

Abstract
Background
Both the six gene signature (6GS: CPA3, DNASE1L3, CLC, IL1B, ALPL, and CXCR2) and T‐helper 2 signature (TH2S: CLCA1, SERPINB2, and POSTN) are proposed as biomarkers in the identification of inflammatory phenotypes of asthma in induced sputum and epithelial brushings, respectively. The aim of this study was to explore patterns of gene expression of known signatures, 6GS and TH2S in endobronchial biopsies.
Methods
This was an exploratory cross‐sectional study of gene expression in endobronchial biopsies of 55 adults with asthma and 9 healthy controls (HC). The expression of the 6GS and TH2S was determined by quantitative polymerase chain reaction. Correlations with clinical and cellular characteristics were performed, and receiver operating characteristic was utilized to assess signatures' ability to predict asthma from HC and inflammatory phenotypes.
Results
Gene expression of DNASE1L3 (P = .045) was upregulated in asthma compared with HC, and IL1B (P = .017) was upregulated in neutrophilic asthma compared with non‐neutrophilic asthma. In asthma, the expression of CPA3 was negatively associated with ICS daily dose (r = −.339; P = .011), IL1B expression was positively associated with bronchial lavage fluid (BLF) total cell count (r = .340; P = .013) and both CLC and POSTN expression were associated with lymphocytes percentage in BLF (r = −.355, P = .009; r = −.300, P = .025, respectively). Both 6GS (area under curve [AUC] = 86.3%; P = .017) and TH2S (AUC = 72.7%; P = .037) could significantly predict asthma from HC. In addition, 6GS can identify neutrophilic (AUC = 93.2%; P = .005) and TH2S identifies eosinophilic (AUC = 62.7%; P = .033) asthma.
Conclusions and Clinical Relevance
There was increased expression of DNASE1L3 in asthma and IL1B in neutrophilic asthma. These results show similar upregulated patterns of expression in two genes of the 6GS in endobronchial biopsies, previously identified in sputum. The upregulation of DNASE1L3 and IL1B suggests that common mechanisms may be at play throughout the airway.
Keywords: asthma, endobronchial biopsy, inflammation, inflammatory phenotypes
Similar patterns of expression of known gene signatures were observed in endobronchial biopsies characterized by the upregulation of DNASE1L3 in asthma and IL1B in neutrophilic asthma suggesting these pathways are relevant in both biopsy and sputum in asthma.

1. INTRODUCTION
Asthma is a heterogeneous disease with multiple clinical and inflammatory manifestations.1 Many genes and environmental factors play a role in pathogenesis,2, 3, 4 and many phenotypes have been described.5, 6, 7 The absence or presence of certain features determine the clinical manifestations, severity of asthma, and the response to treatment.8, 9 The first cases of asthma described were early onset, eosinophilic in nature, and related to allergic immune responses. More recently it is recognized that airway eosinophilia is not present in everyone with asthma and noneosinophilic phenotypes have been described.10, 11 In the majority of patients, asthma symptoms and exacerbations can be controlled by a combination of inhaled corticosteroids (ICS), acting to suppress the inflammation, and a β2 adrenergic agonist, acting to open the constricting bronchial smooth muscle. Although, corticosteroids have a pronounced effect in most patients, 30% to 40% of patients have a poor response to traditional treatment approaches and high sputum neutrophil counts have been linked to reduced responsiveness to corticosteroids as well as more severe asthma.11, 12 The identification of phenotypes of asthma based on the mechanisms of the disease pathogenesis that reflect clinical features offers the promise to better predict clinical outcomes and personalize response to treatment. Such an approach has utilized the identification of gene biomarker signatures that reflects pathogenic mechanisms associated with different airway inflammatory phenotypes of asthma. Previous transcriptomic studies of asthma have led to the identification of gene expression signatures including the six gene expression signature (6GS) in sputum samples7 and the T‐helper 2 signature (TH2S) in epithelial brushings.2 The 6GS comprises the expression of carboxypeptidase A3 (CPA3); deoxyribonuclease I‐like3 (DNASEIL3); Charcot‐Leyden crystal protein (CLC); alkaline phosphatase isozyme (ALPL); IL‐1β (IL1B); and chemokine (C‐X‐C motif) receptor 2 (CXCR2)13 to differentiate inflammatory phenotypes, corticosteroid treatment responsiveness, and future exacerbations.7, 14, 15 The TH2S comprises the expression of calcium‐activated chloride channel regulator 1 (CLCA1), plasminogen activator inhibitor 2 (SERPINB2), periostin (POSTN) for the identification of subjects with a TH2‐high immunity.2
Endobronchial biopsies sample structural cells including epithelial cells, mesenchymal cells, subepithelial matrix components as well as immune cells, which are not sampled in either sputum or epithelial brushings, but are involved in asthma pathogenesis. The aim of this study was to explore patterns of expression of known signatures, 6GS and TH2S in endobronchial biopsies of subjects with asthma compared with healthy, neutrophilic compared with non‐neutrophilic, and eosinophilic with noneosinophilic. We then explored associations between gene expression and clinical and cellular characteristics in participants with asthma, as well as the capacity of the signatures to predict disease status and inflammatory phenotypes.
