

Abstract
Pancreatic cancer is a malignant tumor with the worst prognosis worldwide. This cancer type requires new insight to help with diagnosis and, eventually, treatment. Adenosine deaminases acting on RNA 1 (ADAR1) is reportedly overexpressed in many types of tumors, such as lung, liver, breast, and esophageal cancers. However, the biological significance and specific mechanism of ADAR1 in pancreatic cancer have not been explored. In this study, we reveal that the expression level of ADAR1 is significantly up-regulated in pancreatic cancer tissues. We also find that highly expressed ADAR1 is closely associated with poor prognosis in pancreatic cancer specimens. Overexpressed ADAR1 equally increased the growth activity of pancreatic cancer cells in vivo and in vitro. We further demonstrate that ADAR1 stabilizes c-Myc through AKT signaling, which contributes to cancer cell resistance to BET inhibitors in pancreatic cancer cells. Moreover, we reveal that EZH2 regulates ADAR1 expression, and EZH2 and BET inhibitors show synergistic inhibition in pancreatic cancer. Collectively, these findings suggest that ADAR1 could serve as a new diagnostic and prognostic marker for the treatment of pancreatic cancer.
Keywords: Adenosine deaminases acting on RNA 1 (ADAR1), pancreatic cancer, c-Myc, PI3K/AKT signaling, BET inhibitors, EZH2 inhibitors
Introduction
Pancreatic cancer is a malignant tumor that is challenging to diagnose and treat [1-3]. Various therapeutic strategies have been used to treat pancreatic cancer, including surgery, chemoradiotherapy, and immunotherapy. However, the prognosis of pancreatic cancer remains poor, and the five-year survival rate is less than 5% [4].
Genetic mutation is a common characteristic of tumors that drives cancer initiation and progression [5]. Several genetic alterations have been reported to characterize the highly heterogeneous malignant tumor that is pancreatic cancer [5]. KRAS, TP53, SMAD4, and CDKN2A mutations are associated with the tumorigenesis of pancreatic cancer [6]. Therefore, understanding the role of mutated genes in pancreatic cancer could help identify new candidates for cancer therapy.
Myc has been found amplified in pancreatic cancer patients [6,7] and is also constitutively up-regulated in multiple types of tumors, including breast, colorectal, gastric, and uterine cancers [8]. Increasing evidence indicates that c-Myc alone could drive the initiation of pancreatic cancer and promote the proliferation, invasion, and chemoresistance of tumor cells [9-11]. Also, it has been reported that the aberrant expression of c-Myc contributes to the resistance of cancerous cells to bromodomain and extra-terminal domain (BET) inhibitors in pancreatic cancer cells, but the down-regulation of c-Myc enhances the sensitivity to these cells to inhibitors [12]. Therefore, c-Myc is a proto-oncogenic protein and a key mediator in the resistance to BET inhibitors and is potentially a promising target for the treatment of cancer.
Adenosine deaminases acting on RNA (ADAR) is an RNA-binding protein that converts adenosine (A) to inosine (I) by deamination [13]. ADAR proteins make RNA structures unstable by damaging base pairing between adenosine (A) and uracil (U). Editing leads to codon changes, which may cause changes for the protein-coding sequences and their functions [14]. Three types of ADARs are known to exist, including ADAR1, 2, and 3 [15]. ADAR1 plays a significant role in modulating the processes of apoptosis, proliferation, and differentiation in cancer cells [16-20], but its specific function in pancreatic cancer has not been investigated yet.
In this research, we systematically explore the specific role of ADAR1 in pancreatic cancer to assess it as a potential target for pancreatic cancer diagnosis and treatment. First of all, we study the clinical feature of ADAR1 in pancreatic cancer. Next, we assess ADAR1’s ability to promote pancreatic cancer cell proliferation in vivo and in vitro. Lastly, we investigate EZH2 inhibitors’ (GSK126) ability to decrease ADAR1 expression and show a synergistic anti-tumor effect when combined with BET inhibitors in pancreatic cancer cells.
Materials and methods
Cell lines, cell culture, cell transfection, and infection
Pancreatic cancer cell lines (PANC-1 and BxPC-3) were purchased from the Chinese Academy of Science Cell Bank. All cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, USA) containing 10% fetal bovine serum (FBS) (HyClone, USA). All cell lines were kept in a 37°C incubator at 5% CO2 atmosphere.
For cell transfection, pancreatic cancer cells were seeded in 6-well plates at a density of 2.5a d5 cells per well for 24 hours. Transfections were performed using Lipofectamine 2000 (Thermo Fisher Scientific). For cell infection, a lentivirus-based control and gene-specific shRNAs (Sigma-Aldrich) were employed to interfere with the expression of genes. Firstly, shRNAs were transfected in 293T cells using Opti-MEM medium, and after 24 hours, the medium was replaced by prepared DMEM containing 10% FBS and 1 mM sodium pyruvate. Next, the virus culture solution was gathered after 48 hours of continuous culture and mixed with PANC-1 and BxPC-3 cell lines supplemented with 12 μg/ml of polybrene. 10 μg/ml of puromycin was used to filtrate successfully infected cells 24 hours after mixing. The specific sequence information of shRNAs is provided in Table S1. shADAR1m was a mix of shADAR1 #1 (50%) and #2 (50%).
Plasmids and reagents
Flag-ADAR1s were purchased from Shanghai Genechem Co., LTD. Antibodies used were: ADAR1 (Proteintech, 14330-1-AP, working dilution 1:1000), GAPDH (Cell Signaling Technology, 5174, working dilution 1:5000), EZH2 (Cell Signaling Technology, 5246, working dilution 1:1000), Myc (Abcam, ab168724, working dilution 1:1000), AKT (Cell Signaling Technology, 2920, working dilution 1:1000), and pAKT-S473 (Cell Signaling Technology, 4060, working dilution 1:1000). Reagents used were: Palbociclib (PD0332991) (Cat. No. S1579), JQ1 (Cat. No. S7110), MK2206 (Cat. No. S1078), GSK126 (Cat. No. S7061), Dinaciclib (SCH727965) (Cat. No. S2768), Everolimus (RAD001) (Cat. No. S1120), MK1775 (Cat. No. S1525), p38 MAPK inhibitor (SB203580) (Cat. No. S1076), JSH-23 (Cat. No. S7351), Gemcitabine PD0325901 (Cat. No. S1714), PD0325901 (Cat. No. S1036), LY3214996 (Cat. No. S8534), and MG 132 (Cat. No. S2619), all purchased from Selleckchem.
