

Abstract
Carcinogenic aromatic amines such as 4-aminobiphenyl (ABP) and 2-aminofluorene (AF) require metabolic activation to form electrophilic intermediates that mutate DNA leading to carcinogenesis. Bioactivation of these carcinogens includes N-hydroxylation catalyzed by CYP1A2 followed by O-acetylation catalyzed by arylamine N-acetyltransferase 2 (NAT2). To better understand the role of NAT2 genetic polymorphism in ABP- and AF-induced mutagenesis and DNA damage, nucleotide excision repair-deficient (UV5) Chinese hamster ovary (CHO) cells were stably transfected with human CYP1A2 and either NAT2*4 (rapid acetylator) or NAT2*5B (slow acetylator) alleles. ABP and AF both caused significantly (p<0.001) greater mutagenesis measured at the hypoxanthine phosphoribosyl transferase (hprt) locus in the UV5/CYP1A2/NAT2*4 acetylator cell line compared to the UV5, UV5/CYP1A2, and UV5/CYP1A2/NAT2*5B cell lines. ABP- and AF-induced hprt mutant cDNAs were sequenced and over 80% of the single-base substitutions were at G:C base pairs. DNA damage also was quantified by γH2AX in-cell western assays and by identification and quantification of the two predominant DNA adducts, N-(deoxyguanosin-8-yl)-4-aminobiphenyl (dG-C8-ABP) and N-(deoxyguanosin-8-yl)-2-aminofluorene (dG-C8-AF) by liquid chromatography-mass spectrometry. DNA damage and adduct levels were dose-dependent, correlated highly with levels of hprt mutants, and were significantly (p<0.0001) greater in the UV5/CYP1A2/NAT2*4 rapid acetylator cell line following treatment with ABP or AF as compared to all other cell lines. Our findings provide further clarity on the importance of O-acetylation in CHO mutagenesis assays for aromatic amines. They provide evidence that NAT2 genetic polymorphism modifies aromatic amine-induced DNA damage and mutagenesis that should be considered in human risk assessments following aromatic amine exposures.
Keywords: N-acetyltransferase 2, genetic polymorphism, 4-aminobiphenyl, 2-aminofluorene
INTRODUCTION
As recently reviewed [Wang et al., 2019], more than one in eight known or suspected human carcinogens is an aromatic amine, or a chemical that can convert into one, making them a major class of human carcinogens. Two aromatic amines, 4-aminobiphenyl (ABP) and 2-aminofluorene (AF) are frequently the subject of laboratory research investigations. ABP is a known human carcinogen present in tobacco smoke and dye products whereas N-acetylaminofluorene (a metabolite of AF) is reasonably anticipated to be a human carcinogen [National Toxicology Program, 2016].
Aromatic amine-induced DNA adduct formation and mutagenesis require metabolic activation, and the primary pathway includes N-hydroxylation catalyzed by hepatic CYP1A2 [Schut and Snyderwine, 1999]. The N-hydroxy metabolites formed are weakly reactive but are more likely to undergo further activation catalyzed by phase II enzymes [Snyderwine et al., 2002]. Arylamine N-acetyltransferase 2 (NAT2) activates N-hydroxy metabolites by O-acetylation to N-acetoxy metabolites which break down to electrophilic intermediates that form DNA adducts [Snyderwine, 2002]. In contrast, NAT2-catalyzed N-acetylation of aromatic amines competes with N-hydroxylation and in this role is considered a detoxification pathway.
ABP- and AF-induced DNA-adducts primarily form at the C8 position of deoxyguanosine [Heflich and Neft, 1994; Chen et al., 2018]. Bulky DNA adducts such as these are recognized by the nucleotide excision repair (NER) pathway. Since DNA damage and mutagenesis are a function of both environmental exposure and carcinogen metabolism, they are informative biomarkers to investigate genetic variation in carcinogen metabolism. N-hydroxy-ABP mutagenesis at the hprt and TK loci, and dG-C8-ABP DNA adduct formation have been investigated in human lymphoblastoid cells [Ricicki et al., 2005]. The metabolic activation of AF has been investigated in V79 Chinese hamster cells expressing human CYP1A2 and NAT2 alleles [Scheuenpflug et al., 2005], as well as other strains of CHO cells, by laboratories investigating both mutagenesis and DNA adduct formation [Heflich et al., 1988; Nairn et al., 1988].
Since aromatic amines require metabolic activation to exert their carcinogenic effects, genetic polymorphisms in carcinogen-metabolizing enzymes such as NAT2 may modify cancer risk following exposure [Hein, 2002]. Humans exhibit genetic polymorphism in NAT2 resulting in rapid and slow acetylator phenotypes. The NAT2*4 allele is associated with rapid acetylator phenotype whereas the NAT2*5B allele is associated with slow acetylator phenotype. Higher levels of ABP-DNA adducts have been reported in human breast [Ambrosone et al., 2007] from rapid versus slow NAT2 acetylators, whereas ABP-hemoglobin adducts are higher in slow acetylators [Bartsch et al, 1990]. Epidemiological studies suggest a role for NAT2 genetic polymorphism in susceptibility to various cancers [Hein et al, 2000]. However, laboratory-based experiments are required to support the biological plausibility and the conclusions inferred from these epidemiological studies.
Our laboratory has reported on previous investigations in which DNA repair-deficient CHO cells transfected with CYP1A1 or CYP1A2 and NAT2*4 or NAT2*5B were incubated with heterocyclic amine carcinogens such as 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine (PhIP), 2-amino-3-methylimidazo [4,5-f] quinoline (IQ), and 2-amino-3,8-dimethylimidazo-[4,5-f]quinoxaline (MeIQx). The results differed with the heterocyclic amine carcinogen tested, as rapid acetylator NAT2*4 did not affect levels of mutagenesis and DNA adduct formation following incubation with PhIP [Metry et al., 2007] whereas both mutagenesis and DNA adduct formation were markedly higher in CHO cells possessing rapid acetylator NAT2*4 following incubation with IQ or MeIQx [Metry et al., 2010]. The conclusion that NAT2 is necessary for the DNA damage and mutagenesis induced by MeIQx and IQ but not PhIP was recently confirmed in V79 Chinese hamster cells [Chevereau et al., 2017]. Our laboratory also reported higher levels of ABP-induced hprt mutants and DNA adducts in DNA repair-deficient CHO cells transfected with CYP1A1 and NAT2*4 [Bendaly et al., 2009]. However, a study in V79 Chinese hamster cells with heterologous co-expression of human CYP1A2 and NAT2*4 found lower levels of AF-induced mutants in cells transfected with both CYP1A2 and NAT2*4 compared to transfection with CYP1A2 alone [Scheuenpflug et al., 2005] suggesting that the findings might differ with aromatic amine (ABP vs. AF) or cytochrome P450 (CYP1A1 vs. CYP1A2).
To better understand the role of CYP1A2 and NAT2 in ABP- and AF- induced mutagenesis and DNA damage, NER-deficient CHO cells were stably transfected with human CYP1A2 and either NAT2*4 (rapid acetylator) or NAT2*5B (slow acetylator) alleles. ABP- and AF-induced mutagenesis was measured at the hypoxanthine phosphoribosyl transferase (hprt) locus. DNA damage was quantified by γH2AX in-cell western assays and by identification and quantification of the two predominant DNA adducts, N-(deoxyguanosin-8-yl)-4-aminobiphenyl (dG-C8-ABP) and N-(deoxyguanosin-8-yl)-2-aminofluorene (dG-C8-AF).
