

Abstract
Blocking the biological functions of scaffold proteins and aggregated proteins is a challenging goal. PROTAC proteolysis-targeting chimaera (PROTAC) technology may be the solution, considering its ability to selectively degrade target proteins. Recent progress in the PROTAC strategy include identification of the structure of the first ternary eutectic complex, extra-terminal domain-4-PROTAC-Von-Hippel-Lindau (BRD4-PROTAC-VHL), and PROTAC ARV-110 has entered clinical trials for the treatment of prostate cancer in 2019. These discoveries strongly proved the value of the PROTAC strategy. In this perspective, we summarized recent meaningful research of PROTAC, including the types of degradation proteins, preliminary biological data in vitro and in vivo, and new E3 ubiquitin ligases. Importantly, the molecular design, optimization strategy and clinical application of candidate molecules are highlighted in detail. Future perspectives for development of advanced PROTAC in medical fields have also been discussed systematically.
KEY WORDS: Protein degradation, PROTAC, Ubiquitin−proteasome system, E3 ubiquitin ligase, Target protein, Heterobifunctional molecule
Graphical abstract
Strategies for protein degradation include simulating misfolded proteins through hydrophobic tagging; causing proteins to be hijacked by E3 ubiquitin ligase and ubiquitinated through proteolysis-targeting chimaeras (PROTAC). Stable PROTAC ternary complex, which forms new protein–protein interactions (PPIs) by protein with E3 ligase, is essential for protein degradation.

1. Introduction
Proteins play critical roles in maintaining the life of organisms1, 2, 3. Correct protein folding controls cell health and survival4, 5, 6. However, most proteins are inherently prone to aggregation in their misfolded or partially misfolded state7. In addition, misfolding or misregulation of proteins leads to the development of many diseases, including neurodegenerative diseases, cancers and type 2 diabetes mellitus (T2DM)8, 9, 10. Therefore, cells must constantly adjust their protein composition to maintain normal proteomes11. Misfolded proteins are refolded or degraded by quality control systems12, 13, and elimination of misfolded proteins is critical for maintaining protein homeostasis and cell viability14.
Under physiological conditions a complex network that includes folding enzymes, chaperones, lectins and ATP-driven motors controls the elimination of misfolded proteins. The ubiquitin-proteasome system (UPS) and autophagy are the two major intracellular pathways for protein degradation15, 16, 17, 18. The UPS and autophagy have long been considered as independent degradation pathways with little or no interaction points. In spite of growing evidence of close coordination and complementarity between the two systems19, they are actually different mechanisms: UPS is responsible for the degradation of short-lived proteins and soluble misfolded proteins, whereas autophagy eliminates long-lived proteins, insoluble protein aggregates and even whole organelles (such as mitochondria, peroxisomes), macromolecular compounds, and intracellular parasites (e.g., certain bacteria)20, 21.
In addition, small interfering RNA (siRNA)22 and clustered regularly interspaced short palindromic repeats/associated protein 9 nuclease (CRISPR-Cas9)23 technologies can also down-regulate or eliminate proteins. However, these 2 technologies also have limitations: for example, CRISPR-Cas9 technology has undesired off-target effects and low efficiency, which limit its application in vivo24. Inefficient delivery to target cells in vivo and non-specific immune responses following systemic or local administration are barriers for the clinical application of siRNA. Researchers are still developing various technology platforms to improve in vivo delivery of therapeutic siRNA25.
In addition, heat shock proteins (HSPs) also play important roles in protein kinase degradation26. For example, the level of many oncogenic kinases, such as ERBB2, BRAF-V600E, FGFR-G719S and BCR-ABL, are reported to be tightly coupled to heat shock protein 90 (HSP90)27.
The methods mentioned above for controlling protein degradation are mostly achieved via biomacromolecules. In order to target a broader range of proteins with sufficiently high efficiency for clinical application, in recent years pharmaceutical researchers have developed a series of new strategies for protein degradation using small molecules. One representative strategy is proteolysis-targeted chimera (PROTAC) that degrades proteins by hijacking the UPS28, 29, 30, 31, 32. PROTAC is a bifunctional-hybrid molecule that binds both E3 ubiquitin (U) ligase and target proteins, thereby leading to the exposed lysine on the target protein being ubiquitinated by the E3 ubiquitin ligase complex, followed by UPS-mediated protein degradation33. Theoretically, PROTACs not only provide binding activity, but also have great potential to eliminate protein targets that are “undruggables” by traditional inhibitors or are non-enzymatic proteins34, 35, e.g., transcription factors36, 37. In addition, the PROTAC technique is “event-driven”, which does not require direct inhibition of the functional activity of the target protein. These characteristics make PROTAC technology an attractive strategy for targeting protein degradation (TPD).
In this review we summarize the unique advantages and the core design philosophy of PROTAC through representative examples of PROTAC use in recent years. We also highlight the discovery of E3 ubiquitin ligase, the development and optimization of corresponding ligands, and its application to PROTAC technology. We also introduce rapid synthesis of PROTAC based on “click chemistry” reactions. Finally, we present the opportunities and risks of this emerging PROTAC technology in clinical applications. In summary, this work will provide insights for discovering new E3 ubiquitin ligases and designing PROTACs.
2. Degradation of protein by misfolded protein simulator
2.1. Hydrophobic tagging
The hydrophobic tagging (HyT) technology extends the concept of inducing protein instability to a broader range of protein targets by mimicking protein misfolding38. The HyT consists of a hydrophobic fragment and a ligand fragment of the protein of interest (POI), which is capable of causing degradation of the POI (Fig. 1A)22, 39, 40. One mechanism is that the HyT destabilizes the POI, thereby recruiting an endogenous chaperone protein to the misfolded protein and then degrading the protein by the proteasome; another mechanism is the direct recognition of the HyT by chaperones, mediating the proteasomal degradation of the tagged protein. The hydrophobic marker then is released and the POI can be destroyed in successive rounds22. However, the current literature does not prove the hydrophobic marker is completely released in the protein degradation process induced by HyT, and remains to be further studied.
Figure 1.

The hydrophobic tagging (HyT) technology. (A) The strategy of protein degradation through induced protein misfolding (or mimicking misfolding) with HyT using bifunctional adamantly-taggd molecules. (B) Degradation of misfolded proteins through HSP70/40 chaperones under normal physiological conditions. (C) The chemical structures of representative HyTs.
Protein ubiquitination and degradation can be achieved by recruiting chaperones using lipophilic small molecule tags. For example, HSP70 family members recognize the exposed hydrophobic cores of misfolded proteins to hijack misfolded protein reactions41. HSP70 is highly conserved and ubiquitous in microorganisms, plants and animals42, and is involved in many cellular processes, including protein folding, transmembrane protein translocation and protein degradation regulation43. Proteins with mild or partial misfolding are ubiquitinated by HSP40 and HSP70 and then degraded by HSP70 and 26S proteasomes44 (Fig. 1B).
The HyT technology was further extended to degrade endogenous proteins such as human epidermal growth factor receptor 3 (HER3), a kinase playing important roles in cancer45. The effective ligand of HER346 is coupled to the adamantane moiety via a short linker to form a HyT degrader known as TX2-121-1 (1) (Fig. 1C). Covalent binding of 1 to HER3 resulted in HER3 degradation at 500 nmol/L and induced HER3-dependent cell death at an EC50 of 0.8–1.4 μmol/L45. However, the degradation of HER3 using HyT technology still relies on covalent interactions, which are stoichiometric rather than substoichiometric.
The breast cancer drug fulvestrant was originally designed as a selective estrogen receptor modulator (SERM), but was later found to induce degradation of the estrogen receptor alpha (ERα) receptor47. By inducing a conformational change to the receptor, fulvestrant causes ERα to expose a hydrophobic side chain mimicking the misfolded portion of the ERα protein recognized by the cell housekeeper, resulting in degradation of the ERα protein48. In 2002, fulvestrant was approved by the FDA for treating ER-positive metastatic breast cancer49.
Inspired by the clinical success of fulvestrant, a series of selective androgen receptor degraders (SARD) were designed for high affinity to the androgen receptor (AR) agonist, with a polyethylene glycol (PEG) linker to a hydrophobic degron (an adamantyl group)50. As the first small molecule SARD51 (Fig. 1C), SARD279 (2) has a 50% degradation concentration (DC50) of 2 μmol/L. Researchers believe that HSPs may be involved in the mechanism of SARD-mediated AR degradation. After incubation with the potent HSP90 inhibitor geldanamycin, the level of HSP70 increased in a geldanamycin-dependent manner, which was consistent with the discovery that HSP90 inhibition resulted in the activation of heat shock factor 1 (HSF1) and its target genes (including HSP70)52. This suggests that HSP70 mediated the AR degradation and elevated HSP70 levels were the basis for the increased activities of SARD279 (2) in the context of HSP90 inhibition53.
The early HyT technology was based on the adamantane HyT strategy and has been applied to a broad range of objectives. In addition to adamantyl, tert-butyl carbamate-protected arginine (Boc3-Arg) can also be used as a HyT to induce protein degradation38. Although Boc3-Arg has a higher molecular weight than adamantane, the capability of Boc3-Arg to induce protein degradation was confirmed in 201254. The non-covalent inhibitor trimethoprim (TMP) binds Boc3-Arg to form TMP-B3A (3) (Fig. 2B), which degrades dihydrofolate reductase at a micromolar concentration. Similarly, the covalent inhibitor ethacrynic acid (EA) was ligated with Boc3-Arg to form EA-B3A (4) (Fig. 2B), which degraded Glutathione-S-transferase (GST). In addition, the protein degradation occurred in U- and ATP-independent manners by the proteasome54. Taking the GST protein degradation agent EA as an example, as a recognition fragment of the GST protein (Fig. 2B, the red part of the structure), the double bond of EA can alkylate the Cys residue in the active site of GST. Moreover, Boc3-Arg may bind to the 20S proteasome, which makes the GST protein passively captured and degraded by the 20S proteasome.
Figure 2.

The 20S proteasome. (A) Degradation of POI through 20S proteasome. (B) The representative chemical structures of Boc3-Arg.
The 20S proteasome is a 700 kDa barrel-shaped protein consisting of four loops with two stacked β loops sandwiched between two α loops55. The 20S proteasome is widely distributed throughout the cell and degrades most of the oxidized proteins in U and ATP-independent processes54, 56, 57, 58. The U pathway of the 20S proteasome is required for the degradation of oxidatively damaged proteins59. In addition, protein cofactors such as HSP90 can synergize with the 20S proteasome to promote protein degradation60. The 20S proteasome can also induce POI degradation in combination with HyT (Fig. 2A).
There are three possible mechanisms of Boc3-Arg-mediated degradation: First, the Boc3-Arg portion can enter the proteasome and “drag” the rest of the protein into the proteolytic chamber. Second, the Boc3-Arg group can be embedded in the target protein to expose its hydrophobic surface to interact with the 20S proteasome. Third, Boc3-Arg may interact with other protein factors such as HSP90.
However, how the Boc3-Arg portion targets the protein remains to be elucidated. A direct non-covalent interaction between Boc3-Arg and the 20S proteasome was discovered: Boc3-Arg activated the purified 20S proteasome, indicating that the tag binded directly to the 20S proteasome, and Boc3-Arg targeted the target protein to 20S proteasome61. In addition, the proteasome subunits α7 and β7 were strongly enriched in a resin-binding protein pool linked to Boc3-Arg. The purified 20S proteasome was sufficient to degrade the target protein in a Boc3-Arg-dependent manner in the presence of its respective Boc3-Arg. This was the first example using bifunctional small molecules to target protein directly to the 20S proteasome and degrade protein. The Boc3-Arg portion inhibits the translational machinery mediated by the mammalian target of rapamycin complex 1 (mTORC1) pathway, and the off-target effect may potentially limit the clinical application of Boc3-Arg as a HyT62.
The HyT method has used covalent and non-covalent ligands to initiate protein degradation. However, its physicochemical and pharmacokinetic (PK) properties may be the major challenge for clinical development.
2.2. Fusion-based degron (HaloTag)
Some protein fusion tags have been extensively studied: e.g., fluorescent proteins63, His tag64, FLAG tag65, etc. HaloTag is a modified bacterial dehalogenase enzyme that covalently binds with a hexyl chloride label. HaloTag fusion protein has been widely used as a biological orthogonal marker66, 67, e.g., in vivo molecular imaging, protein purification/transport, high-throughput detection, etc. HaloTag forms stable covalent bonds with compounds containing alkyl chlorides68 via a very simple binding moiety with low molecular weight and reasonable cell permeability69. More importantly, HaloTag has high selectivity and sensitivity70.
The HaloTag-based bifunctional molecule contains an alkyl chain HaloTag and a ligand that binds to the target protein. This bifunctional molecule transfers the fusion domain onto the POI, binds the bacterial HaloTag protein and generates a hydrophobic group on its surface, which is mediated by a chaperone. The protein then becomes unstable and is subsequently degraded by the proteasome (Fig. 3). More importantly, PK studies of adamantane-based HyT in mice indicated that 75% of HyT is still present after 24 h, and that HyT compounds also inhibited 80% of tumor growth in xenograft HaloTag-HRAS-G12V-driven mice70. Immunoprecipitation showed that the addition of the adamantane-labeled HaloTag fusion protein was associated with HSP70, suggesting that the adamantyl-mediated degradation is related to HSP7071.
Figure 3.

HyT strategy developed based on HaloTag fusion protein system for protein degradation.
3. PROTAC technology
Traditional small molecule inhibitors can inhibit the activity of some enzymes and block the function of certain disease-related proteins, but they still have some limitations in applications72, 73. TPD is a new direction in the field of drug discovery. Traditional small molecule inhibitors only block part of the protein's function, while TPD eliminates all the functions of the protein.
As a protein degradation system, UPS is a key post-translational modification process74 and is involved in protein quality control, antigen processing, signal transduction, cell cycle control, cell differentiation and apoptosis. UPS plays a key role in regulating protein homeostasis75, 76. E3 ubiquitin ligase is a specific substrate member of UPS and represents an attractive protein target for drug discovery. The molecular mechanism of proteasome protein degradation is driven by the sequential actions of three enzymes (E1-activation, E2-conjugation and E3-binding)77, 78 (Fig. 4A). First, with ATP as the energy source, the carboxyl group at the end of U glycine is linked to the thiol group of the U-activating enzyme E1 to form a thioester bond between U and E1. Second, E1 transfers the activated U to E2 through a lactide process. Third, E3 binds E2 U to the target protein and releases E2 to leave a specific ubiquitinated protein79. Finally, ubiquitinated proteins are recognized by specific proteasomes and degraded into short peptides or amino acids by proteases80.
Figure 4.

The ubiquitin−proteasome system and PROTAC. (A) Ubiquitin (U) is activated by ubiquitin-activating enzyme (E1). Activated ubiquitin is transferred in thioester linkage from E1 to ubiquitin-conjugating enzyme (E2). Ubiquitin ligase (E3) catalyzes the transfer of ubiquitin from E2 to substrate via lysine residues. (B) The PROTAC bind both the target protein and the E3 ligase simultaneously to induce the formation of a ternary complex. The target protein is then polyubiquitinated and undergoes proteolysis. A PROTAC molecule consists of a ligand for recruiting E3, a linker, and a ligand binding to POI.
In physiological conditions E3 ubiquitin ligase requires a special recognition signal to recruit and ubiquitinate its target protein81. Generally, ubiquitination occurs on the lysine residues of a target protein. PROTAC is a bifunctional-hybrid compound consisting of three parts: one side is the ligand to the target protein, the opposite side is the ligand to the E3 ubiquitin ligase, and the middle is a linker connecting two ligands82. Small molecule PROTAC can bind to both E3 ubiquitin ligase and the target protein and induce the formation of a ternary complex which leads to polyubiquitination and subsequent degradation of the target protein (Fig. 4B). Meanwhile, PROTAC can be recycled for subsequent rounds of degradation83.
3.1. The first generation of peptide PROTAC
In 2001 the first bifunctional molecule compound 5 was reported (Fig. 5B), which induced the degradation of the target protein methionine aminopeptidase-2 (MetAP-2) by recruiting the ubiquitinated protein β-TRCP (F-box protein) in a compound 5-dependent manner84 (Fig. 5A). To overcome the impermeability of SCFα-TRCP binding to the IB phosphopeptide moiety, injection of β-TRCP-based PROTAC into HEK293 cells by microinjection was found to induce AR degradation85. It was the first time that the concept of PROTAC was clearly proposed with a comprehensive validation in vitro. Other small molecule protein-targeting chimeras were also developed to specifically degrade target proteins by recruiting E3 ubiquitin ligase86. However, the first generation of PROTAC were polypeptides that did not have favorable physiochemical properties for therapeutics. Besides a high molecular weight and low activity, the major problem of the peptide-based PROTAC is the poor permeability to intracellular targets87.
Figure 5.

The first PROTAC. (A) A schematic diagram of the first PROTAC. This PROTAC contains a phosphopeptide derived from IκBα to recruit the E3 ubiquitin ligase SCFβ-TRCP, a linker and ovalicin to bind with MetAP-2, which triggers MetAP-2 ubiquitination. (B) Structure of first PROTAC, compound 5. Blue: ligand for target proteins is shown; Red: ligand for recruiting E3 ubiquitin ligase.
3.2. KEAP1-dependent peptide PROTAC
Kelch-like ECH-associated protein 1 (KEAP1) is a Cullin3–RBX1 U ligase substrate adaptor protein88 containing a BTB domain at its N-terminus. The BTB domain mediates the interaction with Cullin3 (Cul3)89. KEAP1 acts as a sensor for reactive oxygen species (ROS) and protects cells from oxidative damage90. One substrate for the KEAP1−Cul3 complex is nuclear factor erythroid 2-related factor 2 (NRF2). NRF2 is an important transcription factor for regulating oxidative/xenobiotic stress response and anti-inflammation91.
Ubiquitination is the most important pathway to regulate the activity of NRF292. Three E3 ligases have been discovered to control the ubiquitination of NRF2, namely KEAP1−Cullins3−RING-box protein 1 (KEAP1−Cul3−RBX1)93, β-TRCP-S-phase kinase-associated protein 1−Cul1−RBX1 (SKP1−Cul1−RBX1)94, and HMG-COA reductase degradation 1 homologue (HRD1)95. Under normal conditions KEAP1 inhibits NRF2 signaling through UPS96. After exposure to electrophiles, ROS, and reactive nitrosated species (RNS) or heavy metals, the cysteine residue modification in KEAP1 results in a conformational change in the complex to impede the proteasome-mediated degradation of NRF2. In this case NRF2 remains stable and accumulates in the nucleus97.
The development of PROTAC with these potent small molecule KEAP1−NRF2 inhibitors may be a better strategy for inhibiting the protein−protein interaction (PPI) of KEAP1−NRF2 (Fig. 6). The KEAP1−NRF2 signaling pathway might be a valuable therapeutic target for the treatment of neurodegenerative diseases98. A peptide-PROTAC which recruits KEAP1−Cul3 U E3 ligase is reported to induce Tau degradation in cells34. The peptide 1 strongly binds to KEAP1 and Tau in vitro with Kd values of 22.8 and 763 nmol/L, respectively. Peptide 1 induces the degradation of Tau in a time- and concentration-dependent manner, suggesting the usage of PROTAC to recruit KEAP1 to induce Tau degradation to treat neurodegenerative diseases.
Figure 6.

