

Abstract
Protein homeostasis (proteostasis) is a result of a dynamic equilibrium between protein synthesis and degradation. It is important for healthy cell/organ functioning and is often associated with diseases such as neurodegenerative diseases and non-Alcoholic Fatty Liver disease. Heavy water metabolic labeling, combined with liquid-chromatography and mass spectrometry (LC-MS), is a powerful approach to study proteostasis in vivo in high throughput. Traditionally, intact peptide signals are used to estimate stable isotope incorporation in time-course experiments. The time-course of label incorporation is used to extract protein decay rate constant (DRC). Intact peptide signals, computed from integration in chromatographic time and mass-to-charge ratio (m/z) domains, usually, provide an accurate estimate of label incorporation. However, sample complexity (co-elution), limited dynamic range, and low signal-to-noise ratio (S/N) may adversely interfere with the peptide signals. These artifacts complicate the DRC estimations by distorting peak shape in chromatographic time and m/z domains. Fragment ions, on the other hand, are less prone to these artifacts and are potentially well suited in aiding DRC estimations. Here, we show that the label incorporation encoded into the isotope distributions of fragment ions reflect the isotope enrichment during the metabolic labeling with heavy water. We explore the label incorporation statistics for devising practical approaches for DRC estimations.
Keywords: in vivo protein turnover, heavy water metabolic labeling, protein half-life, fragment ion quantification from deuterium labeled peptides
Graphical Abstract
Introduction.
The cellular proteome is in a state of dynamic equilibrium – proteins are continuously synthesized and degraded. Protein homeostasis is essential for the proper function of cellular proteins1. Transition to a new equilibrium is observed, for example, during organismal growth, differentiation, or diseases2. Metabolic labeling combined with LC-MS is a powerful tool to study protein turnover in living organisms3. Advancements in resolution, mass accuracy, sensitivity, and scanning speed of modern mass spectrometers4 have made it possible to analyze thousands of proteins in one experiment. Deuterium labeling with heavy water is experimentally simple, as enriched water is provided as the drinking water5. The water rapidly equilibrates throughout the body. There are other metabolic labeling approaches to evaluate dynamic proteome in vivo3,6. Stable isotope labeling of mammals (SILAM) uses metabolic labeling by an essential amino acid (e.g., Lys) to fully label the proteins in pulse phase and monitor their clearance in the chase phase7,8. SILAM is dependent on specific amino acid in a peptide to measure its turnover, and the labeled amino acid diet is expensive. Another metabolic labeling technique9 is to provide 15N labeled diet. The labeling results in complex isotope profiles and relatively low number (1000 or lower) of quantified proteins10. Reaching equilibrium of the labeling precursors in these approaches is an additional issue to consider. A recent study found that the excretion rates of pools of sub-soluble Lys from diet and endogenous proteins are different8. Therefore, a longer period for reaching an equilibrium of precursor labeling (free Lys) is required. In the case of the labeling with heavy water, the equilibrium is rapidly achieved11.
Current bioinformatics techniques to evaluate proteome dynamics from metabolic labeling mostly use the isotope profiles of intact peptides to determine the label incorporation. However, the isotope distributions are prone to distortions due to co-eluting species and other artifacts. The fragment ions in the tandem mass spectra (MS/MS) provide complementary information about the label incorporation and can improve the DRC estimation.
In the context of stable isotope labeling and protein turnover estimations, the fragment ions from MS/MS have previously been used for quantification of labeled amino acid incorporation12,13. 13C6 labeled Leu was used for labeling proteins. The labeled and unlabeled proteins were immunoisolated, digested into peptides, and analyzed using LC-MS/MS. The quantification approach termed SILT(stable isotope labeling and tandem mass spectrometry)14 utilizes labeled and unlabeled fragment ions (from different tandem mass spectra) to estimate the label incorporation. More recently, selected reaction monitoring (SRM) has been applied in several protein turnover studies15–17. SRM spectra are acquired using tandem quadrupole mass spectrometers. Metabolic labeling with labeled Leu (D3-leucine)16,17 or Lys ([13C6]lysine)15 were used in these studies. SRM method is a targeted approach and is well suited for focused studies, where dozens of proteins are quantified, and their dynamics are estimated. The quantification is done using chromatographic profiles of preselected fragment ions.