MATERIALS AND METHODS
Construction of transformed UV5-CHO cell lines
The UV5-CHO cell line, a NER-deficient derivative of the AA8 line [Thompson et al., 1980], was obtained from the ATCC (Catalog number: CRL-1865). Since UV5-CHO lacks NER due to a mutation in the XPD (ERCC2) gene [Weber et al., 1988], it is hypersensitive to bulky adduct mutagens and belongs to the excision repair cross complementation group 2. All cells were grown in alpha-modified minimal essential medium (Cambrex) without L-glutamine, ribosides, and deoxyribosides, supplemented with 10% fetal bovine serum (Hyclone), 100 units/ml penicillin, 100 units/ml streptomycin (Cambrex), and 2 mM L-glutamine (Cambrex) at 37 °C in 5% CO2. Media were supplemented with appropriate selective agents to maintain stable transfectants.
Construction and characterization of the UV5-CHO cell lines stably transfected with CYP1A2 only (UV5/CYP1A2), CYP1A2 plus NAT2*4 (rapid acetylator) (UV5/CYP1A2/NAT2*4), and CYP1A2 plus NAT2*5B (slow acetylator) (UV5/CYP1A2/NAT2*5B) alleles has been described in detail previously [Metry et al., 2007].
N-Acetyltransferase assays
N-acetyltransferase activities for ABP and AF were measured by separation of parent and their N-acetylated metabolite via high performance liquid chromatography (HPLC) as previously described [Leff et al., 1999; Fretland et al., 2002]. In order to isolate cell cytosols, 1 x 106 cells were seeded into 10 cm culture dishes and incubated for 48 hr. The cells were trypsinized and collected. After washing once with PBS, the cells were disrupted by three cycles of freezing and thawing in lysis buffer, and centrifuged for 10 min at 10,000 x g. Lysis buffer consisted of 20 mM sodium phosphate buffer, pH 7.4, containing 1 mM EDTA, 1 mM dithiothreitol, 100 μM phenylmethylsulphonylfluoride (PMSF), 1 μM pepstatin A, and 1 μg/ml aprotinin. Supernatants were collected and used immediately or stored at − 80°C until used. Cell lysates were incubated with 300 μM ABP or AF and 1 mM acetyl-coenzyme A at 37 °C for 10 min. Reactions were terminated by the addition of 1 M perchloric acid. Arylamine and N-acetylated products were separated with a mobile phase of 20 mM sodium perchlorate (pH 2.5) and acetonitrile at a ratio of 91:9. Protein was quantified using the Bio-Rad protein assay kit (Bio-Rad).
Cell survival and hprt mutations
Assays for cell survival and mutagenesis were modified slightly from methods previously described [Wu et al., 1997; Metry et al., 2007] . Briefly, cells were grown for 12 doublings, with selective agents in complete HAT medium (30 μM hypoxanthine, 0.1 μM aminopterin, and 30 μM thymidine). Cells were plated at a density of 5 x 105 cells/T-25 flask and incubated for 24 hr, after which media were changed and the cells treated for 48 hr with various concentrations of AF or ABP dissolved in DMSO or vehicle control (0.5% DMSO). Survival was determined by a colony forming assay and expressed as percent of vehicle control. The remaining cells were replated and subcultured for 7 days of growth. Then, cells were plated for cloning efficiency in complete media and for hprt mutations in complete medium containing 40 μM 6-thioguanine (6-TG) (Sigma, St. Louis, MO). Dishes were seeded with 1 x 105 cells/100 mm dish (10 replicates) and incubated for 7 days in 6-TG media; cloning efficiency dishes were seeded with 100 cells/well/6-well plate in triplicate and incubated for 6 days.
Sequencing of hprt mutants
Sequencing of hprt mutants following incubation with ABP or AF was carried out as previously described [Metry et al., 2010]. Briefly, one viable colony was picked from each 6-TG containing culture dish of 2.0 μM ABP-treated or 1.0 μM AF-treated UV5/CYP1A2/NAT2*4 cells and propagated until one confluent 10-cm dish was obtained. The cells were harvested and pelleted. RNA was extracted immediately using the RNeasy Mini Kit (Qiagen) or pellets were snap-frozen in liquid nitrogen and stored at −80 °C for later use. RNA (1 μg) was reverse transcribed with oligo(dT) primer from the SuperScript III First-Strand Synthesis System (Invitrogen) as described by the manufacturer. The hprt coding region was amplified and the PCR product was purified and sequenced as previously described [Metry et al., 2010].
Identification and quantification of DNA adducts
Cells grown in 15-cm plates were treated with ABP or AF as described above for the cytotoxicity and mutagenesis assays. Cells were harvested after 48 hr of treatment, centrifuged, and the pellet resuspended in 500 μl of 50 mM Tris-HCl, 10 mM EDTA, pH 8.0. DNA samples (200 μg) from ABP- or AF- treated cells were digested and prepared for LC-MS-MS analysis as previously described [Metry et al., 2007]. Samples used for quantitative analysis of N-(deoxyguanosin-8-yl)-4-aminobiphenyl (dG-C8-ABP; Toronto Research Chemicals) were spiked with one ng deuterated internal standard dG-C8-ABP-d5 (Toronto Research Chemicals; >97% purity by NMR spectroscopic analysis, 97.5% pure by mass spectral analysis) before any sample treatment. N-(deoxyguanosin-8-yl)-2-aminofluorene (dG-C8-AF) was generously provided by Dr. Fred Beland from the National Center for Toxicological Research, Jefferson, Arkansas. Samples used for quantitative analysis of dG-C8-AF were spiked with one ng of dG-C8-ABP (97.5% pure by mass spectral analysis) as the type C internal standard. HPLC conditions and MS analysis parameters were set as previously described [Metry et al., 2007], with a few modifications. Electrospray ionization was set at 2.5 kV, cone voltages were 25 V and the collision energy was 25 V for both dG-C8-ABP and dG-C8-AF. Argon was used as the collision gas and set at 2.0e−4 mBar. Collision energy was 20 V set to minimize fragmentation and to cleave the glycosidic bond, producing one major fragment, which was [(M-116) + H]+ for each adduct.
The dG-C8-ABP adduct was monitored using the multiple reaction monitoring (MRM) transition from m/z 435 to 319 m/z and the deuterated internal standard (dG-C8-ABP-d5) was monitored using the transition from m/z 440 to m/z 324 as previously described [Neale et al., 2008]. Similarly, the dG-C8-AF adduct was monitored using the MRM transition from m/z 446 to m/z 330 with the internal standard (dG-C8-ABP) using the transition from m/z 435 to m/z 319.
Quantification of DNA damage
DNA damage was assessed by a γH2AX in-cell western staining protocol as previously described [Audebert et al., 2010]. Cells were grown with selective agents in 10-cm plates. Then 1x105 cells were plated into black/clear bottom 96-well plates (Corning, Corning, NY, USA) and allowed to attach overnight. The next morning media was removed (cell debris and non-adherent cells were washed away and removed) and attached cells washed with PBS and replaced with fresh pre-warmed media containing 0.01–100 μM ABP or AF. Cells were incubated for up to 24 hr, after which media were removed and γH2AX in-cell western staining protocol was performed as follows; cells were fixed to the plate using 3.7% formaldehyde and incubating at room temperature for 20 min. Then, the cells were permeabilized by washing five times with 0.1% Triton X-100 in TBS. After permeabilization, the cells were blocked using FISH Gelatin Blocking Agent (Biotium, Fremont, CA, USA) diluted in TBS for 90 min at room temperature with constant agitation. Primary antibody anti-phospho-histone H2AX (Millipore-Sigma, Burlington, MA, USA) was diluted to 2 μg/mL and added to the cells and then incubated overnight at 4°C. The next morning cells were washed with 0.1% Tween 20 in TBS for 5 min, five times. Secondary antibody IRDye® 800CW goat anti-mouse IgG (H+L) (LI-COR, Lincoln, NE, USA) was used at a 1:1000 dilution and DNA dye RedDot™ 2 diluted to 1X (Biotium, Fremont, CA, USA) to normalize for DNA content. Cells were incubated with this combination for 60 min and washed again with the Tween 20 solution as previously described. DNA and the γH2AX were simultaneously visualized using an Odyssey CLx imaging system (LI-COR, Lincoln, NE, USA) with the 680 nm fluorophore (red) and the 800 nm fluorophore (green). Relative fluorescence units from the scanning allowed a quantitative analysis. Relative fluorescent units for γH2AX per cell (as determined by γH2AX divided by DNA content) were divided by untreated cells to determine percent change in phosphorylation of H2AX levels relative to control.