Cell proliferation and clone formation assay
The MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) assay was used to detect cell viability in vitro. Briefly, PANC-1 and BxPC-3 cells were seeded in 96-well plates at a density of 1 × 103 cells per well containing 100 ul of DMEM medium. 20 ul of MTS was added to each well and incubated for 1 hour in an incubator positioned away from the light. The absorbance of samples was then measured at 490 nm using a microplate reader.
6-well plates were used to conduct clone formation assays, with 500 cells counted and seeded in each well. Media in the plates were replaced every three days, and two weeks later, 4% paraformaldehyde was used to fix cell colonies for 30 minutes, followed by staining of the cell colonies with crystal violet for 20 minutes.
Western blotting of cells and tissues
The Local Ethics Committee (Tongji Medical College, China) authorized our request to collect 12 pairs of cancer and adjacent tissues. The patients signed written informed consent before surgery. Tumor tissues or cells were lysed with RIPA lysis buffer containing 1% of protease inhibitors, after which proteins were extracted, and their concentrations were determined using protein assay kits (Pierce Biotechnology, USA). The extracted proteins were separated using SDS-PAGE and transferred to PVDF membranes (Pierce Biotechnology, USA). The PVDF membranes were then dipped in 5% skimmed milk for 1 hour at room temperature, followed by incubation with primary antibodies overnight at 4°C, washing with 1ght a, incubation with secondary antibodies for 1 hour at room temperature, and exposure of protein bands using an ECL illuminating liquid under the illumination of X-rays.
Real-time RT-PCR
Trizol reagent (Thermo Fisher Scientific, USA) was used to extract total RNA in pancreatic cancer cells. The RNA was reverse transcribed to produce cDNA per the manufacturer’s instructions (PrimeScript™ RT reagent Kit). RT-PCR analysis was carried out to amplify the produced cDNA using a PCR kit (TB Green™ Fast qPCR Mix). The cycle index was normalized to GAPDH, and the 2-ΔCq method was used to describe multiple variations. The forward and reverse primer sequences are provided in Table S2.
Flow cytometry analysis
We utilized the Annexin V-FITC/PI kit (AntGene, China) to mark viable and non-viable apoptotic cells to detect apoptosis rates. According to the protocol of the manufacturer of the kit, the cells were first washed with 1 × PBS, then Annexin V-FITC was added to the resuspended cells using a binding buffer and left to incubate for 10 minutes in the absence of light at room temperature. Finally, PI was added to the cells and analyzed using a Flow Cytometer.
A xenograft transplantation model in nude mice
BALB/c-nude mice (4-5 weeks of age, 18-20 g) were purchased from Vitalriver (Beijing, China) and randomly divided into eleven groups with six mice per group. Mice in each group were subcutaneously inoculated on the left back side with corresponding infective cells (5 × 106/each mouse). The length and width of xenografts were measured using a vernier caliper, and their volumes were determined using the formula (L × W2)/2. All mice were euthanized 21 days after subcutaneous implantation, and all xenografts were excised and weighed. All experimental procedures involving animals were approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology.
Tissue microarray and immunohistochemistry (IHC)
Tissue microarray slides were purchased from Outdo Biobank (Shanghai, China). These slides were immunostained with ADAR1 (Proteintech, working dilution 1:400) and c-Myc (Santa Cruz Biotechnology, 5605P, working dilution 1:100). The staining index (SI) was calculated as the product of the staining intensity score and the proportion of positive tumor cells. The staining intensity was graded according to the following criteria: 1 = weak staining at 100 × magnification but little or no staining at 40 × magnification; 2 = medium staining at 40 × magnification; 3 = strong staining at 40 × magnification. The immunostaining was scored independently by two experienced pathologists.
Statistical analysis
Statistical tests for data analyses were carried out to determine statistical significance using a one-sided or two-sided paired Student’s t-test for a single comparison. The data are expressed as mean ± SD. P < 0.05 was considered statistically significant.
Results
High expression of ADAR1 is associated with poor prognosis in pancreatic cancer
At first, we investigated the clinical feature of ADAR1 in pancreatic cancer using the Gene Expression Profiling Interactive Analysis (GEPIA) web tool and found that ADAR1 increased in pancreatic cancer tissues compared with non-tumor tissues (Figure 1A). The clinical specimens from tissue microarray, including pancreatic non-tumor tissues (n = 25) and pancreatic ductal adenocarcinoma tissues (n = 31), were examined using IHC staining to determine ADAR1 protein levels. The results indicated that ADAR1 was up-regulated in pancreatic cancer tissues compared to non-tumor tissues (Figure 1B and 1C).
Figure 1.
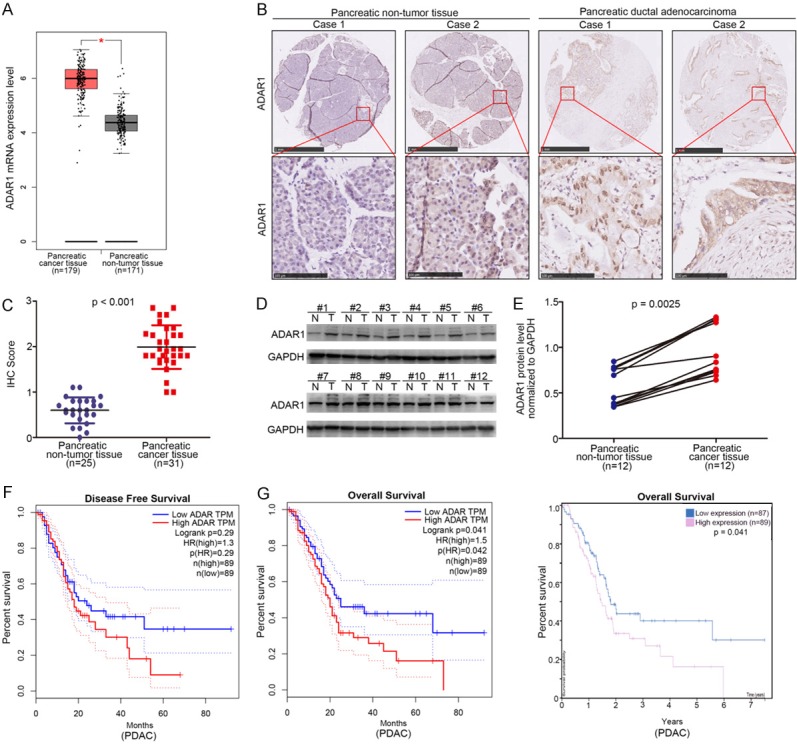
Highly expressed ADAR1 is closely associated with poor prognosis in pancreatic cancer. A. The mRNA expression level analysis of ADAR1 in pancreatic cancer patient specimens (n = 179) and pancreatic non-tumor tissues (n = 171) by using the GEPIA web tool. *, P < 0.05. B. The typical IHC image of ADAR1 in pancreatic tissue microarray (pancreatic non-tumor tissue n = 21, pancreatic cancer n = 31). C. The expression level of ADAR1 determined by IHC in pancreatic tissue microarray (pancreatic non-tumor tissue n = 25, pancreatic cancer n = 31), P < 0.001. D and E. The expression level of ADAR1 analyzed by Western Blotting from 12 pair of pancreatic cancer patient specimens and adjacent non-tumor tissues. P = 0.0025. F. The disease free survival time of pancreatic cancer patients with different expression level of ADAR1 was determined by GEPIA, p values as indicated. G. The overall survival time of pancreatic cancer patients with different expression level of ADAR1 was determined by GEPIA and Human Protein atlas.
