

Abstract
A subset of gliomas has DNA repair defects that lead to hypermutated genomes. While such tumors are resistant to alkylating chemotherapies, they may also express more mutant neoantigens on their cell surfaces, and thus be more responsive to immunotherapies. A fast, inexpensive method of screening for hypermutated gliomas would therefore be of great clinical value. Since immunohistochemistry (IHC) for the DNA mismatch repair (MMR) proteins Msh2, Msh6, Mlh1, and Pms2 is already used to screen for hypermutated colorectal cancers, we sought to determine whether that panel might have similar utility in gliomas. MMR IHC was scored in 100 WHO grade I-IV gliomas (from 96 patients) with known tumor mutation burden (TMB), while blinded to TMB data. Cases included 70 grade IV GBMs, 13 grade III astrocytomas, 4 grade II astrocytomas (3 diffuse astrocytomas and 1 pleomorphic xanthoastrocytoma), 1 grade I pilocytic astrocytoma, 2 grade III oligodendrogliomas, 7 grade II oligodendrogliomas, and 3 grade I glioneuronal tumors. Eight of 100 tumors showed loss of one or more MMR proteins by IHC, and all 8 were hypermutated. Among the remaining 92 gliomas with intact MMR IHC, only one was hypermutated; that tumor had an inactivating mutation in another DNA repair gene, ATM. Overall accuracy, sensitivity, and specificity for DNA MMR IHC compared to the gold standard of TMB were 99, 89, and 100%, respectively. The strongest correlates with hypermutation were prior TMZ treatment, MGMT promoter methylation, and IDH1 mutation. Among the 8 MMR-deficient hypermutated gliomas, 4 (50%) contained both MMR-lost and MMR-retained tumor cells. Together, these data suggest that MMR IHC could be a viable front-line screening test for gliomas in which immunotherapy is being considered. They also suggest that not all cells in a hypermutated glioma may actually be MMR-deficient, a finding that might need to be considered when treating such tumors with immunotherapies.
Keywords: Glioma, Hypermutator, MGMT, IDH1, Temozolomide, Mismatch repair
Introduction
Gliomas are the most common tumors to arise from the brain parenchyma in adults [20]. Standard of care is maximal safe surgical resection. Grade III and IV gliomas are typically treated with radiation and temozolomide (TMZ), a DNA alkylating agent [25]. Diffusely infiltrating gliomas nearly always recur and lose sensitivity to adjuvant therapy.
When gliomas are challenged with TMZ, recurrent subclones often emerge with inactivating mutations in genes encoding DNA mismatch repair (MMR) enzymes, most commonly MSH2, MSH6, MLH1, and PMS2. Loss of function in these genes leads to failure of MMR mechanisms, which is essential for inducing programmed cell death in tumor cells damage by temozolomide, thus contributing to temozolomide resistance in recurrent tumors [6, 12, 30]. Tumor mutation burden (TMB) is normally ~ 1 mutation per megabase (Mb) of DNA [1], but MMR defects can lead to a high mutation burden, which has previously been defined as TMB > 20 mutations/Mb DNA [11–13]. In previously published work, this has been referred to as a “hypermutator” or “hypermutated” phenotype. Hypermutated tumor cells tend to display more mutant proteins on their surfaces, making them potentially more vulnerable to immunotherapies like PD-1 and PD-L1 checkpoint inhibitors [3, 10]. Other forms of hypermutated cancers have shown promising responses to immune checkpoint inhibitors [21], and there is great interest in this strategy for hypermutated gliomas [5, 11].
Next generation sequencing (NGS) is the current gold standard for detecting DNA MMR defects and quantifying TMB. While NGS is a powerful tool, it involves significant cost and turnaround time, compared to routine histopathologic tissue evaluation. Panels which cover over 500 genes, such as Tempus xT, cost several thousand US dollars, and have turnaround times ranging from 10 days to 3 weeks [8]. Smaller, more focused NGS panels, such as Glioseq [19], are less expensive, but often do not cover enough of the genome to reliably determine TMB.
A standardized quartet of IHC stains (Msh6, Msh2, Mlh1, and Pms2) is currently used to detect loss of normal MMR gene expression in colorectal cancers [23]. Because most pathology laboratories already have this MMR IHC panel for routine use, we sought to determine its utility as a screening test for hypermutated gliomas.
