

Abstract
Rationale:
Elastin is an important extracellular matrix protein in large and small arteries. Vascular smooth muscle cells (SMCs) produce the layered elastic laminae found in elastic arteries but synthesize little elastin in muscular arteries. However, muscular arteries have a well-defined internal elastic lamina (IEL) that separates endothelial cells (ECs) from SMCs. The extent to which endothelial cells (ECs) contribute elastin to the IEL is unknown.
Objective:
To use targeted elastin knockout mice to explore the relative contributions of SMCs and ECs to elastic laminae formation in different arteries.
Methods and Results:
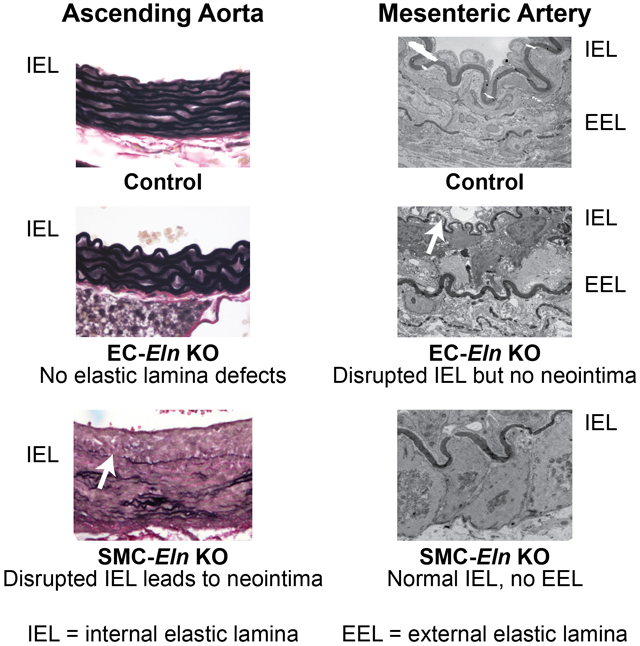
We used SMC- and EC-specific Cre recombinase transgenes with a novel floxed Eln allele to focus gene inactivation in mice. Inactivation of Eln in SMCs using Sm22aCre resulted in depletion of elastic laminae in the arterial wall with the exception of the IEL and SMC clusters in the outer media near the adventitia. Inactivation of elastin in ECs using Tie2Cre or Cdh5Cre resulted in normal medial elastin as well as a typical IEL in elastic arteries. In contrast, the IEL was absent or severely disrupted in muscular arteries. Interruptions in the IEL resulted in neointimal formation in the ascending aorta but not in muscular arteries.
Conclusions:
Combined with lineage-specific fate mapping systems, our knockout results document an unexpected heterogeneity in vascular cells that produce the elastic laminae. SMCs and ECs can independently form an IEL in most elastic arteries, whereas ECs are the major source of elastin for the IEL in muscular and resistance arteries. Neointimal formation at IEL disruptions in the ascending aorta confirms that the IEL is a critical physical barrier between SMCs and ECs in the large, elastic arteries. Our studies provide new information about how SMCs and ECs contribute elastin to the arterial wall and how local elastic laminae defects may contribute to cardiovascular disease.
Keywords: Elastin, elastic fibers, aorta, stenosis, neointima, animal model cardiovascular disease, extracellular matrix
Subject Terms: Animal Models of Human Disease, Basic Science Research, Stenosis, Vascular Biology, Vascular Disease
Graphical abstract
INTRODUCTION
A vital adaptation that made the vertebrate closed circulatory system possible was the appearance of the extracellular matrix (ECM) protein elastin1, which allows vessels to expand and store energy during the cardiac cycle. Elastin is found throughout the arterial system and is organized, predominantly, as protein sheets, or laminae, that separate smooth muscle cell (SMC) layers in large arteries and endothelial cells (ECs) from SMCs in all arteries. Elastin is required for postnatal cardiovascular function, since mice lacking elastin die shortly after birth2, 3.
In developing arteries, mural cells produce elastin soon after their recruitment to the nascent tubular vessel and after their commitment to the SMC lineage4. In large conducting arteries, each SMC layer produces fenestrated elastin sheets and connecting fibers that are designed to distend outward with the surge of blood pressure during cardiac contraction. In muscular and resistance arteries where pulse pressure is low, outward stretch is less important and medial elastin layers are absent. The exceptions are an occasional, and frequently discontinuous, elastin layer between the media and adventitia (the external elastic lamina, EEL) and a layer of elastin that separates intimal ECs from SMCs (the internal elastic lamina, IEL). The IEL is present in most all arteries including muscular and resistance arteries and was assumed to be formed by SMCs, which produce the other elastic structures in the vessel wall. The absence of the IEL was suggested to contribute to aortic occlusion in elastin knockout (Eln−/−) mice2.
In this report, we generated mice with SMC- and EC-specific deletion of Eln to determine how the absence of elastin deposition by vascular wall cells affects structure and function in different arteries. We found an unexpected heterogeneity in elastin production by SMCs and ECs in different arterial beds and documented a contribution of elastin to the IEL by ECs. We also found that areas of malformed IEL in the ascending aorta were associated with neointimal formation by SMCs. These results suggest that elastin production by vascular wall cells is more complicated than heretofore appreciated and that disruption of the IEL correlates with neointimal formation in large arteries.
METHODS
The authors declare that all supporting data are available within the article and its online supplementary files.
Mice and genotyping.
LoxP sites were inserted surrounding the 4th and 29th exons of the Eln gene using CRISPR/Cas9 technology (Sage Labs) (Fig. 1A). Validated Cas9 pairs and donor vectors were microinjected into one cell stage embryos from female C57BL6 embryos. Positive founders were identified by PCR genotyping using the following primer pairs: LoxP1F: CAGGCTTGGTTAAGCCACCA, LoxP1R: CCTACCTTTCTGGGGCCACT [LoxP1 knock-in 455bp, wildtype 415bp]; LoxP2F: CAACTGGTCCCCTTGTCACT, LoxP2R: AGACAGTGTGGGTCTGGCTA [LoxP1 knock-in 583bp, wildtype 543bp]. The directionality of the LoxP sites were confirmed by subjecting the amplicons to partial digestion with SalI, with expected band sizes indicated in Fig. 1B. Sanger sequencing confirmed correct Cas9/sgRNA-mediated genome editing. A single Elnf/+ founder was bred to produce an F1 breeding pair and subsequent Elnf/f mice.
Figure 1. Generation and validation of Eln floxed mice.

(A) Genomic sequence of Eln floxed allele. Red boxes indicate sequences introduced by CRISPR/Cas9 near exons 4 and 29. Green highlighting indicates LoxP sequences. (B) Genomic amplicon partially digested by SalI indicating presence and correct orientation of upstream and downstream LoxP sites (LoxP1 and LoxP2, respectively). Mouse number 57 was confirmed and used as the founder for subsequent studies. (C) VVG staining of P4 Eln+/+ and Elnf/f ascending (AscAo) and descending (DescAo) aorta. Scale bar = 50 μm.
