

Abstract
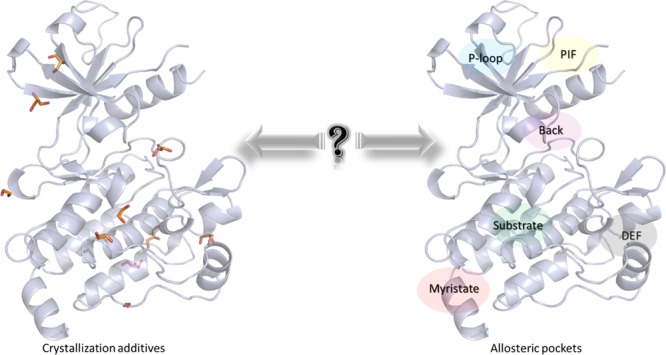
Allosteric effect can modulate the biological activity of a protein. Thus, the discovery of new allosteric sites is very attractive for designing new modulators or inhibitors. Here, we propose an innovative way to identify allosteric sites, based on crystallization additives (CA), used to stabilize proteins during the crystallization process. Density and clustering analyses of CA, applied on protein kinase and nuclear receptor families, revealed that CA are not randomly distributed around protein structures, but they tend to aggregate near common sites. All orthosteric and allosteric cavities described in the literature are retrieved from the analysis of CA distribution. In addition, new sites were identified, which could be associated to putative allosteric sites. We proposed an efficient and easy way to use the structural information of CA to identify allosteric sites. This method could assist medicinal chemists for the design of new allosteric compounds targeting cavities of new drug targets.
Introduction
Proteins are fundamental entities in an organism, controlling normal living cells and disorder processes. These biological entities are divided into families according to their amino acid sequences, three-dimensional (3D) structural motifs, and primary functions. Proteins perform their biological function through interactions with other proteins, nucleic acids, or small ligands. The interactions between a protein and a ligand are often located in a well-defined active site or cavity of the protein, the so-called orthosteric site. These interactions are directly associated to protein function modulation. However, small ligands can also bind to other protein sites, called allosteric sites, distant from the active site. This binding could induce a conformational change on the protein structure, resulting in an increase or decrease of its intrinsic activity.1,2
The term “allostery” was introduced in 19613,4 even if Christian Bohr has already described the process as the “Bohr effect” in the early 20th century related to hemoglobin conformational change. Since then, allostery has progressively evolved to a unified concept5 associated with its main property: the conformational change. It is an integrant part of the protein dynamics and may be present in every protein in the living world.6−8 Not surprisingly, allostery has raised a great interest in pharmaceutical research, especially in identifying allosteric sites in protein and/or developing allosteric drugs. The latter could present some advantages compared to drugs targeting orthosteric sites, such as a greater specificity, fewer side effects, and an easier up- and down-regulation of proteins.9 In some cases, this interest for allosteric approaches in drug discovery has led to successful results. Indeed, in 2004, Cinacalcet was the first allosteric drug approved by the Food and Drug Administration (FDA). This positive allosteric modulator targets the calcium-sensing receptor belonging to the GPCR family for the treatment of hyperparathyroidism.10 Interest in allostery is well described in the protein kinase (PK) family, a major therapeutic target due to its implication in several diseases such as cancer.11,12 Most of protein kinase inhibitors approved by the FDA or under clinical trials are targeting the orthosteric adenosine triphosphate (ATP) binding site.13 However, several allosteric sites have been identified in PKs such as ABL, CK2α, FLT3, or MEK.14−19 Several allosteric kinase inhibitors have been already approved by the FDA such as trametinib, cobimetinib, and bimenitinib in 2013, 2015, and 2018, respectively, for the treatment of patients with metastatic melanoma involving a BRAF V600E or V600K mutation.12,17
Nowadays, some databases20 and benchmarks21 are available for helping in the identification of allosteric cavities through computational approaches. Those approaches developed or adapted specifically for this objective are normal mode analysis, Gaussian network mode,22,23 and binding leverage approach24 and are based on the calculation of protein cavity volumes. A computational mapping protocol, the multiply copy simultaneous search (MCSS) was also published in 1996. In this methodology, thousands of ligands are minimized around a protein structure to identify the main binding sites.25 In this paperwork, we proposed a novel computational approach to identify allosteric cavities in a protein family based on the presence of experimental crystallization additives (CA). Initially, those molecules are present together with ions, buffers, and solvent to facilitate the crystallization process of proteins or protein–ligand complexes.26 Interestingly, those molecules are not always randomly distributed around the structures but seem located in protein hotspots, especially near the binding cavities.27 While they cannot be directly used in FBDD projects, their binding reveals some key points on the interaction of drug-like ligand or fragments.28 Some previous experimental mapping studies (multiple solvent crystal structures; MSCS) on crystalline proteins have demonstrated the ability of those additives to bind into interesting regions of the protein surface.25,29,30 Thus, we decided to evaluate the relationship between the sites where CA are located and the known orthosteric and allosteric sites in a protein family. Here, we focused on two protein families that have been substantially crystallized: the PK and the nuclear receptor (NR) families. In the first step, starting from a dataset built from several databases, CA distribution is evaluated within 3D structures of PKs and NRs, aligned on a unique reference protein, to determine their location sites. Then, we assessed the ability of those CA to be collocated with known allosteric ligands (AL) already identified in PK and NR families through a clustering approach. Interestingly, we identified that the strong presence of CA in cavities of experimentally determined protein structures corresponds to known orthosteric and allosteric sites. Therefore, this study suggests a novel approach to identify allosteric sites.
Results and Discussion
PK Family: A Model for Allostery
PKs constitute one of the most studied protein families, and their large involvements in several diseases (cancer, inflammation, Alzheimer’s disease, etc.) lead to the search of novel therapeutic drugs, targeting the catalytic binding sites or allosteric cavities. In this study, we focused on PKs since this protein family exhibits well-defined orthosteric and allosteric cavities. Indeed, PKs belong to the transferase superfamily, catalyzing the phosphate transfer to a protein substrate. Through the catalytic mechanisms and regulation of PKs, the ATP molecule located in the orthosteric site, bound to the hinge region (Figure 1, orange), will transfer the γ phosphate group to a protein substrate. PK inhibitors designed to target this orthosteric site and to compete with ATP are classified as type I and bind the active conformation of the protein kinase. In addition to this major active site, several other cavities have been described during the past years, distributed all around the kinase structure.16,17 In this study, 18 PDB structures representing various groups and subgroups of the PK family will be considered as reference for the definition of those allosteric sites:
-
(1)
The so-called back pocket (Figure 1, purple) is close to the orthosteric site. This back pocket concerned type II inhibitors, which bind to the orthosteric and back pocket, in the inactive conformation of the kinase and type III inhibitors, which bind exclusively to the back pocket.31 Here, the back pocket is represented by PDB IDs 3O96 (AKT1), 3LW0 (IGF1R), 1S9J (MEK), 4LMN (MEK), 3EQC (MEK), 4ITH (RIPK1), and 4ZJI (PAK1).
-
(2)
The myristate pocket (Figure 1, red) is located at the C-terminal lobe of the ABL protein kinase and is targeted by type IV inhibitors. This pocket is associated to a subfamily of PKs, ABL (PDB IDs 3MS9, 3K5V, and 3PYY).
-
(3)
One additional pocket of interest is the substrate pocket (Figure 1, green), in which the protein that will be phosphorylated by the PK usually binds. Type V inhibitors target this site, and PDB IDs 3JVR (CHK1) and 3F9N (CHK1) contain an allosteric inhibitor bound in this pocket.
