

Abstract
Background
The upper gastrointestinal tract plays a prominent role in human physiology as the primary site for enzymatic digestion and nutrient absorption, immune sampling, and drug uptake. Alterations to the small intestine microbiome have been implicated in various human diseases, such as non-alcoholic steatohepatitis and inflammatory bowel conditions. Yet, the physiological and functional roles of the small intestine microbiota in humans remain poorly characterized because of the complexities associated with its sampling. Rodent models are used extensively in microbiome research and enable the spatial, temporal, compositional, and functional interrogation of the gastrointestinal microbiota and its effects on the host physiology and disease phenotype. Classical, culture-based studies have documented that fecal microbial self-reinoculation (via coprophagy) affects the composition and abundance of microbes in the murine proximal gastrointestinal tract. This pervasive self-reinoculation behavior could be a particularly relevant study factor when investigating small intestine microbiota. Modern microbiome studies either do not take self-reinoculation into account, or assume that approaches such as single housing mice or housing on wire mesh floors eliminate it. These assumptions have not been rigorously tested with modern tools. Here, we used quantitative 16S rRNA gene amplicon sequencing, quantitative microbial functional gene content inference, and metabolomic analyses of bile acids to evaluate the effects of self-reinoculation on microbial loads, composition, and function in the murine upper gastrointestinal tract.
Results
In coprophagic mice, continuous self-exposure to the fecal flora had substantial quantitative and qualitative effects on the upper gastrointestinal microbiome. These differences in microbial abundance and community composition were associated with an altered profile of the small intestine bile acid pool, and, importantly, could not be inferred from analyzing large intestine or stool samples. Overall, the patterns observed in the small intestine of non-coprophagic mice (reduced total microbial load, low abundance of anaerobic microbiota, and bile acids predominantly in the conjugated form) resemble those typically seen in the human small intestine.
Conclusions
Future studies need to take self-reinoculation into account when using mouse models to evaluate gastrointestinal microbial colonization and function in relation to xenobiotic transformation and pharmacokinetics or in the context of physiological states and diseases linked to small intestine microbiome and to small intestine dysbiosis.
Video abstract.
Keywords: Microbial quantification, Metabolomics analyses, Mouse models, Small intestine microbiota, Bile acids, Deconjugation, Coprophagy, Microbial colonization, 16S rRNA gene amplicon sequencing
Background
The small intestine is the primary site for enzymatic digestion and nutrient uptake, immune sampling, and drug absorption in the human gastrointestinal system. Its large surface area vastly exceeds that of the large intestine [1], and thus may serve as a broad interface for host-microbial interactions.
A growing body of scientific evidence highlights the importance of the small intestine microbiome in normal human physiology and response to dietary interventions [2, 3]. Alterations in the small intestine microbiome are implicated in a number of human disorders, such as malnutrition [4, 5], obesity, and metabolic disease [6], inflammatory bowel disease (IBD) and irritable bowel syndrome (IBS) [7–9], and drug side effects [10]. Despite the apparent importance of the small intestine microbiome in human health, it remains understudied and poorly characterized largely because of the procedural and logistical complexities associated with its sampling in humans (methods are too invasive and require specialized healthcare facilities). Moreover, microbial composition tends to differ substantially among the small intestine, large intestine, and stool of the same animal or human subject [11, 12], which highlights the importance of targeted sampling of the small intestine for analyses.
Mice are the predominant animal species of model organisms in the field of microbiome research. Compared with other mammalian models, mice have a lower cost of maintenance, their environment and diet can be easily controlled, they are amenable to genetic manipulation, there are numerous genetic mouse models already available, and propagation using inbred colonies reduces inter-individual variability [13]. Additionally, murine germ-free (GF) and gnotobiotic technologies are well established. Using mouse models enables interrogation of the entire gastrointestinal tract (GIT) and examination of the changes in microbiome and host physiology that occur in response to experimental conditions (e.g., dietary modifications, xenobiotic administration) or microbial colonization (e.g., monocolonization, colonization with defined microbial consortia, human microbiota-associated mice).
Rodent models also have several well-recognized limitations associated with their genetic, anatomical, and physiological differences with humans [13, 14]. Among these limitations is the persistent tendency of rodents to practice gastrointestinal auto- and allo-reinoculation with large intestine microbiota (via fecal ingestion, or coprophagy) in laboratory settings [15–17]. This pervasive behavior has been documented in classical studies using observational techniques in both conventional and GF mice [18], in conventional mice maintained on standard and fortified diets [19], in animals with and without access to food [20], and across different mouse strains [16, 21].
Multiple classical studies have attempted to evaluate the effects of self-reinoculation on the structure of the microbiota in the rodent small intestine [22–24] and large intestine and stool [20, 23, 25, 26] using traditional microbiological techniques, but reported conflicting results [23, 25, 26]. This lack of consensus may be attributed to the use of different methods for preventing coprophagy (some of which are ineffective), non-standardized diets, inter-strain or inter-species differences among the animal models, or other unaccounted for experimental parameters. It has been also suggested that repeated self-exposure in mice via coprophagy can promote microbial colonization of the GIT by “exogenous” microbial species, such as Pseudomonas spp. [27]. All of these observations highlight the importance of considering self-reinoculation in studies of gastrointestinal microbial ecology in murine models. However, the field currently lacks precise and comprehensive evaluations of the effects of self-reinoculation on the spatial, structural, and functional state of the gut microbiome and its effects on murine host physiology. Current microbiome studies in rodents either do not take self-reinoculation into account, or assume it can be eliminated by single housing of animals or housing them on wire mesh floors (also referred to as “wire screens” or “wire grids”) [14]. Despite classical literature suggesting these assumptions can be incorrect [16, 21, 28–32], they have not been tested on mice housed in modern facilities using state-of-the-art quantitative tools.
Here, we explicitly test these assumptions about murine self-reinoculation to answer the following three questions relevant to gastrointestinal microbiome research: (1) Do quantitative 16S rRNA gene amplicon sequencing tools detect differences in small intestine microbial loads between mice known to be coprophagic and non-coprophagic? (2) Does coprophagy impact the microbial composition of the small intestine? (3) Do differences in microbiota density and composition associated with self-reinoculation in mice impact microbial function (e.g., alter microbial metabolite production or modifications) in the small intestine?
To answer these questions, we analyzed gastrointestinal samples from mice under conditions known to prevent coprophagy (fitting with “tail” or “fecal collection” cups [16, 23, 26, 30, 33]) and typical laboratory conditions in which mice are known to be coprophagic (housing in standard cages). We also included samples from single-housed mice in standard and wire-floor cages. We analyzed the quantitative and compositional changes in the microbiome along the entire length of the mouse GIT in response to self-reinoculation, computationally inferred the changes in microbial function, and evaluated the microbial function-related metabolite profiles in the corresponding segments of the gut.
Results
We first performed a pilot study to confirm that preventing coprophagy in mice would result in decreased viable microbial load and altered microbiota composition in the small intestine. We used a most probable number (MPN) assay utilizing anaerobic BHI-S broth medium to evaluate the live (culturable) microbial loads along the entire GIT of mice known to be coprophagic (housed in standard cages in groups, N = 5) and mice known to be non-coprophagic (fitted with tail cups and housed in standard cages in groups, N = 5). Consistent with the published classical literature [20, 24], we found that coprophagic mice had significantly higher loads of culturable microbes in their upper GIT than mice that were non-coprophagic (Additional file 1: Figure S4A). Moreover, the microbial community composition in the proximal GIT, particularly in the stomach, of coprophagic mice more closely resembled the microbial composition of the large intestine (Additional file 1: Figure S4B) as revealed by 16S rRNA gene amplicon sequencing (N = 1 mouse analyzed from each group) and principal component analysis (PCA) of the resulting relative abundance data.
This pilot study confirmed that in our hands, tail cups were effective at preventing the self-reinoculation of viable fecal flora in the upper GIT of mice. These results spurred us to design a rigorous, detailed study (Fig. 1) to answer the three questions posed above using state-of-the-art methods: quantitative 16S rRNA gene amplicon sequencing (to account for both changes in the total microbial load and the unculturable taxa), quantitative functional gene content inference, and targeted bile-acid metabolomics analyses.
Fig. 1.
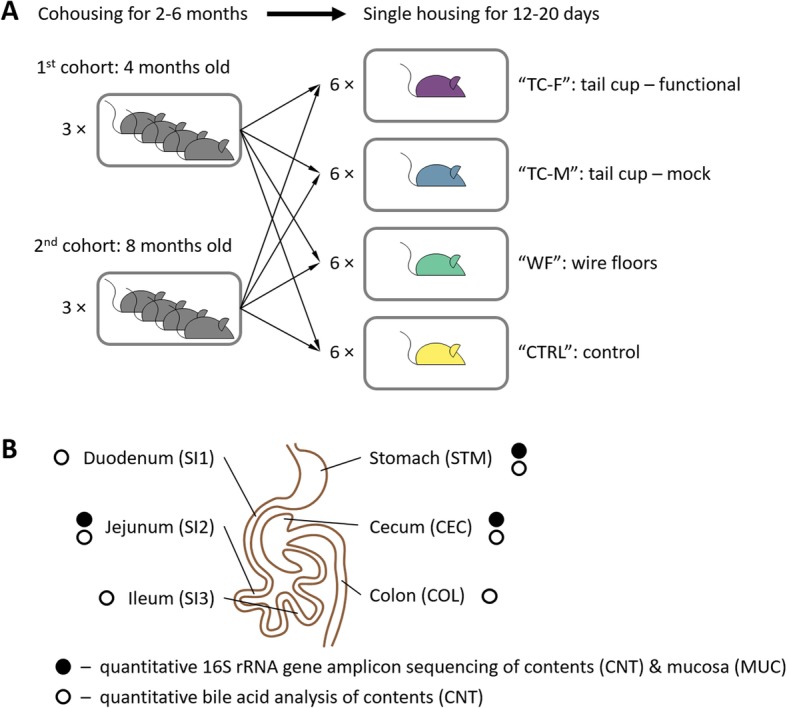
An overview of the study design and timeline. a Mice from two age cohorts (4-month-old and 8-month-old) were raised co-housed (four mice to a cage) for 2–6 months. One mouse from each cage was then assigned to one of the four experimental conditions: functional tail cups (TC-F), mock tail cups (TC-M), housing on wire floors (WF), and controls housed in standard conditions (CTRL). All mice were singly housed and maintained on each treatment for 12–20 days (N = 24, 6 mice per group). b Samples were taken from six sites throughout the gastrointestinal tract. Each sample was analyzed by quantitative 16S rRNA gene amplicon sequencing of lumenal contents (CNT) and mucosa (MUC) and/or quantitative bile-acid analyses of CNT. Panel b is adapted from [13, 34])
The study design (Fig. 1) consisted of six cages of four animals each that were co-housed for 2–6 months and then split into four experimental groups and singly housed for 12–20 days. The four experimental conditions were the following: animals fitted with functional tail cups (TC-F) and singly housed in standard cages, animals fitted with mock tail cups (TC-M) and singly housed in standard cages, animals singly housed on wire floors (WF), and control animals singly housed in standard conditions (CTRL). At the end of the study, gastrointestinal contents and mucosal samples were collected from all segments of the GIT of each animal and we evaluated total microbial loads (entire GIT) and microbiome composition (stomach (STM), jejunum (SI2), and cecum (CEC)).
We chose the cecum segment of the large intestine for quantitative 16S rRNA gene amplicon sequencing because the analysis of the contents of this section can provide a complete snapshot of the large intestine and fecal microbial diversity in response to environmental factors [35–37]. Cecal contents also enabled us to collect a more consistent amount of sample from all animals across all experimental conditions (whereas defecation may be inconsistent among animals at the time of terminal sampling).
Self-reinoculation increases microbial loads in the upper gut
To answer our first question (Can quantitative sequencing tools detect the difference in 16S rRNA gene DNA copy load in the upper GIT of mice known to be coprophagic and non-coprophagic?), we analyzed total quantifiable microbial loads across the GIT using 16S rRNA gene DNA quantitative PCR (qPCR) and digital PCR (dPCR). Preventing self-reinoculation in mice equipped with functional tail cups dramatically decreased the lumenal microbial loads in the upper GIT but not in the lower GIT (Fig. 2a). Total quantifiable microbial loads in the upper GIT were reduced only in mice equipped with functional tail cups. All other experimental groups of singly-housed animals (those equipped with mock tail cups, housed on wire floors, or housed on standard woodchip bedding) that retained access to fecal matter and practiced self-reinoculation had similarly high microbial loads in the upper GIT, as expected from the published literature [16, 21, 28–32].
Fig. 2.

Quantification of microbial loads in lumenal contents and mucosa of the gastrointestinal tracts (GIT) of mice in the four experimental conditions: functional tail cups (TC-F), mock tail cups (TC-M), housing on wire floors (WF), and controls housed in standard conditions (CTRL). a Total 16S rRNA gene DNA copy loads, a proxy for total microbial loads, were measured along the GIT of mice of all groups (STM = stomach; SI1 = upper third of the small intestine (SI), SI2 = middle third of the SI, SI3 = lower third of the SI roughly corresponding to the duodenum, jejunum, and ileum respectively; CEC = cecum; COL = colon). Multiple comparisons were performed using a Kruskal–Wallis test, followed by pairwise comparisons using the Wilcoxon–Mann–Whitney test with false-discovery rate (FDR) correction. Individual data points are overlaid onto box-and-whisker plots; whiskers extend from the quartiles (Q2 and Q3) to the last data point within 1.5 × interquartile range (IQR). b Correlation between the microbial loads in the lumenal contents (per gram total contents) and in the mucosa (per 100 ng of mucosal DNA) of the mid-SI. N = 6 mice per experimental group
Across all test groups, mucosal microbial loads in the mid-small intestine demonstrated high correlation (Pearson’s R = 0.84, P = 2.8 × 10− 7) with the microbial loads in the lumenal contents (Fig. 2b).