2. METHODS
2.1. Participants
Sixty‐four adults recruited from the Respiratory Ambulatory Care Service, John Hunter Hospital, New South Wales, Australia. Adults (>18 years old) with no history of a clinical chest or upper respiratory tract infection in the previous 6 weeks were studied. Participants with asthma (n = 55) had a physician's diagnosis of asthma with previous confirmation of variable airway obstruction defined as either a post bronchodilator change in forced expiratory volume (FEV1) of at least 12% or 200 mL after 400 μg of salbutamol and/or the bronchial hyper‐responsiveness defined as at least 15% decline in FEV1 after inducing bronchial provocation with 4.5% saline solution. Bronchoscopy in healthy controls (HC) (n = 9) was conducted and none of the participants had underlying cardiac or lung disease. All HC had FEV1 percentage predicted more than 80% assessed by spirometry. Current smokers were excluded. Written consent was obtained from all the participating individuals. The study was approved by the Hunter New England Human Research Ethics Committee (05/08/10/3.09).
2.2. Study design
This is an exploratory cross‐sectional study in which clinical characteristics such as age, sex, body mass index (BMI), age of onset, asthma control questionnaire (ACQ) score, smoking history, bronchial lavage fluid (BLF) for cell count and endobronchial biopsies were collected during bronchoscopy.
2.3. Lung function
Spirometry was performed (Easy One Spirometer; Medical Technologies, MA) before bronchoscopy and without withholding usual medications on the day of the test. Percentage predicted for FEV1 and forced vital capacity (FVC) were calculated using The Third National Health and Nutrition Examination Survey (NHANES III).
2.4. BLF and endobronchial biopsies
Flexible bronchoscopy was performed to obtain BLF by wedging into the bronchus of the right middle lobe or lingula and lavaging with 20 to 40 mL of sterile normal saline. BLF was filtered and total cell count (TCC) and viability was measured.16 The BLF was centrifuged and the cell pellet was resuspended in phosphate‐buffered saline to the concentration of 1 × 106/mL and cellular cytospins were prepared. The cytospins were stained with May‐Grünwald Giemsa (Beckman Coulter, Brea, CA) and differential cell count of 400 nonsquamous cells was performed. There were one to three endobronchial biopsies taken using alligator forceps from the second to third generation airways in the right or left lower lobe. Samples were stored in RNA later at −20°C.
2.5. Inflammatory phenotyping
Participants were characterized based on inflammatory phenotypes defined by the BLF cell count and differential determined from slides collected at the time of bronchoscopy. BLF cell count of HC (n = 82) from the cohort was utilized to determine a TTC cutoff for neutrophilic asthma (95th percentile = 0.99 × 106/mL). Phenotypes were defined as follows: eosinophilic asthma as eosinophil greater than or equal to 3% of the TTC; noneosinophilic: less than 3% eosinophils; neutrophilic asthma: if neutrophils greater than or equal to 61% and greater than or equal to 0.99 × 106/mL TTC, and non‐neutrophilic: less than 61% neutrophils of the TTC.17, 18, 19
2.6. RNA preparation
RNA was extracted from endobronchial biopsies using the RNeasy Mini Kit (Qiagen, Hilden, Germany). The quality of the RNA was assessed using Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). RNA from samples had a mean (standard deviation [SD]) RNA integrity number value of 7.2 (SD 1.6).
2.7. Quantitative real‐time polymerase chain reaction
RNA (200 ng) from endobronchial biopsies was reverse‐transcribed to complementary DNA (cDNA) using the high‐capacity cDNA reverse transcription kit (Applied Biosystems, Foster City). Predesigned primers (CPA3: Hs00157019_m1; DNASE1L3: Hs00172840_m1; CLC: Hs00171342_m1; ALPL: Hs01029144_m1; IL1B: Hs01555410_m1; CXCR2: Hs01891184_s1; CLCA1: Hs00976287_m1; SERPINB2: Hs01010736_m1; POSTN: Hs01566750_m1; Applied Biosystems) were combined with Taqman gene expression master mix as per the manufacturer's instructions in duplicate singleplex real‐time PCRs (7500 Real‐Time PCR System; Applied Biosystems). Statistical analysis was performed using ΔC T (cycle of threshold) of target gene subtracting the cycle threshold of housekeeping gene. Eukaryotic 18S ribosomal RNA with a C t < 20 was considered acceptable. Fold change results were calculated by using and scaled (×104).7
2.8. Statistical analysis
Stata 15 (StataCorp, College Station, TX) was used to analyse data. Student t test was used for parametric data and Fishers' exact test for categorical data. For nonparametric data including gene expression we used the Wilcoxon rank‐sum test. Significance determined as P < .05. Data were reported in mean and SD for normally distributed and media and interquartile (IQR) Q1, Q3 for non‐normally distributed data. Spearman's rank correlation measured the association between gene expression and continuous measures of lung function and cell count in BLF. Logistic regression with receiver operating characteristic (ROC) curve analysis was utilized to evaluate gene set's capacity to identify asthma and inflammatory phenotypes.
