

Summary
Despite their important roles in host nutrition and metabolism, and potential to cause disease, our knowledge of the fungal community in the mammalian gut is quite limited. To date, diversity and composition of fungi in swine gut still remains unknown. Therefore, the first internal transcribed spacer of fungi in faecal samples from three breeds of pigs (10 pigs for each breed) was sequenced based on an Illumina HiSeq 2500 platform, and the relationship between the fungal community and the concentrations of main short‐chain fatty acids (SCFAs) was also analysed. Results indicated that Chenghua (local, higher body fat rate), Yorkshire (foreign, higher lean meat and growth rate) and Tibetan (plateau, stronger disease resistance) pigs harboured distinct fungal community. The Basidiomycota and Ascomycota presented as the two predominant phyla, with Loreleia, Russula and Candida as the top three genera in all samples. Network analysis revealed a total of 35 correlations among different fungal genera, with 27 (77.14%) positive and 8 (22.86%) negative pairwise interactions. Canonical correspondence analysis suggested that fungi in the faeces of pigs were more correlated to the concentration of acetate and butyrate rather than propionate. Spearman’s correlation further showed that Tomentella was positively correlated to both acetate and butyrate, and Loreleia was positively correlated to propionate (P < 0.05), while Nephroma and Taiwanofungus were negatively correlated to acetate and propionate (P < 0.05). These findings expanded our knowledge on the intestinal fungi in pigs with different genotypes and phenotypes, indicating that fungi may play an indispensable role during the metabolism of host and the maintenance of intestinal health. The cross‐feeding between fungi and other microorganisms may be crucial during the digestion of dietary carbohydrates and the associated physiological processes, which is worthy to be further studied.
Results of the current study indicateed that Chenghua (local, higher body fat rate), Yorkshire (foreign, higher lean meat and growth rate) and Tibetan (plateau, stronger disease resistance) pigs harbored distinct fungal community. The Basidiomycota and Ascomycota presented as the two predominant phyla, with Loreleia, Russula and Candida as the top three genera in all samples. Fungi in the feces of pigs were more correlated to the concentration of acetate and butyrate rather than propionate. Of those fungal genera, Tomentella was positively correlated to both acetate and butyrate, and Loreleia was positively correlated to propionate, while Nephroma and Taiwano fungus were negatively correlated to acetate and propionate. These findings expanded our knowledge on the intestinal fungi in pigs with different genotypes and phenotypes, indicating that fungi may play an indispensable role during the metabolism of host and the maintenance of intestinal health. The cross‐feeding between fungi and other microorganisms may be crucial during the digestion of dietary carbohydrates and the associated physiological processes, which is worthy to be further studied.
Introduction
The distal gut of humans and monogastric animals harbours a large number of microorganisms including bacteria, archaea and fungi. The fungal population, or the so‐called ‘mycobiota’, makes up at most 0.1% of the microorganisms in the gastrointestinal (GI) tract of human and the role of intestinal fungi has often been ignored in the past a few decades (Arumugam et al., 2011; Koh, 2013; Auchtung et al., 2018). So far most studies on the gut microbiota of humans and animals have focused on the pathogenic microorganisms and commensal bacteria, but several recent studies indicated that fungi might be another vital microbial group that contribute to the metabolism and intestinal health of human and mice (Qin et al., 2010; Hoffmann et al., 2013; Heisel et al., 2017). Moreover, the size of a typical fungal cell is more than 100‐fold larger than a typical bacterial cell (Frey‐klett et al., 2011), indicating that fungi might contribute with important metabolic functions to the intestinal microbiota and provide a substantial surface area for the host‐microbe interactions. Although several studies have explored the fungal communities in human and mice intestines (Suhr and Hallen‐Adams, 2015; Li et al., 2017), studies on the composition and diversity of fungi in the intestines of pigs are still very rare. According to the limited results of deep sequencing, Ascomycota and Basidiomycota have been identified as the two most predominant fungal phyla in the faeces of piglets (Hu et al., 2016; Summers et al., 2019), which is consistent with previous findings in the gut of humans and mice. However, Kazachstania slooffae might be the dominant fungal species in weaning piglets (Summers et al., 2019), while its relative abundance showed extremely low or undetectable in the gut of human and mice (Suhr and Hallen‐Adams, 2015; Li et al., 2017). All the above results indicate that piglets may harbour unique intestinal fungal flora. Accumulating evidence indicates that diet (Filippo et al., 2010), environment (Benson et al., 2010), and host genotypes and phenotypes (Pajarillo et al., 2014) can strongly influence the composition of gut microbiota in various mammals. However, it is unclear whether the fungal community in the GI tract of pigs is also affected by these factors. Our previous research showed that the gut bacterial (Diao et al., 2016) and archaeal (Luo et al., 2012) profile in the gut of pigs among different breeds were very distinct, indicating a potential relationship between the microbial compositions in swine gut and the different genotypes or phenotypes. However, so far no evidence has been shown in regards to the variation of fungal community in the GI tract of these pigs. Therefore, in the current study, pigs from three dominant swine breeds, Chenghua, Yorkshire and Tibetan, in the Southwest of China were selected, and their faecal samples were collected to gain insights into their gut fungal community structure.
