

Abstract
Purpose of review
The purpose of this review is to outline the current understanding of the molecular mechanisms and natural history of osteogenesis imperfecta (OI), and to describe the development of new treatments for this disorder.
Recent findings
The introduction of next-generation sequencing technology has led to better understanding of the genetic etiology of OI and enabled cost-effective and timely diagnosis via expanded gene panels and exome or genome sequencing. Clinically, despite genetic heterogeneity, different forms of OI share similar features that include connective tissue and systemic manifestations in addition to bone fragility. Thus, the goals of treatment in OI extend beyond decreasing the risk of fracture, to include the maximization of growth and mobility, and the management of extra-skeletal complications. The standard of care in pediatric patients is bisphosphonates therapy. Ongoing pre-clinical studies in OI mouse models and clinical studies in individuals with OI have been instrumental in the development of new and targeted therapeutic approaches, such as sclerostin inhibition and TGF beta inhibition.
Summary
OI is a skeletal dysplasia characterized by bone fragility and extra-skeletal manifestations. Better understanding of the mechanisms of OI will enable the development of much-needed targeted therapies to improve the outcome in affected individuals.
Keywords: Osteogenesis imperfecta, bisphosphonates, targeted therapy
Introduction
Osteogenesis imperfecta (OI) is a systemic connective tissue disorder characterized by low bone mass and bone fragility causing significant morbidity due to pain, immobility, skeletal deformities and growth deficiency (1–3). Decreased bone strength leads to low-trauma fractures or fractures in atypical locations (such as olecranon and vertebral compression fractures) (3, 4). Extra-skeletal manifestations may include dental anomalies, blue-gray sclera, hearing loss, joint hypermobility and more rarely muscle weakness, cardiovascular and pulmonary complications (1, 2). The estimated incidence of OI is about 1:10 000 (1). Identification of variants in OI-related genes in mildly affected or apparently asymptomatic individuals supports there being a broader clinical spectrum that ranges from low bone mass and early onset osteoporosis to progressively deforming and perinatal lethal OI (3).
OI is a genetically heterogeneous connective tissue disorder. The majority of OI (about 85–90% of cases) is associated with dominantly-inherited pathogenic variants in COL1A1 or COL1A2 (1–3). The remaining cases are caused by pathogenic variants in non-collagenous genes, encoding proteins involved in collagen biosynthesis, or transcription factors and signaling molecules related to bone cell differentiation and mineralization, and are associated with an autosomal recessive (most commonly), dominant or X-linked inheritance (Table 1) (1–3). With the advent of next-generation sequencing technology, new OI-causing genes and novel pathogenic variants in well-established OI genes continue to be identified increasingly in apparently phenotypically unique families or patients (3, 5).
Table 1:
Genetic classification of osteogenesis imperfecta types and main clinical features.
| OI type | Inheritance | Gene | Severity & unique clinical features |
|---|---|---|---|
| I | AD | COL1A1 | Mild, normal or short stature; little or no deformities |
| II | AD | COL1A1, COL1A2 | Lethal, minimal calvarial mineralization, beaded ribs, long bone deformities |
| III | AD | COL1A1, COL1A2 | Severe, progressively deforming bones |
| IV | AD | COL1A1, COL1A2 | Moderate severity with short stature |
| V | AD | IFITM5 | Variable severity, calcification of interosseous membrane of the forearm, hyperplastic callus formation |
| VI | AR | SERPINF1 | Moderate to severe, accumulation of un-mineralized osteoid; biopsy shows fish-scale pattern of the lamellae |
| VII | AR | CRTAP | Severe to lethal, rhizomelia |
| VIII | AR | LEPRE1 | Severe to lethal, rhizomelia, coxa vara, popcorn metaphyses |
| IX | AR | PPIB | Severe, short bowed femurs with anterior bowing of the tibiae |
| X | AR | SERPINH1 | Severe |
| XI | AR | FKBP10 | Moderate to severe, joint contractures; biopsy shows distorted lamellar structure and a fish scale-like pattern |
| XII | AR | SP7 | Moderate severity |
| XIII | AR | BMP1 | Severe |
| XIV | AR | TMEM38B | Moderate to severe |
| XV | AR | WNT1 | Moderate to severe, also have brain malformations |
| XVI | AR | CREB3L1 | Severe, perinatal fractures, multiple fractured tubular bones with an accordion-like broadened appearance |
| XVII | AR | SPARC | Progressively severe |
| XVIII | AR | FAM64A | Moderate to severe, dysmorphic features, developmental delay |
| XIX | X-linked | MBTPS2 | Moderate to severe, pectus deformity |
| Un-classified | AR | PLOD2 | Moderate to severe, joint contractures |
| Un-classified | X-linked | PLS3 | Osteoporosis with fractures, clinical overlap with OI |
The clinical classification of OI was first established by Sillence and colleagues (6) in 1979 and divided OI into four subgroups: type I with mild non-deforming phenotype, type II associated with perinatal lethality, type III with severe non-lethal progressive deforming phenotype, and type IV with a moderate severity (that is intermediate between types I and III). This classification was later expanded based on distinguishing clinical features, as in the case of OI type V-VII (7), or based on the genetic cause (1, 2). However, regardless of the molecular etiology on which more recent nosology have been based with types ranging up to XIX (Table 1), most forms of OI would fit into one of the Sillence subtypes based on the clinical features and severity (3), and the clinical classification is still most commonly used to determine treatment and prognosis for patients (1, 2). For the purpose of this review, we will use the clinical Sillence classification (6).