Materials and methods
Aims, design, and setting
The specific aims of this study are: 1) Determine the reliability of immunohistochemistry for DNA mismatch repair enzymes as a screening test for hypermutation in gliomas; 2) Determine association of hypermutation with the factors of temozolomide therapy, MGMT methylation status, IDH1 mutation status, tumor histotype, WHO grade, patient age, and patient gender, and to compare these results with previously published data. This study was designed as a blinded case review and comparison of immunohistochemistry against the gold standard of next generation sequencing with TMB. Evaluation of tumor characteristics associated with hypermutation was based on a retrospective case-control model.
Characteristics of participants
The cohort consisted of 100 World Health Organization (WHO) grade I-IV gliomas from the Northwestern Nervous System Tumor Bank with known TMB and MMR gene mutations, as determined by the commercially available targeted NGS panel, Tempus xT, covering approximately 600 genes. Tumors were taken from 96 patients (two patients had two separate tumor resections, and one patient had three separate resections). Summary information for the cohort is contained in Table 1. Gliomas were diagnosed according to 2016 WHO classification. Tumors originally diagnosed prior to the publication of the 2016 WHO updates were re-labeled and classified according to histologic and molecular features per 2016 guidelines. MGMT promoter methylation status was determined by pyrosequencing. Case collection was done under a Northwestern Institutional Review Board-approved protocol.
Table 1.
Cohort characteristics
| Age | mean 49.6 years (range 19–85) |
|---|---|
| Variable | N |
| Sex | |
| Male | 45 |
| Female | 52 |
| WHO grade | |
| I | 4 |
| II | 11 |
| III | 15 |
| IV | 70 |
| Histotype | |
| Astrocytic | 91 |
| Oligodendroglial | 9 |
| IDH1 status | |
| Wild-type | 66 |
| Mutant | 34 |
| MGMT status | |
| Methylated | 45 |
| Unmethylated | 49 |
| Unknown | 6 |
| Recurrent | |
| Yes | 38 |
| No | 62 |
| TMZ treatment | |
| Pre | 67 |
| Post | 33 |
| Hypermutated | |
| Yes | 9 |
| No | 91 |
| TOTAL | 100 |
N = 100 tumors from 96 patients (two females each had two tumors in this cohort, and one male had three tumors). See Additional file 1: Table S1 for further information on each tumor. TMZ temozolomide
Description of materials and processes
IHC was performed using 4 different primary antibodies, including Msh2 (Cell Marque G219–1129, 1:700), Msh6 (Cell Marque 44 (287 M-15), 1:100), Mlh1 (Leica NCL-L-MLH1, 1:100), and Pms2 (Cell Marque MRQ-28 (288 M-15), 1:50). Formalin-fixed, paraffin-embedded 4 μm thick tissue sections were baked at 60 °C for 30–60 min before being deparaffinized and re-hydrated. Antigen retrieval for Msh6, Msh2, and Pms2 was achieved using a Universal Retrieval (Abcam) buffer in a decloaking chamber reaching 110 °C for 5–20 min. Antigen retrieval for Mlh1 used a citrate buffer (pH 6) in a decloaking chamber reaching 110 °C for 10 min. Slides were cooled to room temperature and washed in TBS before neutralizing endogenous peroxidase (Biocare Peroxidase 1). Slides were then treated with a serum-free casein background block (Biocare Background Sniper) before pre-incubation in a 10% goat serum block for 60 min. Primary antibody was added to the slides for overnight incubation at 4 °C. After incubation, slides were washed in 3 5-min washes with TBS-T before incubating in HRP polymer (Biocare MACH 4 Universal HRP Polymer). Reaction products were visualized with DAB (Biocare Betazoid DAB Chromogen Kit). Slides were counterstained with hematoxylin, dehydrated, and mounted with xylene-based mounting media.
Each IHC marker was examined under light microscopy by two independent reviewers (MM and CH) while blinded to NGS data and TMB. Each IHC marker was scored as “retained” or “lost.” In accordance with published data on MMR enzyme immunohistochemistry, cases were scored as “retained” if uniform intact nuclear staining for the protein was observed [23]. Cases were scored as “lost” if nuclear staining was absent in at least some tumor cells that appeared viable and were not near areas of necrosis or thermal artefact. In tumors with lost MMR expression, the pattern (homogeneous versus heterogeneous) was noted. Neoplastic cells within each glioma were identified and differentiated from non-neoplastic cells by variation in size, shape, and density of nuclear chromatin. Nonneoplastic cells were identified by their overall monomorphic appearance and clues within the context of tissue architecture (e.g. small round cells with high nuclear to cytoplasmic ratio were lymphocytes; spindle-shaped cells surrounding lumens with red blood cells were endothelial cells). The non-neoplastic cells served as internal positive controls. Interobserver discrepancies were resolved by reviewing equivocal cases together.