Stomach (STM) and small intestine (SI1, SI2, and SI3) samples from one (out of six) of the TC-F mice showed higher microbial loads compared with the other TC-F mice. The total microbial load in the upper GIT in this TC-F mouse was similar to mice from all other groups (TC-M, WF, CTRL), which emphasizes the crucial importance of performing analyses of both microbial load and composition (discussed below) on the same samples.
Self-reinoculation substantially alters the microbiota composition in the upper gut but has less pronounced effects in the large intestine
To answer our second question (Does self-reinoculation with fecal microbiota impact upper GIT microbial composition?), we performed quantitative 16S rRNA gene amplicon sequencing [38, 39] (Barlow JT, Bogatyrev SR, Ismagilov RF: A quantitative sequencing framework for absolute abundance measurements of mucosal and lumenal microbial communities, submitted) on the stomach (STM), jejunum (SI2), and cecum (CEC) samples. Qualitative sequencing revealed dramatic overall changes in the upper GIT microbiota caused by self-reinoculation (Fig. 3). An exploratory PCA performed on the multidimensional absolute microbial abundance profiles highlights the unique and distinct composition of the upper GIT microbiome of non-coprophagic mice (Fig. 3a). It is noteworthy that the stomach (STM) and small intestine (SI2) microbiota in all coprophagic mice clustered closer to the large intestine microbiota, suggesting the similarity was due to persistent self-reinoculation with the large intestine microbiota (Fig. 3a).
Fig. 3.

Compositional and quantitative 16S rRNA gene amplicon sequencing analysis of the gut microbiota. a Principal component analysis (PCA) of the log10-transformed and standardized (mean = 0, SD = 1) absolute microbial abundance profiles in the stomach, mid-small intestine, and cecum. Loadings of the top contributing taxa are shown for each principal component. b Mean relative and absolute abundance profiles of microbiota in the mid-SI (order level) for all experimental conditions. Functional tail cups (TC-F), mock tail cups (TC-M), housing on wire floors (WF), and controls housed in standard conditions (CTRL). N = 6 mice per experimental group, 4 of which were used for sequencing. c Absolute abundances of microbial taxa (order level) compared between coprophagic and non-coprophagic mice along the mouse GIT. *Chloroplast and *Richettsiales (mitochondria) represent 16S rRNA gene DNA amplicons from food components of plant origin. Multiple comparisons were performed using the Kruskal–Wallis test
Self-reinoculation had differential effects across microbial taxa (Fig. 3c), which could be classified into three main categories depending on the pattern of their change as follows:
“Fecal taxa” (e.g., Clostridiales, Bacteroidales, Erysipelotrichales) that either dropped significantly or disappeared (fell below the lower limit of detection [LLOD] of the quantitative sequencing method [38] (Barlow JT, Bogatyrev SR, Ismagilov RF: A quantitative sequencing framework for absolute abundance measurements of mucosal and lumenal microbial communities, submitted)) in the upper GIT of non-coprophagic mice;
“True small intestine taxa” (e.g., Lactobacillales) that remained relatively stable in the upper GIT in non-coprophagic mice;
Taxa that had lower absolute abundance in the cecum (e.g., Bacteroidales, Erysipelotrichales, Betaproteobacteriales) of non-coprophagic (compared with coprophagic) mice.
Overall, the composition of the small intestine microbiota of coprophagic mice was consistent with that previously reported in literature [35]. The upper GIT microbiota in non-coprophagic mice was dominated by Lactobacilli (Fig. 3c), known to be a prominent microbial taxon in human small intestine microbiota [3, 40, 41]. Importantly, the compositional analysis showed that the single TC-F mouse that had high microbial loads in its stomach and small intestine had a microbial composition in those segments of the GIT similar (i.e., dominated by Lactobacillales) to all other TC-F mice, and very distinct from all coprophagic mice (Fig. 3b, c). The PCA showed that the stomach and mid-small intestine of this mouse clustered with the stomach and mid-small intestine of all other TC-F mice (Fig. 3a).
Changes in the small intestine microbiota lead to differences in inferred microbial functional gene content
We hypothesized that the quantitative and qualitative changes in the small intestine microbiota induced by self-reinoculation may result in altered microbial function [42, 43] and an altered metabolite profile, either indirectly, as a result of functional changes in the microbiota, or directly via re-ingestion of fecal metabolites. To understand how such alterations to microbiota would impact microbial function in the small intestine, we next aimed to predict how the absolute abundances of functional microbial genes would be affected. We coupled the pipeline for microbial functional inference based on the 16S rRNA marker gene sequences (PICRUSt2) [44, 45] with our quantitative 16S rRNA gene amplicon sequencing approach [38] (Barlow JT, Bogatyrev SR, Ismagilov RF: A quantitative sequencing framework for absolute abundance measurements of mucosal and lumenal microbial communities, submitted). We focused our analysis on microbial functions that would be highly relevant to small intestine physiology: microbial conversion of host-derived bile acids and microbial modification of xenobiotics.
We found that the inferred absolute abundances of a number of microbial gene orthologs implicated in enzymatic hydrolysis of conjugated bile acids (bile salt hydrolase, BSH [46–48]) and xenobiotic conjugates (e.g., beta-glucuronidase, arylsulfatase [49, 50]) in the stomach and the small intestine of coprophagic mice were dramatically higher (in some cases by several orders of magnitude) than in non-coprophagic mice (Fig. 4). This difference was not observed in the cecum.
Fig. 4.

Inference of microbial genes involved in bile-acid and xenobiotic conjugate modification along the GIT of coprophagic and non-coprophagic mice. Inferred absolute abundance of the microbial genes encoding (a) bile salt hydrolases (cholylglycine hydrolases), (b) beta-glucuronidases, and (c) arylsulfatases throughout the GIT (STM stomach, SI2 middle third of the small intestine (SI) roughly corresponding to the jejunum, CEC cecum). KEGG orthology numbers are given in parentheses for each enzyme. In all plots, individual data points are overlaid onto box-and-whisker plots; whiskers extend from the quartiles (Q2 and Q3) to the last data point within 1.5 × interquartile range (IQR). Multiple comparisons were performed using the Kruskal–Wallis test; pairwise comparisons were performed using the Wilcoxon–Mann–Whitney test with FDR correction. N = 4 mice per group
Changes in the small intestine microbiota induced by self-reinoculation alter the bile acid profile
Bile acids are a prominent class of host-derived compounds with multiple important physiological functions and effects on the host and its gut microbiota [51, 52]. These host-derived molecules are highly amenable to microbial modification in both the small and large intestine [53]. The main microbial bile acid modifications in the GIT include deconjugation, dehydrogenation, dehydroxylation, and epimerization [52]. Thus, we next performed quantitative bile acid profiling along the entire GIT to evaluate the effects of self-reinoculation on bile acid composition.
The small intestine is the segment of the GIT that harbors the highest levels of bile acids (up to 10 mM) and where they function in lipid emulsification and absorption [54–56]. Given these high concentrations of bile acid substrates, we specifically wished to analyze whether the differences we observed in small intestine microbiota (Figs. 2 and 3) between coprophagic and non-coprophagic mice would result in pronounced effects on microbial deconjugation of bile acids. We also wished to test whether any differences in bile acid deconjugation were in agreement with the differences in the absolute BSH gene content we inferred (Fig. 4a) from the absolute microbial abundances (Fig. 3c).
We first confirmed that in all four experimental groups, total bile acids levels (conjugated and unconjugated; primary and secondary) across all sections of the GIT were highest in the small intestine (Fig. 5a). We then compared the levels of conjugated and unconjugated (Fig. 5b) as well as primary (host-synthesized) and secondary (microbe-modified) bile acids (Additional file 1: Figure S5) between coprophagic and non-coprophagic mice.
Fig. 5.

Bile acid profiles in gallbladder bile and in lumenal contents along the entire GIT. a Total bile acid levels (conjugated and unconjugated; primary and secondary) and b the fraction of unconjugated bile acids in gallbladder bile and throughout the GIT (STM stomach; SI1 upper third of the small intestine (SI), SI2 middle third of the SI, SI3 lower third of the SI roughly corresponding to the duodenum, jejunum, and ileum respectively; CEC cecum; COL colon). In all plots, individual data points are overlaid onto box-and-whisker plots; whiskers extend from the quartiles (Q2 and Q3) to the last data point within 1.5 × interquartile range (IQR). Multiple comparisons were performed using the Kruskal–Wallis test; pairwise comparisons were performed using the Wilcoxon–Mann–Whitney test with FDR correction. N = 6 mice per group
Across all sections of the GIT and in the bile, non-coprophagic mice (TC-F) had significantly lower levels of unconjugated bile acids compared with coprophagic mice (Fig. 5b). Consistent with the computational inference in Fig. 4a (performed on mid-SI samples only), in all three sections of the small intestine of non-coprophagic mice (TC-F), the levels of unconjugated bile acids were substantially lower than in coprophagic mice. Almost 100% of the total bile acid pool remained in a conjugated form in the small intestine of non-coprophagic mice.
In all groups of coprophagic mice (TC-M, WF, and CTRL) the fraction of unconjugated bile acids gradually increased from the proximal to distal end of the small intestine. Gallbladder bile acid profiling (Fig. 5b) confirmed that bile acids were secreted into the duodenum predominantly in the conjugated form in all coprophagic mice. This pattern is consistent with the hypothesis that the exposure of bile acids to microbial deconjugation activity increases as they transit down a small intestine with high microbial loads (Fig. 2a) [54].
In the large intestine, non-coprophagic (TC-F) mice carried a smaller fraction of unconjugated bile acids compared with all coprophagic experimental groups (Fig. 5b).
Bile acid deconjugation in the small intestine of coprophagic mice was uniform for all glyco- and tauro-conjugates of all primary and secondary bile acids measured in our study (Additional file 1: Table S7), suggesting a broad-specificity BSH activity was provided by a complex fecal flora in the small intestine of those animals.
In the gallbladder bile and across all segments of the GIT from the stomach to the cecum, non-coprophagic mice had a statistically significantly lower fraction (but not lower absolute levels) of total secondary bile acids (conjugated and unconjugated) than coprophagic mice (Additional file 1: Figure S5). This change was uniform for the entire secondary bile acid pool of those analyzed (Additional file 1: Table S7). The only segment of the gut in which the difference in the fraction of secondary bile acids was not statistically significant between coprophagic and non-coprophagic mice was the colon. In fact, the differences in the fractions of total unconjugated and total secondary bile acids between coprophagic and non-coprophagic mice would have gone largely undetected had we only analyzed colonic contents or stool. These findings further highlight the importance of the comprehensive spatial interrogation of the complex crosstalk between the microbiota and bile acids in the gastrointestinal tract.
Discussion
In this study, we used modern tools for quantitative microbiota profiling and showed that when self-reinoculation with fecal flora is prevented, the mouse small intestine harbors dramatically lower densities of microbiota and an altered microbial profile. Consistent with published literature [16, 21, 28–32], we confirmed that single housing on wire floors failed to prevent mice from practicing coprophagy and that only functional tail cups reliably prevented the self-reinoculation with fecal flora.
Despite its effectiveness, the tail cup approach has limitations. Tail cups in their current design may not be suitable for female rodents due to anatomical differences leading to urine entering and remaining inside the devices [57]. Animals need to be singly housed to prevent them from gnawing on each other’s tail cups and causing device failure or injury. The tail cup approach may be hard to implement in younger and actively growing mice (e.g., before or around weaning). Some mice in our study developed self-inflicted skin lesions from over-grooming at the location where the tail cups come in contact with the body at the animal’s hind end. Thus, we concluded that the approach in its current implementation is limited to 2–3 weeks in adult animals.
Our device design reduced the risk of tail injury and necrosis described in previous works [33] and allows for emptying the cups only once every 24 h to reduce handling stress. Because host stress can affect the microbiota [58] and other physiological parameters, we included a mock tail cup group. Both TC-F and TC-M mice demonstrated a similar degree of weight loss (Additional file 1: Figure S3A) when compared with the WF and CTRL mice despite similar food intake rates across all four groups (Additional file 1: Figure S3B). Mice fitted with mock tail cups (TC-M) had microbial patterns and bile acid profiles similar to the CTRL mice, thus the effects on the upper GIT microbiota and bile acid profiles that we observed in non-coprophagic (TC-F) mice are not attributable to stress.
We believe that the tail cup approach is implementable in gnotobiotic settings (e.g., flexible film isolators and individually ventilated cages), which can aid studies that involve association of mice with defined microbial communities or with human-derived microbiota.
The non-coprophagic mouse model may be more relevant to humans
Using quantitative microbiota profiling, our study demonstrated that preventing self-reinoculation dramatically reduced the total levels of several prominent taxonomical groups of obligate anaerobes (e.g., Clostridiales, Bacteroidales, Erysipelotrichale) in the upper gastrointestinal microbiota of conventional mice. Despite these differences in taxa, levels of Lactobacillales in the small intestine and cecum, but not in the stomach, remained similar between coprophagic and non-coprophagic animals (Fig. 3c). The physiological significance of the maintained persistent population of Lactobacillales in the upper gastrointestinal tract (e.g., stomach or small intestine) and their overall consistent presence along the entire GIT [14, 59] for the host is not fully understood. However, Lactobacilli colonization in the stomach and small intestine has been shown to promote resistance to colonization by pathogens (reviewed in [60, 61]).
Compared with conventional (coprophagic) mice, the non-coprophagic mice displayed features of the small intestine microbiota and bile acid profiles that are more similar to the patterns seen in the small intestine of humans: orders of magnitude lower microbiota density, reduced abundance of obligate anaerobic flora and dominance of Lactobacillales, and a higher ratio of conjugated bile acids. These findings highlight the need to understand and control self-reinoculation in mouse models used to answer questions relevant to host-microbiota interactions in human health.