The phenotypic severity of COL1A1/COL1A2-related OI is largely dependent on the nature of type I collagen mutations (1–3). Type I collagen is the major component of bone extracellular matrix, and its quantity and integrity determine bone strength (8). The triple helix molecule of type I collagen is composed of two α1 chains and one α2 chain, encoded by COL1A1 and COL1A2, respectively. It is characterized by repeated amino acid sequence, Gly-X-Y, typically with proline and hydroxyproline in positions X and Y. Pathogenic variants that alter the structure of type I collagen (qualitative collagen defects, such as glycine substitutions) are usually associated with a moderate to severe clinical phenotype, with severity generally associated with more carboxyl-terminal glycine substitutions and in glycine substitutions in COL1A1 more than COL1A2 (1–3). Pathogenic variants that cause haploinsufficiency (quantitative collagen defects, such as truncating variants) result in decreased amount of a structurally normal collagen and typically are associated with milder clinical phenotype (1–3). Pathogenic variants in genes that function in collagen biosynthesis (such as members of the prolyl-3 hydroxylase complex CRTAP, P3H1 and PPIB, the chaperone molecule FKBP10 and others) impact bone strength by disrupting the synthesis, post-translation modifications, intra-cellular trafficking, assembly and cross-linking of the collagen molecule and result in a moderate to severe OI phenotype (1, 2, 8). It is important to note that this genotype-phenotype correlation is not absolute, and clinical variability exist even within family members carrying the same variant (1, 9, 10).
Clinical Manifestations of OI
OI is a skeletal dysplasia characterized by bone fragility and high incidence of fractures that may occur with minimal or no trauma. Fractures may involve atypical locations, as compared to the general population (4), for example vertebral fractures occur in about 70% of patients with OI (11). Severe OI may present prenatally by detection of in utero fractures and shortening of long bones on prenatal ultrasound (1). Moderate to severe forms of OI present with progressive bone deformities, including bowing of long bones, scoliosis and rib cage deformities (3, 11, 12). Short stature is an important feature in moderate, and progressively deforming OI (types III-IV), although growth deficiency and reduced growth velocity are also appreciated in the milder OI type I (even in the absence of significant bone deformities) (13, 14). Craniocervical junction anomalies (basilar invagination, basilar impression and platybasia) are rare but potentially serious complications that should be screened for (1, 15).
OI is a systemic connective tissue disorder. Type I collagen is an important component of many tissues and alteration in its structure and synthesis causes an array of extra-skeletal manifestations (1, 2). Joint hypermobility is common (1, 15). Gray or blue scleral hue is a prominent feature in OI type I but may also be noted in other forms of OI, regardless of the clinical severity (1). Dental anomalies may include dentinogenesis imperfecta, clinically presenting as discoloration of the teeth due to defect in dentin, missing permanent teeth, and malocclusion (16, 17). Teeth brittleness and malocclusion are more severe in OI type III (16). Hearing loss, usually with onset at the 2nd-3rd decade of life, may be conductive, sensorineural or mixed and is more prevalent in OI type I (18). Muscle weakness has been reported in patients with OI and correlates with the clinical severity (19, 20). Decreased muscle strength is associated with gross motor delay, easy fatigability and exercise intolerance (19, 20), and together with the fractures, bone deformities and ligamentous laxity may contribute to the limited mobility, especially in severe OI (21). Cardiovascular manifestations have been reported in OI and include aortic root dilatation, valvular dysfunction, and rarely aortic aneurysm and dissection (1, 22). Pulmonary complications are an important cause of morbidity and mortality in OI and may be due to extrinsic factors such as rib fractures, scoliosis, and muscle weakness, as well as increasingly recognized intrinsic lung abnormalities (23). Pulmonary function decreases with age, and across all ages is most significantly impaired in OI type III (23, 24). In addition, a recent study suggests that sleep disordered breathing (obstructive sleep apnea) may be slightly more common in OI than in the general population (25). The physical disability, discomfort, and chronic pain that result from OI complications significantly affect the patients’ quality of life, which as expected is correlated with the phenotypic severity (26–28).