Since the SILT and SRM approaches use amino acid based labeling, they are limited to quantification of peptides that contain the labeling amino acid. Heavy water labeling strategy labels all nonessential amino acids and is applicable to a larger peptide population. SILT quantifies label incorporation by computing fragment ion abundances from MS/MS spectra of labeled and unlabeled peptides. It requires two peptide fragmentation events, one for labeled peptide and the other for unlabeled peptide. At each time point of labeling, our approach quantifies the label incorporation from a single MS/MS. Deuterium incorporation from heavy water into a peptide (its fragments) depletes the relative abundance (RA) of the monoisotopic peak of a fragment ion. The time-course (in metabolic labeling dimension) of the depletion is used to extract DRCs. As it will be seen below, data in this approach presents issues with fluctuations in the isotope distributions (not used in SILT) of fragment ions, spectral accuracy of fragment ions, and the sensitivity of changes in the isotope distributions to the number of exchangeable hydrogens (NEH) in a fragment.
Data Description.
Data generation for proteome dynamics studies includes animal metabolic labeling, measurements of body water enrichment, protein fractionation, LC-MS analysis, and protein identification and quantification18. The data used in this study is fully described in our recent publication19. Briefly, murine liver protein fractionation was done by SDS-PAGE. LC-MS analysis was done by Ultimate 3000 UHPLC (Thermo Scientific, CA) coupled online to Q Exactive™ Plus mass spectrometer (Thermo Scientific, CA). Full mass scans (MS1) were performed at the range of 380–1300 Th (MS) with 70,000 resolution (200 m/z). MS/MS spectra were collected in data-dependent acquisition (DDA) mode for the 12 most abundant product ions. The precursor ion isolation window was 1.4 Th, the offset was 0.3 Th. The resolution in MS/MS was 17,500 at 200 m/z. MS and MS/MS spectra were acquired for 100 ms with the automatic gain control (AGC) target set at 1.0 × 106 and 2.0 × 104 ions for MS and MS/MS scans, respectively. Higher-energy collisional dissociation (HCD) was performed at normalized collision energy (NCE) of 25 eV. Dynamic exclusion was enabled for a duration of 17 s. One microscan was acquired for each tandem mass spectrum. The data is available at ProtemeXchange.
Peptide/Protein identification.
Mascot20 database search engine was used to identify peptides from tandem mass spectra and group them into proteins. The following parameters were used in the searches of tandem mass spectra: precursor mass accuracy was set to 15 ppm; fragment ion mass accuracy was set to 0.6 Da; carbamidomethylation of Cys was the fixed modification; oxidation of Met and acetylation of Lys were set as dynamic modifications. Trypsin specificity of peptides was used, and up to 2 missed cleavages were allowed. The swiss_prot database (downloaded in May 2017) and mouse taxonomy were used. The global false discovery rate (FDR) was controlled using the decoy database approach. The FDR was set at 5%.
The labeling and following LC-MS experiments were done at six time points: 0, 3, 7, 11, 15, 21 days of metabolic labeling with heavy water (resulted in 3% enrichment of body water with deuterium). At each time point, there were two technical replicates. In total, there were twelve experiments. We have focused our analyses in this work on peptides of murine mitochondrial carbamoyl phosphate synthase, CPSM, the urea cycle enzyme. Overall there were 338 distinct peptides quantified by the traditional MS1 quantification (in at least four out of six time points of labeling) in our dataset. We focused on +2 charged peptides only. Of the 227 +2 charged peptides 122 were fragmented at least once in all time points of labeling. However, in some cases, the fragmentation events were centered on non-monoisotope. As it will be seen below, currently our modeling is based on the fragmentation events of the monoisotope, only. Also, for statistics purposes it was required that at each time point there be at least 3 MS/MS spectra for a peptide. These requirements further reduced the number of spectra to 57.
Results and Discussions.