Statistical analyses
Statistical analyses were performed using GraphPad Prism (La Jolla, CA). Differences between measurements in the UV5, UV5/CYP1A2, UV5/CYP1A2/NAT2*5B and UV5/CYP1A2/NAT2*4 cell lines were analyzed for significance by two-way ANOVA followed by Tukey’s multiple comparison tests. Correlations between levels of ABP- and AF-induced DNA adducts and mutants in the UV5, UV5/CYP1A2, UV5/CYP1A2/NAT2*5B and UV5/CYP1A2/NAT2*4 cell lines were analyzed by the Pearson correlation coefficient. Differences or correlations were considered significant if p < 0.05.
RESULTS
N-acetylation capacity in NAT2-transfected cells
ABP and AF N-acetyltransferase activity was measured and found to be greater in the UV5/CYP1A2/NAT2*4 rapid acetylator cell line than the UV5/CYP1A2/NAT2*5B slow acetylator cell line (Figure 1). ABP and AF N-acetyltransferase activity in the UV5 and UV5/CYP1A2 cell lines were below the level of detection (< 20 pmol/min/mg).
Figure 1.
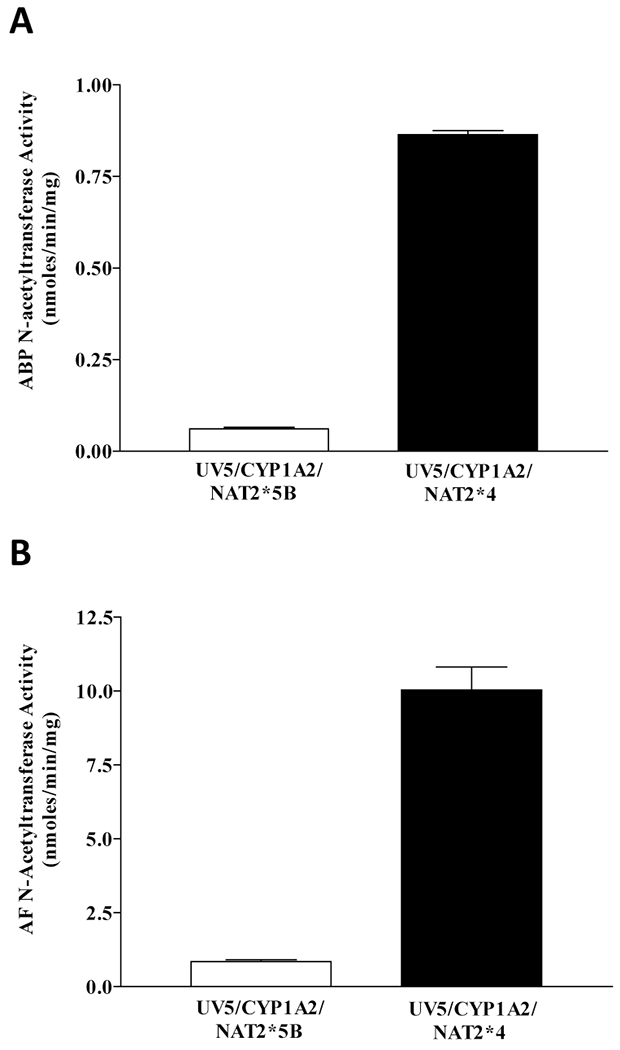
ABP and AF NAT activity in UV5/CYP1A2/NAT2*5B or UV5/CYP1A2/NAT2*4 cells. Each bar represents Mean ± SEM for three independent experiments. A: The difference in ABP NAT activity between the CYP1A2/NAT2*5B and the CYP1A2/NAT2*4 CHO cell lines was significant (p < 0.0001). B: The difference in AF NAT activity between the CYP1A2/NAT2*5B and the CYP1A2/NAT2*4 CHO cell lines was significant (p = 0.0003).
Aromatic amine-induced cytotoxicity and mutagenesis
Only the UV5/CYP1A2/NAT2*4 cells showed concentration-dependent cytotoxicity and hprt mutagenesis following incubation with ABP or AF (Figure 2). There were significantly more ABP- and AF-induced hprt mutants in the UV5/CYP1A2/NAT2*4 rapid acetylator cell line (p < 0.001) than in all other cell lines for all concentrations tested (Figure 2).
Figure 2.
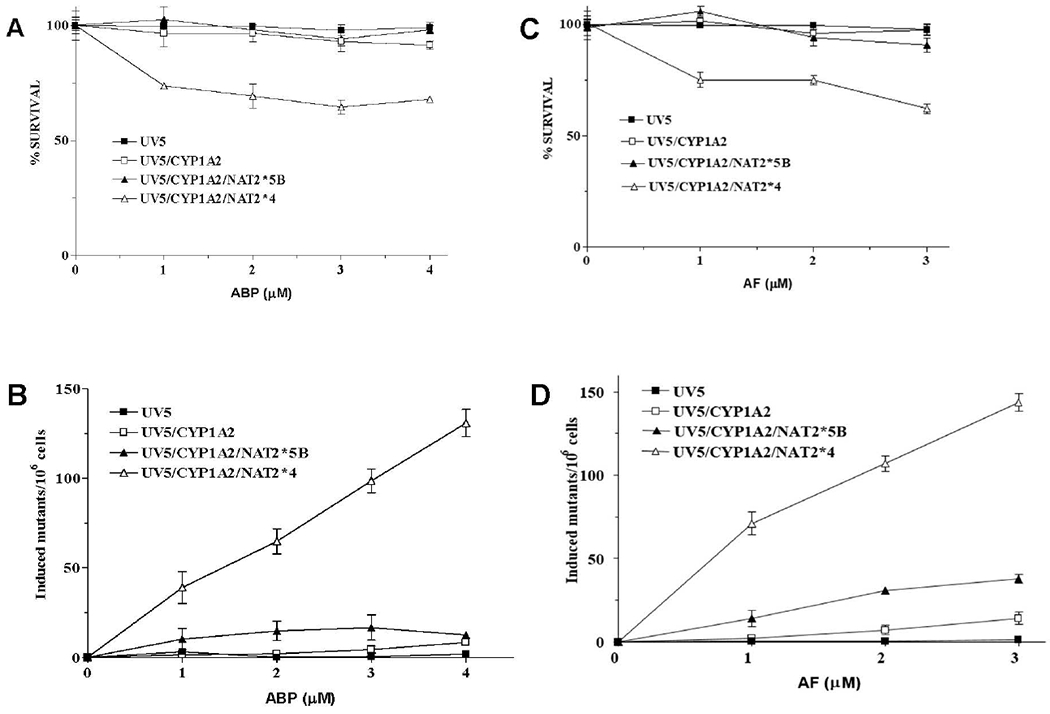
Cytotoxicity and hprt mutants. Percent survival measured by colony forming ability plotted for each of the four UV5 CHO cell lines following treatment with ABP (A) or AF (C). Data represent Mean ± SEM for two independent experiments. hprt mutant fractions following treatment with ABP (B) or AF (D). Data presented as Mean ± SEM for 4 independent experiments. Error bars not shown fall within the symbol. The UV5/CYP1A2/NAT2*4 cells had significantly more (p < 0.001) hprt mutants as compared to all other cell lines following treatment with ABP or AF at each concentration tested. The UV5/CYP1A2/NAT2*5B cells had significantly more hprt mutants than the UV5 and UV5/CYP1A2 cells following treatment with 2.0 μM (p < 0.001) or 3.0 μM (p < 0.01) AF.