Next, we analyzed the protein levels of ADAR1 in 12 cases of pancreatic cancer specimens and corresponding adjacent tissues using Western blotting, revealing that ADAR1 was also overexpressed in pancreatic cancer tissues (P = 0.0025) (Figure 1D and 1E). The Kaplan-Meier survival analysis data, meanwhile, indicated that ADAR1 expression had no effect on prolonging or shortening the disease-free survival time of pancreatic cancer patients (P = 0.29) (Figure 1F), despite GEPIA (P = 0.042) and Human Protein Atlas (P = 0.041) (Figure 1G) showing that the overexpression of ADAR1 in pancreatic cancer patient specimens shortened their overall survival time. Our data suggest that overexpressed ADAR1 results in a lower survival rate in pancreatic cancer.
The aberrant expression of ADAR1 promotes tumor proliferation in pancreatic cancer
With ADAR1 a potential prognostic marker of pancreatic cancer (Figure 1), its specific role in the progression of pancreatic cancer needs to be explored further. To that effect, we knocked down ADAR1 in PANC-1 and BxPC-3 cells using shRNAs (Figure 2A). Both cell proliferation and colony formation assays revealed that knocking down ADAR1 markedly inhibited the growth activity of pancreatic cancer cells (Figure 2B and 2C). Simultaneously, the overexpression of ADAR1 was also performed per the indicated plasmid transfections in pancreatic cancer cells (Figure 2D), resulting in the significant promotion of pancreatic cancer cell growth (Figure 2E and 2F).
Figure 2.

Aberrant expression of ADAR1 promotes tumor proliferation in pancreatic cancer. (A-C) Pancreatic cancer cell lines (PANC-1 and BxPC-3) were infected with indicated plasmids. After 72 h, cells were harvested for Western Blotting analysis (A), cell proliferation assay (B) and colony formation assay (C). Data presented as Means ± SD (n = 3). **, P < 0.01; ***, P < 0.001. (D-F) Pancreatic cancer cell lines (PANC-1 and BxPC-3) were transfected with indicated plasmids. 72 h post-transfection, cells were used for Western Blotting analysis (D), cell proliferation assay (E) and colony formation assay (F). Data presented as Means ± SD (n = 3). **, P < 0.01; ***, P < 0.001. (G-J) PANC-1 cells infected with indicated plasmids. After 72 h, the protein level of ADAR1 was analyzed by Western Blotting (G), then cells were injected subcutaneously into the nude mice for xenografts assay for 21 days. The image of xenografts was shown in (H), the tumor mass and volume of xenografts was determined in (I and J). Data presented as Means ± SD (n = 6). ***, P < 0.001.
Additionally, we employed the xenografts tumor model after knocking down ADAR1 and then rescuing its expression in PANC-1 cells to investigate the growth-promoting effect of ADAR1 in pancreatic cancer in vivo (Figure 2G). Per this experiment, knocking down ADAR1 impeded tumor growth, while rescuing ADAR1 expression diminished the inhibition of ADAR1 expression (Figure 2H-J). Collectively, our results indicate that ADAR1 played a key role in increasing the growth activity of pancreatic cancer cells.
ADAR1 regulates the sensitivity of BET inhibitors in pancreatic cancer cells
To study the role of ADAR1 in pancreatic cancer further, we performed a drug screening assay with 11 sorts of small molecular inhibitors and compared the corresponding drug sensitivity after knocking down ADAR1 in PANC-1 cells (Figure 3A). Intriguingly, knocking down ADAR1 with mixed pool shRNAs of ADAR1 (shADAR1m) increased the sensitivity of cancer cells to BET inhibitors in PANC-1 cells by reducing the IC50 ratio of JQ1, the commonly studied BET inhibitor [21] (Figure 3A). Furthermore, MTS and colony formation assays revealed slower growth rates in both BxPC-3 and PANC-1 cells in the ADAR1 knockdown group when treated with JQ1 compared with the control group treated with JQ1 (Figure 3B and 3C). Consistent with these findings, the expression level of cleaved-Caspase-3 in pancreatic cancer cells was up-regulated in the ADAR1 knockdown group after treatment with JQ1 compared to the control group (Figure 3D), suggesting that the down-regulation of ADAR1 makes pancreatic cancer cells more prone to apoptosis after treatment with JQ1.
Figure 3.

ADAR1 regulates the sensitivity of BET inhibitors in pancreatic cancer cells. (A) PANC-1 cells were transfected with indicated plasmids for 72 h. Cells were treated the different types of small inhibitors for 48 h, the IC50 values was analyzed and IC50 ratio between shControl and shADAR1m group was determined and shown in the panel. (B) PANC-1 and BxPC-3 cells were transfected with indicated with plasmids for 72 h. Then, cells were treated with or without JQ1 (5 uM) for 48 h. cells were harvested for Western Blotting analysis. (C and D) PANC-1 and BxPC-3 cells were transfected with indicated plasmids for 72 h. Then, cells were treated with or without JQ1 (5 uM) for MTS assay and colony formation assay. Data presented as Means ± SD (n = 3). **, P < 0.01; ***, P < 0.001. (E and F) PANC-1 cells were transfected with indicated plasmids for 72 h. Then, cells were treated with or without JQ1 (5 uM) for 48 h. Cells were subjected to Annexin-V/Propidium Iodide assay. Data presented as Means ± SD (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001. (G-J) PANC-1 cells were transfected with indicated plasmids for 72 h. Cell were subcutanousely injected into the nude mice. These mice were treated with or without JQ1 for 27 days (I). These tumors were harvested for photograph (G), weight (H) and caspase 3 analysis by IHC (J). Data presented as Means ± SD (n = 6 for H and I, n = 3 for J). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The Annexin-V/Propidium Iodide assay was also performed through flow cytometry analysis to provide more compelling evidence that ADAR1 knockdown plus JQ1 treatment produced a significant apoptosis rate in PANC-1 cells compared to the control group (Figure 3E and 3F).