-
(4)
The DEF (docking site for ERK, FXF) pocket facilitates substrate recognition32 as presented in the PDB IDs 4E6C (MAPK14) and 3O2M (JNK1).
-
(5)
The last cavities are located at the N-terminal part of PKs: the PIF (PDK1 interacting fragment) pocket33 illustrated in PDB IDs 3PXF (CDK2) and 3HRF (PDK1) and the P-loop pocket, exemplified in PDB IDs 3H30 (CK2a1) and 4CFE (AMPKα1/2).
Figure 1.
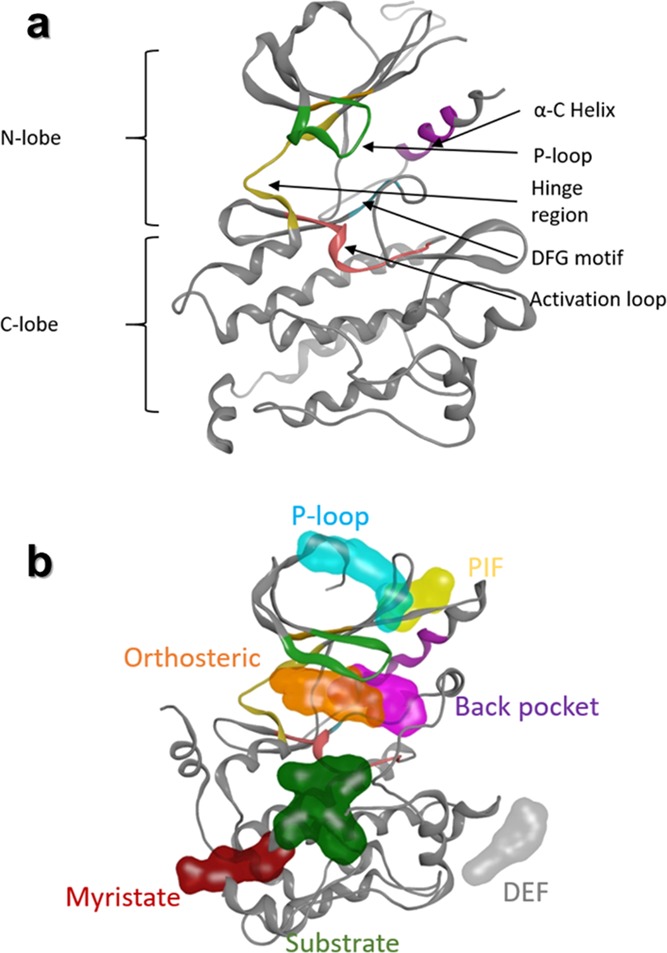
Structural properties of protein kinase family. (a) Structural common motifs in protein kinases, characterizing the active site. (b) Representation of cavities described in the literature (orange, orthosteric cavity; purple, back pocket; cyan, P-loop pocket; yellow, PIF pocket; gray, DEF pocket; red, myristate pocket; and green, substrate pocket).
CA in Kinase Family
Generally, 3D structures of complexes may also contain small molecules such as conventional ligands and/or CA. The latter are involved in the crystallization process, and their coordinates are obtained from the electron density mapping like for protein and ligand atoms. In most in silico studies such as docking, the 3D structural information of CA are ignored during calculations. In this study, we focused on the spatial positions of CA in several crystallographic structures of PKs to identify allosteric sites. Eight hundred forty-five diverse structures of protein–ligand complexes were retrieved from the PDB, and a total of 2459 CA molecules (CA dataset) are present in these complexes. Among these CA, we mainly noticed polar compounds like ethylene glycol or glycerol but also sugars (β-octylglucoside) that present amphipathic properties (an exhaustive list is provided in Table S1). For each CA, we only considered their geometric center (centroid) to simplify the structural analysis. Distribution of CA centroids was evaluated around and inside PK structures (Figure 2). An analysis of the aligned protein structures shows that CA could be placed in three different positions: (i) far away from the protein surface, (ii) at the solvent-exposed protein surface, or (iii) in cavities inside the protein structure (Figure 2a). We analyzed the density of CA distribution around several aligned structures. The distribution of the CA centroids revealed that only two main cavities having more than 80% density of CA centroids are both surface-exposed (details of the methodology section are found in the Supporting Information). The first one is in the C-terminal part of the PK and did not correspond to an allosteric pocket already identified in the literature. The second one points toward the α-E helix (Figure 2b) and is located near the “peptide” pocket. In 2004, Heo et al. published the crystal structure of JNK1 (PDB ID 1UKI), a PK that binds to a peptide of the JIP1 protein, and this interaction plays a role in the JNK1 phosphorylation activity.34 The interface formed by this protein–protein interaction is also an allosteric site.16,34 By lowering the density to 50%, CA are aggregated in three other sites. Two centroids are very close together and are located in the DEF pocket. The last centroid is found near the substrate pocket.
Figure 2.
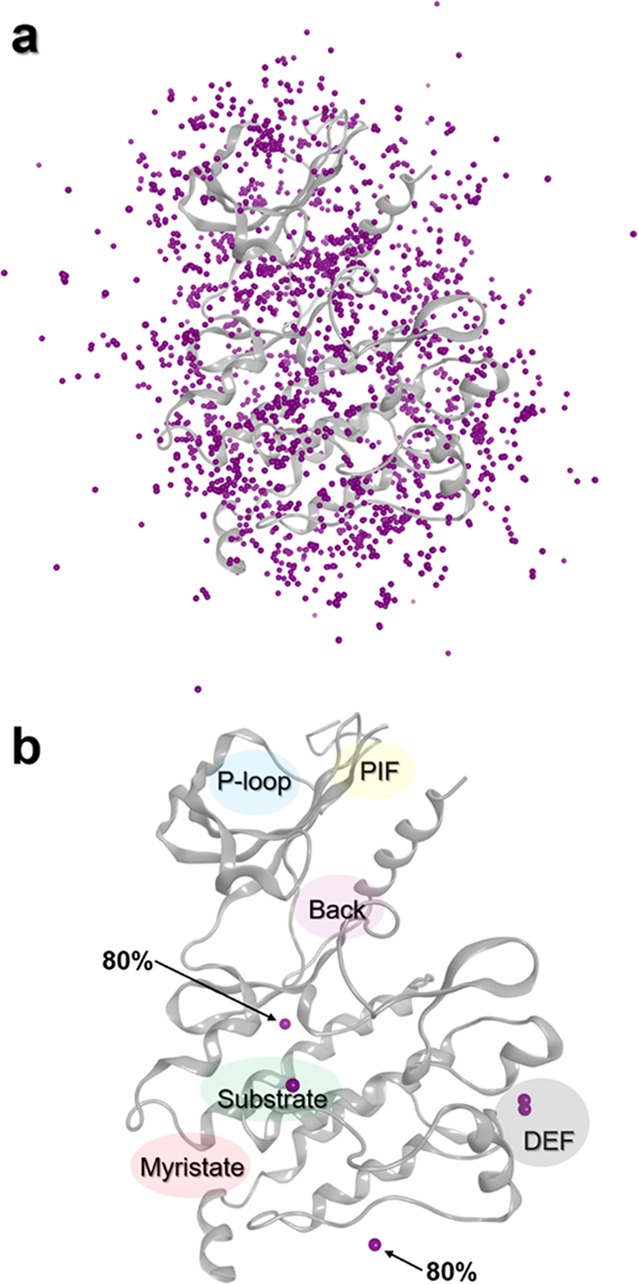
Distribution of CA centroids around a PK (PDB ID 1K5V). (a) Representation of all CA centroids. (b) Position of centroids with densities greater than 80 (centroids indicated by an arrow) and 50% (all purple spheres).