Mutant sequencing
Mutations in hprt cDNAs from 23 independent ABP-induced hprt mutants and 22 independent AF-induced mutants are described in Tables I and II, respectively. For ABP-induced hprt mutants, 74% of the mutants were single base substitutions, 88% of which were transversions. The remaining 26% were exon deletions presumably resulting from splice-site mutations . Half the deletion mutants had exon 5 deleted. Furthermore, 88% of the single base substitutions were at G:C base pairs, with 60% G:C→ T:A, 7% G:C → A:T, and 33% G:C → C:G.
Table I.
ABP-induced hprt* mutations in UV5/CYP1A2/NAT2*4 CHO cells
| Missense/Nonsense Single-Base Substitutions | |||||
|---|---|---|---|---|---|
| Mutant # | Nucleotide | Exon | Type of Mutation† | Surrounding | Amino Acid |
| Position | Sequence | Change | |||
| 1 | 74 | 2 | C:G > G:C | attcctaat | Pro > Arg |
| 2 | 113 | 2 | C:G > A:T | attcctcat | Pro > His |
| 3 | 197 | 3 | G:C > T:A | ctctgtgtg | Cys > Phe |
| 3 | 208 | 3 | G:C > T:A | aaggggggc | Gly > Trp |
| 4 | 209 | 3 | G:C > A:T | aaggggggc | Gly > Glu |
| 5 | 209 | 3 | G:C > T:A | aaggggggc | Gly > Val |
| 6 | 236 | 3 | T:A > G:C | ctgctggat | Leu > Arg |
| 8 | 464 | 6 | T:A > C:G | aacctcaaa | Leu > Pro |
| 9 | 482 | 6 | C:G > A:T | gttgcaagc | Ala > Glu |
| 10 | 482 | 6 | C:G > A:T | gttgcaagc | Ala > Glu |
| 11 | 550 | 8 | C:G > G:C | attccagac | Pro > Ala |
| 12 | 550 | 8 | C:G > G:C | attccagac | Pro > Ala |
| 13 | 575 | 8 | C:G > A:T | tatgccctt | Ala > Asp |
| 14 | 582 | 8 | C:G > G:C | cttgactat | Asp > Glu |
| 15 | 617 | 9 | G:C > T:A | atttgtgtc | Cys > Phe |
| 16 | 628 | 9 | G:C > T:A | agtgaaact | Glu > Stop |
| 17 | 648 | 9 | C:G > G:C | aaatacaaa | Tyr > Stop |
| Deletions†† | |||||
| 18 | 319-384 | Δ4 | Deletion | actgtΔaatgt | Frameshift |
| 19 | 385-402 | Δ5 | Deletion | gaaagΔgacat | Frameshift |
| 20 | 385-402 | Δ5 | Deletion | gaaagΔgacat | Frameshift |
| 21 | 385-402 | Δ5 | Deletion | gaaagΔgacat | Frameshift |
| 22 | 486-532 | Δ7 | Deletion | ctgctΔttgct | Frameshift |
| 23 | 533-609 | Δ8 | Deletion | agactΔcatat | Frameshift |
GenBank sequence accession #J00060.
Mutated or inserted nucleotides are bolded.
The actual single-base substitution in the splice-site region which led to the deletions cannot be determined with certainty because sequencing was performed on mutant cDNAs.
Table II.
AF-induced hprt* mutations in UV5/CYP1A2/NAT2*4 CHO cells
| Missense/Nonsense Single-Base Substitutions | |||||
|---|---|---|---|---|---|
| Mutant # |
Position |
Exon |
Type of Mutation† |
Surrounding Sequence | Amino Acid change |
| 1 | 97 | 2 | G:C > T:A | ttggaaaag | Glu > Stop |
| 2 | 134 | 2 | G:C > T:A | gacaggact | Arg > Met |
| 3 | 134 | 2 | G:C > T:A | gacaggact | Arg > Met |
| 4 | 134 | 2 | G:C > T:A | gacaggact | Arg > Met |
| 5 | 166 | 3 | G:C > A:T | aaagagatg | Glu> Lys |
| 6 | 209 | 3 | G:C > T:A | aaggggggc | Gly > Val |
| 7 | 229 | 3 | G:C > T:A | gctgacctg | Asp > Phe |
| 8 | 292 | 3 | G:C > C:G | gtagatttt | Asp > His |
| 9 | 419 | 6 | G:C > T:A | actggtaaa | Gly > Val |
| 10 | 439 | 6 | C:G > G:C | ctgctttcc | Leu > Val |
| 11 | 464 | 6 | T:A > C:G | aacctcaaa | Leu > Pro |
| 12 | 482 | 6 | C:G > A:T | gttgcaagc | Ala > Glu |
| 13 | 496 | 7 | A:T > G:C | gtgaaaagg | Lys > Glu |
| 14 | 533 | 8 | T:A > C:G | gactttgtt | Phe >Ser |
| 15 | 538 | 8 | G:C > C:G | gttggattt | Gly > Arg |
| 16 | 551 | 8 | C:G > G:C | attccagac | Pro > Arg |
| 17 | 589 | 8 | G:C > T:A | aatgagtac | Glu > Stop |
| 18 | 589 | 8 | G:C > T:A | aatgagtac | Glu > Stop |
| 19 | 599 | 8 | G:C > A:T | ttcagggat | Arg > Lys |
| Deletions†† | |||||
| 20 | 403-485 | Δ6 | Deletion | ttgagΔaatga | Frameshift |
| 21 | 532-609 | Δ8 | Deletion | agactΔcatat | Frameshift |
| 22 | 533-609 | Δ8 | Deletion | agactΔcatat | Frameshift |
GenBank sequence accession #J00060.
Mutated or inserted nucleotides are bolded.
The actual single-base substitution in the splice-site region which led to the deletions cannot be determined with certainty because sequencing was performed on mutant cDNAs.
For AF-induced hprt mutants, 86% of the mutants were single base substitutions, 74% of which were transversions. (The remaining were exon deletions presumably caused by splice-site mutations). Two of the three deletion mutants had exon 8 deleted. Also, of the single-base substitutions, 84% were at GC base pairs, with 62.5% G:C→T:A, 12.5% G:C → A:T, and 25% G:C → C:G. Mutants in untreated cells were not sequenced because the frequency of spontaneous mutants was less than 10% of either ABP- or AF-induced mutants.
In order to compare single-base substitutions induced by ABP and AF, we mapped these mutations in the hprt cDNA sequence (Figure 3). For comparison, we added heterocyclic amine-induced single-base substitutions in the hprt cDNA sequence previously described [Metry et al., 2010].
Figure 3.
