

Abstract
Our recent studies demonstrated that the natural product nobiletin (NOB) served as a promising multidrug resistance (MDR) reversal agent and improved the effectiveness of cancer chemotherapy in vitro. However, low aqueous solubility and difficulty in total synthesis limited its application as a therapeutic agent. To tackle these challenges, NOB was synthesized in a high yield by a concise route of six steps and fourteen derivatives were synthesized with remarkable solubility and efficacy. All the compounds showed improved sensitivity to paclitaxel (PTX) in P-glycoprotein (P-gp) overexpressing MDR cancer cells. Among them, compound 29d exhibited water solubility 280-fold higher than NOB. A drug-resistance A549/T xenograft model showed that 29d, at a dose of 50 mg/kg co-administered with PTX (15 mg/kg), inhibited tumor growth more effective than NOB and remarkably increased PTX concentration in the tumors via P-gp inhibition. Moreover, Western blot experiments revealed that 29d inhibited expression of NRF2, phosphorylated ERK and AKT in MDR cancer cells, thus implying 29d of multiple mechanisms to reverse MDR in lung cancer.
Key words: Nobiletin, Cancer multidrug resistance, Mechanism, Nobiletin, P-gp inhibition, Reversal agents, Solubility, Total synthesis
Abbreviations: Ac2O, acetic anhydride; AcOH, acetic acid; AcONa, sodium acetate; BF3·Et2O, boron trifluoride diethyl etherate; DCM, dichloromethane; DCE, dichloroethane; DMF, N,N-dimethylformamide; DMSO, dimethyl sulfoxide; DOX, doxorubicin; Et3N, triethylamine; Flutax-2, a fluorescent taxol derivative; MDR, multidrug resistance; NOB, nobiletin; NIS, N-iodosuccinimide; P-gp, P-glycoprotein; PI, propidium iodide; PTX, paclitaxel; QND, quinidine; Rho123, rhodamine 123; t-BuOK, potassium tert-butylate; TCA, trichloroacetic acid; THF, tetrahydrofuran; TLC, thin-layer chromatography; SRB, sulforhodamine B; Ver, verapamil
Graphical abstract
Compound 29d exhibited water solubility 280-fold higher than nobiletin (N). In a drug resistance A549/T xenograft model, 29d co-administered with paclitaxel (PTX) inhibited tumor growth more effective than N and remarkably increased PTX concentration in the tumors. Activated NRF2/PI3K/AKT pathways in MDR cancer cells were markedly inhibited by 29d and PTX.

1. Introduction
As the second leading cause of death next to heart disease, cancer is a growing global threat with 18 million cases and 9.6 million deaths in 2018 around the world1. Chemotherapy is commonly used for treatment of many types of cancers, but multidrug resistance (MDR) often causes chemotherapy failure, leading to death of a majority of patients2. Overcoming drug resistance has far-reaching implications for cancer patients and society3. Cellular mechanisms of drug resistance have been classified into transport-based classical and nonclassical MDR phenotypes3. Overexpression of P-glycoprotein (P-gp, P-gp/ABCB1 encoded by the MDR1 gene), one of the ABC-family efflux transporters, is associated with poor prognosis in many types of cancers and is frequently observed in recurrent tumors in clinic4. In the past 20 years, three generations of MDR inhibitors have been developed to target efflux transporters, especially P-gp. Unfortunately, drawbacks of the targeted therapy occurred after treatment for long time4. Recently, studies on reversal of MDR by natural products and their synthesized analogues, such as secalonic acid D, epigallocatechin gallate derivative, WS-10, etc5, 6, 7. have received increasing attention. Among them, several fragments (Murcko, Murcko generic, RECAP and aromatic ring) are found to actively inhibit ABCB16, serving as templates for designing new potent reversal agents to overcome MDR of cancers.
As a polymethoxy flavonoid isolated from tangerines, nobiletin (NOB, 3′,4′,5,6,7,8,-hexamethoxyflavone, Fig. 1) exhibits a good safety profile and a broad spectrum of pharmacological activities, such as anti-oxidative, anti-inflammatory8 and anti-tumor effects9, especially high potency in cocktail therapy for drug-resistant cancers10. However, NOB exists as an aglycone, has poor water solubility (1–5 μg/mL) and low oral bioavailability (<1%), which limit its application as a therapeutic agent11. In addition to the traditional methods of separation and extraction, NOB could be prepared by total synthesis of over eleven steps12. Our recent studies demonstrated that NOB worked as an inhibitor of the MDR-efflux proteins by competing with chemotherapy drugs for the same binding site of P-gp, thus leading to improvement of the effectiveness of cancer chemotherapy in vitro10. NOB showed the MDR reversal ability to make the ovarian tumor cells of A2780/T, which resisted to paclitaxel (PTX), became >400 folds sensitive in comparison with its parent A2780 cells. Although there are lots of synthesized compounds or natural products demonstrated for their MDR reversal abilities, few of them have comparable reversal ability with NOB. In addition, our previous study revealed that NOB not only inhibited the P-gp function but also modulated the NRF2/PI3K/AKT pathways which are the important mechanisms of the MDR. These findings encourage further studies on producing NOB and its derivatives in a simpler and more effective synthetic way to find more potent and soluble products.
Figure 1.
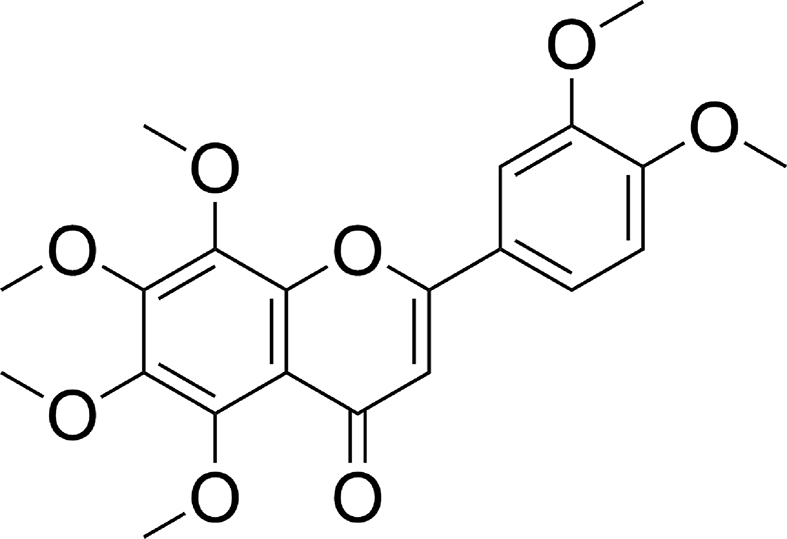
Chemical structure of nobiletin (NOB).
Herein, to overcome the disadvantages and expand the scope of application, NOB was newly generated in high yield by a concise route of six steps and fourteen derivatives with remarkable solubility were synthesized. Further, we analyzed the efficacy of the compounds as MDR reversal agents in P-gp-overexpressing MDR cancer cells in vitro and in an A549/T xenograft model in vivo. We also investigated the underlying mechanism for the MDR reversal effects of NOB and its derivatives.
2. Results and discussion
2.1. Synthesis of the designed compounds
The target compounds were prepared by the synthetic routes reported in Scheme 1, Scheme 2, Scheme 3. First, commercially available 3,4,5-trimethoxyphenol (1) was selected as the starting material and reacted with acetic anhydride to afford the acetylation product 2, followed by Fries rearrangement to provide 3, which was then halogenated with N-iodosuccinimide to obtain compound 4 (Scheme 1). The key intermediate 1-(2-hydroxy-3,4,5,6-tetramethoxyphenyl)ethan-1-one (5) was synthesized by the reaction of 4 with sodium methoxide in the presence of copper chloride13. Compounds 6a‒6c were synthesized by acylation of 5 with the corresponding acyl chlorides14. The target compounds 7a‒7c were obtained by cyclization of 6a‒6c in the presence of triethylsilyl trifluoromethanesulfonate, respectively.
Scheme 1.

Syntheses of 5,6,7,8-tetramethoxy-2-phenyl-4H-chromen-4-ones 7a‒7c. Reagents and conditions: (a) Ac2O, AcOH, 110 °C, 2 h; (b) BF3·Et2O, AcOH, 70 °C, 2 h; (c) NIS, p-toluenesulfonic acid monohydrate, CH3CN, rt, 2 h; (d) 4 mol/L MeONa in MeOH, CuCl, DMF, 90 °C, 30 min; (e) acyl chlorides, Et3N, DCM, 0 °C to rt, 2 h; (f) triethylsilyl trifluoromethanesulfonate, Et3N, DCE, reflux, 2 h.
Scheme 2.

Syntheses of 2-(3,4-dimethoxyphenyl)-5,6,7-trimethoxy-4H-chromen-4-ones 13, 16 and 18. Reagents and conditions: (a) 3,4-dimethoxybenzoyl chloride, Et3N, DCM, 0 °C to rt, 2 h; (b) Br2, AcOH, AcONa, 0 °C to rt, 12 h; (c) HNO3, 0 °C, 10 min; (d) triethylsilyl trifluoromethanesulfonate, Et3N, DCE, reflux, 2 h; (e) 3,4-dimethoxybenzoyl chloride, Et3N, DCM, 0 °C to rt, 2 h; (f) 3,4-dimethoxybenzoyl chloride, Et3N, DCM, 0 °C to rt, 2 h; (g) triethylsilyl trifluoromethanesulfonate, Et3N, DCE, reflux, 2 h; (h) triethylsilyl trifluoromethanesulfonate, Et3N, DCE, reflux, 2 h; (i) Pd/C, H2, THF, rt, 12 h.
Scheme 3.