Elnf/f mice were bred to various Cre lines and maintained in mixed backgrounds. Sm22aCre (Jackson Labs #004746)5, 6, Tie2Cre (Jackson Labs #008863)7, Cdh5Cre (Jackson Labs #006137)8, and ROSA26mT/mG mice (Jackson Labs #007576)9 were described previously. Eln+/− mice were also used2, 10. Experimental mice were compared to littermate controls. Elnf/f and Elnf/+mice were combined as controls in most instances. Males were used for the mechanical testing, as carotid artery diameter varies with sex11. Arterial blood pressure, which also varies with sex12, was measured in male mice before sacrifice for mechanical testing. Both sexes were used for all other studies. All animal studies were approved by the Institutional Animal Care and Use Committee at Washington University. Genotyping was performed by TransnetYX, Inc. The Eln alleles were amplified with the following primers: ElnF: CCATGTGGGTGCTGTAAGCT, ElnR: GCAGTGCTGGCTCCCA, Probe 1, wildtype: CCTGCCTGAGTTCTCA, Probe 2, LoxP floxed: AGGTCGACATAACTTCG. Genotyping for Cre, Sm22aCre, and ROSA26mT/mG alleles were performed with TransnetYX’s proprietary primers.
Vascular casting.
Mice were euthanized by CO2 and their chest wall excised. Blood was drained from the vasculature by injecting phosphate-buffered saline (PBS) through the left ventricular apex after transecting the left common iliac artery. Yellow latex (Ward’s Science) was then injected through the left ventricle13. Mice were fixed in 10% neutral buffered formalin (Thermo Fisher Scientific) at 4°C overnight, followed by fine dissection to reveal the aorta and aortic arch arteries.
Echocardiography.
Mouse echocardiography was performed at the Washington University Mouse Cardiovascular Phenotyping Core facility using a Vevo 2100 Imaging System (FUJIFILM VisualSonics Inc.) and MS700 or MS550 probes. Postnatal day (P) 5 or 10 mice were weighed, anesthetized with 1% isoflurane, and then placed on a heated stage under a heat lamp. 2D and M-mode short axis images of the LV were obtained, as well as 2D and continuous wave Doppler flow images of the aortic arch. LV dimensions and aortic arch velocities were obtained using edge detection software and standard techniques14.
Blood pressure and mechanical testing.
Postnatal day (P) 30 male mice were anesthetized with 1.5% isoflurane and arterial blood pressure was measured with a solid-state catheter (1.2F, Transonic). Mice were then sacrificed by thoracotomy under anesthesia. Images were taken of the left common carotid artery before and after dissection to determine the in vivo axial stretch ratio. The artery was mounted at its in vivo axial stretch ratio in a pressure myograph (110P, Danish Myotechnology) in physiologic saline solution at 37°C. Arteries were inflated from 0 – 175 mmHg in steps of 25 mmHg (12 sec/step) while pressure and outer diameter were recorded. The compliance was calculated as the average change in outer diameter for each pressure step15.
Protein quantification.
Ascending and descending aortic segments from P8 mice were hydrolyzed in 6 N HCl, dried, and resuspended in ultrapure water. Desmosine, an elastin specific crosslink, was measured through a competitive ELISA and normalized to total protein as described in Stoilov et al.16.
Histology and immunostaining.
Harvested tissues were fixed overnight at 4°C in 4% paraformaldehyde in PBS solution (Electron Microscopy Sciences) or 10% neutral buffered formalin (Thermo Fisher Scientific). For frozen sections, fixed samples were embedded in Optimum Cutting Temperature Compound (OCT, Sakura Finetek), flash frozen, and sectioned at 5 μm thickness with a cryostat. For paraffin sections, fixed tissue was dehydrated, paraffinized with a tissue processor, embedded and sectioned at 5 μm thickness using a microtome.
Hematoxylin and eosin (H&E) staining was performed following standard protocols. Verhoeff staining was performed by first overstaining sections with alcoholic hematoxylin-ferric chloride-iodine, followed by differentiation with ferric chloride and then sodium thiosulfate. Verhoeff stained slides were counterstained with van Gieson solution for Verhoeff-Van Gieson (VVG) staining. All histological staining solutions were from American MasterTech.
For immunohistochemistry, paraffin tissue sections were rehydrated, immersed in antigen retrieval solution (10 mM Tris, 1 mM EDTA, pH 9) and then heated for 10 minutes in a pressure cooker. After cooling down to room temperature, the slides were blocked with 0.3% hydrogen peroxide and the M.O.M. blocking reagent (Vector Labs), followed by incubation with the primary antibody for 1 hour at room temp in the M.O.M. diluent. The slides were washed with PBS and incubated with biotin-conjugated secondary antibodies in the M.O.M. diluent. Visualization was accomplished using streptavidin-based ABC Kits (Vector Labs) following the manufacturer’s instructions. Nuclei were counterstained with hematoxylin. Stained slides were dehydrated, cleared, and mounted with Cytoseal 60 (Thermo Fisher Scientific,) or VectaMount Permanent (Vector Labs). Primary antibodies used include mouse anti-smooth muscle α-actin (1A4, #A2547, Sigma-Aldrich), mouse anti-PCNA (PC10, #2586, Cell Signaling Technology), rabbit anti-Myh11 (JA03-35, #NBP2-66967, Novus Biologicals), rabbit anti-calponin (SI67-01, #NBP2-67401, Novus Biologicals), and rabbit anti-Sm22a (ab155272, Abcam). Secondary antibodies used include biotinylated anti-mouse (M.O.M kit) and biotinylated goat anti-rabbit antibody (#14708, Cell Signaling). Concentration of 1:200 was used for primary antibodies and 1:400 for secondary antibodies. Secondary antibody-only staining controls of ascending aorta slides were used for each antibody to ensure the specificity of staining. For fluorescence imaging, nuclei were counterstained with Hoechst 34580 (5 μM, Life Technologies) or DAPI (Sigma-Aldrich). Elastin autofluorescence was obtained by illuminating the slides with an LED bulb and capturing the image under a dark field.
Transmission electron microscopy (TEM).
Arteries used for TEM were fixed in 2.5% glutaraldehyde and 0.1 M sodium cacodylate (both from Electron Microscopy Sciences) at 4°C overnight. They were sent to Washington University’s Center for Cellular Imaging for processing and thin sectioning following standard protocols. Images were taken using a JEOL JEM-1400Plus transmission electron microscope with an Advanced Microscopy Techniques XR111 high-speed, 4000 × 2000–pixel, phosphor-scintillated, 12-bit charge-coupled device (CCD) camera.