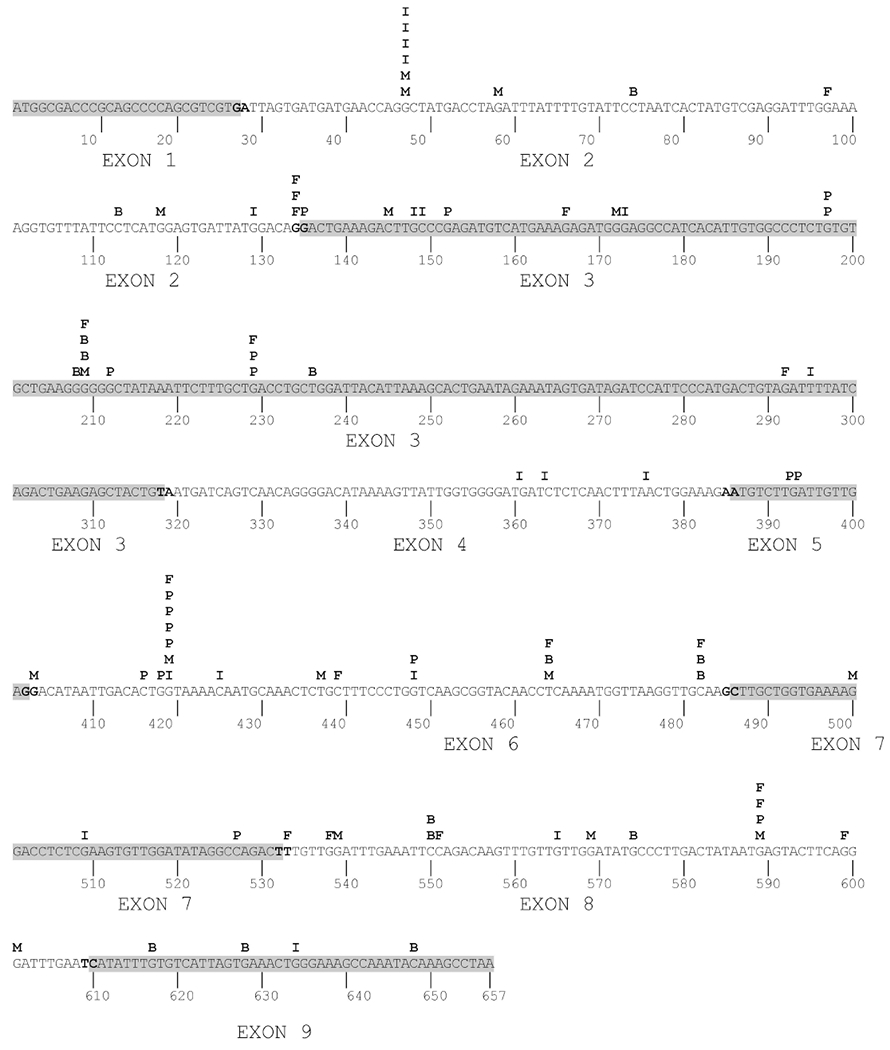
Location of ABP- and AF- induced base substitutions in the mutated hprt cDNAs. Highlighted regions designate deleted exons. Previously identified heterocyclic amine-induced mutations are also shown: B: ABP; F: AF; I: IQ; M: MeIQx; P: PhIP.
Identification and measurement of DNA adducts and DNA double-strand breaks
Under the low energy MRM mass spectrometry scanning conditions employed, one principal deoxyribonucleoside adduct was observed following incubation with ABP or AF. The adduct following incubation of ABP had a molecular mass of 435 reflecting dG-C8-ABP. The adduct following incubation with AF had a molecular mass of 447 reflecting dG-C8-AF.
dG-C8-ABP and dG-C8-AF adduct levels were dose-dependent and significantly (p < 0.001) greater in the UV5/CYP1A2/NAT2*4 rapid acetylator cell line than all other cell lines (Figure 4). As shown in Figure 5, DNA adduct levels correlated highly with induced-hprt mutants in all cell lines following treatment with ABP (r2 = 0.96) or AF (r2 = 0.94). γH2AX-positive DNA double-strand breaks in the CHO cells transfected with CYP1A2 and NAT2 were dose-dependent following incubation with both ABP and AF (Figure 4). The DNA double-strand breaks were at background levels in the UV5 and UV5/CYP1A2 cells. DNA damage was significantly higher in the UV5/CYP1A2/NAT2*4 than the UV5/CYP1A2/NAT2*5B cells (p < 0.0001). As shown in Figure 5, levels of DNA double-strand breaks correlated highly with dG-C8-ABP adducts (r2 = 0.85) following incubation with ABP and dG-C8-AF adducts (r2 = 0.94) following incubation with AF.
Figure 4.
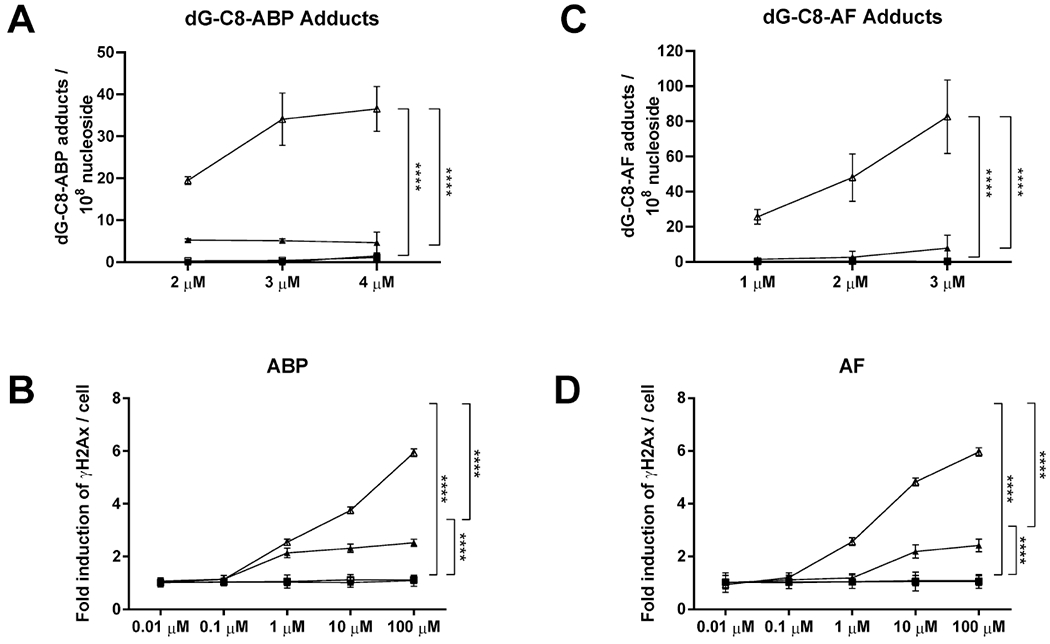
DNA adduct formation and double-strand breaks indicated by gamma-H2AX staining. dG-C8-ABP (A) or dG-C8-AF (B) DNA adduct levels in CHO cells treated with ABP or AF, respectively. Gamma H2AX staining indicating DNA double-strand breaks in CHO cells following incubation with ABP (C) or AF (D), respectively. Data for UV5 cells (closed squares), UV5/CYP1A2 cells (open squares), UV5/CYP1A2/NAT2*5B cells (closed triangles) and UV5/CYP1A2/NAT2*4 cells (open triangles) represent Mean ± SEM for three independent experiments in each of the stably transfected CHO cell lines. dG-C8-ABP (A) and dG-C8-AF (B) DNA adduct levels were significantly greater in the UV5/CYP1A2/NAT2*4 compared to all other cell lines at each concentration tested (p < 0.0001). ****, Differences between the CHO cell lines were significant (p<0.0001) as indicated.
Figure 5.