Also, the xenograft tumor assay revealed that the ADAR1 knockdown group treated with JQ1 had the smallest excised tumor weight, the slowest tumor growth rate, and the highest cleaved-Caspase-3 level (Figure 3G-J), corroborating the finding that ADAR1 expression levels regulated JQ1 sensitivity in a living body. These results, therefore, indicate that ADAR1 could regulate the sensitivity of cancer cells to BET inhibitors in pancreatic ductal adenocarcinoma.
ADAR1 increases the protein level of c-Myc in pancreatic cancer
We have demonstrated that the overexpression of ADAR1 accelerated the growth of pancreatic cancer cells, although the underlying mechanism remains unknown. We have also shown that ADAR1 contributed to cancer cell resistance to BET inhibitors in pancreatic cancer (Figure 3). c-Myc is the key mediator of cancer cell sensitivity to BET inhibitors in pancreatic cancer [12,22,23], and it is also recognized as an oncogenic protein to promote tumor progression [24]. Therefore, we attempted to establish the relationship between ADAR1 and c-Myc in pancreatic cancer. First, we observed that the protein expression level of c-Myc was markedly suppressed after knocking down ADAR1 in pancreatic cancer cells (Figure 4A). In contrast, ADAR1 overexpression increased the protein level of c-Myc in pancreatic cancer (Figure 4C). Interestingly, RT-qPCR analysis showed no significant change in the expression of c-Myc after both the up-regulation and down-regulation of ADAR1 (Figure 4B and 4D).
Figure 4.
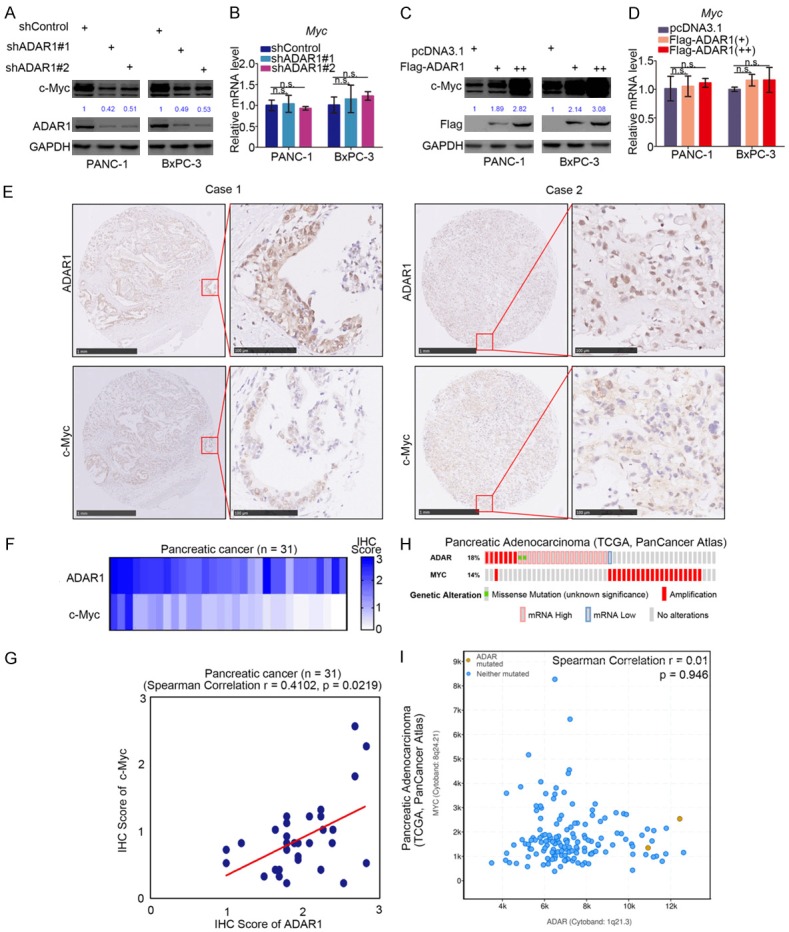
ADAR1 increases the protein level of c-Myc in pancreatic cancer. (A and B) PANC-1 and BxPC-3 cells were infected with indicated plasmids. After 72 h, cells were subjected to Western Blotting analysis (A) and RT-qPCR analysis (B). Data presented as Means ± SD (n = 3). n.s., not significant. (C and D) PANC-1 and BxPC-3 cells were transfected with indicated plasmids for 48 h. Cells were subjected to Western Blotting analysis (C) and RT-qPCR analysis (D). Data presented as Means ± SD (n = 3). n.s., not significant. e, the tissue microarray of pancreatic cancer (n = 31) was stained with ADAR1 and c-Myc respectively. The typical image of ADAR1 and c-Myc was shown in (E), the expression level of ADAR1 and c-Myc was shown in (F) and the correlation of these two proteins was shown in (G). (H and I), the mRNA expression level of ADAR and Myc in TCGA data sets were presented in (H and I).
To ascertain the relationship between ADAR1 and c-Myc further, IHC analysis was performed using the tissue microarray of pancreatic cancer (n = 31) to determine the protein expression level of ADAR1 and c-Myc. The representative images of ADAR1 and c-Myc are displayed in Figure 4E. The IHC scores of ADAR1 and c-Myc tissue microarray (n = 31) were calculated respectively and aggregated into a Heatmap (Figure 4F). We found that there was a positive correlation between ADAR1 and c-Myc protein (Spearman Correlation Coefficient r = 0.4102, P = 0.0219) (Figure 4G). Besides, by analyzing the mRNA levels of ADAR1 and Myc gene alteration from The Cancer Genome Atlas (TCGA) datasets, we found that an increased ADAR1 expression did not accompany the up-regulation of Myc gene expression, and the fluctuation of these two genes did not have any correlativity (Spearman Correlation Coefficient r = 0.01, P = 0.946) (Figure 4H and 4I). Taken together, we demonstrated that ADAR1 increased the c-Myc protein levels in pancreatic cancer cells.