Thus, in the case of PKs, it appears that CA are not always distributed arbitrarily but seem to be attracted to some conserved areas on the protein structure, such as the surface or deep cavities. This observation is in agreement with a very recent paper, proposing that fragments and CA often bind in the same way as drug-like ligands in four proteins (BACE2, CLK2, TYR1, and CAH2).27 Moreover, in some cases, these attractive regions in PKs could be related to allosteric sites such as pocket, substrate, and DEF pockets.
CA: A Kinase Allosteric Identifier
In proteins, orthosteric ligands (OL) and AL bind to well-defined orthosteric and allosteric sites, respectively. Based on the density of centroids, our preliminary results suggest an existence of preferential CA sites. A second analysis was performed to determine whether there is a possible relation between the position of CA molecules on the PK surface and the various sites occupied by OL and AL. A ligand dataset was built from 864 diverse structures of PK-ligand complexes containing exclusively OL and AL (1049 ligands). It is important to note that we have always considered complexes that have at least one AL, and in some cases, an OL was also present in the crystal structures such as in PDB ID 4AN2, where ATP and cobimetinib are bound to the MEK1 crystal structure. This dataset was joined to the CA dataset, guided by an alignment of all PKs to the same reference structure PDB ID 1ATP. By focusing on CA, AL, and OL centroids, two clustering analysis were performed using a density-based algorithm (DBSCAN),35 first on the CA dataset and then on the combined CA and ligand datasets (Figure S1). Parameters of the clustering were controlled by changing gradually the minimal number of points in the cluster (minpts from 1 to 4) and the distance between two points in the cluster (ε from 0.5 to 3 Å). According to the results, we observed three different types of clusters: (i) some clusters contain only centroids of CA from the CA dataset or (ii) some clusters contain only centroids of ligands (AL and OL) from the ligand dataset (those two types of clusters are called homogenous clusters) and (iii) others clusters contain centroids from both datasets (called heterogeneous clusters). Heterogeneous clusters indicate that CA probably occupy the same site than ligands (AL and OL). Heterogeneous cluster IDs are depending on the clustering parameters, and three parameter pairs [minpts - ε] have the most populated clusters ([1 - 2], [2 - 2], and [3 - 2]) (Figure S3). The 18-reference PK complexes containing AL were used to optimize the parameters. Three parameter pairs ([1 - 2], [2 - 2], and [3 - 2]) give at least eight heterogeneous clusters containing both reference ligands and CA. The parameter pair [3 - 2] provides the largest common cluster since there is a large number of CA and ligands in the same sites.
To compare the results obtained from the density analysis and the clustering method, an unsupervised clustering was carried out first on the CA dataset with the parameter [3 - 2]. Under these conditions, about 33% of the CA cannot be clustered. The remaining CA are grouped into 118 clusters, classified by their population, and the most populated cluster is located in the substrate pocket, a site that has been already identified from density analysis with a density threshold of 50%.
Adding the ligand dataset (AL and OL) to the CA dataset in the clustering method provides also 118 different clusters constituted by homogeneous and heterogeneous clusters. Eight hundred fourteen compounds (i.e., 23%), mainly AL, are identified as singletons. We observed that all the known pockets in PK are found in the 20 densest clusters, among 117 clusters in total (see Figure S4). Not surprisingly, OL are in the most populated cluster (∼31% of all compounds) and are grouped with a small number of CA in this heterogeneous cluster (cluster 0; Figure 3a and Figure S4). Thus, the ATP binding site is also a cavity that can accommodate CA, which is consistent with the study of Drwal et al.27 The next most populated (Figure S4) heterogeneous clusters present interesting results. Indeed, we found that almost all the heterogeneous clusters define an allosteric site described in the literature and experimentally identified in crystal structures. Again, a substrate pocket appears as an important attractive region for CA since this pocket contains centroids of cluster 1, the second most populated cluster (Figure 3d and Table S2). Between the two reference ligands present in this pocket, only the centroid of the allosteric CHEK1 inhibitor (PDB ID 3F9N) is present in cluster 1. The other allosteric CHEK1 inhibitor (PDB ID 3JVR) partially occupies this pocket, and its centroid was not detected in the clusters. We also observed that cluster 103, close to cluster 1, is located in the same substrate pocket. Thus, many clusters can point toward the same allosteric site.
Figure 3.
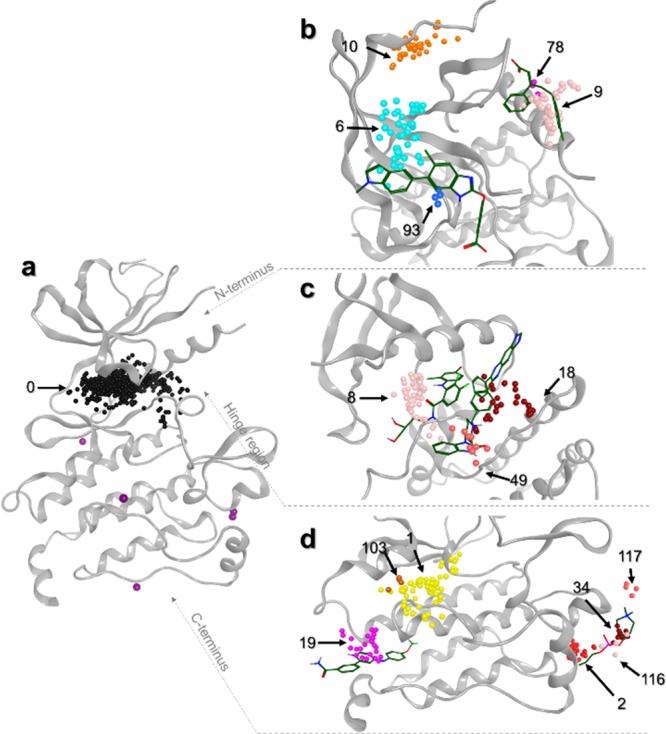
Representation of heterogeneous clusters on a kinase structure (PDB ID 1K5V). (a) Orthosteric site represented by cluster 0 in black. Density points are still present in purple. (b) Heterogeneous clusters are displayed on the N-terminal region of the protein kinase. Reference ligands bound to AMPK (PDB ID 4CFE, left) and PDK1 (PDB ID 3HRF, right) are represented in sticks. (c) Heterogeneous clusters are displayed on the hinge region. Reference ligands bound to MEK1 (PDB ID 3EQC, left) and AKT1 (PDB ID 3O96, right) are represented in sticks. (d) Heterogeneous clusters are displayed on the C-terminal part. Reference ligands bound to ABL1 (PDB ID 3K5V, left) and MAPK14 (PDB ID 4E6C, right) are represented in sticks. Cluster IDs are indicated, and centroids are color-coded based on their cluster IDs.