Analysis of Fragment Ion Isotope Abundances for Label Incorporation
As proteins incorporate stable isotopes, the RA of heavy peptides increases. It is expected that the changes in the isotope distributions of intact peptides will result in changes in isotope distributions of fragment ions. However, how to detect and more importantly, how to model these changes and use them for peptide quantification remains challenging. Here, we first demonstrate the changes in isotope distributions of peptides due to the incorporation of deuterium. We then show that the isotope distribution of a fragment resulting from isolation and fragmentation of an intact peptide is not the same as the natural isotope distribution of the fragment. We then describe an approach to model isotope distributions of fragments in MS/MS. Next, we extend the modeling to heavy water labeled peptides, and their time-course of labeling for extracting peptide DRC.
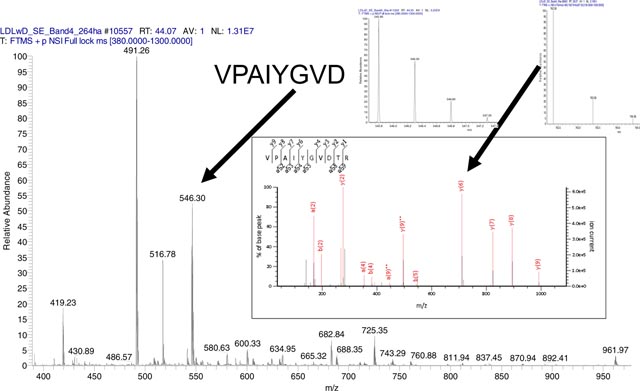
For every peptide identified in MS/MS, there is information about its fragment ions and their isotope distributions. Often these are isotopes of +1 charged fragments with two or three isotopomers. This information can potentially be used in models for estimating the rate of label incorporation. We demonstrate the label incorporation with an example of the peptide sequence, VVAVDCGIK, of CPSM. The tandem mass spectra of the unlabeled and partially labeled (after 21 days of labeling with heavy water) forms of this peptide are shown in Figures 1 A and 1 B. The experimental peaks matching theoretical monoisotopic peaks are shown in red.
Figure 1 A and B.

Annotated MS/MS spectra of VVAVDCGIK.
Fragment ions showed increased in RAs of the heavy isotopes during the labeling. This can be seen in Figure 2, which is the zoomed isotope distributions of y7 fragment ion at the start (0 day), 3 days and the end (21 days) of metabolic labeling. The relevant data for a2 and y5 ions are shown on Figures S1–S2 (Supplementary Materials). Figure S3 is the annotated tandem mass spectrum of the peptide after 3 days of labeling. Not all fragment ions can be used in quantitative analysis. For example, the y5 ion is a low abundance ion (Figure 1). Before the start of the labeling, only its monoisotope is detected, Figure S2. The a2 ion is a high abundance ion, and its first two isotopes are detected before the start of labeling. However, this fragment ion does not contain non-essential amino acids, and its label incorporation is small, Figure S1. The y7 ion (AVDCGIK), on the other hand, contains all non-essential amino acids (Ala, Asp, Cys, and Gly) of the peptide sequence, has a high abundance (Figure 1 A and B), and three measured isotopes at all time points of labeling, Figure 2. It is the most suitable fragment ion for quantifying the label incorporation for the peptide. The RAs (also referred to as fractional abundances) of the monoisotopic peaks have decreased for all three ions upon 21 days of metabolic labeling. For example, the fractional abundance of the y7 ion has reduced from 0.79 (unlabeled peptide) to 0.59 (after 21 days of labeling).
Figure 2.

Isotope distributions of y7 fragment ion of the peptide sequence, VVAVDCGIK.
The peptide, VVAVDCGIK, was fragmented and recorded in several tandem mass spectra in every LC-MS (the number of the fragmentation events per experiment varied between 7 and 15). It is informative to examine the distribution of RAs of monoisotopic peaks in different tandem mass spectra. Figure 3 shows the violin plot of the RAs from all durations of labeling. The corresponding distributions for fragment ions of several other peptides are presented in Figures S4–S12. As is seen from the figures, the medians (white circles in the violin plots) of RAs are mostly monotonic (except for Figure S7), as expected. Figure S7 is the RA of the y1 ion of the peptide, FVHDNYVIR. The figure illustrates isotope incorporation into a single, non-essential amino acid, Arg. Since NEH for this fragment is small (about 3), the results are heavily influenced by technical and other fluctuations. However, the data demonstrate a potential application of this approach to monitor label incorporation into a single amino acid in high throughput experiments. This functionality is not possible in amino acid based labling.