Statistics and animal numbers.
For survival analysis, the day of death was defined as the day the mouse was found dead in the cage. In some instances when no carcass was found, the day of death was defined as the midpoint since the day the mouse was last seen. Those mice euthanized for experiments were censored. Log-rank survival analysis was performed using RStudio version 1.1.463 (RStudio, Inc, Boston, MA) and its “survdiff” function in the “survival” package, with default settings and significance level set at P < 0.05. Kaplan-Meier curves were produced using Prism 8 (GraphPad Software, San Diego, CA). 161 total mice were used for the survival analysis, of which 22 were controls, 39 SM22aCre;Elnf/+, and 84 SM22aCre;Elnf/f. For gross phenotyping approximately 10 mice per group were examined. For echocardiography, 1 SM22aCre;Elnf/f and 1 control were imaged at P5 and 1 SM22aCre;Elnf/f and 3 controls were imaged at P10. For histological analyses, images shown are representative of 4-6 mice examined in each group. For TEM analysis, images shown are representative of 2-3 mice examined in each group. A priori power calculations were utilized to estimate group sizes for quantitative data using estimates of mean differences and variability from our previously published work on elastin insufficient mice3, 15, 17 and assuming a normal distribution. The D’Agostino-Pearson omnibus normality test was used to confirm normality of the mechanical testing data. All other quantitative datasets were too small for formal normality tests. 4-5 arteries per group were analyzed for quantification of elastic layers and protein amounts. Two-way ANOVA with Sidak’s multiple comparisons test was used to determine significant differences between genotypes for each artery type. For the blood pressure and mechanical testing, 6-8 male mice were used for each group. One-way ANOVA with Sidak’s multiple comparisons test was used to determine significant differences for experimental genotypes compared to Eln+/+. For proliferation analysis, 3-4 mice were used for each group. An image of the PCNA staining was captured from a representative section from each ascending aorta. Quantification was done by counting the total number of PCNA-positive cells in tunica media and neointima (in the case of SM22aCre;Elnf/f) divided by the number of total cells in the area. One-way ANOVA with Sidak’s multiple comparisons test was used to determine significant differences for experimental genotypes compared to control. Prism 8 (Graphpad Software) was used for statistical analysis and significance was set to P < 0.05. Researchers were blinded to animal genotypes during experimentation and analyses.
RESULTS
Floxed Eln allele has no adverse effect on aortic wall structure.
CRISPR/Cas9 technology was used to generate an Elnf allele. LoxP sites were inserted near the 4th and 29th exons of Eln, with SalI sites inserted adjacent to both LoxP sites to facilitate genotyping (Fig. 1A). Sanger sequencing confirmed correct Cas9/sgRNA-mediated genome editing (data not shown). Hemizygous offspring of a founder with LoxP sites in the correct location on the same chromosome (Fig. 1B) were bred to produce mice homozygous for the Elnf allele (Elnf/f). Elnf/f mice have no discernable gross phenotype and live a normal lifespan. Histological analysis of the ascending and descending aorta showed normal morphology with the appropriate number of elastic layers (Fig. 1C). These results confirm insertion of the LoxP sites in the Eln gene and show that mice homozygous for the floxed elastin allele have typical vascular wall structure.
Mice heterozygous for SMC-specific deletion of Eln show cardiovascular remodeling similar to Eln+/− mice, but have normal blood pressure.
To inactivate elastin expression in SMCs, we crossed Elnf mice with mice bearing a transgenic Sm22aCre allele5, 6. Sm22aCre;Elnf/+ mice were grossly indistinguishable from Elnf/f littermates, were fertile, and showed no premature death. Similar to mice bearing a global knockout allele (Eln+/−)10, the ascending and descending aorta in Sm22aCre;Elnf/+ mice had a slightly thicker tunica media and more circumferential elastic laminae than control littermates (Figs. 2A and B). Sm22aCre;Elnf/+ mice also had reduced diameter and altered compliance in the carotid artery, which is in agreement with arterial changes in Eln+/− mice18 (Fig. 2C). Unlike Eln+/− mice19, P30 Sm22aCre;Elnf/+ mice did not show systolic hypertension (Fig. 2D).
Figure 2. Mice heterozygous for SMC-specific deletion of Eln show cardiovascular remodeling similar to Eln+/− mice, but have normal blood pressure.

(A) H&E and VVG staining of P18 ascending and (B) P12 descending aorta show the elastic laminae structure. Scale bar = 50 μm. Average number of elastic laminae (EL) are shown below the images. * = P < 0.05 between genotypes for each artery by two-way ANOVA with Sidak’s multiple comparisons test. N = 4-5/group. (C) Reduced arterial diameter (top) and compliance (bottom) in P30 male Sm22aCre;Elnf/+ common carotid artery is similar to Eln+/−. N = 8/group. (D) Unlike Eln+/− mice, Sm22aCre;Elnf/+ mice do not have increased systolic blood pressure. Heart rate is similar across groups. N = 6-8/group. For C and D, * and # indicate P < 0.05 compared to Eln+/+ for Sm22aCre;Elnf/+and Eln+/−, respectively, by one-way ANOVA with Sidak’s multiple comparisons test. Sm22aCre;Eln+/+ were not significantly different from Eln+/+. Mean ± SD.
Homozygous deletion of Eln in SMCs results in extensive heart and vascular changes.
Mice with SMC-specific Eln deletion (Sm22aCre;Elnf/f) were born at the expected Mendelian ratio and were initially similar in appearance to their littermates. By P7-8, however, Sm22aCre;Elnf/f mice were appreciably smaller than control mice (Fig. 3A) and most died between P9-18 (Fig. 3B). Unlike the relatively mild cardiovascular phenotype seen in Sm22aCre;Elnf/+mice, Sm22aCre;Elnf/f mice exhibited a grossly lengthened and thickened ascending aorta, coarctation of the aortic arch, and severe tortuosity of the carotid arteries with 100% penetrance. Their descending aorta, in contrast, was neither tortuous nor aneurysmal (Fig. 3C). Echocardiography of P5 and P10 mice confirmed coarctation of the aortic arch with greatly increased peak flow velocity (peak pressure gradient ~53 mmHg, consistent with moderate-to-severe stenosis) (Fig. 3D). By P5-10, Sm22aCre;Elnf/f mice exhibited cardiomegaly (Fig. 3E) owing to a dilated, eccentrically hypertrophic left ventricle (LV) with compromised fractional shortening (Fig. 3F). Histologically, the Sm22aCre;Elnf/f heart exhibited chronic thrombosis, necrosis, and calcification in the left atrium and ventricle (Online Figs. IA and B) at P9 and P18. By contrast, these histopathological changes were absent in P2 Sm22aCre;Elnf/f hearts (Online Fig. IC), suggesting the cardiomyopathy was likely secondary to pressure overload by aortic coarctation.