3. RESULTS
3.1. Study participants
The characteristics of the participants are summarized in Table 1. Both groups were similar with respect to age, sex, and BMI. Lung function was significantly lower in participants with asthma compared with HC (FEV1% predicted mean (SD): asthma 77 (20.4), HC: 109.8 (13.5), P < .001; and FEV1/FVC mean (SD) asthma: 67.8 (12), HC: 81.4 (4.7); P = .001). The asthma cohort was comprised of predominantly females with adult onset of asthma, with 5 (9%) identified as former smokers with a median of 5 pack year. Total and differential cell count from the BLF are shown in Table 1. Participants with asthma had a significantly increased proportion of eosinophils in comparison to HC.
Table 1.
Clinical and molecular characteristics of participants comparing asthma and healthy controls
| Characteristic | HC | Asthma | P value |
|---|---|---|---|
| N | 9 | 55 | … |
| Age (y), mean (SD) | 50 (14) | 57 (15) | .193 |
| Gender, F, n (%) | 3 (33) | 39 (71) | .028 |
| Age of onset, n (%) | … | ||
| Childhood | N/A | 20 (36) | |
| Adult | N/A | 26 (47) | |
| Unknown | N/A | 9 (16) | |
| FEV1% predicted, mean (SD) | 109.8 (13.5) | 77.0 (20.4)* | <.001 |
| FEV1/FVC, mean (SD) | 81.4 (4.7) | 67.8 (12.0)* | .001 |
| Former smoker, n (%) | 1 (11) | 5 (9) | .847 |
| Pack years, median (IQR) | 5 (5, 5) | 5 (4.5, 5) | .617 |
| BLF | |||
| Total cell count ×106/mL, median (IQR) | 0.1 (0.1, 0.2) | 0.1 (0.1, 0.6) | .324 |
| Viability, median (IQR) | 75 (60, 76) | 78 (57, 91) | .505 |
| Neutrophils %, median (IQR) | 62.3 (15.3, 63.8) | 47.3 (23.8, 78.8) | .481 |
| Eosinophils %, median (IQR) | 0.3 (0, 1.0) | 2.5 (1.0, 8.0)* | <.001 |
| Macrophages %, median (IQR) | 23.5 (17.3, 40.5) | 23.8 (12.8, 42.5) | .562 |
| Lymphocytes %, median (IQR) | 1.0 (0.3, 1.8) | 0.5 (0, 2.0) | .643 |
| Epithelial cells %, median (IQR) | 14.5 (9.3, 20.0) | 5.0 (1, 19.3) | .104 |
| Squamous cells %, median (IQR) | 2.4 (2.2, 4.3) | 1.5 (0, 4.5) | .071 |
| Endobronchial mRNA, median (IQR) | |||
| 6GS | |||
| CPA3 | 0.63 (0.22, 1.71) | 1.58 (0.51, 1.96) | .134 |
| DNASEIL3 | 0.13 (0.06, 0.28) | 0.22 (0.19, 0.25)* | .045 |
| CLC | 0.13 (0.06, 0.32) | 0.07 (0.05, 0.08) | .144 |
| ALPL | 1.75 (0.75, 3.16) | 1.46 (1.10, 1.86) | .628 |
| IL1B | 0.11 (0.05, 0.24) | 0.17 (0.05, 0.19) | .754 |
| CXCR2 | 0.18 (0.10, 0.42) | 0.18 (0.07, 0.48) | .790 |
| TH2S | |||
| CLCA1 | 0.02 (0.01, 0.05) | 0.03 (0.02, 0.07) | .171 |
| SERPINB2 | 0.29 (0.13, 1.06) | 0.19 (0.16, 0.36) | .550 |
| POSTN | 1.04 (0.43, 3.32) | 0.61 (0.56, 0.89) | .124 |
Note: Data was analyzed using Wilcoxon rank or a Student t test depending on the outcome distribution and Fishers' exact test for categorical data.
Abbreviations: BLF, bronchial lavage fluid; FEV, forced expiratory volume; FVC, forced vital capacity; HC, healthy controls; IQR, interquartile range; mRNA, messenger RNA.
Significant P ≥ .05 versus healthy controls.
3.2. Gene expression in asthma and HC
We assessed the expression of individual genes from the 6GS and TH2S signatures in asthma compared with HC. Participants with asthma had higher expression levels of DNASEIL3 (P = .045) compared with HC (Figure 1A). There were no differences for the rest of individual genes in the 6GS and TH2 signatures between asthma and HC. There was no difference in the expression of the genes by sex (data not shown).
Figure 1.
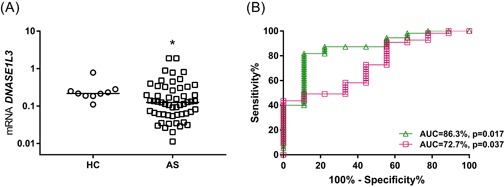
Gene signatures' performance in asthma. A, DNASE1L3 upregulated in asthma compared with healthy controls (*P < .05); (B) ROC curves demonstrate that 6GS (green triangle/line) and TH2S (magenta square/line) can predict asthma from HC. AS, asthma; AUC, area under curve; HC, healthy controls; mRNA, messenger RNA; ROC, receiver operating characteristic
We performed ROC analysis to evaluate the signatures' ability to predict disease status in endobronchial biopsy. The AUC revealed that 6GS (AUC = 86.3%; P = .017) and TH2S (AUC = 72.7%; P = .037) could both significantly predict asthma from HC (Figure 1B).