Figure 3.

Violin plot of the relative isotope abundance of the monoisotopic peak of y7 ion (m/z = 762.3801 Th) of CPSM peptide, VVAVDCGIK, at six time points of metabolic labeling with heavy water. The white circles are the median values of RAs. The vertical profiles are the empirical density functions of RAs. The middle thick bars are 50% quantile of data. The thin bars (in each direction) contain the half of data points outside of the 50% bars (quarters of the overall data).
As is seen from Figures 3 and S4–S12 the variability in the estimation of the RA of the monoisotopic peak is high. Some of the variability is due to the low ion count. It can be seen in Figure 4. The figure is the scatter plot of RAs of the monoisotopic peak of y7 ion of the peptide, VVAVDCGIK, at 0 (black circles), 3 (blue triangles) and 21 (magenta rhomboids) days of labeling, as a function of 10-base logarithm of ion counts. Higher variability is seen for low ion counts. In Supplementary Material 1.csv, we list the RAs, Mascot’s ion scores, ion count, scan numbers, and the raw file information for each tandem mass spectrum of the peptide. As is seen from the table, ion scores do not differentiate the spectra producing outliers of the RA. However, the outliers tend to have low ion counts. Thus, in the tandem mass spectra that produce the largest differences from the median, ion count of the monoisotopic peak is in tens of thousands (0 day of labeling). These are the lower range of recorded ion counts for this fragment. Ion counts reached tens of millions. We have tried abundance-averaged values and median values for modeling label incorporation into fragment ions. The median value of RAs of the monoisotope showed more consistent depletion with the label incorporation. In the computations reported below, the median values are used.
Figure 4.

Scatter plot of the relative abundance of monoisotopic peak of y7 fragment ion (m/z = 762.3815) of peptide, VVAVDCGIK, as a function of log10 transformed ion abundance. Shown are the data for 0 (black circles), 3 days (blue triangles) and 21 days (magenta rhomboids) of labeling.
Figure 5 is the RA of the monoisotopic peak, I0(t), as a function of label incorporation time for the y7 ion of the peptide. The raw data points (medians of relevant distributions at each time point) are shown with triangles. In heavy water metabolic labeling, I0(t) is modeled as:
| Eq. (1) |
where k is the peptide/protein DRCs (to be estimated via a fit to the experimental data), I0asymp is the asymptotically expected RA of the monoisotope, I0(0) is the RA of the monoisotope before the start of the labeling, i.e., the RA of the monoisotope from naturally occurring isotopes. In the data processing for heavy water labeling, I0asymp is obtained from the body water enrichment of the deuterium, p, via the equation18,21:
where is the NEH of the fragment ion. We use the I0(0) value of the y7 ion from the tandem mass spectrum before the start of labeling. The NEH for the y7 ion, AVDCGIK, is 11. The body water enrichment was 0.03. We used the non-linear least-squares fit in R22 to compute the rate constant. The computed rate constant was 0.084 day−1. The corresponding rate constant for the CPSM protein is 0.099 day−1. The rate constants for peptides were obtained via peak detection, integration (in the chromatographic domain), and time-course fit (in the metabolic time domain) using MS1 data. The difference from the protein rate constant obtained in MS/MS is 15%. This is an improvement, as the DRC value for this peptide from the MS1 quantification was 0.1464 day−1 (49% difference from the protein DRC). The elution profile of the precursor ion contained co-eluting contaminants, Supplementary Figure S14, which affected the DRC calculation from MS1.
Figure 5.

Decay curve of RAs of monoisotopic peak of y7 ion, AVDCGIK, of peptide VVAVDCGIK as a function of labeling duration.
Modeling Isotope Distributions of Fragment Ions.