Figure 3. Homozygous deletion of Eln in SMCs results in extensive cardiac and vascular changes.

(A) Pictures of members from a P14 litter with genotypes indicated. (B) Kaplan-Meier curves showing premature death of Sm22aCre;Elnf/f mice. Control indicates Elnf/+ and Elnf/f combined. N = 161 mice total. P = 6 x 10−16 (log-rank test) for Sm22aCre;Elnf/f compared with control (*). (C) Latex angiogram demonstrating elongated ascending aorta, coarctation of the aortic arch (arrow), tortuous carotid arteries (arrowhead), and straight descending aorta in P6 Sm22aCre;Elnf/f mice. Scale bar = 1 mm. (D) 2D echocardiograms of ascending aorta and arch. Continuous wave Doppler blood flow velocity was measured in the arch (small panels). Coarctation is visible in the Sm22aCre;Elnf/f P10 aortic arch (arrow) leading to an increase in peak blood flow velocity (Vpeak). Scale bars = 0.5 mm. (E) Whole mount picture demonstrating cardiomegaly in P9 Sm22aCre;Elnf/f mice. Scale bar = 2 mm. (F) M-mode echocardiograms showing representative measures of the left ventricular inner diameter (LVID) at systole (s) and diastole (d) and the associated fractional shortening (FS) in P10 mice. Scale bars = 0.5 mm.
Both the ascending and descending aorta in Sm22aCre;Elnf/f mice (Fig. 4A) showed a markedly thickened tunica media with SMCs in the descending aorta maintaining their circumferential orientation in the absence of elastin, as did SMCs in the outer part of the ascending aorta. SMCs near the lumen of the ascending aorta, however, showed an axial or random orientation. There were no intact sheets of elastic fibers in the media of either artery, which was instead collagen-laden, as demonstrated by red VVG staining. There were, however, random patches of elastin usually located in the outer half of the media near the adventitia. Amounts of desmosine, an elastin specific crosslink, in Sm22aCre;Elnf/f ascending and descending aorta were 20 and 30% of controls, respectively (Fig. 4B). Elastic fibers within the patches near the outer media were thin and ultrastructurally fragmented (Fig. 4C). Lineage tracing with the ROSA26mT/mG reporter mouse confirmed Sm22aCre recombination in areas of the media where elastin was absent and no recombination where elastin was present (Fig. 4D). However, cells throughout the Sm22aCre;Elnf/f ascending aorta media stained positively for SM22a regardless of the presence or absence of elastin (Fig. 4E).
Figure 4. Analysis of the aorta in mice with homozygous deletion of Eln in SMCs.

(A) H&E and VVG staining of P18 ascending (left) and P12 descending (right) aorta show the elastic laminae structure. Sm22aCre;Elnf/f ascending aorta has a fragmented IEL, while the descending aorta has an intact IEL (arrows). Both arteries have patches of elastin (arrowheads) in the outer half of the media near the adventitia. Littermate control images are shown in Figs. 2A and B. Scale bars = 50 μm. (B) Desmosine levels were expressed as a ratio of total protein in the ascending and descending aorta. Control is Elnf/f and Elnf/+ combined. P values were determined between genotypes for each artery by two-way ANOVA with Sidak’s multiple comparisons test. (C) Electron microscopy images of the outer media show that the patches of elastin present in Sm22aCre;Elnf/f ascending aorta are thin and fragmented. Scale bar = 10 μm. (D) Lineage tracing with the ROSA26mT/mG reporter mouse demonstrates Sm22aCre-mediated recombination (green) in the media corresponding with the absence of elastin, as well as in the neointima (arrow). There are also cells in outer regions of the wall near the patches of elastin that do not show Sm22aCre-mediated recombination (red, arrowhead). Bright field image of the same field is shown below for comparison. Scale bar = 50 μm. (E) Patches of elastin are shown with autofluorescence in P2 Sm22aCre;Elnf/f ascending aorta (arrowhead, top). The medial cells stain positive for SM22a protein even if they are near the patches of elastin associated with nonrecombination (arrowhead, bottom). The border between the media and adventitia is indicated. Scale bar = 50 μm.
IEL is retained in both SMC- and EC-specific Eln knockouts.
An unexpected finding in the SMC-specific elastin knockout mice was the persistence of the IEL, which suggests that ECs contribute to, or solely synthesize, this layer of elastin. The relative contribution of ECs to IEL formation was determined by targeting the floxed elastin gene in ECs using Tie2Cre. Tie2Cre;Elnf/f mice were fertile, grossly similar to control mice and exhibited no early mortality (tracked up to 10 months of age). Histological analysis showed that the IEL and medial lamellae formed normally in Tie2Cre;Elnf/f ascending and descending aorta (Figs. 5A and B). Lineage tracing with the ROSA26mT/mG reporter mouse confirmed recombination in ECs in Tie2Cre;Elnf/f mice (Fig. 5C). Using the EC-specific cadherin gene (Cdh5Cre) to inactivate Eln gave similar results (data not shown).
Figure 5. The IEL appears normal in the aorta despite EC-specific Eln deletion.

H&E and VVG staining of P10 ascending (A) and descending aorta (B) show normal IEL and medial lamellae formation despite EC-specific Eln deletion. (C) Lineage tracing with the ROSA26mT/mG reporter mouse confirms green (recombined) ECs in Elnf/+ and Elnf/f mice expressing Tie2Cre and red (not recombined) ECs in Elnf/+ controls (arrowheads). All scale bars = 50 μm.
We used transmission electron microscopy (TEM) to compare IEL ultrastructure in the ascending and descending aorta. The IEL appeared normal in the ascending and descending aorta of Elnf/f mice (Figs. 6A and C), confirming observations from light microscopy (Fig. 1C) that insertion of the LoxP sites does not affect elastin integrity. Consistent with the light microscopy images in Fig. 4A, the IEL in the ascending aorta of Sm22aCre;Elnf/f mice was disrupted (Figs. 6A and B), while the IEL in the descending aorta was normal (Fig. 6C). The IEL in the ascending aorta of Sm22aCre;Elnf/f mice frequently consisted of small aggregates of elastin that never fused to form a continuous elastin layer (Fig. 6A). Where the IEL was more intact, the elastin had a moth-eaten appearance when viewed at higher magnification (Fig. 6B). In contrast, the IEL in the ascending aorta appeared ultrastructurally normal when elastin was inactivated in ECs (Tie2Cre;Elnf/f) (Fig. 6D). Together, the results from the SMC and EC-specific Eln knockout studies suggest that ECs are capable of contributing elastin to the IEL, but require a contribution from SMCs in the ascending aorta to produce an intact structure. SMCs in the ascending and descending aorta, in contrast, can produce an intact IEL with no elastin contribution from ECs.