The Chenghua pig (CH) is a typical local breed and is widely reared in the southwest of China, including the Sichuan province and Chongqing area, and has a slower growth rate and higher meat quality compared to foreign breeds (Yang et al., 2003). The Yorkshire pig (YS) is an imported commercial breed that is usually used as the male parent during breeding to improve the growth rate and lean meat rate of local breeds (Diao et al., 2016). The Tibetan pig (TB) is widely distributed in the Tibetan Plateau of China and Northwest Plateau of Sichuan Province, and reared mostly through grazing. These pigs are characterized by very low growth rate, enhanced adaptability to the environment and stronger disease resistance than foreign breeds (Yang et al., 2011; Yang et al., 2014). Thus, the different genetic backgrounds, habitats and diets of these three breeds of CH, YS and TB pigs might be related to the differences in the fungal compositions in their GI tract.
In the current study, the fungal community in the faecal samples of CH, YS and TB pigs were analysed and compared using next‐generation sequencing targeting ITS1 of fungi. Furthermore, the interaction between the fungal community and the main metabolites of bacteria, short‐chain fatty acids (SCFAs; Mayu et al., 2015), in the faecal samples were also analysed. The results presented here should aid in the further understanding of the relationship between the host and gut fungal community.
Results
The concentration of SCFAs in the faecal samples from CH, YS and TB pigs
According to the results of gas chromatography, the concentration of acetate, propionate and butyrate in the faecal samples showed significantly different among the CH, YS and TB pigs (P < 0.01, Table 1) respectively. The concentrations of both acetate and propionate in the faecal samples from YS pigs were significantly higher than those from TB and CH pigs (P < 0.01).
Table 1.
Concentrations of acetate, propionate and butyrate in the faecal samples of the three different breeds of pigs.
| Item | CH | YS | TB | P value |
|---|---|---|---|---|
| Acetate (μmol g−1) | 28.69 ± 7.64a | 42.03 ± 12.46b | 27.55 ± 3.67a | 0.002 |
| Propionate (μmol g−1)* | 11.38 ± 3.00b | 14.33 ± 1.22c | 8.40 ± 1.72a | 0.001 |
| Butyrate (μmol g−1) | 6.57 ± 2.65ab | 8.77 ± 1.79b | 4.47 ± 0.79a | 0.005 |
The rest of the data are not normally distributed and presented as median ± interquartile range (IQR). Different alphabetical (a, b, c, ab) superscripts mean significant difference (P < 0.05).
Means the normally distributed data which are presented as means ± standard deviation (SD).
The diversity of fungal community in faecal samples from the three different breeds of pigs
According to the results of de novo OTU picking protocol with a threshold of 97% similarity, the alpha diversity was estimated based on observed species (observed OTUs), ACE, Chao1 (estimated number of OTUs), Goods_coverage and Shannon’s diversity index. The observed species of fungi in the faecal samples of TB pigs was significantly higher than that of CH pigs (P < 0.05, Table 2, Table S1), while other indices showed no difference among the three groups (Table 2; Table S1, Figs S1 and S2).
Table 2.
The similarity and differences of fungal community diversity and richness in the faecal samples of three different breeds of pigs.
| Item | CH | YS | TB | P value |
|---|---|---|---|---|
| Observed species* | 729.60 ± 166.84a | 813.40 ± 195.46ab | 939.20 ± 161.99b | 0.04 |
| ACE* | 905.55 ± 202.50 | 986.47 ± 272.24 | 1143.04 ± 201.81 | 0.08 |
| Chao1* | 893.66 ± 188.65 | 959.64 ± 271.14 | 1121.04 ± 202.31 | 0.08 |
| Goods_coverage* | 0.996 ± 0.08 | 0.996 ± 0.14 | 0.996 ± 0.12 | 0.36 |
| Shannon* | 6.52 ± 0.58 | 6.79 ± 0.43 | 7.04 ± 0.43 | 0.08 |
| Item | CH‐TB | TB‐YS | CH‐YS |
|---|---|---|---|
| A | 0.11 | 0.11 | 0.15 |
| observed‐delta | 0.53 | 0.56 | 0.54 |
| expected‐delta | 0.60 | 0.63 | 0.64 |
| Significance | 0.001 | 0.001 | 0.001 |
The multi‐response permutation procedure (MRPP) method was utilized to analyse the similarity of the fungal community in the samples from the three groups of pigs. When the A value is greater than 0, it means that the difference between groups is greater than intra‐group. On the contrary, it means that the intra‐group difference is greater when the A value is less than 0. The smaller the observed‐delta value, the smaller the difference between groups. The greater the expected‐delta value, the greater the difference between groups. Different alphabetical (a, b, c, ab) superscripts mean significant difference (P < 0.05).
Means the normally distributed data which are presented as means ± standard deviation (SD). The rest of the data are not normally distributed and presented as median ± interquartile range (IQR).
According to the result of principal coordinates analysis (PCoA), the fungal sequences obtained from the faecal samples of CH, YS and TB pigs were clustered and divided into three different types of communities (R = 0.173, P = 0.003, Fig. 1). The non‐metric multidimensional scaling (NMDS) analysis based on the Bray–Curtis similarity indices also showed detectable dissimilarities of fungal community among the three groups (Fig. S3). In addition, according to the result of multi‐response permutation procedure (MRPP), the inter‐groups difference of fungal community showed significantly greater than intra‐group (P < 0.05, Table 2).