3.3. Inflammatory phenotypes of asthma
Characteristics of asthma study participants with different inflammatory phenotypes are presented in Table 2. Participants with eosinophilic asthma (n = 26) were compared with noneosinophilic (n = 29) and also those with neutrophilic asthma (n = 7) were compared with non‐neutrophilic asthma (n = 48). Those with eosinophilic asthma had significantly lower BLF TTC and higher eosinophil percentage compared with noneosinophilic (Table 2). Those with neutrophilic asthma had a significantly lower ACQ score (P = 0.043) compared with non‐neutrophilic. Participants with neutrophilic asthma had significantly higher TTC, viability, percentages of neutrophils compared with non‐neutrophilic, while non‐neutrophilic had higher percentage of eosinophils, macrophages, epithelial cells, and squamous cell count in BLF compared with participants with neutrophilic asthma (Table 2).
Table 2.
Characteristics of participants with eosinophilic and neutrophilic asthma
| Characteristic | Eosinophilic | Noneosinophilic | P value | Neutrophilic | Non‐neutrophilic | P value |
|---|---|---|---|---|---|---|
| N | 26 | 29 | … | 7 | 48 | … |
| Age (y), mean (SD) | 58 (14) | 56 (16) | .478 | 62 (12) | 56 (15) | .369 |
| Gender, F, n (%) | 16 (62) | 23 (79) | .147 | 5 (71) | 34 (71) | .974 |
| BMI (kg/m2), mean (SD) | 30 (7) | 31 (8) | .882 | 24 (2)* | 31 (7) | .017 |
| Age of onset, n (%) | .109 | .103 | ||||
| Childhood | 7 (27) | 13 (45) | 4 (57) | 16 (33) | ||
| Adult | 15 (58) | 11 (38) | 1 (14) | 25 (52) | ||
| Unknown | 4 (15) | 5 (17) | 2 (29) | 7 (15) | ||
| FEV1% predicted, mean (SD) | 77.2 (19.0) | 76.8 (22.0) | .953 | 72.7 (21.3) | 77.6 (20.4) | .558 |
| FEV1/FVC, mean (SD) | 67.8 (11.0) | 67.7 (13.0) | .977 | 62.4 (11.8) | 68.6 (11.9) | .209 |
| Former smoker, n (%) | 3 (12) | 2 (7) | .357 | 0 | 5 (10) | .802 |
| Pack years, media (IQR) | 5 (4, 5) | 5 (5, 5) | .564 | … | 5 (4, 5) | … |
| ACQ score, median (IQR) | 2.3 (0.8, 2.8) (n = 21) | 1.8 (0.9, 2.5) (n = 24) | .710 | 0.3 (0.3, 1) (n = 5)* | 2 (1, 2.8) (n = 40) | .043 |
| ICS use, yes n (%) | 26 (100) | 28 (96.6) | .596 | 7 (100) | 47 (97.9) | .732 |
| ICS dose mcg/d, median (IQR) | 1300 (800, 2000) | 2000 (800, 2000) | .117 | 2000 (2000, 2000) | 1600 (800, 2000) | .100 |
| GINA step 4‐5, n (%) | 22 (85) | 24 (83) | .473 | 6 (86) | 40 (83) | .442 |
| OCS use, yes, n (%) | 3 (11.5) | 2 (6.9) | .550 | 1 (14.3) | 4 (8.3) | .609 |
| OCS dose mcg/day, median (IQR) | 10 (5, 10) | 20 (15, 25) | .076 | 25 (25, 25) | 10 (7.5, 12.5) | .147 |
| BLF | ||||||
| Total cell count, ×106/mL, median (IQR) | 0.11 (0.1, 0.3)** | 0.2 (0.1, 1.1) | .032 | 6.4 (2.3, 6.9)* | 0.1 (0.1, 0.4) | <.001 |
| Viability, median (IQR) | 68.8 (60, 84) | 82 (50, 94) | .224 | 95.4 (94, 96.5)* | 70.3 (50, 84.3) | <.001 |
| Neutrophils %, median (IQR) | 34.1 (24.5, 67.3) | 59.5 (23.8, 84.5) | .158 | 89 (84, 91.5)* | 39 (17.3, 67) | <.001 |
| Eosinophils %, median (IQR) | 8.1 (4.8, 20.8)** | 1 (0.5, 2) | <.001 | 1.3 (0.5, 2.3)* | 3.5 (1.1, 9.1) | .030 |
| Macrophages %, median (IQR) | 22.1 (14.5, 40) | 29.8 (12.8, 48.3) | .312 | 8.8 (7.8, 13)* | 27.6 (15.1, 44) | .003 |
| Lymphocytes %, median (IQR) | 0.5 (0, 1.3) | 0.8 (0, 2) | .672 | 0 (0, 1) | 0.8 (0, 2.3) | .154 |
| Epithelial cells %, median (IQR) | 7.5 (3.3, 24.8) | 2.8 (0.8, 9) | .090 | 0 (0, 0.8)* | 7.3 (2.5, 22) | <.001 |