Figure 6. Electron microscopy of the IEL in SMC- and EC-specific Eln knockouts.

(A) The IEL in the ascending aorta of P8 Elnf/f mice is intact (arrowhead), while in some regions it is composed of small aggregates of elastin that never fused to form a continuous barrier in Sm22aCre;Elnf/f mice (arrow). (B) In regions where the IEL is thicker and more intact in Sm22aCre;Elnf/f ascending aorta, it has a moth eaten appearance (arrow). (C) In contrast, the descending aorta in P8 Elnf/f and Sm22aCre;Elnf/f mice has an intact IEL (arrowheads). (D) The IEL is also intact in the ascending aorta of P10 Elnf/+ and Tie2Cre;Elnf/f mice (arrowheads). All scale bars = 10 μm.
ECs produce the IEL in resistance arteries.
A characteristic of muscular and resistance arteries is the absence of elastic laminae between the medial SMC layers. These arteries do, however, have a well-defined IEL and, in many cases, a complete or partial EEL that separates the media from the adventitia. To see how the loss of cell type-specific elastin expression influences resistance artery structure, we studied second-order mesenterics. Elnf/f mesenterics have an intact IEL and a thin discontinuous EEL in light and electron microscopy (Fig. 7A). Sm22aCre;Elnf/f mesenterics have an intact IEL, but the EEL was not detectable (Fig. 7B), consistent with SMCs making elastin in the outer regions of the wall. When Eln was inactivated in ECs (Tie2Cre;Elnf/f and Cdh5Cre;Elnf/f), however, the IEL was severely disrupted and the EEL appeared normal or sometimes thicker (Figs. 7C and D). These results suggest that SMCs in the second-order mesenteric arteries make small amounts of elastin that contribute partially to the IEL and solely to the EEL, but cannot increase elastin levels enough to compensate for the absence of elastin production from ECs in the IEL.
Figure 7. ECs produce the IEL in resistance arteries.

H&E (upper), VVG (middle), and transmission electron microscopy (TEM) (lower) images of second-order mesenteric arteries from (A) P13 Elnf/f, (B) P8 Sm22aCre;Elnf/f, (C) P10 Tie2Cre;Elnf/f, and (D) P13 Cdh5Cre;Elnf/f mice. Elnf/f and Sm22aCre;Elnf/f arteries have a prominent, intact IEL (A, B, arrows). Elnf/f arteries have a thin, discontinuous EEL (A, arrowhead), while the EEL in Sm22aCre;Elnf/f arteries is undetectable (B, arrowhead at expected location). In contrast, Tie2Cre;Elnf/f and Cdh5Cre;Elnf/f arteries have a thin, fragmented IEL (C, D, arrows) and a normal or slightly thicker EEL (C, D, arrowheads). Scale bars = 50 μm for H&E and VVG and 2 μm for TEM.
Based on our findings of heterogeneous cellular contributions to elastin deposition in the ascending aorta, descending aorta, and second-order mesenteric arteries, we performed a histological survey of different arterial beds in SMC- and EC-specific elastin knockout mice. The aortic root in Sm22aCre;Elnf/f mice had a similar wall structure to the ascending aorta, with a fragmented IEL, neointimal formation, and patches of elastin in the external medial layer (Online Fig. II). Medium-sized elastic and muscular/resistance arteries in Sm22aCre;Elnf/f mice showed wall structures that were similar to the descending aorta; the arterial wall was generally thicker than controls, and all arteries had an intact IEL even through medial elastic lamellae were missing (Online Fig. III). When Eln was inactivated in ECs (Tie2Cre;Elnf/f), the IEL, medial elastic fibers, and overall wall structure were indistinguishable at the histological level from Elnf/f controls in the medium-sized elastic arteries, but the IEL was less prominent and appeared fragmented in the muscular/resistance arteries (Online Fig. IV). The abdominal aorta, carotid artery, subclavian artery, iliac artery, and internal thoracic artery in Tie2Cre;Elnf/f mice had a normal IEL (Online Figs. IVA – E), while the femoral artery, renal artery, and inferior epigastric artery had a thin or fragmented IEL (Online Figs. IVF – H).
Neointima formation in Sm22aCre;Elnf/f ascending aorta correlates with IEL disruptions.
Histological sections of Sm22aCre;Elnf/f ascending aorta frequently showed cells luminal to the IEL in areas where the IEL was absent or severely disrupted. Cells in this neointima express SMC markers including smooth muscle α-actin, calponin, and smooth muscle myosin heavy chain 11, suggesting a smooth muscle identity (Fig. 8A). Lineage tracing using the ROSA26mT/mG reporter allele confirmed that the neointimal cells were derived from the Sm22aCre lineage (Fig. 4D). PCNA staining showed that the percentage of proliferating cells in the neointima was not different from the portion of proliferating cells in the media of control ascending aorta (Fig. 8B), suggesting that neointimal cells arise by a mechanism other than uncontrolled proliferation. Areas of neointimal hyperplasia correlated with local IEL disruption (Fig. 8C). These findings show that the IEL functions as a physical barrier to prevent medial SMCs from migrating into the arterial lumen and forming a neointima in the ascending aorta.
Figure 8. Neointima formation in Sm22aCre;Elnf/f ascending aorta.

(A) Immunostaining revealed SMC-marker positive neointima in the ascending aorta of Sm22aCre;Elnf/f mice. Neointimal cells are positive for smooth muscle α-actin (P18, SMA) (top), calponin (P9, center), and myosin heavy chain 11 (P9, Myh11) (bottom) (arrows). Scale bar = 50 μm. (B) Quantification of PCNA-positive cells in ascending aortic media from mice of indicated genotypes. Quantification of PCNA-positive cells in neointima (only in Sm22aCre;Elnf/f) is also shown. N = 4 for Sm22aCre;Elnf/+ and N = 3 for others. No significant differences for each group compared to control by one-way ANOVA with Sidak’s multiple comparison test. (C) Neointimal formation correlates with IEL disruption. Arrows indicate IEL disruption and neointima formation. Arrowheads indicate intact IEL without neointimal formation. Scale bar = 50 μm.
DISCUSSION
In our previous studies, we identified how global inactivation of one or both elastin alleles influences arterial development and cardiovascular function in mice3, 15, 20. In this report, we use SMC- and EC-targeted Cre drivers to inactive elastin expression in those two vascular cell populations. While there are several characteristics shared between the global and targeted knockouts, being able to specifically limit elastin production to a particular cell type has provided new information about how SMCs and ECs contribute elastin to the arterial wall and how defects in local elastin deposition may lead to cardiovascular disease.