Another example concerns the large DEF pocket, which presents four different cluster IDs (2, 34, 116, and 117). In this case, the reference ligands are correctly retrieved in clusters 34 and 117 (Table S2). It is important to note that these reference ligands are bound to kinases classified in two different PK subgroups: MAPK14 (p38α) and JNK. There are major structural differences in the large DEF pocket between the two crystal structures (PDB ID 4E6C and PDB ID 3O2M), which explain the different binding mode observed for the two allosteric inhibitors (reference ligands). These substrate and DEF pockets were also identified above from the density analysis. Moreover, the clustering analysis allows the detection of additional allosteric pockets. As shown in Figure 3d, cluster 19 indicates the position of the myristate pocket since this cluster is located at the same position than the reference ligand (myristate), crystallized in the ABL subgroup (PDB ID 3K5V). However, other AL of ABL selected as reference ligands (PDB IDs 3MS9 and 3PYY) were not classified and were considered as singletons. This can be explained by the fact that the three ligands are not fully superimposed. Ligand cocrystallized in PDB ID 3K5V has a centroid located at 3.7 and 5.3 Å from ligand centroids of PDB IDs 3MS9 and 3PYY, respectively.
On the N-terminal lobe of the protein kinase (Figure 3b), clusters 6 and 93 are correctly positioned on the P-loop pocket, and clusters 78 and 9 are positioned on the PIF pocket. In fact, reference ligands are included in the different clusters corresponding to known allosteric sites (Table S2). In addition to the PIF and P-loop pockets, we also identified cluster 10, which did not correspond to any reference ligand. This cluster highlighted a new site, which was described very recently in the literature and therefore not yet included in our database.36 This pocket is occupied by ligands involved in an allosteric mechanism of Aurora kinase inhibition and could be a new site of interest for other PKs.
In the case of the back pocket, the cavity volume is large due to structural variations present in different PKs and due to different chemical structures of type II inhibitors. In our study, we detected three different clusters in the back pocket (Figure 3c). Among our reference ligands, the AL of AKT1 (PDB ID 3O96) is detected by the clustering method (cluster 18). This ligand binds in a rather different position than the AL of MEK, RIPK, or PAK, other ligands binding to the back pocket. We also identified cluster 49 in close proximity to cluster 18. The AL of AKT occupies a space defined by the two clusters. Hence, those clusters seem to correctly define the allosteric pocket of the AKT subgroup. The last cluster, cluster 8, is also involved in the back pocket and contains the AL of the MAPK group according to the presence of a ligand recently discovered as an allosteric inhibitor of the ERK5 and MAPK7 proteins.37 Unfortunately, the MEK reference ligands (MAPK subgroup - PDB IDs 1S9J, 4LMN, and 3EQC) were not found in this cluster probably due to a bias induced by the consideration of centroid instead of the whole ligands. The back pocket is in close vicinity of the orthosteric site. During the clustering step, the centroids of the OL are not correctly distinguished from centroids of the AL located in the back pocket. For this reason, AL of MEK (PDB IDs 1S9J, 4LMN, and 3EQC), RIPK1 (4ITH), and PAK (4ZJI), used as references, were classified in the orthosteric cluster (cluster 0). To avoid this problem, OL were removed using a pharmacophore search defined near the hinge region. Clustering of datasets without OL provides more meaningful results since AL present in the back pocket were mainly grouped in cluster 0 and the AL of AKT (PDB ID 3O96) in cluster 19 (Table S2). The other allosteric sites were not modified, and for the DEF pocket, for example, reference AL were correctly identified in clusters 37 and 120.
Homogeneous clusters, formed by CA molecules, represent the largest number of centroids in a cluster (Table S2 and Figure S4). They are concerned with CA that cannot be clustered with ligands (AL nor OL). According to the analysis of heterogeneous clusters, the different allosteric sites were found in clusters containing more than 25 centroids. Beyond this threshold, all known allosteric pockets were identified by the clustering method, meaning that all allosteric sites are retrieved in the top 20 clusters (Figure S4). For homogeneous cluster analysis, those clusters (number ≥ 25 centroids) were also identified in allosteric sites in PKs (Figure 4). The homogeneous cluster 5, located near the α-E helix, represents the peptide pocket, identified above from density analysis. Cluster 11 is similar to the heterogeneous cluster 10, which defined a cavity already identified in the literature as an allosteric site.36 Clusters 3, 4, 7, and 12 revealed three other sites at the C-terminal part of PKs. The first one, cluster 3, is located at the end of the α-E helix, and the second, cluster 12, is located at the C-terminal lobe of PKs. The third site, formed by clusters 4 and 7, is also at the bottom of PKs, and interestingly, this site was identified as an important attractive cavity for CA in PKs using the density analysis. Those three sites are described for the first time by our approach and were not described in our reference structures nor in the literature. They could potentially be considered as novel binding sites of interest.
Figure 4.
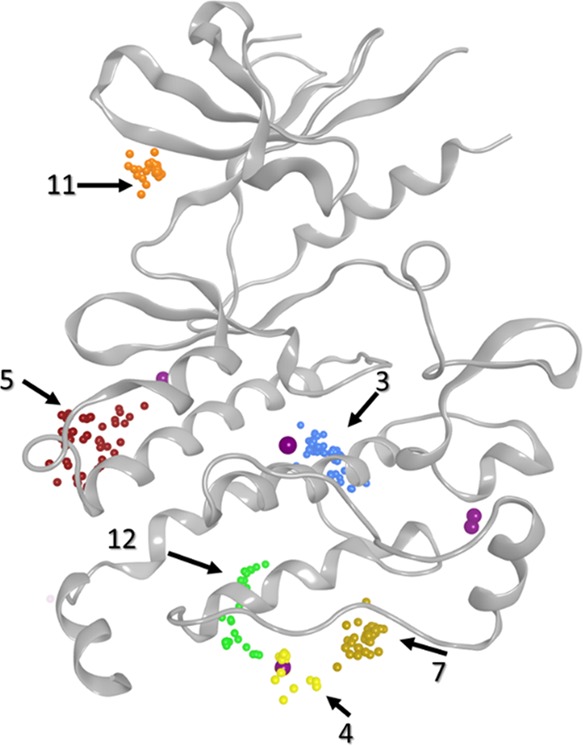
Representation of homogeneous clusters on a protein kinase (PDB ID 1K5V). Cluster IDs are indicated by arrows.
Finally, we evaluated the overlap of CA and OL on one side and the overlap of CA and AL on the other side. In fact, the CA distribution, via density analysis, revealed that CA are preferentially grouped in close proximity to DEF and substrate pockets. The clustering analysis goes further and suggests that CA can be located in the orthosteric site and in the six known allosteric sites, defined by the reference structures. Thus, considering CA positions can help to determine orthosteric cavity but more importantly allosteric sites in PKs. Moreover, both density and clustering analyses highlighted some new sites, for which allosteric properties were not yet studied and determined but could be targeted using de novo approaches for the design of novel allosteric ligands. However, it might be possible that those sites could be an attractive hotspot without any allosteric function. A validation experiment will be needed to assess the computational results and the allosteric regulation of these identified sites. Furthermore, because of the nature of CA (mainly hydrophilic), the detected cavities are hydrophilic, and hydrophobic cavities are missed with our approach.
Application of the CA Approach on NR Family
Considering the interesting results obtained for the PK family, our approach was extended to the NRs. These proteins play an important role in biological events such as cell growth or development and as a pathological regulator in many diseases. NRs share 3D structural motifs: the N-terminal activation function 1 domain (AF1, or A/B domain), the DNA binding domain including two Zn fingers (DBD, also called the C domain), the nuclear localization region (D domain), and the C-terminal ligand-binding domain (LBD, referred to the E domain).38 In general, NRs interact with ligands in an orthosteric pocket inside the LBD, and this interaction results to the cofactor binding regulation and a gene transcription regulation. However, some cavities, away from the orthosteric pocket, have been identified in several studies.39−41 Although these studies suggest that those sites are putative allosteric spots, no conclusion has been drawn on the nature of those cavities.42 Even if the nature of those sites is not yet fully characterized, we applied our approach on NRs to detect the known putative allosteric sites and to identify new ones. Using the same protocol as for PKs, 591 crystal structures were extracted for the CA dataset, and 450 crystal structures were extracted for the ligand dataset (containing also AL and OL). Density analysis with a threshold of 80% showed that CA have the tendency to interact in the AF2 coregulator site,42 a putative allosteric site near the helix H12 (Figure 5a).