| Squamous cells %, median (IQR) | 2 (0.3, 6.5) | 0.7 (0, 3.2) | .115 | 0 (0, 0.3)* | 2 (0.3, 5.2) | .006 |
| 6GS | ||||||
| CPA3 | 0.7 (0.2, 1.7) | 0.6 (0.2, 2.0) | .940 | 0.3 (0.1, 0.7) | 0.7 (0.2, 1.9) | .225 |
| DNASEIL3 | 0.1 (0.1, 0.3) | 0.1 (0.1, 0.4) | .960 | 0.1 (0.1, 0.8) | 0.1 (0.1, 0.2) | .419 |
| CLC | 0.14 (0.09, 0.38) | 0.09 (0.04, 0.24) | .109 | 0.06 (0.05, 0.52) | 0.13 (0.06, 0.31) | .495 |
| ALPL | 1.86 (1.07, 3.34) | 1.73 (0.62, 2.97) | .469 | 1.73 (0.37, 5.88) | 1.79 (0.79, 3.16) | .743 |
| IL1B | 0.10 (0.04, 0.24) | 0.11 (0.06, 0.29) | .337 | 0.38 (0.11, 0.51)* | 0.10 (0.04, 0.23) | .017 |
| CXCR2 | 0.19 (0.09, 0.33) | 0.18 (0.11, 0.46) | .578 | 0.20 (0.10, 0.46) | 0.18 (0.10, 0.39) | .781 |
| TH2S | ||||||
| CLCA1 | 0.02 (0.01, 0.06) | 0.02 (0.01, 0.03) | .363 | 0.01 (0.01, 0.04) | 0.02 (0.01, 0.05) | .363 |
| SERPINB2 | 0.51 (0.16, 1.88) | 0.21 (0.09, 0.46) | .069 | 0.26 (0.05, 0.29) | 0.31 (0.13, 1.29) | .251 |
| POSTN | 1.31 (0.56, 4.39) | 0.70 (0.40, 1.95) | .183 | 0.70 (0.15, 2.99) | 1.10 (0.43, 3.81) | .434 |
Abbreviations: ACQ, Asthma Control Questionnaire; BLF, bronchial lavage fluid; BMI, body mass index; GINA, Global Initiative for Asthma; ICS, inhaled corticosteroid; OCS, oral corticosteroid.
Significant P ≥ .05 versus non‐neutrophilic.
Significant P ≥ .05 versus noneosinophilic.
3.4. Gene signature expression in inflammatory phenotypes of asthma
Gene expression for each of the individual genes from the gene signatures evaluated presented similar expression in eosinophilic asthma compared with noneosinophilic (Table 2). Gene expression for IL1B (P = .017) was significantly higher in those with neutrophilic asthma compared with non‐neutrophilic (Figure 2A).
Figure 2.
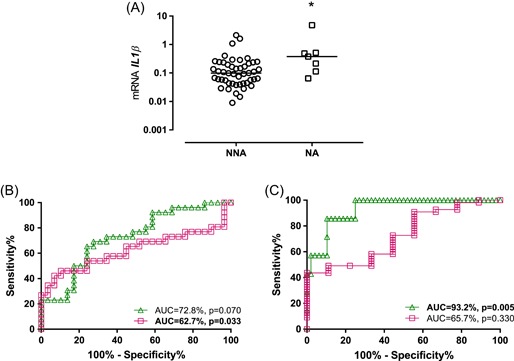
Gene signatures performance in inflammatory phenotypes of asthma. A, IL1B significantly upregulated in neutrophilic compared with non‐neutrophilic asthma (*P < .05). ROC curves demonstrate that (B) 6GS (green triangle/line) could not predict eosinophilic from noneosinophilic asthma, while TH2S (magenta square/line) showed capacity to predict; (C) 6GS but not the TH2S could predict neutrophilic from non‐neutrophilic asthma. AUC, area under curve; NA, neutrophilic asthma; NNA, non‐neutrophilic asthma; ROC, receiver operating characteristic
ROC analysis revealed TH2S could predict eosinophilic from noneosinophilic asthma with a moderate discriminatory capacity (AUC = 62.7%; P = .033) (Figure 2B). Conversely, the 6GS but not the TH2S presented discriminatory capacity (AUC = 93.2%; P = .005) for neutrophilic from non‐neutrophilic asthma (Figure 2C).
3.5. Relationship of gene expression to clinical and cellular characteristics
We explored the associations between gene expression and clinical features presented in Table 1. Bronchial biopsy gene expression for CPA3 was negatively associated with ICS daily dose (r = −.339; P = .011) suggesting higher expression when ICS doses are the lowest. Gene expression for IL1B was significantly and positively associated with BLF TTC (r = .340; P = .013), while both CLC and POSTN were significantly associated with BLF lymphocytes percentage (r = −.335; P = .009 and r = −.300; P = .025, respectively). There were no other associations found between expression of genes and clinical or inflammatory cell characteristics.