The amount of elastin has a significant influence on arterial wall structure and function. With normal elastin levels, the number of SMC layers that are present before the onset of elastin production remains the same throughout the lifetime of the organism, including humans21. However, in response to loss-of-function mutations that reduce elastin levels (e.g., to 50% in Eln+/− mice), additional SMCs are recruited from the adventitia to the media right before birth to form more elastic laminae and normalize wall stress10, 20. Humans with supravalvular aortic stenosis (SVAS) caused by genetic mutations that lead to functional elastin haploinsufficiency show a similar increase in arterial SMC layers and elastic laminae10. Mice heterozygous for the SMC-specific Eln deletion show arterial wall changes remarkably similar to what occurs in Eln+/− mice, including recruitment of additional SMC layers and changes in arterial compliance, implying that the phenotype of Eln+/− mice is largely driven by reduced elastin amounts in the medial layer. Despite the structural and mechanical similarities in the arterial wall, one major difference is the lack of increased systolic blood pressure in P30 Sm22aCre;Elnf/+ mice compared to Eln+/−. Hypertension becomes more significant as Eln+/− mice mature19 and scales with elastin amounts in other elastin deficient mouse models17, so it is possible that Sm22aCre;Elnf/+ may have a delayed hypertensive phenotype due to changes in elastin amounts in the SMC versus global heterozygote. It is also possible that phenotype discrepancies are due to differences in the resistance arteries22 or RAS signaling18 between Sm22aCre;Elnf/+ and Eln+/− mice, which were not investigated in the current study.
The arterial remodeling similarities mostly disappear when elastin inactivation in SMCs (Sm22aCre;Elnf/f) is compared to the global knockout (Eln−/−). A major difference is that Sm22aCre;Elnf/fmice live longer than global knockouts (10-15 days compared to 48 hours, respectively), which allows for a lengthier evaluation of how arterial wall development proceeds without elastin. Eln−/−mice die when SMCs in the aortic media reorient, proliferate, and move into the lumen where they obstruct blood flow2. This is not the case when elastin is inactivated only in SMCs. SMCs in the descending aorta of Sm22aCre;Elnf/fmice remain in the media, most likely because of an intact IEL made by ECs. Also, the characteristic tortuosity in the Eln−/− descending aorta15 is absent in Sm22aCre;Elnf/f mice. The lack of tortuosity in the descending aorta suggests a tempered mechanical remodeling response23, likely a result of the small amount of elastin present in Sm22aCre;Elnf/f arteries compared to Eln−/−. Sm22aCre;Elnf/f mice have approximately the same amount of arterial elastin as mEln−/−hELN+/+ mice that express human elastin in the mouse knockout background17. mEln−/−hELN+/+ mice have hypertension and narrow arteries, but do not have focal aortic stenoses or a reduced lifespan17, 24, indicating that elastin organization throughout the aortic wall must be considered in addition to elastin amounts. The cause of death for Sm22aCre;Elnf/f mice is not clear, but the cardiomegaly, thrombosis, calcification and necrosis found in the heart suggest that cardiomyopathy may be a factor. As the cardiac histopathology did not manifest by P2, it is likely that the cardiomyopathy was secondary to pressure overload caused by aortic coarctation. The fact that Eln SMC knockout mice live for approximately two weeks indicates that medial elastin is not required for early postnatal survival.
The IEL is unique among elastic laminae in that it lies adjacent to the basal lamina of the endothelium, forming a common boundary between ECs in the intima and SMCs in the media. Because SMCs are the major elastin-producing cells in the arterial wall25-27, the assumption has been that they are responsible for producing the IEL. However, the presence of an IEL when elastin production is inactivated in SMCs indicates that ECs are capable of contributing elastin to this first elastic layer. The question of whether ECs contribute some, or all, of the elastin to the IEL was investigated by inactivating Eln in ECs using either Tie2- or Cdh5Cre. In both mouse lines, an IEL was present in the elastic arteries, but was barely detectable or highly fragmented in the muscular/resistance arteries. Together, data from the SMC and EC Eln knockouts suggest that both cell types contribute elastin to the IEL in elastic arteries, but ECs are mainly responsible for producing the IEL in resistance arteries where SMCs normally make little elastin.
In most elastic arteries, the Sm22aCre;Elnf/f IEL was indistinguishable from controls. The exception was the ascending aorta and aortic root where regions of the IEL consisted of a linear arrangement of disconnected small elastin globules as well as regions of more intact elastin with a moth-eaten appearance. These structural irregularities suggest that ECs in the ascending aorta and aortic root can make the necessary elastic fiber building blocks, but are not able to structure a functional elastic lamina without SMC contributions. It is possible that the IEL produced solely by ECs is unable to withstand the unique high systolic and pulse pressure in the ascending aorta, or alternatively that ascending aortic ECs require crosstalk with SMCs to make an intact IEL. Elastin production by cultured ECs has been documented in several studies28-30, but EC contribution to elastic structures in vivo has been difficult to establish. Our results show that ECs play a major role in the production of the IEL in vivo. Elastin production in the arterial wall begins only after mural cells associate with the vascular endothelium suggesting that SMCs provide a signal to ECs to initiate elastin production. Indeed, our previous studies showed that cultured ECs synthesize and secrete soluble elastin only when incubated in medium conditioned by SMCs30.
As discussed above, Eln−/− mice die within 48 hours of birth with an obstructive arterial occlusion resulting from SMC migration into the vascular lumen. The absence of an IEL was offered as an explanation for the unrestricted SMC ingrowth observed in Eln−/− mice2. With only an IEL and no other elastin in the arterial wall, luminal obstruction in Sm22aCre;Elnf/f mice is less pronounced. Neointimal cells were evident, however, around discontinuities in the IEL of the ascending aorta and aortic root in Sm22aCre;Elnf/f mice, and the extent of neointima correlated with local IEL integrity. These findings show that the IEL is an effective barrier against SMC migration into the lumen of the ascending aorta and aortic root. Neointima was not detected in the resistances arteries of TieCre;Elnf/f mice, despite there also being a defective IEL. These results suggest that additional factors beyond IEL integrity, such as hemodynamic forces or SMC developmental origin that vary with artery location, play a role in neointimal formation.
The origin of cells that populate the neointima associated with disease in mature vessels has been a source of debate. Endothelial, mesenchymal, and hematopoietic lineages have been identified in neointima, as have arterial wall-resident stem/progenitor cells expressing Sca-1 and/or c-kit31, 32. In our study, the neointimal cells are of Sm22aCre lineage and are most likely derived from medial SMCs. Due to the constitutively expressed nature of the Cre line, we are unable to distinguish when the cells poised to become neointimal cells turned on smooth muscle genes. An inducible lineage-tracing technique would be better suited to answer such questions.
