [station name]_[sample number/ ID]_[consecutive number of the plastic suspect (starting with 001)]). It is advisable to mark the location of the particle in the sandwich by circling it with a permanent marker on the outside.
If the possibility exists, particles may be directly transferred to the analysis stage of an ATR-FTIR instead of “sandwiching”.
2. Sort in petri dish: When further picking on the sieve is limited (due to the dark background and/or a high number of particles overlaying each other) rinse the sieve content into a petri dish using MPF MQ29 water and continue picking all MP suspects. Ensure to assess all particles, so none remain on the filter.
3. Creation of particle groups: In case of a high number (>>40) of particles of identical morphological characteristics (i.e. colour, size class and surface structure) only transfer a subsample of 40 particles into individual sandwiches, labelled with an additional consecutive index number (e.g.: […].[1–40]). Count all other particles of the same kind (e.g. using a click counter) and place together into an additional glass slide sandwich or petri dish, name accordingly (e.g.: […].rest:[remaining total particle number]).30
New equipment
Within the postulated protocols, the required work effort is reduced and extraction efficiency is increased by the conception and application of new equipment. The technical advancements are explained and validated in the following section. Minor accessory equipment, namely simple and mobile tap water filters is presented in the SI.3, Fig. SI.1 which provides ample MPF water supply in the laboratory and thus contribute to QA measures.
Modified separation funnels – a versatile application for particle-rich samples
The application of glass separation funnels for density separation was applied previously [[17], [18], [19]]. They exhibit many advantages as such that they fulfil all relevant system requirements previously defined (Fig. 1, Fig. 3). Firstly, glass allows for visual control of the status and quality of the separation process. Secondly, by having a steep sloping wall, the Squibb-form separation funnels was chosen in order to prevent particle adherence to walls when rising while still having an acceptable volumetric capacity that allow handling in standard laminar flow benches. Furthermore, separation funnels are off-the-shelf, long-lasting products of little cost. The previous problem of only fitting small amounts of sample material [19,20] is overcome by using the newly developed conical spiral conveyor, presented in the respective section below, which allows for efficient MP extraction despite an enormous overlay of particles. Some modifications on the separation funnels were required for matter-rich samples, for instance, to overcome the problem of clogging [17] when using very fine or very coarse materials. Standard outlet valves of all separation funnels, especially those intended for particle-rich samples, were replaced by larger ones (ø 10 mm) to ease the release of sedimented particles. To satisfy the non-contaminating system requirement, plastic handles and nuts were replaced by stainless steel parts. Furthermore, a widening of the standard neck size (ø 25 mm) was necessary to allow the installation of the larger spiral conveyor (ø 35 mm, Fig. 3). We decided that 50 mm was the optimum size as this also fits the sieves potentially used for particle fractionation (m5). While custom equipment can be costly, these adaptations typically come at a low cost and constitute a one-time investment, fulfilling QuEChERS requirements (Fig. 1). All customisations of glass ware can be provided by a glass blower. Protocol modularity can limit costs, simplify the procedures and equalise extraction effectiveness when working with various sample matrices. Hence, the modular use of equipment is of advantage and is implemented by the universal application of the separation funnels for samples from water, sediments, beaches, soils, waste water, etc.
Fig. 3.

A) Sketch showing the custom-made conical spiral conveyor, which improves the extraction efficiency of MP in a density separation despite being buried by particulate matter. The spiral conveyor is operated within a separation funnel (Squibb form). The particles of the sample are transported upwards and released in a fountain-like manner into the density separation solution above, where they can freely separate. A video (picture sequence over 2 hours) visualising the efficiency of the density separation process by means of the conical spiral conveyor is provided as supplementary material to the paper. Approximately 0.5 g of test MP i.e. fluorescent green polyethylene microspheres (125–150 μm, ρ = 1025 kg m−3) were added initially in the lower part of the sediment fraction (glass particles, 40–70 μm). SPT (ρ = 1800 kg m−3) is added as the density separation solution. When starting the spiral conveyor, it can be observed that the test MP are being transported upwards and partially rise to the top. Some test MP cannot be freed in the first cycle and are covered again by sediments. After several cycles (after ∼2 h) all particles are extracted and float on the top of the density solution (supernatant). B) The technical drawing of the large spiral conveyor (ø 35 mm) with details on the physical dimensions. Other sizes can be up or down-scaled accordingly.