Self-reinoculation and microbial ecology in the mouse GIT
We observed that within the approximately 2-week timeframe of our study, the taxonomical diversity of the mouse large intestine microbiome was stable in the absence of persistent microbial self-reinoculation: all taxonomical groups at the order level observed in the cecum of coprophagic mice were present in the cecum of non-coprophagic mice, and vice versa.
The trending changes in the absolute abundances of several taxa (Bacterodales, Erysipelotrichales, and Betaproteobacteriales) in the large intestine of non-coprophagic mice may be the result of eliminated self-reinoculation and/or the consequence of the altered profile of bile acids entering the cecum from the small intestine, or other undetected changes in the biochemical environment. Additionally, changes in the absolute abundance of some taxa may lead to changes in the absolute abundances of other metabolically coupled taxa. It has been previously suggested that the degree of bile acid deconjugation may alter the microbiota profile [46]. Erysipelotrichales (including Turicibacter spp.) in the mouse ileum and cecum have been shown to be positively correlated with unconjugated ileal and cecal [62] and plasma [63] bile acids. Bacteroidales (including Muribaculum spp.) in the cecum increased upon dietary supplementation of unconjugated cholic and chenodeoxycholic acids [64] and Betaproteobacteriales (including Parasutterella spp.) were positively correlated with unconjugated primary and secondary bile acids [65, 66] in mice. Thus, the decrease in the fraction of unconjugated bile acids in the large intestine of non-coprophagic mice (Fig. 5b) may be responsible for the decreased absolute abundance of these three taxonomic groups in the cecum of non-coprophagic mice. It is of note that most of the published reports describe correlations between bile acid profiles and microbiota composition based on relative abundance data and without accounting for the inherent compositionality of relative abundance data [67], which is known to introduce inaccuracies in the correlation analysis [64, 68]. For improved correlation analysis, our study reports absolute abundances of the taxa, which could lead to discrepancies between such correlations observed in this study and previously published studies.
Stability of complex microbiomes in response to perturbations with and without continuous species reintroduction is an important subject of research in microbial ecology [69, 70]. Eliminating fecal ingestion provides a way to study stability and recovery of the mouse gut microbiota (e.g., in response to dietary change or antibiotic exposure [71]) in a way more relevant to modern humans. Thus, the non-coprophagic mouse model can significantly aid such research.
Self-reinoculation with fecal flora leads to altered bile acid profiles in the GIT
We demonstrated that changes to small intestine microbiota density and composition had pronounced effects on microbial function resulting in increased bile acid deconjugation in that segment of the GIT. Bile acid deconjugation is a microbiota-mediated process that in healthy humans is conventionally believed to take place in the distal small intestine (ileum) and in the large intestine [72] such that sufficient lipid emulsification (with conjugated bile acids) and absorption can take place in the small intestine by the time digesta reaches the ileum [73]. As a result of the much higher bile acid concentrations in the small intestine compared with the large intestine, altered deconjugation of bile acids in the small intestine may have more wide-ranging effects on the entire enterohepatic system. Our data indicate that bile acid deconjugation can take place in any segment of the small intestine of conventional healthy mice as a function of the microbial density and composition (Figs. 2a, 3 and 5b), which is consistent with previous findings in animal models and in humans with small intestinal bacterial overgrowth (SIBO) [74–78].
Strikingly, the very low degree of bile acid deconjugation in the small intestine of non-coprophagic mice in our study resembles profiles seen in germ-free animals [79–81], gnotobiotic animals colonized only with microbes incapable of deconjugating bile acids [82–85], and antibiotic-treated animals [86–88]. Our observations suggest a mechanistic link between the small intestine microbiota density and composition and the bile acid modification in this segment of the GIT. The small intestine of healthy human subjects is believed to harbor bile acids predominantly in the conjugated form [89], which further substantiates that (compared with coprophagic mice) the small intestine of non-coprophagic mice is more similar to the small intestine of a healthy human.
Although microbiota density and composition in the large intestine of coprophagic and non-coprophagic mice were largely similar, non-coprophagic mice had a higher fraction of bile acids that remained in the conjugated form in the large intestine (Fig. 4b), likely as a result of the bile acids entering the large intestine from the ileum predominantly in a conjugated form. Additionally, across all study groups, the total concentrations of bile acids in the small intestine were ~ 10-fold greater than in the large intestine. We therefore infer that in coprophagic mice, a greater absolute amount of bile acids underwent deconjugation in the small intestine than in the large intestine; i.e., in coprophagic mice, the small intestine contaminated with high loads of fecal flora was the primary site of bile acid deconjugation.
Regulation of bile acid deconjugation activity in the gut is considered a potential health-promoting modality in a number of contexts, including lowering blood cholesterol levels (reviewed in [90–92]). BSH-active probiotics can be a promising delivery vehicle for promoting increased bile acid deconjugation in the gut. Our study emphasizes the importance of controlling for self-reinoculation when using mice to study the effects of BSH-active microbial strains or probiotics [48, 93–98] (especially those with high selectivity for particular bile acid conjugates [47, 82, 85]) because conventional (coprophagic) mice already have pronounced BSH activity in their small intestines. A non-coprophagic mouse may be a better animal model in such studies.
Our findings also have implications for the use of conventional (coprophagic) mice in diet studies. Deconjugated bile acids are less effective than conjugated bile acids at lipid emulsification and fat micelle formation [74, 99]. Increased bile acid deconjugation in the small intestine of animals and humans can lead to lipid malabsorption and fat-soluble vitamin deficiency, and in extreme scenarios even to steatorrhea [77, 100]. Previous research has shown that the small intestine microbiota plays an important role in mediating the effect of high-fat diets on the host [101]; our results suggest that future studies of the microbiota-mediated effects of high-fat diets need to consider increased microbial bile acid deconjugation in the mouse intestine due to self-reinoculation with fecal flora.
Bile acid deconjugation is considered to be obligatory [84, 102, 103] before the secondary bile acid metabolism (believed to be predominantly occurring in the large intestine [72]) can take place. These reactions in many cases are carried out by different members of the microbiota. Thus, the reduction of the deconjugation activity in the small intestine of non-coprophagic mice and consequently lower availability of free primary bile acids for further microbial modification can explain the decrease in the secondary bile acid fraction (percentage of all bile acids) in the bile acid pool across the GIT and gallbladder bile of non-coprophagic mice in our study. A similar but more pronounced trend has been observed in rabbits [104]. Reduced oral intake and recycling of fecal secondary bile acids as a result of eliminating coprophagy may also be a contributing factor to the lower fraction of secondary bile acids in the total bile acid pool in the enterohepatic circulation in these animals.
Total bile acid levels in the stomach were similar in coprophagic and non-coprophagic mice (and agree with literature [104, 105]); however, bile acid profiles (including the fraction of total unconjugated and total secondary bile acids) were substantially different. Surprisingly, in all coprophagic mice the fraction of unconjugated bile acids in the stomach appeared to be intermediate between the profiles in the small intestine and in the large intestine (Fig. 5b), suggesting that the bile acids in the stomach of coprophagic mice could be accumulating from bile acids re-ingested in feces and bile acids refluxed from the duodenum. This pattern was not observed in non-coprophagic mice, suggesting that coprophagy may alter the bile acid profile in the upper GIT both directly (via re-ingestion of fecal metabolites) and indirectly (via altered microbiota function).
Inferences about microbial function in bile acid and drug modification
Our quantitative functional gene inference analysis predicted differential absolute abundance of the BSH orthologs between the small intestine of coprophagic and non-coprophagic mice (Fig. 4a). This approach has limitations associated with incomplete gene annotations, limited ability to infer metagenomes from the marker gene sequences when multiple microbial strains with similar 16S rRNA gene sequences exist [44, 45], difficulty to predict the exact gene expression and enzyme activity and specificity. To test our prediction about BSH we employed a targeted bile acid metabolomic analysis of mouse gastrointestinal samples and observed the differences in the small intestine bile acid deconjugation between coprophagic and non-coprophagic mice (Fig. 5b) that were in agreement with the differences in the inferred BSH gene abundances in the small intestine of those two types of animals (Fig. 4a). Interestingly, despite similar inferred BSH gene abundance in the cecum of coprophagic and non-coprophagic mice, the fraction of unconjugated bile acids in the cecum and colon of non-coprophagic mice was statistically significantly lower (Fig. 5b) compared with coprophagic mice.
Michaelis–Menten constants (Km) for many known BSH isoforms are in the range of hundreds of nanomoles [72]—similar to the levels of bile acids observed in the small intestine of all groups of mice in this study (Fig. 5a). Total bile acid levels in the cecum and colon were ~ 10-fold lower than those in the small intestine, and thus they were ~ 10-fold lower than the BSH Km (Fig. 5a). The predominantly conjugated form of the bile acids arriving into the cecum from the small intestine of non-coprophagic mice and their absolute concentration (~ 10-fold lower than BSH Km) can potentially explain the lower degree of bile acid deconjugation in the large intestine of these animals (compared with coprophagic mice) on the timescale of normal gastrointestinal transit.
This highlights the importance of considering functional inference (based on either taxonomy or in silico hidden state prediction [44, 45, 106, 107]) in the context of a variable biochemical environment (e.g., substrate availability) and the host gastrointestinal physiology (secretion, gastrointestinal transit, absorption and transport, etc.) and warrants functional validation (e.g., metabolomics). Additionally, the validity of functional inference based on 16S rRNA gene sequence counts (from next generation sequencing) versus absolute 16S rRNA gene sequence abundances (this study) should be further explored in future work.
We next explored the effects of self-reinoculation on the absolute abundance of microbial gene orthologs implicated in xenobiotic modification [108] in the small intestine, as microbiota-dependent drug modification and toxicity in the small intestine have been previously observed in rodents [109–119]. Many drugs administered to humans and mice both via enteral and parenteral routes after reaching the systemic circulation are transformed by the liver into conjugates (e.g., glucuronic acid, sulphate, or glutathione conjugates) and excreted with bile into the GIT lumen. Such transformations are believed to reduce the small intestine reabsorption of xenobiotics and promote their excretion from the body with stool. Alterations in the small intestine microbiota may also lead to increased hydrolysis of such conjugates by microbial enzymes and promote the local toxicity of the drug and enable its re-uptake from the small intestine (i.e., undergo enterohepatic circulation) [10, 116], resulting in an increase in the xenobiotic flux through the liver [120, 121] and to an overall microbiota-dependent change in drug pharmacokinetics.
As with the inferred differential BSH absolute abundances (correlating activity of which we confirmed with the bile acid deconjugation measurements), our analysis predicted differences in the absolute abundance (Fig. 4b, c) of the microbial gene orthologs responsible for drug conjugate hydrolysis (e.g., beta-glucuronidases, sulfohydrolases) between the small intestine of coprophagic and non-coprophagic mice. If this prediction is further experimentally confirmed, it would imply that self-reinoculation must be controlled for or taken into account when investigating drug pharmacology in mice.
Relevance of self-reinoculation in probiotics research
Many studies on probiotics and their effects on host animal physiology rely on repeated oral administration of live probiotic microorganisms to rodents. Our study suggests that self-reinoculation with live fecal flora in laboratory mice could both interfere with and introduce inconsistencies in live probiotic administration regimens. As has been stated earlier, particular attention should be given to self-reinoculation and its effects on the small intestine bile acid profile in studies aiming to evaluate the health effects of probiotics and other therapeutic modalities [48, 90–98] targeting bile acid deconjugation and metabolism.
Relevance of mouse models in human microbiota research
The role of mouse models in human microbiota research remains a subject of debate [13, 14, 122]. At the same time, the field is recognizing the importance of reproducibility in gut microbiota research that uses mouse models [58, 122]. Several recent studies have highlighted the variability in lab-mouse microbiota related to animal strains and sources of origin [36, 123–127]. Others have attempted to catalog a “normal” or “core” gut microbiome [128, 129] and its spatial organization [35, 36] and function [130] in laboratory and wild mice. Recently, the small intestine microbiome has become the focus of studies conducted in mice in the context of host physiology [101] and disease [4, 131]. Yet, little attention has been given to the impact of self-reinoculation on the gut microbiota spatial structure and function, or to how study outcomes might be affected by controlling (or not controlling) for this experimental parameter in mouse models.
Self-reinoculation in rodents may affect not only their native microbiota, but also individual microbial colonizers [24] (e.g., in gnotobiotic animals) and complex xenomicrobiota (e.g., in human microbiota-associated (HMA) mice). HMA mice have emerged as an important research model for dissecting the mechanistic connection between the gut microbiota and the host phenotype in health and disease, even though the field acknowledges its limitations [132, 133]. Compositional differences between the small intestine and large intestine microbiomes in primates and humans [12, 40, 41] appear to be more substantial than those reported for laboratory mice [35, 130]. Our study emphasizes that the compositional similarity between small and large intestine microbiota in conventional laboratory mice can be a result of self-reinoculation with fecal flora. Thus, the effects of self-reinoculation on the spatial organization and function of human microbiota in HMA mice warrant future exploration.
Conclusions
In conclusion, this study uses modern tools to demonstrate the importance of self-reinoculation in the context of microbial ecology and function within the mammalian gastrointestinal system. Our work highlights the importance of recognizing and properly controlling for self-reinoculation when murine studies analyzing small intestine microbiota, and its function intend to draw parallels with human physiology and pathophysiology. Additionally, spatial interrogation of the gut microbiota and its function in mouse models is important because even dramatic changes in the small intestine microbiome profile, function, and metabolome may be overlooked if only large intestine and stool samples are analyzed.
Methods
Experimental animals
All animal handling and procedures were performed in accordance with the California Institute of Technology (Caltech) Institutional Animal Care and Use Committee (IACUC). C57BL/6 J male specific-pathogen-free (SPF) mice were obtained at the age of 7–8 weeks from Jackson Laboratory (Sacramento, CA, USA) and housed four mice per cage. Two cohorts of animals were used: the first cohort was allowed to acclimate in the Caltech animal facility for 2 months and mice were 4 months old at the start of the study; the second cohort acclimated for 6 months and mice were 8 months old at the start of the study.