Figure 5.

Allosteric sites identification on NR. (a) Superimposition of density points on the reference structure (PDB ID 2PIP) with the orthosteric site represented in green. All purple spheres represent a region with 50% density of presence, and only two spheres have a density greater than 80%. (b) Clustering results obtained with parameter [3 - 2]. (c) Some heterogeneous clusters superimposed on the reference structure indicate the position of defined allosteric ligands (PDB IDs 2PIP, top; 2PIN, middle) and orthosteric ligand (PDB ID 2PIP, bottom) represented in sticks. (d) Some homogeneous clusters superimposed on the reference structure show undefined allosteric sites. Orthosteric ligand (PDB ID 2PIP) is represented in sticks. Cluster ID and some protein helices are indicated.
This site was also detected using the clustering analysis. In fact, as shown in Figure 5c, heterogeneous clusters 5 and 6 are located in the same pocket than the allosteric reference ligand, which is usually associated with a thyroid hormone receptor (PDB ID 2PIN).43 A surface called binding function 3 or BF344 has been identified as a putative allosteric site in an NR-like androgen receptor.45 This site was exclusively found in cluster 9 using the clustering analysis. Thus, these first results confirm the existence of these two putative allosteric sites in NRs. Moreover, a density analysis at 50% threshold also revealed an additional cavity (Figure 5a) filled by homogeneous clusters 1, 2, and 4 constituted only by CA (Figure 5b,d). The homogeneous cluster 7 also points a surface-exposed cavity. These two cavities are still untargeted since no AL reference ligand was identified in NRs.
Regarding the NRs, the combination of both methods (density and clustering) showed that the CA are located into validated-known sites described in the literature. Consequently, our approach is not limited to PKs but can be successfully extended to other protein superfamilies like NRs. Considering the size of those superfamilies, further study could be performed on each different subfamily or subgroup.
Conclusions
Nowadays, allostery is a fundamental concept in protein regulation and reveals great interest to modulate the activity of a biological target in the context of drug discovery. Here, we proposed a novel and efficient computational way to detect allosteric sites, using crystallization additives (CA), instead of protein cavity volume and their overlap with orthosteric and allosteric ligands. CA are often present in 3D structures but generally unexploited in the Computational Methods. We put forward that CA are not randomly distributed around protein structures but seem to be attracted by hotspots. We demonstrate, with unsupervised classification and density analysis, that CA tend to be attracted by orthosteric and allosteric sites of protein kinases and nuclear receptors. Indeed, all those cavities of interest have been detected by one or both of those methods. This leads to the conclusion that the location of CA in crystal structures can be used to identify new cavities of interest and putative allosteric sites. Finally, this method has been effectively applied on two protein families and could be applied on other therapeutic targets for which new allosteric cavities are still unexplored. Thus, our method could be used for designing new allosteric drugs for the treatment of human diseases.
Computational Methods
General Process
Protein kinase (PK) and nuclear receptor (NR) families, for which allosteric modulators have been already described, were treated individually along the study following the procedure shown in Figure S1.
Datasets
Two structural datasets were generated for this study, one dataset containing the crystallographic additives (CA) in complex with the proteins (Figure S2) and the second dataset containing proteins cocrystallized with allosteric ligands (AL) and orthosteric ligands (OL). We described below the procedure to generate those datasets:
-
(1)
CA dataset contains molecules that are involved in the crystallographic process (polyols, sugars, and buffer molecules) excluding water molecules and salts. Crystal structures of protein–ligand complexes, with a resolution of less than 3 Å and with less than 10,000 residues, were retrieved from the RCSB protein data bank (PDB).46 The right protein family was selected using the annotations of the PFAM protein family database47 (PF00069 and PF07714 for protein kinases and PF00104 for nuclear receptors). PDB files containing multiple chains were split into individual chains, and each chain is considered as a unique entity. AL and OL were removed according to the presence of aromatic rings in the ligand structures. Then, crystal structures of apo or holo proteins containing CA compounds were conserved, resulting in 845 PK and 591 NR structures. The definition of the CA used in our approach is detailed in Table S1.
-
(2)
Ligand dataset contains AL and OL small molecule modulators. To ensure the presence of AL in this dataset, a search was made using different sources: the allosteric database (ASD),20 inventorying all protein structures containing AL until 2015, the ASbench,21 which provides a list of allosteric sites, and finally, a manual search in the PDB website using “allosteric” or “allostery” keywords. These crystal structures of protein–ligand complexes obtained from the PDB site are then filtered based on the structure resolution (<3 Å), on the number of residues in the protein sequence (<10,000 residues), and on their Pfam annotations. In this dataset, only protein–ligand complexes were kept, and other molecules such as solvent molecules, CA, and counter-ions were removed. This dataset contains 864 PK and 450 NR complexes. Among them, some complexes with well-studied AL were considered as a reference to validate the approach: PDB IDs 4E6C (p38α),483O2M (JNK1),493O96 (AKT1),503LW0 (IGF1R),511S9J (MEK),524LMN (MEK),533EQC (MEK),544ITH (RIPK1),554ZJI (PAK1),563MS9 (ABL),573K5V (ABL),583PYY (ABL),593PXF (CDK2),603HRF (PDK1),613JVR (CHK1),623F9N (CHK1),633H30 (CK2a1),64 and 4CFE (AMPKα1/2)65 (Table S2).
Sequence and Structure Alignment
For each dataset, protein sequences were annotated with conserved residues, and multiple sequence alignments were performed using the MOE software.66 Based on these sequence alignments, Cα atoms of each protein were superimposed to the Cα atoms of the chain E of PDB ID 1ATP(67) for all PKs and the chain A of PDB ID 1IE9(68) for all NRs.
Density Analysis
To evaluate the distribution of CA in protein families, we calculated the geometric center (centroid) of each CA molecule with the RDKit package.69 Because of the large differences in molecular size and chemical structure for the ligands in the datasets, we consider the centroid of the ligands for the analysis.70 Then, using the cpptraj program available in the AMBER suite, we generated a grid of density of centroids. Spacing (1 Å) between the bins of the grid is considered on the three coordinates of the box. The dimension of the box is considered to be 70 × 70 × 70 Å3 to encompass all the centroids of CA. The number of centroids is enumerated within a box, and a histogram of population is created. Only points that have a density greater than the 50 and 80% threshold, compared to the maximum value, were recorded for subsequent analysis.
Unsupervised Classification
Two datasets were studied for the unsupervised classification, the CA dataset alone and the combined CA and ligands (OL and AL) datasets to validate the overlap of CA and AL molecules in a proximal space. Each molecule (OL, AL, and CA) was also converted into one geometric point corresponding to their centroid using RDKit. We carried out multiple clustering based on the Cartesian centroid through the DBSCAN algorithm,35 implemented in the AMBER cpptraj module. Two parameters, the distance between two points in a cluster (ε in Å) and the minimal number of points present in a cluster (minpts), were modified for clustering optimization. So, different pairs of parameters [minpts - ε] were evaluated ([1 - 0.5], [1 - 1], [1 - 2], [2 – 0.5], [2 - 1], [2 - 2], [3 - 2], [3 - 3], [4 - 2], [4 - 3]).
