

Abstract
Background
Gelsemium elegans (G. elegans) (2n = 2x = 16) is genus of flowering plants belonging to the Gelsemicaeae family.
Method
Here, a high-quality genome assembly using the Oxford Nanopore Technologies (ONT) platform and high-throughput chromosome conformation capture techniques (Hi-C) were used.
Results
A total of 56.11 Gb of raw GridION X5 platform ONT reads (6.23 Gb per cell) were generated. After filtering, 53.45 Gb of clean reads were obtained, giving 160 × coverage depth. The de novo genome assemblies 335.13 Mb, close to the 338 Mb estimated by k-mer analysis, was generated with contig N50 of 10.23 Mb. The vast majority (99.2%) of the G. elegans assembled sequence was anchored onto 8 pseudo-chromosomes. The genome completeness was then evaluated and 1338 of the 1440 conserved genes (92.9%) could be found in the assembly. Genome annotation revealed that 43.16% of the G. elegans genome is composed of repetitive elements and 23.9% is composed of long terminal repeat elements. We predicted 26,768 protein-coding genes, of which 84.56% were functionally annotated.
Conclusion
The genomic sequences of G. elegans could be a valuable source for comparative genomic analysis in the Gelsemicaeae family and will be useful for understanding the phylogenetic relationships of the indole alkaloid metabolism.
KEY WORDS: Gelsemium elegans, Nanopore sequencing, Genome assembly, Hi-C, Genome annotation, Monoterpene indole alkaloid
Graphical abstract
Here, we report a high-quality Gelsemium elegans genome assembly using the ONT platform and Hi-C. The de novo genome assemblies 335.13 Mb, was generated with contig N50 of 10.23 Mb. The vast majority (99.2%) of the G. elegans assembled sequence was anchored onto the 8 pseudo-chromosomes.
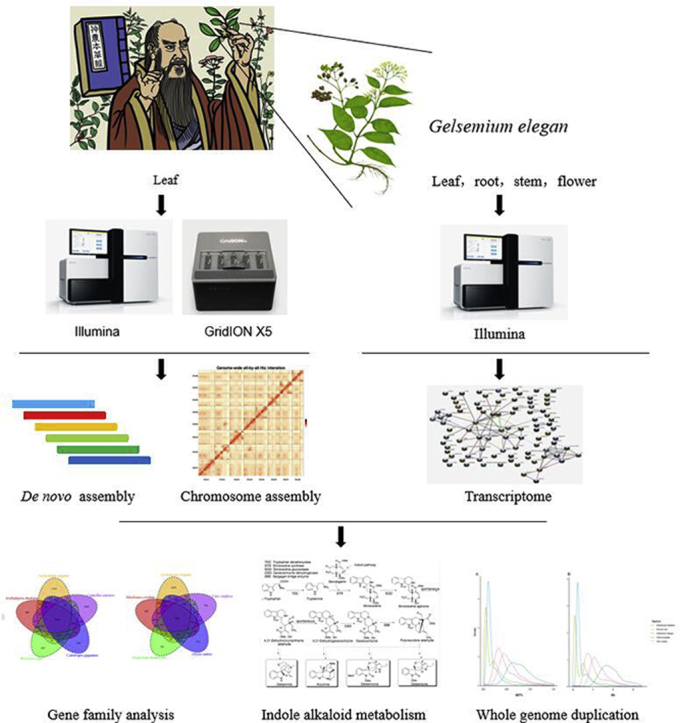
1. Introduction
Gelsemium is a genus of flowering plants belonging to the Gelsemicaeae family. The genus comprises 3 species: the Asian Gelsemium elegans (G. elegans) and two North American species, Gelsemium sempervirens and Gelsemium rankinii. G. elegans (National Center for Biotechnology Information Taxonomy ID: 427660) is also known as Gou Wen, Da Cha Yao or Duan Chang Cao in Chinese1, 2. This herb is extremely famous for murdered the Yan Emperor of China who was also called Shen Nong in the Chinese mythology “Shen Nong tastes hundreds of grasses”. In this myth, Shen Nong diligently tasted all manner of flora for people to eat or used for medicine. But one day, he tasted Duan Chang Cao which has the yellow flower, and this poison was so terrible that he died quickly. He sacrificed himself to save humanity, so people call him the “Bodhisattva of medicine”, and people forever commemorate him. Just as the myth described, this species is widely distributed in the Fujian, Guangxi, Hunan and Guizhou provinces of China and in southeastern Asia (Fig. 1). It has been used as an herbal medicine for the treatment of rheumatoid arthritis, neuropathic pain, spasticity, skin ulcers and cancer for many years3, 4 and the whole plant has been widely added to animal feed for livestock. To date, more than 200 compounds, including indole alkaloids, iridoids, and steroids, have been isolated and identified from G. elegans5, 6.
Figure 1.

Example of the Gelsemium elegans (Gou Wen or Duan Chang Cao). (A) Natural habitat of Gelsemium elegans (image from Qi Tang). (B) Gelsemium elegans (image from Yisong Liu). (C) The flower of Gelsemium elegans (image from Qi Tang).