All animals were maintained on chow diet (PicoLab Rodent Diet 20 5053, LabDiet, St. Louis, MO, USA) and autoclaved water ad lib and subjected to a daily 13:11 light:dark cycle during acclimation and throughout the entire study. Mice were given measured amounts of food, and food intake during the experiment was measured by weighing the food during weekly cage changes and at the end time point for each animal. Body weight was measured at the start of the experiment, during weekly cage changes, and at the end time point.
Animal housing conditions
During the experiment, all mice were singly housed in autoclaved cages (Super Mouse 750, Lab Products, Seaford, DE, USA). The mice in the control (CTRL), mock tail cup (TC-M), and functional tail cup (TC-F) treatments were housed on heat-treated hardwood chip bedding (Aspen Chip Bedding, Northeastern Products, Warrensburg, NY, USA) and provided with tissue paper (Kleenex, Kimberly-Clark, Irving, TX, USA) nesting material. The mice in the WF treatment were housed on raised wire floors with a mesh size of 3 × 3 per square inch (#75016, Lab Products) and provided with floorless paper huts (#91291, Shepherd Specialty Papers, Watertown, TN, USA). A thin layer of woodchip bedding was added under the wire floors to absorb liquid waste from the animals (Additional file 1: Figure S1D).
Tail cup design and mounting
We designed the tail cups based on published literature [30, 134–137], including the locking mechanism [30]. Each cup was locked in place around the hind end of animals by anchoring to a tail sleeve designed with a perpendicular groove. Such tail sleeves allow for the cup to be held snugly against the animal so that the total weight of the tail cup is distributed along a large surface area of the tail skin, which minimizes complications. When mounted, the tail cups can freely rotate along the longitudinal axis, which ensures the locking mechanism does not strangulate the tail.
We hand-made the tail cups from 20 mL syringes (#4200.000 V0 Norm-Ject 20 mL Luer-Lock, Henke-Sass Wolf GmbH, Tuttlingen, Germany) as depicted on Additional file 1: Figure S1A-C. Multiple perforations were designed to accelerate desiccation of the captured fecal pellets. Lateral slits allowed for increasing the diameter of the locking edge; pressing on the slits with two fingers allowed tail cups to be quickly unfastened from tail sleeves. Mock tail cups were modified with wide gaps in the walls to allow the fecal pellets to fall out of the cup.
To prevent mice from gnawing on the plastic parts of the tail cups (which could create a jagged edge and lead to a subsequent injury), they were reinforced with metal flared rings made from stainless steel grommets (#72890, SS-4, C.S. Osborne, Harrison, NJ, USA) that were modified to reduce their size and weight. Metal rings were attached to tail cups using 4-mm-wide rubber rings cut from latex tubing (Amber Latex Rubber Tubing #62996–688, 1/2″ ID, 3/4″ OD; VWR, Radnor, PA, USA).
Tail sleeves were made from high-purity silicone tubing (HelixMark 60–411–51, 1/8“ ID, 1/4” OD; Helix Medical, Carpinteria, CA, USA). The tubing was split longitudinally, and a 2.0-mm-wide longitudinal strip of the wall was removed to accommodate for variable tail diameters among animals, and along the length of the tail to prevent uneven tail compression, and to facilitate uniform application of the tissue adhesive. The perpendicular tail cup mounting groove was made using a rotary tool (Craftsman #572.610530, Stanley Black & Decker, New Britain, CT, USA) equipped with a cutting disc (RD1, Perma-Grit Tools, Lincolnshire, UK). Each tail cup and sleeve together weighed approximately 4.12 g empty.
Before mounting the tail cups, animals were anesthetized for 10 min with isoflurane and placed on a heating pad to maintain body temperature. Sleeves were de-greased on the inside using 70% ethanol, and a veterinary tissue adhesive (GLUture Topical Adhesive #32046, Abbott Laboratories, Lake Bluff, IL, USA) was applied to the tail base. The adhesive was allowed to cure for 5 min, and then tail cups were mounted. Mice were returned back to their cages and allowed to recover from the anesthesia and ambulate.
Tail cups were emptied of fecal pellets daily at 08:00 AM. Mice were prompted to enter a restrainer [138] made from a black polypropylene 50-mL conical tube (TB5000 LiteSafe, Cole-Parmer, Vernon Hills, IL, USA), and the tail cups were unclipped and quickly emptied. Any residue on the tail cup was cleaned using a paper towel and Rescue solution (Virox Technologies, Oakville, ON, Canada) prior to the cups being remounted. Animals fitted with the mock tail cups were subjected to the identical procedure to match the handling conditions.
Tail cups were mounted on animals for a duration of between 12 and 20 days. All TC-F animals were time-matched with TC-M animals, (i.e., each animal from the TC-F group had a time-matched animal from the TC-M group handled and euthanized at the same time).
Sample collection and treatment
All mice were euthanized as approved by the Caltech IACUC in accordance with the American Veterinary Medical Association Guidelines on Euthanasia [139]. Mice were euthanized while under isoflurane anesthesia (delivered via a calibrated gas vaporizer in an induction chamber followed by maintenance on a nose cone) via cardiac puncture followed by cervical dislocation. Blood was collected using a 1-mL syringe (#309659, Becton Dickinson) and 21G × 1″ needle (#26414, EXELINT International, Redondo Beach, CA, USA).
Blood was immediately placed into K2EDTA plasma separation tubes (MiniCollect 450480, Greiner Bio-One GmbH, Kremsmünster, Austria), gently mixed, and stored on ice for up to 1 h prior to centrifugation. Bile and urine were collected directly from the gall and urinary bladders respectively using a 1-mL syringe (#4010.200 V0 Norm-Ject 1-mL Tuberculin Luer, Henke-Sass Wolf GmbH) and 27G × 1/2″ needle (#26400, EXELINT International) and stored on ice.
Fecal samples were collected if present at the time of euthanasia. The entire gastrointestinal tract was excised from the gastroesophageal junction to the anal sphincter and stored on ice during processing.
Plasma separation
Blood samples were centrifuged in the plasma separation tubes at 2000 RCF for 5 min at 4 °C. Plasma was separated and stored at − 80 °C.
Processing of GIT contents
To prepare samples for the main experimental analyses (Figs. 2, 3, and 4), each mouse GIT was split into stomach, three-equal-length thirds of the small intestine, cecum, and colon. Contents from each segment of the GIT were flushed out using 2–5 mL of cold (4 °C) sterile autoclaved saline solution (0.9% NaCl (#S5886, Sigma-Aldrich) in ultrapure water (Milli-Q, MilliporeSigma, Burlington, MA, USA) followed by very gentle squeezing with tweezers to avoid mucosal damage. All samples were stored on ice during processing.
An aliquot of each sample diluted in saline was concentrated by centrifugation at 25,000 RCF for 10 min at 4 °C. The supernatant was removed and the pellet was reconstituted in 9 volumes of 1× DNA/RNA Shield (DRS) solution (R1100-250, Zymo Research, Irvine, CA, USA), mixed by vortexing and stored at – 80 °C for future DNA extraction. Separate aliquots of each sample were stored at − 80 °C for the metabolomic (bile acid) analysis.
Preparation of GIT contents for the MPN-based microbial quantification and 16S rRNA gene amplicon sequencing (pilot study; Additional file 1: Figure S4B) was the same as above, but conducted inside a vinyl anaerobic chamber (Coy Laboratory Products, Grass Lake, MI, USA) in an atmosphere of 5% hydrogen, 10% carbon dioxide, and 85% nitrogen. All samples were maintained on ice and immediately processed for the culture-based assay.
Preparation of GIT mucosa
After flushing its contents, each segment of the GIT was gently rinsed in sterile cold (~ 4 °C) saline, cut longitudinally, and placed flat on a glass slide. The mucosa was scraped from the tissue gently using a second clean glass slide. Glass slides (VistaVision #16004-422, VWR) were sterilized by dry heat sterilization at 200 °C for at least 2 h. Mucosal scrapings were collected and combined with 9 volumes of DRS solution, mixed by vortexing, and stored at − 80 °C in preparation for DNA and RNA extraction.
Most probable number (MPN) assay
For the pilot study (Additional file 1: Figure S4A), the MPN assays (adapted from [140–144]) were performed on each GIT section (stomach, three sub-sections of the small intestine, cecum, and colon) from five mice fitted with functional tail cups and five control mice. The growth medium was brain heart infusion broth (Bacto BHI, #237500, Becton Dickinson, Franklin Lakes, NJ, USA), prepared in ultrapure water (Milli-Q), sterilized by autoclaving, allowed to cool to room temperature, and supplemented with 1.0 mg/L vitamin K1 (#L10575, Alfa Aesar, Haverhill, MA, USA), 5 mg/L hematin (#H3281, Sigma-Aldrich St. Louis, MO, USA), and 0.25 g/L l-cysteine (#168149, Sigma-Aldrich). The medium was allowed to equilibrate inside the anaerobic chamber for at least 24 h before use.
MPN assays were performed in clear, sterile, non-treated polystyrene 384-well plates (Nunc 265202, Thermo Fisher Scientific, Waltham, MA, USA). Two series of eight consecutive 10-fold serial dilutions were prepared from each sample in sterile autoclaved saline solution (equilibrated inside the anaerobic chamber for at least 24 h) on clear sterile non-treated polystyrene 96-well plates (Corning Costar 3370, Corning, NY, USA). We injected 10 μL of each serial dilution from each series into four (eight total per dilution) culture-medium replicates (wells) filled with 90 μL of the BHI-S broth medium.
Plates were sealed with a breathable membrane (Breath-Easy BEM-1, Diversified Biotech) and incubated for 5 days at 37.0 °C inside the anaerobic chamber. The plates were lidless for the first 24 h to facilitate uniform gas equilibration, then from 24 h to the end of the incubation period (120 h), a plastic lid was kept over the plates.
At the end of the incubation, the plates were scanned using a flatbed scanner (HP ScanJet 8250, Hewlett-Packard, Palo Alto, CA, USA) in the reflective mode with black background at 300 dpi resolution. The positive wells (replicates) were called by visually observing each acquired high-resolution image. The MPN for each sample was calculated using Microsoft Excel with the “Calc_MPN” macro [145].
DNA extraction
DNA was extracted from thawed GIT contents and mucosal sample aliquots preserved in DRS solution with the ZymoBIOMICS DNA Miniprep Kit (D4300, Zymo Research) according to the manufacturer’s instructions. Samples were homogenized on a bead beater (MiniBeadBeater-16, Model 607, Bio Spec Products, Bartlesville, OK, USA) for 5 min at the default speed of 3450 RPM. Quantitative recovery of DNA across multiple orders of microbial loads in the samples was previously verified in [38, 39].
DNA yield and purity in the extracts was evaluated via light absorbance (NanoDrop 2000c, Thermo Fisher Scientific) and via a fluorometric assay (Qubit dsDNA HS Assay Kit Q32854, Thermo Fisher Scientific) on a fluorometer (Invitrogen Qubit 3, Thermo Fisher Scientific).
Quantitative PCR (qPCR) for 16S rRNA gene DNA copy enumeration
The qPCR reactions were set up in triplicates for each DNA sample. A single replicate reaction volume of 15 μL contained 1.5 μL of the DNA extracts combined with the qPCR master mix (SsoFast EvaGreen Supermix, #172-5200, Bio-Rad Laboratories, Hercules, CA, USA), forward and reverse primers (synthesized by Integrated DNA Technologies, San Diego, CA, USA; Additional file 1: Table S1) at a final concentration of 500 nM, and ultrapure water (Invitrogen UltraPure DNase/RNase-Free Distilled Water 10977-015, Thermo Fisher Scientific). Reactions were set up in white 96-well PCR plates (#HSP9655, Bio-Rad Laboratories) sealed with a PCR tape (#MSB1001, Bio-Rad Laboratories).
The standard curve was built for each qPCR run based on the included series of 10-fold dilutions of the “standard” SPF mouse fecal DNA extract (with the quantified absolute concentration of 16S rRNA gene copies using digital PCR).
Amplification was performed with real-time fluorescence measurements (CFX96 Real-Time PCR Detection System, Bio-Rad Laboratories). Thermocycling conditions were used according to Additional file 1: Table S2. The qPCR data files were analyzed using Bio-Rad CFX Manager 3.1 (#1845000, Bio-Rad Laboratories) and the Cq data were exported to Microsoft Excel for further processing.
Digital PCR (dPCR) for absolute 16S rRNA gene DNA copy enumeration
Droplet digital PCR (ddPCR) reactions were set up according to [38, 39]. A single replicate reaction volume of 20 μL contained 2.0 μL of the DNA extracts combined with the ddPCR master mix (QX200 ddPCR EvaGreen Supermix, #1864033, Bio-Rad Laboratories), forward and reverse primers (synthesized by Integrated DNA Technologies; Additional file 1: Table S1) at final concentration of 500 nM each, and ultrapure water (Thermo Fisher Scientific).
Droplets were generated using DG8 cartriges (#1864008, Bio-Rad Laboratories), droplet generation oil (#1864006, Bio-Rad Laboratories), and DG8 gaskets (#1863009, Bio-Rad Laboratories) on a QX200 droplet generator (#1864002, Bio-Rad Laboratories) and analyzed using a QX200 Droplet Digital PCR System (#1864001, Bio-Rad Laboratories) using droplet reader oil (#1863004, Bio-Rad Laboratories). The ddPCR data files were analyzed using QuantaSoft Software (#1864011, Bio-Rad Laboratories), and the raw data were exported to Microsoft Excel for further processing.
Thermocycling conditions were used according to [38, 39] and Additional file 1: Table S3. Amplification was performed in PCR plates (#0030133374, Eppendorf, Hauppauge, NY, USA) sealed with pierceable heat seals (#1814040, Bio-Rad Laboratories) using PCR plate sealer (PX1, #1814000, Bio-Rad Laboratories) on a 96-deep well thermocycler (C1000 Touch, #1841100, Bio-Rad Laboratories).