Conical spiral conveyor – a gentle way to separate particles
During density separation, it has to be ensured that particles get in contact with the liquid phase of the density separation solution in order to prevent trapping of particles by overlaying material. Different approaches were applied in previous studies, such as shaking or stirring the sample (e.g. [4,13]). However, shaking (by hand or machine) is efficient only on relatively small sample amounts and stirring blades, such as those implemented in the MicroPlastic Sediment Separator (MPSS; [4]), are prone to disintegrate MP particles by grinding sample matter directly against a hard (i.e. stainless steel) surface and thus falsify results [21]. To increase the extraction efficiency of MP from particle-rich samples whilst overcoming these potential limitations, we developed a custom-made spiral conveyor that allows for gentle and sufficient separation of the particles (Fig. 3A ). Instead of uncontrolled mixing, particles from the bottom of the separation vessel are gently transported upwards and released in a fountain-like manner to the liquid separation phase above. Once freely suspended, particles can separate based on their physical features and either float to the top, stay in suspension or settle again to the sedimented solid phase. A side effect of the gentle movement is the disintegration of material conglomerates which contributes to an efficient separation process. The conical shape reduces the bottom-facing area of the rotating spiral conveyor to just a fine tip. Compared to other stirring procedures described in the literature which use flat rotor blades or rods (e.g. Imhof, 2012 [4]) this is an important improvement, as it minimises the area and thus the possibility of grinding and squeezing particles between the rotating element and the vessel walls.
The principal functioning of the spiral conveyor is based on the widely-used industrial screw conveyors. In the lower 80 mm, the spiral conveyor fits the conical shape of the Squibb form separation funnels with a 5 mm wide gap in between. As different sample volumes are required depending on the sample type and research question, two different conveyor sizes were built that allow sufficient mobilisation of the particle phase. The smaller one (ø 20 mm) is designed for the application in 250 ml separation funnels, the larger one (ø 35 mm) for 500–1000 ml separation funnels. The spatial dimensions of the larger spiral conveyor are given in Fig. 3B. Other sizes could be simply scaled up or down or adapted to other types of separation vessels. It is important to note, that the screw conveyor part is designed to protrude into the separation solution above the sedimented particles for an efficient separation process to work. The shaft fits into a drill chuck of a typical electrical overhead stirrer for quick (dis)mounting. Due to the corrosive nature of most separation media, stainless steel is considered the optimal material. In the case of the larger conveyor, brass was used due to limitations of the available lathe machine. The presented set up for particle-rich matrices has a volumetric capacity of up to 1 l, appropriated to fit approximately 500 ml of particulate matter. By experience, this is a reasonable amount to achieve robust MP numbers. This volumetric capacities compares to other methods like the elutriation approach [22], though requiring less space and less steps involved in the separation process to reach reasonable extraction efficiencies. There are few, such as the MPSS, that have a larger sample capacity (∼6 kg, [4]), however, most other separators hold substantially smaller volumes [17,20].
Method validations and comparison
Recovery and reduction rates
A first semi-quantitative validation test was given by the video sequence shown above (link, Fig. 3A) demonstrating the efficiency of the density separation process by means of the conical spiral conveyor. Additionally, we performed recovery tests as a measure of extraction efficiency of the entire MP extraction procedure based on beach sediment samples (n = 7, d50 ∼ 300 μm). The recovery tests followed the respective pathway though the protocol tree including the operation of the spiral conveyor to separate particle-rich matrices. Test particles were visually identified using a microscope by their specific morphological characteristics. Polyamide particles (PA-6) of 450 μm (n = 20) in size were used and resulted in recovery rate of 95 ± 6%. This value is within the higher margin of other available recovery rates of other MP extraction methods, e.g. modified decanting, elutriation, MP sediment separator, sequential density extraction, oil extraction (see Table 1 in [17] or [19,23] for comparison). However, recovery rates are known to decrease with MP size [20] and most studies lack sufficient tests [24], especially in the size range below 300 μm (longest dimension). We determined the recovery rate of smaller MP using PE fluorescent particles of 125–150 μm (n = 60–80) in size resulting in 78 ± 6%. In this size class, however, reasonable comparisons to other studies are difficult as insufficient data is available. Not all study designs in the available literature (keyword search Web of Science: microplastics, recovery, sediment/soil/sludge) were