Current Treatment in OI
Goals of treatment in OI include decreasing fracture incidence, improving pain, and promoting growth, mobility and functional independence. Given the many skeletal and extra-skeletal manifestations, multi-disciplinary care should be provided, combining medical genetics, orthopedic surgery, physical therapy, dental expertise and other subspecialties based on the symptoms and complications (29). Orthopedic care includes the treatment of fractures and operative management of long bone and spinal deformities (1, 12). Physical therapy and rehabilitation is essential to promote motor development, increase endurance, relieve pain, gain independence, and to assist with recovery from surgical procedures (1, 29, 30). From a nutritional standpoint, maintaining adequate levels of vitamin D and calcium is important to support bone health, and for improving the efficacy and minimizing the adverse effects of bisphosphonate use but dose should be adjusted to prevent hypercalciuria (1, 2).
Growth Hormone Therapy.
Growth hormone has been proposed as a treatment in OI to address the growth deficiency and due to its potential anabolic effect in bone. An early study suggested that a subset of patients, mostly with OI type IV, may respond to growth hormone treatment by increasing linear growth rate with a mild improvement in cancellous bone density (31). Another study compared the combination of growth hormone and bisphosphonate therapy to bisphosphonates alone, and showed a synergistic effect on growth velocity but no effect on fracture risk (32). Overall, growth hormone treatment appears to be less efficient in the more severe OI type III. There is not enough data to support its benefit in the treatment of OI, and currently this approach is not in standard clinical use.
Anti-resorptive treatments.
The nitrogen-containing bisphosphonates (Pamidronate, Alendronate, Risedronate and Zoledronic acid) are currently the mainstay of pharmacological care in pediatric patients with OI. Bisphosphonates were shown to improve bone mass and architecture, and to some extent, reduce fracture risk (1, 2, 33, 34). They exert their effect via inhibition of osteoclast activity and induction of osteoclast apoptosis (34). Bisphosphonates can be administered orally or by intravenous infusion. Recent studies suggest a superior effect for intravenous bisphosphonates over oral bisphosphonates in improving bone mineral density (BMD) (35), and reducing fracture rate (36). Cyclic bisphosphonate infusion appears to improve chronic pain (37). Additional advantages of intravenous bisphosphonates include lower risk of gastrointestinal side effects, better bioavailability, adjustable dosing, and increased patient compliance. Following administration bisphosphonates are concentrated in the bone matrix, where they may reside for months to years. This raises a concern regarding their long-term effect on depressed bone turnover (2, 34). As bone formation and resorption are coupled, suppression of resorption by bisphosphonates always leads to a secondary inhibition of bone formation. In the pediatric setting, the robust bone formation associated with growth may compensate for this effect. This may underlie the anecdotal reports of less effectiveness of bisphosphonates in adults with OI. Bisphosphonates are generally well-tolerated, and most common side effects include gastrointestinal irritation when taken orally and an acute phase infusion reaction (hyperpyrexia, myalgia, and weakness), likely immune-mediated, that typically occurs during the first infusion. Transient hypocalcemia can be prevented by pre-treatment with calcium and vitamin D. Osteonecrosis of the jaw (a known side effect in adults often with high dose treatment in the context of cancer) has never been reported in children.
Meta-analyses which investigated the efficacy of bisphosphonates in OI concluded that treatment significantly improved BMD (38), and was associated with a moderate decrease in fracture rate (39). The effect of bisphosphonates on the occurrence and progression of scoliosis was inconclusive, although recent cohort study suggests a decrease in scoliosis probability at least in the milder OI type I (35). Overall, the effects of bisphosphonate therapy in OI are less robust compared to patients with osteoporosis, and they may be less effective in adults (39, 40).
Denosumab (Prolia, Amgen Inc. [Thousand Oaks, CA, USA]) is a human monoclonal antibody that targets the receptor activator of nuclear factor kappa-B ligand (RANKL), a cytokine that mediates osteoclastogenesis and osteoclast survival (41). Thus, the mechanism of action is similar to bisphosphonates in that it inhibits osteoclast activity to suppress bone resorption. Preclinical studies in animal models showed an additional effect to allow long term periosteal bone formation, suggesting a potential advantage over bisphosphonates (42). Denosumab is given via subcutaneous injections (which may be more convenient for patients and families compared to intravenous bisphosphonates). In contrast to bisphosphonates, it has a short half-life of approximate 30 days. This can induce a rebound of increased bone turnover and hypercalcemia upon discontinuation of treatment, an effect that appears to be especially strong in children (41). Other reported side effects include rare hypocalcemia, osteonecrosis of the jaw, and atypical hip fractures (41).