16S rRNA gene DNA amplicon barcoding for next-generation sequencing (NGS)
PCR reactions were set up according to [38, 39], in triplicates for each DNA sample. Single-replicate reaction volumes of 30 μL contained 3 μL of the DNA extracts combined with the PCR master mix (5PRIME HotMasterMix, #2200400, Quantabio, Beverly, MA, USA), DNA intercalating dye (EvaGreen, #31000, Biotium, Fremont, CA, USA) at the concentration suggested by the manufacturer (× 1), barcoded forward and reverse primers (synthesized by Integrated DNA Technologies; Additional file 1: Table S1) at a final concentration of 500 nM each, and ultrapure water (Thermo Fisher Scientific). Reactions were set up in 0.2-mL white PCR tubes (#TLS0851) with flat optical caps (#TCS0803, Bio-Rad Laboratories). Thermocycling conditions were used according to [38, 39] and Additional file 1: Table S4. Amplification was performed with real-time fluorescence measurements (CFX96 Real-Time PCR Detection System, Bio-Rad Laboratories) and samples were amplified for a variable number of cycles until the mid-exponential (logarithmic) phase to maximize the amplicon yield and minimize artifacts related to over-amplification [146].
Digital PCR (dPCR) for Illumina library quantification
Single replicate reaction volume of 20 μL contained 2.0 μL of the diluted amplicon sample ligated with the Illumina adapters, 10 μL of ddPCR master mix (QX200 ddPCR EvaGreen Supermix, #186-4033, Bio-Rad Laboratories), forward and reverse primers (synthesized by Integrated DNA Technologies; Additional file 1: Table S1) targeting the Illumina P5 and P7 adapters respectively at the final concentration of 125 nM each, and ultrapure water (Invitrogen). Thermocycling conditions were used according to Additional file 1: Table S5. PCR amplification and droplet analysis were performed as above.
Barcoded sample quantification, pooling, library purification, and quality control
Triplicates of each barcoded amplicon sample were combined. Each sample was diluted × 105–107-fold and the molar concentration of barcoded amplicons was quantified using a home-brew ddPCR library quantification assay and KAPA SYBR FAST Universal qPCR Library Quantification Kit (#KK4824, Kapa Biosystems, Wilmington, MA, USA) according to the manufacturer’s instructions (the qPCR reaction was set up same as above).
Barcoded samples were pooled in equimolar amounts. The pooled library was purified using Agencourt AMPure XP beads (#A63880, Beckman Coulter, Brea, CA, USA) according to the manufacturer’s instructions and eluted with ultrapure water (Invitrogen).
The purified library was confirmed to have the 260 nm to 280 nm light absorbance ratio of > 1.8 using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific). The average amplicon size of approximately ~ 400 bp was confirmed with a High Sensitivity D1000 ScreenTape System (#5067-5584 and #5067-5585, Agilent Technologies, Santa Clara, CA, USA) using a 2200 TapeStation instrument (Agilent Technologies) and the Agilent 2200 TapeStation Software A.02.01 (Agilent Technologies).
The molar concentration of the pooled library was measured using the ddPCR and KAPA qPCR assays, and the library was submitted for NGS with the sequencing primers described in Additional file 1: Table S1.
Next-generation sequencing
The library was sequenced on a MiSeq instrument (Illumina, San Diego, CA, USA) in a 300-base-paired-end mode using a MiSeq Reagent Kit v3 (#MS-102-3003, Illumina). PhiX control spike-in was added at 15%.
PCR primer oligonucleotides (Additional file 1: Table S1)
The same universal microbial 16S rRNA gene V4 primers (modified from [147, 148] and validated in [38, 39] (Barlow JT, Bogatyrev SR, Ismagilov RF: A quantitative sequencing framework for absolute abundance measurements of mucosal and lumenal microbial communities, submitted)) targeting the V4 region of the 16S rRNA gene from the 519 to 806 positions were used for 16S rRNA gene DNA copy quantification and multiplexed microbial community profiling based on 16S rRNA gene amplicon sequencing. Reverse barcoded primers for 16S rRNA gene DNA amplicon barcoding were according to [148].
Primers targeting the P5 and P7 Illumina adapters for barcoded amplicon and pooled library quantification using the ddPCR assay were according to [147–151].
Sequencing read processing
Demultiplexed 2 × 300 reads were processed using the Qiime2-2019.01 pipeline [152]. DADA2 plugin [153] was used to filter (forward trimming, 5; forward truncation, 230; reverse trimming, 5; reverse truncation, 160), denoise, merge the paired-end sequences, and remove the chimeras. Taxonomic sequence (amplicon sequence variant, ASV) classification was performed using the classifier (available for download from [154]) trained [155] on the V4 515-806 bp regions of 16S rRNA gene sequences from the Silva rRNA reference database, release 132 [156] (available for download from [157]).
Functional gene inference analysis with PICRUSt2 [44, 45] was performed on the ASVs within the Qiime2 environment. Absolute and relative abundances of ASVs were normalized using the inferred 16S rRNA gene DNA copy counts. Obtained predicted metagenome data were used to calculate the normalized relative and absolute abundances of the gene orthologs of interest using Python tools (described below).
Sequencing data processing
Data handling, calculations, and statistical analyses were performed using Microsoft Excel with the Real Statistics Resource Pack [158], and the Python packages NumPy [159], Pandas [160], SciPy [161], and Statsmodels [162]. Plotting was performed with Matplotlib [163] and Seaborn [164]. All Python packages were run using IPython [165] within Jupyter notebooks [166] distributed with the Anaconda environment [167].
Frequency data for the 16S rRNA gene ASVs assigned to taxa in each sample were converted to relative abundances for each sample. Relative abundances then were converted to absolute abundances using the corresponding values of total 16S rRNA gene DNA loads obtained from the qPCR and ddPCR assays for each sample.
Absolute abundance data were then collapsed to the genus (Fig. 3a) or order (Fig. 3b, c) taxonomical levels using a custom-made Python function (confirmed to yield identical results to the “collapse” method of the Qiime2 “Taxa” plugin [152]). We defined contaminating taxa (from sample handling during collection or from the DNA extraction kit or PCR reagents) using two methods: taxa that were not present in at least 1 out of 16 cecum contents samples (4 mice out of 6 from each group × 4 groups), and taxa identified with a frequency-based contaminant identification [168] implemented by us in Python. Data for chloroplasts and mitochondria of plant origin (likely from the chow diet) were kept in the dataset for Fig. 3a and c and removed for Fig. 3b. Mean absolute abundances of taxa for each group were calculated, converted to relative abundances, and plotted in Fig. 3b.
Principal component analysis (PCA) of the relative abundance data (Additional file 1: Figure S4B) was performed on centered log ratio (CLR)-transformed [169, 170] (after a pseudocount equal to the minimal non-zero sequence count in the dataset was added to all zero values) genus-level relative abundance data using the Python Scikit-learn package [171].
PCA of the absolute abundance data (Fig. 3a) was performed on log10-transformed and centered standardized (converted to normally distributed data with mean = 0 and standard deviation = 1) [172] genus-level absolute abundance data using the Python Scikit-learn package [171].
Bile acid analysis
Reagents
TαMCA, TβMCA, TωMCA, THCA, αMCA, βMCA, ωMCA, HCA, HDCA, MCA, GCDCA, GDCA, and GCA (Additional file 1: Table S6) were obtained from Steraloids (Newport, RI, USA).
TCA, CA, DCA, TCDCA, TDCA, TUDCA, TLCA, CDCA, UDCA, LCA, D4-TCA, D4-DCA, D4-CA, D4-TDCA, D4-GLCA, D4-GUDCA, D4-GCDCA, D4-GCA, and D4-GDCA (Additional file 1: Table S6) were obtained from Isosciences (Ambler, PA, USA).
LC/MS grade acetonitrile (#A955-500), water (#W6500), and formic acid (#A117-50) were obtained from Thermo Fisher Scientific.
Sample preparation
To overcome sample buffering (pH issues), samples were extracted (using a protocol adapted and modified from [83–85]) in 9 volumes of ethanol with 0.5% formic acid and 9 different heavy isotope (D4) internal standards at 5 μM. D4 internal standards were taurocholic acid (TCA), cholic acid (CA), deoxycholic acid (DCA), taurodeoxycholic acid (TDCA), glycocholic acid (GCA), glycolithocholic acid (GLCA), glycoursodeoxycholic acid (GUDCA), glycochenodeoxycholic acid (GCDCA), and glycodeoxycholic acid (GDCA). Samples were heated for 1 h at 70 °C with orbital shaking at 900 RPM. Solids were precipitated by centrifugation at 17,000 RCF for 15 min at 4 °C. Supernatants were decanted as 10% of the original sample (e.g., 100 μL of a 1-mL extraction sample) and evaporated at approximately 100 mTorr at RT on a rotovap (Centrivap Concentrator #7810016, Labconco, Kansas City, MO, USA). The evaporated samples were reconsistuted at 100 × dilution from the original sample (e.g., 100 μL decanted solution is resuspended at 1 mL) in 20% acetonitrile, 80% water with 0.1% formic acid.
Due to small volumes, gall bladder bile samples were first diluted in 10 volumes of 100% ethanol (#3916EA, Decon Labs, King of Prussia, PA, USA). The ethanol-based dilutions were combined with 9 volumes of ultrapure water (Invitrogen) and subjected to extraction as above.
Each 10-μL extracted and reconsistuted sample injection was analyzed on a Waters Acquity UPLC coupled to a Xevo-qTOF Mass Spectrometer (Waters, Manchester, UK) using an Acquity UPLC HSS T3 1.8 micron, 2.1 × 10-mm column (# 186003539) and Acquity UPLC HSS T3 1.8 micron Guard Column (# 186003976). Needle wash was two parts isopropanol, one part water, and one part acetonitrile. Purge solvent was 5% acetonitrile in water. A pooled quality control sample was run every eight injections to correct for drift in response.
Mass spectrometer instrument parameters were as follows: Capillary Voltage 2.4 kV, Collision Energy 6.0 eV, Sampling Cone 90 V, Source Offset 40 V, Source 120 °C, desolvation gas temperature 550 °C, cone gas 50 L/h, and desolvation Gas 900 L/h. Time-of-flight mass spectra were collected in resolution mode, corresponding to 30,000 m/Δm. The mass axis was calibrated with sodium formate clusters and locked using leucine enkephalin.
A seven point external calibration curve was collected three times within the run from 0.05 to 30 μM of the bile acid standards (0.05, 0.1, 0.5, 1, 5, 10, and 30 μM). External standards were taurocholic acid (TCA), tauro-alpha-muricholic acid (TαMCA), tauro-beta-muricholic acid (TβMCA), tauro-omega-muricholic acid (TωMCA), tauro-hyocholic acid (THCA), tauro-deoxycholic acid (TDCA), tauro-ursodeoxycholic acid (TUDCA), tauro-chenodeoxycholic acid (TCDCA), taurolithocholic acid (TLCA), glyco-cholic acid (GCA), glyco-hyocholic acid (GHCA), glyco-deoxycholic acid (GDCA), glyco-hyodeoxycholic acid (GHDCA), cholic acid (CA), alpha-muricholic acid (αMCA), beta-muricholic acid (βMCA), omega-muricholic acid (ωMCA), hyocholic acid (HCA, also known as γ-muricholic acid), deoxycholic acid (DCA), chenodeoxycholic acid (CDCA), ursodeoxycholic acid (UDCA), hyodeoxycholic acid (HDCA), murocholic acid (murideoxycholic acid, MDCA), lithocholic acid (LCA), glycolithocholic acid (GLCA), glycourosodeoxycholic acid (GUDCA), and glycochenodeoxycholic acid (GCDCA). It was not possible to resolve UDCA and HDCA; so the sum was reported.
Integrated areas of extracted ion chromatograms were obtained using QuanLynx (Waters, Milford, MA, USA) and a mass extraction window of 10 mDa. Final corrections accounting for drift in instrumental sensitivity were performed in Microsoft Excel.
Elution gradient
Samples were eluted using the following gradient of water with 0.1% formic acid (“A”) and balance of acetonitrile with 0.1% formic acid:
0 min, 0.55 mL/min at 68% A
2 min, 0.55 mL/min at 60% A, 10 curve
5 min, 0.55 mL/min at 40% A, 5 curve
6 min, 1.1 mL/min at 0% A, 10 curve
6.2 min, 1.2 mL/min at 0% A, 6 curve
6.5 min, 1.47 mL/min at 0% A, 6 curve
8.9 min, 1.5 mL/min at 0% A, 6 curve
9.0 min, 0.9 mL/min at 68% A, 6 curve
10 min, 0.55 mL/min at 68% A, 6 curve
Bile acid data processing
Bile acid data analysis was performed using the tools described in “Sequencing data processing.”
Supplementary information
Additional file 1: Figure S1. Tail cup design and experimental setup for preventing coprophagy. Figure S2. Mounting of functional tail cups onto mice. Figure S3. Body weight changes across all groups of mice in relation to food intake over the course of the study. Figure S4. Quantification of the culturable microbial load and microbiota profile along the entire GIT of mice fitted with functional tail cups (TC-F) and control mice (CTRL). Figure S5. Bile acid profiles in gallbladder bile and in lumenal contents along the entire GIT. Table S1. Primer oligonucleotide sequences used in the study. Table S2. Thermocycling parameters for the quantitative PCR (qPCR) assay for 16S rRNA gene DNA copy quantification. Table S3. Thermocycling parameters for the digital PCR (dPCR) assay for absolute 16S rRNA gene DNA copy quantification. Table S4. Thermocycling parameters for the 16S rRNA gene DNA amplicon barcoding PCR reaction for next generation sequencing (NGS). Table S5. Thermocycling parameters for the digital PCR (dPCR) assay for barcoded amplicon and Illumina NGS library quantification. Table S6. Reagents and chemical standards used in the bile acid metabolomics assay. Table S7. Bile acid concentrations in gallbladder bile and in lumenal contents along the entire GIT.