ADAR1 stabilizes c-Myc through AKT signaling
Since we have demonstrated that ADAR1 regulated the protein level but not the mRNA level of c-Myc in pancreatic cancer cell lines (Figure 4A-D), we explored whether ADAR1 modulates the protein stability of c-Myc in pancreatic cancer. Firstly, we showed that the knockdown of ADAR1 in pancreatic cancer cells using mixed shADAR1 (shADAR1m) decreased the expression of c-Myc, but the proteasome inhibitor (MG132) halted the process (Figure 5A). Moreover, the knockdown of ADAR1 in PANC-1 cells decreased the protein half-life of c-Myc, but the ectopic overexpression of ADAR1 prolonged the protein half-life of c-Myc (Figure 5B and 5C).
Figure 5.

ADAR1 stabilizes c-Myc through AKT signaling. A. PANC-1 and BxPC-3 cells were infected with indicated plasmids for 48 h. Cells were collected for Western Blotting analysis after treated with or without MG132 for 8 h. B. PANC-1 cells were infected with indicated plasmids. After 72 h, cells were treated with Cycloheximide (CHX) and cells were collected for Western Blotting analysis at different time points. C. PANC-1 cells were transfected with indicated plasmids. After 24 h, cells were treated with Cycloheximide (CHX) and cells were collected for Western Blotting analysis at different time points. D. PANC-1 cells were transfected with indicated plasmids. 24 h post-transfection, cells were collected for Western Blotting analysis after treated with MG132 for 8 h. E. PANC-1 cells were infected with indicated with indicated plasmids. After 72 h, cells were collected for Western Blotting analysis after treated with MG132 for 8 h. F. PANC-1 and BxPC-3 cells were infected with indicated plasmids. After 72 h, cells were harvested for Western Blotting analysis. G. PANC-1 and BxPC-3 cells were transfected with indicated plasmids. After 24 h, cells were treated with or without MK2206 (5 uM) for other 24 h. Cells were harvested for Western Blotting analysis. H. PANC-1 and BxPC-3 cells were transfected with indicated plasmids. After 48 h, cells were treated with or without MK2206 (5 uM) for other 24 h. Cells were harvested for Western Blotting analysis. I. PANC-1 and BxPC-3 cells were knocked down ADAR1 alone, c-Myc alone, or both ADAR1 and c-Myc concurrently for Western Blotting analysis. J. PANC-1 and BxPC-3 cells, knocked down ADAR1 alone, c-Myc alone, or both ADAR1 and c-Myc concurrently, were treated with JQ1and subjected to MTS assay. ***, P < 0.001.
Protein ubiquitin modification is one of the leading causes of the degradation of target proteins by the ubiquitin-proteasome system in cells [25]. Here, our result demonstrated that ADAR1 decreased the polyubiquitination of c-Myc in PANC-1 cells (Figure 5D and 5E).
Because ADAR1 does not belong to the E3 ligase protein family, the underlying mechanism of how ADAR1 modulates the stability of c-Myc needs to be investigated further. The ADAR1/FGFR2/AKT signaling pathway reportedly promotes tumor cell proliferation [26]. Hence, we examined whether the knockdown of ADAR1 inhibits the phosphorylation of AKT at Ser-473 sites (Figure 5F). The PI3K/AKT pathway, meanwhile, is reported to inhibit the ubiquitination and degradation of c-Myc [27]. Our data demonstrated that the effect of increasing or decreasing c-Myc in pancreatic cancer cell lines induced by the overexpression or knockdown of ADAR1 was diminished by blocking the activation of the AKT pathway using AKT inhibitors (MK2206) (Figure 5G and 5H). Therefore, our results suggest that ADAR1 stabilizes c-Myc protein through PI3K/AKT signaling in pancreatic cancer.
To further determine whether ADAR1 regulated cancer cell resistance to BET inhibitors via c-Myc, we knocked down ADAR1 alone, c-Myc alone, or both ADAR1 and c-Myc concurrently in pancreatic cancer cells (Figure 5I), treated the cells with JQ1, and subjected them to the MTS assay (Figure 5J). Our data revealed that the co-knockdown of ADAR1 and c-Myc did not increase the sensitivity of cancer cells to JQ1 more than the knockdown of c-Myc alone did in both PANC-1 and BxPC-3 cells, which suggested that c-Myc is a key mediator for ADAR1-induced resistance to BET inhibitors.
Synergistic inhibition of pancreatic adenocarcinoma with EZH2 and BET inhibitors
Given that ADAR1 stabilized the protein levels of c-Myc via the PI3K/AKT pathway and impacted cancer cell resistance to BET inhibitors in pancreatic cancer, we regarded ADAR1 as a promising therapeutic target for pancreatic cancer and, thus, needed to test new drugs or inhibitors that can suppress the expression of ADAR1. To do that, we treated PANC-1 and BxPC-3 cells with 9 different inhibitors, including, PD0332991 (5 uM), MK2206 (5 uM), AZD1775 (0.5 uM), GSK126 (10 uM), SB203580 (0.5 uM), RAD001 (5 nM), SCH727965 (5 nM), PD0325901 (1 nM), and JQ1 (1 uM) for 48 h. We then subjected the treated cells to Western Blotting and RT-qPCR analyses to estimate the expression of ADAR1 and found that the EZH2 inhibitor (GSK126) suppressed the expression of ADAR1 compared to the DSMO treatment group (Figure 6A and 6B).
Figure 6.

Synergistic inhibition of pancreatic adenocarcinoma with the EZH2 inhibitors and the BET inhibitors. (A and B) PANC-1 and BxPC-3 cells were treated with DMSO, PD0332991 (5 uM), MK2206 (5 uM), AZD1775 (0.5 uM), GSK126 (10 uM), SB203580 (0.5 uM), RAD001 (5 nM), SCH727965 (5 nM), PD0325901 (1 nM), JQ1 (1 uM) for 48 h. Cells were harvested for RT-qPCR analysis (A) and Western Blotting analysis (B). Data presented as Means ± SD (n = 3). **, P < 0.01. (C and D) PANC-1 and BxPC-3 cells treated with different dose of GSK126 for 48 h. Cells were harvested for Western Blotting analysis (C) and RT-qPCR analysis (D). Data presented as Means ± SD (n = 3). ***, P < 0.001. (E and F) PANC-1 and BxPC-3 cells treated with GSK126 (10 uM) for 0, 3 and 5 days. Cells were harvested for Western Blotting analysis (E) and RT-qPCR analysis (F). Data presented as Means ± SD (n = 3). ***, P < 0.001. (G and H) PANC-1 and BxPC-3 cells were infected with indicated plasmids. After 72 h, cells were harvested for Western Blotting analysis (G) and RT-qPCR analysis (H). Data presented as Means ± SD (n = 3). ***, P < 0.001. (I) the spearman correlation of the mRNA expression level of ADAR1 and EZH2 in pancreatic cancer (P = 4.1e-62, r = 0.74). (J-M) PANC-1 cells were treated with indicated drugs. Cells were collected for MTS assay (J), colony formation assay and xenografts assay (I and M). Data presented as Means ± SD (n = 3 for I and K, n = 6 for I and M). **, P < 0.01;***, P < 0.001.