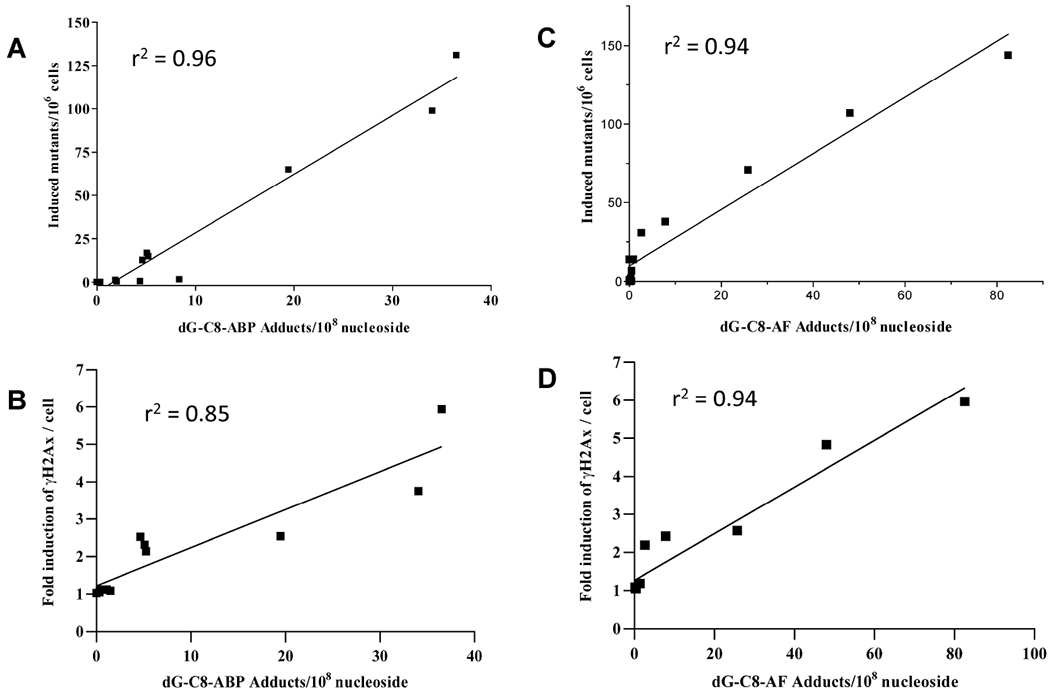
Relationships of hprt mutants and DNA double strand breaks to dG-C8-adduct levels. A. Correlation of ABP-induced hprt mutants to dG-C8-ABP adduct levels in all four cell lines (r2 = 0.96). B. Correlation of ABP-induced DNA double strand breaks to dG-C8-ABP adduct levels in all four cell lines (r2 = 0.85). C. Correlation of ABP-induced hprt mutants to dG-C8- AF adduct levels in all four cell lines (r2 = 0.94). D. Correlation of AF-induced DNA double strand breaks to dG-C8- AF adduct levels in all four cell lines (r2 = 0.94). Data from Figures 2 and 4 have been combined to produce the correlation plots.
DISCUSSION
Carcinogenic aromatic amines such as ABP and AF require metabolic activation to form electrophilic intermediates that mutate DNA leading to carcinogenesis. Bioactivation includes N-hydroxylation catalyzed by CYP1A2 followed by O-acetylation catalyzed by NAT2. NER-deficient CHO cells were stably transfected with human CYP1A2 and either NAT2*4 (rapid acetylator) or NAT2*5B (slow acetylator) alleles. ABP and AF both caused significantly (p<0.001) greater mutagenesis measured at the hprt locus in the UV5/CYP1A2/NAT2*4 compared to the UV5, UV5/CYP1A2, and UV5/CYP1A2/NAT2*5B cell lines. Both ABP- and AF-induced hprt mutant cDNAs were sequenced and over 80% of the mutations were single-base missense or nonsense substitutions at G:C base pairs. dG-C8-ABP and dG-C8-AF adduct levels were dose-dependent and significantly greater in the rapid NAT2*4 acetylator cell line following treatment with AF or ABP as compared to all other cell lines. DNA adduct levels correlated very highly with hprt mutagenesis for both ABP and AF and were in the following order: UV5 < UV5/CYP1A2 < UV5/CYP1A2/NAT2*5B < UV5/CYP1A2/NAT2*4. Similar findings have been reported following administration of heterocyclic amines MeIQx and IQ [Metry et al., 2010] but not PhIP [Metry et al., 2007]. More recently, it was confirmed that transfection of human NAT2 into V79 Chinese hamster cells increases mutagenicity of MeIQx and IQ, but not PhIP [Chevereau et al., 2017].
Consistent with the CHO cell data described above, dG-C8-MeIQx, but not dG-C8-PhIP adducts are higher in liver from rapid NAT2 acetylator than slow acetylator congenic rats following administration of MeIQx [Metry et al., 2009]. The role of N-acetyltransferase in levels of DNA adduct formation following administration of ABP and AF also has been investigated in animal models. Higher levels of DNA damage were observed following incubation with AF [McQueen et al., 1982] but not ABP [McQueen et al., 1983] in hepatocytes from rapid than slow acetylator rabbits. Levels of the major DNA adduct dG-C8-AF were higher in urinary bladders of rapid versus slow acetylator Syrian hamsters [Flammang et al., 1992] as well as in Syrian hamsters congenic for NAT2 [Feng et al., 1996]. Hepatic DNA adduct levels were higher in rapid than slow acetylator mice [Levy and Weber, 1989] and Syrian hamsters [Flammang et al, 1992] following administration of AF. In contrast, rapid and slow acetylator mice did not differ in levels of hepatic DNA adducts following administration of ABP [McQueen et al., 2003]. Hepatic dG-C8-ABP levels were significantly higher in neonatal double NAT knockout mice compared to wildtype mice administered ABP [Sugamori et al., 2012]. Hepatic levels of dG-C8-ABP were lower in adult mice than in neonatal mice and did not differ significantly in adult double NAT knockout compared to wildtype mice [Sugamori et al., 2012]. Perhaps these findings are influenced by the important role of sulfotransferases in the metabolic activation of ABP in mice [Li et al., 2018].
CHO cells have negligible endogenous phase I and II enzymes, and our present findings showed that both ABP and AF induced highest levels of mutagenesis and DNA adduct formation in the cell line expressing both human CYP1A2 and rapid acetylator NAT2*4. A study in V79 Chinese hamster cells with heterologous co-expression of human CYP1A2 and rapid or slow NAT2 alleles found that AF induced mutants in the CYP1A2 cell line with negligible mutants in the CYP1A2/NAT2*4 cell line [Scheuenpflug et al., 2005]. These results differ from those obtained in our experiments in which we found highest levels in the UV5/CYP1A2/NAT2*4 cell line. The reason(s) for this difference is unknown, but based on our results, it does not appear to be related to aromatic amine or cytochrome P450. While the results in V79 Chinese hamster cells [Scheuenpflug et al., 2005] are consistent with N-acetylation as a deactivation pathway for AF mutagenesis, our results with both AF and ABP suggest that rapid acetylators have increased mutations as compared to slow acetylators because O-acetylation is required to form DNA adducts leading to mutagenesis. Although we have investigated NAT2 only in combination with CYP1A1 [Bendaly et al., 2009] and CYP1A2, it is likely that other cytochrome P450s activate aromatic amines depending upon the cell and tissue. For example, a recent report showed that CYP2A13 expressed in the urinary bladder bioactivates ABP to form DNA adducts [Bellamri et al., 2019]. The ability of recombinant human NAT2 allozymes to catalyze metabolic activation of N-hydroxy-ABP and N-hydroxy-AF to DNA adducts via O-acetyltransferase is markedly lower when catalyzed by NAT2 5B as compared to the NAT2 4 [Hein et al., 1995; Hein, 2002]. Similar results towards the metabolic activity of N-hydroxy-ABP have been reported in COS-1 cells transfected with NAT2*4 and NAT2*5B [Zang et al., 2007] and in rapid and slow acetylator cryopreserved human hepatocytes [Doll et al., 2010]. Previous studies in our laboratory quantified ABP-induced hprt mutations and DNA adducts in NER-deficient UV5 cells transfected with CYP1A1 and NAT2 [Bendaly et al., 2009]. ABP-induced hprt mutants and DNA adduct levels were negligible in UV5 cells transfected with CYP1A1 alone. However, whether co-transfected with CYP1A1 [Bendaly et al., 2009] or CYP1A2 (current study), ABP-induced hprt mutant and DNA adduct levels were higher following transfection with the NAT2*5B slow acetylator allele, and much higher following transfection with the NAT2*4 rapid acetylator allele.