Figure 1.
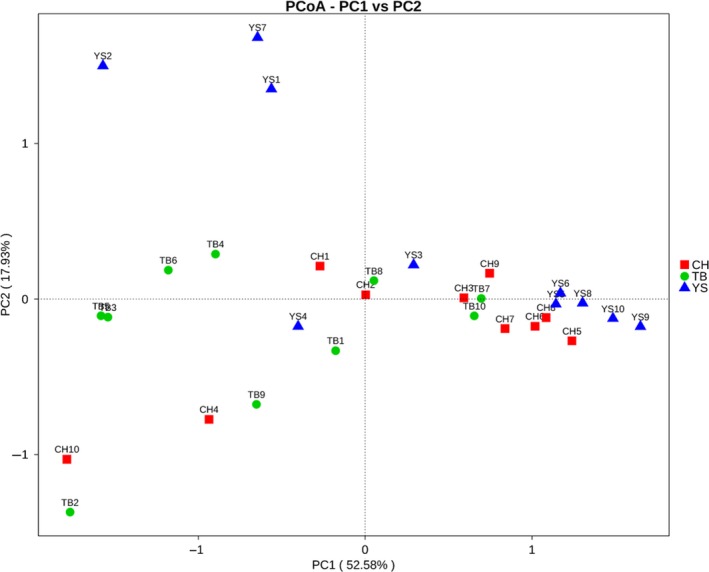
Principal coordinate analysis (PCoA) of fungal community structures of the faecal microbiota of three groups based on the weighted Unifrac distance metric. The fungal communities from the faecal samples of pigs in the three different groups are significantly different (P = 0.001). Each symbol represents one sample, and symbols with same colour mean samples from the same group.
The community and composition of fungi in faecal samples from the three different breeds of pigs
After chimera checking, the total of 30 faecal samples yielded 2 304 968 sequences with an average of 76 832 ± 1706 sequences per sample. The filtered sequences were clustered into 6942 fungal OTUs (Fig. S4). A total of 399 OTUs were only found in the faecal samples from CH pigs, whereas 711 and 821 OTUs were specifically identified in the faecal samples from TB and YS pigs respectively. A number of 1011 fungal OTUs were shared by CH, YS and TB pigs. Based on pairwise comparisons, 1117 and 531 OTUs were uniquely found in the faecal samples from TB and CH pigs, respectively, while 1372 OTUs were shared by pigs in these two groups. There were 1043 and 982 unique OTUs in the faecal samples from YS and TB pigs, respectively, and 1507 OTUs were shared by pigs in the two groups. In addition, 1227 and 580 unique OTUs were found in the faecal samples from YS and CH pigs, respectively, and 1323 OTUs were shared by pigs in the two groups.
According to the species annotations, a total of six identified fungal phyla, Basidiomycota, Ascomycota, Chytridiomycota, Zygomycota, Glomeromycota and Neocallimastigomycota, were detected in the faeces of all pigs (Table 3). The most predominant two OTUs in the faeces of pigs in the three groups were identified as Basidiomycota (73.94%) and Ascomycota (24.33%) for CH pigs, Basidiomycota (67.77%) and Ascomycota (25.56%) for YS pigs, as well as Basidiomycota (44.86%) and Ascomycota (38.13%) for TB pigs respectively. The relative abundance of other OTUs all showed less than 1%. Of those, Chytridiomycota accounted for 0.52% and 0.62% of all OTUs in the faeces of CH and TB pigs, respectively, while Zygomycota accounted for 0.38% in the faeces of YS pigs (Table 3, Fig. 2). In addition, the relative abundance of sequences belonging to phylum Zygomycota was 0.20%, 0.38% and 0.49% in the faeces of CH, YS and TB pigs respectively, which was significantly different between groups (P < 0.05). The abundance of Glomeromycota in the faeces of YS pigs (0.12%) was significantly higher than that in the faeces of TB (0.07%) and YS (0.03%) pigs (P < 0.05).
Table 3.
The relative abundance (%) of the main fungal phyla and genera in the faecal samples from the three different pig breeds.