Denosumab therapy has been trialed in pediatric patients with OI type VI (cause by PEDF mutations) who responded poorly to bisphosphonate therapy (43–45). In these studies the effect on BMD was variable, perhaps not surprising given the underlying osteomalacia component of this disease. Hypercalcemia and hypercalciuria were reported both during (between doses) and after treatment with denosumab, and there was concern for rapid bone loss post treatment (41, 45). Consolidation with bisphosphonates has been proposed to alleviate this effect. Another study in ten patients with OI showed a significant increase in lumbar spine BMD with no significant change in spine morphology or in mobility (46). Currently, data is limited regarding the clinical use of denosumab in OI (47) and a multi-center clinical trial is ongoing to address this question (ClinicalTrials.gov Identifiers: , https://clinicaltrials.gov/ct2/show/study/NCT02352753 and , https://clinicaltrials.gov/ct2/show/NCT03638128).
Anabolic treatments.
Osteo-anabolic therapies are targeted to increase osteoblast activity and bone formation (48). Currently they are only approved for use in adults with osteoporosis, and the experience in OI is limited. Teriparatide (recombinant human PTH 1–34, Forteo, Eli Lilly & Co. [Indianapolis, IN, USA]) is a parathyroid hormone analogue that was the first anabolic therapy approved for use in osteoporosis. Pre-clinical studies in rats that received high-dose PTH 1–34 treatment raised concerns for risk of osteosarcoma (49). Because of this its clinical use is limited for a duration of 24 months, and it has not been used in pediatric patients. Randomized, placebo controlled clinical trials of teriparatide therapy in adult OI patients showed a significant increase in BMD in OI type I, although therapy does not seem to be as effective in moderate to severe OI types III and IV (50, 51). Abaloparatide (a synthetic analogue of PTH related peptide, Tymlos, Radius Health, Inc [Waltham, MA, USA]) has demonstrated a potent effect on bone anabolic activity (52) and may be considered in the future for treatment in OI.
Sclerostin-inhibitory antibodies (Scl-Ab/Romosozumab, Amgen Inc. [Thousand Oaks, CA, USA]; Blosozumab, Eli Lilly & Co. [Indianapolis, IN, USA]; BSP804, Novartis [Basel, Switzerland]) target sclerostin, a glycoprotein that inhibits bone formation via inhibition of the canonical Wnt signaling pathway in osteoblasts (34). Romosozumab was recently approved for use in osteoporosis. Pre-clinical studies in OI animal models demonstrated beneficial effects of sclerostin inhibition therapy on bone formation and BMD (2, 34, 53). Data is not available yet regarding the safety and efficacy in OI patients although a different formulation is in clinical trial (ClinicalTrials.gov Identifier: , https://clinicaltrials.gov/ct2/show/NCT03216486).
Anti-TGFβ antibody.
Anti-resorptive and anabolic treatments improve bone mass; however, they do not target the mechanistic basis of type I collagen and matrix abnormality that underlie the bone fragility in OI. Studies in animal models suggest that continuous activation of TGF beta signaling leads to low bone mass and bone fragility in OI related to collagen mutation and post-translational modification (54, 55). Based on this evidence, a clinical trial is currently ongoing to evaluate the safety and efficacy of TGF beta inhibition in adult OI patients (ClinicalTrials.gov Identifier: , https://clinicaltrials.gov/ct2/show/NCT03064074).