Parameters [1 - 2] and [3 - 2], which provide a maximum number of clusters containing both AL and CA (heterogeneous cluster), were conserved for successive analysis. For PKs, a partial search using a pharmacophore was also applied on the combined datasets to detect and remove the OL bound to the hinge region of the ATP binding site using the MOE software.66 Based on the chain E of PDB ID 1ATP,67 we built the partial pharmacophoric feature on the adenosine moiety of ATP constituted by an acceptor feature (with a radius of 1.7 Å), a donor feature (1.7 Å), and a heavy atom (3 Å). Then, a second clustering using parameters [1 - 2] and [3 - 2] was applied without considering OL.
Acknowledgments
The authors wish to thank the Région Centre Val de Loire for financial support (project Drug4Arth). S.A.-S. and P.B. are supported by LABEX SynOrg (ANR-11-LABX-0029).
Glossary
ABBREVIATIONS
- NR
nuclear receptor
- PK
protein kinase
- CA
crystallization additives
- OL
orthosteric ligands
- AL
allosteric ligands
- PIF
PDK1 interacting fragment
- DEF
docking site for ERK, FXF
- FDA
Food and Drug Administration
- AF
activation function
- BF
binding function 3
- DBD
DNA binding domain
- LBD
ligand binding domain
- ATP
adenosine triphosphate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b02697.
Details of the general workflow (Figure S1), list of crystallographic additives (Table S1), dataset representation (Figure S2), cluster population (Figure S3), position of the reference ligands in clusters (Table S2), and histograms of the clusters (Figure S4) (PDF)
Author Present Address
± Present address: School of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife KY16 9ST, United Kingdom
Author Present Address
† Present address: Biologie intégrée du globule rouge, UMR_S 1134 Inserm - Université Paris 7, Paris Diderot, Institut National de la transfusion Sanguine, 6 rue Alexandre Cabanel, 75015 Paris Cedex 15, France
Author Contributions
‡ J.F. and J.D. contributed equally to the work.
The authors declare no competing financial interest.
Supplementary Material
References
- Wenthur C. J.; Gentry P. R.; Mathews T. P.; Lindsley C. W. Drugs for Allosteric Sites on Receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 165–184. 10.1146/annurev-pharmtox-010611-134525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Zhou H. X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 6503–6515. 10.1021/acs.chemrev.5b00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F.; Monod J. Genetic Regulatory Mechanisms in the-Synthesis. J. Mol. Biol. 1961, 3, 318–356. 10.1016/S0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- Monod J.; Jacob F. General Conclusions: Teleonomic Mechanisms in Cellular Metabolism, Growth, and Differentiation. Cold Spring Harbor Symp. Quant. Biol. 1961, 26, 389–401. 10.1101/SQB.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- Tsai C.-J.; Nussinov R. A Unified View of How Allostery Works. PLoS Comput. Biol. 2014, 10, e1003394 10.1371/journal.pcbi.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh H. N.; Wrabl J. O.; Li J.; Hilser V. J. The Ensemble Nature of Allostery. Nature 2014, 508, 331–339. 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suplatov D.; Švedas V. Study of Functional and Allosteric Sites in Protein Superfamilies. Acta Nat. 2015, 7, 34–54. 10.32607/20758251-2015-7-4-34-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrabl J. O.; Gu J.; Liu T.; Schrank T. P.; Whitten S. T.; Hilser V. J. The Role of Protein Conformational Fluctuations in Allostery, Function, and Evolution. Biophys. Chem. 2011, 159, 129–141. 10.1016/j.bpc.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peracchi A.; Mozzarelli A. Exploring and Exploiting Allostery: Models, Evolution, and Drug Targeting. Biochim. Biophys. Acta, Proteins Proteomics 2011, 922–933. 10.1016/j.bbapap.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Wild C.; Cunningham K. A.; Zhou J. Allosteric Modulation of G Protein-Coupled Receptors: An Emerging Approach of Drug Discovery. Austin J. Pharmacol. Ther. 2014, 2, 1101. [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A.; Diharce J.; Schröder M.; Foucourt A.; Leblond B.; Casagrande A.-S.; Désiré L.; Bonnet P.; Knapp S.; Besson T. An Unusual Binding Model of the Methyl 9-Anilinothiazolo[5,4-f] quinazoline-2-carbimidates (EHT 1610 and EHT 5372) Confers High Selectivity for Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases. J. Med. Chem. 2016, 59, 10315–10321. 10.1021/acs.jmedchem.6b01083. [DOI] [PubMed] [Google Scholar]
- Fabbro D.; Cowan-Jacob S. W.; Moebitz H. Ten Things You Should Know about Protein Kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. 10.1111/bph.13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carles F.; Bourg S.; Meyer C.; Bonnet P. PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules 2018, 23, 908. 10.3390/molecules23040908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott J. A.; Noble M. E. M.; Johnson L. N. The Structural Basis for Control of Eukaryotic Protein Kinases. Annu. Rev. Biochem. 2012, 81, 587–613. 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Fang Z.; Grütter C.; Rauh D. Strategies for the Selective Regulation of Kinases with Allosteric Modulators: Exploiting Exclusive Structural Features. ACS Chem. Biol. 2013, 8, 58–70. 10.1021/cb300663j. [DOI] [PubMed] [Google Scholar]
- Mobitz H.; Jahnke W.; Cowan-Jacob S. Expanding the Opportunities for Modulating Kinase Targets with Allosteric Approaches. Curr. Top. Med. Chem. 2017, 17, 59–70. 10.2174/1568026616666160719165314. [DOI] [PubMed] [Google Scholar]