technically able to extract the full range of polymer types (e.g. ρ = 900–1400 kg m−3). Of those that were, only very few studies tested the distinct size range of small MP from real, particle-rich environmental matrices (e.g. soils, sediments, sludge), whilst applying particle-based analytics and reporting sufficient information on the recovery test to allow a comparison. For instance, Imhof et al. [4] and Claessens et al. [22] tested particles between 40–309 μm and ∼250 μm respectively, resulting in a recovery rate above 95%. However, a comparison of our recovery rates to the two mentioned is in question for several reasons. These studies used sediments that underwent several cleaning steps (sequential density separation, elutriation) before performing recover tests which ultimately results in a test matrix of very low conglomeration potential and thus not a realistic sample matrix. It is expected that several factors such as the sediment composition, i.e. grain size and/ or organic matter content influence the extraction efficiency, potentially to a significant degree. This is a current knowledge gap that needs to be investigated in detail in a separate study. Further, when testing such small MP it is important to ensure that the size remained unchanged after the treatment as some techniques are prone to disintegrate MP [21] and thus potentially falsely increase the MP number. This remained unclear in some cases [4]. Hengstmann et al. [25] performed tests in a comparable size range (63–200 μm) to our study acquiring smaller recovery rates of 66 ± 17%. However, they used MP test particles with a higher density which often show reduced recovery rates compared to lighter once. Inconsistencies between recovery test designs exist as some studies tested the entire MP extraction pipeline (present, [23]), while others determined the recovery rates of the separation step solely [4,25]. The latter is likely to achieve higher values. The use of large MP size class ranges (e.g. 100–500 μm, [20]) without determining the size distribution (e.g. median, mean value) complicates a comparison to more narrow size ranges, such as those used in the present study, as the size-dependency of extraction efficiency remains disregarded. For a reasonable comparison of extraction methods, standards on the test material and procedure are needed. To date, a comparison of recovery rates of small MP (<500 μm) between different studies is not reliable due to the listed reasons.
Another measure of method validation is the mass reduction, i.e. the weight difference between the initial and treated dry weight sample. The mass reduction of exemplary sediment samples, ranging from beach sand (low in organics, coarse sediment) to estuary sediments (high in organics, fine sediment) was 99.98 ± 0.03% (n = 6) on average (SI.4, Table SI.1). Neither the initial sample mass nor the different sample matrix complexities could entirely account for the variability in the amount of material left in a sample after treatment. It can be assumed that the final number of particles also depends on the instruments and procedures used as well as on the operator.
Process time
While selecting the best methodological steps is critical for final accuracy of results, improvements must also be time-efficient. The time consuming nature of MP extraction was often reported in the past (e.g. Zarfl, 2019 [1]), however, without documentation of the concrete time measurements [24]. We report estimated procedural time for exemplary protocol pipelines, conducted by an experienced person, for better comparability of methods and planning of treatment procedures (SI.5, Table SI.2).
This paper presents a compilation of all methods necessary to fully process any given environmental sample ready for automated surface chemistry analysis. These methods are presented in a modular way which can be adapted to what a special sample requires. New equipment has also been described which, combined with the presented methods, significantly improve the methods for extracting both large (>0.5 mm) and small (<0.5 mm) MP from environmental samples.
Funding
This work resulted from the BONUS MICROPOLL project supported by BONUS (Art 185), funded jointly by the European Union and Federal Ministry of Education and Research (BMBF) (03F0775A), the BMBF project MicroCatch_Balt (03F0788A) and the BMBF project PLASTRAT (02WPL1446 J).
Acknowledgements
We thank the IOW workshop for the construction of the spiral conveyor and the clean bench setup, and Dr. Klaus-Jochen Eichhorn (Leibniz-Institute for Polymer Research), as well as Jakob Strand (Aarhus University) for the helpful advice on the development of the pentane protocol for improved spectroscopic analysis. Thanks also to Franziska Fischer and Dieter Fischer for their assistance in method validation.
Acknowledgments
Declaration of Competing Interest
The authors declare that there are no conflicts of interest.
Footnotes
Note: This document does not include a complete health and safety evaluation of the laboratory procedures. Please carry out a risk assessment for all field and lab work activities and adhere to the health and safety guidelines at your institute or organisation.
It was observed that changing the power resulted in a more efficient removal of matter.