Syntheses of 2-(3,4-dimethoxyphenyl)-6,7,8-trimethoxy-4H-chromen-4-ones 23 and 29a‒29d. Reagents and conditions: (a) Ac2O, AcOH, 110 °C, 2 h; (b) BF3·Et2O, AcOH, 70 °C, 2 h; (c) 3,4-dimethoxybenzoyl chloride, Et3N, DCE, 0 °C to rt, 2 h; (d) triethylsilyl trifluoromethanesulfonate, Et3N, DCE, reflux, 2 h. (e) iodomethane, K2CO3, DMF, rt, 1 h; (f) HNO3, 0 °C, 10 min; (g) Pd/C, H2, THF, rt, 12 h; (h) 3,4-dimethoxybenzoyl chloride, Et3N, DCM, 0 °C to rt 2 h; (i) t-BuOK, THF, reflux, 12 h; (j) iodomethane, K2CO3, DMF, chloroalkanes, NaH, DMF, 90 °C, 2.5 h.
As outlined in Scheme 2, compounds 11 and 12 were obtained from 3 by treatment with bromine and nitric acid, respectively. Aryl esters 10, 14 and 15 were synthesized by acylation of 1-(2-hydroxyphenyl)ethan-1-ones with the corresponding acyl chlorides, which were treated with triethylsilyl trifluoromethanesulfonate to provide the corresponding flavones 13, 16 and 17. Subsequently, compound 17 was reduced by catalytic hydrogenation with Pd/C to afford 8-amino-2-(3,4-dimethoxyphenyl)-5,6,7-trimethoxy-4H-chromen-4-one (18).
Furthermore, commercially available 2,3-dimethoxyphenol (19a) or 2,3,4-trimethoxyphenol (19b) were selected as the starting materials, and the same procedures as in Scheme 1, Scheme 2 were used to obtain compounds 23a‒23d (Scheme 3). The intermediate 21b was treated with iodomethane and K2CO3 in the presence of N,N-dimethylformamide (DMF) to obtain 24, followed with nitric acid to afford compound 25. After 25 was reduced by catalytic hydrogenation with Pd/C to yield compound 26, it was treated with 3,4-dimethoxybenzoyl chloride to obtain 27. Finally, 27 was treated with t-BuOK in a solution of tetrahydrofuran (THF) to produce 2-(3,4-dimethoxyphenyl)-5,6,7,8-tetramethoxyquinolin-4(1H)-one (28)15. Compounds 29a‒29d were synthesized by the reaction of 28 and corresponding alkyl halides in the presence of sodium hydride.
To avoid the interference of false positive compounds in our subsequent study, Pan Assay Interference Compounds (PAINS) screening of the designed compounds was carried out via an online program “PAINS Remover” (http://www.cbligand.org/PAINS/), and all of the compounds passed the filter and were retained.
2.2. NOB and its derivatives reverse ABCB1-mediated MDR
The IC50 values of NOB and its derivatives against PTX-resistant HCT8/T cells (without adding 0.94 μmol/L PTX to the culture medium) were listed in Table 1. These compounds showed anti-tumor effects against resistant human intestine and lung cancer cell lines, and their cytotoxicity are much lower than that of PTX (5.89 μmol/L in HCT8/T).
Table 1.
Nobiletin (NOB) and its derivatives reduce the IC50 of paclitaxel (PTX) for multidrug resistant HCT8/T cancer cells.
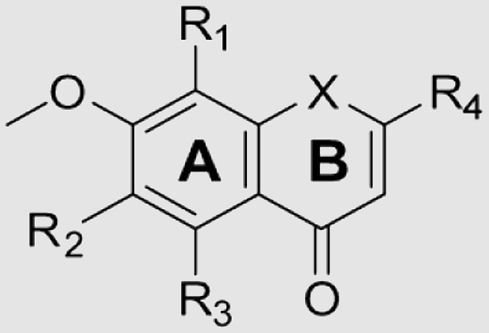
| Compd. | R1 | R2 | R3 | R4 | X | Cytotoxicity IC50 (μmol/L)a | Cytotoxicity of combination IC50 (μmol/L)b | Reversal foldc |
|---|---|---|---|---|---|---|---|---|
| PTX | – | – | – | – | – | 5.89 ± 0.24 | 5.89 ± 0.24 | 1 |
| 7a | OMe | OMe | OMe | 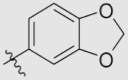 |
O | >100 | 0.14 ± 0.01 | 43 |
| 7b (NOB) | OMe | OMe | OMe | 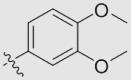 |
O | 44.67 ± 3.83 | 0.030 ± 0.002 | 185 |
| 7c | OMe | OMe | OMe | 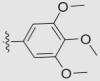 |
O | 38.46 ± 3.30 | 0.080 ± 0.004 | 81 |
| 13 | H | OMe | OMe | |
O | 74.99 ± 4.04 | 0.090 ± 0.005 | 66 |
| 16 | Br | OMe | OMe | |
O | >100 | 0.15 ± 0.01 | 40 |
| 18 | NH2 | OMe | OMe | |
O | 66.83 ± 3.99 | 0.29 ± 0.05 | 21 |
| 23a | OMe | H | H | |
O | 66.83 ± 2.73 | 0.29 ± 0.01 | 21 |
| 23b | OMe | H | H | 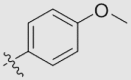 |
O | 69.98 ± 6.78 | 0.44 ± 0.04 | 14 |
| 23c | OMe | H | H | 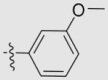 |
O | 39.36 ± 4.41 | 0.36 ± 0.02 | 17 |
| 23d | OMe | OMe | H | |
O | 66.83 ± 2.73 | 0.13 ± 0.01 | 47 |
| 28 | OMe | OMe | OMe | |
NH | >100 | 0.23 ± 0.02 | 27 |
| 29a | OMe | OMe | OMe | |
NMe | 26.61 ± 2.63 | 0.11 ± 0.01 | 55 |
| 29b | OMe | OMe | OMe | |
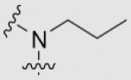 |
21.13 ± 1.77 | 0.15 ± 0.01 | 40 |
| 29c | OMe | OMe | OMe | |
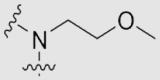 |
59.57 ± 3.62 | 0.24 ± 0.01 | 26 |
| 29d | OMe | OMe | OMe | |
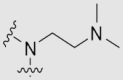 |
>100 | 0.070 ± 0.008 | 87 |
‒Not applicable.
Intrinsic cytotoxicity of target compounds or PTX alone to HCT8/T cells.
Values of IC50 for cytotoxicity of combination (P-gp inhibitor 10 μmol/L + PTX) were as mean ± SD of at least three independent experiments.
The reversal fold was calculated as a ratio of IC50 (PTX) to IC50 (P-gp inhibitor 10 μmol/L + PTX).
The long-term colony formation assays further ascertained the enhanced anti-proliferative effect of NOB and its derivatives on A549/T and HCT8/T cells, as shown in Fig. 2A and B. We noticed that NOB, 13 and 29d could inhibit MDR cancer cell colony formation in a dose-dependent manner. However, there is no difference in the inhibition of the colony formation among 13, 29d and NOB at their lowest effect dosage, indicating the similar reversal effects of NOB and its derivatives after long-time treatment.
Figure 2.

NOB and its derivatives reverse the resistance of paclitaxel (PTX) to P-gp-overexpressing MDR cancer cells. 29d, 13 and NOB enhanced the anti-proliferative effects of PTX in two drug-resistant cancer cell lines, HCT8/T (A) and A549/T (B), in a dose-dependent manner according to the colony formation assay. The cells were treated with PTX (0.24 μmol/L for A549/T and 0.94 μmol/L for HCT8/T for maintaining the resistance of cancer cells) in the presence or absence of 29d, 13 and NOB at the indicated concentrations.
As shown in Table 1, NOB and its derivatives enhanced the anti-tumor effects of PTX by decreasing its IC50 against the resistant HCT8/T cells. Specifically, the IC50 values of PTX in the presence of NOB, 7c, 13 and 29d were reduced by 185-, 81-, 66- and 87-fold, respectively. However, NOB, 7c, 13 and 29d did not affect the IC50 of PTX for the sensitive HCT8 cells (data not shown). In view of the relatively stronger cytotoxicity (38.46 μmol/L) of 7c than that of NOB, only 13, 29d and NOB were selected for further studies.
2.3. Structure–activity relationships (SARs) of NOB and its derivatives
When a methoxy group was added at the C-5 position (7c) relative to 7b (NOB), the affinity decreased since there was insufficient space for accommodation of another methoxy group. The presence of 1,3-benzodioxole (7a) was unfavorable to affinity due to steric hindrance. For inhibitors 23a (one methoxy group), 23d (two methoxy groups), 13 (two methoxy groups) and 7b (three methoxy groups) with the same R4 group, we found that more methoxy groups on the benzene ring A was propitious to better inhibitory potency, whereas the opposite phenomenon was observed on the R4 position for 7b (two methoxy groups) and 7c (three methoxy groups). In addition, for inhibitors 13, 16 and 18, the bulky bromine atom of 16 or the NH2 group of 18 at the R1 group decreased the inhibitory affinity in comparison with the hydrogen atom of 1316, 17, 18.
On the basis of these results, we decided to retain the methoxy groups on the A and B rings and change the side chain at the X position. The dimethylaminoethyl sidechain of 29d formed hydrophobic and van der Waals interactions with the residues of P-gp, which significantly differs from NOB. Therefore, other modifications such as replacement of methylethyldimethylamine with 2-methoxyethyl (29c), H (28), Me (29a) or n-propyl (29b), were failed to improve their binding affinities. Thus, NOB, 29d and 13 were selected for subsequent study.
Moreover, the solubility data of NOB, 13 and 29d were measured as 5.58 ± 1.17, 3.67 ± 0.52 and 1.58 ± 0.13 mg/mL in sodium phosphate buffer (0.35 mol/L, pH = 7.3) at 25 °C, respectively, using the high-performance liquid chromatography (HPLC) approach. The presence of the dimethylaminoethyl sidechain of 29d possibly accounts for the notable improvement in the water solubility of 29d over NOB and 13 (280-fold higher), predicting that 29d possibly has better performance in vivo.