In addition to an intact IEL in most Sm22aCre;Elnf/f arteries, another unexpected finding was clusters of elastin-producing cells in the outer part of the media. Lineage tracing using the ROSA26mT/mG reporter allele and the Sm22aCre transgene confirmed that these elastin-producing cells did not undergo recombination with Sm22aCre. However, immunostaining with an antibody to SM22a protein established that cells within the elastin-positive clusters expressed SM22a, suggesting that Cre expression driven by the SM22a promoter in the transgene does not faithfully recapitulate expression of the native SM22a gene in these cells or that there are subtle differences in cell populations that produce differences in recombination efficiency. It has been shown that cells expressing the promoter gene in a transient or submaximal fashion can have lower Cre recombination efficiency33. Evidence that the cells are a unique cell population in terms of ECM production include: (1) the unrecombined cells are not randomly distributed across the arterial wall but, for the most part, localize to the outer aspect of the media in both ascending and descending aorta in all mice examined, and (2) very few unrecombined cells are detected in SMC22αCre;Elnf/+ aorta, and (3) elastic structures made by the cells are abnormal. SM22a is expressed around E9.5 in mouse vascular SMCs shortly after expression of smooth muscle α-actin, and expression continues into adulthood34. An intriguing possibility is that the clusters of elastin-producing cells are derived from adventitial SMC progenitors. Passman et al35 have previously shown a population of adventitial Sca1+ cells capable of becoming SMCs. Our lineage tracing analysis using the ROSA26mT/mG reporter showed that most adventitial cells in the aorta of SM22aCre;Elnf/f mice did not recombine. While the majority of these cells may be myofibroblasts, those at the medial-adventitial boarder are in the correct environmental niche to be SMC progenitor cells35, 36. These cells may migrate into the media of SM22aCre;Elnf/f arteries due to the absence of defined elastic laminae that normally hamper their transmural movement.
Heterogeneity within the vascular population of elastin-producing SMCs has been observed in previous studies. In the developing avian vascular system, elastin production is first detected in the aorta near the aortic root and then expands outward along the SMCs of the ascending aorta. Interestingly, the onset of elastogenesis coincides with the loss of smooth muscle α-actin staining, which reappears in later development. Arteries that develop a muscular phenotype, however, never lose smooth muscle α-actin expression even though they express elastin37, 38. Other complex patterns of elastin production have been observed, including radial gradients of elastin expression that also differed between elastic and muscular arteries39, 40. In these studies, the highest levels of elastin mRNA in the developing ascending aorta were detected in the outer layers of the media, which is the location of the cell clusters observed in our study. Clearly, different populations of SMCs in the vasculature exhibit distinct ECM phenotypes. Future studies to unravel the mechanisms driving heterogeneity in cellular ECM expression may assist in repairing ECM defects in cardiovascular disease.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Elastin is arranged in layers within the arterial wall that are critical for cardiovascular function.
The internal elastic lamina (IEL) separates endothelial cells (ECs) from smooth muscle cells (SMCs).
The relative contributions of ECs and SMCs to elastic fiber formation in vivo are unknown.
What New Information Does This Article Contribute?
SMCs and ECs can independently form the IEL in most elastic arteries.
ECs are the major source of elastin in the IEL for muscular and resistance arteries.
Lack of SMC elastin in the ascending aortic IEL leads to disruptions that correlate with neointimal formation.
Elastin is arranged in concentric layers in the arterial wall and the number of layers decreases with arterial size. Almost all arteries have at least one layer, the IEL, which separates ECs from SMCs. In elastin knockout mice (Eln−/−) the lack of an IEL may contribute to aortic stenosis and eventual occlusion. We used EC- and SMC-Eln knockout mice to determine how each cell type contributes to elastin deposition and cardiovascular function. In EC-Eln knockout mice, the IEL was severely disrupted in small arteries that have 1–2 elastin layers, but was normal in large arteries with multiple elastin layers. In SMC-Eln knockout mice, the IEL was normal in all arteries except the ascending aorta where there were IEL disruptions that correlated with neointimal formation. The mid-wall elastin layers in large arteries were absent in SMC-Eln knockout mice and the mice died around 2 weeks of age from cardiomyopathy secondary to severe aortic stenosis. Our results show heterogeneity in elastin deposition depending on cell type and arterial location that was previously unappreciated. Future studies to unravel mechanisms driving this heterogeneity may assist in repairing elastin defects that lead to cardiovascular diseases such as aortic stenosis.
ACKNOWLEDGEMENTS
The Washington University Mouse Cardiovascular Phenotyping Core performed the echocardiography studies. We acknowledge Thomas Broekelmann for performing the protein quantification studies.
SOURCES OF FUNDING
This study was partially funded by NSF grant 1662434 (J. Wagenseil), and NIH grants HL-115560 (J. Wagenseil), HL-53325 (R. Mecham), and HL-105314 (R. Mecham and J. Wagenseil). C.-J. Lin was supported by T32-HL007081. Funds were also provided to R. Mecham by the Ines Mandl Research Foundation.