When a small isolation window for fragmenting a precursor ion is used, the isotope distribution of the resulting fragment is not the same as that of the natural fragment. Figure S14 shows a model of isotope distribution for the peptide VVAVDCGIK. Isolation window of 1.4 Th with an offset of 0.3 Th (from the monoisotope) was used in the LC-MS experiments. This window size (for +2 charged peptides) would theoretically cover three isotopes: the monoisotope (M0), the first (M1) and (part of) the second (M2) heavy isotopes. Fragment ion isotope distributions will differ from the natural isotope distributions, as peptide’s full isotope envelope is not fragmented. To account for this effect, we remodel the isotope distributions of fragments to include only the peptide isotopes that were in in the isolation window. Assume a peptide consists of two complementary b- and y- ions at a particular peptide bond. Each of the fragments has its own isotope distribution, p(Xb) and p(Xy), respectively. Xb and Xy are random variables whose values correspond to the number of heavy isotopes in a fragment with the probability mass functions, p(Xb) and p(Xy), determined from their atomic compositions23. The isotope distribution of the peptide, p(X), is the convolution of the probability density functions of Xb and Xy. Assuming that only three isotopes (X = 0,1, 2) are isolated in the isolation window for fragmentation by mass spectrometry, the basic probabilities for the expected isotope distributions of the fragments are determined from the following three equations:
| (Eq. 2) |
The probabilities of the first three isotopes (M0, M1, and M2) of (for example, y-ion) are:
| (Eq.3) |
In the last line the first equation normalizes the sum of the abundances for the three isotopes. The second identity defines the value of α, which is the proportion of the M2 isotope of the precursor ion covered by the isolation window. We used Eq.(3) for computing the isotope distributions of fragment ions. The RA of the monoisotope of the y7 ion (AVDCGIK) in the nature is 62%. However, from the isotope distribution of the fragment in MS/MS, the median value was higher, 73%. The experimental observation is different from the theoretically expected value (referred to as spectral discrepancy24). This was true for all peptides, as shown in Figure S15. In most cases, the experimental observation overestimated the theoretically expected value. The results for three isotopes (M0, M1, M2) from all fragment ions (one for each of 122 peptides) are summarized in a bar plot in Figure S16. The bar plot shows the absolute difference between theoretical and observed RAs as the percentage of the theoretical RA. As is seen from the plot, the largest absolute difference percentage is observed for M2. This is expected, given that the offset and the isolation width in the experiments did not cover M2 isotope completely, Figure S14.
To account for the observed discrepancy, we have optimized the value of a in Eq. (3) to minimize the residual sum of the squares between the experimental and theoretical observations:
| Eq. (4) |
where is the experimentally observed (normalized) abundance of the nth isotope, Mn is the theoretically expected abundance of the nth isotope. The latter is dependent on the proportion, a, of the second heavy isotope covered by the fragmentation:
where p(Xy = j) is defined in Eq. (3). We believe, this is the first study to model the fragment isotopes to account for the limited width of the isolation window during peptide fragmentation. Many work25,26 has observed discrepancy between the experimental and expected isotope distribution, but no attempt has been made to account for it.
The above observations are not surprising as it is known that for fragment ions generated via HCD in Q Exactive, there are differences between the expected isotope distributions of the fragment ions and their natural isotope distributions26. The main effects contributing to this discrepancy are thought to be isolation width26 and NCE27. For the NCE of 20 eV, the isotope distributions of the fragment ions faithfully represented the expected distributions, in the case of the 4 Th of the isolation width26.
It should be noted that the discrepancy (the lack of the spectral accuracy) between the theoretically predicted isotope distributions and the distributions observed in the mass profiles obtained via Orbitrap mass spectrometers has been well documented for not only fragment ions, but for intact ions as well24.
Results for other CPSM peptides
Metabolic labeling with heavy water is cost-effective. It labels all non-essential amino acids. The label incorporation into peptides (proteins) is measured via the enrichment of heavy isotopes in high throughput LC-MS. However, since the labeling is incomplete (3% body water enrichment in the data used in this study), bioinformatics analysis of mass spectral data is complicated. Metrics, such as the correlation between the experimental and fitted data, averaged residual sum of squares, are used to select high-quality results. Large portions, more than 70%, of spectral data may be filtered out21. The resulting number of peptides and spectra are still comparable to those from the other labeling approaches8. For example, in amino acid based labeling, only peptides that contain the labeling amino acid can be used in quantification. One of the reasons affecting the quality of the quantification of label incorporation into peptide is the complexity of peak detection and integration of isotope clusters in MS1 of LC-MS. In this work, we study MS/MS of peptides for quantifying the isotope incorporation.