4. DISCUSSION
This study explored the expression of the gene signatures 6GS and TH2S previously reported in sputum and epithelial brushings in endobronchial.2, 13 We observed upregulation of two genes of the 6GS, an upregulation of DNASE1L3 in asthma compared with HC, and upregulation of IL1B in neutrophilic asthma compared with non‐neutrophilic asthma. In addition, we evaluated the overall capacity of these signatures to identify disease status and inflammatory phenotypes, we observed that 6GS was able to predict asthma from HC, and neutrophilic from non‐neutrophilic asthma; and TH2S was able to predict asthma from HC, and eosinophilic from noneosinophilic asthma.
In an era of personalized medicine, the search and development of biomarkers that identify asthma and those that are more likely to benefit from a targeted therapeutic approach is an urgent unmet need. The majority of recent advances in asthma therapies have targeted TH2 mechanisms,20 however, with more than half of those with severe asthma exhibiting no evidence of active TH2 inflammation,21 there is a need to continue to explore the inflammatory profile and mechanisms in asthma. Our results support this importance, identifying genes not classically associated with a type 2 signature to be upregulated in the setting of treatment with ICS. Molecular phenotyping of well‐characterized people with asthma offers the hope that we will be able to identify and target new pathogenic mechanisms that will lead to novel therapies.22
DNASE1L3 is an endonuclease that mediates the breakdown of DNA during apoptosis.23 Transcriptomic studies described it as an eosinophilic gene and responsive to ICS treatment in induced sputum of subjects with asthma.7, 13, 24 However, there is no further evidence that identifies specific roles of DNASE1L3 in the pathogenesis of asthma. Our study showed upregulation of DNASE1L3 in participants with asthma compared with healthy controls but no differences between inflammatory phenotypes of asthma. DNASE1L3 is increased during apoptosis and plays an important role in fragmentation of DNA from the apoptotic vesicles; our results might be reflective of an overall increase of cellular apoptosis in participants with asthma.
When examining gene expression in neutrophilic compared with non‐neutrophilic, we observed a significant increase in IL1B in participants with neutrophilic asthma. Previous studies by our group have recognized IL1B gene expression associated with neutrophilic inflammation in induced sputum.7, 13, 25 It is well known that neutrophils are recruited from the proximal to the distal part of the airway to reside in the airway epithelium and submucosal glands, this process is mediated by IL‐8, IL‐1β, TNF‐α, and leukotriene B4.26 IL‐1β is produced mainly by macrophages, cultured bronchial epithelial cells, and neutrophils.27, 28 In those with asthma, the presence of neutrophilia has been associated with frequency of exacerbations,28 poor response to ICS,29, 30, 31 and disease severity.32 Simpson et al33 observed elevated expression of IL‐1β in subjects with neutrophilic asthma. It has also been reported that the inhibition of NLRP3 prevents neutrophilia and decreases airway hyper‐responsiveness.34
CPA3 is a metalloexopeptidase specifically expressed in a particular subtype of mast cells in combination with tryptase.35 Expression of CPA3 has been associated with TH2‐high asthma in sputum and epithelial brushings of steroid naïve asthma.36, 37 Berthon et al15 reported reduction of CPA3 expression following treatment with oral corticosteroid, suggesting responsiveness to treatment. In addition, the number of mast cells containing tryptase and CPA3 decreased following ICS treatment.24 In this study, we found a relationship between CPA3 expression levels and ICS daily dose pattern that is consistent with what has previously been reported in induced sputum.
The discrepancies found in the expression of other genes investigated in this study and other studies and therefore, sample type, may be reflective of the compartmentalization of inflammation and variability of mast cells subtypes and eosinophils in the lung tissue compared with sputum and epithelial brushings.38 While application of gene signatures in biopsy samples is not a practical approach in distinguishing asthma from healthy controls and phenotypes, it may be helpful in determining common mechanisms with measureable activity in different compartments of the airways. ROC analysis demonstrated that 6GS when applied to endobronchial biopsies performed well in predicting asthma from HC, as well as neutrophilic from non‐neutrophilic asthma. Baines et al7 identified the 6GS by performing a transcriptomic analysis in induced sputum from a diverse group of subjects with asthma with varying treatment dose, clinical severity, and airway inflammatory phenotypes.7 This is quite similar to the population presented in this study. The TH2S was derived from epithelial brushings, so largely from a single structural cell type.2, 5 TH2S was found associated with eosinophilic airway inflammation, subepithelial basement membrane thickening, and response to ICS in a cohort with mild asthma who were steroid‐naïve.2, 5 This group was quite different from the cohort presented in this study comprised predominantly participants on high daily doses of ICS as described. Despite these differences we observed capacity of the TH2S to predict asthma from HC, and eosinophilic from noneosinophilic asthma.