| CH | YS | TB | P value | |
|---|---|---|---|---|
| Phylum | ||||
| Basidiomycota | 73.94% ± 24.45% | 67.77% ± 34.69% | 44.86% ± 20.31% | 0.21 |
| Ascomycota | 24.33% ± 22.92% | 25.56% ± 35.70% | 38.13% ± 36.04% | 0.58 |
| Chytridiomycota | 0.52% ± 0.22% | 0.27% ± 0.35% | 0.62% ± 0.69% | 0.054 |
| Zygomycota | 0.20% ± 0.27% a | 0.38% ± 0.23% ab | 0.49% ± 0.25% b | 0.04 |
| Neocallimastigomycota | 0.01% ± 0.02% | 0.02% ± 0.30% | 0.03% ± 0.12% | 0.36 |
| Glomeromycota | 0.03% ± 0.04%a | 0.12% ± 0.22%b | 0.07% ± 0.08%ab | 0.046 |
| Others | 1.57% ± 2.17%a | 6.62% ± 6.50%ab | 8.67% ± 15.50%b | 0.01 |
| Genus | ||||
| Loreleia | 21.03% ± 10.29%ab | 18.95% ± 18.02%b | 7.15% ± 4.27%a | 0.02 |
| Russula | 12.27% ± 8.31% | 16.88% ± 11.77% | 12.85% ± 13.20% | 0.60 |
| Nephroma | 5.55% ± 13.61%b | 0.06% ± 4.73%a | 10.92% ± 14.95%c | 0.01 |
| Candida | 6.59% ± 11.35% | 9.03% ± 37.54% | 7.51% ± 14.20% | 0.51 |
| Metschnikowia | 2.89% ± 2.59%ab | 7.61% ± 6.79%b | 1.97% ± 1.45%a | 0.01 |
| Hydnocystis | 2.61% ± 1.62%b | 0.57% ± 0.32%a | 0.53% ± 0.42%a | 0.004 |
| Bullera * | 1.21% ± 0.71% | 1.09% ± 0.47% | 1.37% ± 0.80% | 0.65 |
| Taiwanofungus | 0.14% ± 0.32% | 0.15% ± 0.95% | 0.60% ± 1.62% | 0.08 |
| Microidium | 0.05% ± 0.04% | 0.08% ± 0.11% | 0.15% ± 1.03% | 0.12 |
| Ceraceomyces | 0a | 0.001% ± 0.01%ab | 0.02% ± 0.43%b | 0.02 |
| Others* | 44.68% ± 6.70% | 36.48% ± 6.14% | 46.51% ± 13.67% | 0.06 |
All of the known fungal phyla and the top 10 known genera are presented. Different alphabetical (a, b, c, ab) superscripts mean significant difference (P < 0.05).
Means the normally distributed data which are presented as means ± standard deviation (SD). The rest of the data are not normally distributed and presented as median ± interquartile range (IQR).
Figure 2.
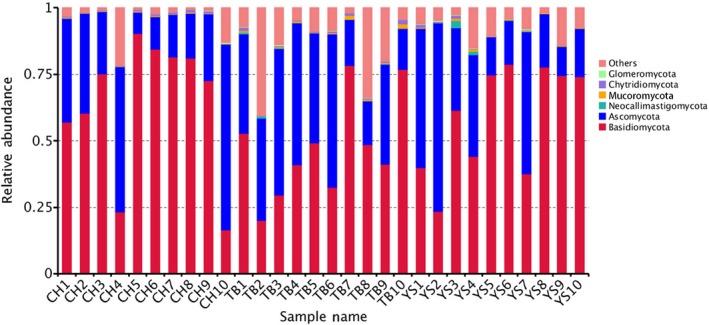
Fungal composition at the phylum level in the faecal samples from each pig in the three different groups. Each bar represents the relative abundance of each fungal taxon in a sample.
A total number of 148 known fungal genera were detected in all faecal samples, and Loreleia, Russula, Nephroma, Candida, Metschnikowia, Hydnocystis, Bullera, Taiwanofungus, Microidium and Ceraceomyces were found as the top 10 genera (Table 3). Of these genera, Loreleia (21.03%), Russula (12.27%), Candida (6.59%), Nephroma (5.55%) and Metschnikowia (2.89%) affiliated with the two phyla, Basidiomycota and Ascomycota, were identified as the five predominant genera in the faecal samples of CH pigs, which together constituted 48.33% of all sequences in these pigs. For YS pigs, the predominant genera were annotated as Loreleia (18.95%), Russula (16.88%), Candida (9.03%), Metschnikowia (7.61%) and Bullera (1.09%), and these genera together comprised 53.56% of all sequences. Meanwhile, the genera Russula (12.85%), Nephroma (10.92%), Candida (7.51%), Loreleia (7.15%) and Metschnikowia (1.97%) constituted 40.40% of all sequences in the faecal samples of TB pigs. Additionally, the relative abundance of genus Loreleia in the faeces of TB pigs was significantly lower than that of CH and YS pigs (P < 0.05, Table 3, Fig. S5, Fig. 3), while the abundance of Nephroma in the faeces of TB pigs was significantly higher than that of CH and YS pigs (P < 0.05). Moreover, the abundance of Metschnikowia in the faeces of YS pigs was significantly higher than that of CH and TB pigs (P < 0.01), and the abundance of Hydnocystis in the faeces of CH pigs was significantly higher than that of YS and TB pigs (P < 0.01).
Figure 3.
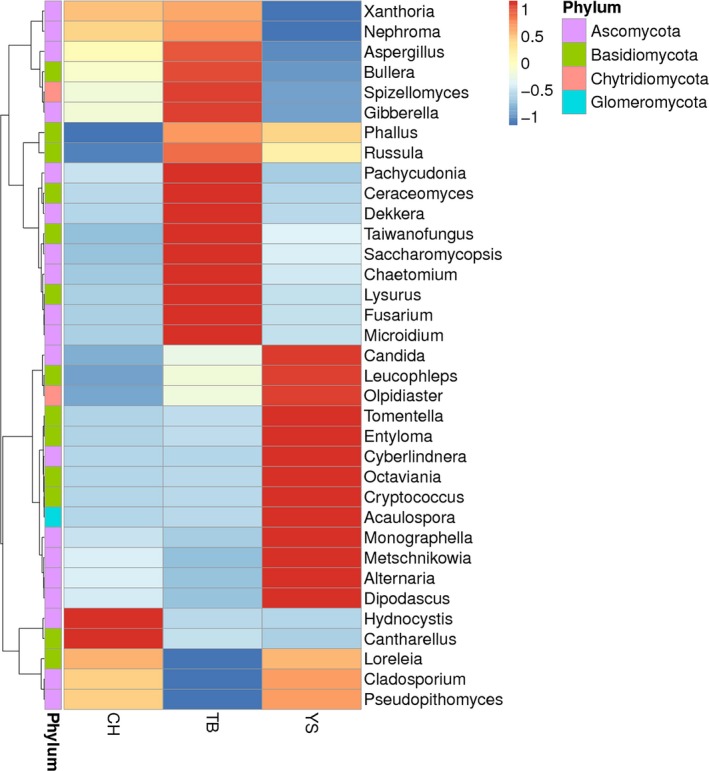
Heatmap distribution of OTUs for samples in the three groups. OTUs are arranged in rows and are clustered on the vertical axis (y‐axis). Samples are arranged vertically and are on the horizontal axis (x‐axis). Clustering was done using Phylotrac’s heatmap option with Pearson correlations and complete lineage algorithms.