Cell and gene therapy aim to address this issue by correcting the genetic defect, although these interventions are associated with ethical and safety concerns and are still considered experimental (2, 56), and their broad distributed targeting remains a challenge.
A summary of the pharmacologic interventions described above is provided in Figure 1.
Figure 1:
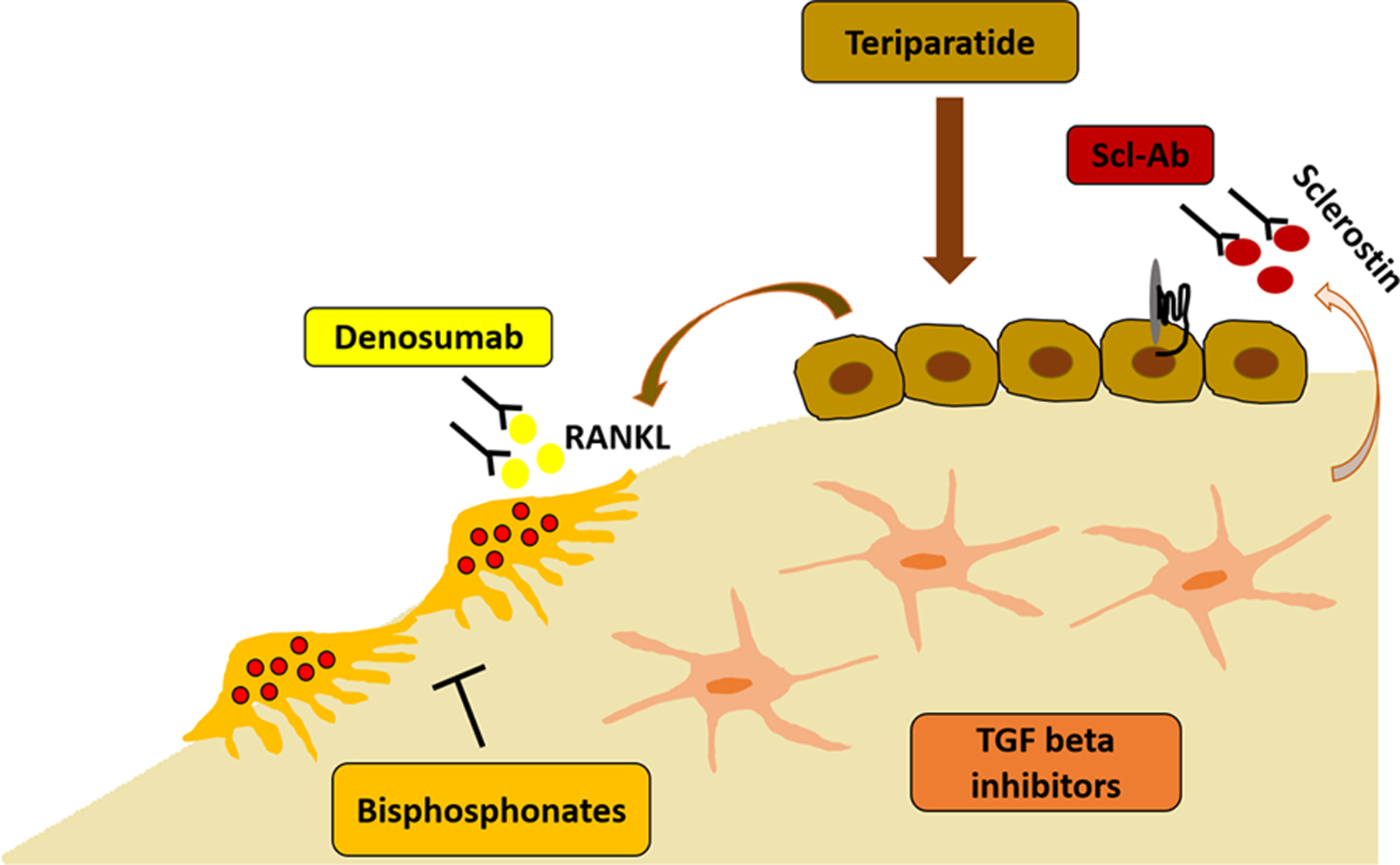
Pharmacologic interventions in OI and their target within the bone. Anti-resorptive agents include the bisphosphonates that reside in the bone matrix and inhibit osteoclast activity, and denosumab which targets the receptor activator of nuclear factor kappa-B ligand (RANKL that stimulates osteoclast formation and function). Anabolic therapies include teriparatide (a PTH analogue) that is promoting osteoblast differentiation and activity, and sclerostin inhibitory antibodies (Scl-Ab) that increase bone formation via Wnt signaling. TGF beta inhibition is a new approach designed to target the excessive activation of TGF beta signaling within the bone matrix which is thought to contribute to the bone fragility.
Conclusion
OI is a multi-systemic connective tissue disorder. Recent discoveries in the field have expanded our knowledge on the etiologies and mechanism of OI, as well as the natural history and clinical management. The genetic studies have also identified factors that regulate bone mass and quality in children and adults. Current therapeutic approaches were mostly designed for the treatment of osteoporosis, hence it is not surprising that they seem to be comparatively less effective in OI. Studies are warranted to develop personalized, genotype-based treatment strategies that will target the specific molecular pathophysiology of OI.
Key Points.
OI is a systemic connective tissue disorder. Primary skeletal features include recurrent fractures, bone deformities and short stature. Extra-skeletal manifestations include joint hypermobility, dentinogenesis imperfecta, blue sclera, hearing loss, and more rarely muscle weakness, pulmonary and cardiovascular complications.
OI is a genetically heterogeneous disorder. The majority of OI is associated with pathogenic variants in COL1A1/COL1A2 with an autosomal-dominant inheritance. Other OI-causing genes encode for proteins that are involved in collagen synthesis, trafficking and post-translational modifications, or for factors related to bone cell differentiation and mineralization.
OI requires multi-disciplinary care, including orthopedic management, rehabilitation, nutritional care, dental care and other subspecialties in addition to pharmacologic therapy.
Current pharmacologic therapies in OI may improve bone mass and architecture but do not target the matrix abnormality, which is the primary cause of bone fragility. Research is ongoing to design therapies that will address the genetic defect and mechanism of OI.
Acknowledgements
This work was supported by NIH P01 HD070394 (BL) and NIH U54 AR068069 (BL). This work was also supported by the Rolanette and Berdon Lawrence Bone Disease Program of Texas (RM).
Footnotes
Conflicts of interests
None.