Development of PROTAC based on KEAP1 as E3 ubiquitin ligase. (A) Under physiological conditions, endogenous substance NRF2 is recognized by KEAP1, ubiquitinated, and degraded by the proteasome. (B) KEAP1 inhibitors block the interaction of KEAP1−NRF2, leading to stabilization and activation of NRF2. (C) PROTAC developed based on KEAP1 inhibitors is used for the degradation of POI.
Subsequent research into PROTAC has focused on the development of small molecule PROTAC technology with in vivo stability.
4. Small molecule PROTAC
There has been great progress in the discovery of E3 ubiquitin ligases: more than 600 human genome-encoded E3 ubiquitin ligases have been identified99. Von-Hippel-Lindau (VHL), murine double minute 2 (MDM2), cell inhibitor of apoptosis protein (cIAP) and CRBN (cereblon), have been utilized successfully for small molecule PROTAC, and these molecules are likely to become therapeutic candidates100. However, which other E3 ligases can be recruited to PROTAC needs further study.
4.1. MDM2-based PROTAC
MDM2 is a key oncogenic protein that contributes to cell growth, survival, invasion and chemotherapy resistance in cancer101. As a negative regulator of the tumor suppressor P53, MDM2 directly binds to the transactivation domain (TAD) of P53 protein and blocks P53 transcriptional activity102, 103. The P53 protein upregulates MDM2 gene expression levels, while MDM2 promotes P53 export from the nucleus and promotes its proteasome-mediated degradation104. In addition, MDM2 also acts as an E3 ubiquitin ligase to reduce P53 levels105, 106. Since P53 regulates many important processes in cells, including DNA repair, cell cycle arrest and apoptosis, and tumor therapy107, 108, it is important to maintain an appropriate amount of P53 in the nucleus. Therefore, inhibiting the interaction between P53 and MDM2 to restore normal P53 levels may be a good strategy109, 110.
In recent years, several small molecule inhibitors of the P53−MDM2 interaction have been developed111. One class are cis-imidazoline derivatives called nutlins112. In this class, compound 6 (nutlin-3) had the best inhibitory activity (Fig. 7A). In addition, compound 6 also inhibits cancer cell growth, cell migration and induces apoptosis113, 114. Compound 6 act as a ligand for MDM2 to recruit E3 ubiquitin ligase115. On this basis, a new generation of molecules were developed, including RG7112 (7)116 and RG7388 (8)117 (Fig. 7A). Their inhibitory efficiency is significantly higher than that of nutlin-3. The development of PROTACs based on MDM2 as E3 ubiquitin ligase is shown in Fig. 7B.
Figure 7.

Structures of MDM2 ligands and development of PROTAC based on MDM2. (A) Structures of MDM2 ligands (nutlin-3, 6) and a new generation of nutlin-like molecules (7 and 8). (B) Development of PROTAC based on MDM2 as E3 ubiquitin ligase. Under normal physiological conditions, endogenous substance P53 is recognized by MDM2, ubiquitinated and degraded by the proteasome. MDM2 inhibitors block the interaction of MDM2−P53, leading to stabilization and activation of P53. PROTAC developed based on MDM2 inhibitors is used for the degradation of POI.
In 2008 researchers reported the first all-small molecule PROTAC, compound 9115, which targeted the AR in HeLa cells. Compound 9 was a combination of a non-steroidal AR ligand and an imidazoline derivative known to bind to MDM2 via a PEG linker. The soluble PEG linker provides compound 9 with an acceptable level of cell permeability. After incubation of HeLa cells with compound 9 significant degradation of AR can be achieved at micromolar concentrations, with a DC50 value of 10 μmol/L.
Poly(ADP-ribose) polymerases (PARPs) are post-translational modification enzymes that play an important role in DNA repair118, 119. Among them, PARP1 is a valid target for cancer treatment120. Recently, an alkyne analogue of niraparib and the compound 6 derivative were linked by “click chemistry” to develop the PARP1 degradation inducer compound 10121. Compound 10 selectively induced significant PARP1 degradation in MDA-MB-231 cell line with a potency 5-fold higher than that of niraparib, olaparib and veliparib. The maximal level of degradation (Dmax) of 10 was 70% with a DC50=4–6 μmol/L. This indicates that this PARP1-targeting PROTAC has great potential for treating the MDA-MB-231 cell-like subtype of triple negative breast cancer (TNBC).
Recently a PROTAC, A1874 (11)122 targeting BRD4 was developed by linking a bromodomain and an extra-terminal (BET) inhibitor and a MDM2 inhibitor with a 13-atom long PEG linker. Different concentrations of 11 were applied to the colon cancer cell line HCT116, and the Dmax=98% at 100 nmol/L, which yielded a dose-dependent degradation of BRD4. In addition, the P53 level in HCT116 cells also showed dose-dependent stabilization. In particular, its potency (DC50=32 nmol/L) is significantly higher than that of 9 (DC50=10 μmol/L). The MDM2-based PROTAC can lead to oncoprotein degradation, indicating that these PROTACs have great potential as cancer therapeutics.
Structures and protein degradation activities of MDM2-based PROTACs 9–11 are shown in Fig. 8 and Table 1.
Figure 8.

Structures of PROTACs 9–11 based on MDM2 as E3.
Table 1.
PROTACs 9–11 with the ability to degrade target proteins developed based on MDM2 as E3a.
| Compd. | Target | Degradation in cell lines |
Ref. No. (Year) | |
|---|---|---|---|---|
| DC50 | Dmax (%) | |||
| 9 | AR | 10 μmol/L | NA | 115 (2008) |
| 10 | PARP1 | 4–6 μmol/L | 70 | 121 (2019) |
| 11 | BRD4 | 32 nmol/L | NA | 122 (2019) |
DC50: the concentration at which 50% degradation was observed. Dmax: the maximal level of degradation. NA: not available.
4.2. cIAP1-based PROTAC
“Inhibitor of apoptosis” proteins (IAPs) were first discovered in baculovirus123 and play an important role in maintaining cellular homeostasis. They also control a range of biological processes such as the inflammatory response, cell death, cell division, cell proliferation and cell differentiation124, 125. Among them, XIAP, cIAP1, cIAP2, LIVIN/ML-IAP and IAP-like protein 2 have a conserved RING domain at their C-terminus, and the RING domain binds E2 U-conjugating enzymes (UBCs). This allows the RING-containing protein to become an E2 U-binding enzyme that catalyzes the recruitment of U into the target protein. The RING domain is endowed with E3 ubiquitin ligase activity, which acts as an E3 ubiquitin ligase126, 127, 128.
cIAP1 is overexpressed in many cancers and down-regulating cIAP1 expression may be a promising approach for cancer treatment129. Bestatin is an aminopeptidase inhibitor with immunomodulatory activity and is approved for the treatment of acute non-lymphocytic leukemia in Japanese adults130. Studies have shown that bestatin-methyl ester MEBS (12) selectively down-regulate cIAP1 (Fig. 9A). 12 directly interacts with the cIAP1-BIR3 domain, promoting self-ubiquitination depending on its RING domain and subsequent proteasomal degradation of cIAP1131. In 2007, researchers reported that a new class of small molecule IAP antagonists binds to the BIR domain of IAP proteins, leading to the rapid ubiquitination and proteasomal degradation of cIAP proteins132. To elucidate the mechanism of IAP antagonism, a structure-based design was used to develop a novel monovalent MV1 (13) targeting the IAP protein, yielding a pan-antagonist of all XIAP, cIAP1 and cIAP2133 (Fig. 9A).
Figure 9.

cIAP ligands and PROTACs. (A) Development of cIAP ligands 12–14. (B) Structures of PROTACs 15–23 based on cIAP as E3.
Both 12 and 13 can induce self-ubiquitination and proteasomal degradation of cIAP1 E3 ligase131, 133, which may limit protein knockdown efficacy. Based on this, a number of IAP antagonists have been developed, and some have been evaluated as anti-tumor drugs in clinical phase studies134, 135. These IAP antagonists have higher affinity for IAP than 12, and they have been used to develop a compound with potent protein knockdown activity. This LCL 161 derivative (14) (Fig. 9A) has been used as an IAP ligand136.
Coupling 12 and all-trans retinoic acid (ATRA) with a spacer yielded “specific and non-genetic IAP-dependent protein eraser 4” (SNIPER (4)) (15)137, which selectively induced degradation of cellular retinoic acid-binding proteins-I and II (CRABP-I and –II). At the concentrations of 10 and 30 μmol/L 15 significantly reduced cell migration by about 75% and 95%, respectively. However, the ester type 15 had poor selectivity to its target protein, and the ester group could be easily hydrolyzed in cells. Therefore, an amide-type SNIPER (6) (16) capable of overcoming these problems was developed: 16 selectively induced degradation of CRABP-II but did not induce IAP138. Later, researchers developed compound 17 by combining ATRA and 12 through a linker moiety, which could induce the degradation of cIAP1 and CRABP-II in a proteasome-dependent manner at a concentration of 1 μmol/L. It also effectively inhibited the proliferation of IMR32 cells139.
In 2013 the Naito140 team used 12 and 4-hydroxy tamoxifen (4-OHT) to synthesize compound 18, which significantly reduced ERα and cIAP1 at 30 μmol/L. In order to increase the target protein degradation efficiency, they replaced 12 with the IAP antagonist 13, which had a higher affinity for IAP than bestatin. A series of SNIPER(ER) compounds containing different ER ligands or different linker lengths were developed with improved activities as compared to 18. Compound 19 was found to be more effective in reducing ER levels than 13. ER concentration was reduced by 50% at concentrations as low as 3 nmol/L, and maximum activity was observed at about 100 nmol/L, with a Dmax value of 70%136. In 2018 the Ohoka team141 reported that the derivatization of the IAP ligand module produced SNIPER(ER) with excellent ERα activity, preferentially recruiting XIAP instead of cIAP1. This improved SNIPER(ER) showed higher binding affinity to IAP and more efficient degradation of ERα than compound 19, with a maximum degradation of 70%. Of these SNIPER(ER)s, compound 20 (DC50=1–10 nmol/L, Dmax>80%) was more effective in inhibiting the growth of MCF-7 tumor xenografts in mice when compared to the previously characterized compound 19.
Transforming acidic-coiled coil 3 (TACC3) is a spindle regulatory protein that is overexpressed in many cancers, including ovarian cancer, breast cancer, squamous cell carcinoma, cell carcinoma and lymphoma142, 143, 144, indicating that TACC3 is a molecular target for anticancer drugs. Compound 21 significantly reduced TACC3 levels in human fibrosarcoma HT1080 cells (DC50=10–30 μmol/L, Dmax>50%)145.
In 2017 PROTAC bound to the allosteric site of the oncoprotein BCR-ABL was first reported146. SNIPER(ABL)-062 (22) had a relatively good binding affinity for ABL1, cIAP1/2 and XIAP. BCR-ABL protein could be efficiently and selectively degraded at 30 nmol/L (DC50=30–100 nmol/L, Dmax>70%) of 22. The method of degrading target proteins by binding ligands to the allosteric sites of target proteins provides new approaches to future protein degradation146.
The Naito147 team developed a novel SNIPER that induces proteasome degradation of the AR, which recruits IAP U ligase to degrade target proteins. Hybrid optimization strategies include: using multiple AR antagonists and E3 ligase ligands, and altering the linker and junction sites.
First, the bestatin moiety is replaced by 2 specific IAP antagonists, and a series of compounds are further derivatized by linking different linkers, such as amides, ethers, acetylenes or alkyl groups. Among them, SNIPER(AR)-51 (23) was developed by linking 14 and an AR antagonist with a PEG linker to effectively degrade AR. Based on compound 23, the effect of different attachment sites of the IAP ligand on the activity of SNIPER was further explored. The m-analogue showed the same ability to reduce the level of AR protein as the p-analogue, but the o-analogue showed no activity against AR protein at a concentration of 1 or 3 μmol/L. In evaluating the effect of the type of the linker on the activity of the SNIPER-degrading protein, the optimal protein degradation activity was found in the SNIPER containing the flexible PEG-linked chain. In summary, 23 presented the best performance in reducing AR protein compared with other compounds.
Structures and protein degradation activities of cIAP-based PROTACs 15–23 are shown in Fig. 9B and Table 2.
Table 2.
PROTACs 15–23 with the ability to degrade target proteins developed based on cIAP as E3a.
| Compd. | Target | Degradation in cell lines |
Ref. No. (Year) | |
|---|---|---|---|---|
| DC50 | Dmax (%) | |||
| 15 | CRABP-I, –II | NA | NA | 137 (2010) |
| 16 | CRABP-II | NA | NA | 138 (2011) |
| 17 | cIAPI, CRABP-II | NA | NA | 139 (2012) |
| 18 | ER | NA | NA | 140 (2013) |
| 19 | ER | 1–3 nmol/L | >70 | 136 (2017) |
| 20 | ER | 1–0 nmol/L | >80 | 141 (2018) |
| 21 | TACC3 | 10–30 μmol/L | >50 | 145 (2014) |
| 22 | BCR-ABL | 30–100 nmol/L | >70 | 146 (2017) |
| 23 | AR | 1–3 μmol/L | >80 | 147 (2018) |
DC50: the concentration at which 50% degradation was observed. Dmax: the maximal level of degradation. NA: not available.
4.3. VHL-based PROTAC
The VHL gene has been referred as a tumor suppressor148. Its germline mutations are associated with the inherited VHL cancer syndrome149. VHL is a component of the multi-subunit E3 ligase containing ELONGINB (ELOB), ELONGINC (ELOC), Cul2 and RBX1 (VBCCR complex)150. The major substrate for VHL is hypoxia inducible factor-1α (HIF-1α), a transcription factor that upregulates many proteins151, e.g., pro-angiogenic factors, vascular endothelial growth factor (VEGF), glucose transporters, GLUT1, etc. VHL recognizes and labels the prolyl-hydroxylated transcription factor HIF-1α and marks it to be degraded by the proteasome152, 153. HIF-1α is recruited to the CRL2VHL complex via the β domain of pVHL, which recognizes post-translational modifications of hydroxyproline. Once bound to CRL2VHL, HIF-1α is ubiquitinated and then undergoes proteasome-mediated targeted degradation154.
Fragment screening is widely used to find an attractive starting point in drug design. Ciulli et al.155 used it to study the interaction of pVHL: HIF1α, which was roughly divided into three sub-regions: the left side (LHS), the central core, and the right side (RHS). Compound 24 (hydroxyproline diamide 13) (Fig. 10A) had a fairly modest 0.01 nmol/L affinity, but it provided the greatest free energy for initial inhibitory binding to be a useful starting point for constructing promising small molecules. Later, they found (3R,4S)–F-Hyp (25) (Fig. 10B) acts as the best VHL ligand fragment to target protein degradation156.
Figure 10.

The development of VHL ligands. (A) 24 is a useful starting point for constructing VHL small molecule ligands. (B) 25 acts as the best VHL ligand fragment. (C) Development of VHL ligands. LE: ligand efficiency.
Based on these studies, researchers used the HIF-1α peptide structure as a starting point to design VHL ligands, and the first small molecule VHL ligand compound 26 was developed. However, its binding affinity to pVHL is not high (Kd=5.3 μmol/L), and its lipophilicity is poor157, 158. Later, a second VHL small molecule ligand 27 was developed with an affinity to VHL of Kd=1.5 μmol/L, and lipophilicity (LE=0.25), which are improved compared to the first VHL ligand159. Based on the structural design, the second-generation VHL small molecule ligand VHL-1 (28) was obtained (Kd=185 nmol/L and clogP=1.71). This ligand inhibitor is more active than the 10-mer HIF-1α peptide model, verifying that VHL was an excellent starting point as a drug target160.
The small molecule inhibitor compound 28 has limited activity and cannot induce intracellular HIF accumulation. One optimization strategy is to retain the carbonyl group on the LHS to maintain the hydrogen bond between the bag and the structured water, and replace the three hydrogens on the LHS with electron-withdrawing groups such as cyano and cyclopropyl, yielding VH298 (29). VH298 is the primary target for VHL with Kd=185 nmol/L and clogP=1.71, indicating that 29 can permeate the cell membrane more efficiently161, 162. The graphical representation of the discovery, development, and optimization of the VHL ligands discussed above is shown in Fig. 10C. VHL is widely used as a PROTAC-conjugated E3 ligase ligand for TPD, and the development of PROTACs based on VHL as an E3 ubiquitin ligase is shown in Fig. 11.
Figure 11.