Previous studies on the crude and purified alkaloids of G. elegans have demonstrated that this species possesses anti-inflammatory4, immunomodulating7, analgesic4, anxiolytic, anti-tumor8, 9, and neuropathic pain-relieving properties10. Indole alkaloids such as gelsemine, koumine, humantenine, gelsemicine and gelsenicine are the major active components of Gelsemium. Gelsemine and koumine are the principal alkaloids in G. elegans, and their toxicity is relatively weak11. Gelsenicine {[LD50 = 0.128 mg/kg, mice (i.p.); 0.26 mg/kg, rat (i.p.); 0.15 mg/kg, rat (i.v.)], which was found in a lesser amount, was the most toxic alkaloid in G. elegans. Gelsenicine was also the most toxic compound in G. sempervirens12, 13. The typical symptoms of gelsenicine intoxication include chest distress, asphyxia, dizziness, tonic convulsions, limb paralysis, and difficulty breathing. Severe gelsenicine poisoning can cause multiple organ failure leading to death14. Therefore, the actual bioactive components of G. elegans have attracted attention from chemists, pharmacologists and toxicologists due to their complex structural features and multiple biological effects.
Despite the considerable pharmaceutical importance of G. elegans, the genomic information available for this species is limited, which has hindered its utilization. Former results suggest that the Oxford Nanopore Technologies (ONT) can be used to quickly and cost-effectively generate informative assemblies15, 16, and a combination of sequencing and mapping data often leads to improved assemblies and is potentially more cost effective than sequencing alone. For example, the cottons17 and human18 genomes were assembled using a combination of long reads and Hi-C (high-throughput chromosome conformation capture techniques)-based data, have remarkably high quality with long contig (contig N50 of 18.7 and 26.8 Mb, respectively), chromosome length scaffolds (scaffold N50 of 87 and 60.0 Mb) and nearly 100% sequence fidelity. Here we report a high-quality reference genome for G. elegans using ONT technology and Hi-C map to cluster the majority of the assembled contig onto 8 pseudo-molecules, which is expected to facilitate and expand its use.
2. Materials and methods
2.1. Sampling and sequencing
All samples were collected from Liucheng city, Guangxi Province, China (N24°39′15.96″, E109°14′25.37″). Genomic DNA was extracted from leaves of a single plant using the Plant Genomic DNAkit (Qiagen, San Diego, CA, USA). Genomic DNA sample was further purified for ONT sequencing with the Zymo Genomic DNA Clean and Concentrator-10 column (Zymo Research, Irvine, CA, USA). The purified DNA was then prepared for sequencing following the protocol in the genomic sequencing kit SQK-LSK108 (ONT, Oxford, UK). Single-molecule real-time sequencing of long reads was conducted on a GridION X5 platform (Oxford Nanopore Technology, OX4 4DQ, Oxford, UK) with 9 flow cells19. A total of 56.11 Gb of genomic data (6.23 Gb per cell) with an average read length of 14.59 kb was generated after quality filtering, from which the longest reading is 153.6 kb (Supporting Information Table S1). Compared with other sequencing platforms, Nanopore platform reading length has more advantages. In addition, a separate paired-end (PE) DNA library with an insert size of 400 bp (amplification by 8 PCR cycles) was constructed and sequenced using the Illumina platform (PE150) to enable a genome survey, and a total of 53.2 Gb of raw data was collected (Supporting Information Table S2).
For RNA-seq, total RNA from 12 samples were extracted from leaf, root, stem and flower of one G. elegans, using the QIAGEN RNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). The cDNA library was prepared using the TruSeq Sample Preparation Kit (Illumina, CA, USA), and paired-end sequencing with 150 bp was conducted on a HiSeq X Ten platform (Illumina, CA, USA). A total of 135.9 Gb clean data were obtained (Supporting Information Table S3).
2.2. Genome size and heterozygosity estimation
The genome size of G. elegans was estimated by the k-mer method20 using sequencing data from the Illumina DNA library. Quality-filtered reads were subjected to 17-mer frequency distribution analysis using the Jellyfish program20. The genome size (G) of G. elegans was estimated using the following formula: G = k-mer number/average k-mer depth, where k-mer number = total k-mers-abnormal k-mers. The count distribution of 17-mers followed a Poisson distribution, with the highest peak occurring at a depth of 120 (Supporting Information Table S4 and Fig. S1). The estimated genome size was 338,031,359 bp, and the heterozygosity rate of the G. elegans genome was approximately 0.38%.