To MPF-clean glass-fibre filters, transfer them to a clean petri dish and cover it with aluminium foil. Bake all filters at 500 °C for 5 h. Only remove the filters from the petri dish inside the bench.
Check filter after rinsing step under a binocular microscope for remaining material.
The use (or avoidance) of an ultra-sonification should be conducted in a systematic way within one study.
Remaining solutes can influence spectroscopic results.
Cooling the extracted MP sample prevents new biofilms to establish, which would hamper spectroscopic analysis.
If the sample is in a glass container, there is a risk of the glassware breaking as a function of the increasing volume. Placing the sample at a <45° angle can improve matters as this increases the expandable surface area. However, the use of non-breakable MPF containers, such as stainless steel or aluminium, is an ideal alternative for large water volumes.
Mixing times need to be adjusted depending on sample volume and container.
Dry sieving should only be applied if unavoidable as it comes at the risk of losing small particles either during the transfer processes or due to adherence on larger particle surfaces. If applied, use it consistently for the entire data set.
None of the chemicals proposed here are listed under “severely hazardous” (class 3) in the water hazard class (German Water Hazard Classification).
Bubbles indicate that the oxidation of the organic matter started (CO2 is formed as a result of the chemical reaction between H2O2 and organic C). Bubble formation can be delayed by several minutes to hours after addition.
This recommendation aims to provide H2O2 in excess to the available digestible OM.
To estimate the required amount of digestion solution measure the wet weight of the samples.
Droplets of fat or oily remains may or may not be visible, but fatty acids, their salts or alcohol combinations can be identified by spectroscopic analysis if present in the sample.
Based on theoretical chemical considerations, it is not expected that the protocol could impact the integrity, numbers or spectral recognisability of common plastic polymers. However, a detailed test on actual MP is yet to be completed.
Pentane has a boiling point of 36 °C, pressure may build up in the tightly closed containers.
The pentane phase should not contain any MP due to a lower density (630 kg m−3) than that of plastics and low surface tension. This was checked by observing the behaviour of standard fluorescent PE spheres mixed in a water/pentane dispersion.
Low density MP particles might float directly in the phase boundary.
Maximum filling level of the separation funnels should not exceed 2/3 of the total volume. If samples with high matter content (e.g. sediment, soils, etc.) are treated and the application of the spiral conveyor is needed, use the appropriate separation funnels equipped with a wider neck and wider outlet valve (see section: Modified separation funnels - a versatile application for particle-rich samples).
The here-presented set up also allows for sieving without a funnel, as the neck of the separation funnel fits the size of the small sieves applied. However, using a funnel potentially prevents loss of material.
Be careful when adding H2O2 to the density separation. Always add only a few droplets to prevent the risk of strong foam formation and loss of sample material. Consider that the addition of H2O2 dilutes the density of the solution, therefore adjust or use an initially higher density.
The time reported here is based on sufficient extraction results based on test MP particles (125–150 μm) in very fine sediments (40–70 μm) in a 500 ml separation funnel (see section: Conical spiral conveyor - a gentle way to separate particles). Larger volumes or more complex sample compositions may require longer time. If in doubt, test the specific sediment matrix with recovery particles.
In order to recover potential MP that have become trapped close to the outlet of the separation funnel.
The required rising time (waiting time) is calculated based on a spherical 10 μm particle of a density of 1.7 g cm−3 that travels a certain distance measured between the bottom to the top of the separation phase in SPT of 1.8 g cm−3 (Stokes law). Example: For a distance of 13.5 cm (maximum height experienced by the authors) this particle needs 13.7 h to rise to the top (1.1 h for 1 cm).
The pore size of the filter used here is 1 mm, which is based on the experiences made by the authors especially when treating Manta net samples. However, this needs to be evaluated case by case and filters can be changed or substituted with additional filters depending on the specific sample compositions.
The step of flushing and rinsing the filters/sieves used to perform size-compartmentalisation of the sample must be executed exhaustively in order to prevent smaller particles remaining within the larger size fractions as they may otherwise be lost for analysis.
The pre-analysis of plastic suspects should be executed in a conservative manner, which increases the number of false-positives, however, this also prevents false-negative particles and thus the loss of particles. False-positives will be identified and eliminated in the later chemical analysis.