A strength of this study is that it is the first to investigate these gene signatures in endobronchial biopsies in a well‐characterized cohort of adults with asthma and HC and to consider compartmentalization of the inflammation in asthma for mechanisms related to these signatures.18, 39 This exploratory study has particular limitations. Our study is cross‐sectional with a small sample size and does not consider different time points in participants with different inflammatory phenotypes and it is not clear how the expression of these genes may change over time or in response to known treatments. The discrepancies found in the expression of other genes investigated in this study compared with previous studies may be reflective of the compartmentalization of inflammation, variability of cells subtypes in the lung tissue compared with sputum and epithelial brushings.38 The correlations observed in this study are weak and a larger study is needed to explore complex interrelationships between gene expression and clinical features. The use of cell count cutoffs from induced sputum to identify eosinophilic and neutrophilic asthma in BLF is another limitation of this study. There is evidence that supports BLF eosinophils count resemble closely those found in induced sputum, however, the neutrophils cell counts are more variable and there is less agreement as to what should constitute a cutoff for neutrophilic asthma.18, 40 Furthermore, the HC group in this study presented high variability in percentage of BLF neutrophils. Studies on a larger cohort are needed to address these limitations.
In conclusion, our study sought to explore patterns of gene expression in endobronchial biopsies and to assess compartmentalization of inflammation in asthma by comparing our results to previously published studies. We have demonstrated upregulation of DNASE1L3 in asthma compared with HC and IL1B in neutrophilic compared with non‐neutrophilic asthma. We confirmed the ability of 6GS to predict asthma from HC, and neutrophilic from non‐neutrophilic asthma in endobronchial biopsies, while the TH2S exhibited capacity to predict asthma from HC, as well as eosinophilic from noneosinophilic asthma. This study highlights the need for more research in larger cohorts to investigate further mechanisms of pathogenesis that may be reflected in endobronchial biopsies of subjects with different inflammatory phenotypes of asthma.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
All authors designed the study. JLS supervised and coordinated the study. SS‐O performed the experiments and wrote the manuscript which was further refined and edited by JLS, PAW, KJB, and DB. PAW performed the bronchoscopy, KJB supervised the laboratory analysis, and DB supervised the statistical analysis. All authors approved the final manuscript for submission.
ACKNOWLEDGMENTS
We acknowledge technical support from Heather McDonald, Kristy Nichol, Kavita Prabeja, Naomi Fibbens, Kellie Fakes, Bridgette Donati and the clinical support from Lorissa Hopkins and Douglas Dorahy of the Priority Research Centre for Healthy Lungs, Hunter Medical Research Institute, Newcastle, Australia.
Sánchez‐Ovando S, Joanne Baines K, Barker D, Wark PA, Louise Simpson J. Six gene and TH2 signature expression in endobronchial biopsies of participants with asthma. Immun Inflamm Dis. 2020;8:40–49. 10.1002/iid3.282
DATA AVAILABILITY STATEMENT
The data of this study is available from the corresponding author upon request.
REFERENCES
- 1. Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183‐192. [DOI] [PubMed] [Google Scholar]
- 2. Woodruff PG, Boushey HA, Dolganov GM, et al. Genome‐wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104(40):15858‐15863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mersha TB, Martin LJ, Biagini Myers JM, et al. Genomic architecture of asthma differs by sex. Genomics. 2015;106(1):15‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. von Mutius E. Gene‐environment interactions in asthma. J Allergy Clin Immunol. 2009;123(1):3‐11. [DOI] [PubMed] [Google Scholar]
- 5. Woodruff PG, Modrek B, Choy DF, et al. T‐helper type 2‐driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180(5):388‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18(5):716‐725. [DOI] [PubMed] [Google Scholar]
- 7. Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol. 2011;127(1):153‐160. [DOI] [PubMed] [Google Scholar]
- 8. Low K, Bardin PG. Targeted therapy for severe asthma: identifying the right patients. Mol Diagn Ther. 2017;21:235‐247. [DOI] [PubMed] [Google Scholar]
- 9. Opina MT, Moore WC. Phenotype‐driven therapeutics in severe asthma. Curr Allergy Asthma Rep. 2017;17(2):10. [DOI] [PubMed] [Google Scholar]
- 10. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16(1):45‐56. [DOI] [PubMed] [Google Scholar]
- 11. Pavord ID, Brightling CE, Woltmann G, Wardlaw AJ. Non‐eosinophilic corticosteroid unresponsive asthma. Lancet. 1999;353(9171):2213‐2214. [DOI] [PubMed] [Google Scholar]
- 12. Heaney LG, Djukanovic R, Woodcock A, et al. Research in progress: Medical Research Council United Kingdom Refractory Asthma Stratification Programme (RASP‐UK). Thorax. 2016;71(2):187‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baines KJ, Simpson JL, Wood LG, et al. Sputum gene expression signature of 6 biomarkers discriminates asthma inflammatory phenotypes. J Allergy Clin Immunol. 2014;133(4):997‐1007. [DOI] [PubMed] [Google Scholar]