Development of PROTAC based on VHL as E3 ubiquitin ligase. (A) Under normal physiological conditions, endogenous substance HIF-1α is recognized by VHL, ubiquitinated, and degraded by the proteasome. (B) VHL inhibitors block the interaction of VHL−HIF-1α, leading to stabilization and activation of HIF-1α. (C) PROTAC developed based on VHL inhibitors used for the degradation of POI.
In 2015, PROTAC_ERRα (30) and PROTAC_RIPK2 (31) which almost completely degrade estrogen-related receptor-α (ERRα) and RIPK2 with nanomolar activity were developed163. The ERRα levels were dose-dependently reduced in MCF-7 breast cancer cells after incubation with 30 (Dmax=86%, DC50 ∼100 nmol/L). After concentration escalations of 31, human THP-1 monocytes showed a stable and dose-dependent RIPK2 degradation (Dmax>95% at a concentration of 10 nmol/L and DC50=1.4 nmol/L). Furthermore, 30 reduced ERRα in mouse hearts, kidneys, and MDA-MB-231 tumors by approximately 44%, 44% and 39%, respectively. This was the first evidence that the small molecule VHL-based PROTACs have substoichiometric activity in a mouse model.
BET protein families, including BRD2, BRD3, BRD4 and testicular-specific BRDT, play an important role in cancers164. A VHL-based PROTAC ARV-771 (32) was developed165, which showed<5 nmol/L potency of BRD2/3/4 degradation in several prostate cancer cell lines (DC50<1 nmol/L, Dmax>90%). In addition, unlike BET inhibitors, 32 leads to inhibition of AR signaling and AR levels, and such an anti-proliferative effective is 500 times more active than other BET inhibitors in these cell lines. 32 also leads to tumor regression in castration-resistant prostate cancer (CRPC) mouse xenograft models. It not only validates BET protein degradation as a promising clinical strategy for anti-metastatic CRPC, but also demonstrates the feasibility of using PROTACs to treat small tumor-mediated protein-degrading solid tumor malignancies.
The dysregulation of BET protein, especially BRD4, is closely related to cancer and inflammations, making BET protein an attractive drug target166. JQ1 was linked to VHL to form PROTAC MZ1 (33)167, and more than 90% of the BET protein was degraded at a concentration as low as 1 μmol/L. The degradation of BRD4 (DC50<100 nmol/L, Dmax>90%) was stronger than BRD2 and BRD3. Researchers further developed AT1 (34) based on the crystal structure of 33168. After treatment with 34, BRD4 was significantly degraded (DC50=10–100 nmol/L, Dmax>90%), however, 34 had no effect on the levels of BRD2 and BRD3. VZ185 (35) induced a rapid and effective degradation of BRD7 and BRD9 within a few hours (BRD9: DC50=4 nmol/L; BRD7: DC50=34 nmol/L, Dmax=95%)169.
The VHL-based PROTAC 36 mediated the degradation of c-ABL170. TANK-binding kinase 1 (TBK1) is an atypical member of the IKK family of serine/threonine kinases involved in the development of innate immune responses, tumor and various cellular functions171, 172. Subsequently, researchers reported a proteasome-dependent PROTAC 37173. It was a potent TBK1 degrader (DC50=12 nmol/L, Dmax=96%), completely degraded TBK1 at the concentration of 100 nmol/L in several cancer cell lines, and had excellent selectivity over the related kinase IKK.
Using VHL-based PROCTACs to induce degradation of the active receptor tyrosine kinase (RTK) proved feasible174. Compound 38 targeted EGFR (epidermal growth factor receptor) (DC50=215.8 nmol/L); compound 39 targeted c-Met, contrary to inhibition of kinase activity, which was a strategy for degrading RTK. Recently, an FMS-like tyrosine kinase 3 (FLT-3) PROTAC 40175 was developed by linking the clinical candidate quizartinib176 (AC220) to the VHL ligand via an optimized linker. It effectively induced degradation of FLT-3 ITD protein in MV4-11 cells and MOLM-14 cells at nanomolar concentrations. In addition, 40 inhibited cell growth more effectively than a single warhead.
TRIM24 is a multidomain protein extensively characterized as a transcriptional co-regulator177. dTRIM24 (41) was synthesized using VHL and TRIM24178. Dose- and time-dependent degradation of TRIM24 was observed in 293FT cells (DC50=2.5–5 μmol/L and Dmax=70% at 5 μmol/L). As an emerging cancer-dependent chemical probe, 41 provides a good approach of utilizing selective but ineffective ligands of POI. The anaplastic lymphoma kinase (ALK)-PROTAC TD-004 (42) consists of ceritinib and VHL ligands to degrade the ALK fusion protein, such as NMP−ALK or EML4−ALK179. Compound 42 effectively inhibited cell proliferation of the SU−DHL-1 cancer cell line that expresses ALK-fusion protein (IC50=60 μmol/L, H3122, IC50=180 μmol/L). However, 42 did not inhibit the growth of the A549 cell line, which does not express the ALK-fusion protein (IC50>0.01 nmol/L). Focal adhesion kinase (FAK) is a key player in tumor invasion and metastasis180. Approved kinase inhibitor drugs have limited inhibition of FAK181. In contrast, the degradation of FAK may greatly reduce FAK signaling. Thus, researchers reported a selective and potent FAK-degrader, compound 43182 (DC50=3.0 nmol/L, Dmax=99%). It is regarded as a drug candidate for certain tumor diseases.
Recently, Shaomeng Wang group developed a group of potential PROTAC AR degraders using different linkers to link VHL ligands with different AR antagonists. The solubility of PROTAC depends on the linker, thus a pyridyl group directly linking the ethynyl group led to a good solubility of PROTAC. Among these PROTACs, ARD-69 (44) appeared to be the most effective AR degrader183. The DC50 values of 44 in the LNCaP and VCaP cell lines were 0.86 and 0.76 nmol/L respectively, and the Dmax>95% is at a concentration of 10 nmol/L. In 22Rv1 cells, the DC50=10.4 nmol/L, and 1 μmol/L of 44 almost completely degraded AR. 44 effectively inhibited the expression of the prostate specific antigen (PSA), TMPRSS2 and FKBP5 genes in LNCaP and prostate vertebral body cancer (VCaP) cell lines in a dose-dependent manner. It also reduced mRNA levels of the PSA and TMPRSS2 genes. In addition, 44 is 100 times more potent than the AR antagonist in inhibiting AR-regulated gene transcription in LNCaP and VCaP cells. AR signaling drives cell growth of AR-positive prostate cancer cells. Compound 44 strongly inhibited the growth of LNCaP, VCaP, and 22Rv1 cell lines and the IC50 values were 0.25, 0.34 and 183 nmol/L respectively, which were >100-fold more potent than the 2 AR antagonists. More importantly, 44 effectively reduced AR and PSA proteins in mouse VCaP xenograft tumor tissues and such an effect was sustained for at least for 48 h. Further optimization of 44 may lead to drug candidates for the treatment of metastatic castration-resistant prostate cancer (mCRPC).
PROTAC can lead to degradation of different target proteins. However, for closely related protein families, controlling the selectivity of degradation is still a challenge. In order to develop isoform-selective PROTACs selectively targeting p38α and p38δ in the p38 MAPK family, researchers used different linkers to link a single warhead (foreinib) with VHL184, as well as changing the orientation of the VHL recruiting molecule. Researchers found that PROTAC consisting of different attachment sites of VHL ligands yields selectivity to degrade different proteins. SJFα (45) (t-butyl position of VHL ligand) degraded the p38α in human breast cancer MDA-MB-231 cells (DC50=7.16 nmol/L, Dmax=97.4%), but the efficiency of degrading p38δ was significantly lowered (DC50=299 nmol/L, Dmax=18%). In addition, other p38 isoforms were not degraded (β and γ) at concentrations up to 2.5 μmol/L, indicating that 45 can selectively degrade p38α. However, SJFδ (46) (phenyl position of VHL ligand) is capable of degrading p38δ (DC50=46.17 nmol/L, Dmax=99.4%) but has no effect on the level of p38α, β or γ. This indicated that such PROTACs can selectively target protein members even in closely related protein families.
Interleukin-1 receptor-associated kinase 4 (IRAK4) is a serine/threonine kinase involved in toll-like receptors (TLRs) and white blood cells185. The transduction pathway stimulated by the interleukin-1 (IL-1) receptor family further leads to the activation of IRAK4, which is associated with various diseases such as psoriasis, rheumatoid arthritis and cancer186. Therefore, it is important to develop drugs that can promote IRAK4 protein degradation. Recently, the protein degradation drug development team of GlaxoSmithKline (GSK) Drug Research Center187 reported the results of a PROTACs study on IRAK4 protein. Compound 47 was most effective in PBMC cells (DC50=151 nmol/L). Furthermore, compound 47 was able to induce a decrease in IRAK4 protein levels in cells of human skin fibroblasts (DC50=36 nmol/L), but did not inhibit IL-6 secretion. Therefore, PROTAC-targeted degradation of IRAK4 may open up opportunities to develop new therapeutic drugs for the treatment of inflammatory and neoplastic diseases.
PROTACs promise to expand the druggable proteome as degradation is not limited to the protein domain which is functionally responsible for the disease. The ATP-dependent activities of the BAF (SWI/SNF) affect the localization of nucleosomes on DNA, thereby affecting many cellular processes related to chromatin structure, including transcription, DNA repair and decatenation mutant cancers, etc188. The BAF complex is mutated in approximately 20% of human cancers, and SMARCA2 and SMARCA4 are 2 mutant subunits of which. These mutants have different roles in different cancers; SMARCA4 inhibits cancer in solid tumors and promotes cancer cell growth in acute myeloid leukemia (AML)189. Therefore, targeting the above proteins is crucial for cancer treatment. The Ciulli team190 showed how structure-based PROTAC design can identify potent degraders of SMARCA2 and SMARCA4 with anticancer activity.
They first designed the PROTAC 1 (48) molecule based on SMARCA bromodomain inhibitors. By observing the eutectic structure of the high resolution SMARTA2BD:PROTAC 1:VCB (VHL−ELONGINC−ELONGINB complex) ternary complex, researchers performed a reasonable structural optimization of PROTAC. This operation is mainly to insert benzene on the linker which can form pi−π stacking with Tyr98 of VCB. The optimized PROTAC 2 (49) was found to have better molecular recognition ability and synergism of SMARCA than 48, as well as improved stability of the ternary complex. Next, the researchers observed the eutectic structure of the second-generation ternary complex SMARTA2BD:PROTAC 2:VCB, and added an oxygen to the linker to increase the length of the linker and the permeability of the molecule to obtain the PROTAC molecule ACBI1. The results showed that PROTAC ACBI1 (50) has good cell permeability and can induce the degradation of SMARCA2 (DC50=6 nmol/L) or SMARCA4 (DC50=11 nmol/L) and PBRM1 (DC50=32 nmol/L) rapidly, effectively, and specifically. Further, 50 can induce anti-proliferative and apoptotic effects in acute myeloid leukemia cells, which provides a new possibility for treating cancers that are sensitive to BAF complex loss.
Structures and protein degradation activities of VHL-based PROTAC 30–50 are shown in Fig. 12 and Table 3.
Figure 12.

Structures of PROTACs 30–50 based on CRBN as E3.
Table 3.
PROTACs 30–47 with the ability to degrade target proteins developed based on VHL as E3a.
| Compd. | Target | Degradation in cell lines |
Ref. No. (Year) | |
|---|---|---|---|---|
| DC50 | Dmax (%) | |||
| 30 | ERRα | 100 nmol/L | 86 | 163 (2015) |
| 31 | RIPK2 | 1.4 nmol/L | >95 | 163 (2015) |
| 32 | BRD2/3/4 | <5 nmol/L/<1 nmol/L for BRD2/3/4 | >50 | 165 (2016) |
| 33 | BRD4 | <100 nmol/L | >90 | 167 (2015) |
| 34 | BRD4 | 10–100 nmol/L | >90 | 168 (2017) |
| 35 | BRD9/7 | 4 nmol/L for BRD9 | 95 | 169 (2018) |
| 34.5 nmol/L for BRD7 | ||||
| 36 | C-ABL | NA | NA | 170 (2016) |
| 37 | TBK1 | 12 nmol/L | 96 | 173 (2018) |
| 38 | EGFR/HER2 | 215.8 nmol/L | 79.1 | 174 (2018) |
| 39 | c-Met | NA | NA | 174 (2018) |
| 40 | FLT-3 | 43 nmol/L for FLT-3 ITD 36 nmol/L for FLT-3 WT |
NA | 175 (2018) |
| 41 | TRIM24 | 2.5–5 μmol/L | 70 | 178 (2018) |
| 42 | ALK | NA | NA | 179 (2018) |
| 43 | FAK | 3.0 nmol/L | 99 | 182 (2018) |
| 44 | AR | <1 nmol/L | 95 | 183 (2018) |
| 45 | p38α | 7.16 nmol/L | 97.4 | 184 (2019) |
| 46 | p38δ | 299 nmol/L | 99.4 | 184 (2019) |
| 47 | IRAK4 | 151 nmol/L | NA | 187 (2019) |
DC50: the concentration at which 50% degradation was observed. Dmax: the maximal level of degradation. NA: not available.
In this section, we discuss how VHL acts as an E3 ligase and its interaction with HIF-1α. We also highlight the optimization of VHL ligands and the development of VHL-based PROTAC. We hope to provide a reference to find more VHL ligands and the development of more VHL-based PROTACs in the future.
4.4. CRBN-based PROTAC
The immunomodulatory drugs (IMiDs) based on thalidomide (51) as a lead compound have been successfully repurposed for erythema nodosum leprosum, multiple myeloma (MM) and myelodysplasia. Thalidomide was found to bind to CRBN, leading to the identification of more drug-like E3 ligase binders. CRBN was the primary teratogenic target of thalidomide (51)191. The C-terminal domain of CRBN, named CULT, has been defined as a binding site of cellular ligands and thalidomide (51). The crystal structures of the DDB1−CRBN complex bound to thalidomide (51), lenalidomide (52) and pomalidomide (53) (Fig. 13A) have been established and provided the molecular basis for teratogenicity192. Unexpectedly, evidence demonstrated that 51 and its derivatives 52 and 53 exerted immunomodulatory and anti-proliferative activities by reducing protein levels of the anti-apoptotic protein. Selective ubiquitination and degradation of specific targets including transcription factors, Ikaros (IKZF1) and Aiolos (IKZF3), casein kinase 1α through hijacking U ligase CRBN provided a novel mechanism of therapeutic activity for proteins193, 194, 195.
Figure 13.

CRBN (cereblon) ligands 51–53 and CRBN PROTACs 54–64. (A) Development of CRBN ligands. (B) Structures of PROTACs 54–64 based on CRBN as E3.
CRBN is ubiquitously expressed in physiologic and pathophysiologic tissues, but CRBN modulators may exert tissue-specific effects. Inspired by the retrieval of CRBN using 51, a series of bifunctional PROTACs have been rationally designed. Especially, experiments have demonstrated that the aryl ring of 53 can tolerate chemical substitution in PROTAC. The phthalimide (53)-based conjugated ligands have been widely used to develop the libraries of CRBN-targeting PROTACs, which can be easily converted into multiple PROTAC precursors196. Recently, several CRBN modulators have been reported, which were demonstrated to mediate antitumor effects through the ubiquitination and degradation of the translation termination factor G1 to S phase transition 1 protein (GSPT1)/Ikaros and Aiolos. Studies have revealed that glutarimide mediates substrate binding on the surface of CRBN via a PPI197, 198, 199.
ARV-825 (54) was the first CRBN based PROTAC generated by Crews group200, which mediates the degradation of the oncoprotein BRD4 in a substoichiometric but dose-dependent manner. BRD4 plays a pivotal role in regulating essential oncogene expression, including c-MYC, BCL-xL and BCL-6200, 201. BRD4 is a promising therapeutic target in multiple cancer types, e.g., CRPC202 and pancreatic cancer203. Compound 54 completely degraded BRD4 at 10 nmol/L within 6 h (DC50<1 nmol/L). In a word, 54 can efficiently lead to degradation of pathogenic proteins.
Another small molecule BRD4 degrader, dBET1 (55), was developed using JQ1 and thalidomide (51) derivatives, resulting in the chemical recruitment of BRD4. Treatment with increasing concentrations of 55 in human AML cell lines (MV4; 11) for 18 h yielded good results (DC50=430 nmol/L, Dmax>95% for BRD4)41. Comparing the 2 PROTACs (54 and 55), they have similar binding moieties but different linkers. Interestingly, the degradation activity of 54 was 10-fold greater than that of 55, suggesting that careful design of the “linker” region may improve PROTAC's selectivity and affinity204.
CRBN-based PROTAC 56 not only retains the ability to induce c-ABL degradation (>85% degradation in 1 μmol/L), but also induces BCR-ABL degradation (>60% degradation in 1 μmol/L)170. Compound dBRD9 (57) is a PEG-linked 53 conjugate that promotes rapid degradation of BRD9 over a wide range of concentrations205.
Cyclin-dependent kinase 9 (CDK9) is a member of the CDK family. CDK9 inhibitors have therapeutic effects on hematological malignancies and HIV infection206, 207. Compound 58 selectively leads to degradations of CDK9 in HCT116 cells208, without affecting other CDK family members. In addition, a series of wogonin-based PROTACs were developed by “click chemistry” to link the target protein to CRBN209. Among these compounds, a compound having a triazole linker was more effective than a compound having an alkane chain, and a compound 59 having a linker of 10 atoms length exhibited an optimum CDK9 degrading activity and could selectively down-regulate CDK9 levels in a concentration-dependent manner. CDK9 was inhibited at submicromolar concentrations (IC50=523±12 nmol/L), while the CDK2, CDK4, CDK5, CDK7 and CDK8 levels remained unchanged. Compound 59 inhibited the proliferation of MCF-7 cells at a concentration (IC50=17±1.9 μmol/L) lower than that of wogonin (IC50=30±3.5 μmol/L), indicating that 59 is a potent inducer of apoptosis. In conclusion, this wogonin-based PROTAC is highly selective in CDK9 high expression cell lines, and will be a useful tool for further study of CDK9-dependent effects in cancer cells. THAL-SNS-032 (60)210 was also a selective CDK9 degrader, consisted of a SNS-032 ligand and a thalidomide (51) derivative. Compound 60 induced rapid degradation of CDK9 without affecting the levels of other SNS-032 targets (DC50<250 nmol/L). Compound 60 has improved degradation efficiency compared to the previously reported degradation of CDK-producing PROTAC.
In the CDK family, CDK4/6 can regulate G1-S cell cycle transition by phosphorylating retinoblastoma tumor suppressor protein, further triggering the gene expression process that promotes the entry of S phase. Therefore, CDK4/6 is a very important target for cancer therapy. Some ATP-competitive CDK4/6 inhibitors have been reported to show significant clinical activity211, but they cannot specifically recognize CDK4 and CDK6. On this basis, researchers developed a series of bifunctional small molecules that jointly or selectively target CDK4/6. Among them, a pomalidomide (53)-based degrader BSJ-03-123 (BSJ) (61)212 uses the protein interface to selectively degrade CDK6 within the proteome range. In addition, BSJ-02-162 (62) induces the degradation of both CDK4 and CDK6, whereas BSJ-01-187 (63) selectively targets CDK4, and YKL-06-102 (64) targets CDK6213.
Compound 65 was developed by the cycloaddition reaction which linked a thiramide-derived azide to an alkynylation inhibitor214. Compound 65 induced isoform-selective Sirtuin 2 (SIRT2) degradation (IC50=0.25±0.02 μmol/L). Recently, 66 was designed by combining an indazole-based BET inhibitor and a 51 analog215. It effectively induced degradation of BRD2-4 at concentrations as low as 0.1–0.3 nmol/L in the RS4-11 acute leukemia cell lines (IC50=51 pmol/L). The second-generation BRD inhibitor BETd-246 (67)216 has good selectivity and anti-tumor activity. It induced selective degradation of BET protein in TNBC cells at low nanomolar concentrations, and showed excellent cytotoxicity in most TNBC cell lines. In a word, 66 and 67 have potent therapeutic activity against acute leukemia and TNBC. In 2018, compound 68 was developed by combining a BET inhibitor and CRBN ligands to target the BET protein36. After incubation with 68, the levels of BRD2, BRD3 and BRD4 in leukemia cell lines were effectively reduced at concentrations of 30–100 pmol/L. The 3 proteins were completely degraded in the RS4-11 cell line (DC50<0.5 nmol/L, Dmax ∼100%). Furthermore, 68 was 1000-fold more potent than dBET1 and at least 10-fold more potent than 54 in inducing protein degradation. 68 inhibited cell growth in the MV4-11, RS4-11, and MOLM-13 human leukemia cell lines with IC50 values of 8.3, 62 and 32 pmol/L, respectively. This suggested that 68 provided a more powerful strategy for the treatment of acute leukemia.
Researchers have developed some potential multi-kinase degraders by combining highly promiscuous kinase inhibitors with CRBN ligands. Among them, TL13-149 (69)217 and DD-04-015 (70)217 selectively target FLT-3 and Bruton's tyrosine kinase (BTK). A small molecule PROTAC MT-802 (71) targets the wild-type and C481S mutant BTK218. The experimental results showed that 72 had good BTK degradation activity for wild-type BTK (DC50=14.6 nmol/L, Dmax>99%) and C481S mutant BTK (DC50=14.9 nmol/L, Dmax>99%), which indicated that 71 is a therapeutic candidate for targeting mutant BTK. In addition, compound 72 also has good activity to reduce BTK levels (DC50 ∼10 nmol/L)219.
Although the above multi-kinase degraders can target many kinases, some pathogenic kinases such as ALK still cannot be effectively degraded. ALK is a tyrosine kinase receptor and involved in the development of a variety of human cancers220. Some researchers have reported a novel PROTAC for ALK. Considering the high potency and selectivity of ceritinib for ALK, ceritinib was selected as the ALK binding moiety. The X-ray crystal structure of ALK in the complex with ceritinib (PDB ID:4MKC) indicated that the piperidine group is located in the solvent-exposed zone. Therefore, the researchers accessed different lengths and types of linkers from the piperidine position of ceritinib to the ligand of the CRBN221. Among these PROTAC molecules, MS4078 (73) showed high affinity for ALK with a Kd value of 19±3 nmol/L in SU-DHL-1 cells. 73 effectively reduce the cellular level of oncogenic activity of ALK fusion protein in SU-DHL-1 lymphoma (DC50 ∼11 nmol/L, Dmax=90%) and NCI–H2228 lung cancer cells (DC50=59 nmol/L) in a concentration- and time-dependent manner. In addition, 73 effectively inhibited the proliferation of SU-DHL-1 cells. More importantly, 73 showed good plasma exposure in mouse PK studies. Therefore, 73 can be a useful chemical tool for in vivo efficacy studies, laying the foundation for the development of the next generation of ALK PROTAC.
P300/CBP-associated factor (PCAF) and general control non-repressed protein 5 (GCN5) are closely related epigenetic proteins222, and PCAF can produce some inflammatory factors223. In THP1 cells, GSK983 (74)224 induced the concentration-dependent degradation of PCAF (DC50=1.5 nmol/L, Dmax=80%). Similarly, it also degraded GCN5 (DC50=3 nmol/L, Dmax=80%). Thus, 74 may provide a new anti-inflammation strategy.
Casein kinase 2 (CK2) is a highly pleiotropic active serine/threonine protein kinase overexpressed in many cancers. CK2 inhibitors are linked to porphyrins by a “click chemistry” reaction to form several PROTAC targeting CK2225. Among them, compound 75 induced CK2 degradation in a dose- and time-dependent manner, and had the best inhibitory effect at 10 μmol/L, maintaining CK2 at a low basal level through UPS. Recently, compound 76, a B-cell lymphoma 6 (BCL6) PROTAC was reported226. In a range of diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL) cell lines, 76 effectively induced BCL6 degradation at a concentration of 1 μmol/L. MDM2 is a key oncogenic protein that contributes to cell growth, survival, invasion and therapeutic resistance in cancer101. Using the potent MDM2 inhibitor and CRBN ligand lenalidomide (52), researchers successfully obtained an effective PROTAC MD-224 (77)227. Even at concentrations<1 nmol/L, 77 still induced rapid degradation of MDM2 protein in human leukemia cells. 77 was 10–50-fold more potent than MDM2 inhibitors in inducing P53 activation and inhibiting cell growth in RS4-11 and MV4. PK data showed that a single dose of 77 continuously degraded MDM2 and up-regulated P53 over 24 h.
Recently, a new method for preparing PROTAC conjugates using solid phase organic synthesis (SPOS) has been reported228. A pre-loaded resin, composed of a thalidomide moiety and an ethylene-oxyl linker, can simply and rapidly synthesize PROTAC. Compound 78 prepared by this method induced BTK degradation in a dose-dependent manner, reducing BTK levels to 15% at 2000 μmol/L concentration, IC50=0.16±0.04 μmol/L228. Of course, this thalidomide-prepackaged resin (TPR) can also be applied to other proteins that can be obtained with suitable inhibitors/regulators/ligands, suggesting this method has useful versatility. In addition, researchers also developed the next generation of BTK degrader, L18I (79), a molecule that combined ibrutinib and lenalidomide (52) via PEG linkers229. Compound 79 efficiently degraded C481S BTK in HBL-1 cells (DC50=29 nmol/L). More importantly, 79 had significant anti-tumor effects in a mouse xenograft model inoculated with C481S BTK HBL-1 cells. These results indicate that 79 has great potential in the treatment of drug-resistant cancers.
A new generation of multifunctional histone deacetylase 6 (HDAC6) degraders was reported by combining the selective HDAC6 inhibitor Nexturastat A with the CRBN ligand230. These new degraders can synergize with HDAC6 degradation for the antiproliferation of MM. By optimizing the linker length and the position of the linker, compound 80 has the best potency and selectivity for degrading HDAC6 (DC50=1.6 nmol/L, Dmax=86%). More importantly, due to the multifunctionality of the degrader, 80 also has significantly more potent antiproliferative effects over HDAC6 inhibitor alone, IMiD alone, or its combination in MM cancer cell lines. These multifunctional HDAC6 degraders may provide a novel strategy to therapy MM.
The small molecule ARV-110, an orally available PROTAC protein degrader, binds specifically to AR and mediates AR degradation231. ARV-110 completely degraded AR in all tested cell lines (DC50<1 nmol/L). Recently, Arvinas company announced that ARV-110 has been administered to patients in Phase I clinical trials232.
In preclinical studies, the AR degradation agent ARV-110 showed promising activity233. In the enzalutamide-sensitive model, ARV-110 was able to significantly reduce PSA levels at low doses and was superior to enzalutamide. In an in vivo model of acquired and intrinsic resistance to enzalutamide, the ARV-110 pair showed 70% and 100% tumor inhibition rates, respectively. ARV-110 degrades 95%–98% of AR in various cell lines typically used in prostate cancer research. In VCaP, ARV-110 can reduce AR in a time-dependent manner, where AR was degraded almost completely within 4 h after administration. In a VCaP xenograft mouse model, ARV-110 was able to reduce PSA in plasma at lower doses and was superior to enzalutamide. In an in vivo model of acquired enzalutamide resistance, daily and orally delivered ARV-110 (3 mpk) inhibited the tumor growth by up to 70%. In a PDX patient model, orally delivered ARV-110 (10 mpk) significantly inhibited the growth of enzalutamide-insensitive tumors. ARV-110 is currently undergoing Phase I clinical trials to evaluate its safety and tolerability in patients with mCRPC who have progressed on standard treatment options.
Structures and protein degradation activities of CRBN-based PROTACs 54–80 are shown in as Figure 13, Figure 14 and Table 4.
Figure 14.