Nonstandard Abbreviations and Acronyms:
- Eln
elastin
- EC
endothelial cell
- EEL
external elastic laminae
- ECM
extracellular matrix
- IEL
internal elastic laminae
- P
postnatal day
- SMC
smooth muscle cell
- TEM
transmission electron microscopy
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Sage H, Gray WR. Studies on the evolution of elastin--i. Phylogenetic distribution. Comparative biochemistry and physiology. B, Comparative biochemistry. 1979;64:313–327 [DOI] [PubMed] [Google Scholar]
- 2.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280 [DOI] [PubMed] [Google Scholar]
- 3.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. Reduced vessel elasticity alters cardiovascular structure and function in newborn mice. Circ Res. 2009;104:1217–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLean SE, Mecham BH, Kelleher CM, Mariani TJ, Mecham RP. Extracellular matrix gene expression in the developing mouse aorta. Advances in Developmental Biology. 2005;15:81–128 [Google Scholar]
- 5.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-a prevents the acute but not chronic effects of anp on blood pressure. Proc Natl Acad Sci U S A. 2002;99:7142–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. Lrp: Role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332 [DOI] [PubMed] [Google Scholar]
- 7.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242 [DOI] [PubMed] [Google Scholar]
- 8.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela-Arispe ML. Ve-cadherin-cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Developmental dynamics : an official publication of the American Association of Anatomists. 2006;235:759–767 [DOI] [PubMed] [Google Scholar]
- 9.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent cre reporter mouse. Genesis. 2007;45:593–605 [DOI] [PubMed] [Google Scholar]
- 10.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102:1783–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferruzzi J, Bersi MR, Uman S, Yanagisawa H, Humphrey JD. Decreased elastic energy storage, not increased material stiffness, characterizes central artery dysfunction in fibulin-5 deficiency independent of sex. J Biomech Eng. 2015;137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barsha G, Denton KM, Mirabito Colafella KM. Sex- and age-related differences in arterial pressure and albuminuria in mice. Biol Sex Differ. 2016;7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, van Erp C, Gerber EE, Parker SJ, Sauls K, Judge DP, Cooke SK, Lindsay ME, Rouf R, Myers L, ap Rhys CM, Kent KC, Norris RA, Huso DL, Dietz HC. Angiotensin ii-dependent tgf-beta signaling contributes to loeys-dietz syndrome vascular pathogenesis. J Clin Invest. 2014;124:448–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le VP, Wagenseil JE. Echocardiographic characterization of postnatal development in mice with reduced arterial elasticity. Cardiovasc Eng Technol. 2012;3:424–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagenseil JE, Nerurkar NL, Knutsen RH, Okamoto RJ, Li DY, Mecham RP. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol. 2005;289:H1209–1217 [DOI] [PubMed] [Google Scholar]
- 16.Stoilov I, Starcher BC, Mecham RP, Broekelmann TJ. Measurement of elastin, collagen, and total protein levels in tissues. Methods Cell Biol. 2018;143:133–146 [DOI] [PubMed] [Google Scholar]
- 17.Hirano E, Knutsen RH, Sugitani H, Ciliberto CH, Mecham RP. Functional rescue of elastin insufficiency in mice by the human elastin gene: Implications for mouse models of human disease. Circ Res. 2007;101:523–531 [DOI] [PubMed] [Google Scholar]
- 18.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest. 2003;112:1419–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le VP, Knutsen RH, Mecham RP, Wagenseil JE. Decreased aortic diameter and compliance precedes blood pressure increases in postnatal development of elastin-insufficient mice. Am J Physiol Heart Circ Physiol. 2011;301:H221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. The importance of elastin to aortic development in mice. Am J Physiol Heart Circ Physiol. 2010;299:H257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolinsky H, Glagov S. A lamellar unit of aortic medial structure and function in mammals. Circ Res. 1967;20:99–111 [DOI] [PubMed] [Google Scholar]
- 22.Osei-Owusu P, Knutsen RH, Kozel BA, Dietrich HH, Blumer KJ, Mecham RP. Altered reactivity of resistance vasculature contributes to hypertension in elastin insufficiency. Am J Physiol Heart Circ Physiol. 2014;306:H654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Humphrey JD, Eberth JF, Dye WW, Gleason RL. Fundamental role of axial stress in compensatory adaptations by arteries. J Biomech. 2009;42:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiao Y, Li G, Korneva A, Caulk AW, Qin L, Bersi MR, Li Q, Li W, Mecham RP, Humphrey JD, Tellides G. Deficient circumferential growth is the primary determinant of aortic obstruction attributable to partial elastin deficiency. Arterioscler Thromb Vasc Biol. 2017;37:930–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giro MG, Hill KE, Sandberg LB, Davidson JM. Quantitation of elastin production in cultured vascular smooth muscle cells by a sensitive and specific enzyme-linked immunoassay. Coll Relat Res. 1984;4:21–34 [DOI] [PubMed] [Google Scholar]
- 26.Faris B, Tan OT, Toselli P, Franzblau C. Long-term neonatal rat aortic smooth muscle cell cultures: A model for the tunica media of a blood vessel. Matrix. 1992;12:185–188 [DOI] [PubMed] [Google Scholar]
- 27.Ruckman JL, Luvalle PA, Hill KE, Giro MG, Davidson JM. Phenotypic stability and variation in cells of the porcine aorta: Collagen and elastin production. Matrix Biol. 1994;14:135–145 [DOI] [PubMed] [Google Scholar]
- 28.Carnes WH, Abraham PA, Buonassisi V. Biosynthesis of elastin by an endothelial cell culture. Biochemical and biophysical research communications. 1979;90:1393–1399 [DOI] [PubMed] [Google Scholar]
- 29.Cantor JO, Keller S, Parshley MS, Darnule TV, Darnule AT, Cerreta JM, Turino GM, Mandl I. Synthesis of crosslinked elastin by an endothelial cell culture. Biochemical and biophysical research communications. 1980;95:1381–1386 [DOI] [PubMed] [Google Scholar]
- 30.Mecham RP, Madaras J, McDonald JA, Ryan U. Elastin production by cultured calf pulmonary artery endothelial cells. J Cell Physiol. 1983;116:282–288 [DOI] [PubMed] [Google Scholar]
- 31.Psaltis PJ, Simari RD. Vascular wall progenitor cells in health and disease. Circ Res. 2015;116:1392–1412 [DOI] [PubMed] [Google Scholar]
- 32.Xie Y, Fan Y, Xu Q. Vascular regeneration by stem/progenitor cells. Arterioscler Thromb Vasc Biol. 2016;36:e33–40 [DOI] [PubMed] [Google Scholar]
- 33.Klinger M, Chmura SA, Killeen N. Reporter alleles that inform on differences in cre recombinase expression. J. Immunol 2010;184:6170–6176 [DOI] [PubMed] [Google Scholar]
- 34.Li L, Miano JM, Cserjesi P, Olson EN. Sm22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ Res. 1996;78:188–195 [DOI] [PubMed] [Google Scholar]
- 35.Passman JN, Dong XR, Wu SP, Maguire CT, Hogan KA, Bautch VL, Majesky MW. A sonic hedgehog signaling domain in the arterial adventitia supports resident sca1+ smooth muscle progenitor cells. Proc Natl Acad Sci U S A. 2008;105:9349–9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Majesky MW, Horita H, Ostriker A, Lu S, Regan JN, Bagchi A, Dong XR, Poczobutt J, Nemenoff RA, Weiser-Evans MC. Differentiated smooth muscle cells generate a subpopulation of resident vascular progenitor cells in the adventitia regulated by klf4. Circ Res. 2017;120:296–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergwerff M, DeRuiter MC, Poelmann RE, Gittenberger-de Groot AC. Onset of elastogenesis and downregulation of smooth muscle actin as distinguishing phenomena in artery differentiation in the chick embryo. Anatomy and embryology. 1996;194:545–557 [DOI] [PubMed] [Google Scholar]
- 38.Rosenquist TH, McCoy JR, Waldo KL, Kirby ML. Origin and propagation of elastogenesis in the developing cardiovascular system. The Anatomical record. 1988;221:860–871 [DOI] [PubMed] [Google Scholar]
- 39.Selmin O, Volpin D, Bressan GM. Changes of cellular expression of mrna for tropoelastin in the intraembryonic arterial vessels of developing chick revealed by in situ hybridization. Matrix. 1991;11:347–358 [DOI] [PubMed] [Google Scholar]
- 40.Holzenberger M, Lievre CA, Robert L. Tropoelastin gene expression in the developing vascular system of the chicken: An in situ hybridization study. Anatomy and embryology. 1993;188:481–492 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.