2.4. Anti-tumor effects in A549/T xenograft model
We next evaluated whether synthesized NOB and its derivatives could overcome PTX resistance in the A549/T xenograft model. Based on previous study, PTX was administered via intraperitoneal (i.p.) route at an effective dose (15 mg/kg) for normal tumor19. As expected, PTX alone exhibited no effect on treating MDR tumors, as shown by similar tumor progression between the PTX treated group and vehicle control group, indicating resistance (Fig. 3). However, for the cocktail 29d and PTX administration, 29d at both 25 mg/kg and 50 mg/kg dosages significantly reduced the tumor volumes (Fig. 3B‒D) compared with PTX alone, indicating considerable therapeutic efficacy. Moreover, there was no apparent weight loss observed in all nude mice (Fig. 3A), suggesting that the combination regimen did not bring additional toxicity. Notably, 29d (50 mg/kg) with PTX greatly reduced the volumes of tumors by 57% in the A549/T xenograft model, which is more effective than NOB at the same dosage (P < 0.05). Similarly, NOB derivative 13 also enhanced the therapeutic efficacy of PTX in the A549/T xenograft model (Fig. 4). However, the tumor growth inhibition effect of 13 is similar to that of NOB.
Figure 3.

NOB (N) and its derivative 29d enhanced the anticancer effect of PTX in the PTX-resistance A549/T cell nude mouse xenograft model. The changes in body weight (A) and tumor growth curves (B) were recorded after A549/T cell implantation. The excised tumors were photographed (C) and weighed (D) on day 30 after implantation. The data were shown as mean ± SD for each group (n = 6), ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. PTX alone; #P < 0.05 vs. administered PTX and NOB. CTR, the control group.
Figure 4.

NOB (N) and its derivative 13 enhanced the anticancer effect of PTX in the PTX resistance A549/T cell nude mouse xenograft model. The changes in body weight (A) and tumor growth curves (B) were recorded after A549/T cell implantation. The excised tumors were photographed (C) and weighed (D) on day 30 after implantation. The data were shown as mean ± SD for each group (n = 6), ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. PTX alone.
2.5. Both 29d and 13 potentiate apoptosis induced by PTX
Consistent with the effects of NOB, 29d and 13 significantly potentiated the apoptosis induced by PTX, as shown in Fig. 5A and B, while PTX (5.89 μmol/L), 29d or 13 (9 μmol/L) alone did not induce cell apoptosis. Moreover, cell cycle progressions were evaluated by flow cytometry in asynchronously growing HCT8/T cells and sensitive HCT8 cells after treatment with PTX in the absence or presence of 29d or 13. When 0.94 μmol/L PTX was added to the culture medium of HCT8/T for maintaining resistance, 71.9% of cells in G1 phase and 17.1% of cells in G2 phase after incubation were observed, which is similar to the phase ratios in the vehicle group, as shown in Fig. 5C and D. However, PTX (0.94 μmol/L) treated with 29d or 13 (9 μmol/L) for 48 h resulted in significant G2M arrest (>80%) in HCT8/T cells. Therefore, 29d or 13 significantly enhanced the cell growth inhibition, apoptosis and G2M cell cycle arrest induced by 0.94 μmol/L PTX, although HCT8/T cells were remarkably resistant to 0.94 μmol/L PTX.
Figure 5.

The effects of 29d, 13 and NOB (N) on the apoptosis and cell cycle of HCT8/T induced by PTX. (A) 29d, 13 and NOB at 9 μmol/L increased the proportion of apoptosis induced by 0.94 μmol/L PTX. (B) Addition of 29d, 13 or NOB to medium containing 0.94 μmol/L PTX increased the percentage of cells in G2M phase, as exhibited by flow cytometry. The data were obtained from three different experiments and shown as mean ± SD (n = 3). ∗∗P < 0.01, ∗∗∗P < 0.001 vs. absence of 29d, 13 or N.
2.6. 29d and 13 inhibit the P-gp function
We also evaluated the modulating effects of synthesized NOB and its derivatives on P-gp by evaluating the accumulation of P-gp substrates, including doxorubicin (DOX) and flutax-2 (a fluorescent PTX derivative), using fluorescence microscopy and flow cytometry analysis. Enhanced intracellular accumulations of DOX or flutax-2 by NOB and its derivatives were confirmed by flow cytometry analysis, as shown in Fig. 6A and C. Moreover, stronger fluorescence signals of DOX and flutax-2 were observed by adding NOB, 29d, 13 and quinidine (QND, positive control), as shown in Fig. 6B and D. The effects of 29d on the intracellular accumulation were stronger than those of 13 and NOB.
Figure 6.

Effects of 29d, 13 and NOB (N) on intracellular accumulation of DOX and Flutax-2 in HCT8T cells. HCT8/T cells were treated with 5 μmol/L DOX (A) and (B) or 1 μmol/L Flutax-2 (C) and (D) for 6 h in the absence or presence of 9 μmol/L 29d, 13, NOB and 20 μmol/L QND as indicated. Intracellular DOX or Flutax-2 were observed with a fluorescence microscope (B) and (D) and evaluated by measuring fluorescence with flow cytometry (A) and (C). The experiments were repeated 3 times, and representative images were presented.
LC‒MS/MS was used to detect the contents of PTX, 29d and NOB in the tumors growing in the A549T xenograft model after treatment. Compared with the PTX alone group, NOB enhanced the content of PTX in tumors by 2.93-fold, while 29d enhanced by 4.82-fold, which was approximately 1.65-fold that of NOB (Fig. 7A). Moreover, the concentration of 29d in tumor tissues was significantly increased by 1.72-fold in the PTX+29d (50 mg/kg) group compared with the 29d (50 mg/kg) group (Fig. 7B), indicating that 29d and PTX were competing for binding on P-gp. All of these results suggested that synthesized NOB and its derivatives could inhibit P-gp function in MDR cancer cells which led to increased intracellular chemotherapeutic agents to kill the MDR cancer, and 29d exhibited a stronger inhibition effect on the function of P-gp than 13 and NOB.
Figure 7.

The concentrations of PTX (A) and 29d (B) in MDR tumors after treatment with PTX alone or the combination of 29d and NOB (N). The concentrations of PTX or 29d in the tumor were evaluated with HPLC‒MS, as described in contents. The data were shown as mean ± SD for each group (n = 6), ∗∗P < 0.01, ∗∗∗P < 0.001 vs. PTX alone or 29d alone; ##P < 0.01 vs. PTX + NOB.
2.7. Putative binding pattern of 29d to the pocket of P-gp
To show the putative binding pattern, 29d was docked by the Surflex-dock module embedded in Tripos Sybyl X 2.0 (St. Louis, USA) to the crystal structure of P-gp in complex with QZ59-RRR (PDB ID: 4M2S). The selected binding pose of 29d fitted the binding pocket well, with Surflex docking score (Total-Score) of 6.2274. In this binding modes (Fig. 8 and Supporting Information Section 1), several kinds of specific interactions between 29d and P-gp were observed. Inhibitor 29d formed two H-bonds with Tyr306 and Gln721, respectively. Strong π‒π and π‒σ interactions were also observed for 29d with residues Phe979, Phe339, Phe724 and Phe728. In addition to van der Waals interactions and hydrophobic interactions with residues of Phe299, Gln986, Ile302 and Ser725, the dimethylaminoethyl sidechain of 29d formed extra interactions with Leu335 and Phe331, which significantly differed from NOB10.
Figure 8.

Binding patterns of 29d (A‒D, yellow in C and D) and NOB (E and F, grey) with P-gp (cyan) by docking and MD simulation methods. (A) Ribbon diagram of P-gp with docked 29d (green). (B) Two-dimensional interaction mode between docked 29d and P-gp. The interactions between 29d and P-gp (green sticks). Hydrogen bonds are depicted by dashed blue. Hydrogen bonds and π‒π/σ‒π interactions are shown by green lines. Green and purple bubbles represent hydrophobic and polar amino acid residues, respectively. (C) and (E) refer to the binding patterns by docking method while (D) and (F) refer to the binding patterns by MD method. Therein, hydrogen bonds are depicted by dashed blue lines.
Subsequently, molecular dynamics (MD) simulations were applied for a more precise prediction of binding patterns using AMBER 14. As a result, the predicted binding free energies of the P-gp/29d and P-gp/NOB complexes were −43.99 ± 3.12 and −39.47 ± 2.62 kcal/mol, respectively. From the MD simulation results, apparent hydrophobic interactions with residues Phe299, Phe724, Phe728, Phe979 and Leu336 in P-gp were observed by 29d and NOB. In addition, 29d and NOB formed hydrogen bonds with Gln721 and Tyr306, respectively. Nevertheless, dimethylaminoethyl chain of 29d formed extra van der Waal interactions with Phe339 and Ile302, which might account for its more negative binding free energy compared with NOB.
2.8. NOB and its derivatives overcome MDR via AKT/ERK/NRF2 pathway
Multiple targets are characteristics of natural products and ultimately offer clinical benefits in comparison with the three generations of MDR inhibitors20. To explore the underlying mechanisms for functions of NOB and its derivatives, we evaluated the signaling pathways (Fig. 9) of NRF2/PI3K/AKT and ERK, which had been shown to be closely associated with resistance to chemotherapy drugs16. Interestingly, we found that Nrf2, phosphorylated AKT and ERK were significantly reduced in HCT8/T cells after administration of NOB, 29d or 13, while the expression of total AKT and ERK remained the same (Fig. 9). These results indicated that the MDR reversal effects of 29d and 13 to chemotherapy agents were produced by the inhibition of Nrf2/PI3K/AKT pathways, the same as NOB.