Structures of PROTACs 65–80 based on CRBN as E3.
Table 4.
PROTACs 54–80 with the ability to degrade target proteins developed based on CRBN as E3a.
| Compd. | Target | Degradation in cell lines |
Ref. No. (Year) | |
|---|---|---|---|---|
| DC50 | Dmax (%) | |||
| 54 | BRD2/3/4 | <1 nmol/L for BRD4 | ∼100 | 200 (2015) |
| 55 | BRD2/3/4 | 430 nmol/L for BRD4 | >95 | 41 (2015) |
| 56 | BCR-ABL& c-ABL | NA | NA | 170 (2016) |
| 57 | BRD9 | NA | NA | 205 (2017) |
| 58 | CDK9 | NA | NA | 208 (2017) |
| 59 | CDK9 | NA | NA | 209 (2018) |
| 60 | CDK9 | <250 nmol/L | ∼100 | 210 (2018) |
| 61 | CDK6 | NA | NA | 212 (2018) |
| 62 | CDK4/6 | NA | NA | 213 (2019) |
| 63 | CDK4 | NA | NA | 213 (2019) |
| 64 | CDK6 | NA | NA | 213 (2019) |
| 65 | SIRT2 | 0.2–1 μmol/L | 90 | 214 (2018) |
| 66 | BRD2/3/4 | <0.03 nmol/L for BRD4 | ∼100 | 215 (2018) |
| 67 | BET | NA | ∼100 | 216 (2017) |
| 68 | BET | <0.5 nmol/L | ∼100 | 36 (2018) |
| 69 | FLT-3 | NA | NA | 217 (2018) |
| 70 | BTK | NA | NA | 217 (2018) |
| 71 | BTK | 14.6 nmol/L for wild-type BTK; 14.9 nmol/L for C481S BTK | >99 | 218 (2018) |
| 72 | BTK | ∼10 nmol/L | NA | 219 (2018) |
| 73 | ALK | 11 nmol/L for NPM-ALK | >90 | 221 (2018) |
| 59 nmol/L for EML4-ALK | ||||
| 74 | PCAF/GCN5 | 1.5 nmol/L for PCAF | 80 | 224 (2018) |
| 3 nmol/L for GCN5 | ||||
| 75 | CK2 | NA | NA | 225 (2018) |
| 76 | BCL | NA | NA | 226 (2018) |
| 77 | MDM2 | <1 nmol/L | NA | 227 (2018) |
| 78 | BTK | NA | 85 | 228 (2019) |
| 79 | BTK | 29 nmol/LM | NA | 229 (2019) |
| 80 | HDAC6 | 1.6 nmol/L | 86 | 230 (2019) |
DC50: the concentration at which 50% degradation was observed. Dmax: the maximal level of degradation. NA: not available.
Small molecule PROTACs have been successfully developed to target MDM2, cIAP, VHL, CRBN. More importantly, a series of patent applications were filed reporting the synthesis of these PROTACs, such as PROTAC 33234, PROTAC 37235, ARV-825 (54)236. The patent applications also claimed that these PROTACs can degrade different proteins and provide a good strategy for treating diseases such as cancer.
5. The advantages of PROTACs
Traditional small molecule inhibitors have many limitations. The selection pressure of small molecule kinase inhibitors often results in drug resistance in cancer cells. For instance, long-term clinical application of gefitinib leads to EGFR mutations237. Moreover, some protein structures do not support high-affinity inhibitors, and low-affinity inhibitors at high concentrations may cause unfavorable off-target effects238. The successful PROTAC targeting RTK suggests that the PROTAC degradation strategy may be superior to inhibitors in several key aspects. First, PROTACs may effectively induce protein degradation even at low concentrations. Second, PROTACs result in more effective and durable signal transduction inactivation and growth inhibition, without the concern about rewiring in the kinase group. Finally, PROTACs may induce degradation of mutant proteins to prevent them from “disengaging”174. We will thoroughly discuss the advantages of PROTACs in the following paragraphs.
5.1. Targeted degradation of undruggable proteins
The FDA has approved drugs for ∼400 human proteins248, but there are about 3000 proteins associated with disease. There are several reasons for the fact that most disease-related proteins have no corresponding drugs: first, some proteins may have multiple functions and catalytic domain structures, thus blocking just one function may not be sufficient. Second, some drug targets have no specific active catalytic sites. Therefore, the traditional protein inhibitor approach cannot be applied to these disease-related proteins83. Not all molecular targets are enzymes or receptors with druggable “hot spots” accessible to site-directed inhibitors239. For these challenging drug targets, PROTAC-induced protein degradation can provide a new solution. PROTAC ligands can be designed to bind target proteins specifically, even if no traditional “hot spot” is available.
The highly selective and potent FAK degrader compound 43182 (IC50=6.5 nmol/L, DC50=3.0 nmol/L, Dmax=99%) (Fig. 15A) has been developed based on the most potent clinical FAK inhibitor defactinib. Inducing FAK degradation not only alters kinase-dependent signal transduction, but also affects its kinase-independent signal transduction due to the lack of FAK itself. These results indicate that 43 has great potential in expanding the space for regulating protein function.
Figure 15.

The advantages of PROTACs. (A) PROTAC is advantageous over inhibitors for degrading both kinase-dependent and non-dependent protein. (B) A stable ternary complex between E3 and a potential substrate is required for degradation.
5.2. Eliminating the accumulation of drug targets
When inhibitor drugs bind targets, the accumulation of drug targets can be observed even within a short period of time. On the one hand, the accumulation of targets can stabilize proteins and prolong their half-life; this phenomenon can be seen in many inhibitors, such as the BRD4 inhibitor JQ1200. In general, the accumulation of drug targets may limit the efficacy of the drug or induce drug resistance. Therefore, target protein degradation induced by PROTAC may be a valuable method for these proteins that may escape by protein stabilization or compensatory upregulation. For example, BRD4 inhibitors rapidly lose efficacy due to BRD4 regulation; compound 54200, a heterologous PROTAC, can rapidly and efficiently prolong the degradation of BRD4 in BL cells, suggesting that 52 is particularly suitable for overcoming BRD4's escape from sensitivity to inhibitors by protein stabilization or overexpression.
5.3. Specificity
Developing highly selective molecules is the main goal for many pharmaco-chemical researchers. Good selectivity can be achieved by rational design based on the structure of the compound, and experimental data generation in parallel. It is reported that an amide type SNIPER selectively degraded CRABP-II but had no effect on IAPs139. Ideally, some small molecule inhibitors only inhibit pathogenic proteins but have no effect on other proteins. For example, a small molecule inhibitor of BRAFV600E kinase selectively targets the melanoma with the mutant BRAFV600E gene or other mutant BRAF protein changed at codon 600240, but has no effect on cell lines expressing wild-type BRAF. However, this ideal is not easy to achieve due to limited differences between disease-related proteins and other proteins in the same family.
PROTAC targeting degradable proteins provide a good strategy for high selectivity (Fig. 15B). Compound 36 induces degradation of c-ABL but not BCR-ABL degradation, whereas compound 56 not only retains the ability to induce c-ABL degradation >85% degradation in 1 μmol/L), but also induces BCR-ABL degradation (>85% degradation in 1 μmol/L), indicating that 36 may have higher selectivity170. In addition, some PROTACs were developed to induce selective degradation of the target protein among closely related proteins184. For instance, compound 45 can selectively degrade p38α in MDA-MB-231 human breast cancer cells (DC50=7.16 nmol/L, Dmax=97.4%), and it has little influence on p38β, γ and δ. Similarly, compound 46 can selectively induce degradation of p38δ instead of p38α, β or γ.
5.4. Substoichiometric catalytic activity
Another feature of PROTACs is their substoichiometric catalytic activity, which reduces the need for target engagement and occupation of traditional inhibitors163. Traditional small molecule protein inhibitors regulate protein function in manners highly dependent on concentrations. However, even low concentrations of PROTACs are sufficient to degrade proteins to basal levels and maintain this effect to achieve a desirable pharmacological effect, especially effective for some slowly-synthesized proteins. In addition, even if a PROTAC is exhausted or metabolized, the recovery of the target protein may need hours to days. It may be feasible to achieve sustained release of PROTACs via certain formulations, e.g., tablets, subcutaneous or intramuscular injection, etc., to treat chronic diseases that need long-term medication241.
Catalytic degradation induced by PROTAC is usually highly time-dependent and consumes more proteins over time. In addition, this kind of catalysis also has other advantages: PROTAC concentration can be observed in the target protein degradation, and it is far lower than the high level of E3 ligase inhibition.
For example, compounds 9 and 22 can respectively induce AR and BCR-ABL ubiquitination and degradation at micromolar concentrations242. More importantly, PROTACs at lower concentrations also produce less off-target toxicity than traditional small-molecule inhibitors, likely leading to better therapeutic indices.
5.5. Others
PROTACs also have several other advantages apart those mentioned above. Recent studies have shown that PROTACs have potent in vitro and in vivo activities. Some examples show that PROTACs, based on the E3 ligand design of VHL and CRBN, have promising medicinal properties, and BET-PROTAC is a typical example of these small molecules. Using BET-PROTAC as a model system for long-term administration, cancer cells showed resistance to PROTAC containing VHL and CRBN ligands. However, unlike other targeted therapeutics (e.g., kinase inhibitors), BET-PROTAC resistance is not due to PROTAC-bound target protein mutations, but due to VHL and CRBN-based BET-PROTAC243. It is interesting that the resistance to BET-PROTAC is related to the composition of the E3 ligase. In addition, PROTACs provide rapid pharmacological effects on target proteins by Cmax-driven pharmacodynamic effects and competitive binding sites on target proteins to recruit U ligase83.
Largely, PROTACs have the advantage of traditional small molecule inhibitors in selectivity, as well as the advantage of siRNA in downregulating the target proteins. Further development of PROTAC technology would include improving cell activity, oral bioavailability and safety.
6. Other options for target protein ligands
The PROTACs we discussed in previous sections of this article were constructed by linking E3 ligase and target ligands with various linkers. However, some molecules are constructed by other methods, e.g., using “recognition-cleavage” strategy or developing a phosphate-dependent proteolytic-targeting chimera (phosphoPROTAC).
6.1. “Recognition-cleavage” strategy
The “identification-cutting” strategy is another method to regulate protein level. The PROTAC molecule consists of two parts, the recognition group that selectively binds to the target protein, and the cutting group to cut the target protein. Cyclen's Co(III) complex [Co(III)cyclen] is the catalytic center for targeting a selective artificial protease with certain hydrolytic ability. Based on this, a Cu(II)cyclen complex with a recognition group was developed, which was used as the cutting group of a target protein244.
Compound 81 (Fig. 16) was developed using selective amyloid-β (Aβ) and identifies several groups (KLVFF or curcumin). KLVFF (Aβ residues of 16–20) can be captured with Aβ of copper, using Cu(II) to replace Co(III). It can degrade Aβ and effectively reduce the aggregation of Aβ245. In addition, 2 hydrolytic enzyme molecules containing “Tau recognition motif” were reported, e.g., cycln-hybrid artificial “hydrolytic enzyme” i1-Cu(II) (82) that can cleave Tau protein in vitro, and cell-permeable “hydrolytic enzyme” i2-Cu(II) (83) that can also cleave Tau protein246 (Fig. 16). The soluble oligomer of human islet amyloid polypeptide (h-IAPP) induces the apoptosis of sxin-cells, which leads to the occurrence and development of T2DM247. Apo-cyclen is attached to the specific hIAPP recognition motif (NYGAIL) to form the cyclen−NYGAIL–copper complex248. The benzothiazole-aniline (BTA) derivative is combined with the new Cu(II) cleavage agent to develop a new molecule249. Both can interfere with hIAPP aggregation and cleave hIAPP, providing a good strategy for the treatment of T2DM. Many tissues express the cellular prion protein PrPC, especially in the central nervous system. Proteolytic cleavage of the cellular prion protein PrPC provides a strategy to treat prion diseases250.
Figure 16.

Structures of compounds 81, 82 and 83.
6.2. PhosphoPROTAC
Apart from the “recognition-cleavage” strategy, molecules with a protein phosphorylation sequence can also recognize and degrade proteins.
Protein phosphorylation is one of the most common post-translational modifications (PTMs) and plays an important role in cellular signal transductions251, such as regulating protein activity and subcellular localization252. However, an abnormal phosphorylation process can also cause some serious diseases. For example, several tyrosine residues of RTK are phosphorylated upon activation. The domains of Src homology 2 (SH2) and phosphotyrosine binding (PTB) structures recognize the phosphorylated tyrosine in RTK253, recruiting and phosphorylating its downstream signaling proteins, further activating the cascade of carcinogenic signaling254. HER2 is produced by this activation process and is highly expressed in some breast cancer tissues255. Despite significant advances in the development of small molecule inhibitors and monoclonal antibodies for RTK, targeting the RTK signaling pathway remains a challenge in the cancer field256.
Inducing the degradation of proteins with PTB or SH2 domains may shut down tyrosine kinase signaling, and therefore inhibit tumor cell proliferation. Based on this idea, researchers developed a phosphate-dependent proteolytic-targeting chimera (phosphoPROTAC) which consists of four parts (Fig. 17A): the RTK phosphorylation sequence, the peptide VHL binding fragment, the linker, and the Poly-d-Arg sequence that increases cell permeability. The activated RTK can phosphorylate the Tyr residue of the RTK, then recruit and activate proteins with PTB or SH2 domains. After the hydroxylation of proline residues, the peptide proline-binding fragment recruits E3 ubiquitin ligase CRL2VHL to induce polyubiquitination, leading to degradation of proteins with PTB or SH2 domains. These phosphoPROTACs only inhibit the activated RTK signaling occurring in cancer cells, and have little toxicity on normal cells29.
Figure 17.

Chemical structures of phosphoPROTAC and HaloPROTAC-3 (86). (A) The PhosphoPROTAC is composed of an RTK phosphorylation sequence, VHL-binding sequence and a Poly-d-Arg sequence connected via a linker. (B) PhosphoPROTACs: compound 84 and 85. Green: RTK phosphorylation sequence; Red: ligand to recruit E3 ubiquitin ligase is shown in red color; Blue: poly-arginine. (C) Chemical structure of compound HaloPROTAC-3 (86).
Researchers have reported 2 PhosphoPROTACs: TrkAPPFRS2α and ERBB2PPPI3K257 (Fig. 17B). TrkAPPFRS2α is formed by conjugating the phosphorylation sequence TrkA (pre-myosin receptor kinase A) to the peptide ligand VHL. The neuregulin receptor ERBB2 (erythropoietin gene B2) was conjugated to the peptide ligand VHL to form ERBB2PPPI3K257. These phosphoPROTAC recruited neurotrophic signaling effectors, including fibroblast growth factor receptor substrate 2α (FRS2α) and PI3K, which were activated and phosphorylated by these 2 phosphoPROTACs. After phosphorylation, FRS2α and PI3K were ubiquitinated and degraded. TrkAPPFRS2α (84) reduced FRS2α protein in a dose- and time-dependent manner. After treating PC12 cells with nerve growth factor (NGF) and 84 for 60 min, the FRS2α protein was reduced by approximately 90%, indicating the high efficiency of this PROTAC. In addition, the mouse xenograft model showed that the tumor size was significantly reduced by 40% after treatment with ERBB2PPPI3K (85) daily, indicating that this phosphoPROTAC had good therapeutic efficacy in vivo. PhosphoPROTAC can target and inactivate RTK signaling, but the molecular weight of these two phosphoPROTACs is too high for ideal cell permeability. For this reason, phosphoPROTACs need further improvements on pharmacological properties.
Several PROTACs containing a small molecule VHL ligand were developed to induce the degradation of HaloTag7 fusion protein. Among them, HaloPROTAC-3 (86) was one of the most effective PROTACs (Fig. 17C) to induce GFP-Halotag degradation (DC50=19 nmol/L, Dmax=90%)67. These PROTACs with specific protein recognition motifs demonstrate that this is a good protein degradation strategy.
7. Outlook
7.1. PROTAC application in aggregated protein (degradation of Tau, etc.)
The amyloid beta protein plaques and the phosphorylated Tau neurofibrillary tangles (NFTs) are the major pathological signs of some neurodegenerative disease, e.g., frontotemporal dementia (FTD), Alzheimer's disease (AD) and Parkinson's disease (PD)258. Many approaches to regulating Aβ and Tau levels have been reported, including UPS, autophagy lysosomal degradation, proteasome degradation259, 260, and microglia phagocytosis261. More importantly, inhibitors of the protein refolding pathway can prevent the accumulation of abnormal proteins by enhancing the activity of the associated proteasome (26S proteasome, HSP90)262. PROTACs may provide a possible way to selectively remove these protein aggregates263.
A series of multifunctional PROTACs have been reported: they contain Tau recognition sequence and VHL E3 ligase binding ligand to induce Tau degradation and therefore prevent cytotoxicity35 (Fig. 18A). Meanwhile, these PROTACs effectively induced Tau degradation in primary nerve cells and 3xTg-AD mice. In addition, Lu et al.34 synthesized some peptide PROTACs to recruit Tau into KEAP1−Cul3 U E3 ligase complex for ubiquitination and subsequent proteasome-mediated Tau degradation (Fig. 18B). Among them, peptide 1 strongly bound to KEAP1 and Tau in vitro and has Kd values of 22.8 and 763 nmol/L respectively. Using PROTAC to recruit KEAP1 to induce Tau degradation instead of inhibiting NRF2 may provide a new strategy to treat neurodegenerative diseases. This demonstrates the capability of PROTACs to prevent the aggregates of Aβ, Tau and other proteins.
Figure 18.