- 14. Fricker M, Gibson PG, Powell H, et al. A sputum 6‐gene signature predicts future exacerbations of poorly controlled asthma. J Allergy Clin Immunol. 2019;144:51‐60. [DOI] [PubMed] [Google Scholar]
- 15. Berthon BS, Gibson PG, Wood LG, MacDonald‐Wicks LK, Baines KJ. A sputum gene expression signature predicts oral corticosteroid response in asthma. Eur Respir J. 2017;49(6):1700180. [DOI] [PubMed] [Google Scholar]
- 16. Sukkar MB, Wood LG, Tooze M, et al. Soluble RAGE is deficient in neutrophilic asthma and COPD. Eur Respir J. 2012;39(3):721‐729. [DOI] [PubMed] [Google Scholar]
- 17. Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology. 2006;11(1):54‐61. [DOI] [PubMed] [Google Scholar]
- 18. Grootendorst DC, Sont JK, Willems LN, et al. Comparison of inflammatory cell counts in asthma: induced sputum vs bronchoalveolar lavage and bronchial biopsies. Clin Exp Allergy. 1997;27(7):769‐779. [PubMed] [Google Scholar]
- 19. Agusti A, Bel E, Thomas M, et al. Treatable traits: toward precision medicine of chronic airway diseases. Eur Respir J. 2016;47(2):410‐419. [DOI] [PubMed] [Google Scholar]
- 20. Grainge CL, Maltby S, Gibson PG, Wark PA, McDonald VM. Targeted therapeutics for severe refractory asthma: monoclonal antibodies. Expert Rev Clin Pharmacol. 2016;9(7):927‐941. [DOI] [PubMed] [Google Scholar]
- 21. Wenzel SE. Emergence of biomolecular pathways to define novel asthma phenotypes. Type‐2 immunity and beyond. Am J Respir Cell Mol Biol. 2016;55(1):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gibson PG. Inflammatory phenotypes in adult asthma: clinical applications. Clin Respir J. 2009;3(4):198‐206. [DOI] [PubMed] [Google Scholar]
- 23. Sisirak V, Sally B, D'Agati V, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. 2016;166(1):88‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang G, Baines KJ, Fu JJ, et al. Sputum mast cell subtypes relate to eosinophilia and corticosteroid response in asthma. Eur Respir J. 2016;47(4):1123‐1133. [DOI] [PubMed] [Google Scholar]
- 25. Baines KJ, Simpson JL, Bowden NA, Scott RJ, Gibson PG. Differential gene expression and cytokine production from neutrophils in asthma phenotypes. Eur Respir J. 2010;35(3):522‐531. [DOI] [PubMed] [Google Scholar]
- 26. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459‐489. [DOI] [PubMed] [Google Scholar]
- 27. Arend WP, Palmer G, Gabay C. IL‐1, IL‐18, and IL‐33 families of cytokines. Immunol Rev. 2008;223(1):20‐38. [DOI] [PubMed] [Google Scholar]
- 28. Hastie AT, Everts KB, Cho SK, et al. IL‐1 beta release from cultured bronchial epithelial cells and bronchoalveolar lavage cells from allergic and normal humans following segmental challenge with ragweed. Cytokine. 1996;8(9):730‐738. [DOI] [PubMed] [Google Scholar]
- 29. Green RH, Brightling CE, Woltmann G, et al. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax. 2002;57:875‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Green BJ, Wiriyachaiporn S, Grainge C, et al. Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PLOS One. 2014;9(6):e100645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wenzel SE, Schwartz LB, Langmack EL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. 1999;160(3):1001‐1008. [DOI] [PubMed] [Google Scholar]
- 32. Moore WC, Hastie AT, Li X, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol. 2014;133(6):1557‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J. 2014;43(4):1067‐1076. [DOI] [PubMed] [Google Scholar]
- 34. Rossios C, Pavlidis S, Hoda U, et al. Sputum transcriptomics reveal upregulation of IL‐1 receptor family members in patients with severe asthma. J Allergy Clin Immunol. 2018;141:560‐570. [DOI] [PubMed] [Google Scholar]
- 35. Balzar S, Fajt ML, Comhair SA, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am J Respir Crit Care Med. 2011;183(3):299‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dougherty RH, Sidhu SS, Raman K, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2‐high asthma. J Allergy Clin Immunol. 2010;125(5):1046‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV. Measures of gene expression in sputum cells can identify TH2‐high and TH2‐low subtypes of asthma. J Allergy Clin Immunol. 2014;133(2):388‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Balzar S, Chu HW, Strand M, Wenzel S. Relationship of small airway chymase‐positive mast cells and lung function in severe asthma. Am J Respir Crit Care Med. 2005;171(5):431‐439. [DOI] [PubMed] [Google Scholar]
- 39. Arafah MA, Raddaoui E, Al Kassimi F, et al. Endobronchial biopsy in the final diagnosis of chronic obstructive pulmonary disease and asthma: a clinicopathological study. Ann Saudi Med. 2018;38(2):118‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Balbi B, Pignatti P, Corradi M, et al. Bronchoalveolar lavage, sputum and exhaled clinically relevant inflammatory markers: values in healthy adults. Eur Respir J. 2007;30(4):769‐781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data of this study is available from the corresponding author upon request.