Statistical analysis
Linear regression and Spearman correlation analyses were performed using GraphPad Prism 8.3.0.
Results
Characteristics of tumors and patients in the cohort
The cohort consisted of 100 gliomas, diagnosed in 96 patients (44 male, 52 female) with NGS and TMB data. The cohort included 70 grade IV GBMs, 13 grade III astrocytomas (all anaplastic astrocytomas), 4 grade II astrocytomas (including 3 diffuse astrocytomas and 1 pleomorphic xanthoastrocytoma), 2 grade III oligodendrogliomas; 7 grade II oligodendrogliomas; and 4 grade I gliomas, including 3 grade I gangliogliomas and 1 pilocytic astrocytoma. Nine tumors were hypermutated by NGS.
Of the 100 gliomas, 96 were located in the cortex (53 left-sided and 43 right-sided). There were 41 frontal, 11 parietal, 35 temporal, 4 occipital, 1 fronto-parietal, 2 fronto-temporal, 1 temporo-parietal, and 1 insular. Of the 4 non-cortical gliomas, 2 originated in the corpus callosum, 1 in the 3rd ventricle, and 1 in the brainstem. A total of 66 gliomas were IDH1 wild type, while 34 were IDH1 mutant. A total of 94 gliomas had known MGMT promoter methylation status (45 methylated, 49 unmethylated). A total of 62 gliomas were primary (prior to any therapy), while 38 were recurrent post-therapy (surgery, chemotherapy, or radiation) (Table 1). Of the 38 recurrent gliomas, 33 were recurrent post-temozolomide, and 5 were recurrent post-surgery, with or without radiation. A total of 67 gliomas were temozolomide-naïve. Available information on temozolomide dosage and cycles is included in Additional file 1: Table S1. Of the 38 recurrent gliomas, 32 were treated with radiation, 6 were treated with CCNU, and 5 were treated with bevacizumab prior to re-resection.
Regarding patients with multiple resections, one patient had three resections, one primary and two recurrent (Additional file 1: Table S1). Only the third resection (second recurrence) showed hypermutation. Another patient had re-resection of a recurrent, non-hypermutated IDH1-mutant anaplastic astrocytoma, which recurred again as a hypermutated GBM on the subsequent re-resection. The third patient had two re-resections of two separate recurrences of a GBM; both tumor samples were hypermutated. One patient, a 31 year old female diagnosed with a non-hypermutated IDH1-wild type GBM (tumor #81 in Additional file 1: Table S1), underwent germline DNA sequencing and was found to have a TP53 mutation, diagnostic of Li-Fraumeni syndrome. No other patients had germline DNA sequenced. None had previously been diagnosed with Lynch syndrome.
DNA mismatch repair enzyme immunohistochemistry is an accurate screening test
As previously stated, 9 of 100 gliomas were hypermutated by NGS (TMB > 20/Mb). Two of them were separate recurrences from the same patient (Table 2, Additional file 1: Table S1). Eight of 100 gliomas (8%), showed loss of at least one MMR protein by IHC (Fig. 1, Table 2). All 8 gliomas with MMR loss were hypermutators, and all 8 had mutations and/or copy number losses in corresponding MMR genes (Table 2). Of the remaining 92 gliomas with intact MMR IHC, only one was hypermutated (TMB = 29.5/Mb). This glioma did not have mutations in MSH2, MSH6, MLH1, or PMS2, but instead contained an inactivating splice site mutation in ATM (Table 2). Thus, 8 out of 9 hypermutators tested positive on IHC. Sensitivity, specificity, and overall accuracy of MMR IHC for identifying hypermutated gliomas in this cohort was 89, 99, and 98%, respectively. In 4 of 8 gliomas with lost MMR expression, the pattern of loss was clearly heterogeneous, as some tumor cells retained all MMR enzymes, while other cells lost expression of one or more MMR enzymes (Fig. 2).
Table 2.