A series of multifunctional PROTAC molecules hijacking Tau. (A) Design of the peptide PROTAC contain Tau recognition part and VHL E3 ligase binding part to enhance Tau degradation. (B) Design of the peptide PROTAC (peptide 1) contain Tau recognition sequence and KEAP1 E3 ligase binding ligand to enhance Tau degradation. (C) Design of the hydrophobic tagging-conjugated peptide induces degradation of Tau. (D) Chemical structures of QC-01-175 (87).
Hydrophobic tag-conjugated peptide (HyT−Tau−CPP)264 (Fig. 18C) was reported to selectively accelerate Tau degradation in a concentration- and time-dependent manner. 5(6)-Carboxyfluorescein-hydrophobic tag-conjugated peptide (CF−HyT−Tau−CPP) specifically binds Tau (Kd=0.77±0.19 μmol/L). HyT−Tau−CPP specifically interacted with Tau and permeates the cell membrane in vitro. HyT−Tau−CPP also reduced the level of Tau in the brain of AD mouse models. In conclusion, a hydrophobic marker-conjugated peptide may be a potential therapeutic strategy for AD by degrading Tau. In addition to these methods, the “Recognition-Cleavage” strategy may also effectively degrade aggregate proteins such as Aβ and Tau244. Most of these molecules use either selective Aβ recognition groups (KLVFF or curcumin) as recognition groups245 or contain 2 “Tau recognition” motifs246. Recently, Silva et al.265synthesized a small molecule Tau protein degrader using the Tau positron emission tomography (PET) tracer as Tau protein ligand. Among them, QC-01-175 (87) (Fig. 18D) exhibited superior Tau protein scavenging effects. Compound 87 was able to promote Tau clearance in FTD neurons expressing Tau-A152T or Tau-P301L in a concentration-dependent manner, subsequently rescued Tau-mediated neuronal stress vulnerability. When there was only about 50% of Tau expressed with variants, the effect of 87 in A152T and P301L heterozygous neurons would result in more than 70% and 60% Tau clearance respectively on average. It is worth noting that 87 can remove Tau from FTD patient-derived neuronal cell models specifically, with minimal effect on Tau of healthy controls. The affected neurons provide an unprecedented advantage for targeting pathologically related Tau species.
In summary, the application of PROTACs is very extensive. PROTACs not only induce degradation of certain tumor proteins, but other aggregates such as Aβ and Tau as well. However, in clinical settings for treating neurodegenerative diseases such as AD, PROTACs need to be delivered into the central neutral system. Many of the reported PROTACs have large molecular weights, which are unfavorable for permeability through the blood–brain barrier (BBB). The “Lipinski's Rule of Five” indicates that membrane permeability of a compound is related to its molecular weight, lipophilicity, polar surface area, hydrogen bonding and charge266.
For effective central nervous system drugs: 1) Lipophilicity is generally higher, with logP between 2 and 5267. Also, molecular weight is usually less than 450 Da268. In addition, most are neutral or weakly basic molecules with a pKa between 7.5 and 10.5269. 2) Lower molecular polar surface area is beneficial, generally less than 90 Å268. A low hydrogen bond donor number, generally less than 3268; what's more, lower hydrogen bonding capacity, ΔlogP is usually less than 2.3) Most are spherical molecules, and increasing the branching will reduce the ability to cross the BBB268. In addition, molecular flexibility is lower, and with fewer spin-fast bonds268.
Therefore, to ensure permeability across BBB, an ideal PROTAC should have good lipophilicity, a small polar surface area, a suitable number of hydrogen bonds, and a molecular weight of not more than 450 Da270.
In the future development of PROTACs attempts can be made to combine the PROTAC with some drug delivery systems (such as nanocarriers), which not only improve the drugability of PROTACs, but also deliver those PROTACs that degrade aggregated proteins into brain.
7.2. Homo-PROTAC (dimers), degradation tag (dTAG) systems and endosome targeting chimeras (ENDTACs)
E3 ubiquitin ligase is a key player in the U−proteasome pathway and it appears as an attractive drug target, particularly in cancer271. Ciulli's team envisions using the PROTAC method, and E3 ligases themselves hijack each other, thereby inducing E3 ligase degradation rather than blocking E3. In 2017, Ciulli's team272 described Homo-PROTAC, a small molecular approach that effectively dimerizes E3 ubiquitin ligase and induces its own degradation. Based on this assumption, they link the same ligand of the ubiquitously expressed VHL protein through a linker. This bifunctional small molecule induces VHL dimerization, which causes ubiquitination and subsequent degradation of the VHL protein. Among them, the symmetric homo-PROTAC, CM11 (88) (Fig. 19), has ∼20-fold higher affinity (cooperative), resulting in effective, complete and prolonged degradation of VHL in different cell lines.
Figure 19.

Chemical structures of Homo-PROTACs 88–90.
Subsequently, Krönke, Gütschow and their colleagues reported a homologous PROTAC of CRBN ligase273, which showed that homo-PROTAC 89 (Fig. 19) can reduce intracellular CRBN levels for a long time even at low concentrations. In addition, PROTAC 89 specifically induced CRBN degradation, had little effect on the new pomalidomide substrates IKZF1 and IKZF3, and had no effect on other members of the CRL4 ligase family. In 2019, Ciulli's team274 extended the homo-PROTAC method by combining 2 different E3 ligases using a heterobifunctional PROTAC to form VHL-CRBN heterodimeric PROTAC. The results showed that CRBN was preferentially degraded, and the most potent PROTAC compound 90 (Fig. 19) induced CRBN degradation with high potency (DC50=200 nmol/L) and at a high level (Dmax>98%) and rapidly (within 1 h of treatment). In summary, the homo-PROTAC may provide a good strategy for the degradation of E3 ligases.
In addition, the degradation tag (dTAG) protein degradation technology can be used as an effective tool for target discovery and confirmation. This technology uses CRISPR to fuse a variant non-native protein FKBP12 (F36V) to a target protein, and then uses a series of PROTAC compounds (dTAG compounds) that have a good membrane permeability and can selectively bind to FKBP12 (F36V) and CRBN to degrade this fusion protein275.
The dTAG technology uses non-endogenous proteins that do not theoretically affect proteins other than the target protein. Once this highly active, highly selective ligand is found, the tagged protein can be fused to many target proteins to degrade these target proteins. This single dTAG system can be used as a broad-spectrum, universal protein degradation tool. PROTAC and dTAG are 2 more broad technologies, a combination of bifunctional molecules and chemically induced proximity (CIP) technology. Although dTAG cannot be used as a drug, the significance of highly selective target confirmation techniques is noteworthy.
PROTACs can only degrade the proteins in cells by means of UPS. Dr. Crews' team276 developed a new method to degrade extracellular proteins through receptor-mediated endosome pathway internalization and degradation, using a heterodimeric molecule of “endosome targeting chimera” (ENDTAC). Both ends of the ENDTAC molecule bind to extracellular target proteins and receptors, which mediate cell surface endocytosis. ENDTAC can trigger the endocytic process by attaching an extracellular POI to receptors on the cell surface, which are subsequently degraded by lysosomes. Compared to PROTAC, ENDTAC technology provides a strategy to degrade disease-associated extracellular proteins, such as cytokines.
7.3. Choices of PROTAC warheads and linkers
Currently, the design and subsequent optimization of PROTACs has been focused on the binary interactions between the “warhead” and the respective target protein. This requires a large amount of crystal structures of binary complexes derived from the Protein Date Bank (PDB). However, in order to develop a PROTAC with protein degradation capability, the following aspects need to be noted:
1): Type of linkers (Table 5): The linker has an important influence on the biological activities and physicochemical properties of the PROTAC molecule. Researchers initially used peptide linkers, and in the development of PROTACs, it was found that the linker preferably has a certain flexibility, thus varying lengths of PEG and (un)saturated alkane chains are commonly used. Of course, there are also some relatively rigid linkers. The groups on the linker, such as oxygen atoms, may also interact with the protein to stabilize the orientation of the protein. In order to increase the solubility of PROTACs, a piperidine group is also a good choice as a linker183. Amide bonds, ether bonds, alkylamines, carbon–carbon single bonds, and carbon–carbon triple bonds are also generally used in PROTACs. In addition, the chemical bonds described above are linked to an alkyne or azide linkage and then joined by a “click chemistry” reaction.
Table 5.
The common linker for construction of PROTACs and lead-out position of E3 ligase ligand.
| Example of linkers | E3 (VHL-CRBN) ligand | ||||
|---|---|---|---|---|---|
| Alkyne PEG linkers | Flexible linkers | Relatively rigid linkers | Triazole linkers | 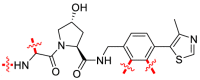 |
|
 |
 |
 |
 |
 |
 |
The cyclic addition reaction between 1,1,3-dipole acetylene and azide was first proposed by Huisgen in the 1960s277. Sharpless and others subsequently developed the concept of “click chemistry” in this reaction, which has a high yield and simple reaction conditions278. In order to achieve high molecular weight, lipophilicity and permeability, researchers explored the application of this bio-orthogonal click chemistry for rapid synthesis of PROTAC.
Currently, the latest applications of “click chemistry” are focused on copper-catalyzed azide–alkyne cycloaddition (CuAAC) and the Diels–Alder (DA) reaction (Fig. 20A and B). However, PROTACs based on “click chemistry” are mostly synthesized by CuAAC reaction.
Figure 20.

General strategy using “click chemistry” for PROTAC synthesis. (A) Cu(I)-cataylzed alkyne–azide cycloaddition (CuAAC). (B) Diels–Alder (DA) cycloadditions between tetrazines and stained alkenes. (C) Chemical structures of the in-cell click-formed proteolysis targeting chimeras (CLIPTACs), 91 and 92.
A Cu(I) catalyzed azide and an alkynization inhibitor from thalidomide are linked by a “click chemistry” reaction to form a PROTAC and the highly effective inhibition of SIRT2 was detected with an IC50 value of 0.25±0.02 μmol/L214. In addition, researchers designed a tetrazine-labeled thalidomide derivative that reacts rapidly with the trans-cyclooctene-labeled ligand of the target protein in the cell to form a CRBN-based PROTAC279. The in-cell click-formed proteolysis targeting chimeras (CLIPTACs) (compounds 91 and 92) were successfully used to reduce two key targets of oncology, BRD4 and ERK1/2, respectively (Fig. 20C). “Click chemistry” reactions provide a new strategy for the selection of synthetic PROTAC connection chains and lay a solid foundation for the development of PROTACs.
2): Length of linkers: the length of the linker also plays a key role in the degradation of the targeted protein. In fact, the optimization process of PROTAC molecules mainly focuses on the study of the structure–activity relationship of the length of linkers. First, if the linker is too short, the 2 ligands cannot simultaneously bind to their targets due to steric effects, resulting in the failure of ternary complex formation. Conversely, if the linker is too long, the 2 proteins cannot be pulled in to form PPI, nor will the target protein be ubiquitinated.
3): Site of attachment (Table 5): The site of attachment is typically based on analysis of solvent-exposed areas on ligand-protein binding structures. Generally, the end of the ligand is in the solvent-exposed area of the POI target. If the linker of the PROTAC has no flexibility for extension, the E3 ligase and the protein surface of the link site cannot form an effective PPI. Recent studies have also shown that increased affinity does not necessarily increase target protein degradation, most likely because E3 ubiquitin ligase, target protein and ligand did not form a suitable ternary complex. In a recent crystal structure of PROTAC 33167, Gadd et al. elucidated that 33 is “sandwiched” between VHL and the BRD4BD2 bromodomains, inducing a wide range of new protein−protein and proteins–ligand interactions, with hydrophobic and electrostatic properties. This indicates that the PROTAC not only binds to the target protein and E3, but also promotes the interaction between E3 and the target protein. Furthermore, the isoform-specific cooperativity of ternary complex formation is determined by the surface complementarity between E3 and the target protein. More importantly, a highly stable ternary E3–PROTAC-target complex is critical for PROTAC's ability to degrade proteins, especially for ligands with inherently weak binary binding affinities.
4): Type of E3 ubiquitin ligase: Most of the current studies are targeted to E3 ligases CRBN, IAP and VHL for PROTAC development, but these E3 ligases show restricted substrate specificities217, 280. Crews used different E3 ligands and 3 target protein ligands, as well as four different linkages to design chemically spatially diverse PROTACs with the goal of degrading the fusion protein BCR-ABL146. The results indicate that an imatinib-based PROTAC cannot induce degradation of ABL or BCR-ABL; a bosutinib-based PROTAC can induce degradation of ABL, while a dasatinib-based PROTAC can induce degradation of BCR-ABL. In addition, E3 ligase types also determine protein degradation efficiency. CRBN can simultaneously degrade BCR-ABL, while VHL can only degrade ABL. There is no convincing explanation for these observations, and we believe that E3 ligases can only recognize specific types of target proteins. To discover additional ligandable E3 ligases, Cravatt's team281 presented a chemical proteomics approach to discover E3 ligases. This strategy leverages cysteine-directed electrophilic fragments (chloroacetamide and acrylamide) in order to couple the target protein ligands with the screen which was designed for heterobifunctional degrader compounds operated by covalent adduction of E3 ligases. This approach identified the E3 ligase subunit DCAF16 as a target of electrophilic PROTACs that promote the nuclear-restricted degradation of proteins. Compound KB02-SLF (93) (Fig. 21) promoted a substantial reduction in nuclear FKBP12, which was sustained across a 4–72 h time frame. In addition, DCAF16 could also support the degradation of the nuclear protein BRD4. KB02-JQ1 (94) (Fig. 21) bifunctional compound induced degradation of BRD4 in HEK293T cells in a concentration-dependent manner. More importantly, the study underscores the value of broadly reactive electrophilic fragments as tools to discover ligandable sites on poorly characterized proteins such as DCAF16.
Figure 21.