Figure 9.

Effects of the combination treatment of PTX with 29d, 13 and NOB (N) on expressions of P-gp, AKT, ERK and NRF2 in HCT8/T cells. Cells were treated with 9 μmol/L 29d, 13 and N for 48 h. Equal amounts of total lysate were used for Western blot. Combination treatment of PTX with 29d, 13 or NOB did not influence P-gp expression, but reduced the level of NRF2 as well as the phosphorylation of AKT/ERK. The experiments were repeated three times. The data were shown as mean ± SD. ∗P < 0.05 and ∗∗P < 0.01.
Change of sensitivity of the MDR cancer cells to drugs could be due to reduction of ABCB1 expression, competitive inhibition of the ABCB1 transporter, or both. Therefore, we evaluated the effects of synthesized NOB, 29d and 13 on the expression of ABCB1. Interestingly, none of them at the reversal concentrations alter the protein level of ABCB1 in HCT8/T cells (Fig. 9). In addition, NOB and its derivatives did not affect the expression of BCRP and MDR1 in A549/T cells (Fig. 10) which are two major efflux transporters related to the PTX resistance. Thus, synthesized NOB and its derivatives likely inhibited ABCB1 transporter function but did not affect the expression of ABCB1.
Figure 10.

The expression of efflux transporters P-gp, MRP2 and BCRP in A549/T and HCT8/T cells, and the effects of 29d and NOB (N) on their expression by comparing with sensitive A549 or HCT8 cells (red frame). Cells were treated with 9 μmol/L 29d or N for 48 h. Equal amounts of total lysate were loaded and detected by Western blot.
Several metabolites of NOB including 3′-demethylnobiletin, 4′-demethylnobiletin, 3′,4′-didemethylnobiletin (DTF), 5-demethylnobiletin, 5,3′-didemethylnobiletin, 5,4′-didemethylnobiletin and 5,3′,4′-tridemethylnobiletin have been identified as the major metabolites from the urine of mouse by the optimized HPLC method21, 22. Anti-inflammatory effects of 4′-demethylnobiletin on TPA-induced mice ear inflammation have been shown through inhibition of PI3K/AKT/ERK phosphorylation22. Therefore, the metabolites of NOB derivatives possibly have inhibitory activity on P-gp function via the NRF2/PI3K/AKT pathways.
3. Conclusions
In short, NOB was produced with a high yield by total synthesis of six steps and fourteen derivatives were synthesized to discover potent compounds with remarkable solubility and efficacy as MDR reversal agents. Among them, 29d showed 280-fold higher water solubility than NOB and 13. As a result, 29d significantly increased PTX concentration in the tumor in vivo and showed a stronger tumor growth inhibition than NOB and 13 after co-administered with PTX. Moreover, our studies demonstrated that the activated NRF2/PI3K/AKT pathways in MDR cancer cells were remarkably inhibited by the conjunction of 29d and PTX.
4. Experimental
4.1. Chemicals and reagents
PTX, DOX, verapamil (Ver), QND, rhodamine 123 (Rho123), dimethyl sulfoxide (DMSO), propidium iodide (PI), trichloroacetic acid (TCA), crystal violet, sulforhodamine B (SRB) and other chemicals were purchased from Sigma–Aldrich (St. Louis, USA). The flutax-2, RPMI 1640 medium, fetal bovine serum, penicillin and streptomycin were obtained from Life Technologies, Inc. (Grand Island, USA). P-gp antibodies were purchased from Calbiochem (cat. No. 517310). ERK1/2 and actin antibodies were purchased from Santa Cruz Biotechnology, USA. AKT, P-AKT and P-ERK1/2 were purchased from Cell Signaling Technology, Inc. (Boston, USA).
All starting materials and reagents were purchased from commercial suppliers (Sigma–Aldrich, Adamas, Energy, Bide, ShuYa, J&K and Meryer, Shanghai, China) and used directly without further purification. Chemical HG/T2354-92 silica gel (200–300 mesh, Haiyang®, Qingdao, China) was used for chromatography, and silica gel plates with fluorescence F254 (0.25 mm, Huanghai®, Qingdao, China) were used for thin-layer chromatography (TLC) analysis. Reactions requiring anhydrous conditions were performed under argon or with a calcium chloride tube. 1H NMR and 13C NMR spectra were recorded at room temperature (rt) on a Bruker AVANCE III 400 instrument (Germany) with tetramethylsilane (TMS) as an internal standard. The following abbreviations are used: s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet) and m (multiplet). Coupling constants were reported in Hz. High-resolution mass spectra (HRMS) were recorded on SHIMADZU LCMS-IT-TOF (Kyoto, Japan). The purities of compounds were determined by reverse-phase HPLC analysis, confirming purity to exceed 95%. HPLC instrument: SHIMADZU LC-20AT (column: Hypersil BDS C18, 5.0 μm, 150 mm × 4.6 mm (Elite); detector: SPD-20A UV/VIS detector, UV detection at 254 nm; elution, MeOH in water (80%, v/v); T = 25 °C; flow rate = 1.0 mL/min; Kyoto, Japan).
4.1.1. 3,4,5-Trimethoxyphenyl acetate (2)
A mixture of 1 (9.2 g, 50 mmol) and sodium acetate (8.2 g, 100 mmol) in acetic anhydride (47 mL, 500 mmol) was heated at 110 °C for 2 h. TLC analysis (petroleum ethe/ethyl acetate = 7:3) showed completion of the reaction. Then the reaction mixture was concentrated under reduced pressure, diluted with water and extracted with ethyl acetate thrice. The organic phase was washed with brine, dried over anhydrous sodium sulfate. The organic layer was concentrated to afford the product 2 (11.2 g) as a white solid, which was used directly in the next step without further purification. Yield: 91%. 1H NMR (CDCl3, 400 MHz) δ 6.34 (s, 2H), 3.83 (s, 9H), 2.29 (s, 3H).
4.1.2. 1-(6-Hydroxy-2, 3, 4-trimethoxyphenyl) ethanone (3)
Boron trifluoride etherate (ca. 48% BF3, 250 mL) was added to a solution of 2 (11.2 g, 50 mmol) in glacial acetic acid (37.5 mL). The reaction mixture was stirred at 70 °C for 2 h. TLC analysis (petroleum ether/ethyl acetate = 7:3) showed completion of reaction. Then the reaction mixture was quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 10:1) to afford product 3 (10.2 g) as a yellow oil. Yield: 91%. 1H NMR (CDCl3, 500 MHz): δ 13.44 (s, 1H), 6.24 (s, 1H), 3.99 (s, 3H), 3.89 (s, 3H), 3.78 (s, 3H), 2.66 (s, 3H).
4.1.3. 1-(2-Hydroxy-3-iodo-4,5,6-trimethoxyphenyl) ethanone (4)
Adding NIS (1.4 g, 6.0 mmol) and p-toluenesulfonic acid monohydrate (950 mg, 5.0 mmol) to a stirred solution of 3 (1.1 g, 5.0 mmol) in CH3CN (50 mL). The mixture was stirred at rt for 2 h until the start material disappeared as monitored by TLC. After evaporated under vacuum, the reaction mixture was extracted with ethyl acetate. The organic phase was washed with saturated aqueous sodium thiosulfate and brine, and dried over anhydrous sodium sulfate. The solvent was evaporated to afford product 4 as a yellow‒green solid, which was used directly in the next step without further purification. Yield: 73%. 1H NMR (500 MHz, CDCl3) δ 14.00 (s, 1H), 4.02 (d, J = 4.6 Hz, 3H), 4.01 (s, 3H), 3.80 (s, 3H), 2.70 (s, 3H).
4.1.4. 1-(2-Hydroxy-3,4,5,6-tetramethoxyphenyl) ethanone (5)
Copper chloride (359 mg, 3.6 mmol) was added to a solution of 4 (3.6 g, 10.2 mmol) in DMF (30 mL) under an atmosphere of Ar at rt. Then freshly prepared 4.0 mol/L solution of sodium methoxide in methanol (25 mL, 102 mmol) was added to the mixture at 90 °C for 20 min. After cooling, the solution was poured into ice and acidified with 5.0 mol/L HCl. The organic layer was extracted with ethyl acetate. The combined organic extracts were washed with brine and dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 10:1) to afford product 5 (1.5 g) as a light yellow-green liquid. Yield: 59%. 1H NMR (500 MHz, CDCl3) δ 13.16 (s, 1H), 4.08 (s, 3H), 3.94 (s, 3H), 3.86 (s, 3H), 3.81 (s, 3H), 2.68 (s, 3H).
4.1.5. General procedure for synthesis of compounds 6a−6c
To a solution of 5 (128 mg, 0.5 mmol) in dichloromethane (2.5 mL) was added triethylamine (208 μL, 1.5 mmol) and acyl chlorides (0.65 mmol) at 0 °C. The mixture was stirred at rt for 2 h. Then the reaction mixture was quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 5:1) to afford the product as white solids.
4.1.5.1. 2-Acetyl-3,4,5,6-tetramethoxyphenyl benzo[d][1,3]dioxole-5-carboxylate (6a)
White solid. Yield: 85%. 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.7 Hz, 1H), 7.56 (s, 1H), 6.91 (d, J = 4.8 Hz, 1H), 6.07 (s, 2H), 3.98 (s, 3H), 3.93 (s, 3H), 3.90 (s, 3H), 3.81 (s, 3H), 2.47 (s, 3H).
4.1.5.2. 2-Acetyl-3,4,5,6-tetramethoxyphenyl 3,4-dimethoxybenzoate (6b)
White solid. Yield: 80%. 1H NMR (400 MHz, CDCl3) δ 7.82 (dd, J = 8.4, 2.0 Hz, 1H), 7.63 (d, J = 2.0 Hz, 1H), 6.94 (d, J = 8.5 Hz, 1H), 3.98 (s, 3H), 3.97 (s, 3H), 3.95 (s, 3H), 3.94 (s, 3H), 3.90 (s, 3H), 3.81 (s, 3H), 2.47 (s, 3H).