Acknowledgements
We thank Karen Lencioni, Janet Baer, the Caltech Office of Laboratory Animal Resources, veterinary technicians at the Church Animal Facility for experimental resources. We thank Liang Ma for the introduction to 16S rRNA gene amplicon sequencing, Heidi Klumpe for her assistance with the preliminary MPN experiments, and Justin Bois for the introduction to data analysis in Python. S.R.B. would like to thank Kimberly Zhou for the personal feedback on the project and inspiration. This project benefited from the use of instrumentation made available by the Caltech Environmental Analysis Center and technical support from Nathan Dalleska. We thank Natasha Shelby for contributions to writing and editing this manuscript.
Authors’ contributions
SRB Conception, mouse tail cup development, animal study execution, animal study sample processing for quantitative 16S rRNA gene amplicon sequencing, quantitative 16S rRNA gene amplicon sequencing and data analysis, animal study sample processing for metabolomic analysis, bile acid metabolomics data analysis, manuscript preparation. JCR Metabolomics method development and validation, animal study sample processing for metabolomic analysis, UPLC-MS instrument setup and sample analysis, chromatography and mass spectra data analysis. RFI Project supervision and administration, acquisition of funding, manuscript review and editing. All authors read and approved the final manuscript.
Funding
This work was supported in part by a Kenneth Rainin Foundation Innovator Award (2018–1207), Army Research Office (ARO) Multidisciplinary University Research Initiative (MURI) contract #W911NF-17-1-0402, and the Jacobs Institute for Molecular Engineering for Medicine. National Science Foundation (NSF) Emerging Frontiers in Research and Innovation Award Grant 1137089. The funders had no role in the design of the study, collection, analysis or interpretation of data, nor in the writing of the manuscript.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. Sequencing data (paired end reads in FASTQ) and a manifest file for analysis in Qiime2 are available under a CC-BY license via CaltechDATA: 10.22002/D1.1295. Supplementary Information includes a zip file containing all sequencing sample metadata, numerical microbial quantification data (16S copies from the main study + MPN from the pilot study), Qiime2 sequencing output data, PICRUSt2 output data, numerical bile acid analysis data, numerical body weight data, numerical food intake data, and analytical scripts (iPython Notebooks) for all figures and statistical analyses in the manuscript.
Ethics approval and consent to participate
All animal handling and procedures were performed in accordance with the California Institute of Technology (Caltech) Institutional Animal Care and Use Committee (IACUC).
Consent for publication
Not applicable
Competing interests
The contents of this article are the subject of a patent application filed by Caltech.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s40168-020-0785-4.
References
- 1.Helander HF, Fändriks L. Surface area of the digestive tract - revisited. Scand J Gastroenterol. 2014;49:681–689. doi: 10.3109/00365521.2014.898326. [DOI] [PubMed] [Google Scholar]
- 2.El Aidy S, van den Bogert B, Kleerebezem M. The small intestine microbiota, nutritional modulation and relevance for health. Curr Opin Biotechnol. 2015;32:14–20. doi: 10.1016/j.copbio.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Saffouri GB, Shields-Cutler RR, Chen J, Yang Y, Lekatz HR, Hale VL, et al. Small intestinal microbial dysbiosis underlies symptoms associated with functional gastrointestinal disorders. Nat Commun. 2019;10:2012. doi: 10.1038/s41467-019-09964-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown EM, Wlodarska M, Willing BP, Vonaesch P, Han J, Reynolds LA, et al. Diet and specific microbial exposure trigger features of environmental enteropathy in a novel murine model. Nat Commun. 2015;6:7806. doi: 10.1038/ncomms8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donowitz JR, Petri WA. Pediatric small intestine bacterial overgrowth in low-income countries. Trends Mol Med. 2015;21:6–15. doi: 10.1016/j.molmed.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ierardi E, Losurdo G, Sorrentino C, Giorgio F, Rossi G, Marinaro A, et al. Macronutrient intakes in obese subjects with or without small intestinal bacterial overgrowth: an alimentary survey. Scand J Gastroenterol. 2016;51:277–280. doi: 10.3109/00365521.2015.1086020. [DOI] [PubMed] [Google Scholar]
- 7.Giamarellos-Bourboulis EJ, Pyleris E, Barbatzas C, Pistiki A, Pimentel M. Small intestinal bacterial overgrowth is associated with irritable bowel syndrome and is independent of proton pump inhibitor usage. BMC Gastroenterol. 2016;16:67. doi: 10.1186/s12876-016-0484-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah A, Morrison M, Holtmann GJ. Gastroduodenal “Dysbiosis”: a new clinical entity. Curr Treat Options Gastroenterol. 2018;16:591–604. doi: 10.1007/s11938-018-0207-x. [DOI] [PubMed] [Google Scholar]
- 9.Shah A, Morrison M, Burger D, Martin N, Rich J, Jones M, et al. Systematic review with meta-analysis: the prevalence of small intestinal bacterial overgrowth in inflammatory bowel disease. Aliment Pharmacol Ther. 2019;49:624–635. doi: 10.1111/apt.15133. [DOI] [PubMed] [Google Scholar]
- 10.Muraki M, Fujiwara Y, Machida H, Okazaki H, Sogawa M, Yamagami H, et al. Role of small intestinal bacterial overgrowth in severe small intestinal damage in chronic non-steroidal anti-inflammatory drug users. Scand J Gastroenterol. 2014;49:267–273. doi: 10.3109/00365521.2014.880182. [DOI] [PubMed] [Google Scholar]
- 11.Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y. Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol. 2005;54(Pt 11):1093–1101. doi: 10.1099/jmm.0.45935-0. [DOI] [PubMed] [Google Scholar]
- 12.Yasuda K, Oh K, Ren B, Tickle TL, Franzosa EA, Wachtman LM, et al. Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host Microbe. 2015;17:385–391. doi: 10.1016/j.chom.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen TLA, Vieira-Silva S, Liston A, Raes J. How informative is the mouse for human gut microbiota research? Dis Model Mech. 2015;8:1–16. doi: 10.1242/dmm.017400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hugenholtz F, de Vos WM. Mouse models for human intestinal microbiota research: a critical evaluation. Cell Mol Life Sci. 2018;75:149–160. doi: 10.1007/s00018-017-2693-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenagy GJ, Hoyt DF. Reingestion of feces in rodents and its daily rhythmicity. Oecologia. 1979;44:403–409. doi: 10.1007/BF00545245. [DOI] [PubMed] [Google Scholar]
- 16.Ebino KY. Studies on coprophagy in experimental animals. Jikken Dobutsu. 1993;42:1–9. doi: 10.1538/expanim1978.42.1_1. [DOI] [PubMed] [Google Scholar]
- 17.Soave O, Brand CD. Coprophagy in animals: a review. Cornell Vet. 1991;81:357–364. [PubMed] [Google Scholar]
- 18.Ebino KY, Amao H, Suwa T, Kuwabara Y, Saito TR, Takahashi KW. Coprophagy in the germfree mouse. Jikken Dobutsu. 1987;36:33–37. doi: 10.1538/expanim1978.36.1_33. [DOI] [PubMed] [Google Scholar]
- 19.Ebino KY, Suwa T, Kuwabara Y, Saito TR, Takahashi KW. Analyses of constituents of feces and the effect of a vitamin B12 fortified diet on coprophagy in the mouse. Jikken Dobutsu. 1986;35:381–386. doi: 10.1538/expanim1978.35.4_381. [DOI] [PubMed] [Google Scholar]
- 20.Smith HW. Observations on the flora of the alimentary tract of animals and factors affecting its composition. J Pathol Bacteriol. 1965;89:95–122. doi: 10.1002/path.1700890112. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi KW, Ebino KY, Saito TR, Imamichi T. Strain difference in Coprophagous behavior in laboratory mice (Mus musculus) Zool Sci. 1985;2:p249–p255. [Google Scholar]
- 22.Klaasen HLBM, Koopman JP, Scholten PM, Van Den Brink ME, Theeuwes AGM. Effect of preventing Coprophagy on colonisation by segmented filamentous Bacteria in the small bowel of mice. Microb Ecol Health Dis. 1990;3:99–103. doi: 10.3109/08910609009140123. [DOI] [Google Scholar]
- 23.Lev M, Alexander RH, Levenson SM. Stability of the Lactobacillus population in feces and stomach contents of rats prevented from coprophagy. J Bacteriol. 1966;92:13–16. doi: 10.1128/JB.92.1.13-16.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Syed SA, Abrams GD, Freter R. Efficiency of various intestinal bacteria in assuming normal functions of enteric flora after association with germ-free mice. Infect Immun. 1970;2:376–386. doi: 10.1128/IAI.2.4.376-386.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitzgerald RJ, Gustafsson BE, McDaniel EG. Effects of coprophagy prevention on intestinal microflora in rats. J Nutr. 1964;84:155–160. doi: 10.1093/jn/84.2.155. [DOI] [PubMed] [Google Scholar]
- 26.Gustafsson BE, Fitzgerald RJ. Alteration in intestinal microbial flora of rats with tail cups to prevent coprophagy. Exp Biol Med. 1960;104:319–322. doi: 10.3181/00379727-104-25821. [DOI] [PubMed] [Google Scholar]
- 27.George SE, Kohan MJ, Claxton LD, Walsh DB. Acute colonization study of polychlorinated biphenyl-degrading pseudomonads in the mouse intestinal tract: comparison of single and multiple exposures. Environ Toxicol Chem. 1989;8:123–131. doi: 10.1002/etc.5620080203. [DOI] [Google Scholar]
- 28.Roscoe MH. Spontaneous Cures in Rats reared upon a Diet devoid of Vitamin B and Antineuritic Vitamin. J Hyg (Lond) 1927;27:103–107. doi: 10.1017/S0022172400031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnes RH, Fiala G. Effects of the prevention of coprophagy in the rat. I Growth studies J Nutr. 1958;64:533–540. doi: 10.1093/jn/64.4.533. [DOI] [PubMed] [Google Scholar]
- 30.Barnes RH, Fiala G, McGehee B, Brown A. Prevention of Coprophagy in the rat. J Nutr. 1957;63:489–498. doi: 10.1093/jn/63.4.489. [DOI] [PubMed] [Google Scholar]
- 31.Barnes RH. Nutritional implications of coprophagy. Nutr Rev. 1962;20:289–291. doi: 10.1111/j.1753-4887.1962.tb04498.x. [DOI] [PubMed] [Google Scholar]
- 32.Hörnicke H, Björnhag G. Coprophagy and related strategies for digesta utilization. In: Ruckebusch Y, Thivend P, editors. Digestive physiology and metabolism in ruminants: proceedings of the 5th international symposium on ruminant physiology, held at Clermont --- Ferrand, on 3rd--7th September, 1979. Dordrecht: Springer Netherlands; 1980. pp. 707–730. [Google Scholar]
- 33.Ebino KY, Yoshinaga K, Saito TR, Takahashi KW. A simple method for prevention of coprophagy in the mouse. Lab Anim. 1988;22:1–4. doi: 10.1258/002367788780746548. [DOI] [PubMed] [Google Scholar]
- 34.Zmora N, Zilberman-Schapira G, Suez J, Mor U, Dori-Bachash M, Bashiardes S, et al. Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell. 2018;174:1388–1405.e21. doi: 10.1016/j.cell.2018.08.041. [DOI] [PubMed] [Google Scholar]
- 35.Gu S, Chen D, Zhang J-N, Lv X, Wang K, Duan L-P, et al. Bacterial community mapping of the mouse gastrointestinal tract. PLoS One. 2013;8:e74957. doi: 10.1371/journal.pone.0074957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki TA, Nachman MW. Spatial heterogeneity of gut microbial composition along the gastrointestinal tract in natural populations of house mice. PLoS One. 2016;11:e0163720. doi: 10.1371/journal.pone.0163720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ericsson AC, Gagliardi J, Bouhan D, Spollen WG, Givan SA, Franklin CL. The influence of caging, bedding, and diet on the composition of the microbiota in different regions of the mouse gut. Sci Rep. 2018;8:4065. doi: 10.1038/s41598-018-21986-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bogatyrev SR, Ismagilov RF. Quantitative microbiome profiling in lumenal and tissue samples with broad coverage and dynamic range via a single-step 16S rRNA gene DNA copy quantification and amplicon barcoding. 2020. bioRxiv. https://www.biorxiv.org/content/10.1101/2020.01.22.914705v1.
- 39.Bogatyrev, SR. Development of analytical tools and animal models for studies of small-intestine dysbiosis. Dissertation (Ph.D.), California Institute of Technology. 2020. 10.7907/VJDZ-7B52.
- 40.Seekatz AM, Schnizlein MK, Koenigsknecht MJ, Baker JR, Hasler WL, Bleske BE, et al. Spatial and temporal analysis of the stomach and small intestinal microbiota in fasted healthy humans. mSphere. 2019;4. 10.1128/mSphere.00126-19. [DOI] [PMC free article] [PubMed]
- 41.Vuik F, Dicksved J, Lam SY, Fuhler GM, van der Laan L, van de Winkel A, et al. Composition of the mucosa-associated microbiota along the entire gastrointestinal tract of human individuals. United Eur Gastroenterol J. 2019;7:897–907. doi: 10.1177/2050640619852255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Z, Malmer D, Langille MGI, Way SF, Knight R. Which is more important for classifying microbial communities: who’s there or what they can do? ISME J. 2014;8:2357–2359. doi: 10.1038/ismej.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Contijoch EJ, Britton GJ, Yang C, Mogno I, Li Z, Ng R, et al. Gut microbiota density influences host physiology and is shaped by host and microbial factors. Elife. 2019;8. 10.7554/eLife.40553. [DOI] [PMC free article] [PubMed]
- 44.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Douglas GM, Maffei VJ, Zaneveld J, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv. 2019:672295. 10.1101/672295.