Key characteristics of hypermutated gliomas
| Tumor # | pre vs post TMZ | IDH1 status | TMB | Msh2 | Msh6 | Mlh1 | Pms2 | MMR mutations (AF) | pattern of MMR IHC loss |
|---|---|---|---|---|---|---|---|---|---|
| 1 | post | WT | > 50.0 | lost | lost | retained | retained |
MSH2 stop gain (14.8%) MSH2 stop gain (9.7%) |
homogeneous |
| 2 | post | WT | 67.8 | lost | lost | retained | retained | MSH2 stop gain (33.0%) | homogeneous |
| 3 | post | mut | 108.7 | retained | lost | retained | retained |
MSH6 stop gain (26.1%) MSH6 frameshift (16.6%) |
homogeneous |
| 4 | post | mut | 65.0 | retained | retained | lost | lost |
PMS2 missense (7.2%) MLH1 copy loss |
heterogeneous |
| 5 | post | mut | 27.0 | lost | lost | retained | retained |
MSH2 splice variant (37.8%) MSH2 stop gain (7.7%) |
homogeneous |
| 6 | post | WT | 58.0 | lost | lost | retained | retained |
MSH6 stop gain (27.5%) MSH6 missense (29.4%) |
heterogeneous |
| 7 | post | mut | 70.5 | lost | lost | retained | retained | MSH2 stop gain (74.0%) | heterogeneous |
| 8 | post | mut | 20.8 | lost | lost | retained | retained |
PMS1 missense (32.5%) MSH6 missense (11.2%) |
heterogeneous |
| 9 | post | WT | 29.5 | retained | retained | retained | retained | ATM splice variant (10.5%) | none |
Tumor # corresponds to each specimen listed in Additional file 1: Table S1. TMB tumor mutation burden; AF allelic fraction; IHC immunohistochemistry
Fig. 1.
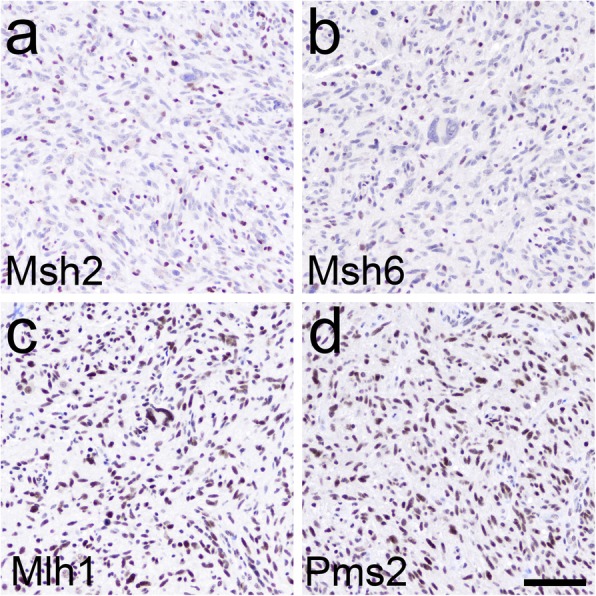
MMR IHC in Tumor 1. The tumor was a recurrent GBM, post-TMZ therapy, in a 57 year-old woman (Table 2). Tumor cells showed loss of Msh2 (a) and Msh6 (b), and retention of Mlh1 (c) and Pms2 (d). Note the normal immunostaining within nonneoplastic cells scattered throughout the tumor in (a) and (b). Scale bar = 100 μm
Fig. 2.