The tandem mass spectra are collected during DDA and are available for the use in quantification. Their quantification is very fast. However, quantification from MS/MS requires statistical filtering as well. We required at least 3 MS/MS spectra per time point of labeling. For the all of the fragments (one for each of the 57 peptides), we provide a full list of spectral and statistical information, and results of data processing in the Supplementary Material 2.csv. The corrected (using Eq. (4)) RAs of the monoisotope are shown in Supplementary Figure S17. The relationship between the experimental and modeled data is closer to the line of unity.
Only six fragments out of 57 showed correlation (between the experimental and predicted time course of RA) less than 0.85. Figure 6 shows the density function of DRCs after filtering out data of the six fragments. As is seen from the figure, the mode of the distribution is close to 0.1 day−1, which is the value of CPSM protein DRC computed from MS1, 0.099 day−1. However, the overall median was 0.066 day−1. The difference between the medians is most likely due to the lack of the spectral accuracy observed in data produced by Orbitrap mass spectrometers for fragment ions. The requirement of higher correlation between the experimental and theoretical fit reduces the difference between the medians. Thus, if we required correlation of 0.9 or higher (35 fragment ions passed the criterium), the median value of the DRCs from the fragment quantification was 0.07 day−1. For fragment ions with correlation higher than 0.95 (12 fragment ions passed the criterium), the median of the DRCs was 0.08 day−1. It is noteworthy that all peptides that passed the 0.9 correlation coefficient threshold have NEH numbers higher than 11.
Figure 6.

Density plot of DRCs of all peptides calculated from the time course of the RA of fragment monoisotope. At least three tandem mass spectra per time point of labeling were required. From the resulting 57 peptides, 6 were excluded because of the low (less than 0.85) correlation between the experimental data and theoretical fit.
In summary, we have analyzed 57 peptides from CPSM_MOUSE protein for label incorporation in metabolic labeling with heavy water. For all peptides, we have observed label incorporation (decreased RA of the monoisotope) into fragment ions. However, not all observations are suited for consistent quantification. We have found that peptides with multiple fragmentation events (many tandem mass spectra per time point) are, generally, better suited for quantification of DRCs. Some of the effects of variability of isotope distributions are attributed to the ion count. Fragment ions with low ion count showed higher variability. The high statistics of ion fragments (multiple fragmentation events of a peptide) alleviated this issue. For extracting protein degradation rates, we used median values of the monoisotope RA at each labeling time point from all spectra of a peptide.
In a future work, we will study experimental and data processing approaches that will address the issues outlined above. We will design ways of modeling of isotope incorporations into fragment ions to extract quantitative information about the rate constants. The approach will be automated to be applicable to larger datasets to gain statistical significance of obtained results. In this study, we excluded MS/MS spectra that were collected from non-monoisotope of precursors. In a future work, we will analyze these spectra and model them for use in DRC calculations. In addition, we will look into the effects of MS settings (NCE, number of microscans for each MS/MS, position of the offset, the length of the isolation window) on the fragment ion isotope distributions.
Conclusion.
The quantification of label incorporation from MS1 scans involves peak detection and integration in the chromatographic time and m/z domains19,28. Comparatively, the quantification in MS/MS is simpler. In addition, if a peptide has been chosen for fragmentation multiple times in DDA, the ion abundances can be averaged or the median value can be used (similar to this study). In the subsequent data processing, peptides that have been chosen for quantification pass several filtering criteria such as identifications in multiple time points of incorporation with a certain, small FDR29. These criteria result in the selection of peptides with multiple fragment ions in MS/MS. Therefore, in MS/MS there are potentially several fragment ions to measure the label incorporation.