References
- 1.Marini JC, Forlino A, Bachinger HP, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. [DOI] [PubMed] [Google Scholar]
- 2.Marom R, Lee YC, Grafe I, Lee B. Pharmacological and biological therapeutic strategies for osteogenesis imperfecta. Am J Med Genet C Semin Med Genet. 2016;172(4):367–83. [DOI] [PubMed] [Google Scholar]
- 3.Robinson ME, Rauch F. Mendelian bone fragility disorders. Bone. 2019. [DOI] [PubMed] [Google Scholar]; ** This is an up-to-date summary of the current knowledge on the genetics, phenotype and pathomechanisms of OI and related disorders.
- 4.Peddada KV, Sullivan BT, Margalit A, Sponseller PD. Fracture Patterns Differ Between Osteogenesis Imperfecta and Routine Pediatric Fractures. J Pediatr Orthop. 2018;38(4):e207–e12. [DOI] [PubMed] [Google Scholar]; * This study compared fracture patterns in a cohort of OI patients and controls, and identified OI-specific fracture characteristics that are of diagnostic value.
- 5.Lu JT, Campeau PM, Lee BH. Genotype-phenotype correlation--promiscuity in the era of next-generation sequencing. N Engl J Med. 2014;371(7):593–6. [DOI] [PubMed] [Google Scholar]
- 6.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16(2):101–16. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study was the first to describe and establish the clinical classification of OI which still holds most use from a clinical perspective.
- 7.Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363(9418):1377–85. [DOI] [PubMed] [Google Scholar]
- 8.Eyre DR, Weis MA. Bone collagen: new clues to its mineralization mechanism from recessive osteogenesis imperfecta. Calcif Tissue Int. 2013;93(4):338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben Amor IM, Glorieux FH, Rauch F. Genotype-phenotype correlations in autosomal dominant osteogenesis imperfecta. J Osteoporos. 2011;2011:540178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maioli M, Gnoli M, Boarini M, et al. Genotype-phenotype correlation study in 364 osteogenesis imperfecta Italian patients. Eur J Hum Genet. 2019;27(7):1090–100. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study expands the knowledge on the genotype-phenotype correlation in a large cohort of patients with COL1A1/COL1A2-related OI.
- 11.Castelein RM, Hasler C, Helenius I, et al. Complex spine deformities in young patients with severe osteogenesis imperfecta: current concepts review. J Child Orthop. 2019;13(1):22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This paper outlines the natural history of spine deformities in OI from an orthopedic perspective.
- 12.Franzone JM, Shah SA, Wallace MJ, Kruse RW. Osteogenesis Imperfecta: A Pediatric Orthopedic Perspective. Orthop Clin North Am. 2019;50(2):193–209. [DOI] [PubMed] [Google Scholar]; * This is a comprehensive review of the orthopedic management of OI.
- 13.Jain M, Tam A, Shapiro JR, et al. Growth characteristics in individuals with osteogenesis imperfecta in North America: results from a multicenter study. Genet Med. 2019;21(2):275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This is the largest cohort study to date of growth characteristics in OI, and the first to demonstrate that growth is affected in all OI subtypes, including the milder OI type I, although growth deficiency is more significant in more severe forms of OI.
- 14.Barber LA, Abbott C, Nakhate V, et al. Longitudinal growth curves for children with classical osteogenesis imperfecta (types III and IV) caused by structural pathogenic variants in type I collagen. Genet Med. 2019;21(5):1233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study suggests OI-specific growth curves for OI types III and IV that can serve as a baseline reference for clinical monitoring of patients.
- 15.Arponen H, Makitie O, Waltimo-Siren J. Association between joint hypermobility, scoliosis, and cranial base anomalies in paediatric Osteogenesis imperfecta patients: a retrospective cross-sectional study. BMC Musculoskelet Disord. 2014;15:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Najirad M, Ma MS, Rauch F, et al. Oral health-related quality of life in children and adolescents with osteogenesis imperfecta: cross-sectional study. Orphanet J Rare Dis. 2018;13(1):187. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study reports the primary dental manifestations in OI and their effect on the patients’ quality of life in a cohort of pediatric and adolescent patients.
- 17.Andersson K, Dahllof G, Lindahl K, et al. Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta - A retrospective cohort study. PLoS One. 2017;12(5):e0176466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carre F, Achard S, Rouillon I, et al. Hearing impairment and osteogenesis imperfecta: Literature review. Eur Ann Otorhinolaryngol Head Neck Dis. 2019. [DOI] [PubMed] [Google Scholar]; * This is a current review of the incidence, age of onset and types of hearing loss in OI.
- 19.Veilleux LN, Trejo P, Rauch F. Muscle abnormalities in osteogenesis imperfecta. J Musculoskelet Neuronal Interact. 2017;17(2):1–7. [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips CL, Jeong Y. Osteogenesis Imperfecta: Muscle-Bone Interactions when Bi-directionally Compromised. Curr Osteoporos Rep. 2018;16(4):478–89. [DOI] [PubMed] [Google Scholar]; * This is an up-to-date review of the current understanding of muscle-bone interactions and muscle weakness in OI.