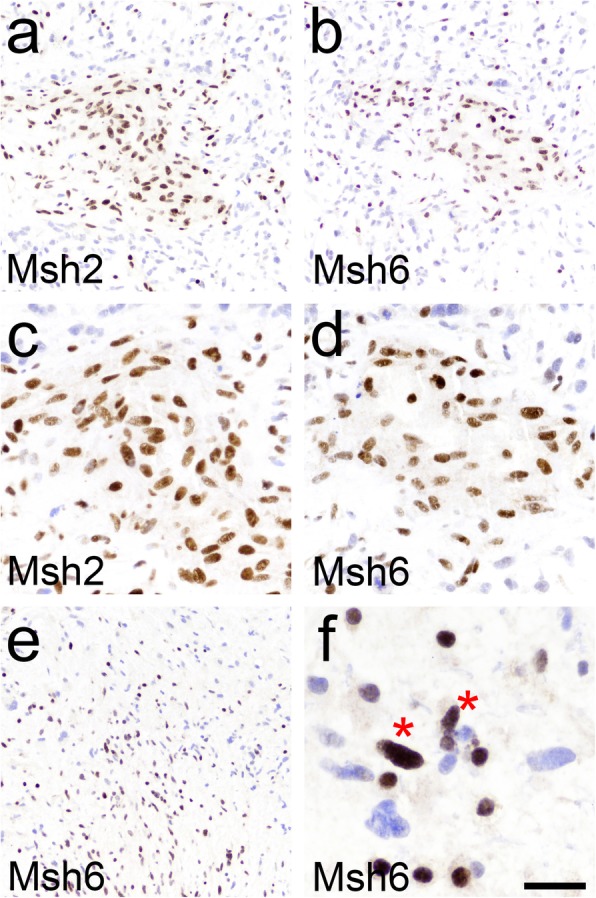
Heterogeneous MMR IHC in hypermutated gliomas. In tumor 7, which was a post-TMZ IDH1 mutant GBM in a 43 year-old woman (Table 2), clusters of tumor cells retained Msh2 and Msh6 positivity, but were surrounded by Msh2/6-deficient cells (a, b). Tumor 6 was a post-TMZ IDH1 wild-type GBM in a 65 year-old woman (Table 2). Msh6 was lost in many glioma cells (c), but under high power, it was apparent that a subset of cells with identical tumor nuclear morphology retained Msh6 (d, red asterisk). Also note the smaller rounded nuclei in (d), which are most likely lymphocytes and/or oligodendrocytes. Scale bar = 100 μm in (a, b, e), 50 μm in (c, d) and 25 μm in (f)
Variables associated with Hypermutation
All 9 hypermutated gliomas had MGMT promoter methylation and were post-TMZ, and 5/9 (56%) were IDH1 mutant (Table 2). Correlation analyses showed that the variables most strongly and significantly associated with hypermutation were prior treatment with TMZ, recurrence, and MGMT promoter methylation (Fig. 3, Additional file 1: Table S2). On multivariate regression, which weighs the relative strengths of each variable concurrently with other the variables in determining hypermutation likelihood, IDH1 mutation was the only variable to reach significance, even though prior TMZ treatment had the highest regression coefficient (Table 3).
Fig. 3.

Correlation matrix. Heatmap showing Spearman ρ (rho) correlation coefficients, with 1 = perfect direct correlation, 0 = no correlation, and − 1 = perfect inverse correlation. N = 93 tumors (cases without MGMT data were excluded, as well as hypermutated tumor #2 since it came from the same patient as tumor #1, see Additional file 1: Tables S1 and S2). TMZ = temozolomide
Table 3.
Multiple regression results
| variable | regression coefficient | 95% CI | t statistic | P |
|---|---|---|---|---|
| post-TMZ | 0.22 | −0.070 to 0.51 | 1.5 | 0.13 |
| IDH1mutant | 0.15 | 0.0020 to 0.30 | 2 | 0.047 |
| WHO grade | 0.097 | −0.036 to 0.23 | 1.5 | 0.15 |
| MGMTmethylated | 0.072 | −0.051 to 0.19 | 1.2 | 0.25 |
| astrocytic | 0.031 | −0.22 to 0.29 | 0.24 | 0.81 |
| male | 0.017 | −0.091 to 0.13 | 0.32 | 0.75 |
| age | 0.00021 | −0.0041 to 0.0045 | 0.099 | 0.92 |
| recurrent | −0.038 | −0.32 to 0.24 | 0.27 | 0.79 |
Variables are listed from largest to smallest regression coefficients, relative to hypermutation. N = 93 tumors (cases without MGMT data were excluded, as well as hypermutated tumor #2 since it came from the same patient as tumor #1, see Additional file 1: Tables S1 and S2). TMZ temozolomide. Multiple R2 = 0.26, df = 84
Discussion
Despite their generally aggressive behavior, gliomas tend to have low TMB relative to most other kinds of cancer [1]. However, gliomas that are hypermutated, either at initial presentation or recurrence, may be ideal targets for immunotherapy. Such gliomas usually show increased numbers of infiltrating CD8+ cytotoxic T cells [18, 29], which is consistent with the postulate that hypermutated gliomas are more immunogenic.