We have analyzed a potential usability of fragment ions for measuring proteome dynamics from metabolic labeling with heavy water and LC-MS/MS. The fragment ions reflected the stable isotope incorporation in their isotope profiles. However, the variability of isotope distribution measurements for fragment ions in Q Exactive is rather high. For data collected in this instrument, the isotope distribution of fragment ions are known to be influenced by the isolation width and NCE26. We show that the experimentally observed isotope distributions consistently overestimated the monoisotopic peak abundance of fragment ions. One contribution to this overestimation is the narrow isolation width used in data collection. The overestimation can be alleviated by analyzing each isotope included in the isolation width of the precursor. However, there is still unresolved overestimation of the monoisotopic peak which we believe is due to the NCE. We have studied the use of normalization factor to correct for the discrepancy in the spectral accuracy.
Supplementary Material
Highlights:
In vivo Proteome dynamics estimation using fragment ions
Natural and fragment ion isotope distributions
Spectral accuracy correction
Time-course modeling
Heavy Water Stable Isotope Metabolic Labeling
Acknowledgements.
Research reported in this publication was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM112044. AB is partly supported by a training fellowship from the Gulf Coast Consortia, on the NLM Training Program in Biomedical Informatics and Data Science T15LM007093. WH was supported by UTMB High School Summer Biomedical Research Program.
Abbreviations:
- DDA
data-dependent acquisition
- DRC
decay rate constant
- HCD
higher-energy collisional dissociation
- LC-MS
liquid chromatography, mass spectrometry
- NCE
normalized collision energy
- RA
relative abundance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- (1).Balch WE; Morimoto RI; Dillin A; Kelly JW Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [DOI] [PubMed] [Google Scholar]
- (2).Baiceanu A; Mesdom P; Lagouge M; Foufelle F Endoplasmic reticulum proteostasis in hepatic steatosis. Nat Rev Endocrinol 2016, 12, 710–722. [DOI] [PubMed] [Google Scholar]
- (3).Claydon AJ; Beynon R Proteome dynamics: revisiting turnover with a global perspective. Mol. Cell Proteomics 2012, 11, 1551–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Scheltema RA; Hauschild JP; Lange O; Hornburg D; Denisov E; Damoc E; Kuehn A; Makarov A; Mann M The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol Cell Proteomics 2014, 13, 3698–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Price JC; Holmes WE; Li KW; Floreani NA; Neese RA; Turner SM; Hellerstein MK Measurement of human plasma proteome dynamics with (2)H(2)O and liquid chromatography tandem mass spectrometry. Anal. Biochem 2012, 420, 73–83. [DOI] [PubMed] [Google Scholar]
- (6).Hsieh EJ; Shulman NJ; Dai DF; Vincow ES; Karunadharma PP; Pallanck L; Rabinovitch PS; MacCoss MJ Topograph, a software platform for precursor enrichment corrected global protein turnover measurements. Mol. Cell Proteomics 2012, 11, 1468–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rauniyar N; McClatchy DB; Yates JR 3rd. Stable isotope labeling of mammals (SILAM) for in vivo quantitative proteomic analysis. Methods 2013, 61, 260–268. [DOI] [PubMed] [Google Scholar]
- (8).Fornasiero EF; Mandad S; Wildhagen H; Alevra M; Rammner B; Keihani S; Opazo F; Urban I; Ischebeck T; Sakib MS; Fard MK; Kirli K; Centeno TP; Vidal RO; Rahman RU; Benito E; Fischer A; Dennerlein S; Rehling P; Feussner I; Bonn S; Simons M; Urlaub H; Rizzoli SO Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat Commun 2018, 9, 4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Park SK; Venable JD; Xu T; Yates JR III. A quantitative analysis software tool for mass spectrometry-based proteomics. Nat. Methods 2008, 5, 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Price JC; Guan S; Burlingame A; Prusiner SB; Ghaemmaghami S Analysis of proteome dynamics in the mouse brain. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Busch R; Kim YK; Neese RA; Schade-Serin V; Collins M; Awada M; Gardner JL; Beysen C; Marino ME; Misell LM; Hellerstein MK Measurement of protein turnover rates by heavy water labeling of nonessential amino acids. Biochim. Biophys. Acta 2006, 1760, 730–744. [DOI] [PubMed] [Google Scholar]