2.3. Genome assembly
Genome assembly was performed on full ONT long reads using Canu v1.7.121 and WTDBG v1.2.822. Because of a high error rate of Nanopore reads, we first corrected reads by the error correction module of Canu (canu -nanopore-raw -correct -fast genome size = 300 m). Then, the corrected reads independently assembled with WTDBG (wtdbg-1.2.8 --tidy-reads 8000 -k 0 -p 17 -S 2 --rescue-low-cov-edges;wtdbg-cns -k 13 -c 3). Finally, the preliminary genome assembly was approximately 331.8 Mb in size with a contig N50 size of 10.14 Mb (Supporting Information Table S5). Nanopolish calibration uses the Burrow-Wheeler Aligner (BWA, v0.7.12-r1039) default parameter to compare the quality-controlled Nanopore data to the assembled genome23. The second-generation data are then compared to the Nanopolish-corrected genome using the BWA default parameter, and the Pilon iteration is used to correct it two times24. The ultimate version of genome assembly was approximately 335.13 Mb in size with a contig N50 size of 10.23 Mb (Supporting Information Table S6). A guanine-cytosine (GC) depth analysis was conducted to assess the potential contamination during sequencing and the coverage of the assembly, revealing that the genome had an average GC content of 37% and a unimodal GC content distribution (Supporting Information Fig. S2). The GC depth as well as the sequencing depth of the genome assembly suggested that there was no contamination from other species (Supporting Information Fig. S3). G. elegans genome were performed with mitochondrial database in NCBI, the results showed that the coverage of some sequences was nearly 1, but the identity was low (Supporting Information Table S7). As mitochondrion was cyclic, the sequences might be short after the process of DNA extraction and library construction. When we used the long reads to assemble, the very short sequences were filtered which might include the mitochondrial sequence. So nearly all the assembled genome sequences were nuclear genome sequences.
2.4. Chromosome assembly using Hi-C data
Hi-C technology enables the generation of genome-wide 3D proximity maps and is an efficient and low-cost strategy for sequences cluster, ordered, and orientation for pseudomolecule construction25. This technology has been successfully applied in recent complex genome projects, including goat26, Tartary buckwheat27, wild emmer28, and barely29. To generate a chromosomal-level assembly of the G. elegans genome, Hi-C fragment libraries were constructed. The Hi-C library was prepared followed by a procedure30 with an improved modification. In brief, freshly harvested leaves were cut into 2 cm pieces and vacuum infiltrated in nuclei isolation buffer supplemented with 2% formaldehyde. Crosslinking was stopped by adding glycine and additional vacuum infiltration. Fixed tissue was frozen in liquid nitrogen and grounded to powder before re-suspending in nuclei isolation buffer to obtain a suspension of nuclei. The purified nuclei were digested with 100 units of HindIII and marked by incubating with biotin-14-dCTP. Biotin-14-dCTP from non-ligated DNA ends was removed owing to the exonuclease activity of T4 DNA polymerase. The ligated DNA was sheared into 300–600 bp fragments, and then was blunt-end repaired and A-tailed, followed by purification through biotin-streptavidin-mediated pull down. Finally, the Hi-C libraries were quantified and sequenced using the Illumina Hiseq platform (Illumina, San Diego, CA, USA). In total, 370 million paired-end reads were generated from the libraries. Then, quality controlling of Hi-C raw data were performed using Hi-C-Pro (v2.8.0) as former research25. Firstly, low-quality sequences (quality scores < 20), adaptor sequences and sequences shorter than 30 bp were filtered out using fastp v0.12.6 (fastp, RRID:SCR_016962)31, and then the clean paired-end reads were mapped to the draft assembled sequence using bowtie2 (v2.3.2) (bowtie2, RRID:SCR_005476) to get the unique mapped paired-end reads32. As a result, 107 million uniquely mapped pair-end reads were generated, of which 76.28% were valid interaction pairs. Combined with the valid Hi-C data, we subsequently used the LACHESIS (ligating adjacent chromatin enables scaffolding in situ) de novo assembly pipeline to produce chromosome-level scaffolds. As shown in Fig. 2, the assembled sequence was anchored onto the 8 pseudo-chromosomes with lengths ranging from 36.08 to 52.33 Mb. The total length of pseudo-chromosomes accounted for 99.2% of the genome sequences, with scaffold N50 values of 40.47 Mb (Supporting Information Table S8).
Figure 2.

Genome-wide Hi-C map of Gelsemium elegans. Interaction frequency distribution of Hi-C links among chromosomes shows in color key of heatmap ranging from light yellow to dark red indicated the frequency of Hi-C interaction links from low to high (0–10).
2.5. Evaluation of the completeness of the genome assembly gene space
To evaluate the coverage of the assembly, we randomly selected the RNAseq reads aligned against the G. elegans genome assembly using HISAT2 (hierarchical indexing for spliced alignment of transcripts2)33 with default parameters. The percentage of aligned reads ranged from 91.57% to 92.10% (Table S2). We then used Benchmarking Universal Single-Copy Orthologs (BUSCO, RRID:SCR 015008)34 to search the annotated genes in the assembly for the 1308 single-copy genes conserved among all embryophytes. About 92.9% of the complete BUSCOs were found in the assembly (Supporting Information Table S9). These results suggested that the genome assembly was complete and robust.
2.6. Genome annotation
The repeat sequences in the genome consisted of simple sequence repeats (SSRs), moderately repetitive sequences, and highly repetitive sequences. The microsatellite identification tool (MISA)35 was used to search for SSR motifs in the G. elegans genome, with default parameters. A total of SSRs were identified in this way: 134,047, 29,668, 9336, 1557, 409, and 524 mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide repeats, respectively (Supporting Information Table S10).