Hypermutated gliomas have been a subject of intense investigation for some time, though the reported frequencies of hypermutation vary markedly due to differences in cohort selection. In our screening of over 660 untreated sporadic grade II-IV TCGA gliomas in GlioVis [4], only 15 had detectable mutations in DNA repair enzymes (not shown). But a study by the TCGA consortium showed that 7/19 (36%) TMZ-treated GBMs were hypermutated [26]. Johnson et al. reported hypermutation patterns in 6/10 (60%) post-TMZ tumors, and suggested that most of the acquired mutations were likely directly induced by TMZ [15]. In a separate study of 114 matched pre- and post-treatment GBMs, 17 (15%) showed a hypermutator profile at recurrence; among those 17 cases, 16 had mutations in MMR genes, and showed enrichment for MGMT methylation and IDH1 mutation [28]. Others have verified the association between IDH1 mutations and hypermutation after TMZ [15, 27, 32]. Among 157 pediatric gliomas, only 9 (6%) were hypermutated, and 7 contained mutations in DNA repair genes [14]. In our own cohort, 9/100 gliomas were hypermutated, all 9 had been previously treated with TMZ, all 9 had MGMT promoter methylation, and 5/9 were IDH1 mutant (Table 2). Screening for hypermutation-associated MMR defects therefore appears to be of greatest value in recurrent, post-TMZ gliomas, especially MGMT-methylated and/or IDH1 mutant tumors (Fig. 3, Table 3, Additional file 1: Table S2).
Although the Msh2, Msh6, Mlh1, and Pms2 IHC panel is used to screen colorectal cancer, mutations in other DNA repair genes have also been reported in post-TMZ hypermutated gliomas, including MSH4, MSH5, MLH3, PMS1, POLE, and POLD1 [4, 11, 14, 28]. In our own cohort, we found a hypermutated glioma with an inactivating mutation in yet another gene associated with DNA repair, ATM (Table 2) [2]. The MMR IHC panel designed for colorectal cancer detected hypermutated gliomas with good sensitivity and excellent specificity and accuracy. Further studies with a panel using more IHC markers, such as Atm, Msh4, Msh5, or other proteins which may be altered in hypermutated gliomas, could potentially improve the sensitivity further.
Interpretation of MMR IHC in gliomas is relatively straightforward, especially since nonneoplastic cells within the glioma serve as a reliable positive control (Fig. 1). The process is similar to evaluation of ATRX staining in gliomas [7]. MMR staining is lost in areas of necrosis and thermal artifact (not shown), so such regions should not be scored.
Thus far, results from immune checkpoint inhibitors in gliomas have been mixed [5, 16, 22, 31]. While at least partial responses to immune checkpoint inhibitors have been observed in patients whose sporadic gliomas had elevated TMB [9, 17], the best responses have mostly been in glioma patients with an inherited defect in an MMR gene, where 100% of the glioma cells have MMR deficiency [3, 13, 24]. Our data showing frequent heterogeneity of MMR loss in hypermutated gliomas (Fig. 2 and Table 2) underscores the fact that TMB is just a mathematical average of the specimen that was submitted for NGS, and that non-hypermutated subclones of cells may exist in “hypermutated” gliomas [12]. Conversely, hypermutated subclones could potentially exist in tumors whose overall TMB has not yet reached the widely accepted cutoff of 20 mutations/Mb, although we did not see this in our own cohort (not shown).
In sum, DNA MMR enzyme IHC can serve as a rapid, low-cost method of screening for hypermutated gliomas. Highest yield for screening includes recurrent post-TMZ gliomas with MGMT promoter methylation and/or IDH1 mutations. While the current panel used for colorectal cancers has very good sensitivity and excellent specificity, adding more DNA repair IHC markers would further enhance its value. Finally, the observation of heterogeneous loss of staining in some cases is interesting. Future work could elucidate a potential relationship between the heterogeneity of MMR loss of function and degree of tumor response to immune checkpoint inhibitor therapy.
Conclusions
This study indicates that DNA MMR IHC can serve as a front-line screen for hypermutated gliomas. Focusing on post-TMZ recurrent gliomas that have MGMT promoter methylation and/or IDH1 mutations increases the per-case value of such screening. MMR loss within hypermutated gliomas is often heterogeneous, which could help explain why some hypermutated tumors respond to immunotherapies more than others.