Our findings confirm previous conclusions [Heflich et al., 1988] that proposed a major difference in the metabolism of N-hydroxy arylamines in CHO versus bacterial mutagenesis assays to account for different sensitivities towards aromatic amines. The bacterial mutagenicity of AF is highly dependent upon bacterial O-acetylase [McCoy et al., 1982; 1983]. N-hydroxy-AF is about 10-fold more mutagenic in Salmonella typhimurium strain TA98 than it is in strain TA98/1,8-DNP6 [McCoy et al., 1983] which is a derivative deficient in endogenous O-acetylase activity [McCoy et al., 1982; 1983]. Transformation of TA98/1,8-DNP6 with human NAT increases sensitivity to AF-induced mutagenesis [Grant et al., 1992].
The levels of hprt mutants and DNA adducts correlated very highly following administration of either ABP or AF. The majority of induced mutations for both aromatic amines were missense/nonsense single-base substitutions, in which over 80% were at G:C base pairs. There were a few locations in the hprt cDNA where both aromatic amines had induced mutations such as within a string of G’s at position 208-209, 482 and 550-551. Furthermore, when adding previously published heterocyclic amine-induced mutations [Metry et al., 2010], positions 134-135 (GpG), 208-209 (string of G’s), 418-419 (GpG) and 589 were frequent mutation locations. It is also important to note that for both ABP and AF–induced hprt mutants, over 60% of the single-base substitutions at G:C base pairs were G to T transversions and about 30% were G to C transversions. These results are similar to our studies for PhIP, IQ, and MeIQx [Metry et al., 2010] and to previous studies with PhIP and IQ [Wu et al., 1997] . Also, in a study with transgenic Big Blue rats dosed with N-hydroxy-2-acetylaminoflourene, G:C to T:A transversions predominated [Chen et al., 2001].
In conclusion, CHO cells stably transfected with CYP1A2 and NAT2 exhibited dose-dependent increases in mutagenesis and DNA damage following incubation with ABP or AF. The levels of hprt mutants, dG-C8 adducts, and double-strand breaks correlated very highly for both ABP and AF. The levels were dose-dependent and consistently highest in the UV5/CYP1A2/NAT2*4 rapid acetylator cell line. Our findings provide further clarity on the importance of O-acetylation in CHO mutagenesis assays for aromatic amines. Since more than one in eight known or suspected human carcinogens is an aromatic amine, or a chemical that can convert into one [Wang et al., 2019], mutagenesis assays for aromatic amines or chemicals that can be converted to aromatic amines should be conducted in CHO cells stably transfected with human CYP1A2 and NAT2. Furthermore, the findings provide further evidence that the NAT2 genetic polymorphism modifies aromatic amine-induced mutagenesis and DNA damage and should be considered in human risk assessments following aromatic amine exposures.
Acknowledgments
Preliminary aspects of this work were included in the dissertation of KJB submitted for the PhD in pharmacology and toxicology at the University of Louisville.
Supported in part by grant [R01-CA034627] from the National Cancer Institute and by grant [P20-GM113226) from the National Institutes of Health.
References
- Ambrosone CB, Abrams SM, Gorlewska-Roberts K, Kadlubar FF. 2007. Hair dye use, meat intake, and tobacco exposure and presence of carcinogen-DNA adducts in exfoliated breast ductal epithelial cells. Arch Biochem Biophys 464(2):169–75 [DOI] [PubMed] [Google Scholar]
- Audebert M, Riu A, Jacques C, et al. 2010. Use of the gammaH2AX assay for assessing the genotoxicity of polycyclic aromatic hydrocarbons in human cell lines. Toxicol Lett 199(2):182–92 [DOI] [PubMed] [Google Scholar]
- Bartsch H, Caporaso N, Coda M, et al. 1990. Carcinogen hemoglobin adducts, urinary mutagenicity, and metabolic phenotype in active and passive cigarette smokers. J Natl Cancer Inst 82(23):1826–31 [DOI] [PubMed] [Google Scholar]
- Bellamri M, Yao L, Bonala R, Johnson F, Von Weymarn LB, Turesky RJ 2019. Bioactivation of the tobacco carcinogens 4-aminobiphenyl (4-ABP) and 2-amino-9H-pyrido[2,3-b]indole (AαC) in human bladder RT4 cells. Arch Toxicol 93(7):1893–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendaly J, Doll MA, Millner LM, et al. 2009. Differences between human slow N-acetyltransferase 2 alleles in levels of 4-aminobiphenyl-induced DNA adducts and mutations. Mutat Res 671(1-2):13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Mittelstaedt RA, Shelton SD, et al. 2001. Gene- and tissue-specificity of mutation in Big Blue rats treated with the hepatocarcinogen N-hydroxy-2-acetylaminofluorene. Environ Mol Mutagen 37(3):203–14 [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhang Y, Vouros P. 2018. Recent technical and biological development in the analysis of biomarker N-deoxyguanosine-C8-4-aminobiphenyl. J Chromatogr B Analyt Technol Biomed Life Sci 1087-1088:49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevereau M, Glatt H, Zalko D, Cravedi JP, Audebert M. 2017. Role of human sulfotransferase 1A1 and N-acetyltransferase 2 in the metabolic activation of 16 heterocyclic amines and related heterocyclics to genotoxicants in recombinant V79 cells. Arch Toxicol 91(9):3175–3184 [DOI] [PubMed] [Google Scholar]
- Doll MA, Zang Y, Moeller T, Hein DW. 2010. Codominant expression of N-acetylation and O-acetylation activities catalyzed by N-acetyltransferase 2 in human hepatocytes. J Pharmacol Exp Ther 334(2):540–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Jiang W, Hein DW. 1996. 2-Aminofluorene-DNA adduct levels in tumor-target and nontarget organs of rapid and slow acetylator Syrian hamsters congenic at the NAT2 locus. Toxicol Appl Pharmacol 141(1):248–55 [DOI] [PubMed] [Google Scholar]
- Flammang TJ, Yerokun T, Bryant MS, et al. 1992. Hemoglobin adduct and hepatic- and urinary bladder-DNA adduct levels in rapid and slow acetylator Syrian inbred hamsters administered 2-aminofluorene. J Pharmacol Exp Ther 260(2):865–71 [PubMed] [Google Scholar]
- Fretland AJ, Doll MA, Zhu Y, Smith L, Leff MA, Hein DW. 2002. Effect of nucleotide substitutions in N-acetyltransferase-1 on N-acetylation (deactivation) and O-acetylation (activation) of arylamine carcinogens: implications for cancer predisposition. Cancer Detect Prev 26(1):10–4 [DOI] [PubMed] [Google Scholar]
- Grant DM, Josephy PD, Lord HL, Morrison LD. 1992. Salmonella typhimurium strains expressing human arylamine N-acetyltransferases: metabolism and mutagenic activation of aromatic amines. Cancer Res 52(14):3961–4 [PubMed] [Google Scholar]
- Heflich RH, Djuric Z, Zhuo Z, Fullerton NF, Casciano DA, Beland FA. 1988. Metabolism of 2-acetylaminofluorene in the Chinese hamster ovary cell mutation assay. Environ Mol Mutagen 11(2):167–81 [DOI] [PubMed] [Google Scholar]
- Heflich RH, Neft RE. 1994. Genetic toxicity of 2-acetylaminofluorene, 2-aminofluorene and some of their metabolites and model metabolites. Mutat Res 318(2):73–114 [DOI] [PubMed] [Google Scholar]