To identify known transposable elements (TEs) in the G. elegans genome, RepeatMasker (RRID:SCR 012954)36 was used to screen the assembled genome against the Repbase (v22.11)37 and Mips-REdat libraries38. In addition, de novo evolved annotation was performed using RepeatModeler v1.0.11 (RRID:SCR 015027)36. The combined results of the homology-based and de novo predictions indicated that repeated sequences account for 43.16% of the G. elegans genome assembly (Supporting Information Table S11), with long terminal repeats accounting for the greatest proportion of 23.9% (Supporting Information Table S12). The de novo and repbase RepeatMasker analysis of the G. elegans genome assembly are shown in Supporting Information Fig. S4.
Homology-based ncRNA annotation was performed by mapping plant rRNA, miRNA, and snRNA genes from the Rfam database (release 13.0)39 to the G. elegans genome using BLASTN40 (E-value ≤ 1 × 10−5). tRNAscan-SE v1.3.1 (tRNAscan-SE, RRID:SCR 010835)41 was used (with default parameters for eukaryotes) for tRNA annotation. RNAmmer v1.242 was used to predict rRNAs and their subunits. These analyses identified 208 miRNAs, 531tRNAs, 279rRNAs, and 1257 snRNAs (Supporting Information Table S13).
The homology-based, de novo based, and RNA sequences-based gene prediction methods were used to annotate protein coding genes. For homology-based predictions, protein sequences from 6 species (Arabidopsis thaliana, Calotropis gigantea, Camellia sinensis, Nicotiana tabacum, Olea europaea and Oryza sativa) (Supporting Information Table S14) were mapped onto the G. elegans genome; the aligned sequences and the corresponding query proteins were then filtered and passed to GeneWise v2.4.1 (GeneWise, RRID:SCR 015054)43 to search for accurately spliced alignments. For the de novo predictions, we first randomly selected 1000 full-length genes from the homology-based predictions to train model parameters for Augustus v3.0 (RRID:SCR 008417)44, GeneID v1.4.445, GlimmerHMM (RRID:SCR 002654)46, and SNAP47. Augustus v3.044, GeneID v1.4.445, GlimmerHMM46, and SNAP47 were then used to predict genes based on the training set. Further, G. elegans RNA-seq data and Iso-seq data were used for gene prediction by PASA (v2.0.2, RRID:SCR 014656)48. Finally, EVidenceModeler v1.1.148 was used to integrate the predicted genes and generate a consensus gene set (Table S14). Genes with TEs were discarded using the TransposonPSI49 package. Low quality genes consisting of fewer than 50 amino acids and/or exhibiting premature termination were also removed from the gene set, yielding a final set of 26,768 genes. The final sets average transcript length, average CDS length, average exon number per gene, average exon length and average intron length were 3961.71 bp, 1088.1 bp, 4.98, 218.63 bp and 722.54 bp, respectively (Supporting Information Table S15 and Fig. S5).
The annotations of the predicted genes of G. elegans were screened for homology against the Uniprot (release 2017/10) and KEGG (release 84.0) databases using Blastall40 and KAAS50. Then, the InterProScan (release 5.2–45.0)51 package was used to annotate the predicted genes using the InterPro (5.21–60.0) database. In total, 22,636 of the total 26,768 genes (84.56%) were annotated with potential functions (Supporting Information Table S16).
After all the above prediction we used Benchmarking Universal Single-Copy Orthologs (BUSCO, RRID:SCR 015008)34 again to search the predicted genes in the assembly for the 1375 single-copy genes conserved among all embryophytes. About 95.5% of the complete BUSCOs were found in the assembly (Supporting Information Table S17). These results suggested that the genome prediction was complete and robust.
2.7. Phylogenetic tree construction and divergence time estimation
To investigate the evolutionary position of G. elegans, we compared its genome to the genome sequences of 8 other plants, which included 3 plants in special order or can produce alkaloids (C. gigantea52, C. sinensis53, and Macleaya cordata54), 3 plants from different orders in the same Eudicots clade (A. thaliana55, Brassica rapa56 and Vitis vinifera57), and 2 monocotyledons (O. sativa58 and Oropetium thomaeum59) as an outgroup. We used the OrthoMCL (v2.0.9) pipeline (OrthoMCL DB: Ortholog Groups of ProteinSequences, RRID:SCR 007839)60 (BLASTP E-value ≤ 1 × 10−5) to identify potentially orthologous gene families within these genomes.
Gene family clustering identified 13,792 gene families containing 20,755 genes in G. elegans (Fig. 3). Of these, 903 gene families were unique to G. elegans (Supporting Information Table S18).
Figure 3.

Venn diagram of shared gene families between Gelsemium elegans and 8 other plants. Each number represents a gene family number.