Supplementary information
Additional file 1: Table S1. Detailed clinical and pathologic data for the full cohort of 100 gliomas. Tumors 1–9 are hypermutated and are also shown in Table S1. of the main manuscript. The following tumors are from the same patients: 1 and 2; 5, 11, and 12; 7 and 94. Pre-TMZ gliomas were designated as N/A in the TMZ dose and duration columns. Records on temozolomide dosage and cycles were incomplete for many post-TMZ patients, because they had been treated at outside institutions prior to re-resection at Northwestern. “D Astro” = diffuse astrocytoma; “A Astro” = anaplastic astrocytoma; “A Oligo” = anaplastic oligodendroglioma; N/A = not available or not applicable. Table S2. Correlation matrix. N = 93 tumors (cases without MGMT data were excluded, as well as hypermutated tumor #2 since it came from the same patient as tumor #1, see Fig. 2 and Additional file 1: Table S1). Each value represents Spearman ρ (rho) correlation coefficients, with 1 = perfect direct correlation, 0 = no correlation, and − 1 = perfect inverse correlation. *P < 0.05. TMZ = temozolomide.
Acknowledgements
The authors thank Kimberley Liggett and Juan Cuellar for their assistance with slide retrieval.
Abbreviations
- IDH1
Isocitrate dehydrogenase 1
- IHC
Immunohistochemistry
- MGMT
O-6-methylguanine-DNA methyltransferase
- MMR
Mismatch repair
- NGS
Next generation sequencing
- TCGA
The Cancer Genome Atlas
- TMB
Tumor mutation burden
- TMZ
Temozolomide
Authors’ contributions
CH and MM designed the project, performed data collection, and scored all cases. KM, RJ, and KK collected the cases, and AS performed MMR IHC. MM and CH wrote the manuscript.
Funding
This work was supported by National Institutes of Health grant R01NS102669 (C.H.) and by the Northwestern SPORE in Brain Cancer P50CA221747. MM was supported by an institutional grant from the Northwestern University Department of Pathology Resident Research Committee.
Availability of data and materials
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
Ethics approval and consent to participate
All patients provided written informed consent for research, under a Northwestern University Institutional Review Board-approved protocol, through the Northwestern Nervous System Tumor Bank.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s40478-020-0892-2.
References
- 1.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 3.Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, et al. Immune checkpoint inhibition for Hypermutant Glioblastoma Multiforme resulting from Germline Biallelic mismatch repair deficiency. J Clin Oncol. 2016;34:2206–2211. doi: 10.1200/JCO.2016.66.6552. [DOI] [PubMed] [Google Scholar]
- 4.Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology. 2017;19:139–141. doi: 10.1093/neuonc/now247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–486. doi: 10.1038/s41591-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniel P, Sabri S, Chaddad A, Meehan B, Jean-Claude B, Rak J, Abdulkarim BS. Temozolomide induced Hypermutation in Glioma: evolutionary mechanisms and therapeutic opportunities. Front Oncol. 2019;9:41. doi: 10.3389/fonc.2019.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebrahimi A, Skardelly M, Bonzheim I, Ott I, Muhleisen H, Eckert F, Tabatabai G, Schittenhelm J. ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol Commun. 2016;4:60. doi: 10.1186/s40478-016-0331-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong J, Pan K, Fakih M, Pal S, Salgia R. Value-based genomics. Oncotarget. 2018;9:15792–15815. doi: 10.18632/oncotarget.24353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorsi HS, Malicki DM, Barsan V, Tumblin M, Yeh-Nayre L, Milburn M, Elster JD, Crawford JR. Nivolumab in the treatment of recurrent or refractory pediatric brain tumors: a single institutional experience. J Pediatr Hematol Oncol. 2019;41:e235–e241. doi: 10.1097/MPH.0000000000001339. [DOI] [PubMed] [Google Scholar]
- 10.Gubin MM, Schreiber RD. Cancer. Odds Immunother Success Sci. 2015;350:158–159. doi: 10.1126/science.aad4140. [DOI] [PubMed] [Google Scholar]