- 46.Foley MH, O’Flaherty S, Barrangou R, Theriot CM. Bile salt hydrolases: gatekeepers of bile acid metabolism and host-microbiome crosstalk in the gastrointestinal tract. PLoS Pathog. 2019;15:e1007581. doi: 10.1371/journal.ppat.1007581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong Z, Lee BH. Bile salt hydrolases: structure and function, substrate preference, and inhibitor development. Protein Sci. 2018;27:1742–1754. doi: 10.1002/pro.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Joyce SA, Shanahan F, Hill C, Gahan CGM. Bacterial bile salt hydrolase in host metabolism: potential for influencing gastrointestinal microbe-host crosstalk. Gut Microbes. 2014;5:669–674. doi: 10.4161/19490976.2014.969986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Role of the Gut Flora in Toxicity and Cancer. 1st ed. London: Academic Press Limited; 1988. 10.1016/B978-0-12-599920-5.X5001-8.
- 50.Klaassen CD, Cui JY. Review: mechanisms of how the intestinal microbiota alters the effects of drugs and bile acids. Drug Metab Dispos. 2015;43:1505–1521. doi: 10.1124/dmd.115.065698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schneider KM, Albers S, Trautwein C. Role of bile acids in the gut-liver axis. J Hepatol. 2018;68:1083–1085. doi: 10.1016/j.jhep.2017.11.025. [DOI] [PubMed] [Google Scholar]
- 52.Wahlström A, Sayin SI, Marschall H-U, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 53.Ridlon JM, Bajaj JS. The human gut sterolbiome: bile acid-microbiome endocrine aspects and therapeutics. Acta Pharm Sin B. 2015;5:99–105. doi: 10.1016/j.apsb.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Northfield TC, McColl I. Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut. 1973;14:513–518. doi: 10.1136/gut.14.7.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tso P, Balint JA, Bishop MB, Rodgers JB. Acute inhibition of intestinal lipid transport by Pluronic L-81 in the rat. Am J Phys. 1981;241:G487–G497. doi: 10.1152/ajpgi.1981.241.6.G487. [DOI] [PubMed] [Google Scholar]
- 56.Phan CT, Tso P. Intestinal lipid absorption and transport. Front Biosci. 2001;6:D299–D319. doi: 10.2741/Phan. [DOI] [PubMed] [Google Scholar]
- 57.Metta V. Chalam, Nash Louis, Johnson B. Connor. A Tubular Coprophagy-Preventing Cage for the Rat. The Journal of Nutrition. 1961;74(4):473–476. doi: 10.1093/jn/74.4.473. [DOI] [Google Scholar]
- 58.Franklin CL, Ericsson AC. Microbiota and reproducibility of rodent models. Lab Anim (NY). 2017;46:114–122. doi: 10.1038/laban.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tannock GW. The lactic acid Bacteria volume 1. Boston: Springer US; 1992. The lactic microflora of pigs, mice and rats; pp. 21–48. [Google Scholar]
- 60.Wang Z-K, Yang Y-S. Upper gastrointestinal microbiota and digestive diseases. World J Gastroenterol. 2013;19:1541–1550. doi: 10.3748/wjg.v19.i10.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liévin-Le Moal V, Servin AL. Anti-infective activities of lactobacillus strains in the human intestinal microbiota: from probiotics to gastrointestinal anti-infectious biotherapeutic agents. Clin Microbiol Rev. 2014;27:167–199. doi: 10.1128/CMR.00080-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Theriot CM, Bowman AA, Young VB. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere. 2016;1. 10.1128/mSphere.00045-15. [DOI] [PMC free article] [PubMed]
- 63.Kemis JH, Linke V, Barrett KL, Boehm FJ, Traeger LL, Keller MP, et al. Genetic determinants of gut microbiota composition and bile acid profiles in mice. PLoS Genet. 2019;15:e1008073. doi: 10.1371/journal.pgen.1008073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morton JT, Sanders J, Quinn RA, McDonald D, Gonzalez A, Vázquez-Baeza Y, et al. Balance trees reveal microbial niche differentiation. mSystems. 2017;2. 10.1128/mSystems.00162-16. [DOI] [PMC free article] [PubMed]
- 65.Dey N, Wagner VE, Blanton LV, Cheng J, Fontana L, Haque R, et al. Regulators of gut motility revealed by a gnotobiotic model of diet-microbiome interactions related to travel. Cell. 2015;163:95–107. doi: 10.1016/j.cell.2015.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G, et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut. 2016;65:830–839. doi: 10.1136/gutjnl-2015-310585. [DOI] [PubMed] [Google Scholar]
- 67.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome datasets are compositional: and this is not optional. Front Microbiol. 2017;8:2224. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis. 2015;26:27663. doi: 10.3402/mehd.v26.27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coyte KZ, Schluter J, Foster KR. The ecology of the microbiome: networks, competition, and stability. Science. 2015;350:663–666. doi: 10.1126/science.aad2602. [DOI] [PubMed] [Google Scholar]
- 71.Ng KM, Aranda-Diaz A, Tropini C, Frankel MR, Van Treuren WW, Laughlin C, et al. Recovery of the gut microbiota after antibiotics depends on host diet and environmental reservoirs. bioRxiv. 2019. 10.1101/717686.
- 72.Ridlon JM, Kang D-J, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 73.Enright EF, Griffin BT, Gahan CGM, Joyce SA. Microbiome-mediated bile acid modification: role in intestinal drug absorption and metabolism. Pharmacol Res. 2018;133:170–186. doi: 10.1016/j.phrs.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 74.Kim YS, Spritz N, Blum M, Terz J, Sherlock P. The role of altered bile acid metabolism in the steatorrhea of experimental blind loop. J Clin Invest. 1966;45:956–962. doi: 10.1172/JCI105411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tabaqchali S, Hatzioannou J, Booth CC. Bile-salt deconjugation and steatorrhoea in patients with the stagnant-loop syndrome. Lancet (London, England) 1968;2:12–16. doi: 10.1016/s0140-6736(68)92888-2. [DOI] [PubMed] [Google Scholar]
- 76.Northfield TC. Intraluminal precipitation of bile acids in stagnant loop syndrome. Br Med J. 1973;2:743–745. doi: 10.1136/bmj.2.5869.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Donaldson RM. Studies on the pathogenesis of steatorrhea in the blind loop syndrome. J Clin Invest. 1965;44:1815–1825. doi: 10.1172/JCI105289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shindo K, Machida M, Fukumura M, Koide K, Yamazaki R. Omeprazole induces altered bile acid metabolism. Gut. 1998;42:266–271. doi: 10.1136/gut.42.2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 80.Madsen D, Beaver M, Chang L, Bruckner-Kardoss E, Wostmann B. Analysis of bile acids in conventional and germfree rats. J Lipid Res. 1976;17:107–111. [PubMed] [Google Scholar]
- 81.Claus SP, Tsang TM, Wang Y, Cloarec O, Skordi E, Martin F-P, et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol. 2008;4:219. doi: 10.1038/msb.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yao L, Seaton SC, Ndousse-Fetter S, Adhikari AA, DiBenedetto N, Mina AI, et al. A selective gut bacterial bile salt hydrolase alters host metabolism. Elife. 2018;7. 10.7554/eLife.37182. [DOI] [PMC free article] [PubMed]
- 83.Narushima S, Itoha K, Miyamoto Y, Park S-H, Nagata K, Kuruma K, et al. Deoxycholic acid formation in gnotobiotic mice associated with human intestinal bacteria. Lipids. 2006;41:835–843. doi: 10.1007/s11745-006-5038-1. [DOI] [PubMed] [Google Scholar]
- 84.Narushima S, Itoh K, Takamine F, Uchida K. Absence of cecal secondary bile acids in gnotobiotic mice associated with two human intestinal bacteria with the ability to dehydroxylate bile acids in vitro. Microbiol Immunol. 1999;43:893–897. doi: 10.1111/j.1348-0421.1999.tb01224.x. [DOI] [PubMed] [Google Scholar]
- 85.Chikai T, Nakao H, Uchida K. Deconjugation of bile acids by human intestinal bacteria implanted in germ-free rats. Lipids. 1987;22:669–671. doi: 10.1007/bf02533948. [DOI] [PubMed] [Google Scholar]
- 86.Gustafsson BE, Gustafsson J, Carlstedt-Duke B. Prolonged induction of germfree bile acid pattern in conventional rats by antibiotics. Acta Med Scand. 1977;201:155–160. doi: 10.1111/j.0954-6820.1977.tb15673.x. [DOI] [PubMed] [Google Scholar]
- 87.Feighner SD, Dashkevicz MP. Subtherapeutic levels of antibiotics in poultry feeds and their effects on weight gain, feed efficiency, and bacterial cholyltaurine hydrolase activity. Appl Environ Microbiol. 1987;53:331–336. doi: 10.1128/AEM.53.2.331-336.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuribayashi H, Miyata M, Yamakawa H, Yoshinari K, Yamazoe Y. Enterobacteria-mediated deconjugation of taurocholic acid enhances ileal farnesoid X receptor signaling. Eur J Pharmacol. 2012;697:132–138. doi: 10.1016/j.ejphar.2012.09.048. [DOI] [PubMed] [Google Scholar]
- 89.Tabaqchali S, Okubadejo OA, Neale G, Booth CC. Influence of abnormal bacterial flora on small intestinal function. Proc R Soc Med. 1966;59:1244–1246. doi: 10.1177/003591576605901209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Begley M, Hill C, Gahan CGM. Bile salt hydrolase activity in probiotics. Appl Environ Microbiol. 2006;72:1729–1738. doi: 10.1128/AEM.72.3.1729-1738.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jones ML, Tomaro-Duchesneau C, Martoni CJ, Prakash S. Cholesterol lowering with bile salt hydrolase-active probiotic bacteria, mechanism of action, clinical evidence, and future direction for heart health applications. Expert Opin Biol Ther. 2013;13:631–642. doi: 10.1517/14712598.2013.758706. [DOI] [PubMed] [Google Scholar]
- 92.Geng W, Lin J. Bacterial bile salt hydrolase: an intestinal microbiome target for enhanced animal health. Anim Health Res Rev. 2016;17:148–158. doi: 10.1017/S1466252316000153. [DOI] [PubMed] [Google Scholar]
- 93.Tannock GW, Dashkevicz MP, Feighner SD. Lactobacilli and bile salt hydrolase in the murine intestinal tract. Appl Environ Microbiol. 1989;55:1848–1851. doi: 10.1128/AEM.55.7.1848-1851.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Joyce SA, MacSharry J, Casey PG, Kinsella M, Murphy EF, Shanahan F, et al. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc Natl Acad Sci U S A. 2014;111:7421–7426. doi: 10.1073/pnas.1323599111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jeun J, Kim S, Cho S-Y, Jun H-J, Park H-J, Seo J-G, et al. Hypocholesterolemic effects of Lactobacillus plantarum KCTC3928 by increased bile acid excretion in C57BL/6 mice. Nutrition. 2010;26:321–330. doi: 10.1016/j.nut.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 96.Li C, Nie S-P, Ding Q, Zhu K-X, Wang Z-J, Xiong T, et al. Cholesterol-lowering effect of Lactobacillus plantarum NCU116 in a hyperlipidaemic rat model. J Funct Foods. 2014;8:340–347. doi: 10.1016/j.jff.2014.03.031. [DOI] [Google Scholar]
- 97.Guan X, Xu Q, Zheng Y, Qian L, Lin B. Screening and characterization of lactic acid bacterial strains that produce fermented milk and reduce cholesterol levels. Braz J Microbiol. 2017;48:730–739. doi: 10.1016/j.bjm.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang G, Huang W, Xia Y, Xiong Z, Ai L. Cholesterol-lowering potentials of Lactobacillus strain overexpression of bile salt hydrolase on high cholesterol diet-induced hypercholesterolemic mice. Food Funct. 2019;10:1684–1695. doi: 10.1039/c8fo02181c. [DOI] [PubMed] [Google Scholar]
- 99.Hofmann AF. Clinical implications of physicochemical studies on bile salts. Gastroenterology. 1965;48:484–494. doi: 10.1016/S0016-5085(65)80010-5. [DOI] [PubMed] [Google Scholar]
- 100.Dawson AM. Bile salts and fat absorption. Gut. 1967;8:1–3. doi: 10.1136/gut.8.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, et al. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe. 2018;23:458–469.e5. doi: 10.1016/j.chom.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Batta AK, Salen G, Arora R, Shefer S, Batta M, Person A. Side chain conjugation prevents bacterial 7-dehydroxylation of bile acids. J Biol Chem. 1990;265:10925–10928. [PubMed] [Google Scholar]
- 103.Lambert JM, Siezen RJ, de Vos WM, Kleerebezem M. Improved annotation of conjugated bile acid hydrolase superfamily members in gram-positive bacteria. Microbiology. 2008;154(Pt 8):2492–2500. doi: 10.1099/mic.0.2008/016808-0. [DOI] [PubMed] [Google Scholar]
- 104.Yahiro K, Setoguchi T, Katsuki T. Effect of coprophagy on bile acid metabolism in the rabbit. Gastroenterol Jpn. 1979;14:545–552. doi: 10.1007/BF02773713. [DOI] [PubMed] [Google Scholar]
- 105.Hellström K, Sjövall J. Turnover of deoxycholic acid in the rabbit. J Lipid Res. 1962;3:397–404. [Google Scholar]
- 106.Iwai S, Weinmaier T, Schmidt BL, Albertson DG, Poloso NJ, Dabbagh K, et al. Piphillin: improved prediction of metagenomic content by direct inference from human microbiomes. PLoS One. 2016;11:e0166104. doi: 10.1371/journal.pone.0166104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aßhauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015;31:2882–2884. doi: 10.1093/bioinformatics/btv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wilson ID, Nicholson JK. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res. 2017;179:204–222. doi: 10.1016/j.trsl.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.LoGuidice A, Wallace BD, Bendel L, Redinbo MR, Boelsterli UA. Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J Pharmacol Exp Ther. 2012;341:447–454. doi: 10.1124/jpet.111.191122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Seitz S, Boelsterli UA. Diclofenac acyl glucuronide, a major biliary metabolite, is directly involved in small intestinal injury in rats. Gastroenterology. 1998;115:1476–1482. doi: 10.1016/S0016-5085(98)70026-5. [DOI] [PubMed] [Google Scholar]