- Hein DW. 2002. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res 506-507:65–77 [DOI] [PubMed] [Google Scholar]
- Hein DW, Doll MA, Fretland AJ, et al. 2000. Molecular genetics and epidemiology of the NAT1 and NAT2 acetylation polymorphisms. Cancer Epidemiol Biomarkers Prev 9(1):29–42 [PubMed] [Google Scholar]
- Hein DW, Doll MA, Rustan TD, Ferguson RJ. 1995. Metabolic activation of N-hydroxyarylamines and N-hydroxyarylamides by 16 recombinant human NAT2 allozymes: effects of 7 specific NAT2 nucleic acid substitutions. Cancer Res 55(16):3531–6 [PubMed] [Google Scholar]
- Leff MA, Epstein PN, Doll MA, et al. 1999. Prostate-specific human N-acetyltransferase 2 (NAT2) expression in the mouse. J Pharmacol Exp Ther 290(1):182–7 [PubMed] [Google Scholar]
- Levy GN, Weber WW. 1989. 2-Aminofluorene-DNA adduct formation in acetylator congenic mouse lines. Carcinogenesis 10(4):705–9 [DOI] [PubMed] [Google Scholar]
- Li Y, Chen Z, Paonessa JD, et al. 2018. Strong impact of sulfotransferases on DNA adduct formation by 4-aminobiphenyl in bladder and liver in mice. Cancer Medicine 7(11):5604–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy EC, Anders M, Rosenkranz HS. 1983. The basis of the insensitivity of Salmonella typhimurium strain TA98/1,8-DNP6 to the mutagenic action of nitroarenes. Mutat Res 121(1):17–23 [DOI] [PubMed] [Google Scholar]
- McCoy EC, McCoy GD, Rosenkranz HS. 1982. Esterification of arylhydroxylamines: evidence for a specific gene product in mutagenesis. Biochem Biophys Res Commun 108(3):1362–7 [DOI] [PubMed] [Google Scholar]
- McQueen CA, Maslansky CJ, Glowinski IB, Crescenzi SB, Weber WW, Williams GM. 1982. Relationship between the genetically determined acetylator phenotype and DNA damage induced by hydralazine and 2-aminofluorene in cultured rabbit hepatocytes. Proc Natl Acad Sci USA 79(4):1269–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueen CA, Maslansky CJ, Williams GM. 1983. Role of the acetylation polymorphism in determining susceptibility of cultured rabbit hepatocytes to DNA damage by aromatic amines. Cancer Res 43(7):3120–3 [PubMed] [Google Scholar]
- McQueen CA, Chau B, Erickson RP, Tjalkens RB, Philbert MA. 2003. The effects of genetic variation in N-acetyltransferases on 4-aminobiphenyl genotoxicity in mouse liver. Chem Biol Interact 146(1):51–60 [DOI] [PubMed] [Google Scholar]
- Metry KJ, Neale JR, Bendaly J, Smith NB, Pierce WM, Hein DW. 2009. Effect of N-acetyltransferase 2 polymorphism on tumor target tissue DNA adduct levels in rapid and slow acetylator congenic rats administered 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) or 2-amino-3,8- dimethylimidazo- [4,5-f] quinoxaline (MeIQx). Drug Metab Dispos 37 (11): 2123–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metry KJ, Neale JR, Doll MA, et al. 2010. Effect of rapid human N-acetyltransferase 2 haplotype on DNA damage and mutagenesis induced by 2-amino-3-methylimidazo-[4,5-f]quinoline (IQ) and 2-amino-3,8-dimethylimidazo-[4,5-f]quinoxaline (MeIQx). Mutat Res 684(1-2):66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metry KJ, Zhao S, Neale JR, et al. 2007. 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine-induced DNA adducts and genotoxicity in Chinese hamster ovary (CHO) cells expressing human CYP1A2 and rapid or slow acetylator N-acetyltransferase 2. Mol Carcinog 46(7):553–63 [DOI] [PubMed] [Google Scholar]
- Nairn RS, Tang MS, Wang RM, Adair GM, Humphrey RM. 1988. Processing of 2-aminofluorene and 2-acetylaminofluorene DNA adducts in Chinese hamster ovary cells. Carcinogenesis 9(8):1369–75 [DOI] [PubMed] [Google Scholar]
- Neale JR, Smith NB, Pierce WM, Hein DW. 2008. Methods for aromatic and heterocyclic amine carcinogen-DNA adduct analysis by liquid chromatography-tandem mass spectrometry Polycycl Aromat Compd 28(4-5):402–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NTP (National Toxicology Program). 2016. Report on Carcinogens, Fourteenth Edition.; Research Triangle Park, NC: U.S. Department of Health and Human Services, Public Health Service; https://ntp.niehs.nih.gov/go/roc14 [Google Scholar]
- Ricicki EM, Soglia JR, Teitel C, Kane R, Kadlubar F, Vouros P. 2005. Detection and quantification of N-(deoxyguanosin-8-yl)-4-aminobiphenyl adducts in human pancreas tissue using capillary liquid chromatography-microelectrospray mass spectrometry. Chem Res Toxicol 18(4):692–9 [DOI] [PubMed] [Google Scholar]
- Scheuenpflug J, Krebsfanger N, Doehmer J. 2005. Heterologous co-expression of human cytochrome P450 1A2 and polymorphic forms of N-acetyltransferase 2 for studies on aromatic amines in V79 Chinese hamster cells. Alternatives to laboratory animals: ATLA 33(6):561–77 [DOI] [PubMed] [Google Scholar]
- Schut HA, Snyderwine EG. 1999. DNA adducts of heterocyclic amine food mutagens: implications for mutagenesis and carcinogenesis. Carcinogenesis 20(3):353–68 [DOI] [PubMed] [Google Scholar]
- Snyderwine EG. 2002. Mammary gland carcinogenesis by food-derived heterocyclic amines: metabolism and additional factors influencing carcinogenesis by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Environ Mol Mutagen 39(2-3):165–70 [DOI] [PubMed] [Google Scholar]
- Snyderwine EG, Venugopal M, Yu M. 2002. Mammary gland carcinogenesis by food-derived heterocyclic amines and studies on the mechanisms of carcinogenesis of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Mutat Res 506-507:145–52 [DOI] [PubMed] [Google Scholar]
- Sugamori KS, Brenneman D, Sanchez O, et al. 2012. Reduced 4-aminobiphenyl-induced liver tumorigenicity but not DNA damage in arylamine N-acetyltransferase null mice. Cancer Lett 318(2):206–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LH, Rubin JS, Cleaver JE, Whitmore GF, Brookman K. 1980. A screening method for isolating DNA repair-deficient mutants of CHO cells. Somatic Cell Genet 6(3):391–405 [DOI] [PubMed] [Google Scholar]
- Wang S, Hanna D, Sugamori KS, Grant DM. 2019. Primary aromatic amines and cancer: Novel mechanistic insights using 4-aminobiphenyl as a model carcinogen. Pharmacol Ther 200:179–189 [DOI] [PubMed] [Google Scholar]
- Weber CA, Salazar EP, Stewart SA, Thompson LH. 1988. Molecular cloning and biological characterization of a human gene, ERCC2, that corrects the nucleotide excision repair defect in CHO UV5 cells. Mol Cell Biol 8(3):1137–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RW, Tucker JD, Sorensen KJ, Thompson LH, Felton JS. 1997. Differential effect of acetyltransferase expression on the genotoxicity of heterocyclic amines in CHO cells. Mutat Res 390(1-2):93–103 [DOI] [PubMed] [Google Scholar]
- Zang Y, Doll MA, Zhao S, States JC, Hein DW. 2007. Functional characterization of single-nucleotide polymorphisms and haplotypes of human N-acetyltransferase 2. Carcinogenesis 28(8):1665–71 [DOI] [PMC free article] [PubMed] [Google Scholar]