- 21.Kruger KM, Caudill A, Rodriguez Celin M, et al. Mobility in osteogenesis imperfecta: a multicenter North American study. Genet Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This is a multi-center study that reports mobility limitations in a large cohort of OI patients, and contributes to our understanding of the natural history, prognosis and rehabilitation needs in this disorder.
- 22.Balasubramanian M, Verschueren A, Kleevens S, et al. Aortic aneurysm/dissection and osteogenesis imperfecta: Four new families and review of the literature. Bone. 2019;121:191–5. [DOI] [PubMed] [Google Scholar]; * This paper highlights a rare, but potentially serious complication in OI.
- 23.Tam A, Chen S, Schauer E, et al. A multicenter study to evaluate pulmonary function in osteogenesis imperfecta. Clin Genet. 2018;94(6):502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This is a large cohort, multi-center study of pulmonary function in OI and the clinical correlation with OI severity.
- 24.LoMauro A, Fraschini P, Pochintesta S, et al. Ribcage deformity and the altered breathing pattern in children with osteogenesis imperfecta. Pediatr Pulmonol. 2018;53(7):964–72. [DOI] [PubMed] [Google Scholar]
- 25.Leotard A, Taytard J, Aouate M, et al. Diagnosis, follow-up and management of sleep-disordered breathing in children with osteogenesis imperfecta. Ann Phys Rehabil Med. 2018;61(3):135–9. [DOI] [PubMed] [Google Scholar]
- 26.Tosi LL, Floor MK, Dollar CM, et al. Assessing disease experience across the life span for individuals with osteogenesis imperfecta: challenges and opportunities for patient-reported outcomes (PROs) measurement: a pilot study. Orphanet J Rare Dis. 2019;14(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song Y, Zhao D, Li L, et al. Health-related quality of life in children with osteogenesis imperfecta: a large-sample study. Osteoporos Int. 2019;30(2):461–8. [DOI] [PubMed] [Google Scholar]; * This is a single-center cohort study that correlated the genotype, phenotype and quality of life measurements in OI patients and performed comparison between OI types, and between OI patients and healthy controls.
- 28.Swezey T, Reeve BB, Hart TS, et al. Incorporating the patient perspective in the study of rare bone disease: insights from the osteogenesis imperfecta community. Osteoporos Int. 2019;30(2):507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study describes the patient perspective on health-related issues, pain and other concerns, and highlights gaps in knowledge and avenues for future research that will improve patient outcome and quality of life.
- 29.Marr C, Seasman A, Bishop N. Managing the patient with osteogenesis imperfecta: a multidisciplinary approach. J Multidiscip Healthc. 2017;10:145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueller B, Engelbert R, Baratta-Ziska F, et al. Consensus statement on physical rehabilitation in children and adolescents with osteogenesis imperfecta. Orphanet J Rare Dis. 2018;13(1):158. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This is a literature review and summary of expert opinion regarding the goals of physical therapy and rehabilitation in OI, and the recommended management.
- 31.Marini JC, Hopkins E, Glorieux FH, et al. Positive linear growth and bone responses to growth hormone treatment in children with types III and IV osteogenesis imperfecta: high predictive value of the carboxyterminal propeptide of type I procollagen. J Bone Miner Res. 2003;18(2):237–43. [DOI] [PubMed] [Google Scholar]
- 32.Antoniazzi F, Monti E, Venturi G, et al. GH in combination with bisphosphonate treatment in osteogenesis imperfecta. Eur J Endocrinol. 2010;163(3):479–87. [DOI] [PubMed] [Google Scholar]
- 33.Glorieux FH, Bishop NJ, Plotkin H, et al. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. 1998;339(14):947–52. [DOI] [PubMed] [Google Scholar]; * This is the first study to assess the efficacy of bisphosphonate treatment in OI. It provided the foundation for use of bisphosphonates in OI, which continues to be the standard of care to date.
- 34.Morello R Osteogenesis imperfecta and therapeutics. Matrix Biol. 2018;71–72: 294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This is an up-to-date review of the pathogenesis of OI and current therapeutic approaches.
- 35.Bains JS, Carter EM, Citron KP, et al. A Multicenter Observational Cohort Study to Evaluate the Effects of Bisphosphonate Exposure on Bone Mineral Density and Other Health Outcomes in Osteogenesis Imperfecta. JBMR Plus. 2019;3(5):e10118. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This is a large cohort study that evaluated the outcome of bisphosphonate treatment in OI. It adds to the literature by showing a significant difference between oral and intravenous bisphosphonates, and by showing beneficial effect on fracture risk, scoliosis probability and mobility.