Phylogenetic analysis was performed using 2989 single-copy orthologous genes from common gene families found by OrthoMCL60 (Supporting Information Fig. S6). We codon-aligned each gene family using MUSCLE (MUSCLE, RRID:SCR 011812)61 and curated the alignments with Gblocks v0.91b62. Phylogeny analysis was performed using RAxML (RAxML, RRID:SCR 006086) v8.2.1163 with the GTRGAMMA model and 100 bootstrap replicates.
We then used MCMCTREE as implemented in PAML v4.9e (PAML, RRID:SCR 014932)64 to estimate the divergence times of G. elegans from the other plants. The parameter settings of MCMCTREE were as follows: clock = 2, RootAge <1.93, model = 7, BDparas = 110, kappa gamma = 62, alpha gamma = 11, rgene gamma = 23.18, and sigma2 gamma = 14.5. In addition, the divergence times of O. sativa (148–173 Mya), V. vinifera (110–124 Mya), and A. thaliana (53–82 Mya) were used for fossil calibration.
The phylogenetic analysis showed that G. elegans is more closely related to C. gigantea than to C. sinensis (Supporting Information Fig. S7), which supports the well-established hypothesis of a close relationship between G. elegans and C. gigantea65, 66. The estimated divergence time of G. elegans and C. sinensis was 97.45 Mya, while that of G. elegans and C. gigantea was about 50.69 Mya (Fig. 4).
Figure 4.

Inferred phylogenetic tree across 9 plant species. The estimated divergence time (Mya) is shown at each node.
2.8. Genes under positive selection
Studies on the crude and purified alkaloids of G. elegans have demonstrated that this species possesses anti-inflammatory4, immunomodulating7, analgesic, anxiolytic, anti-tumor8, 9, and neuropathic pain-relieving properties10. The ratio of nonsynonymous substitution rate (Ka) and synonymous substitution rate (Ks) of protein coding genes can be used to identify genes that show signatures of natural selection. We calculated average Ka/Ks values and conducted the branch-site likelihood ratio test using Codeml implemented in the PAML package64 to identify positively selected genes in the G. elegans lineage. The parameter settings of Codeml were as follows: Model A: model = 2, NS sites = 2, fix_omega = 0; Model A1: model = 2, NSsites = 2, fix_omega = 1, omega = 1.
These genes might contribute to the secondary metabolites of adaption to unfavorable environments. 94 Genes with signatures of positive selection were identified (P ≤ 0.05), of which 77 genes could be annotated with potential functions in the Swissprot database (Supporting Information Table S19). One gene is homologous required for transport of secretory proteins from the Golgi complex, which catalyzes the transfer of phosphatidylinositol and phosphatidylcholine between membranes in vitro67. This gene could potentially contribute to the adaption of G. elegans to the secondary metabolites of environment. While literature reports are rare, other identified genes might also be associated with the adaption of G. elegans. It should be noted that this is just a preliminary analysis of the functions of these genes, and further studies would be needed to clarify their roles.
2.9. Whole-genome duplication and gene family expansion analysis
We used 4-fold synonymous third-codon transversion (4DTv) and Ks estimation to detect whole genome duplication (WGD) events in the G. elegans genome. To this end, paralogous sequences of G. elegans, A. thaliana, Glycine max, O. europaea, and V. vinifera were identified with OrthoMCL60. Then, protein sequences for each of these plants were aligned against each other with Blastp40 (using an E-value threshold of ≤1 × 10−5) to identify conserved paralogs in each species. Finally, potential WGD events in each genome were evaluated based on their 4DTv and Ks distribution. The WGD analysis suggested that G. max and O. europaea may have experienced modern WGD events, and G. max have gone through 2 times of whole genome duplications, while the G. elegans has no modern WGD event, and only experience done ancient whole genome duplications (Fig. 5).
Figure 5.

Whole-genome duplication (WGD) events of 5 plants (Gelsemium elegans, Arabidopsis thaliana, Glycine max, Olea europaea and Vitis vinifera) inferred by 4-fold synonymous third-codon transversion (4DTv) estimations. (A) 4DTv; (B) Ks.
The OrthoMCL gene family analysis results were analyzed further by using Computational Analysis of Gene Family Evolution v3.068 to detect expanded gene families. This approach revealed 509 expanded gene families and 1013 contracted gene families in the G. elegans lineage (Supporting Information Fig. S8).
3. Results and discussion
The current study shows that blueprint of an organism is encoded in its genome and genome mining has become a powerful strategy for botanical studying69. To investigate the evolutionary history of the indole alkaloid gene cluster, we performed 2 rounds of synteny analysis with either the “all BLASTp” result as input of blocks with distant homology or the default “top 5 BLASTp” result for blocks with close homology. The top ranked syntenic block for the indole alkaloid (e.g. koumine and gelsemine) pathway genes is found (Supporting Information Table S20). Key steps in monoterpene indole alkaloid (MIA) biosynthesis are shown in Scheme 1, catalyzed by the enzymes tryptophan decarboxylase (TDC), strictosidine synthase (STR), strictosidine glucosidase (SDG), geissoschizine dehydrogenase (GSD) and sarpagan bridge enzyme (SBE)70. The MIAs comprise approximately 3000 compounds which have different chemical scaffolds. This enormous chemical complexity stand in contrast with only 3 sequenced MIA producers, namely Catharanthus roseus, Rhazya stricta (Apocynaceae, order Gentianales) and Camptotheca acuminate (Nyssaceae, order Cornales)71. Nevertheless, only a small number of the genes primarily from C. roseus related to the enormous diversity of MIAs are known so far, as a result, the genomic context of MIA biosynthesis is largely unknown and has only been systematically investigated in C. roseus. Given the vast chemical diversity of MIAs, we wondered whether these gene clusters would be conserved in MIA producing plants with different chemical profiles, and whether they might potentially be useful for accelerating biosynthetic gene discovery. As a case study, we selected the MIA producer G. elegans, which produces a wide variety of indole and oxindole alkaloids.