- Wu P.; Nielsen T. E.; Clausen M. H. FDA-Approved Small-Molecule Kinase Inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Rivat C.; Sar C.; Mechaly I.; Leyris J.-P.; Diouloufet L.; Sonrier C.; Philipson Y.; Lucas O.; Mallié S.; Jouvenel A.; et al. Inhibition of Neuronal FLT3 Receptor Tyrosine Kinase Alleviates Peripheral Neuropathic Pain in Mice. Nat. Commun. 2018, 9, 1042. 10.1038/s41467-018-03496-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestgen B.; Krimm I.; Kufareva I.; Kamal A. A. M.; Seetoh W.-G.; Abell C.; Hartmann R. W.; Abagyan R.; Cochet C.; Le Borgne M.; et al. 2-Aminothiazole Derivatives as Selective Allosteric Modulators of the Protein Kinase CK2. 1. Identification of an Allosteric Binding Site. J. Med. Chem. 2019, 62, 1803–1816. 10.1021/acs.jmedchem.8b01766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Zhu L.; Cao Y.; Wu G.; Liu X.; Chen Y.; Wang Q.; Shi T.; Zhao Y.; Wang Y.; et al. ASD: A Comprehensive Database of Allosteric Proteins and Modulators. Nucleic Acids Res. 2011, 39, D663–D669. 10.1093/nar/gkq1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Wang G.; Shen Q.; Liu X.; Lu S.; Geng L.; Huang Z.; Zhang J. ASBench: Benchmarking Sets for Allosteric Discovery: Fig. 1. Bioinformatics 2015, 31, 2598–2600. 10.1093/bioinformatics/btv169. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Wang S.; Hu Q.; Gao S.; Ma X.; Zhang W.; Shen Y.; Chen F.; Lai L.; Pei J. CavityPlus: A Web Server for Protein Cavity Detection with Pharmacophore Modelling, Allosteric Site Identification and Covalent Ligand Binding Ability Prediction. Nucleic Acids Res. 2018, 46, W374–W379. 10.1093/nar/gky380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X.; Meng H.; Lai L. Motions of Allosteric and Orthosteric Ligand-Binding Sites in Proteins Are Highly Correlated. J. Chem. Inf. Model. 2016, 56, 1725–1733. 10.1021/acs.jcim.6b00039. [DOI] [PubMed] [Google Scholar]
- Goncearenco A.; Mitternacht S.; Yong T.; Eisenhaber B.; Eisenhaber F.; Berezovsky I. N. SPACER: Server for Predicting Allosteric Communication and Effects of Regulation. Nucleic Acids Res. 2013, 41, W266–W272. 10.1093/nar/gkt460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K. N.; Bellamacina C. R.; Ding X.; Jeffery C. J.; Mattos C.; Petsko G. A.; Ringe D. An Experimental Approach to Mapping the Binding Surfaces of Crystalline Proteins †. J. Phys. Chem. 1996, 100, 2605–2611. 10.1021/jp952516o. [DOI] [Google Scholar]
- Vera L.; Czarny B.; Georgiadis D.; Dive V.; Stura E. A. Practical Use of Glycerol in Protein Crystallization. Cryst. Growth Des. 2011, 11, 2755–2762. 10.1021/cg101364m. [DOI] [Google Scholar]
- Drwal M. N.; Jacquemard C.; Perez C.; Desaphy J.; Kellenberger E. Do Fragments and Crystallization Additives Bind Similarly to Drug-like Ligands?. J. Chem. Inf. Model. 2017, 57, 1197–1209. 10.1021/acs.jcim.6b00769. [DOI] [PubMed] [Google Scholar]
- Drwal M. N.; Bret G.; Perez C.; Jacquemard C.; Desaphy J.; Kellenberger E. Structural Insights on Fragment Binding Mode Conservation. J. Med. Chem. 2018, 61, 5963–5973. 10.1021/acs.jmedchem.8b00256. [DOI] [PubMed] [Google Scholar]
- English A. C.; Groom C. R.; Hubbard R. E. Experimental and Computational Mapping of the Binding Surface of a Crystalline Protein. Protein Eng., Des. Sel. 2001, 14, 47–59. 10.1093/protein/14.1.47. [DOI] [PubMed] [Google Scholar]
- Liepinsh E.; Otting G. Organic Solvents Identify Specific Ligand Binding Sites on Protein Surfaces. Nat. Biotechnol. 1997, 15, 264–268. 10.1038/nbt0397-264. [DOI] [PubMed] [Google Scholar]
- Bosc N.; Wroblowski B.; Aci-Sèche S.; Meyer C.; Bonnet P. A Proteometric Analysis of Human Kinome: Insight into Discriminant Conformation-Dependent Residues. ACS Chem. Biol. 2015, 10, 2827–2840. 10.1021/acschembio.5b00555. [DOI] [PubMed] [Google Scholar]
- Tzarum N.; Komornik N.; Ben Chetrit D.; Engelberg D.; Livnah O. DEF Pocket in P38α Facilitates Substrate Selectivity and Mediates Autophosphorylation. J. Biol. Chem. 2013, 288, 19537–19547. 10.1074/jbc.M113.464511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowsky J. D.; Burlingame M. A.; Wolan D. W.; McClendon C. L.; Jacobson M. P.; Wells J. A. Turning a Protein Kinase on or off from a Single Allosteric Site via Disulfide Trapping. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6056–6061. 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo Y.-S.; Kim S.-K.; Seo C. I.; Kim Y. K.; Sung B.-J.; Lee H. S.; Lee J. I.; Park S.-Y.; Kim J. H.; Hwang K. Y.; et al. Structural Basis for the Selective Inhibition of JNK1 by the Scaffolding Protein JIP1 and SP600125. EMBO J. 2004, 23, 2185–2195. 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester M.; Kriegel H.-P.; Sander J.; Xu X.. A Density-Based Algorithm for Discovering Clusters a Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining; AAAI Press: 1996, 226–231.
- McIntyre P. J.; Collins P. M.; Vrzal L.; Birchall K.; Arnold L. H.; Mpamhanga C.; Coombs P. J.; Burgess S. G.; Richards M. W.; Winter A.; et al. Characterization of Three Druggable Hot-Spots in the Aurora-A/TPX2 Interaction Using Biochemical, Biophysical, and Fragment-Based Approaches. ACS Chem. Biol. 2017, 12, 2906–2914. 10.1021/acschembio.7b00537. [DOI] [PubMed] [Google Scholar]
- Chen L. S.; Redkar S.; Bearss D.; Wierda W. G.; Gandhi V. Pim Kinase Inhibitor, SGI-1776, Induces Apoptosis in Chronic Lymphocytic Leukemia Cells. Blood 2009, 114, 4150–4157. 10.1182/blood-2009-03-212852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billas I.; Moras D. Allosteric Controls of Nuclear Receptor Function in the Regulation of Transcription. J. Mol. Biol. 2013, 425, 2317–2329. 10.1016/j.jmb.2013.03.017. [DOI] [PubMed] [Google Scholar]
- Hughes T. S.; Giri P. K.; de Vera I. M. S.; Marciano D. P.; Kuruvilla D. S.; Shin Y.; Blayo A.-L.; Kamenecka T. M.; Burris T. P.; Griffin P. R.; et al. An Alternate Binding Site for PPARγ Ligands. Nat. Commun. 2014, 5, 3571. 10.1038/ncomms4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris T.; Solt L.; Wang Y.; Crumbley C.; Banerjee S.; Griffett K.; Lundasen T.; Hughes T.; Kojetin D. Nuclear Receptors and Their Selective Pharmacologic Modulators. Pharmacol. Rev. 2013, 65, 710–778. 10.1124/pr.112.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T. W.; Mayne C. G.; Katzenellenbogen J. A. Minireview: Not Picking Pockets: Nuclear Receptor Alternate-Site Modulators (NRAMs). Mol. Endocrinol. 2010, 24, 683–695. 10.1210/me.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux J.-P.; Christopoulos A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 2016, 166, 1084–1102. 10.1016/j.cell.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Estébanez-Perpiñá E.; Arnold L. A.; Jouravel N.; Togashi M.; Blethrow J.; Mar E.; Nguyen P.; Phillips K. J.; Baxter J. D.; Webb P.; et al. Structural Insight into the Mode of Action of a Direct Inhibitor of Coregulator Binding to the Thyroid Hormone Receptor. Mol. Endocrinol. 2007, 21, 2919–2928. 10.1210/me.2007-0174. [DOI] [PubMed] [Google Scholar]
- Buzón V.; Carbó L. R.; Estruch S. B.; Fletterick R. J.; Estébanez-Perpiñá E. A Conserved Surface on the Ligand Binding Domain of Nuclear Receptors for Allosteric Control. Mol. Cell. Endocrinol. 2012, 348, 394–402. 10.1016/j.mce.2011.08.012. [DOI] [PubMed] [Google Scholar]