- 11.Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, Huse JT, de Groot J, Li S, Overwijk WW, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro-Oncology. 2017;19:1047–1057. doi: 10.1093/neuonc/nox026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, Greenman C, Edkins S, Bignell G, Davies H, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006;66:3987–3991. doi: 10.1158/0008-5472.CAN-06-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, Uppaluri R, Ferguson C, Schmidt RE, Dahiya S, et al. Immunogenomics of Hypermutated Glioblastoma: a patient with Germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer discovery. 2016;6:1230–1236. doi: 10.1158/2159-8290.CD-16-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, Daniel S, Covert M, Frampton GM, Hsu S, et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade Gliomas reveals genomic drivers, tumor mutational burden, and Hypermutation signatures. Oncologist. 2017;22:1478–1490. doi: 10.1634/theoncologist.2017-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurz SC, Cabrera LP, Hastie D, Huang R, Unadkat P, Rinne M, Nayak L, Lee EQ, Reardon DA, Wen PY. PD-1 inhibition has only limited clinical benefit in patients with recurrent high-grade glioma. Neurology. 2018;91:e1355–e1359. doi: 10.1212/WNL.0000000000006283. [DOI] [PubMed] [Google Scholar]
- 17.Lukas RV, Rodon J, Becker K, Wong ET, Shih K, Touat M, Fasso M, Osborne S, Molinero L, O'Hear C, et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neuro-Oncol. 2018;140:317–328. doi: 10.1007/s11060-018-2955-9. [DOI] [PubMed] [Google Scholar]
- 18.Mackay A, Burford A, Molinari V, Jones DTW, Izquierdo E, Brouwer-Visser J, Giangaspero F, Haberler C, Pietsch T, Jacques TS, et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade Glioma from the HERBY phase II randomized trial. Cancer Cell. 2018;33:829–842. doi: 10.1016/j.ccell.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nikiforova MN, Wald AI, Melan MA, Roy S, Zhong S, Hamilton RL, Lieberman FS, Drappatz J, Amankulor NM, Pollack IF, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro-Oncology. 2016;18:379–387. doi: 10.1093/neuonc/nov289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro-Oncology. 2017;19:v1–v88. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, Inoges S, de Andrea C, Lopez-Diaz de Cerio A, Tejada S, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25:470–476. doi: 10.1038/s41591-018-0339-5. [DOI] [PubMed] [Google Scholar]
- 23.Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I The utility of immunohistochemistry. J Mol Diagn. 2008;10:293–300. doi: 10.2353/jmoldx.2008.080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder A, Wolchok JD. Successful treatment of a patient with Glioblastoma and a Germline POLE mutation: where next? Cancer Discov. 2016;6:1210–1211. doi: 10.1158/2159-8290.CD-16-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 26.TCGA Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Thuijl HF, Mazor T, Johnson BE, Fouse SD, Aihara K, Hong C, Malmstrom A, Hallbeck M, Heimans JJ, Kloezeman JJ, et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol. 2015;129:597–607. doi: 10.1007/s00401-015-1403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S, Abate F, Liu Z, Elliott O, Shin YJ, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48:768–776. doi: 10.1038/ng.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, de Carvalho AC, Lyu S, Li P, Li Y, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell. 2017;32:42–56. doi: 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, Louis DN. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15:4622–4629. doi: 10.1158/1078-0432.CCR-08-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25:462–469. doi: 10.1038/s41591-019-0349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng S, Alfaro-Munoz K, Wei W, Wang X, Wang F, Eterovic AK, Shaw KRM, Meric-Bernstam F, Fuller GN, Chen K, et al. Prospective clinical sequencing of adult Glioma. Mol Cancer Ther. 2019;18:991–1000. doi: 10.1158/1535-7163.MCT-18-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Detailed clinical and pathologic data for the full cohort of 100 gliomas. Tumors 1–9 are hypermutated and are also shown in Table S1. of the main manuscript. The following tumors are from the same patients: 1 and 2; 5, 11, and 12; 7 and 94. Pre-TMZ gliomas were designated as N/A in the TMZ dose and duration columns. Records on temozolomide dosage and cycles were incomplete for many post-TMZ patients, because they had been treated at outside institutions prior to re-resection at Northwestern. “D Astro” = diffuse astrocytoma; “A Astro” = anaplastic astrocytoma; “A Oligo” = anaplastic oligodendroglioma; N/A = not available or not applicable. Table S2. Correlation matrix. N = 93 tumors (cases without MGMT data were excluded, as well as hypermutated tumor #2 since it came from the same patient as tumor #1, see Fig. 2 and Additional file 1: Table S1). Each value represents Spearman ρ (rho) correlation coefficients, with 1 = perfect direct correlation, 0 = no correlation, and − 1 = perfect inverse correlation. *P < 0.05. TMZ = temozolomide.
Data Availability Statement
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.