Scheme 1.

Key steps in monoterpene indole alkaloid (MIA) biosynthesis, catalyzed by the enzymes tryptophan decarboxylase (TDC), strictosidine synthase (STR), strictosidine glucosidase (SDG), geissoschizine dehydrogenase (GSD) and sarpagan bridge enzyme (SBE). The pathway diverges after strictosidine aglycone and leads to very different alkaloids in the plants Gelsemium elegans. Several representative alkaloids for Gelsemium elegans are shown.
4. Conclusions
This paper reports the sequencing, assembly, and annotation of the G. elegans genome along with details of its evolutionary history and alkaloids metabolism. Thus, we generate a significantly improved genome sequence than another Gelsemium family plant G. sempervirens that is 244 Mb with an N50 scaffold size of 411,072 bp70. The genomic data generated in this work will be a valuable resource for further genetic improvement and effective use of the G. elegans.
Availability of supporting data
The raw data from our genome project was deposited in the SRA (Sequence Read Archive) database of National Center for Biotechnology Information with Bioproject ID PRJNA505365 (Biosample ID from SAMN11089884 to SAMN11089892). We also upload the Hi-C results with the same with Bioproject ID PRJNA505365 (Biosample ID from SAMN12083642 to SAMN12083645). Versions and main parameters of the software used in this study are provided in Supporting Information Table S21.
Acknowledgments
This study was financially supported by Hunan Provincial Natural Science Foundation of China (grant 2017JJ1017), National Key R&D Program of China (grant 2017YFD0501403), National Natural Science Foundation of China (grant 31400275), and Hunan Provincial Natural Science Foundation of China (2018JJ2172).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.apsb.2019.08.004.
Contributor Information
Zhiliang Sun, Email: sunzhiliang1965@aliyun.com.
Zhaoying Liu, Email: liu_zhaoying@hunau.edu.cn.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- 1.Ornduff R. The systematics and breeding system of Gelsemium (Loganiaceae) J Arnold Arbor. 1970;51:1–17. [Google Scholar]
- 2.Sun C.K., Kimura T., But P.P., Guo J.X. World Scientific; London: 1998. International collation of traditional and folk medicine, Northeast Asia, part III. [Google Scholar]
- 3.Rujjanawate C., Kanjanapothi D., Panthong A. Pharmacological effect and toxicity of alkaloids from Gelsemium elegans Benth. J Ethnopharmacol. 2003;89:91–95. doi: 10.1016/s0378-8741(03)00267-8. [DOI] [PubMed] [Google Scholar]
- 4.Xu Y., Qiu H.Q., Liu H., Liu M., Huang Z.Y., Yang J. Effects of koumine, an alkaloid of Gelsemium elegans Benth., on inflammatory and neuropathic pain models and possible mechanism with allopregnanolone. Pharmacol Biochem Behav. 2012;101:504–514. doi: 10.1016/j.pbb.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y.C., Li L., Pi C., Sun Z.L., Wu Y., Liu Z.Y. Fingerprint analysis of Gelsemium elegans by HPLC followed by the targeted identification of chemical constituents using HPLC coupled with quadrupole-time-of-flight mass spectrometry. Fitoterapia. 2017;121:94–105. doi: 10.1016/j.fitote.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y.C., Xiao S., Yang K., Ling L., Sun Z.L., Liu Z.Y. Comprehensive identification and structural characterization of target components from Gelsemium elegans by high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry based on accurate mass databases combined with MS/MS spectra. J Mass Spectrom. 2017;52:378–396. doi: 10.1002/jms.3937. [DOI] [PubMed] [Google Scholar]
- 7.Xu Y.K., Liao S.G., Na Z., Hu H.B., Li Y., Luo H.R. Gelsemium alkaloids, immunosuppressive agents from Gelsemium elegans. Fitoterapia. 2012;83:1120–1124. doi: 10.1016/j.fitote.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Lu J.M., Qi Z.R., Liu G.L., Shen Z.Y., Tu K.C. Effect of Gelsemium elegans Benth injection on proliferation of tumor cells. Chin J Cancer. 1990;9 472–474, 477. [Google Scholar]
- 9.Cai J., Lei L.S., Chi D.B. Antineoplastic effect of koumine in mice bearing H22 solid tumor. J South Med Univ. 2009;29 1851–1852, 1856. [PubMed] [Google Scholar]
- 10.Zhang J.Y., Wang Y.X. Gelsemium analgesia and the spinal glycine receptor/allopregnanolone pathway. Fitoterapia. 2015;100:35–43. doi: 10.1016/j.fitote.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L.L., Wang Z.R., Huang C.Q., Zhang Z.Y., Lin J.M. Extraction and separation of koumine from Gelsemium alkaloids. J First Mil Med Univ. 2004;24:1006–1008. [PubMed] [Google Scholar]
- 12.Liu M., Shen J., Liu H., Xu Y., Su Y.P., Yang J. Gelsenicine from Gelsemium elegans attenuates neuropathic and inflammatory pain in mice. Biol Pharm Bull. 2011;34:1877–1880. doi: 10.1248/bpb.34.1877. [DOI] [PubMed] [Google Scholar]