Results of linear discriminant analysis (LDA) and LefSe showed that the relative abundance of 15 fungal taxa was significantly different among the three groups (LDA score > 4.0, P < 0.05, Fig. S6a). Seven taxa, including Loreleia postii and Metschnikowia pulcherrima, were significantly enriched in the faecal samples from YS pigs (P < 0.05). Another seven taxa, including Nephroma helveticum and unidentified families of Pezizales and Peltigerales, showed significantly enriched in the faeces of TB pigs (P < 0.05). Only one taxon, an unidentified order of Wallemiomycetes, was significantly enriched in the faeces of CH pigs (P < 0.05, Fig. S6b).
The network between different fungal genera in the faecal samples from the three different breeds of pigs
Based on the analysis of all obtained fungal sequences, a total of 26 nodes (genera) belonging to 4 phylum and 35 edges (correlations) with a mean of 1.35 edges per node, which were highly connected and formed a clustered topology, were presented as the intestinal fungal network of pigs (Fig. S7). Of these 35 correlations, a total of 27 (77.14 %) interactions were positive and 8 (22.86 %) were negative. Of the 26 genera, the genus Loreleia was found at the centre of the network and showed the most correlations with other genera. Specifically, Loreleia was negatively correlated with the genera Nephroma, Monographella, Taiwanofungus, Pachycudonia, Phallus and Fusarium, and positively correlated with Metschnikowia and Russula. The genus Candida was positively correlated with Acaulospora and Taiwanofungus, while the genus Russula was positively associated with Loreleia and Bullera, and negatively correlated with Cantharellus. Some genera, such as Saccharomycopsis, Cryptococcus, Aspergillus, Leucophleps and Alternaria, were correlated with only one or two of other genera.
For the sequences obtained from the faeces of CH pigs (Fig. S8), the genus Loreleia was positively correlated with Russula and Hydnocystis, while negatively correlated with Candida. The genus Nephroma was negatively correlated with Bullera and Metschnikowia. For the sequences obtained from the faeces of YS pigs (Fig. S9), Loreleia was positively correlated with Russula, Metschnikowia and Bullera, while the genus Candida was negatively correlated with Metschnikowia. For those sequences obtained from the faeces of TB pigs (Fig. S10), Russula was positively correlated with Loreleia, Bullera and Metschnikowia, while Candida was positively correlated with Taiwanofungus.
The interaction between the fungal community and concentrations of SCFAs in the faecal samples from pigs in different groups
Canonical correspondence analysis (CCA) was used to investigate whether there was a relationship between fungal community and the concentration of the main bacterial metabolites (SCFAs) in the faecal samples of pigs in the three groups (Fig. 4). According to the result, the two first discriminant functions (axis1 and 2) contributed 85.1% of the total variation, with 46.6% for the first discriminant function and 38.5% for the second. All samples obtained from CH pigs were found to be distributed in the second quadrant, while samples from YS pigs were distributed in both the first and fourth quadrant, and those from the TB pigs distributed in both third and fourth quadrant. Considering the length of the arrow lines, the correlation of these environmental factors with fungal community structure in the faeces of pigs could be in the following order: acetate > TSCFAs (Total SCFAs) > butyrate.
Figure 4.
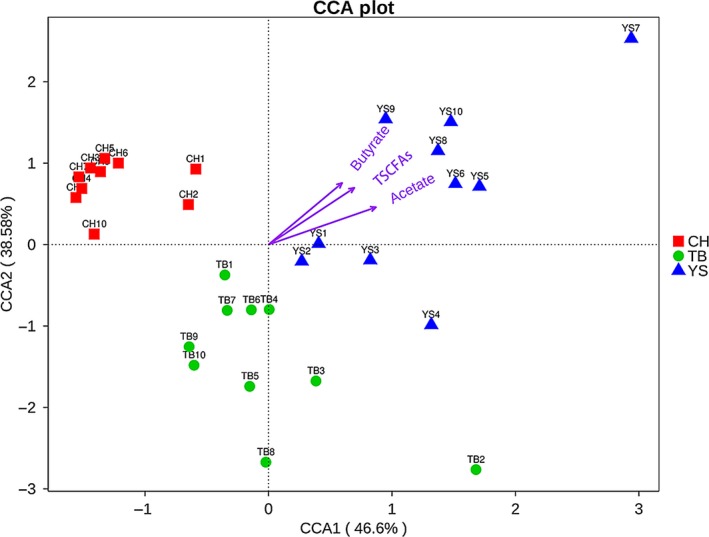
The relationship between the fungal community and the environmental factor in the faecal samples of pigs calculated using the canonical correspondence analysis (CCA). Each symbol represents one group. Each of the arrow line in the CCA chart represents the corresponding environmental factor (acetate, propionate and butyrate), and the length of each arrow line represents the degree of correlation between each environmental factor and the fungal community. An acute angle between two arrow lines indicates a positive correlation between the two corresponding environmental factors, conversely an obtuse angle indicates a negative correlation. Axis 1 and 2 contributed 85.1% of the total variation, with 46.6% for CCA1 and 38.5% for CCA2. All samples obtained from CH pigs distribute in the fourth quadrant, while samples from YS pigs distribute in both the first and second quadrant, and those from the TB pigs distribute in both the second and third quadrant. The correlation of each SCFA with the fungal community structure in the faeces of the animals shows in the following order: acetate > butyrate > propionate.