- 111.Saitta KS, Zhang C, Lee KK, Fujimoto K, Redinbo MR, Boelsterli UA. Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica. 2014;44:28–35. doi: 10.3109/00498254.2013.811314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Blackler RW, Motta J-P, Manko A, Workentine M, Bercik P, Surette MG, et al. Hydrogen sulphide protects against NSAID-enteropathy through modulation of bile and the microbiota. Br J Pharmacol. 2015;172:992–1004. doi: 10.1111/bph.12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fiorucci S, Mencarelli A, Cipriani S, Renga B, Palladino G, Santucci L, et al. Activation of the farnesoid-X receptor protects against gastrointestinal injury caused by non-steroidal anti-inflammatory drugs in mice. Br J Pharmacol. 2011;164:1929–1938. doi: 10.1111/j.1476-5381.2011.01481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mayo Sara A., Song Ye K., Cruz Melissa R., Phan Tri M., Singh Kavindra V., Garsin Danielle A., Murray Barbara E., Dial Elizabeth J., Lichtenberger Lenard M. Indomethacin injury to the rat small intestine is dependent upon biliary secretion and is associated with overgrowth of enterococci. Physiological Reports. 2016;4(6):e12725. doi: 10.14814/phy2.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liang X, Bittinger K, Li X, Abernethy DR, Bushman FD, FitzGerald GA. Bidirectional interactions between indomethacin and the murine intestinal microbiota. Elife. 2015;4:e08973. doi: 10.7554/eLife.08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wallace JL, Syer S, Denou E, de Palma G, Vong L, McKnight W, et al. Proton pump inhibitors exacerbate NSAID-induced small intestinal injury by inducing dysbiosis. Gastroenterology. 2011;141:1314–1322. doi: 10.1053/j.gastro.2011.06.075. [DOI] [PubMed] [Google Scholar]
- 117.Satoh H, Guth PH, Grossman MI. Role of bacteria in gastric ulceration produced by indomethacin in the rat: cytoprotective action of antibiotics. Gastroenterology. 1983;84:483–489. doi: 10.1016/0016-5085(83)90071-9. [DOI] [PubMed] [Google Scholar]
- 118.Scarpignato C. NSAID-induced intestinal damage: are luminal bacteria the therapeutic target? Gut. 2008;57:145–148. doi: 10.1136/gut.2007.134502. [DOI] [PubMed] [Google Scholar]
- 119.Zhong Z-Y, Sun B-B, Shu N, Xie Q-S, Tang X-G, Ling Z-L, et al. Ciprofloxacin blocked enterohepatic circulation of diclofenac and alleviated NSAID-induced enteropathy in rats partly by inhibiting intestinal β-glucuronidase activity. Acta Pharmacol Sin. 2016;37:1002–1012. doi: 10.1038/aps.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.LARSEN G.L. Role of the Gut Flora in Toxicity and Cancer. 1988. Deconjugation of Biliary Metabolites by Microfloral β-glucuronidases, Sulphatases and Cysteine Conjugate β-lyases and their Subsequent Enterohepatic Circulation; pp. 79–107. [Google Scholar]
- 121.Hofmann AF. Targeting drugs to the enterohepatic circulation: lessons from bile acids and other endobiotics. J Control Release. 1985;2:3–11. doi: 10.1016/0168-3659(85)90028-8. [DOI] [Google Scholar]
- 122.Turner PV. The role of the gut microbiota on animal model reproducibility. Anim Model Exp Med. 2018;1:109–115. doi: 10.1002/ame2.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Buhnik-Rosenblau K, Danin-Poleg Y, Kashi Y. Predominant effect of host genetics on levels of Lactobacillus johnsonii bacteria in the mouse gut. Appl Environ Microbiol. 2011;77:6531–6538. doi: 10.1128/AEM.00324-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Snijders AM, Langley SA, Kim Y-M, Brislawn CJ, Noecker C, Zink EM, et al. Influence of early life exposure, host genetics and diet on the mouse gut microbiome and metabolome. Nat Microbiol. 2016;2:16221. doi: 10.1038/nmicrobiol.2016.221. [DOI] [PubMed] [Google Scholar]
- 126.Xiao L, Feng Q, Liang S, Sonne SB, Xia Z, Qiu X, et al. A catalog of the mouse gut metagenome. Nat Biotechnol. 2015;33:1103–1108. doi: 10.1038/nbt.3353. [DOI] [PubMed] [Google Scholar]
- 127.Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell. 2017;171:1015–1028.e13. doi: 10.1016/j.cell.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shin J, Lee S, Go M-J, Lee SY, Kim SC, Lee C-H, et al. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing. Sci Rep. 2016;6:29681. doi: 10.1038/srep29681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Treweek JB, Chan KY, Flytzanis NC, Yang B, Deverman BE, Greenbaum A, et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nat Protoc. 2015;10:1860–1896. doi: 10.1038/nprot.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yamamoto Yuri, Nakanishi Yumiko, Murakami Shinnosuke, Aw Wanping, Tsukimi Tomoya, Nozu Ryoko, Ueno Masami, Hioki Kyoji, Nakahigashi Kenji, Hirayama Akiyoshi, Sugimoto Masahiro, Soga Tomoyoshi, Ito Mamoru, Tomita Masaru, Fukuda Shinji. A Metabolomic-Based Evaluation of the Role of Commensal Microbiota throughout the Gastrointestinal Tract in Mice. Microorganisms. 2018;6(4):101. doi: 10.3390/microorganisms6040101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Llorente C, Jepsen P, Inamine T, Wang L, Bluemel S, Wang HJ, et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat Commun. 2017;8:837. doi: 10.1038/s41467-017-00796-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149:1578–1593. doi: 10.1016/j.cell.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Arrieta M-C, Walter J, Finlay BB. Human microbiota-associated mice: a model with challenges. Cell Host Microbe. 2016;19:575–578. doi: 10.1016/j.chom.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 134.Schaarschmidt K, Müller G, Heinze A, Ruprecht L, Störmann J, Stratmann U, et al. Improved model of a fecal collection device for the prevention of coprophagia in the rat. J Exp Anim Sci. 1991;34:67–71. [PubMed] [Google Scholar]
- 135.Smyth RE. Fecal cup for collection of feces in male rats. Lab Anim Sci. 1979;29:677–678. [PubMed] [Google Scholar]
- 136.Frape DL, Wilkinson J, Chubb LG. A simplified metabolism cage and tail cup for young rats. Lab Anim. 1970;4:67–73. doi: 10.1258/002367770781036517. [DOI] [PubMed] [Google Scholar]
- 137.Ryer FH, Walker DW. An anal cup for rats in metabolic studies involving radioactive materials. Lab Anim Sci. 1971;21:942–943. [PubMed] [Google Scholar]
- 138.Hoff J. Methods of blood collection in the mouse. Lab Anim (NY) 2000;29:47–53. [Google Scholar]
- 139.AVMA Guidelines for the Euthanasia of Animals: 2013 ed. Schaumburg, IL: American Veterinary Medical Association; 2013.
- 140.Oblinger JL, Koburger JA. Understanding and teaching the most probable number technique. J Milk Food Technol. 1975;38:540–545. doi: 10.4315/0022-2747-38.9.540. [DOI] [Google Scholar]
- 141.Rowe R, Todd R, Waide J. Microtechnique for most-probable-number analysis. Appl Environ Microbiol. 1977;33:675–680. doi: 10.1128/AEM.33.3.675-680.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kuai L, Nair AA, Polz MF. Rapid and simple method for the most-probable-number estimation of arsenic-reducing bacteria. Appl Environ Microbiol. 2001;67:3168–3173. doi: 10.1128/AEM.67.7.3168-3173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen CY, Nace GW, Irwin PL. A 6 x 6 drop plate method for simultaneous colony counting and MPN enumeration of Campylobacter jejuni, Listeria monocytogenes, and Escherichia coli. J Microbiol Methods. 2003;55:475–479. doi: 10.1016/S0167-7012(03)00194-5. [DOI] [PubMed] [Google Scholar]
- 144.Ozkanca R, Saribiyik F, Isik K, Sahin N, Kariptas E, Flint KP. Resuscitation and quantification of stressed Escherichia coli K12 NCTC8797 in water samples. Microbiol Res. 2009;164:212–220. doi: 10.1016/j.micres.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 145.Blodgett R. BAM appendix 2: Most probable number from serial dilutions. 2010. [Google Scholar]
- 146.Polz MF, Cavanaugh CM. Bias in template-to-product ratios in multitemplate PCR. Appl Environ Microbiol. 1998;64:3724–3730. doi: 10.1128/AEM.64.10.3724-3730.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108(Suppl):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Aigrain L, Gu Y, Quail MA. Quantitation of next generation sequencing library preparation protocol efficiencies using droplet digital PCR assays - a systematic comparison of DNA library preparation kits for Illumina sequencing. BMC Genomics. 2016;17:458. doi: 10.1186/s12864-016-2757-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.KAPA Library Quantification Technical Guide v2.19. 2019. https://www.n-genetics.com/products/1104/1024/17225.pdf. Accessed 25 Sept 2019.
- 151.Illumina Adapter Sequences 1000000002694 v11. 2019. https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/experimentdesign/illumina-adapter-sequences-1000000002694-11.pdf. Accessed 25 Sept 2019.
- 152.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Taxonomy classifiers for use with q2-feature-classifier. https://docs.qiime2.org/2019.1/data-resources/#taxonomy-classifiers-for-use-with-q2-feature-classifier. Accessed 6 Aug 2019
- 155.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.SILVA rRNA database project. https://www.arb-silva.de/download/archive/qiime.
- 158.Zaiontz C. Real statistics resource pack (Release 6.2) 2019. [Google Scholar]
- 159.van der Walt S, Colbert SC, Varoquaux G. The NumPy array: a structure for efficient numerical computation. Comput Sci Eng. 2011;13:22–30. doi: 10.1109/MCSE.2011.37. [DOI] [Google Scholar]
- 160.McKinney W. Data structures for statistical computing in Python. In: van der Walt S, Millman J, editors. Proceedings of the 9th Python in Science Conference. 2010. pp. 51–56. [Google Scholar]
- 161.Vertanen P, Gommers R, Olphant TE, et al. SciPy 1.0--Fundamental algorithms for scientific computing in Python. 2019. arXiv. https://arxiv.org/abs/1907.10121. [DOI] [PMC free article] [PubMed]
- 162.Seabold S, Perktold J. 9th Python in Science Conference. 2010. Statsmodels: econometric and statistical modeling with python. [Google Scholar]
- 163.Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 164.Waskom M, Botvinnik O, O’Kane D, Hobson P, Ostblom J, Lukauskas S, et al. mwaskom/seaborn: v0.9.0 (July 2018) 2018. [Google Scholar]
- 165.Perez F, Granger BE. IPython: a system for interactive scientific computing. Comput Sci Eng. 2007;9:21–29. doi: 10.1109/MCSE.2007.53. [DOI] [Google Scholar]
- 166.Kluyver T, Ragan-Kelley B, Pérez F, Granger BE, Bussonnier M, Frederic J, et al. ELPUB. 2016. Jupyter notebooks-a publishing format for reproducible computational workflows; pp. 87–90. [Google Scholar]
- 167.Anaconda . Anaconda Software Distribution. Version 4.7.11. 2019. [Google Scholar]
- 168.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gloor GB, Wu JR, Pawlowsky-Glahn V, Egozcue JJ. It’s all relative: analyzing microbiome data as compositions. Ann Epidemiol. 2016;26:322–329. doi: 10.1016/j.annepidem.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 170.Lovell D, Pawlowsky-Glahn V, Egozcue JJ, Marguerat S, Bähler J. Proportionality: a valid alternative to correlation for relative data. PLoS Comput Biol. 2015;11:e1004075. doi: 10.1371/journal.pcbi.1004075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. 2012;12:2825–2830. [Google Scholar]
- 172.van den Berg RA, Hoefsloot HCJ, Westerhuis JA, Smilde AK, van der Werf MJ. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics. 2006;7:142. doi: 10.1186/1471-2164-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Tail cup design and experimental setup for preventing coprophagy. Figure S2. Mounting of functional tail cups onto mice. Figure S3. Body weight changes across all groups of mice in relation to food intake over the course of the study. Figure S4. Quantification of the culturable microbial load and microbiota profile along the entire GIT of mice fitted with functional tail cups (TC-F) and control mice (CTRL). Figure S5. Bile acid profiles in gallbladder bile and in lumenal contents along the entire GIT. Table S1. Primer oligonucleotide sequences used in the study. Table S2. Thermocycling parameters for the quantitative PCR (qPCR) assay for 16S rRNA gene DNA copy quantification. Table S3. Thermocycling parameters for the digital PCR (dPCR) assay for absolute 16S rRNA gene DNA copy quantification. Table S4. Thermocycling parameters for the 16S rRNA gene DNA amplicon barcoding PCR reaction for next generation sequencing (NGS). Table S5. Thermocycling parameters for the digital PCR (dPCR) assay for barcoded amplicon and Illumina NGS library quantification. Table S6. Reagents and chemical standards used in the bile acid metabolomics assay. Table S7. Bile acid concentrations in gallbladder bile and in lumenal contents along the entire GIT.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files. Sequencing data (paired end reads in FASTQ) and a manifest file for analysis in Qiime2 are available under a CC-BY license via CaltechDATA: 10.22002/D1.1295. Supplementary Information includes a zip file containing all sequencing sample metadata, numerical microbial quantification data (16S copies from the main study + MPN from the pilot study), Qiime2 sequencing output data, PICRUSt2 output data, numerical bile acid analysis data, numerical body weight data, numerical food intake data, and analytical scripts (iPython Notebooks) for all figures and statistical analyses in the manuscript.