Next, we showed that not only the dosage (Figure 6C and 6D) but also the treatment time (Figure 6E and 6F) could affect the inhibitory effect of GSK126 on ADAR1 expression in pancreatic cancer. GSK126 manifested inhibitory effects on the expression of ADAR1 in a dose- and time-dependent manner (Figure 6C-F).
Because GSK126 is one of the specific inhibitors of EZH2, we pondered whether EZH2 regulated ADAR1 expression in pancreatic cancer. Knocking down EZH2 in PANC-1 and BxPC-3 cells using specific shRNAs decreased the protein and mRNA levels of ADAR1 (Figure 6G and 6H).
Furthermore, we calculated the Spearman Correlation Coefficient of the mRNA expression level of ADAR1 and EZH2 using the GEPIA web tool to determine any apparent positive correlation between the two genes (r = 0.74, P = 4.1e-62) (Figure 6I). These data indicated that EZH2 regulated the expression of ADAR1, but the underlying mechanism needs further elucidation.
Since ADAR1 was responsible for cancer cell resistance to BET inhibitors (JQ1) and EZH2 inhibitors (GSK126) repressed ADAR1 expression in pancreatic cancer cells, we sought to know whether the combination of GSK126 and JQ1 would show more profound anti-tumor effects than applying the drugs separately. We divided PANC-1 cells into four different treatment groups, including DMSO, GSK126, JQ1, and GSK126+JQ1 groups. Findings from the MTS and colony formation assays suggested that the combination treatment of GSK126 and JQ1 had a more inhibitory effect on tumor cell proliferation compared with each of GSK126 or JQ1 treatment alone (Figure 6J and 6K).
Similarly, in vivo experiments revealed that the tumor in the GSK126 plus JQ1 co-treatment group had the slowest growth rate (Figure 6L and 6M). Collectively, our results indicated that GSK126 and JQ1 had synergistic effects in inhibiting tumor proliferation in pancreatic cancer.
Discussion
ADAR1 is essential for the healthy development of mammals through A-to-I editing [28]. Pathologically, the overexpression of ADAR1 promotes the proliferation, metastasis, and invasion of tumor cells in liver cancer, esophageal cancer, lung cancer, and chronic myelogenous leukemia [29]. In this study, we revealed that ADAR1 was overexpressed in pancreatic cancer patient specimens and acted as a prognostic biomarker for pancreatic cancer. We also observed a growth-promoting effect of ADAR1 in pancreatic cancer cells in vivo and in vitro. Furthermore, we revealed that ADAR1 contributed to cancer cell resistance to BET inhibitors, AKT inhibitors, and mTOR inhibitors in PANC-1 cells (Figure 3A). Our objective was to emphasize ADAR1’s key role in the tumorigenesis of pancreatic cancer.
The abnormal expression of c-Myc occurs in diverse types of tumors, and it usually causes poor prognosis in advanced malignancies [30]. c-Myc functions as a transcriptional factor, forming a complex with MAX to mediate DNA binding and heterodimerization [31]. c-Myc is up-regulated and is regarded as a hub and central effector of oncogenic signaling in pancreatic cancer [32]. It has been reported that c-Myc alone could drive the initiation and progression of pancreatic cancer [33]. The bromodomain and extra-terminal domain (BET) family proteins are considered to enhance gene transcription by recognizing acetylated lysine residues at the tail of histones and the recruitment of transcriptional regulatory complexes [34].
As a BET bromodomain inhibitor, JQ1 can suppress specific important oncogenes, such as c-Myc, to achieve significant therapeutic effects [35-39]. However, resistance to BET inhibitors exists in many types of tumors [40,41]. Kumar K et al. found JQ1-resistant cells to be dependent on the oncogene c-Myc, with the down-regulation of c-Myc re-sensitizing the resistant cells to JQ1 [12], a finding that could help determine the mechanism of tumor cell resistance to BET inhibitors. We showed that ADAR1 stabilized the c-Myc protein and that the process was mediated through the PI3K/AKT pathway in pancreatic cancer cells. We also established that there was a significant positive correlation between ADAR1 and c-Myc in pancreatic cancer specimens (Figure 4). These results provided a reasonable mechanism to explain the effect of ADAR1 on tumor growth-promoting and resistance to BET inhibitors in pancreatic cancer cells.
The reason for the up-regulation of the expression of ADAR1 in pancreatic cancer cells also needed further characterization. George CX et al. had reported that the expression of the ADAR1 protein in adult mice was driven by multiple promoters [42]. Promoter replacement could affect the gene expression related to developmental regulation and tissue specificity [43]. Markle D et al. also found that the ADAR1 KCS element had a significant inconsistency when selectively binding factors in vitro and could not regulate the activity of the constitutive and IFN-induced ADAR1 PI promoter [44]. It can, hence, be seen that the mechanism of increased ADAR1 expression in tumor cells is very complicated.
Given the above important role of ADAR1 in the biological behavior of a tumor, it was extraordinarily significant to investigate the mechanism of increased ADAR1 in various cancers further. In this research, we demonstrated that EZH2 increased ADAR1 expression. Also, the inhibitory effect of EZH2 inhibitors (GSK126) on the expression of ADAR1 indicated that GSK126 could overcome the resistance to BET inhibitors in pancreatic cancer. Strikingly, this speculation is not only uttered by us but was also suggested in another research on glioma [45].
However, our research had some limitations. Firstly, we could not elucidate the mechanism further that EZH2 increased ADAR1 expression. Secondly, the synergistic effect combining JQ1 with EZH2 changed the dynamic of resistance to JQ1, which could not be explained by our existing research results.
In this study, we systematically investigated the clinical feature and biological role of ADAR1 in pancreatic cancer. Our data demonstrate that ADAR1 was overexpressed in pancreatic cancer tissues and might serve as a prognostic marker for pancreatic cancer patients. Moreover, we revealed that c-Myc played a significant role in mediating ADAR1-induced pancreatic cancer cell proliferation and resistance to BET inhibitors (Figure 7). Finally, after the small molecular screening, the EZH2 inhibitor (GSK126) was found to repress the expression of ADAR1. We further showed that GSK126 and JQ1 had synergistic effects in inhibiting tumor proliferation in pancreatic cancer (Figure 7). Collectively, our results indicate that the aberrant expression of ADAR1 increased pancreatic cancer proliferation via PI3K/AKT/c-Myc signaling, and EZH2 inhibitors regulated the expression of ADAR1 and overcame the resistance to BET inhibitors in pancreatic cancer.