MPF MQ is recommended here, to dilute and wash out solutes in and on the picked particles, as they are subjected to spectroscopic analysis thereafter.
Based on the subsample of 40 particles which undergo chemical analysis, the remaining particles will be extrapolated for plastic/non-plastic origin, as well as polymer type distribution. If the analysed fraction of particles is too diverse, a larger subsample can be compiled.
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.mex.2020.100784.
Contributor Information
Kristina Enders, Email: kristina.enders@io-warnemuende.de.
Robin Lenz, Email: robin.lenz@io-warnemuende.de.
Juliana A. Ivar do Sul, Email: juliana.ivardosul@io-warnemuende.de.
Alexander S. Tagg, Email: alexander.tagg@io-warnemuende.de.
Matthias Labrenz, Email: matthias.labrenz@io-warnemuende.de.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- 1.Zarfl C. Promising techniques and open challenges for microplastic identification and quantification in environmental matrices. Anal. Bioanal. Chem. 2019;411:3743–3756. doi: 10.1007/s00216-019-01763-9. [DOI] [PubMed] [Google Scholar]
- 2.Cole M., Webb H., Lindeque P.K., Fileman E.S., Halsband C., Galloway T.S. Isolation of microplastics in biota-rich seawater samples and marine organisms. Sci. Rep. 2014;4:4528. doi: 10.1038/srep04528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dehaut A., Hermabessiere L., Duflos G. Current frontiers and recommendations for the study of microplastics in seafood. TrAC Trends Anal. Chem. 2019;116:346–359. [Google Scholar]
- 4.Imhof H.K., Schmid J., Niessner R., Ivleva N.P., Laforsch C. A novel, highly efficient method for the separation and quantification of plastic particles in sediments of aquatic environments. Limnol. Oceanogr. Methods. 2012;10:524–537. [Google Scholar]
- 5.Lusher A.L., Welden N.A., Sobral P., Cole M. Sampling, isolating and identifying microplastics ingested by fish and invertebrates. Anal. Methods. 2017;9:1346–1360. [Google Scholar]
- 6.Fischer D. Leibniz-Institute for Polymer Research (IPFDD); 2019. Personal Communication. [Google Scholar]
- 7.Alcântara D.B., Fernandes T.S.M., Nascimento H.O., Lopes A.F., Menezes M.G.G., Lima A.C.A., Carvalho T.V., Grinberg P., Milhome M.A.L., Oliveira A.H.B., Becker H., Zocolo G.J., Nascimento R.F. Diagnostic detection systems and QuEChERS methods for multiclass pesticide analyses in different types of fruits: an overview from the last decade. Food Chem. 2019;298 doi: 10.1016/j.foodchem.2019.124958. [DOI] [PubMed] [Google Scholar]
- 8.Anastassiades M., Lehotay S.J., Štajnbaher D., Schenck F.J. 2003. Fast and Easy Multiresidue Method Employing Acetonitrile Extraction/Partitioning and “Dispersive Solid-Phase Extraction” for the Determination of Pesticide Residues in Produce 21. [PubMed] [Google Scholar]
- 9.Buede D.M., Miller W.D. Wiley; 2016. The Engineering Design of Systems: Models and Methods, Wiley Series in Systems Engineering and Management. [Google Scholar]
- 10.Marshall R., Leaney P.G. A systems engineering approach to product modularity. Proc. Inst. Mech. Eng. Part B: J. Eng. Manuf. 1999;213:847–851. [Google Scholar]
- 11.Hidalgo-Ruz V., Gutow L., Thompson R.C., Thiel M. Microplastics in the marine environment: a review of the methods used for identification and quantification. Environ. Sci. Technol. 2012;46:3060–3075. doi: 10.1021/es2031505. [DOI] [PubMed] [Google Scholar]
- 12.Enders K., Lenz R., Beer S., Stedmon C.A. Extraction of microplastic from biota: recommended acidic digestion destroys common plastic polymers. ICES J. Mar. Sci. 2017;74:326–331. [Google Scholar]
- 13.Strand J., Tairova Z. Aarhus University, DCE – Danish Centre for Environment and Energy; 2015. Microplastic Particles in North Sea Sediments (No. No. 178) [Google Scholar]