4.1.5.3. 2-Acetyl-3,4,5,6-tetramethoxyphenyl 3,4,5-trimethoxybenzoate (6c)
White solid. Yield: 90%. 1H NMR (400 MHz, CDCl3) δ 7.41 (s, 1H), 7.39 (s, 1H), 3.99 (s, 4H), 3.96 (s, 3H), 3.95 (s, 3H), 3.94 (s, 3H), 3.93 (s, 6H), 3.90 (s, 3H), 3.83 (s, 3H), 2.49 (s, 3H).
4.1.6. General procedure for synthesis of compounds 7a‒7c
To a solution of 6 (0.4 mmol) in dichloroethane (2.4 mL) was added triethylamine (166 μL, 1.2 mmol) and trimethylsilyl trifluoromethanesulfonate (434 μL, 2.4 mmol). The mixture was stirred at 95 °C for 2 h, then quenched with CH3OH, and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 2:1) to afford the product.
4.1.6.1. 2-(Benzo[d][1,3]dioxol-5-yl)-5,6,7,8-tetramethoxy-4H-chromen-4-one (7a)
White solid. Yield: 75%. Purity: 99%. 1H NMR (400 MHz, MeOD-d4) δ 7.62 (dd, J = 8.3, 1.6 Hz, 1H), 7.47 (d, J = 1.6 Hz, 1H), 7.02 (d, J = 8.3 Hz, 1H), 6.66 (s, 1H), 6.11 (s, 2H), 4.12 (s, 3H), 4.03 (s, 3H), 3.94 (s, 3H), 3.90 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 177.33, 160.86, 151.46, 150.51, 148.50, 148.38, 147.67, 144.13, 138.07, 125.54, 121.15, 114.86, 108.85, 107.12, 106.12, 101.90, 62.28, 62.07, 61.8, 61.68. HR-MS (ESI) m/z Calcd. C20H19O8+ [M+H]+ 387.1074, Found 387.1073.
4.1.6.2. 2-(3,4-Dimethoxyphenyl)-5,6,7,8-tetramethoxy-4H-chromenone (7b)
White solid. Yield: 78%. Purity: 99%. 1H NMR (500 MHz, CDCl3) δ 7.57 (d, J = 8.2 Hz, 1H), 7.42 (s, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.62 (s, 1H), 4.11 (s, 3H), 4.03 (s, 3H), 3.98 (s, 3H), 3.97 (s, 3H), 3.96 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 177.35, 161.04, 151.92, 151.43, 149.28, 148.42, 147.72, 144.08, 138.01, 124.00, 119.62, 114.85, 111.22, 108.53, 106.88, 62.27, 61.98, 61.84, 61.69, 56.09, 55.97. HR-MS (ESI) m/z Calcd. C21H23O8+ [M+H]+ 403.1389, Found 403.1389.
4.1.6.3. 5,6,7,8-Tetramethoxy-2-(3,4,5-trimethoxyphenyl)-4H-chromen-4-one (7c)
White solid. Yield: 80%. Purity: 96%. 1H NMR (400 MHz, CDCl3) δ 7.17 (s, 2H), 6.64 (s, 1H), 4.11 (s, 3H), 4.03 (s, 3H), 3.96 (s, 12H), 3.93 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 177.31, 160.80, 153.59, 151.55, 148.44, 147.70, 144.16, 141.07, 137.98, 126.69, 114.82, 107.63, 103.67, 103.39 × 2, 62.26, 61.89, 61.81, 61.67, 61.03, 56.24 × 2. HR-MS (ESI) m/z Calcd. C22H25O9+ [M+H]+ 433.1493, Found 433.1483.
4.1.7. 2-Acetyl-3,4,5-trimethoxyphenyl 3,4-dimethoxybenzoate (10)
To a solution of 3 (904 mg, 4.0 mmol) in dichloromethane (16 mL) was added triethylamine (1.47 mL, 12 mmol) and 3,4-dimethoxybenzoyl chloride (1.04 g, 5.2 mmol) at 0 °C. The mixture was stirred at rt for 2 h, then quenched with water, and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 5:1) to afford product 10 as a white solid. Yield: 85%. 1H NMR (400 MHz, CDCl3) δ 7.81 (dd, J = 8.4, 2.0 Hz, 1H), 7.63 (d, J = 2.0 Hz, 1H), 6.96 (d, J = 8.5 Hz, 1H), 6.59 (s, 1H), 3.98 (s, 3H), 3.97 (s, 6H), 3.91 (s, 3H), 3.90 (s, 3H), 2.51 (s, 3H).
4.1.8. 1-(3-Bromo-2-hydroxy-4,5,6-trimethoxyphenyl) ethanone (11)
Sodium acetate (1.77 g, 21.6 mmol) was added to a solution of 3 (4.436 g, 19.6 mmol) in glacial acetic acid (15 mL). Then Br2 in glacial acetic acid (1.5 mL, 29.4 mmol) was added dropwise to the reaction mixture at 0 °C. The mixture was stirred at rt for 12 h and poured into ice water (150 mL). The solid was filtered and washed with water to yield product 11 as a yellow solid, which was used directly in the next step without further purification. Yield: 81%. 1H NMR (400 MHz, CDCl3) δ 13.75 (s, 1H), 4.05 (s, 3H), 4.04 (s, 3H), 3.83 (s, 3H), 2.72 (s, 3H).
4.1.9. 1-(2-Hydroxy-4,5,6-trimethoxy-3-nitrophenyl) ethanone (12)
Nitric acid (ca. 70% HNO3, 621 μL, 14 mmol) was added dropwise to 3 (1.58 g, 7 mmol) over 15 min at 0 °C. Then the reaction mixture was quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 5:1) to afford product 12 as a yellow solid. Yield: 79%. 1H NMR (400 MHz, CDCl3) δ 13.56 (s, 1H), 4.10 (s, 3H), 4.07 (s, 3H), 3.81 (s, 3H), 2.69 (s, 3H).
4.1.10. 2-(3,4-Dimethoxyphenyl)-5,6,7-trimethoxy-4H-chromen-4-one (13)
To a solution of 10 (1.17 g, 3.0 mmol) in dichloroethane (18 mL) was added triethylamine (1.2 mL, 9.0 mmol) and trimethylsilyl trifluoromethanesulfonate (3.3 mL, 18 mmol). The mixture was then stirred at 95 °C for 2 h, quenched with CH3OH, and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 2:1) to afford product 13 (905 mg) as a brown solid. Yield: 81%. Purity: 99%. 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 8.4 Hz, 1H), 7.33 (s, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.81 (s, 1H), 6.60 (s, 1H), 4.00 (s, 6H), 3.99 (s, 3H), 3.97 (s, 3H), 3.93 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 177.28, 161.17, 157.68, 154.51, 152.57, 151.79, 149.25, 140.35, 124.08, 119.59, 112.85, 111.10, 108.60, 107.37, 96.26, 62.21, 61.57, 56.33, 56.12, 56.08. HR-MS (ESI) m/z Calcd. C20H21O7+ [M+H]+ 373.1282, Found 373.1285.
4.1.11. 2-Acetyl-6-bromo-3,4,5-trimethoxyphenyl 3,4-dimethoxybenzoate (14)
To a solution of 11 (1.2 g, 4.0 mmol) in dichloromethane (20 mL) was added triethylamine (1.4 mL, 12 mmol) and 3,4-dimethoxybenzoyl chloride (1.0 g, 5.2 mmol) at 0 °C. The mixture was stirred at rt for 2 h, quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 5:1) to afford product 14 as a yellow solid. Yield: 75%. 1H NMR (400 MHz, CDCl3) δ 7.85 (dd, J = 8.4, 2.0 Hz, 1H), 7.64 (d, J = 2.0 Hz, 1H), 6.95 (d, J = 8.5 Hz, 1H), 3.97 (s, 3H), 3.96 (s, 3H), 3.95 (s, 3H), 3.95 (s, 3H), 3.94 (s, 3H), 2.49 (s, 3H).
4.1.12. 2-Acetyl-3,4,5-trimethoxy-6-nitrophenyl 3,4-dimethoxybenzoate (15)
To a solution of 12 (1.2 g, 5.0 mmol) in dichloromethane (25 mL) was added triethylamine (1.8 mL, 15 mmol) and 3,4-dimethoxybenzoyl chloride (1.2 g, 6.0 mmol) at 0 °C. The mixture was stirred at rt for 2 h, quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 5:1) to afford product 15 as a beige solid. Yield: 90%. 1H NMR (500 MHz, CDCl3) δ 7.73 (dd, J = 8.5, 2.0 Hz, 1H), 7.52 (d, J = 2.0 Hz, 1H), 6.92 (d, J = 8.5 Hz, 1H), 4.06 (s, 3H), 4.02 (s, 3H), 3.96 (s, 3H), 3.95 (s, 3H), 3.93 (s, 3H), 2.52 (s, 3H).
4.1.13. 8-Bromo-2-(3,4-dimethoxyphenyl)-5,6,7-trimethoxy-4H-chromen-4-one (16)
To a solution of 14 (1.2 g, 2.6 mmol) in dichloroethane (15 mL) was added triethylamine (1.0 mL, 7.5 mmol) and trimethylsilyl trifluoromethanesulfonate (2.7 mL, 15 mmol). The mixture was stirred at 95 °C for 2 h, then quenched with CH3OH and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 1:1) to afford product 16 (900 mg) as a white solid. Yield: 79%. Purity: 99%. 1H NMR (400 MHz, CDCl3) δ 7.64 (dd, J = 8.5, 2.1 Hz, 1H), 7.53 (d, J = 2.1 Hz, 1H), 7.00 (d, J = 8.5 Hz, 1H), 6.75 (s, 1H), 4.09 (s, 3H), 4.00 (s, 3H), 3.99 (s, 3H), 3.97 (s, 3H), 3.96 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 177.00, 161.45, 155.77, 152.70, 152.07, 150.43, 149.28, 144.61, 123.50, 119.94, 115.81, 111.21, 108.78, 106.53, 101.43, 62.34, 61.84, 61.58, 56.10, 56.00. HR-MS (ESI) m/z Calcd. C20H20O7Br+ [M+H]+ 451.0387, Found 451.0392.