- 13.Yi J.E., Yuan H. Research and development on enterotoxin of Gelsemium elegans benth. J Hunan Environ-Biol Polytech. 2003;9:26–30. [Google Scholar]
- 14.Tan J., Qiu C., Zhen L. Analgesic effect and no physical dependence of Gelsemium elegans benth. Pharmacol Clin Chin Mater Med. 1988;4:24–28. [Google Scholar]
- 15.Jain M., Koren S., Miga K.H., Quick J., Rand A.C., Sasani T.A. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36:338–345. doi: 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deschamps S., Zhang Y., Llaca V., Ye L., Sanyal A., King M. A chromosome-scale assembly of the Sorghum genome using nanopore sequencing and optical mapping. Nat Commun. 2018;9:4844. doi: 10.1038/s41467-018-07271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang M., Tu L., Yuan D., Zhu D., Shen C., Li J. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat Genet. 2019;51:224–229. doi: 10.1038/s41588-018-0282-x. [DOI] [PubMed] [Google Scholar]
- 18.Ghurye J., Pop M., Koren S., Bickhart D., Chin C.S. Scaffolding of long read assemblies using long range contact information. BMC Genomics. 2017;18:527. doi: 10.1186/s12864-017-3879-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senol Cali D., Kim J.S., Ghose S., Alkan C., Mutlu O. Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief Bioinform. 2018:1–18. doi: 10.1093/bib/bby017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marçais G., Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.WTDBG package. https://github.com/ruanjue/wtdbg2 Available from: [accessed 10.01.18]
- 23.Loman N.J., Quick J., Simpson J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods. 2015;12:733–735. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 24.Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burton J.N., Adey A., Patwardhan R.P., Qiu R., Kitzman J.O., Shendure J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bickhart D.M., Rosen B.D., Koren S., Sayre B.L., Hastie A.R., Chan S. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat Genet. 2017;49:643–650. doi: 10.1038/ng.3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L., Li X., Ma B., Gao Q., Du H., Han Y. The tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Mol Plant. 2017;10:1224–1237. doi: 10.1016/j.molp.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 28.Avni R., Nave M., Barad O., Baruch K., Twardziok S.O., Gundlach H. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science. 2017;357:93–97. doi: 10.1126/science.aan0032. [DOI] [PubMed] [Google Scholar]
- 29.Mascher M., Gundlach H., Himmelbach A., Beier S., Twardziok S.O., Wicker T. A chromosome conformation capture ordered sequence of the barley genome. Nature. 2017;544:427–433. doi: 10.1038/nature22043. [DOI] [PubMed] [Google Scholar]
- 30.Belton J.M., McCord R.P., Gibcus J.H., Naumova N., Zhan Y., Dekker J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 35.Thiel T., Michalek W., Varshney R., Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.) Theor Appl Genet. 2003;106:411–422. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- 36.Tarailo-Graovac M., Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;25 doi: 10.1002/0471250953.bi0410s25. 4.10.1–14. [DOI] [PubMed] [Google Scholar]
- 37.Bao W., Kojima K.K., Kohany O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile DNA. 2015;6:11. doi: 10.1186/s13100-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nussbaumer T., Martis M.M., Roessner S.K., Pfeifer M., Bader K.C., Sharma S. MIPS PlantsDB: a database framework for comparative plant genome research. Nucleic Acids Res. 2013;41:D1144–D1151. doi: 10.1093/nar/gks1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalvari I., Argasinska J., Quinones-Olvera N., Nawrocki E.P., Rivas E., Eddy S.R. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018;46:D335–D342. doi: 10.1093/nar/gkx1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lagesen K., Hallin P., Rødland E.A., Staerfeldt H.H., Rognes T., Ussery D.W. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Birney E., Durbin R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 2000;10:547–548. doi: 10.1101/gr.10.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanke M., Steinkamp R., Waack S., Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32:W309–W312. doi: 10.1093/nar/gkh379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blanco E., Parra G., Guigó R. Using geneid to identify genes. Curr Protoc Bioinformatics. 2007;18 doi: 10.1002/0471250953.bi0403s18. 4.3.1–28. [DOI] [PubMed] [Google Scholar]
- 46.Majoros W.H., Pertea M., Salzberg S.L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 47.Bromberg Y., Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haas B.J., Salzberg S.L., Zhu W., Pertea M., Allen J.E., Orvis J. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.TransposonPSI an application of PSI-Blast to mine (Retro-)transposon ORF homologies. http://transposonpsi.sourceforge.net Available from:
- 50.Moriya Y., Itoh M., Okuda S., Yoshizawa A.C., Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quevillon E., Silventoinen V., Pillai S., Harte N., Mulder N., Apweiler R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoopes G.M., Hamilton J.P., Kim J., Zhao D., Wiegert-Rininger K., Crisovan E. Genome assembly and annotation of the medicinal plant Calotropis gigantea, a producer of anticancer and antimalarial cardenolides. G3 Genes Genom Genet. 2018;8:385–391. doi: 10.1534/g3.117.300331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei C., Yang H., Wang S., Zhao J., Liu C., Gao L. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc Natl Acad Sci U S A. 2018;115:E4151–E4158. doi: 10.1073/pnas.1719622115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu X., Liu Y., Huang P., Ma Y., Qing Z., Tang Q. The genome of medicinal plant Macleaya cordata provides new insights into benzylisoquinoline alkaloids metabolism. Mol Plant. 2017;10:975–989. doi: 10.1016/j.molp.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 55.Zapata L., Ding J., Willing E.M., Hartwig B., Bezdan D., Jiao W.B. Chromosome-level assembly of Arabidopsis thaliana Ler reveals the extent of translocation and inversion polymorphisms. Proc Natl Acad Sci U S A. 2016;113:E4052–E4060. doi: 10.1073/pnas.1607532113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X., Wang H., Wang J., Sun R., Wu J., Liu S. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011;43:1035–1039. doi: 10.1038/ng.919. [DOI] [PubMed] [Google Scholar]
- 57.Jaillon O., Aury J.M., Noel B., Policriti A., Clepet C., Casagrande A. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 58.Mahesh H.B., Shirke M.D., Singh S., Rajamani A., Hittalmani S., Wang G.L. Indica rice genome assembly, annotation and mining of blast disease resistance genes. BMC Genomics. 2016;17:242. doi: 10.1186/s12864-016-2523-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.VanBuren R., Bryant D., Edger P.P., Tang H., Burgess D., Challabathula D. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature. 2015;527:508–511. doi: 10.1038/nature15714. [DOI] [PubMed] [Google Scholar]
- 60.Li L., Stoeckert C.J., Jr., Roos D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Talavera G., Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 63.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 65.Sen S., Sahu N.P., Mahato S.B. Flavonol glycosides from Calotropis gigantea. Phytochemistry. 1992;31:2919–2921. doi: 10.1016/0031-9422(92)83668-o. [DOI] [PubMed] [Google Scholar]
- 66.Wang H.T., Yang Y.C., Mao X., Wang Y., Huang R. Cytotoxic gelsedine-type indole alkaloids from Gelsemium elegans. J Asian Nat Prod Res. 2017;20:321–327. doi: 10.1080/10286020.2017.1342637. [DOI] [PubMed] [Google Scholar]
- 67.Mo P., Zhu Y., Liu X., Zhang A., Yan C., Wang D. Identification of two phosphatidylinositol/phosphatidylcholine transfer protein genes that are predominately transcribed in the flowers of Arabidopsis thaliana. J Plant Physiol. 2007;164:478–486. doi: 10.1016/j.jplph.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 68.De Bie T., Cristianini N., Demuth J.P., Hahn M.W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22:1269–1271. doi: 10.1093/bioinformatics/btl097. [DOI] [PubMed] [Google Scholar]
- 69.Seberg O., Droege G., Barker K., Coddington J.A., Funk V., Gostel M. Global genome biodiversity network: saving a blueprint of the tree of life — a botanical perspective. Ann Bot. 2016;118:393–399. doi: 10.1093/aob/mcw121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Franke J., Kim J., Hamilton J.P., Zhao D., Pham G.M., Wiegert-Rininger K. Gene discovery in Gelsemium highlights conserved gene clusters in monoterpene indole alkaloid biosynthesis. Chembiochem. 2019;20:83–87. doi: 10.1002/cbic.201800592. [DOI] [PubMed] [Google Scholar]
- 71.Stavrinides A.K., Tatsis E.C., Dang T.T., Caputi L., Stevenson C.E., Lawson D.M. Discovery of a short-chain dehydrogenase from Catharanthus roseus that produces a new monoterpene indole alkaloid. Chembiochem. 2018;19:940–948. doi: 10.1002/cbic.201700621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data from our genome project was deposited in the SRA (Sequence Read Archive) database of National Center for Biotechnology Information with Bioproject ID PRJNA505365 (Biosample ID from SAMN11089884 to SAMN11089892). We also upload the Hi-C results with the same with Bioproject ID PRJNA505365 (Biosample ID from SAMN12083642 to SAMN12083645). Versions and main parameters of the software used in this study are provided in Supporting Information Table S21.