- 36.Lv F, Liu Y, Xu X, et al. Zoledronic Acid Versus Alendronate in the Treatment of Children with Osteogenesis Imperfecta: A 2-Year Clinical Study. Endocr Pract. 2018;24(2):179–88. [DOI] [PubMed] [Google Scholar]
- 37.Garganta MD, Jaser SS, Lazow MA, et al. Cyclic bisphosphonate therapy reduces pain and improves physical functioning in children with osteogenesis imperfecta. BMC Musculoskelet Disord. 2018;19(1):344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2014;7:CD005088. [DOI] [PubMed] [Google Scholar]
- 39.Shi CG, Zhang Y, Yuan W. Efficacy of Bisphosphonates on Bone Mineral Density and Fracture Rate in Patients With Osteogenesis Imperfecta: A Systematic Review and Meta-analysis. Am J Ther. 2016;23(3):e894–904. [DOI] [PubMed] [Google Scholar]
- 40.Hald JD, Evangelou E, Langdahl BL, Ralston SH. Bisphosphonates for the prevention of fractures in osteogenesis imperfecta: meta-analysis of placebo-controlled trials. J Bone Miner Res. 2015;30(5):929–33. [DOI] [PubMed] [Google Scholar]
- 41.Boyce AM. Denosumab: an Emerging Therapy in Pediatric Bone Disorders. Curr Osteoporos Rep. 2017;15(4):283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ominsky MS, Libanati C, Niu QT, et al. Sustained Modeling-Based Bone Formation During Adulthood in Cynomolgus Monkeys May Contribute to Continuous BMD Gains With Denosumab. J Bone Miner Res. 2015;30(7):1280–9. [DOI] [PubMed] [Google Scholar]
- 43.Ward L, Bardai G, Moffatt P, et al. Osteogenesis Imperfecta Type VI in Individuals from Northern Canada. Calcif Tissue Int. 2016;98(6):566–72. [DOI] [PubMed] [Google Scholar]
- 44.Hoyer-Kuhn H, Netzer C, Koerber F, et al. Two years’ experience with denosumab for children with osteogenesis imperfecta type VI. Orphanet J Rare Dis. 2014;9:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trejo P, Rauch F, Ward L. Hypercalcemia and hypercalciuria during denosumab treatment in children with osteogenesis imperfecta type VI. J Musculoskelet Neuronal Interact. 2018;18(1):76–80. [PMC free article] [PubMed] [Google Scholar]; * This study focused on the effect of denosumab therapy on calcium homeostasis in pediatric patients with OI type VI.
- 46.Hoyer-Kuhn H, Franklin J, Allo G, et al. Safety and efficacy of denosumab in children with osteogenesis imperfecta - a first prospective trial. J Musculoskelet Neuronal Interact. 2016;16(1):24–32. [PMC free article] [PubMed] [Google Scholar]
- 47.Li G, Jin Y, Levine MAH, et al. Systematic review of the effect of denosumab on children with osteogenesis imperfecta showed inconsistent findings. Acta Paediatr. 2018;107(3):534–7. [DOI] [PubMed] [Google Scholar]
- 48.Haas AV, LeBoff MS. Osteoanabolic Agents for Osteoporosis. J Endocr Soc. 2018;2(8):922–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vahle JL, Sato M, Long GG, et al. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1–34) for 2 years and relevance to human safety. Toxicol Pathol. 2002;30(3):312–21. [DOI] [PubMed] [Google Scholar]
- 50.Orwoll ES, Shapiro J, Veith S, et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest. 2014;124(2):491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leali PT, Balsano M, Maestretti G, et al. Efficacy of teriparatide vs neridronate in adults with osteogenesis imperfecta type I: a prospective randomized international clinical study. Clin Cases Miner Bone Metab. 2017;14(2):153–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller PD, Hattersley G, Lau E, et al. Bone mineral density response rates are greater in patients treated with abaloparatide compared with those treated with placebo or teriparatide: Results from the ACTIVE phase 3 trial. Bone. 2019;120:137–40. [DOI] [PubMed] [Google Scholar]
- 53.Cardinal M, Tys J, Roels T, et al. Sclerostin antibody reduces long bone fractures in the oim/oim model of osteogenesis imperfecta. Bone. 2019;124:137–47. [DOI] [PubMed] [Google Scholar]; * This is a pre-clinical study that shows a significant reduction in fracture risk, in addition to improved bone mineral density following treatment with anti-sclerostin antibody in a mouse model of OI.
- 54.Sagar R, Gotherstrom C, David AL, Westgren M. Fetal stem cell transplantation and gene therapy. Best Pract Res Clin Obstet Gynaecol. 2019. [DOI] [PubMed] [Google Scholar]; * This paper reviews current experience, and the limitations of cell and gene therapy in OI.
- 55.Grafe I, Yang T, Alexander S, et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014;20(6):670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim J, Grafe I, Alexander S, Lee B. Genetic causes and mechanisms of Osteogenesis Imperfecta. Bone. 2017;102:40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]