- 14.Albalá-Hurtado S., Pascual-Sastre L.E., Vidal-Carou M.C., Mariné-Font A., Permanyer-Fàbregas J. Comparison of two fat extraction methods in powdered infant milks. J. Food Compos. Anal. 1999;12:333–337. [Google Scholar]
- 15.Dieffenbacher A., Lüthi B. Die Direkte Kolorimetrische Bestimmung der Peroxidzahl (POZ) in Milchprodukten. Mitteilungen aus dem Gebiete der Lebensmitteluntersuchung und Hygiene. 1986;77:544–553. [Google Scholar]
- 16.Fischer M., Scholz-Böttcher B.M. Simultaneous trace identification and quantification of common types of microplastics in environmental samples by pyrolysis-gas chromatography-mass spectrometry. Environ Sci Technol. 2017;51(9):5052–5060. doi: 10.1021/acs.est.6b06362. [DOI] [PubMed] [Google Scholar]
- 17.Coppock R.L., Cole M., Lindeque P.K., Queirós A.M., Galloway T.S. A small-scale, portable method for extracting microplastics from marine sediments. Environ. Pollut. 2017;230:829–837. doi: 10.1016/j.envpol.2017.07.017. [DOI] [PubMed] [Google Scholar]
- 18.Fries E., Dekiff J.H., Willmeyer J., Nuelle M.-T., Ebert M., Remy D. Identification of polymer types and additives in marine microplastic particles using pyrolysis-GC/MS and scanning electron microscopy. Environ. Sci. Process. Impacts. 2013;15:1949–1956. doi: 10.1039/c3em00214d. [DOI] [PubMed] [Google Scholar]
- 19.Mani T., Frehland S., Kalberer A., Burkhardt-Holm P. Using castor oil to separate microplastics from four different environmental matrices. Anal. Methods. 2019;11:1788–1794. [Google Scholar]
- 20.Nakajima R., Tsuchiya M., Lindsay D.J., Kitahashi T., Fujikura K., Fukushima T. A new small device made of glass for separating microplastics from marine and freshwater sediments. PeerJ. 2019;7:e7915. doi: 10.7717/peerj.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zobkov M.B., Esiukova E.E. Evaluation of the Munich Plastic Sediment Separator efficiency in extraction of microplastics from natural marine bottom sediments. Limnol. Oceanogr. Methods. 2017;15:967–978. [Google Scholar]
- 22.Claessens M., Van Cauwenberghe L., Vandegehuchte M.B., Janssen C.R. New techniques for the detection of microplastics in sediments and field collected organisms. Mar. Pollut. Bull. 2013;70:227–233. doi: 10.1016/j.marpolbul.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Hurley R.R., Lusher A.L., Olsen M., Nizzetto L. Validation of a method for extracting microplastics from complex, organic-rich, environmental matrices. Environ. Sci. Technol. 2018;52:7409–7417. doi: 10.1021/acs.est.8b01517. [DOI] [PubMed] [Google Scholar]
- 24.Miller M.E., Kroon F.J., Motti C.A. Recovering microplastics from marine samples: a review of current practices. Mar. Pollut. Bull. 2017;123:6–18. doi: 10.1016/j.marpolbul.2017.08.058. [DOI] [PubMed] [Google Scholar]
- 25.Hengstmann E., Tamminga M., vom Bruch C., Fischer E.K. Microplastic in beach sediments of the Isle of Rügen (Baltic Sea)—implementing a novel glass elutriation column. Mar. Pollut. Bull. 2018;126:263–274. doi: 10.1016/j.marpolbul.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 26.Liebezeit G., Dubaish F. Microplastics in beaches of the East Frisian islands Spiekeroog and Kachelotplate. Bull. Environ. Contam. Toxicol. 2012;89:213–217. doi: 10.1007/s00128-012-0642-7. [DOI] [PubMed] [Google Scholar]
- 27.Moore C.J., Moore S.L., Weisberg S.B., Lattin G.L., Zellers A.F. A comparison of neustonic plastic and zooplankton abundance in southern California’s coastal waters. Mar. Pollut. Bull. 2002;44:1035–1038. doi: 10.1016/s0025-326x(02)00150-9. [DOI] [PubMed] [Google Scholar]
- 28.Thompson R.C., Olsen Y., Mitchell R.P., Davis A., Rowland S.J., John A.W.G., McGonigle D., Russell A.E. Lost at sea: where is all the plastic? Science. 2004;304:838. doi: 10.1126/science.1094559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.