A hierarchical cluster‐based heat map of OTUs based on Spearman correlations was then used to show the relationship between main fungal genus and the concentration of each SCFA (Fig. 5, Table S2). The genus Metschnikowia was positively correlated with the concentrations of all SCFAs (Acetate, Propionate, Butyrate and TSCFAs; P < 0.05), and Tomentella was positively correlated with the concentration of acetate (P < 0.01), butyrate and TSCFAs (P < 0.05) respectively. The genus Loreleia also showed positively correlated to the concentration of propionate (P < 0.05), while the genera Nephroma and Taiwanofungus were negatively correlated with the concentration of acetate and propionate (P < 0.05) respectively.
Figure 5.
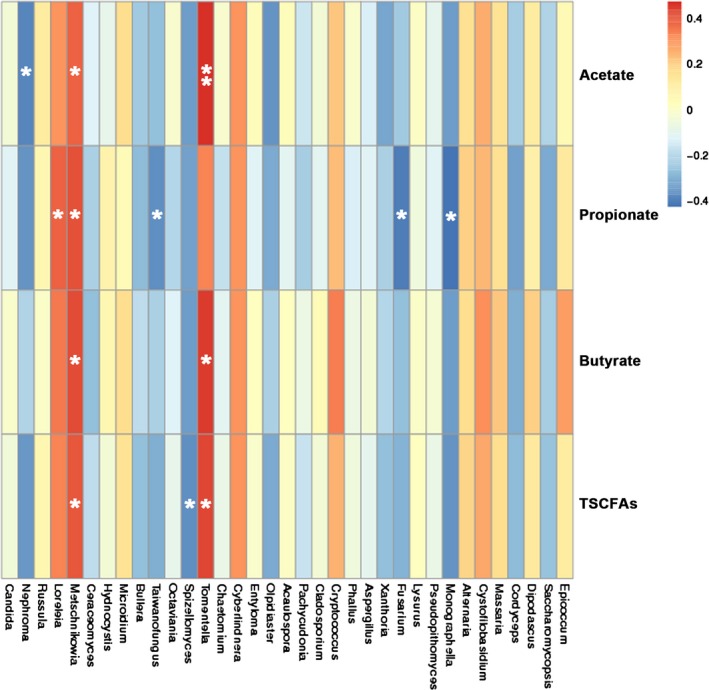
The heat map of correlation between fungal genera and concentrations of SCFAs. Heat map was created according to the result of Spearman’s correlation analysis. Positive correlations are represented in red, while negative correlations are represented in blue. The significant correlations are presented as asterisks (*, P < 0.05; **, P < 0.01).
Discussion
Gut microbes mediate various physiological processes including energy metabolism (Tremaroli and Bäckhed, 2012), intestinal barrier function and immune system of the host (Cerfbensussan and Gaboriaurouthiau, 2010). In addition to the commensal bacteria, according to limited studies, fungi are regarded as another microbial group in the gut of human and mice that may play a critical role in the metabolism and intestinal health of the host (Li et al., 2017). To the best of our knowledge, this study is the first to report that pigs from different breeds harbour distinct gut fungal profiles.
The high coverage, Q20 and Q30 values (Table S3), suggested a high quality of the fungal sequences obtained in the current study. Analysis targeting the community and discrepant species revealed that the fungal compositions at the phylum and genus level were widely different in the faeces of Chenghua, Yorkshire and Tibetan pigs. Our results indicated that genetic background as well as related phenotypes might be important factors influencing the diversity of fungi in swine gut. We also found that the fungal richness (observed species) in the faecal samples of Tibetan pigs was higher than in Chenghua pigs faecal samples, indicating a more diverse fungal community in the gut of Tibetan pigs.