4.1.14. 2-(3,4-Dimethoxyphenyl)-5,6,7-trimethoxy-8-nitro-4H-chromen-4-one (17)
To a solution of 15 (1.9 g, 4.3 mmol) in dichloroethane (26 mL) was added triethylamine (1.7 mL, 13 mmol) and trimethylsilyl trifluoromethanesulfonate (4.6 mL, 256 mmol). The mixture was stirred at 95 °C for 2 h, then quenched with CH3OH and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 1:1) to afford product 17 (1.5 g) as a brown solid. Yield: 84%. 1H NMR (400 MHz, CDCl3) δ 7.42 (dd, J = 8.5, 2.1 Hz, 1H), 7.28 (d, J = 2.1 Hz, 1H), 6.96 (d, J = 8.6 Hz, 1H), 6.63 (s, 1H), 4.17 (s, 3H), 4.05 (s, 3H), 3.97 (s, 3H), 3.96 (s, 3H), 3.96 (s, 3H).
4.1.15. 8-Amino-2-(3,4-dimethoxyphenyl)-5,6,7-trimethoxy-4H-chromen-4-one (18)
To a solution of 17 (140 mg, 0.33 mmol) in tetrahydrofuran (25 mL) was added Pd/C catalyst (21 mg, 15%) under an atmosphere of H2 at 40 °C for 15 h. After cooling to rt, the mixture was filtered with diatomite and washed with ethyl acetate. The organic layer was evaporated under vacuum to afford a residue, which was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 2:1) to yield product 18 (121 mg) as a yellow solid. Yield: 95%. Purity: 99%. 1H NMR (400 MHz, CDCl3) δ 7.50 (dd, J = 8.4, 2.0 Hz, 1H), 7.30 (d, J = 2.0 Hz, 1H), 6.98 (d, J = 8.5 Hz, 1H), 6.57 (s, 1H), 4.04 (s, 3H), 3.97 (s, 3H), 3.97 (s, 3H), 3.96 (s, 3H), 3.91 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 177.80, 160.99, 151.86, 149.31, 144.13, 143.75, 143.40, 141.82, 126.01, 124.35, 119.56, 114.93, 111.17, 108.72, 107.33, 62.35, 61.61, 60.91, 56.10 × 2. HR-MS (ESI) m/z Calcd. C20H22NO7+ [M+H]+ 388.1391, Found 388.1391.
4.1.16. 2,3,4-Trimethoxyphenyl acetate (20b)
A mixture of 19b (5.5 g, 30 mmol) and sodium acetate (4.9 g, 60 mmol) in acetic anhydride (28 mL, 300 mmol) was heated at 110 °C for 2 h. TLC analysis (petroleum ether/ethyl acetate = 9:1) showed completion of the reaction. Then the reaction mixture was concentrated under reduced pressure, diluted with water and extracted with ethyl acetate thrice. The organic phase was washed with brine, dried over anhydrous sodium sulfate. The organic layer was concentrated to afford product 20b (6.4 g) as a brown yellow oil, which was used directly in the next step without further purification. Yield: 94%. 1H NMR (500 MHz, CDCl3) δ 6.74 (d, J = 9.0 Hz, 1H), 6.62 (d, J = 9.0 Hz, 1H), 3.89 (s, 3H), 3.88 (s, 3H), 3.85 (s, 3H), 2.31 (s, 3H).
4.1.17. 1-(2-Hydroxy-3,4-dimethoxyphenyl) ethan-1-one (21a)
A mixture of 19a (1.5 g, 10 mmol) and sodium acetate (1.6 g, 20 mmol) in acetic anhydride (9.4 mL, 100 mmol) was heated at 110 °C for 2 h. TLC analysis (petroleum ether/ethyl acetate = 9:1) showed completion of the reaction. Then the reaction mixture was concentrated under reduced pressure, diluted with water and extracted with ethyl acetate thrice. The organic phase was washed with brine and dried over anhydrous sodium sulfate. The organic layer was concentrated to afford product 20a (1.9 g) as a red brown oil, which was used directly in the next step without further purification. Then boron trifluoride etherate (ca. 48% BF3, 48.5 mL) was added dropwise to a solution of compound 20a (1.9 g, 9.7 mmol) in glacial acetic acid (7.5 mL). The reaction mixture was stirred at 70 °C for 2 h. TLC analysis (petroleum ether/ethyl acetate = 7:3) showed completion of reaction. Then the reaction mixture was quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 10:1) to afford product 21a (1.8 g) as a brown solid. Yield: 95%. 1H NMR (400 MHz, CDCl3) δ 12.67 (s, 1H), 7.09 (s, 1H), 6.48 (s, 1H), 3.94 (s, 3H), 3.89 (s, 3H), 2.59 (s, 3H).
4.1.18. 1-(2-Hydroxy-3,4,5-trimethoxyphenyl) ethan-1-one (21b)
Boron trifluoride etherate (ca. 48% BF3, 140 mL) was added to a solution of 20b (6.4 g, 28 mmol) in glacial acetic acid (21 mL). The reaction mixture was stirred at 70 °C for 2 h. TLC analysis (petroleum ether/ethyl acetate = 7:3) showed completion of reaction. Then the reaction mixture was quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 10:1) to afford product 21b (6.0 g) as a yellow solid. Yield: 95%. 1H NMR (500 MHz, CDCl3) δ 12.44 (s, 1H), 6.93 (s, 1H), 4.04 (s, 3H), 3.92 (s, 3H), 3.85 (s, 3H), 2.59 (s, 3H).
4.1.19. General procedure for synthesis of compounds 22a‒22d
To a solution of compound 21a−21d (196 mg, 1.0 mmol) in dichloromethane (5 mL) was added triethylamine (418 μL, 3.0 mmol) and acyl chloride (1.5 mmol) at 0 °C. The mixture was stirred at rt for 2 h, then quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 5:1) to afford the products 22a‒22d as white solids.
4.1.19.1. 6-Acetyl-2,3-dimethoxyphenyl 3,4-dimethoxybenzoate (22a)
Yield: 78%. 1H NMR (400 MHz, CDCl3) δ 7.90 (dd, J = 8.4, 2.0 Hz, 1H), 7.69 (d, J = 1.8 Hz, 1H), 7.44 (s, 1H), 6.99 (d, J = 8.5 Hz, 1H), 6.69 (s, 1H), 3.99 (s, 3H), 3.97 (s, 3H), 3.93 (s, 3H), 2.51 (s, 3H).
4.1.19.2. 6-Acetyl-2,3-dimethoxyphenyl 3-methoxybenzoate (22b)
Yield: 82%. 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 7.7 Hz, 1H), 7.72 (s, 1H), 7.48–7.41 (m, 2H), 7.21 (dd, J = 8.3, 2.7 Hz, 1H), 6.68 (s, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.89 (s, 3H), 2.51 (s, 3H).
4.1.19.3. 6-Acetyl-2,3-dimethoxyphenyl 4-methoxybenzoate (22c)
Yield: 82%. 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.8 Hz, 2H), 7.44 (s, 1H), 7.02 (d, J = 8.8 Hz, 2H), 6.67 (s, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 3.91 (s, 3H), 2.50 (s, 3H).
4.1.19.4. 6-Acetyl-2,3,4-trimethoxyphenyl 3,4-dimethoxybenzoate (22d)
Yield: 82%. 1H NMR (400 MHz, CDCl3) δ 7.93 (dd, J = 8.4, 1.9 Hz, 1H), 7.72 (d, J = 1.9 Hz, 1H), 7.23 (s, 1H), 7.00 (d, J = 8.5 Hz, 1H), 4.01 (s, 3H), 3.99 (s, 3H), 3.99 (s, 3H), 3.94 (s, 3H), 3.87 (s, 3H), 2.53 (s, 3H).
4.1.20. General procedure for synthesis of compounds 23a−23d
To a solution of 22 (0.8 mmol) in dichloroethane (4.8 mL) was added triethylamine (332 μL, 2.4 mmol) and trimethylsilyl trifluoromethanesulfonate (868 μL, 4.8 mmol). The mixture was stirred at 95 °C for 2 h, then quenched with CH3OH and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 2:1) to afford the products 23a−23d.
4.1.20.1. 2-(3,4-Dimethoxyphenyl)-7,8-dimethoxy-4H-chromen-4-one (23a)
Light yellow solid. Yield: 82%. Purity: 99%. 1H NMR (400 MHz, CDCl3) δ 7.57 (s, 1H), 7.54 (dd, J = 8.5, 1.8 Hz, 1H), 7.37 (d, J = 1.8 Hz, 1H), 6.98 (d, J = 9.3 Hz, 2H), 6.73 (s, 1H), 4.03 (s, 3H), 3.99 (s, 6H), 3.97 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 177.63, 162.83, 154.37, 152.21, 151.85, 149.29, 147.61, 124.49, 119.72, 117.29, 111.16, 108.73, 106.07, 104.45, 99.79, 56.51, 56.38, 56.12, 56.09. HR-MS (ESI) m/z Calcd. C19H19O6+ [M+H]+ 343.1176, Found 343.1166.