Chemical structures of KB02-SLF (93) and KB02-JQ1 (94).
Ciulli's team168 has analyzed and elaborated the crystal structure of the ternary complex BRD4−MZ1−VHL, and structure-based-designed compound 34 exhibits highly BRD4 degradation activity. Structure-based drug design (SBDD) can provide a reference for the length of the linker chain, which rationalizes the PROTAC molecular design, thus accelerating the discovery of active PROTAC molecules. The use of HADDOCK for protein−ligand−protein simulation docking will provide a selection basis for the molecular link chain, and preliminary prediction of different solvent exposure areas. However, this may not reflect the mode of action of the protein complex, not to mention the presence of an artificial ligand. For the rapid synthesis of PROTACs, in addition to “click chemistry”, a new method for preparing PROTAC conjugates has recently been reported. The desired PROTACs are rapidly synthesized by linking TPR with a kinase inhibitor or its compact derivative having a reactive amino function through an ethylene-oxy linker. This method is highly versatile. In addition to protein kinases, it can be applied to other proteins that can be used to conjugate inhibitors/modulators/ligands.
8. Conclusions and future perspectives
Although PROTACs have many unique advantages, they still have several inherent flaws. For example, the pharmacokinetic parameters of the PROTAC molecule may not be particularly desirable, although some studies have confirmed good metabolic stability and tissue distribution. Recently, GlaxoSmithKline reported a review of the in vivo study of PROTAC, linking in vitro cell experiments with in vivo experiments to better guide the design and development of PROTAC animal and clinical trials282. Cellular permeability, tissue distribution, and metabolism can be improved by optimizing the molecular structure of the linker region of the PROTAC. However, due to the high molecular weight and complex structure of PROTACs, their distribution and metabolism within the tissue must be limited by the transmembrane permeability of the PROTAC.
To date, from peptides to small molecule PROTACs, there have been significant improvements in the stability, cell permeability, solubility and tissue distribution of PROTACs. A variety of synthetic techniques, such as click chemistry, further enriches the structural diversity of PROTACs. In just three years since the discovery of E3 ubiquitin ligase-specific small molecule ligands, PROTACs have entered a period of rapid development, creating a new direction in biopharmaceutical industry. Arvinas, founded by Professor Crews, and C4 founded by Dr. Bradner, have entered co-development with major pharmaceutical companies such as Merck, Pfizer and Roche to study the pharmacological properties of PROTACs. Among them, Arvinas' ARV110, a PROTAC molecule targeting AR, has been approved by the FDA for clinical research. Further, Arvinas has announced that its new ER drug-targeting agent ARV-471 for the treatment of locally advanced or metastatic ER-positive/HER2-negative breast cancer has received FDA approval. Phase I clinical trials of ARV-471 are scheduled to begin in the third quarter of 2019283, 284. Kymera's KYM-001 is a class of orally effective IRAK4 selective degrading agent that can cause tumor retraction in the MYD88 mutant ABC DLBCL model285. The degradation of KYM-001 can simultaneously remove the kinase activity and scaffold function of IRAK4, so its efficacy may be better than the IRAK4 kinase inhibitor. KYM-001 is expected to be tested clinically in 2019. Meanwhile, several pharmaceutical companies, including Novartis and AstraZeneca, launched PROTAC-related research projects.
Acknowledgment
We gratefully thank the support from grants (Nos. 81573281) of National Natural Science Foundation of China. We also thank the support from Double First-Class initiative Innovation team project of China Pharmaceutical University (Nos. CPU2018GF11 and CPU2018GY34, China).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Contributor Information
Wenyuan Liu, Email: liuwenyuan@163.com.
Haopeng Sun, Email: sunhaopeng@163.com.
References
- 1.Miklos G.L., Maleszka R. Protein functions and biological contexts. Proteomics. 2001;1:169–178. doi: 10.1002/1615-9861(200102)1:2<169::AID-PROT169>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 2.Seo M.H., Kim P.M. The present and the future of motif-mediated protein−protein interactions. Curr Opin Struct Biol. 2018;50:162–170. doi: 10.1016/j.sbi.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Bastola P., Oien D.B., Cooley M., Chien J. Emerging cancer therapeutic targets in protein homeostasis. AAPS J. 2018;20:94. doi: 10.1208/s12248-018-0254-1. [DOI] [PubMed] [Google Scholar]
- 4.Eisele F., Wolf D.H. Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett. 2008;582:4143–4146. doi: 10.1016/j.febslet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 5.Setiawan D., Brender J., Zhang Y. Recent advances in automated protein design and its future challenges. Expert Opin Drug Discov. 2018;13:587–604. doi: 10.1080/17460441.2018.1465922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finkelstein A.V., Badretdin A.J., Galzitskaya O.V., Ivankov D.N., Bogatyreva N.S., Garbuzynskiy S.O. There and back again: two views on the protein folding puzzle. Phys Life Rev. 2017;21:56–71. doi: 10.1016/j.plrev.2017.01.025. [DOI] [PubMed] [Google Scholar]
- 7.Roberts C.J. Therapeutic protein aggregation: mechanisms, design, and control. Trends Biotechnol. 2014;32:372–380. doi: 10.1016/j.tibtech.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komar A.A. Unraveling co-translational protein folding: concepts and methods. Methods. 2018;137:71–81. doi: 10.1016/j.ymeth.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costes S. Targeting protein misfolding to protect pancreatic beta-cells in type 2 diabetes. Curr Opin Pharmacol. 2018;43:104–110. doi: 10.1016/j.coph.2018.08.016. [DOI] [PubMed] [Google Scholar]
- 10.Cheng B., Li Y., Ma L., Wang Z., Petersen R.B., Zheng L. Interaction between amyloidogenic proteins and biomembranes in protein misfolding diseases: mechanisms, contributors, and therapy. Biochim Biophys Acta Biomembr. 2018;1860:1876–1888. doi: 10.1016/j.bbamem.2018.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Powers E.T., Morimoto R.I., Dillin A., Kelly J.W., Balch W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 12.Comyn S.A., Mayor T. A method to monitor protein turnover by flow cytometry and to screen for factors that control degradation by fluorescence-activated cell sorting. In: Mayor T., Kleiger G., editors. The ubiquitin proteasome system: methods and protocols. Humana Press; New York, NY: 2018. pp. 137–153. [DOI] [PubMed] [Google Scholar]
- 13.Schneider K.L., Nyström T., Widlund P.O. Studying spatial protein quality control, proteopathies, and aging using different model misfolding proteins in S. cerevisiae. Front Mol Neurosci. 2018;11:249. doi: 10.3389/fnmol.2018.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J., Xu Y., Zhang T., Cui L., Saidi L., Ye Y. Secretion of misfolded cytosolic proteins from mammalian cells is independent of chaperone-mediated autophagy. J Biol Chem. 2018;293:14359–14370. doi: 10.1074/jbc.RA118.003660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bustamante H.A., Gonzalez A.E., Cerda-Troncoso C., Shaughnessy R., Otth C., Soza A. Interplay between the autophagy−lysosomal pathway and the ubiquitin−proteasome system: a target for therapeutic development in Alzheimer's disease. Front Cell Neurosci. 2018;12:126. doi: 10.3389/fncel.2018.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciechanover A., Kwon Y.T. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dissmeyer N., Rivas S., Graciet E. Life and death of proteins after protease cleavage: protein degradation by the N-end rule pathway. New Phytol. 2018;218:929–935. doi: 10.1111/nph.14619. [DOI] [PubMed] [Google Scholar]
- 18.Wu S.Y., Lan S.H., Wu S.R., Chiu Y.C., Lin X.Z., Su I.J. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology. 2018;68:141–154. doi: 10.1002/hep.29781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korolchuk V.I., Menzies F.M., Rubinsztein D.C. Mechanisms of cross-talk between the ubiquitin−proteasome and autophagy−lysosome systems. FEBS Lett. 2010;584:1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 20.Kocaturk N.M., Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin−proteasome system. Front Cell Dev Biol. 2018;6:128. doi: 10.3389/fcell.2018.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navid F., Layh-Schmitt G., Sikora K.A., Cougnoux A., Colbert R.A. The role of autophagy in the degradation of misfolded HLA-B27 heavy chains. Arthritis Rheum. 2018;70:746–755. doi: 10.1002/art.40414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai A.C., Crews C.M. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim H., Ham S., Jo M., Lee G.H., Lee Y.S., Shin J.H. CRISPR-Cas9 mediated telomere removal leads to mitochondrial stress and protein aggregation. Int J Mol Sci. 2017;18:2093. doi: 10.3390/ijms18102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlgren M., Simoff I., Keiser M., Oswald S., Artursson P. CRISPR-Cas9: a new addition to the drug metabolism and disposition tool box. Drug Metab Dispos. 2018;46:1776–1786. doi: 10.1124/dmd.118.082842. [DOI] [PubMed] [Google Scholar]
- 25.Lee K., Jang B., Lee Y.R., Suh E.Y., Yoo J.S., Lee M.J. The cutting-edge technologies of siRNA delivery and their application in clinical trials. Arch Pharm Res. 2018;41:867–874. doi: 10.1007/s12272-018-1069-4. [DOI] [PubMed] [Google Scholar]
- 26.Polier S., Samant R.S., Clarke P.A., Workman P., Prodromou C., Pearl L.H. ATP-competitive inhibitors block protein kinase recruitment to the Hsp90−Cdc37 system. Nat Chem Biol. 2013;9:307–312. doi: 10.1038/nchembio.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neckers L., Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deshaies R.J. Protein degradation: prime time for PROTACs. Nat Chem Biol. 2015;11:634–635. doi: 10.1038/nchembio.1887. [DOI] [PubMed] [Google Scholar]
- 29.Gu S., Cui D., Chen X., Xiong X., Zhao Y. PROTACs: an emerging targeting technique for protein degradation in drug discovery. Bioessays. 2018;40:1700247. doi: 10.1002/bies.201700247. [DOI] [PubMed] [Google Scholar]
- 30.Zou Y., Ma D., Wang Y. The PROTAC technology in drug development. Cell Biochem Funct. 2019;37:21–30. doi: 10.1002/cbf.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Itoh Y. Chemical protein degradation approach and its application to epigenetic targets. Chem Rec. 2018;18:1681–1700. doi: 10.1002/tcr.201800032. [DOI] [PubMed] [Google Scholar]
- 32.Paiva S.L., Crews C.M. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. 2019;50:111–119. doi: 10.1016/j.cbpa.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toure M., Crews C.M. Small-molecule PROTACS: new approaches to protein degradation. Angew Chem Int Ed Engl. 2016;55:1966–1973. doi: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- 34.Lu M., Liu T., Jiao Q., Ji J., Tao M., Liu Y. Discovery of a keap1-dependent peptide PROTAC to knockdown tau by ubiquitination−proteasome degradation pathway. Eur J Med Chem. 2018;146:251–259. doi: 10.1016/j.ejmech.2018.01.063. [DOI] [PubMed] [Google Scholar]
- 35.Chu T.T., Gao N., Li Q.Q., Chen P.G., Yang X.F., Chen Y.X. Specific knockdown of endogenous Tau protein by peptide-directed ubiquitin−proteasome degradation. Cell Chem Biol. 2016;23:453–461. doi: 10.1016/j.chembiol.2016.02.016. [DOI] [PubMed] [Google Scholar]
- 36.Qin C., Hu Y., Zhou B., Fernandez-Salas E., Yang C.Y., Liu L. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J Med Chem. 2018;61:6685–6704. doi: 10.1021/acs.jmedchem.8b00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winter G.E., Buckley D.L., Paulk J., Roberts J.M., Souza A., Dhe-Paganon S. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neklesa T.K., Crews C.M. Chemical biology: greasy tags for protein removal. Nature. 2012;487:308–309. doi: 10.1038/487308a. [DOI] [PubMed] [Google Scholar]
- 39.Collins I., Wang H., Caldwell J.J., Chopra R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin−proteasome pathway. Biochem J. 2017;474:1127–1147. doi: 10.1042/BCJ20160762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tae H.S., Sundberg T.B., Neklesa T.K., Noblin D.J., Gustafson J.L., Roth A.G. Identification of hydrophobic tags for the degradation of stabilized proteins. Chembiochem. 2012;13:538–541. doi: 10.1002/cbic.201100793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thirunavukarasu D., Shi H. Aptamer-enabled manipulation of the Hsp70 chaperone system suggests a novel strategy for targeted ubiquitination. Nucleic Acid Ther. 2016;26:20–28. doi: 10.1089/nat.2015.0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu A., Li P., Tang T., Wang J., Chen Y., Liu L. Roles of Hsp70s in stress responses of microorganisms, plants, and animals. BioMed Res Int. 2015;2015:510319. doi: 10.1155/2015/510319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kampinga H.H., Craig E.A. The Hsp70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shiber A., Breuer W., Brandeis M., Ravid T. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Mol Biol Cell. 2013;24:2076–2087. doi: 10.1091/mbc.E13-01-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie T., Lim S.M., Westover K.D., Dodge M.E., Ercan D., Ficarro S.B. Pharmacological targeting of the pseudokinase Her3. Nat Chem Biol. 2014;10:1006–1012. doi: 10.1038/nchembio.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lim S.M., Xie T., Westover K.D., Ficarro S.B., Tae H.S., Gurbani D. Development of small molecules targeting the pseudokinase Her3. Bioorg Med Chem Lett. 2015;25:3382–3389. doi: 10.1016/j.bmcl.2015.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDonnell D.P., Wardell S.E., Norris J.D. Oral selective estrogen receptor downregulators (SERDs), a breakthrough endocrine therapy for breast cancer. J Med Chem. 2015;58:4883–4887. doi: 10.1021/acs.jmedchem.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lebraud H., Heightman T.D. Protein degradation: a validated therapeutic strategy with exciting prospects. Essays Biochem. 2017;61:517–527. doi: 10.1042/EBC20170030. [DOI] [PubMed] [Google Scholar]
- 49.Wu Y.L., Yang X., Ren Z., McDonnell D.P., Norris J.D., Willson T.M. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell. 2005;18:413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 50.Teutsch G., Goubet F., Battmann T., Bonfils A., Bouchoux F., Cerede E. Non-steroidal antiandrogens: synthesis and biological profile of high-affinity ligands for the androgen receptor. J Steroid Biochem Mol Biol. 1994;48:111–119. doi: 10.1016/0960-0760(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 51.Gustafson J.L., Neklesa T.K., Cox C.S., Roth A.G., Buckley D.L., Tae H.S. Small-molecule-mediated degradation of the androgen receptor through hydrophobic tagging. Angew Chem Int Ed Engl. 2015;54:9659–9662. doi: 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim H.R., Kang H.S., Kim H.D. Geldanamycin induces heat shock protein expression through activation of HSF1 in K562 erythroleukemic cells. IUBMB Life. 1999;48:429–433. doi: 10.1080/713803536. [DOI] [PubMed] [Google Scholar]
- 53.McDonough H., Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Long M.J., Gollapalli D.R., Hedstrom L. Inhibitor mediated protein degradation. Chem Biol. 2012;19:629–637. doi: 10.1016/j.chembiol.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pickering A.M., Davies K.J. Degradation of damaged proteins: the main function of the 20S proteasome. Prog Mol Biol Transl Sci. 2012;109:227–248. doi: 10.1016/B978-0-12-397863-9.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jung T., Höhn A., Grune T. The proteasome and the degradation of oxidized proteins: part II-protein oxidation and proteasomal degradation. Redox Biol. 2014;2:99–104. doi: 10.1016/j.redox.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davies K.J. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 58.Demasi M., Da Cunha F.M. The physiological role of the free 20S proteasome in protein degradation: a critical review. Biochim Biophys Acta Gen Subj. 2018;1862:2948–2954. doi: 10.1016/j.bbagen.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 59.Medicherla B., Goldberg A.L. Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J Cell Biol. 2008;182:663–673. doi: 10.1083/jcb.200803022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whittier J.E., Xiong Y., Rechsteiner M.C., Squier T.C. Hsp90 enhances degradation of oxidized calmodulin by the 20S proteasome. J Biol Chem. 2004;279:46135–46142. doi: 10.1074/jbc.M406048200. [DOI] [PubMed] [Google Scholar]
- 61.Shi Y., Long M.J., Rosenberg M.M., Li S., Kobjack A., Lessans P. Boc3Arg-linked ligands induce degradation by localizing target proteins to the 20S proteasome. ACS Chem Biol. 2016;11:3328–3337. doi: 10.1021/acschembio.6b00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coffey R.T., Shi Y., Long M.J., Marr M.T., II, Hedstrom L. Ubiquilin-mediated small molecule inhibition of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem. 2016;291:5221–5233. doi: 10.1074/jbc.M115.691584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giepmans B.N., Adams S.R., Ellisman M.H., Tsien R.Y. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 64.Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- 65.Waugh D.S. Making the most of affinity tags. Trends Biotechnol. 2005;23:316–320. doi: 10.1016/j.tibtech.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 66.England C.G., Luo H., Cai W. HaloTag technology: a versatile platform for biomedical applications. Bioconjug Chem. 2015;26:975–986. doi: 10.1021/acs.bioconjchem.5b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buckley D.L., Raina K., Darricarrere N., Hines J., Gustafson J.L., Smith I.E. HaloPROTACS: use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chem Biol. 2015;10:1831–1837. doi: 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Los G.V., Encell L.P., McDougall M.G., Hartzell D.D., Karassina N., Zimprich C. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 69.Tomoshige S., Naito M., Hashimoto Y., Ishikawa M. Degradation of HaloTag-fused nuclear proteins using bestatin-halotag ligand hybrid molecules. Org Biomol Chem. 2015;13:9746–9750. doi: 10.1039/c5ob01395j. [DOI] [PubMed] [Google Scholar]
- 70.Neklesa T.K., Tae H.S., Schneekloth A.R., Stulberg M.J., Corson T.W., Sundberg T.B. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat Chem Biol. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neklesa T.K., Noblin D.J., Kuzin A.P., Lew S., Seetharaman J., Acton T.B. A bidirectional system for the dynamic small molecule control of intracellular fusion proteins. ACS Chem Biol. 2013;8:2293–2300. doi: 10.1021/cb400569k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tomoshige S., Hashimoto Y., Ishikawa M. Efficient protein knockdown of halotag-fused proteins using hybrid molecules consisting of IAP antagonist and HaloTag ligand. Bioorg Med Chem. 2016;24:3144–3148. doi: 10.1016/j.bmc.2016.05.035. [DOI] [PubMed] [Google Scholar]
- 73.Bondeson D.P., Crews C.M. Targeted protein degradation by small molecules. Annu Rev Pharmacol Toxicol. 2017;57:107–123. doi: 10.1146/annurev-pharmtox-010715-103507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schwartz A.L., Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- 75.Mofers A., Pellegrini P., Linder S., D'Arcy P. Proteasome-associated deubiquitinases and cancer. Cancer Metastasis Rev. 2017;36:635–653. doi: 10.1007/s10555-017-9697-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Warang P., Homma T., Pandya R., Sawant A., Shinde N., Pandey D. Potential involvement of ubiquitin−proteasome system dysfunction associated with oxidative stress in the pathogenesis of sickle cell disease. Br J Haematol. 2018;182:559–566. doi: 10.1111/bjh.15437. [DOI] [PubMed] [Google Scholar]
- 77.Bulatov E., Zagidullin A., Valiullina A., Sayarova R., Rizvanov A. Small molecule modulators of RING-type E3 ligases: MDM and cullin families as targets. Front Pharmacol. 2018;9:450. doi: 10.3389/fphar.2018.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Poirson J., Biquand E., Straub M.L., Cassonnet P., Nominé Y., Jones L. Mapping the interactome of HPV E6 and E7 oncoproteins with the ubiquitin−proteasome system. FEBS J. 2017;284:3171–3201. doi: 10.1111/febs.14193. [DOI] [PubMed] [Google Scholar]
- 79.Randow F., Lehner P.J. Viral avoidance and exploitation of the ubiquitin system. Nat Cell Biol. 2009;11:527–534. doi: 10.1038/ncb0509-527. [DOI] [PubMed] [Google Scholar]
- 80.Lip P.Z., Demasi M., Bonatto D. The role of the ubiquitin proteasome system in the memory process. Neurochem Int. 2017;102:57–65. doi: 10.1016/j.neuint.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 81.Xi M., Chen Y., Yang H., Xu H., Du K., Wu C. Small molecule protacs in targeted therapy: an emerging strategy to induce protein degradation. Eur J Med Chem. 2019;174:159–180. doi: 10.1016/j.ejmech.2019.04.036. [DOI] [PubMed] [Google Scholar]
- 82.Carmony K.C., Kim K.B. PROTAC-induced proteolytic targeting. In: Dohmen R.J., Scheffner M., editors. Ubiquitin family modifiers and the proteasome: reviews and protocols. Humana Press; New York: 2012. pp. 627–638. [Google Scholar]