- (12).Sato C; Barthelemy NR; Mawuenyega KG; Patterson BW; Gordon BA; Jockel-Balsarotti J; Sullivan M; Crisp MJ; Kasten T; Kirmess KM; Kanaan NM; Yarasheski KE; Baker-Nigh A; Benzinger TLS; Miller TM; Karch CM; Bateman RJ Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 97, 1284–1298e1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bateman RJ; Munsell LY; Chen X; Holtzman DM; Yarasheski KE Stable isotope labeling tandem mass spectrometry (SILT) to quantify protein production and clearance rates. J Am Soc Mass Spectrom 2007, 18, 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Elbert DL; Mawuenyega KG; Scott EA; Wildsmith KR; Bateman RJ Stable isotope labeling tandem mass spectrometry (SILT): integration with peptide identification and extension to data-dependent scans. J Proteome Res 2008, 7, 4546–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Holman SW; Hammond DE; Simpson DM; Waters J; Hurst JL; Beynon RJ Protein turnover measurement using selected reaction monitoring-mass spectrometry (SRM-MS). Philos Trans A Math Phys Eng Sci 2016, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tomazela DM; Patterson BW; Hanson E; Spence KL; Kanion TB; Salinger DH; Vicini P; Barret H; Heins HB; Cole FS; Hamvas A; MacCoss MJ Measurement of human surfactant protein-B turnover in vivo from tracheal aspirates using targeted proteomics. Anal Chem 2010, 82, 2561–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lee AY; Yates NA; Ichetovkin M; Deyanova E; Southwick K; Fisher TS; Wang W; Loderstedt J; Walker N; Zhou H; Zhao X; Sparrow CP; Hubbard BK; Rader DJ; Sitlani A; Millar JS; Hendrickson RC Measurement of fractional synthetic rates of multiple protein analytes by triple quadrupole mass spectrometry. Clin Chem 2012, 58, 619–627. [DOI] [PubMed] [Google Scholar]
- (18).Kasumov T; Ilchenko S; Li L; Rachdaoui N; Sadygov RG; Willard B; McCullough AJ; Previs S Measuring protein synthesis using metabolic (2)H labeling, high-resolution mass spectrometry, and an algorithm. Anal. Biochem 2011, 412, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sadygov RG; Avva J; Rahman M; Lee K; Ilchenko S; Kasumov T; Borzou A d2ome, Software for in Vivo Protein Turnover Analysis Using Heavy Water Labeling and LC-MS, Reveals Alterations of Hepatic Proteome Dynamics in a Mouse Model of NAFLD. J Proteome Res 2018, 17, 3740–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Perkins DN; Pappin DJ; Creasy DM; Cottrell JS Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- (21).Lau E; Cao Q; Ng DC; Bleakley BJ; Dincer TU; Bot BM; Wang D; Liem DA; Lam MP; Ge J; Ping P A large dataset of protein dynamics in the mammalian heart proteome. Sci Data 2016, 3, 160015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Team RDC: R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2010. [Google Scholar]
- (23).Sadygov RG Poisson Model To Generate Isotope Distribution for Biomolecules. J Proteome Res 2018, 17, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Su X; Lu W; Rabinowitz JD Metabolite Spectral Accuracy on Orbitraps. Anal Chem 2017, 89, 5940–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Goldford JE; Libourel IG Unsupervised Identification of Isotope-Labeled Peptides. Anal Chem 2016, 88, 6092–6099. [DOI] [PubMed] [Google Scholar]
- (26).Allen DK; Evans BS; Libourel IG Analysis of isotopic labeling in peptide fragments by tandem mass spectrometry. PLoS One 2014, 9, e91537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Allen DK; Goldford J; Gierse JK; Mandy D; Diepenbrock C; Libourel IG Quantification of peptide m/z distributions from 13C-labeled cultures with high-resolution mass spectrometry. Anal Chem 2014, 86, 1894–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Naylor BC; Porter MT; Wilson E; Herring A; Lofthouse S; Hannemann A; Piccolo SR; Rockwood AL; Price JC DeuteRater: a tool for quantifying peptide isotope precision and kinetic proteomics. Bioinformatics 2017, 33, 1514–1520. [DOI] [PubMed] [Google Scholar]
- (29).Benjamini Y; Hochberg Y Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. Journal of Royal Statistical Society 1995, 57, 289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.