In the present study, a total of 6942 OTUs were observed in the faeces from pigs, which was greater than the average of 3883 OTUs estimated in the hindgut of humans (Parfrey et al., 2014). All the results of heat map, PCoA and NMDS revealed that the composition of fungi in the faeces of pigs in the three groups was different. Our results showed that Basidiomycota and Ascomycota were the two most predominant fungal phyla in the faeces of pigs, which is consistent with previous findings in the gut of humans and mice (Scupham et al., 2006; Hoffmann et al., 2013; Hallen‐Adams et al., 2015). It is noteworthy that the relative abundance of Zygomycota in the faeces of Tibetan pigs was the highest in all three breeds. Species of Zygomycota are widely found to be distributed in soils, food, plant and animal excreta, and may utilize simple carbohydrates (Tauktornisielo et al., 2007). Thus, the high abundance of Zygomycota in the GI tract of Tibetan pigs may be associated with their habitats and feeding habits, such as grazing and intake of large amount of green forage. At the genus level, Loreleia, Russula, Nephroma, Candida and Metschnikowia were found as the most predominant taxa in all faecal samples, while in the gut of humans and mice, the relative abundance of these fungal genera, except for Candida, was extremely low or undetectable (Scupham et al., 2006; Hoffmann et al., 2013; Hallen‐Adams et al., 2015). In addition, the abundance of several fungal genera in the faecal samples was found to be remarkably different among the three groups. As compared to Chenghua and Yorkshire pigs, the Tibetan pigs harboured lowest abundance of Loreleia. Loreleia is a genus of brightly coloured agarics that belong to Hymenochaetales (Redhead et al., 2002). They inhabit mosses or liverworts on soil in temperate regions of the Northern Hemisphere (Bresinsky and Schötz, 2006). In the present study, Loreleia was observed as the most abundant fungal genus in the faeces of Chenghua and Yorkshire pigs. Due to the limitations of the current study, whether this fungal genus is an original inhabitant or alien in the GI tract, as well as the relatively low abundance of this genus in the gut of Tibetan pigs needs further investigation. On the other hand, the relative abundance of Nephroma was found to be remarkably higher in the faeces of Tibetan pigs than Yorkshire pigs. Similarly, the abundance of Nephroma was also observed to be higher in the faecal samples of local Chenghua pigs than the foreign Yorkshire pigs. It has been reported that Nephroma is a genus of medium to large foliose lichens which have been widely found on the mossy ground, rocks or trees (Colhoun, 2008). The high proportion of this fungal genus in the intestine of Tibetan and local pigs may be associated with their feeding behaviour. Limited studies in humans and mice suggested a possible shaping of food and environmental factors to the fungal community structures in the GI tract (Hoffmann et al., 2013; Heisel et al., 2017). Unlike humans and model animals, pigs in captivity or grazing have more opportunities to contact the soil, and the habit of digging earth with their snouts (Li et al., 2016) may be another way for the fungi in soil to enter the GI tract of pigs. Many species belonging to Aspergillus can survive under physiological temperature such as 37°C, but are much more widely reported in environmental (soil, air or plant) samples. Some fungal species such as Saccharomyces cerevisiae also has been found in the intestine of humans, indicating that they may play a role in the gut, regardless of preferred niche (Hallen‐Adams and Suhr, 2016). Mucor has already been found in the gut of human (Borges et al., 2018), and can utilize some types of carbohydrates (Tauktornisielo et al., 2007). But other commonly detected foodborne fungi including multiple Penicillium species may not grow at 37°C (Hallen‐Adams and Suhr, 2016). Generally, the ability of fungal strains to grow under animal body temperature and the opportunity for repeated introduction render them as the most commonly detected fungi in the faecal samples of animals, which in turn may likely contribute to gut microbial ecology. However, with the results of the current study, we still cannot explain whether these fungi from the environment have truly colonized the intestine of the pigs or they are excreted as transients, as well as the real function of these microbes. Further studies to investigate the interactions between feed, environment and the diversity of fungi in the gut of pigs are necessary.
As an important symbiotic fungus, Candida is widely distributed in the intestines of humans and mice, and has also been reported as an opportunistic pathogen (Scupham et al., 2006; Hallen‐Adams et al., 2015). The high abundance of Candida was also frequently observed on the intestinal mucosa of patients with ulcer and inflammatory bowel diseases (IBD; Qiu et al., 2015; Sokol et al., 2016). In the current study, the relative abundance of Candida was observed to be the highest in the faeces of Yorkshire pigs. Compared to local breeds, pigs of foreign breeds are indeed reported to be more susceptible to some pathogens (Kang et al., 2016), which might be associated with the high abundance of Candida in the gut of Yorkshire pigs rather than Tibetan and Chenghua pigs. However, whether there is a relationship between the higher ratio of Candida and the disease susceptibility of pigs still needs to be discussed. Interestingly, another genus, Metschnikowia, a genus belonging to the family Metschnikowiaceae (Sisti and Savini, 2014), also showed a higher abundance in the faeces of Yorkshire pigs than the other two breeds. Some species of Metschnikowia, such as Metschnikowia pulcherrima (anamorphic state: Candida pulcherrima), have been proved to display a broad and an effective antimicrobial action (Oro et al., 2014; Sian et al., 2016). In this regard, further experiments will be needed to determine the significance of the high proportion of the genus Metschnikowia in the gut of Yorkshire pigs.