- 83.Neklesa T.K., Winkler J.D., Crews C.M. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174:138–144. doi: 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- 84.Sakamoto K.M., Kim K.B., Kumagai A., Mercurio F., Crews C.M., Deshaies R.J. Protacs: chimeric molecules that target proteins to the Skp1−cullin−F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sakamoto K.M., Kim K.B., Verma R., Ransick A., Stein B., Crews C.M. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteom. 2003;2:1350–1358. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 86.Ottis P., Toure M., Cromm P.M., Ko E., Gustafson J.L., Crews C.M. Assessing different E3 ligases for small molecule induced protein ubiquitination and degradation. ACS Chem Biol. 2017;12:2570–2578. doi: 10.1021/acschembio.7b00485. [DOI] [PubMed] [Google Scholar]
- 87.Raina K., Crews C.M. Targeted protein knockdown using small molecule degraders. Curr Opin Chem Biol. 2017;39:46–53. doi: 10.1016/j.cbpa.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang D.D., Lo S.C., Cross J.V., Templeton D.J., Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Furukawa M., Xiong Y. BTB protein keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Darbandi M., Darbandi S., Agarwal A., Baskaran S., Sengupta P., Dutta S. Oxidative stress-induced alterations in seminal plasma antioxidants: is there any association with keap1 gene methylation in human spermatozoa? Andrologia. 2019;51:e13159. doi: 10.1111/and.13159. [DOI] [PubMed] [Google Scholar]
- 91.Kobayashi E.H., Suzuki T., Funayama R., Nagashima T., Hayashi M., Sekine H. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624. doi: 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jiang Z.Y., Lu M.C., You Q.D. Nuclear factor erythroid 2-related factor 2 (Nrf2) inhibition: an emerging strategy in cancer therapy. J Med Chem. 2019;62:3840–3856. doi: 10.1021/acs.jmedchem.8b01121. [DOI] [PubMed] [Google Scholar]
- 93.Kobayashi A., Kang M.I., Okawa H., Ohtsuji M., Zenke Y., Chiba T. Oxidative stress sensor keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rada P., Rojo A.I., Chowdhry S., McMahon M., Hayes J.D., Cuadrado A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a keap1-independent manner. Mol Cell Biol. 2011;31:1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu T., Zhao F., Gao B., Tan C., Yagishita N., Nakajima T. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708–722. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cullinan S.B., Gordan J.D., Jin J., Harper J.W., Diehl J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y. An Nrf2/small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 98.Joshi G., Johnson J.A. The Nrf2−ARE pathway: a valuable therapeutic target for the treatment of neurodegenerative diseases. Recent Pat CNS Drug Discov. 2012;7:218–229. doi: 10.2174/157488912803252023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li W., Bengtson M.H., Ulbrich A., Matsuda A., Reddy V.A., Orth A. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS One. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Buckley D.L., Crews C.M. Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew Chem Int Ed Engl. 2014;53:2312–2330. doi: 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saadatzadeh M.R., Elmi A.N., Pandya P.H., Bijangi-Vishehsaraei K., Ding J., Stamatkin C.W. The role of MDM2 in promoting genome stability versus instability. Int J Mol Sci. 2017;18:2216. doi: 10.3390/ijms18102216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Oliner J.D., Pietenpol J.A., Thiagalingam S., Gyuris J., Kinzler K.W., Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor P53. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 103.Cao D., Ng T.K., Yip Y.W., Young A.L., Pang C.P., Chu W.K. P53 inhibition by MDM2 in human pterygium. Exp Eye Res. 2018;175:142–147. doi: 10.1016/j.exer.2018.06.021. [DOI] [PubMed] [Google Scholar]
- 104.Levav-Cohen Y., Goldberg Z., Tan K.H., Alsheich-Bartok O., Zuckerman V., Haupt S. The P53-MDM2 loop: a critical juncture of stress response. In: Deb S.P., Deb S., editors. Mutant P53 and MDM2 in cancer. Springer; Dordrecht: 2014. pp. 161–166. [DOI] [PubMed] [Google Scholar]
- 105.De Stephanis L., Mangolini A., Servello M., Harris P.C., Dell'Atti L., Pinton P. MicroRNA501-5p induces P53 proteasome degradation through the activation of the mTOR/MDM2 Pathway in ADPKD Cells. J Cell Physiol. 2018;233:6911–6924. doi: 10.1002/jcp.26473. [DOI] [PubMed] [Google Scholar]
- 106.Honda R., Tanaka H., Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor P53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 107.Pei D., Zhang Y., Zheng J. Regulation of P53: a collaboration between Mdm2 and MdmX. Oncotarget. 2012;3:228–235. doi: 10.18632/oncotarget.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Qian Y., Chen X. Senescence regulation by the P53 protein family. In: Galluzzi L., Vitale I., Kepp O., Kroemer G., editors. Cell senescence: methods and protocols. Humana Press; Totowa: 2013. pp. 37–61. [Google Scholar]
- 109.Soares J., Pereira N.A., Monteiro Â., Leão M., Bessa C., Dos Santos D.J. Oxazoloisoindolinones with in vitro antitumor activity selectively activate a P53-pathway through potential inhibition of the P53−MDM2 interaction. Eur J Pharm Sci. 2015;66:138–147. doi: 10.1016/j.ejps.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 110.Azer S.A. MDM2−P53 interactions in human hepatocellular carcinoma: what is the role of nutlins and new therapeutic options? J Clin Med. 2018;7:64. doi: 10.3390/jcm7040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kamal A., Mohammed A.A., Shaik T.B. P53–Mdm2 inhibitors: patent review (2009 2010) Expert Opin Ther Pat. 2012;22:95–105. doi: 10.1517/13543776.2012.656593. [DOI] [PubMed] [Google Scholar]
- 112.Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z. In vivo activation of the P53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 113.Endo S., Yamato K., Hirai S., Moriwaki T., Fukuda K., Suzuki H. Potent in vitro and in vivo antitumor effects of MDM2 inhibitor nutlin-3 in gastric cancer cells. Cancer Sci. 2011;102:605–613. doi: 10.1111/j.1349-7006.2010.01821.x. [DOI] [PubMed] [Google Scholar]
- 114.Pechackova S., Burdova K., Benada J., Kleiblova P., Jenikova G., Macurek L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget. 2016;7:14458–14475. doi: 10.18632/oncotarget.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schneekloth A.R., Pucheault M., Tae H.S., Crews C.M. Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg Med Chem Lett. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vu B., Wovkulich P., Pizzolato G., Lovey A., Ding Q., Jiang N. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett. 2013;4:466–469. doi: 10.1021/ml4000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ding Q., Zhang Z., Liu J.J., Jiang N., Zhang J., Ross T.M. Discovery of RG7388, a Potent and Selective P53−MDM2 inhibitor in clinical development. J Med Chem. 2013;56:5979–5983. doi: 10.1021/jm400487c. [DOI] [PubMed] [Google Scholar]
- 118.Amé J.C., Spenlehauer C., De Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 119.Beck C., Robert I., Reina-San-Martin B., Schreiber V., Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res. 2014;329:18–25. doi: 10.1016/j.yexcr.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 120.Jagtap P., Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 121.Zhao Q., Lan T., Su S., Rao Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem Commun (Camb) 2019;55:369–372. doi: 10.1039/c8cc07813k. [DOI] [PubMed] [Google Scholar]
- 122.Hines J., Lartigue S., Dong H., Qian Y., Crews C.M. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of P53. Cancer Res. 2019;79:251–262. doi: 10.1158/0008-5472.CAN-18-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sato S., Tetsuhashi M., Sekine K., Miyachi H., Naito M., Hashimoto Y. Degradation-promoters of cellular inhibitor of apoptosis protein 1 based on bestatin and actinonin. Bioorg Med Chem. 2008;16:4685–4698. doi: 10.1016/j.bmc.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 124.Dubrez L., Rajalingam K. IAPs and cell migration. Semin Cell Dev Biol. 2015;39:124–131. doi: 10.1016/j.semcdb.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 125.Berthelet J., Dubrez L. Regulation of apoptosis by inhibitors of apoptosis (IAPs) Cells. 2013;2:163–187. doi: 10.3390/cells2010163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vaux D.L., Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6:287–297. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- 127.Dueber E.C., Schoeffler A.J., Lingel A., Elliott J.M., Fedorova A.V., Giannetti A.M. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science. 2011;334:376–380. doi: 10.1126/science.1207862. [DOI] [PubMed] [Google Scholar]
- 128.Gyrd-Hansen M., Meier P. IAPs: From caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat Rev Cancer. 2010;10:561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- 129.Che X., Yang D., Zong H., Wang J., Li X., Chen F. Nuclear cIAP1 overexpression is a tumor stage- and grade-independent predictor of poor prognosis in human bladder cancer patients. Urol Oncol. 2012;30:450–456. doi: 10.1016/j.urolonc.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 130.Talmadge J.E., Lenz B.F., Pennington R., Long C., Phillips H., Schneider M. Immunomodulatory and therapeutic properties of bestatin in mice. Cancer Res. 1986;46:4505–4510. [PubMed] [Google Scholar]
- 131.Sekine K., Takubo K., Kikuchi R., Nishimoto M., Kitagawa M., Abe F. Small molecules destabilize cIAP1 by activating auto-ubiquitylation. J Biol Chem. 2008;283:8961–8968. doi: 10.1074/jbc.M709525200. [DOI] [PubMed] [Google Scholar]
- 132.Sato S., Aoyama H., Miyachi H., Naito M., Hashimoto Y. Demonstration of direct binding of cIAP1 degradation-promoting bestatin analogs to BIR3 domain: synthesis and application of fluorescent bestatin ester analogs. Bioorg Med Chem Lett. 2008;18:3354–3358. doi: 10.1016/j.bmcl.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 133.Varfolomeev E., Blankenship J.W., Wayson S.M., Fedorova A.V., Kayagaki N., Garg P. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 134.Cohen P., Tcherpakov M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell. 2010;143:686–693. doi: 10.1016/j.cell.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 135.Fulda S., Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 136.Ohoka N., Okuhira K., Ito M., Nagai K., Shibata N., Hattori T. In vivo knockdown of pathogenic proteins via specific and nongenetic inhibitor of apoptosis protein (IAP)-dependent protein erasers (SNIPERs) J Biol Chem. 2017;292:4556–4570. doi: 10.1074/jbc.M116.768853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Itoh Y., Ishikawa M., Naito M., Hashimoto Y. Protein knockdown using methyl bestatin−ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc. 2010;132:5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- 138.Itoh Y., Ishikawa M., Kitaguchi R., Sato S., Naito M., Hashimoto Y. Development of target protein-selective degradation inducer for protein knockdown. Bioorg Med Chem. 2011;19:3229–3241. doi: 10.1016/j.bmc.2011.03.057. [DOI] [PubMed] [Google Scholar]
- 139.Itoh Y., Ishikawa M., Kitaguchi R., Okuhira K., Naito M., Hashimoto Y. Double protein knockdown of cIAP1 and CRABP-II using a hybrid molecule consisting of ATRA and IAPs Antagonist. Bioorg Med Chem Lett. 2012;22:4453–4457. doi: 10.1016/j.bmcl.2012.04.134. [DOI] [PubMed] [Google Scholar]
- 140.Okuhira K., Demizu Y., Hattori T., Ohoka N., Shibata N., Nishimaki-Mogami T. Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci. 2013;104:1492–1498. doi: 10.1111/cas.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ohoka N., Morita Y., Nagai K., Shimokawa K., Ujikawa O., Fujimori I. Derivatization of inhibitor of apoptosis protein (IAP) ligands yields improved inducers of estrogen receptor α degradation. J Biol Chem. 2018;293:6776–6790. doi: 10.1074/jbc.RA117.001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hood F.E., Royle S.J. Pulling it together: the mitotic function of TACC3. BioArchitecture. 2011;1:105–109. doi: 10.4161/bioa.1.3.16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jacquemier J., Ginestier C., Rougemont J., Bardou V.J., Charafe-Jauffret E., Geneix J. Protein expression profiling identifies subclasses of breast cancer and predicts prognosis. Cancer Res. 2005;65:767–779. [PubMed] [Google Scholar]
- 144.Lauffart B., Vaughan M.M., Eddy R., Chervinsky D., DiCioccio R.A., Black J.D. Aberrations of TACC1 and TACC3 are associated with ovarian cancer. BMC Women's Health. 2005;5:8. doi: 10.1186/1472-6874-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ohoka N., Nagai K., Hattori T., Okuhira K., Shibata N., Cho N. Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin−proteasome pathway. Cell Death Dis. 2014;5:e1513. doi: 10.1038/cddis.2014.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shimokawa K., Shibata N., Sameshima T., Miyamoto N., Ujikawa O., Nara H. Targeting the allosteric site of oncoprotein BCR-ABL as an alternative strategy for effective target protein degradation. ACS Med Chem Lett. 2017;8:1042–1047. doi: 10.1021/acsmedchemlett.7b00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shibata N., Nagai K., Morita Y., Ujikawa O., Ohoka N., Hattori T. Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J Med Chem. 2018;61:543–575. doi: 10.1021/acs.jmedchem.7b00168. [DOI] [PubMed] [Google Scholar]
- 148.Hao J., Chen X., Fu T., Liu J., Yu M., Han W. The expression of VHL (Von Hippel-Lindau) after traumatic spinal cord injury and its role in neuronal apoptosis. Neurochem Res. 2016;41:2391–2400. doi: 10.1007/s11064-016-1952-7. [DOI] [PubMed] [Google Scholar]
- 149.Wang S., Xia W., Qiu M., Wang X., Jiang F., Yin R. Atlas on substrate recognition subunits of CRL2 E3 ligases. Oncotarget. 2016;7:46707–46716. doi: 10.18632/oncotarget.8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Min J.H., Yang H., Ivan M., Gertler F., Kaelin W.G., Jr., Pavletich N.P. Structure of an HIF-1α−pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 151.Yang Z., Yang Z., Xiong L., Huang S., Liu J., Yang L. Expression of VHL and HIF-1α and their clinicopathologic significance in benign and malignant lesions of the gallbladder. Appl Immunohistochem Mol Morphol. 2011;19:534–539. doi: 10.1097/PAI.0b013e318212f001. [DOI] [PubMed] [Google Scholar]
- 152.Semenza G.L. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med. 2001;7:345–350. doi: 10.1016/s1471-4914(01)02090-1. [DOI] [PubMed] [Google Scholar]
- 153.Kaelin W.G., Jr. The von Hippel–Lindau Tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 154.Cardote T.A., Gadd M.S., Ciulli A. Crystal structure of the Cul2-Rbx1-EloBC-VHL ubiquitin ligase complex. Structure. 2017;25:901–911. doi: 10.1016/j.str.2017.04.009. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Van Molle I., Thomann A., Buckley D.L., So E.C., Lang S., Crews C.M. Dissecting fragment-based lead discovery at the von Hippel-Lindau protein: hypoxia inducible factor 1α protein−protein interface. Chem Biol. 2012;19:1300–1312. doi: 10.1016/j.chembiol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Testa A., Lucas X., Castro G.V., Chan K.H., Wright J.E., Runcie A.C. 3-Fluoro-4-hydroxyprolines: synthesis, conformational analysis, and stereoselective recognition by the VHL E3 ubiquitin ligase for targeted protein degradation. J Am Chem Soc. 2018;140:9299–9313. doi: 10.1021/jacs.8b05807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Buckley D.L., Van Molle I., Gareiss P.C., Tae H.S., Michel J., Noblin D.J. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J Am Chem Soc. 2012;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Buckley D.L., Gustafson J.L., Van Molle I., Roth A.G., Tae H.S., Gareiss P.C. Small-molecule inhibitors of the interaction between the E3 ligase VHL and Hif1α. Angew Chem Int Ed Engl. 2012;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dias D.M., Van Molle I., Baud M.G., Galdeano C., Geraldes C.F., Ciulli A. Is NMR fragment screening fine-tuned to assess druggability of protein−protein interactions? ACS Med Chem Lett. 2014;5:23–28. doi: 10.1021/ml400296c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Galdeano C., Gadd M.S., Soares P., Scaffidi S., Van Molle I., Birced I. Structure-guided design and optimization of small molecules targeting the protein−protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J Med Chem. 2014;57:8657–8663. doi: 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Frost J., Galdeano C., Soares P., Gadd M.S., Grzes K.M., Ellis L. Potent and selective chemical probe of hypoxic signalling downstream of HIF-α hydroxylation via VHL inhibition. Nat Commun. 2016;7:13312. doi: 10.1038/ncomms13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Soares P., Gadd M.S., Frost J., Galdeano C., Ellis L., Epemolu O. Group-based optimization of potent and cell-active inhibitors of the von Hippel-Lindau (VHL) E3 ubiquitin ligase: structure-activity relationships leading to the chemical probe (2S,4R)-1-((S)-2-(1-cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298) J Med Chem. 2018;61:599–618. doi: 10.1021/acs.jmedchem.7b00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bondeson D.P., Mares A., Smith I.E., Ko E., Campos S., Miah A.H. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gallenkamp D., Gelato K.A., Haendler B., Weinmann H. Bromodomains and their pharmacological inhibitors. ChemMedChem. 2014;9:438–464. doi: 10.1002/cmdc.201300434. [DOI] [PubMed] [Google Scholar]
- 165.Raina K., Lu J., Qian Y., Altieri M., Gordon D., Rossi A.M. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2016;113:7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Belkina A.C., Denis G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zengerle M., Chan K.H., Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gadd M.S., Testa A., Lucas X., Chan K.H., Chen W., Lamont D.J. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13:514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zoppi V., Hughes S.J., Maniaci C., Testa A., Gmaschitz T., Wieshofer C. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J Med Chem. 2019;62:699–726. doi: 10.1021/acs.jmedchem.8b01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lai A.C., Toure M., Hellerschmied D., Salami J., Jaime-Figueroa S., Ko E. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew Chem Int Ed Engl. 2016;55:807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yu T., Yang Y., Yin D.Q., Hong S., Son Y.J., Kim J.H. TBK1 inhibitors: a review of patent literature (2011–2014) Expert Opin Ther Pat. 2015;25:1385–1396. doi: 10.1517/13543776.2015.1081168. [DOI] [PubMed] [Google Scholar]
- 172.Clark K., Plater L., Peggie M., Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IκB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem. 2009;284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Crew A.P., Raina K., Dong H., Qian Y., Wang J., Vigil D. Identification and characterization of von Hippel-Lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-binding kinase 1. J Med Chem. 2018;61:583–598. doi: 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- 174.Burslem G.M., Smith B.E., Lai A.C., Jaime-Figueroa S., McQuaid D.C., Bondeson D.P. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem Biol. 2018;25:67–77. doi: 10.1016/j.chembiol.2017.09.009. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Burslem G.M., Song J., Chen X., Hines J., Crews C.M. Enhancing antiproliferative activity and selectivity of a FLT-3 inhibitor by proteolysis targeting chimera conversion. J Am Chem Soc. 2018;140:16428–16432. doi: 10.1021/jacs.8b10320. [DOI] [PubMed] [Google Scholar]
- 176.Zarrinkar P.P., Gunawardane R.N., Cramer M.D., Gardner M.F., Brigham D., Belli B. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML) Blood. 2009;114:2984–2992. doi: 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Le Douarin B., Zechel C., Garnier J.M., Lutz Y., Tora L., Pierrat P. The N-terminal part of TIF1, a putative mediator of the ligand-dependent activation function (AF-2) of nuclear receptors, is fused to B-raf in the oncogenic protein T18. EMBO J. 1995;14:2020–2033. doi: 10.1002/j.1460-2075.1995.tb07194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gechijian L.N., Buckley D.L., Lawlor M.A., Reyes J.M., Paulk J., Ott C.J. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat Chem Biol. 2018;14:405–412. doi: 10.1038/s41589-018-0010-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kang C.H., Lee D.H., Lee C.O., Du Ha J., Park C.H., Hwang J.Y. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC) Biochem Biophys Res Commun. 2018;505:542–547. doi: 10.1016/j.bbrc.2018.09.169. [DOI] [PubMed] [Google Scholar]
- 180.Lee B.Y., Timpson P., Horvath L.G., Daly R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther. 2015;146:132–149. doi: 10.1016/j.pharmthera.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 181.Béraud C., Dormoy V., Danilin S., Lindner V., Béthry A., Hochane M. Targeting FAK scaffold functions inhibits human renal cell carcinoma growth. Int J Cancer. 2015;137:1549–1559. doi: 10.1002/ijc.29522. [DOI] [PubMed] [Google Scholar]
- 182.Cromm P.M., Samarasinghe K.T., Hines J., Crews C.M. Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J Am Chem Soc. 2018;140:17019–17026. doi: 10.1021/jacs.8b08008. [DOI] [PubMed] [Google Scholar]
- 183.Han X., Wang C., Qin C., Xiang W., Fernandez-Salas E., Yang C.Y. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J Med Chem. 2019;62:941–964. doi: 10.1021/acs.jmedchem.8b01631. [DOI] [PubMed] [Google Scholar]