Figure 7.
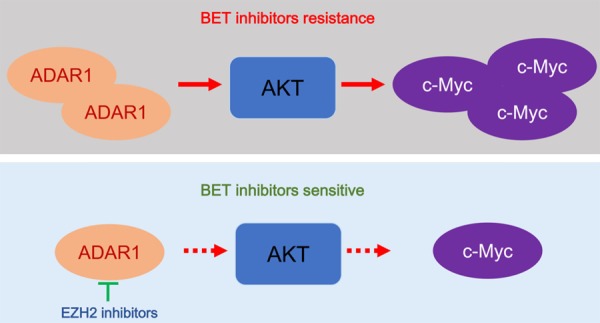
A hypothetical model depicting that overexpressed ADAR1 increased cell proliferation and induced BET inhibitors resistance via AKT/c-Myc signaling in pancreatic cancer cells, but this process could be attenuated by the EZH2 inhibitors through repressing the expression of ADAR1.
Acknowledgements
This work was supported by grants from the Chinese National Natural Science Foundation Grant No. 81702374. The study was conducted in accordance with the principles of the Declaration of Helsinki principles. It was approved by the Animal Use and Care Committees at Tongji Medical College, Huazhong University of Science and Technology.
Disclosure of conflict of interest
None.
Abbreviations
- ADAR1
Adenosine deaminases acting on RNA 1
- EZH2
Enhancer Of Zeste 2 Polycomb Repressive Complex 2
- IHC
immunohistochemistry
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- TMA
tissue microarray
- IP
immunoprecipitation
- CHX
Cycloheximide
- shRNA
short hairpin RNA
Supporting Information
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252–271. doi: 10.3322/caac.21235. [DOI] [PubMed] [Google Scholar]
- 3.Malvezzi M, Carioli G, Bertuccio P, Rosso T, Boffetta P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2016 with focus on leukaemias. Ann Oncol. 2016;27:725–731. doi: 10.1093/annonc/mdw022. [DOI] [PubMed] [Google Scholar]
- 4.Hidalgo M, Cascinu S, Kleeff J, Labianca R, Lohr JM, Neoptolemos J, Real FX, Van Laethem JL, Heinemann V. Addressing the challenges of pancreatic cancer: future directions for improving outcomes. Pancreatology. 2015;15:8–18. doi: 10.1016/j.pan.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 5.Notta F, Hahn SA, Real FX. A genetic roadmap of pancreatic cancer: still evolving. Gut. 2017;66:2170–2178. doi: 10.1136/gutjnl-2016-313317. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, Omberg L, Wolf DM, Shriver CD, Thorsson V Cancer Genome Atlas Research Network. Hu H. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416. e411. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gearhart J, Pashos EE, Prasad MK. Pluripotency redux--advances in stem-cell research. N Engl J Med. 2007;357:1469–1472. doi: 10.1056/NEJMp078126. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, McGee J, Chen X, Doman TN, Gong X, Zhang Y, Hamm N, Ma X, Higgs RE, Bhagwat SV, Buchanan S, Peng SB, Staschke KA, Yadav V, Yue Y, Kouros-Mehr H. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One. 2014;9:e98293. doi: 10.1371/journal.pone.0098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Li X, Zhu S, Zhang JG, Yang M, Qin Q, Deng SC, Wang B, Tian K, Liu L, Niu Y, Wang CY, Zhao G. Ectopic expression of miR-494 inhibited the proliferation, invasion and chemoresistance of pancreatic cancer by regulating SIRT1 and c-Myc. Gene Ther. 2015;22:729–738. doi: 10.1038/gt.2015.39. [DOI] [PubMed] [Google Scholar]
- 10.Zang Y, Zhu L, Li T, Wang Q, Li J, Qian Y, Wei L, Xie M, Tang WH, Liu X, Zhu Y, Wang L. EI24 suppresses tumorigenesis in pancreatic cancer via regulating c-Myc. Gastroenterol Res Pract. 2018;2018:2626545. doi: 10.1155/2018/2626545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez-Arevalo Lobo VJ, Fernandez LC, Carrillo-de-Santa-Pau E, Richart L, Cobo I, Cendrowski J, Moreno U, Del Pozo N, Megias D, Breant B, Wright CV, Magnuson M, Real FX. c-Myc downregulation is required for preacinar to acinar maturation and pancreatic homeostasis. Gut. 2018;67:707–718. doi: 10.1136/gutjnl-2016-312306. [DOI] [PubMed] [Google Scholar]
- 12.Kumar K, Raza SS, Knab LM, Chow CR, Kwok B, Bentrem DJ, Popovic R, Ebine K, Licht JD, Munshi HG. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci Rep. 2015;5:9489. doi: 10.1038/srep09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samuel CE. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate immune responses. J Biol Chem. 2019;294:1710–1720. doi: 10.1074/jbc.TM118.004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savva YA, Rieder LE, Reenan RA. The ADAR protein family. Genome Biol. 2012;13:252. doi: 10.1186/gb-2012-13-12-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.XuFeng R, Boyer MJ, Shen H, Li Y, Yu H, Gao Y, Yang Q, Wang Q, Cheng T. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc Natl Acad Sci U S A. 2009;106:17763–17768. doi: 10.1073/pnas.0903324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez HD, Jasavala RJ, Hinkson I, Fitzgerald LD, Trimmer JS, Kung HJ, Wright ME. RNA editing of androgen receptor gene transcripts in prostate cancer cells. J Biol Chem. 2008;283:29938–29949. doi: 10.1074/jbc.M800534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laxminarayana D, Khan IU, O’Rourke KS, Giri B. Induction of 150-kDa adenosine deaminase that acts on RNA (ADAR)-1 gene expression in normal T lymphocytes by anti-CD3-epsilon and anti-CD28. Immunology. 2007;122:623–633. doi: 10.1111/j.1365-2567.2007.02709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan TH, Lin CH, Qi L, Fei J, Li Y, Yong KJ, Liu M, Song Y, Chow RK, Ng VH, Yuan YF, Tenen DG, Guan XY, Chen L. A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma. Gut. 2014;63:832–843. doi: 10.1136/gutjnl-2012-304037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shoshan E, Mobley AK, Braeuer RR, Kamiya T, Huang L, Vasquez ME, Salameh A, Lee HJ, Kim SJ, Ivan C, Velazquez-Torres G, Nip KM, Zhu K, Brooks D, Jones SJ, Birol I, Mosqueda M, Wen YY, Eterovic AK, Sood AK, Hwu P, Gershenwald JE, Robertson AG, Calin GA, Markel G, Fidler IJ, Bar-Eli M. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat Cell Biol. 2015;17:311–321. doi: 10.1038/ncb3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma A, Larue RC, Plumb MR, Malani N, Male F, Slaughter A, Kessl JJ, Shkriabai N, Coward E, Aiyer SS, Green PL, Wu L, Roth MJ, Bushman FD, Kvaratskhelia M. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc Natl Acad Sci U S A. 2013;110:12036–12041. doi: 10.1073/pnas.1307157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin Y, Sun M, Zhan X, Wu C, Geng P, Sun X, Wu Y, Zhang S, Qin J, Zhuang Z, Liu Y. EGFR signaling confers resistance to BET inhibition in hepatocellular carcinoma through stabilizing oncogenic MYC. J Exp Clin Cancer Res. 2019;38:83. doi: 10.1186/s13046-019-1082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia PL, Miller AL, Kreitzburg KM, Council LN, Gamblin TL, Christein JD, Heslin MJ, Arnoletti JP, Richardson JH, Chen D, Hanna CA, Cramer SL, Yang ES, Qi J, Bradner JE, Yoon KJ. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene. 2016;35:833–845. doi: 10.1038/onc.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang CV. Web of the extended Myc network captures metabolism for tumorigenesis. Cancer Cell. 2015;27:160–162. doi: 10.1016/j.ccell.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Leestemaker Y, Ovaa H. Tools to investigate the ubiquitin proteasome system. Drug Discov Today Technol. 2017;26:25–31. doi: 10.1016/j.ddtec.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Jiang Y, Wang Z, Chen X, Wang W, Wang X. ADAR1 silencing-induced HUVEC apoptosis is mediated by FGFR2 under hypoxia stress. Drug Des Devel Ther. 2018;12:4181–4189. doi: 10.2147/DDDT.S181312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dominguez-Caceres MA, Garcia-Martinez JM, Calcabrini A, Gonzalez L, Porque PG, Leon J, Martin-Perez J. Prolactin induces c-Myc expression and cell survival through activation of Src/Akt pathway in lymphoid cells. Oncogene. 2004;23:7378–7390. doi: 10.1038/sj.onc.1208002. [DOI] [PubMed] [Google Scholar]
- 28.Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun HJ, Pineda G, Morris SR, Mason CN, Geron I, Barrett C, Goff DJ, Wall R, Pellecchia M, Minden M, Frazer KA, Marra MA, Crews LA, Jiang Q, Jamieson CHM. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell. 2016;19:177–191. doi: 10.1016/j.stem.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fritzell K, Xu LD, Lagergren J, Ohman M. ADARs and editing: the role of A-to-I RNA modification in cancer progression. Semin Cell Dev Biol. 2018;79:123–130. doi: 10.1016/j.semcdb.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 30.Cichon MA, Moruzzi ME, Shqau TA, Miller E, Mehner C, Ethier SP, Copland JA, Radisky ES, Radisky DC. MYC is a crucial mediator of TGFβ-induced invasion in basal breast cancer. Cancer Res. 2016;76:3520–3530. doi: 10.1158/0008-5472.CAN-15-3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez-Olivares M, Trento A, Rodriguez-Acebes S, Gonzalez-Acosta D, Fernandez-Antoran D, Roman-Garcia S, Martinez D, Lopez-Briones T, Torroja C, Carrasco YR, Mendez J, Moreno de Alboran I. Functional interplay between c-Myc and Max in B lymphocyte differentiation. EMBO Rep. 2018;19 doi: 10.15252/embr.201845770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ji S, Qin Y, Liang C, Huang R, Shi S, Liu J, Jin K, Liang D, Xu W, Zhang B, Liu L, Liu C, Xu J, Ni Q, Chiao PJ, Li M, Yu X. FBW7 (F-box and WD repeat domain-containing 7) negatively regulates glucose metabolism by targeting the c-Myc/TXNIP (thioredoxin-binding protein) axis in pancreatic cancer. Clin Cancer Res. 2016;22:3950–3960. doi: 10.1158/1078-0432.CCR-15-2380. [DOI] [PubMed] [Google Scholar]
- 33.Wirth M, Mahboobi S, Kramer OH, Schneider G. Concepts to target MYC in pancreatic cancer. Mol Cancer Ther. 2016;15:1792–1798. doi: 10.1158/1535-7163.MCT-16-0050. [DOI] [PubMed] [Google Scholar]
- 34.Qi J. Bromodomain and extraterminal domain inhibitors (BETi) for cancer therapy: chemical modulation of chromatin structure. Cold Spring Harb Perspect Biol. 2014;6:a018663. doi: 10.1101/cshperspect.a018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ, Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, Vakoc CR. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, Giotopoulos G, Lugo D, Jeffrey P, Lee SC, Carpenter C, Gregory R, Ramsay RG, Lane SW, Abdel-Wahab O, Kouzarides T, Johnstone RW, Dawson SJ, Huntly BJ, Prinjha RK, Papenfuss AT, Dawson MA. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525:538–542. doi: 10.1038/nature14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, Stover DG, Ekram MB, Brown J, D’Santos C, Krop IE, Dillon D, McKeown M, Ott C, Qi J, Ni M, Rao PK, Duarte M, Wu SY, Chiang CM, Anders L, Young RA, Winer E, Letai A, Barry WT, Carroll JS, Long H, Brown M, Liu XS, Meyer CA, Bradner JE, Polyak K. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413–417. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.George CX, Wagner MV, Samuel CE. Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J Biol Chem. 2005;280:15020–15028. doi: 10.1074/jbc.M500476200. [DOI] [PubMed] [Google Scholar]
- 43.George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci U S A. 1999;96:4621–4626. doi: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Markle D, Das S, Ward SV, Samuel CE. Functional analysis of the KCS-like element of the interferon-inducible RNA-specific adenosine deaminase ADAR1 promoter. Gene. 2003;304:143–149. doi: 10.1016/s0378-1119(02)01200-3. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Dong W, Zhu J, Wang L, Wu X, Shan H. Combination of EZH2 inhibitor and BET inhibitor for treatment of diffuse intrinsic pontine glioma. Cell Biosci. 2017;7:56. doi: 10.1186/s13578-017-0184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.