- Estébanez-Perpiñá E.; Arnold L. A.; Nguyen P.; Rodrigues E. D.; Mar E.; Bateman R.; Pallai P.; Shokat K. M.; Baxter J. D.; et al. A Surface on the Androgen Receptor That Allosterically Regulates Coactivator Binding. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16074–16079. 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D.; Bateman A.; Clements J.; Coggill P.; Eberhardt R. Y.; Eddy S. R.; Heger A.; Hetherington K.; Holm L.; Mistry J.; et al. Pfam: The Protein Families Database. Nucleic Acids Res. 2014, 42, D222–D230. 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzarum N.; Eisenberg-Domovich Y.; Gills J. J.; Dennis P. A.; Livnah O. Lipid Molecules Induce P38α Activation via a Novel Molecular Switch. J. Mol. Biol. 2012, 424, 339–353. 10.1016/j.jmb.2012.10.007. [DOI] [PubMed] [Google Scholar]
- Comess K. M.; Sun C.; Abad-Zapatero C.; Goedken E. R.; Gum R. J.; Borhani D. W.; Argiriadi M.; Groebe D. R.; Jia Y.; Clampit J. E.; et al. Discovery and Characterization of Non-ATP Site Inhibitors of the Mitogen Activated Protein (MAP) Kinases. ACS Chem. Biol. 2011, 6, 234–244. 10.1021/cb1002619. [DOI] [PubMed] [Google Scholar]
- Wu W.-I.; Voegtli W. C.; Sturgis H. L.; Dizon F. P.; Vigers G. P. A.; Brandhuber B. J. Crystal Structure of Human AKT1 with an Allosteric Inhibitor Reveals a New Mode of Kinase Inhibition. PLoS One 2010, 5, e12913 10.1371/journal.pone.0012913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich T.; Grädler U.; Böttcher H.; Blaukat A.; Shutes A. Allosteric IGF-1R Inhibitors. ACS Med. Chem. Lett. 2010, 1, 199–203. 10.1021/ml100044h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohren J. F.; Chen H.; Pavlovsky A.; Whitehead C.; Zhang E.; Kuffa P.; Yan C.; McConnell P.; Spessard C.; Banotai C.; et al. Structures of Human MAP Kinase Kinase 1 (MEK1) and MEK2 Describe Novel Noncompetitive Kinase Inhibition. Nat. Struct. Mol. Biol. 2004, 11, 1192–1197. 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G.; Haling J. R.; Chen H.; Song K.; Price S.; Heald R.; Hewitt J. F. M.; Zak M.; Peck A.; Orr C.; et al. Mechanism of MEK Inhibition Determines Efficacy in Mutant KRAS- versus BRAF-Driven Cancers. Nature 2013, 501, 232–236. 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- Fischmann T. O.; Smith C. K.; Mayhood T. W.; Myers J. E.; Reichert P.; Mannarino A.; Carr D.; Zhu H.; Wong J.; Yang R. S.; et al. Crystal Structures of MEK1 Binary and Ternary Complexes with Nucleotides and Inhibitors. Biochemistry 2009, 48, 2661–2674. 10.1021/bi801898e. [DOI] [PubMed] [Google Scholar]
- Xie T.; Peng W.; Liu Y.; Yan C.; Maki J.; Degterev A.; Yuan J.; Shi Y. Structural Basis of RIP1 Inhibition by Necrostatins. Structure 2013, 21, 493–499. 10.1016/j.str.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Karpov A. S.; Amiri P.; Bellamacina C.; Bellance M. H.; Breitenstein W.; Daniel D.; Denay R.; Fabbro D.; Fernandez C.; Galuba I.; et al. Optimization of a Dibenzodiazepine Hit to a Potent and Selective Allosteric PAK1 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 776–781. 10.1021/acsmedchemlett.5b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnke W.; Grotzfeld R. M.; Pellé X.; Strauss A.; Fendrich G.; Cowan-Jacob S. W.; Cotesta S.; Fabbro D.; Furet P.; Mestan J.; et al. Binding or Bending: Distinction of Allosteric Abl Kinase Agonists from Antagonists by an NMR-Based Conformational Assay. J. Am. Chem. Soc. 2010, 132, 7043–7048. 10.1021/ja101837n. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Adrián F. J.; Jahnke W.; Cowan-Jacob S. W.; Li A. G.; Iacob R. E.; Sim T.; Powers J.; Dierks C.; Sun F.; et al. Targeting Bcr-Abl by Combining Allosteric with ATP-Binding-Site Inhibitors. Nature 2010, 463, 501–506. 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Campobasso N.; Biju M. P.; Fisher K.; Pan X. Q.; Cottom J.; Galbraith S.; Ho T.; Zhang H.; Hong X.; et al. Discovery and Characterization of a Cell-Permeable, Small-Molecule c-Abl Kinase Activator That Binds to the Myristoyl Binding Site. Chem. Biol. 2011, 18, 177–186. 10.1016/j.chembiol.2010.12.013. [DOI] [PubMed] [Google Scholar]
- Betzi S.; Alam R.; Martin M.; Lubbers D. J.; Han H.; Jakkaraj S. R.; Georg G. I.; Schönbrunn E. Discovery of a Potential Allosteric Ligand Binding Site in CDK2. ACS Chem. Biol. 2011, 6, 492–501. 10.1021/cb100410m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindie V.; Stroba A.; Zhang H.; Lopez-Garcia L. A.; Idrissova L.; Zeuzem S.; Hirschberg D.; Schaeffer F.; Jørgensen T. J. D.; Engel M.; et al. Structure and Allosteric Effects of Low-Molecular-Weight Activators on the Protein Kinase PDK1. Nat. Chem. Biol. 2009, 5, 758–764. 10.1038/nchembio.208. [DOI] [PubMed] [Google Scholar]
- Vanderpool D.; Johnson T. O.; Ping C.; Bergqvist S.; Alton G.; Phonephaly S.; Rui E.; Luo C.; Deng Y. L.; Grant S.; et al. Characterization of the CHK1 Allosteric Inhibitor Binding Site. Biochemistry 2009, 48, 9823–9830. 10.1021/bi900258v. [DOI] [PubMed] [Google Scholar]
- Converso A.; Hartingh T.; Garbaccio R. M.; Tasber E.; Rickert K.; Fraley M. E.; Yan Y.; Kreatsoulas C.; Stirdivant S.; Drakas B.; et al. Development of Thioquinazolinones, Allosteric Chk1 Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1240–1244. 10.1016/j.bmcl.2008.12.076. [DOI] [PubMed] [Google Scholar]
- Raaf J.; Brunstein E.; Issinger O.-G.; Niefind K. The CK2α/CK2β Interface of Human Protein Kinase CK2 Harbors a Binding Pocket for Small Molecules. Chem. Biol. 2008, 15, 111–117. 10.1016/j.chembiol.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Xiao B.; Sanders M. J.; Carmena D.; Bright N. J.; Haire L. F.; Underwood E.; Patel B. R.; Heath R. B.; Walker P. A.; Hallen S.; et al. Structural Basis of Ampk Regulation by Small Molecule Activators. Nat. Commun. 2013, 4, 3017. 10.1038/ncomms4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2014.09; Chemical Computing Group Inc. 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. Chemical Computing Group Inc. 2014.
- Zheng J.; Trafny E. A.; Knighton D. R.; Xuong N.; Taylor S. S.; Ten Eyck L. F.; Sowadski J. M. 2.2 Å Refined Crystal Structure of the Catalytic Subunit of CAMP-Dependent Protein Kinase Complexed with MnATP and a Peptide Inhibitor. Acta Cryst. 1993, 49, 362–365. 10.1107/S0907444993000423. [DOI] [PubMed] [Google Scholar]
- Tocchini-Valentini G.; Rochel N.; Wurtz J. M.; Mitschler A.; Moras D. Crystal Structures of the Vitamin D Receptor Complexed to Superagonist 20-Epi Ligands. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 5491–5496. 10.1073/pnas.091018698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RDKit, Open-Source Cheminformatics. Http://Www.Rdkit.Org.
- AMBER; University of California: San Francisco, 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.