- 184.Smith B.E., Wang S.L., Jaime-Figueroa S., Harbin A., Wang J., Hamman B.D. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun. 2019;10:131. doi: 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Akira S., Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 186.Chiang E.Y., Yu X., Grogan J.L. Immune complex-mediated cell activation from systemic lupus erythematosus and rheumatoid arthritis patients elaborate different requirements for IRAK1/4 kinase activity across human cell types. J Immunol. 2011;186:1279–1288. doi: 10.4049/jimmunol.1002821. [DOI] [PubMed] [Google Scholar]
- 187.Nunes J., McGonagle G.A., Eden J., Kiritharan G., Touzet M., Lewell X. Targeting IRAK4 for degradation with PROTACs. ACS Med Chem Lett. 2019;10:1081–1085. doi: 10.1021/acsmedchemlett.9b00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ribeiro-Silva C., Vermeulen W., Lans H. SWI/SNF: complex complexes in genome stability and cancer. DNA Repair (Amst) 2019;77:87–95. doi: 10.1016/j.dnarep.2019.03.007. [DOI] [PubMed] [Google Scholar]
- 189.Hodges C., Kirkland J.G., Crabtree G.R. The many roles of BAF (mSWI/SNF) and PBAF complexes in cancer. Cold Spring Harb Perspect Med. 2016;6:a026930. doi: 10.1101/cshperspect.a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Farnaby W., Koegl M., Roy M.J., Whitworth C., Diers E., Trainor N. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol. 2019;15:672–680. doi: 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 192.Fischer E.S., Böhm K., Lydeard J.R., Yang H., Stadler M.B., Cavadini S. Structure of the DDB1−CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lopez-Girona A., Mendy D., Ito T., Miller K., Gandhi A.K., Kang J. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26:2326–2335. doi: 10.1038/leu.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Krönke J., Udeshi N.D., Narla A., Grauman P., Hurst S.N., McConkey M. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Petzold G., Fischer E.S., Thoma N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature. 2016;532:127–130. doi: 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- 196.Lohbeck J., Miller A.K. Practical synthesis of a phthalimide-based cereblon ligand to enable PROTAC development. Bioorg Med Chem Lett. 2016;26:5260–5262. doi: 10.1016/j.bmcl.2016.09.048. [DOI] [PubMed] [Google Scholar]
- 197.Hansen J.D., Condroski K., Correa M., Muller G., Man H.W., Ruchelman A. Protein degradation via CRL4CRBN ubiquitin ligase: discovery and structure-activity relationships of novel glutarimide analogs that promote degradation of Aiolos and/or GSPT1. J Med Chem. 2018;61:492–503. doi: 10.1021/acs.jmedchem.6b01911. [DOI] [PubMed] [Google Scholar]
- 198.Matyskiela M.E., Zhang W., Man H.W., Muller G., Khambatta G., Baculi F. A cereblon modulator (CC-220) with improved degradation of ikaros and aiolos. J Med Chem. 2018;61:535–542. doi: 10.1021/acs.jmedchem.6b01921. [DOI] [PubMed] [Google Scholar]
- 199.Matyskiela M.E., Lu G., Ito T., Pagarigan B., Lu C.C., Miller K. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature. 2016;535:252–257. doi: 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- 200.Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lovén J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Coleman D.J., Gao L., Schwartzman J., Korkola J.E., Sampson D., Derrick D.S. Maintenance of MYC expression promotes de novo resistance to BET bromodomain inhibition in castration-resistant prostate cancer. Sci Rep. 2019;9:3823. doi: 10.1038/s41598-019-40518-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shi W., Zhang C., Ning Z., Hua Y., Li Y., Chen L. Long non-coding RNA LINC00346 promotes pancreatic cancer growth and gemcitabine resistance by sponging miR-188-3P to derepress BRD4 expression. J Exp Clin Cancer Res. 2019;38:60. doi: 10.1186/s13046-019-1055-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wurz R.P., Dellamaggiore K., Dou H., Javier N., Lo M.C., McCarter J.D. A “click chemistry platform” for the rapid synthesis of bispecific molecules for inducing protein degradation. J Med Chem. 2018;61:453–461. doi: 10.1021/acs.jmedchem.6b01781. [DOI] [PubMed] [Google Scholar]
- 205.Remillard D., Buckley D.L., Paulk J., Brien G.L., Sonnett M., Seo H.S. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew Chem Int Ed Engl. 2017;56:5738–5743. doi: 10.1002/anie.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Yeh Y.Y., Chen R., Hessler J., Mahoney E., Lehman A.M., Heerema N.A. Up-regulation of CDK9 kinase activity and Mcl-1 stability contributes to the acquired resistance to cyclin-dependent kinase inhibitors in leukemia. Oncotarget. 2015;6:2667–2679. doi: 10.18632/oncotarget.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Boffo S., Damato A., Alfano L., Giordano A. CDK9 inhibitors in acute myeloid leukemia. J Exp Clin Cancer Res. 2018;37:36. doi: 10.1186/s13046-018-0704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Robb C.M., Contreras J.I., Kour S., Taylor M.A., Abid M., Sonawane Y.A. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC) Chem Commun (Camb) 2017;53:7577–7580. doi: 10.1039/c7cc03879h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Bian J., Ren J., Li Y., Wang J., Xu X., Feng Y. Discovery of Wogonin-based PROTACs against Cdk9 and capable of achieving antitumor activity. Bioorg Chem. 2018;81:373–381. doi: 10.1016/j.bioorg.2018.08.028. [DOI] [PubMed] [Google Scholar]
- 210.Olson C.M., Jiang B., Erb M.A., Liang Y., Doctor Z.M., Zhang Z. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018;14:163–170. doi: 10.1038/nchembio.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.O'Leary B., Finn R.S., Turner N.C. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417–430. doi: 10.1038/nrclinonc.2016.26. [DOI] [PubMed] [Google Scholar]
- 212.Brand M., Jiang B., Bauer S., Donovan K.A., Liang Y., Wang E.S. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem Biol. 2019;26:300–306.e9. doi: 10.1016/j.chembiol.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Jiang B., Wang E.S., Donovan K.A., Liang Y., Fischer E.S., Zhang T. Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew Chem Int Ed Engl. 2019;58:6321–6326. doi: 10.1002/anie.201901336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Schiedel M., Herp D., Hammelmann S., Swyter S., Lehotzky A., Robaa D. Chemically induced degradation of Sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals) J Med Chem. 2018;61:482–491. doi: 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- 215.Zhou B., Hu J., Xu F., Chen Z., Bai L., Fernandez-Salas E. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J Med Chem. 2018;61:462–481. doi: 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bai L., Zhou B., Yang C.Y., Ji J., McEachern D., Przybranowski S. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017;77:2476–2487. doi: 10.1158/0008-5472.CAN-16-2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Huang H.T., Dobrovolsky D., Paulk J., Yang G., Weisberg E.L., Doctor Z.M. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem Biol. 2018;25:88–99.e6. doi: 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Buhimschi A.D., Armstrong H.A., Toure M., Jaime-Figueroa S., Chen T.L., Lehman A.M. Targeting the C481S ibrutinib-resistance mutation in Bruton's tyrosine kinase using PROTAC-mediated degradation. Biochemistry. 2018;57:3564–3575. doi: 10.1021/acs.biochem.8b00391. [DOI] [PubMed] [Google Scholar]
- 219.Zorba A., Nguyen C., Xu Y., Starr J., Borzilleri K., Smith J. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci U S A. 2018;115 doi: 10.1073/pnas.1803662115. E7285−92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Holla V.R., Elamin Y.Y., Bailey A.M., Johnson A.M., Litzenburger B.C., Khotskaya Y.B. ALK: a tyrosine kinase target for cancer therapy. Cold Spring Harb Mol Case Stud. 2017;3 doi: 10.1101/mcs.a001115. a001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhang C., Han X.R., Yang X., Jiang B., Liu J., Xiong Y. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK) Eur J Med Chem. 2018;151:304–314. doi: 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nagy Z., Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 2007;26:5341–5357. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
- 223.De Jong R.C., Ewing M.M., De Vries M.R., Karper J.C., Bastiaansen A.J., Peters H.A. The epigenetic factor PCAF regulates vascular inflammation and is essential for intimal hyperplasia development. PLoS One. 2017;12 doi: 10.1371/journal.pone.0185820. e0185820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Bassi Z.I., Fillmore M.C., Miah A.H., Chapman T.D., Maller C., Roberts E.J. Modulating PCAF/GCN5 immune cell function through a PROTAC approach. ACS Chem Biol. 2018;13:2862–2867. doi: 10.1021/acschembio.8b00705. [DOI] [PubMed] [Google Scholar]
- 225.Chen H., Chen F., Liu N., Wang X., Gou S. Chemically induced degradation of CK2 by proteolysis targeting chimeras based on a ubiquitin−proteasome pathway. Bioorg Chem. 2018;81:536–544. doi: 10.1016/j.bioorg.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 226.McCoull W., Cheung T., Anderson E., Barton P., Burgess J., Byth K. Development of a novel B-cell lymphoma 6 (BCL6) PROTAC to provide insight into small molecule targeting of BCL6. ACS Chem Biol. 2018;13:3131–3141. doi: 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
- 227.Li Y., Yang J., Aguilar A., McEachern D., Przybranowski S., Liu L. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J Med Chem. 2019;62:448–466. doi: 10.1021/acs.jmedchem.8b00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Krajcovicova S., Jorda R., Hendrychova D., Krystof V., Soural M. Solid-phase synthesis for thalidomide-based proteolysis-targeting chimeras (PROTAC) Chem Commun (Camb) 2019;55:929–932. doi: 10.1039/c8cc08716d. [DOI] [PubMed] [Google Scholar]
- 229.Sun Y., Ding N., Song Y., Yang Z., Liu W., Zhu J. Degradation of Bruton's tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-hodgkin lymphomas. Leukemia. 2019 doi: 10.1038/s41375-019-0440-x. [DOI] [PubMed] [Google Scholar]
- 230.Wu H., Yang K., Zhang Z., Leisten E.D., Li Z., Xie H. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J Med Chem. 2019 doi: 10.1021/acs.jmedchem.9b00516. [DOI] [PubMed] [Google Scholar]
- 231.Neklesa T., Snyder L.B., Willard R.R., Vitale N., Raina K., Pizzano J. Abstract 5236: ARV-110: an androgen receptor PROTAC degrader for prostate cancer. Cancer Res. 2018;78:5236. [Google Scholar]
- 232.Neklesa T., Snyder L.B., Willard R.R., Vitale N., Pizzano J., Gordon D.A. ARV-110: an oral androgen receptor PROTAC degrader for prostate cancer. J Clin Oncol. 2019;37:259. [Google Scholar]
- 233.Arvinas. ARV-110: targeting the androgen receptor [Internet]. [updated 2019 Mar 25]. Available from: http://arvinas.com/therapeutic-programs/androgen-receptor/.
- 234.Ciulli A., Zengerle M., Chan K.H. 2018 Feb 22. Derivatives of 1-[(Cyclopentyl or 2-pyrrolidinyl)carbonylaminomethyl]-4-(1,3-thiazol-5-yl) benzene which are useful for the treatment of proliferative, autoimmune or inflammatory diseases. United States Patent US 20180050021A1. [Google Scholar]
- 235.Crew A.P., Wang J., Dong H., Qian Y., Crews C.M. 2018 May 31. Tank-binding kinase-1 PROTACs and associated methods of use. United States Patent US 20180147202. [Google Scholar]
- 236.Crew A.P., Crews C., Dong H., Wang J., Qian Y., Siu K. 2016 Mar 3. Imide-based modulators of proteolysis and associated methods of use. United States Patent US 20160058872. [Google Scholar]
- 237.Kobayashi S., Boggon T.J., Dayaram T., Jänne P.A., Kocher O., Meyerson M. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 238.An S., Fu L. Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine. 2018;36:553–562. doi: 10.1016/j.ebiom.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Moon S., Lee B.H. Chemically induced cellular proteolysis: an emerging therapeutic strategy for undruggable targets. Mol Cells. 2018;41:933–942. doi: 10.14348/molcells.2018.0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Yang H., Higgins B., Kolinsky K., Packman K., Go Z., Iyer R. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010;70:5518–5527. doi: 10.1158/0008-5472.CAN-10-0646. [DOI] [PubMed] [Google Scholar]
- 241.Churcher I. PROTAC-induced protein degradation in drug discovery: breaking the rules or just making new ones? J Med Chem. 2018;61:444–452. doi: 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- 242.Demizu Y., Shibata N., Hattori T., Ohoka N., Motoi H., Misawa T. Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg Med Chem Lett. 2016;26:4865–4869. doi: 10.1016/j.bmcl.2016.09.041. [DOI] [PubMed] [Google Scholar]
- 243.Zhang L., Riley-Gillis B., Vijay P., Shen Y. Acquired resistance to BET-PROTACs (proteolysis-targeting chimeras) caused by genomic alterations in core components of E3 ligase complexes. Mol Cancer Ther. 2019;18:1302–1311. doi: 10.1158/1535-7163.MCT-18-1129. [DOI] [PubMed] [Google Scholar]
- 244.Suh J., Yoo S.H., Kim M.G., Jeong K., Ahn J.Y., Kim M.S. Cleavage agents for soluble oligomers of amyloid β peptides. Angew Chem Int Ed Engl. 2007;46:7064–7067. doi: 10.1002/anie.200702399. [DOI] [PubMed] [Google Scholar]
- 245.Wu W.H., Lei P., Liu Q., Hu J., Gunn A.P., Chen M.S. Sequestration of copper from β-amyloid promotes selective lysis by cyclen-hybrid cleavage agents. J Biol Chem. 2008;283:31657–31664. doi: 10.1074/jbc.M804722200. [DOI] [PubMed] [Google Scholar]
- 246.Chu T.T., Li Q.Q., Qiu T., Sun Z.Y., Hu Z.W., Chen Y.X. Clearance of the intracellular high level of the tau protein directed by an artificial synthetic hydrolase. Mol BioSyst. 2014;10:3081–3085. doi: 10.1039/c4mb00508b. [DOI] [PubMed] [Google Scholar]
- 247.Suh J., Chei W.S., Lee T.Y., Kim M.G., Yoo S.H., Jeong K. Cleavage agents for soluble oligomers of human islet amyloid polypeptide. J Biol Inorg Chem. 2008;13:693–701. doi: 10.1007/s00775-008-0354-y. [DOI] [PubMed] [Google Scholar]
- 248.Jeong K.H., Chung W.Y., Kye Y.S., Kim D.W., Song S.U. Cu(II) cyclen cleavage agent with BTA-derived binding group for H-IAPP. Bull Korean Chem Soc. 2011;32:1751–1753. [Google Scholar]
- 249.Hu J., Yu Y.P., Cui W., Fang C.L., Wu W.H., Zhao Y.F. Cyclen-hybrid compound captures copper to protect INS-1 cells from islet amyloid polypeptide cytotoxicity by inhibiting and lysing effects. Chem Commun. 2010;46:8023–8025. doi: 10.1039/c0cc02555k. [DOI] [PubMed] [Google Scholar]
- 250.Klein A.N., Corda E., Gilch S. Peptide aptamer-mediated modulation of prion protein α-cleavage as treatment strategy for prion and other neurodegenerative diseases. Neural Regen Res. 2018;13:2108–2110. doi: 10.4103/1673-5374.241460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Xue Y., Gao X., Cao J., Liu Z., Jin C., Wen L. A summary of computational resources for protein phosphorylation. Curr Protein Pept Sci. 2010;11:485–496. doi: 10.2174/138920310791824138. [DOI] [PubMed] [Google Scholar]
- 252.Nishi H., Shaytan A., Panchenko A.R. Physicochemical mechanisms of protein regulation by phosphorylation. Front Genet. 2014;5:270. doi: 10.3389/fgene.2014.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Schlessinger J., Lemmon M.A. SH2 and PTB domains in tyrosine kinase signaling. Sci STKE. 2003;2003:re12. doi: 10.1126/stke.2003.191.re12. [DOI] [PubMed] [Google Scholar]
- 254.Tan A.C., Vyse S., Huang P.H. Exploiting receptor tyrosine kinase co-activation for cancer therapy. Drug Discov Today. 2017;22:72–84. doi: 10.1016/j.drudis.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Asanuma H., Torigoe T., Kamiguchi K., Hirohashi Y., Ohmura T., Hirata K. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005;65:11018–11025. doi: 10.1158/0008-5472.CAN-05-0491. [DOI] [PubMed] [Google Scholar]
- 256.Regad T. Targeting RTK signaling pathways in cancer. Cancers (Basel) 2015;7:1758–1784. doi: 10.3390/cancers7030860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Hines J., Gough J.D., Corson T.W., Crews C.M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc Natl Acad Sci U S A. 2013;110:8942–8947. doi: 10.1073/pnas.1217206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Chatterjee S., Mudher A. Alzheimer's disease and type 2 diabetes: a critical assessment of the shared pathological traits. Front Neurosci. 2018;12:383. doi: 10.3389/fnins.2018.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Chesser A.S., Pritchard S.M., Johnson G.V. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front Neurol. 2013;4:122. doi: 10.3389/fneur.2013.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Vilchez D., Saez I., Dillin A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun. 2014;5:5659. doi: 10.1038/ncomms6659. [DOI] [PubMed] [Google Scholar]
- 261.Katsumoto A., Takeuchi H., Takahashi K., Tanaka F. Microglia in Alzheimer's disease: risk factors and inflammation. Front Neurol. 2018;9:978. doi: 10.3389/fneur.2018.00978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Leestemaker Y., De Jong A., Witting K.F., Penning R., Schuurman K., Rodenko B. Proteasome activation by small molecules. Cell Chem Biol. 2017;24:725–736.e7. doi: 10.1016/j.chembiol.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 263.Gao N., Chen Y.X., Zhao Y.F., Li Y.M. Chemical methods to knock down the amyloid proteins. Molecules. 2017;22:916. doi: 10.3390/molecules22060916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Gao N., Chu T.T., Li Q.Q., Lim Y.J., Qiu T., Ma M.R. Hydrophobic tagging-mediated degradation of Alzheimer's disease related tau. RSC Adv. 2017;7:40362–40366. [Google Scholar]
- 265.Silva M.C., Ferguson F.M., Cai Q., Donovan K.A., Nandi G., Patnaik D. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. eLife. 2019;8:e45457. doi: 10.7554/eLife.45457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 267.Hitchcock S.A., Pennington L.D. Structure−brain exposure relationships. J Med Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 268.Ghose A.K., Herbertz T., Hudkins R.L., Dorsey B.D., Mallamo J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem Neurosci. 2012;3:50–68. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Palmer A.M., Alavijeh M.S. Translational CNS medicines research. Drug Discov Today. 2012;17:1068–1078. doi: 10.1016/j.drudis.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 270.Fischer H., Gottschlich R., Seelig A. Blood-brain barrier permeation: molecular parameters governing passive diffusion. J Membr Biol. 1998;165:201–211. doi: 10.1007/s002329900434. [DOI] [PubMed] [Google Scholar]
- 271.Galdeano C. Drugging the undruggable: targeting challenging E3 ligases for personalized medicine. Future Med Chem. 2017;9:347–350. doi: 10.4155/fmc-2017-0009. [DOI] [PubMed] [Google Scholar]
- 272.Maniaci C., Hughes S.J., Testa A., Chen W., Lamont D.J., Rocha S. Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun. 2017;8:830. doi: 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Steinebach C., Lindner S., Udeshi N.D., Mani D.C., Kehm H., Köpff S. Homo-PROTACs for the chemical knockdown of cereblon. ACS Chem Biol. 2018;13:2771–2782. doi: 10.1021/acschembio.8b00693. [DOI] [PubMed] [Google Scholar]
- 274.Girardini M., Maniaci C., Hughes S.J., Testa A., Ciulli A. Cereblon versus VHL: hijacking E3 ligases against each other using PROTACs. Bioorg Med Chem. 2019;27:2466–2479. doi: 10.1016/j.bmc.2019.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A. The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Nalawansha D.A., Paiva S.L., Rafizadeh D.N., Pettersson M., Qin L., Crews C.M. Targeted protein internalization and degradation by ENDosome TArgeting chimeras (ENDTACs) ACS Cent Sci. 2019;5:1079–1084. doi: 10.1021/acscentsci.9b00224. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 277.Huisgen R., Szeimies G., Möbius L. 1.3-Dipolare cycloadditionen, XXXII. Kinetik der Additionen organischer Azide a CC-Mehrfachbindungen. Chem Ber. 1967;100:2494–2507. [Google Scholar]
- 278.Amblard F., Cho J.H., Schinazi R.F. Cu(I)-catalyzed huisgen azide−alkyne 1,3-dipolar cycloaddition reaction in nucleoside, nucleotide, and oligonucleotide chemistry. Chem Rev. 2009;109:4207–4220. doi: 10.1021/cr9001462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Lebraud H., Wright D.J., Johnson C.N., Heightman T.D. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent Sci. 2016;2:927–934. doi: 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Bondeson D.P., Smith B.E., Burslem G.M., Buhimschi A.D., Hines J., Jaime-Figueroa S. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem Biol. 2018;25:78–87.e5. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Zhang X., Crowley V.M., Wucherpfennig T.G., Dix M.M., Cravatt B.F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol. 2019;15:737–746. doi: 10.1038/s41589-019-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Watt G.F., Scott-Stevens P., Gaohua L. Targeted protein degradation in vivo with proteolysis targeting chimeras: current status and future considerations. Drug Discov Today Technol. 2019;31:69–80. doi: 10.1016/j.ddtec.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 283.Arvinas presents preclinical data on protein degrader, ARV-471, at the 2018 San Antonio breast cancer symposium (SABCS) [Internet]. [updated 2018 Dec 9]. Available from: http://www.firstwordpharma.com/node/1610303?tsid=17#axzz5tROLEB8T.
- 284.Arvinas receives authorization to proceed for ARV-471, a PROTAC protein degrader to treat patients with locally advanced or metastatic ER+/HER2- breast cancer [Internet]. [updated 2019 Jun 25]. Available from: http://www.firstwordpharma.com/node/1649497?Tsid=4#axzz5rv2bsalg.
- 285.Kelleher J., Campbell V., Chen J., Gollob J., Ji N., Kamadurai H. KYM-001, a first-in-class oral IRAK4 protein degrader, induces tumor regression in xenograft models of MYD88-mutant ABC DLBCL alone and in combination with BTK inhibition. Hematol Oncol. 2019;37:129. [Google Scholar]