4.1.20.2. 7,8-Dimethoxy-2-(3-methoxyphenyl)-4H-chromen-4-one (23b)
Breen solid. Yield: 82%. Purity: 99%. 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.44 (dd, J = 9.0, 6.8 Hz, 2H), 7.08 (dd, J = 8.0, 2.3 Hz, 1H), 7.01 (s, 1H), 6.79 (s, 1H), 4.04 (s, 3H), 4.01 (s, 3H), 3.91 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 177.66, 162.57, 160.01, 154.52, 152.30, 147.71, 133.31, 130.10, 118.53, 117.37, 116.88, 111.59, 107.32, 104.38, 99.81, 56.51, 56.39, 55.49. HR-MS (ESI) m/z Calcd. C18H17O5+ [M+H]+ 313.1071, Found 313.1072.
4.1.20.3. 7,8-Dimethoxy-2-(4-methoxyphenyl)-4H-chromen-4-one (23c)
Brick red solid. Yield: 82%. Purity: 99%. 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 8.8 Hz, 2H), 7.56 (s, 1H), 7.02 (d, J = 8.7 Hz, 2H), 6.99 (s, 1H), 6.71 (s, 1H), 4.02 (s, 3H), 3.99 (s, 3H), 3.89 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 177.68, 162.85, 162.18, 154.31, 152.20, 147.55, 127.76 × 2, 124.27, 117.28, 114.43 × 2, 105.76, 104.45, 99.76, 56.48, 56.39, 55.51. HR-MS (ESI) m/z Calcd. C18H17O5+ [M+H]+ 313.1071, Found 313.1067.
4.1.20.4. 2-(3,4-Dimethoxyphenyl)-6,7,8-trimethoxy-4H-chromen-4-one (23d)
White solid. Yield: 83%. Purity: 96%. 1H NMR (500 MHz, MeOD-d4) δ 7.68 (d, J = 8.4 Hz, 1H), 7.56 (s, 1H), 7.36 (s, 1H), 7.13 (d, J = 8.5 Hz, 1H), 6.82 (s, 1H), 4.12 (s, 3H), 4.04 (s, 3H), 3.97 (s, 6H), 3.94 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 177.72, 162.82, 152.00, 151.16, 149.32, 147.38, 145.75, 142.03, 124.40, 119.79 × 2, 111.25, 108.69, 105.64, 100.08, 62.02, 61.52, 56.30, 56.11, 55.99. HR-MS (ESI) m/z Calcd. C20H21O7+ [M+H]+ 373.1282, Found 373.1275.
4.1.21. 1-(2,3,4,5-Tetramethoxyphenyl)-ethan-1-one (24)
To a solution of 21b (2.3 g, 10 mmol) in DMF was added potassium carbonate (2.1 g, 15 mmol) and iodomethane (679 μL, 11 mmol). The reaction mixture was stirred at rt for 1 h and then extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and concentrated to afford product 24 (2.3 g) as a brown yellow oil, which was used directly in the next step without further purification. Yield: 96%. 1H NMR (500 MHz, CDCl3) δ 7.07 (s, 1H), 3.96 (s, 3H), 3.92 (s, 3H), 3.92 (s, 3H), 3.86 (s, 3H), 2.64 (s, 3H).
4.1.22. 1-(2,3,4,5-Tetramethoxy-6-nitrophenyl) ethan-1-one (25)
Nitric acid (ca. 70% HNO3, 798 μL, 18 mmol) was added dropwise to 24 (2.2 g, 9.0 mmol) over 15 min at 0 °C. Then the reaction mixture was quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 5:1) to afford product 25 (1.9 g) as a light yellow solid. Yield: 74%. 1H NMR (400 MHz, CDCl3) δ 4.00 (s, 3H), 3.97 (s, 3H), 3.95 (s, 3H), 3.88 (s, 3H), 2.59 (s, 3H).
4.1.23. 1-(2-Amino-3,4,5,6-tetramethoxyphenyl) ethan-1-one (26)
To a solution of 25 (1.9 g, 6.7 mmol) in tetrahydrofuran (134 mL) was added Pd/C catalyst (285 mg, 15%) under an atmosphere of H2 at 40 °C for 15 h. After cooling to rt, the mixture was filtered with diatomite and washed with ethyl acetate. The organic layer was evaporated under vacuum to afford a residue, which was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 2:1) to yield product 26 (1.6 g) as a brown yellow oil. Yield: 95%. 1H NMR (400 MHz, CDCl3) δ 4.12 (s, 3H), 4.03 (s, 3H), 4.02 (s, 3H), 3.88 (s, 3H), 2.83 (s, 3H).
4.1.24. N-(2-Acetyl-3,4,5,6-tetramethoxyphenyl)-3,4-dimethoxybenzamide (27)
To a solution of 26 (570 mg, 2.2 mmol) in dichloromethane (11 mL) was added triethylamine (915 μL, 6.6 mmol) and 3,4-dimethoxybenzoyl chloride (883 mg, 4.4 mmol) at 0 °C. The mixture was stirred at rt for 2 h, then quenched with water and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (petroleum ether/ethyl acetate = 5:1) to afford product 27 (738 mg) as a white solid. Yield: 80%. 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.46 (d, J = 1.9 Hz, 1H), 7.44 (dd, J = 8.2, 2.1 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 3.97 (s, 3H), 3.95 (s, 3H), 3.95 (s, 3H), 3.93 (s, 3H), 3.90 (s, 3H), 3.84 (s, 3H), 2.65 (s, 3H).
4.1.25. 2-(3,4-Dimethoxyphenyl)-5,6,7,8-tetramethoxyquinolin-4(1H)-one (28)
To a solution of 27 (738 mg, 1.8 mmol) in anhydrous tetrahydrofuran (60 mL) was added potassium tert-butoxide (606 mg, 5.4 mmol) under an atmosphere of Ar at 75 °C for 18 h. After cooled to rt, the solution was poured into water and acidified by addition of 2.0 mol/L aqueous HCl. The resulting solution was extracted with portion of ethyl acetate. The combined organic extracts were dried over anhydrous sodium sulfate and concentrated to give a crude, which was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 3:1) to yield product 28 (592 mg) as a white solid. Yield: 82%. Purity: 99%. 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 6.6 Hz, 2H), 7.02 (d, J = 8.9 Hz, 1H), 6.72 (s, 1H), 4.12 (s, 3H), 4.09 (s, 3H), 4.03 (s, 3H), 4.00 (s, 6H), 3.98 (s, 3H). 13C NMR (126 MHz, MeOD-d4) δ 176.98, 151.67, 150.93, 149.56, 149.38, 147.53, 143.88, 137.77, 132.72, 125.99, 120.52, 114.51, 111.48, 110.73, 107.50, 61.46, 60.91, 60.88, 60.61, 55.25, 55.14. HR-MS (ESI) m/z Calcd. C21H24NO7+ [M+H]+ 402.1547, Found 402.1542.
4.1.26. 2-(3,4-Dimethoxyphenyl)-5,6,7,8-tetramethoxy-1-methylquinolin-4(1H)-one (29a)
To a solution of 28 (80 mg, 0.2 mmol) in DMF was added potassium carbonate (41 mg, 0.3 mmol) and iodomethane (14 μL, 0.22 mmol). The reaction mixture was stirred at rt for 1 h and then extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and concentrated to give a crude, which was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 3:1) to yield product 29a (62 mg) as a light yellow oil. Yield: 75%. Purity: 99%. 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 2.0 Hz, 1H), 7.65 (dd, J = 8.4, 2.0 Hz, 1H), 7.14 (s, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.20 (s, 3H), 4.13 (s, 3H), 4.11 (s, 3H), 4.04 (s, 3H), 4.01 (s, 3H), 3.96 (s, 3H), 3.91 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 163.70, 155.98, 150.38, 149.21, 148.14, 145.20, 144.52, 144.45, 142.28, 132.78, 119.75, 112.38, 110.83, 110.45, 97.27, 62.33, 62.21, 61.76 × 2, 56.01, 55.96 × 2. HR-MS (ESI) m/z Calcd. C22H26NO7+ [M+H]+ 416.1704, Found 416.1710.
4.1.27. General procedure for synthesis of compounds 29b−29d
To a solution of 28 (0.2 mmol) in anhydrous DMF (2.0 mL) was added sodium hydride (48 mg, 2.0 mmol). The mixture was stirred at 90 °C for 30 min and chloralkanes (0.6 mmol) was added. Then the reaction mixture was further stirred at 90 °C for 2 h. After cooled to rt, the solution was poured into water and acidified by the addition of 2.0 mol/L aqueous HCl. The resulting solution was extracted with portion of ethyl acetate. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, and concentrated to give crude, which was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 3:1) to yield the oily products.
4.1.27.1. 2-(3,4-Dimethoxyphenyl)-5,6,7,8-tetramethoxy-1-propylquinolin-4(1H)-one (29b)
A light yellow oil. Yield: 62%. Purity: 99%. 1H NMR (400 MHz, MeOD-d4) δ 7.89 (d, J = 2.1 Hz, 1H), 7.68 (dd, J = 8.4, 2.1 Hz, 1H), 7.24 (s, 1H), 7.06 (d, J = 8.4 Hz, 1H), 4.25 (t, J = 6.4 Hz, 2H), 4.11 (s, 3H), 4.06 (s, 3H), 3.97 (s, 3H), 3.96 (s, 3H), 3.90 (s, 3H), 3.88 (s, 3H), 2.01 (dd, J = 13.9, 6.5 Hz, 2H), 1.19 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 167.39, 160.83, 154.56, 153.11, 152.03, 149.02, 148.70, 148.09, 145.85, 136.38, 124.05, 116.32, 115.07, 114.73, 102.11, 74.21, 65.30, 65.23, 64.67 × 2, 59.04, 58.99, 26.17, 13.65. HR-MS (ESI) m/z Calcd. C24H30NO7+ [M+H]+ 444.2017, Found 444.2019.
4.1.27.2. 2-(3,4-Dimethoxyphenyl)-5,6,7,8-tetramethoxy-1-(2-methoxyethyl)-quinolin-4(1H)-one (29c)
A light yellow oil. Yield: 70%. Purity: 99%. 1H NMR (400 MHz, MeOD-d4) δ 7.90 (d, J = 2.1 Hz, 1H), 7.71 (dd, J = 8.4, 2.1 Hz, 1H), 7.28 (s, 1H), 7.07 (d, J = 8.4 Hz, 1H), 4.43 (t, J = 4.4 Hz, 2H), 4.11 (s, 3H), 4.07 (s, 3H), 3.97 (s, 3H), 3.97 (s, 3H), 3.93 (t, J = 4.4 Hz, 2H), 3.91 (s, 3H), 3.89 (s, 3H), 3.49 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 167.05, 160.80, 154.60, 153.12, 152.10, 149.12, 148.63, 148.11, 145.86, 136.30, 124.09, 116.27, 115.08, 114.74, 102.22, 74.50, 71.91, 65.37, 65.32, 64.69, 64.67, 61.80, 59.05, 59.00. HR-MS (ESI) m/z Calcd. C24H30NO8+ [M+H]+ 460.1966, Found 460.1966.
4.1.27.3. 2-(3,4-Dimethoxyphenyl)-1-(2-(dimethylamino)-ethyl)-5,6,7,8-tetramethoxyquinolin-4(1H)-one (29d)
A brown oil. Yield: 71%. Purity: 97.1%. 1H NMR (500 MHz, MeOD-d4) δ 7.94 (s, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.32 (s, 1H), 7.08 (d, J = 8.4 Hz, 1H), 4.44 (t, J = 5.6 Hz, 2H), 4.13 (s, 3H), 4.08 (s, 3H), 3.99 (s, 6H), 3.92 (s, 3H), 3.90 (s, 3H), 3.03 (t, J = 5.5 Hz, 2H), 2.48 (s, 6H). 13C NMR (126 MHz, MeOD-d4) δ 162.92, 156.80, 150.69, 149.19, 148.18, 145.14, 144.69, 144.14, 141.93, 132.32, 120.17, 112.25, 111.13, 110.81, 98.38, 66.81, 61.39, 61.31, 60.73, 60.71, 57.46, 55.12, 55.06, 44.81 × 2. HR-MS (ESI) m/z Calcd. C25H33N2O7+ [M+H]+ 473.2282, Found 473.2286.
4.2. Molecular docking and simulation for P-gp and 29d
The detailed methods for molecular docking and simulation are shown in Supporting Information Section 1.
4.3. Cell viability assay, colony formation assay and animal models
The detailed methods for cell viability assay, colony formation assay and animal models are shown in Supporting Information Section 2.
Acknowledgments
This work was supported by National Key R&D Program of China (2017YFB0202600), Macao Science and Technology Development Fund, Macau Special Administrative Region (003/2017/A1 to Ying Xie, China), National Natural Science Foundation of China (21572279, 21877134, 81602955 and 81703341), Guangdong Province Higher Vocational Colleges & Schools Pearl River Scholar Funded Scheme (2016, China).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.apsb.2019.07.007.
Contributor Information
Ying Xie, Email: yxie@must.edu.mo.
Hai-Bin Luo, Email: luohb77@mail.sysu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Alfarouk K.O., Stock C.M., Taylor S., Walsh M., Muddathir A.K., Verduzco D. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int. 2015;15:71. doi: 10.1186/s12935-015-0221-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elshimali Y.I., Wu Y., Khaddour H., Wu Y., Gradinaru D., Sukhija H. Optimization of cancer treatment through overcoming drug resistance. J Cancer Res Oncobiol. 2018;1:107. doi: 10.31021/jcro.20181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Callaghan R., Luk F., Bebawy M. Inhibition of the multidrug resistance p-glycoprotein: time for a change of strategy?. Drug Metab Dispos. 2014;42:623–631. doi: 10.1124/dmd.113.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H., Huang L., Tao L., Zhang J., Wang F., Zhang X. Secalonic acid D induces cell apoptosis in both sensitive and ABCG2-overexpressing multidrug resistant cancer cells through upregulating c-Jun expression. Acta Pharm Sin B. 2019;9:516–525. doi: 10.1016/j.apsb.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang L., Xiao M., Yang L., Wang S., Wang S.Q., Bender A. Discovery of a non-toxic [1,2,4]triazolo[1,5-a]pyrimidin-7-one (WS-10) that modulates ABCB1-mediated multidrug resistance (MDR) Bioorg Med Chem. 2018;26:5974–5985. doi: 10.1016/j.bmc.2018.10.027. [DOI] [PubMed] [Google Scholar]
- 7.Wen Y., Zhao R., Gupta P., Fan Y., Zhang Y., Huang Z. The epigallocatechin gallate derivative Y6 reverses drug resistance mediated by the ABCB1 transporter both in vitro and in vivo. Acta Pharm Sin B. 2019;9:316–323. doi: 10.1016/j.apsb.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L., Zhang X., Zhang C., Bai X., Zhang J., Zhao X. Nobiletin promotes antioxidant and anti-inflammatory responses and elicits protection against ischemic stroke in vivo. Brain Res. 2016;1636:130–141. doi: 10.1016/j.brainres.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Murakami A., Nakamura Y., Torikai K., Tanaka T., Koshiba T., Koshimizu K. Inhibitory effect of citrus nobiletin on phorbol ester-induced skin inflammation, oxidative stress, and tumor promotion in mice. Cancer Res. 2000;60:5059–5066. [PubMed] [Google Scholar]
- 10.Ma W., Feng S., Yao X., Yuan Z., Liu L., Xie Y. Nobiletin enhances the efficacy of chemotherapeutic agents in ABCB1 overexpression cancer cells. Sci Rep. 2015;5:18789. doi: 10.1038/srep18789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onoue S., Nakamura T., Uchida A., Ogawa K., Yuminoki K., Hashimoto N. Physicochemical and biopharmaceutical characterization of amorphous solid dispersion of nobiletin, a citrus polymethoxylated flavone, with improved hepatoprotective effects. Eur J Pharm Sci. 2013;49:453–460. doi: 10.1016/j.ejps.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 12.Asakawa T., Hiza A., Nakayama M., Inai M., Oyama D., Koide H. PET Imaging of nobiletin based on a practical total synthesis. Chem Commun. 2011;47:2868–2870. doi: 10.1039/c0cc04936k. [DOI] [PubMed] [Google Scholar]
- 13.Guo Y., Ji S.Z., Chen C., Liu H.W., Zhao J.H., Zheng Y.L. A ligand-free, powerful, and practical method for methoxylation of unactivated aryl bromides by use of the CuCl/HCOOMe/MeONa/MeOH system. Res Chem Intermed. 2015;41:8651–8664. [Google Scholar]
- 14.Yoshida M., Saito K., Fujino Y., Doi T. A concise synthesis of 3-aroylflavones via lewis base 9-azajulolidine-catalyzed tandem acyl transfer-cyclization. Chem Commun. 2012;48:11796–11798. doi: 10.1039/c2cc37015h. [DOI] [PubMed] [Google Scholar]
- 15.Rocha D.H., Pinto D.C., Silva A.M. Synthesis and cyclisation studies of (E)-2-Aryl-1-methyl-3-styrylquinolin-4(1H)-ones. Tetrahedron. 2015;71:7717–7721. [Google Scholar]
- 16.Wesołowska O., Wiśniewski J., Środa-Pomianek K., Bielawska-Pohl A., Paprocka M., Duś D. Multidrug resistance reversal and apoptosis induction in human colon cancer cells by some flavonoids present in Citrus plants. J Nat Prod. 2012;75:1896–1902. doi: 10.1021/np3003468. [DOI] [PubMed] [Google Scholar]
- 17.Martins I.L., Charneira C., Gandin V., Da Silva J.L., Justino G.C., Telo J.P. Selenium-containing chrysin and quercetin derivatives: attractive scaffolds for cancer therapy. J Med Chem. 2015;58:4250–4265. doi: 10.1021/acs.jmedchem.5b00230. [DOI] [PubMed] [Google Scholar]
- 18.Hadjeri M., Barbier M., Ronot X., Mariotte A.M., Boumendjel A., Boutonnat J. Modulation of P-glycoprotein-mediated multidrug resistance by flavonoid derivatives and analogues. J Med Chem. 2003;46:2125–2131. doi: 10.1021/jm021099i. [DOI] [PubMed] [Google Scholar]
- 19.Vassileva V., Grant J., De Souza R., Allen C., Piquette-Miller M. Novel biocompatible intraperitoneal drug delivery system increases tolerability and therapeutic efficacy of paclitaxel in a human ovarian cancer xenograft model. Cancer Chemother Pharmacol. 2007;60:907–914. doi: 10.1007/s00280-007-0449-0. [DOI] [PubMed] [Google Scholar]
- 20.De Oliveira Júnior R.G., Christiane Adrielly A.F., Da Silva Almeida J.R., Grougnet R., Thiéry V., Picot L. Sensitization of tumor cells to chemotherapy by natural products: a systematic review of preclinical data and molecular mechanisms. Fitoterapia. 2018;129:383–400. doi: 10.1016/j.fitote.2018.02.025. [DOI] [PubMed] [Google Scholar]
- 21.Huang H., Li L., Shi W., Liu H., Yang J., Yuan X. The multifunctional effects of nobiletin and its metabolites in vivo and in vitro. Evid Based Complement Alternat Med. 2016;2016:2918796. doi: 10.1155/2016/2918796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu X., Song M., Rakariyatham K., Zheng J., Wang M., Xu F. Inhibitory effects of 4'-demethylnobiletin, a metabolite of nobiletin, on 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced inflammation in mouse ears. J Agric Food Chem. 2015;63:10921–10927. doi: 10.1021/acs.jafc.5b05156. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.