Considering the possible interaction between gut bacteria and fungi, the relationship between fungal species and SCFAs may reflect the function of fungi to some extent. Our results of CCA indicated that fungi in the hindgut of pigs were mostly linked to the concentration of acetate. Acetate is the most predominant metabolite of bacteria with complex carbohydrates such as dietary fibres, and also the main substrate for the synthesis of cholesterol (Maciejewska et al., 2018). Specific to genus level, Metschnikowia and Tomentella may be the two fungal genera which are most likely associated with the produce of acetate in the gut of pigs, suggesting that the intestinal fungi of pigs might be involved in the metabolism of dietary polysaccharides. Butyrate is another metabolites produced by intestinal bacteria using complex carbohydrates (Mcorist et al., 2011). It is the most important energy source of colonic and caecal epithelial cells, and plays a key role in promoting cell differentiation and maturation and maintaining the stability of intestinal environment (Hamer et al., 2010; Leonel and Alvarez‐Leite, 2012). Our results showed that although the relative abundance of genus Tomentella was very low (less than 0.1% of total OTUs), it was the only one genus that was most likely associated with the production of butyrate in the gut of pigs, indicating a possible function involved in the intestinal health of fungi with low abundance, indicating a potential linkage between Tomentella and the intestinal health of pigs. Besides, the levels of most nutrients in the diets for pigs in the three groups were similar (Table S4), indicating that the influence of nutrition level of diet on the community of the faecal fungi of these pigs in the current study might be limited.
In summary, the composition of fungi in the GI tract of pigs may be completely different from the known fungal communities. Our results showed that Chenghua, Yorkshire and Tibetan pigs harboured distinct fungal profile, which might be influenced by their phenotype and genotype, as well as their different feeding habitat. A correlation between fungi and SCFAs found in the current study indicates a close relationship between fungi and the metabolism of bacteria in the gut of pigs (Fig. 6). Although our study was only a preliminary exploration, the findings would be helpful to enhance our understanding for the role of fungi in the GI tract of pigs gut.
Figure 6.
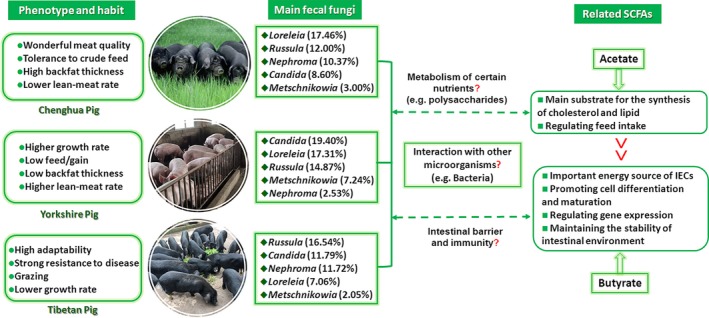
The possible linkage between the genotypes and phenotypes of host, environment and habits, the community of intestinal fungi, as well as the environmental factors (such as the metabolites of other microorganisms). The habitats and habits of animals may largely influence the composition of intestinal fungi. During feeding, fungi in the surrounding environment may be introduced into the GI tract through contact with soil, pasture (such as grazing Tibetan pig), green forage (such as Chenghua pig) or licking the housing facilities. This may also be one of the reasons why fungi that are widely found in the environment are also observed in abundance in the gut of pigs in the current study. In addition, the community of fungi in the gut of pigs exhibited a close relationship with some main metabolites of bacteria, such as acetate and propionate, suggesting that intestinal fungi may be involved in the regulation of metabolic process of some nutrients (e.g. complex carbohydrates) and the health of the intestine (e.g. the maintenance the function of intestinal epithelial) of pigs. Thus, further research should focus on the interaction between fungi and bacteria in the GI tract. Although the population of fungi is relatively small in the gut of monogastric animals (e.g. pig), which has resulted in less attention devoted to intestinal fungi in previous studies, our results show that fungi in the gut should not be ignored in future studies.
Experimental procedures
Experimental design and animal management
In the current study, faecal sample from each of ten healthy pigs of Chenghua (age from 200 to 210 days with an average bodyweight of 90 kg), Yorkshire (age from 140 to 150 days with an average bodyweight of 60 kg) and Tibetan (one year old with an average bodyweight of 50 kg) gilts/sows were collected respectively. All the selected pigs were sexually and physically mature, and were at the same physiological stage. The sows were raised at three different commercial farms in Sichuan Province, Southwest of China (Fig. S11). A corn‐soybean diet was formulated according to the Requirements of Swine of National Research Council Nutrient (NRC, 2012) and Chinese Feeding Standard of Swine (2004). In addition, as Tibetan pigs selected in the current study were captive, an ad libitum access to silage grass was conducted to meet the roughage requirement of the animals. The diets for all pigs were antibiotics and growth promoters free, and no fungal growth promoters or additives were supplemented.
To collect faecal samples, the characteristics of faeces from each selected animal were visually inspected and scored according to the method earlier described (Sudipta et al., 2010). Faecal scores of all pigs were visually assessed using a subjective score on a five‐point scale ranging from 1 to 5: 1 = hard faeces, 2 = firm well formed, 3 = soft and partially formed faeces, 4 = loose, semi‐liquid faeces, and 5 = watery faeces (Table S5). Approximately, 2 g faeces of each pig were collected into a 2‐mL sterilized centrifuge tube by slightly pressing the anus and stored at −80°C until further analysis. The levels of principal nutrients, such as ash, dry matter (DM), crude fat, crude fibre, acid detergent fibre (ADF), crude protein, dietary energy, calcium and phosphorus, in the diets of these animals were determined using the AOAC method (AOAC, 1990; Table S4).
The detection of short‐chain fatty acids (SCFAs) concentrations
In the hindgut of pigs, acetate, propionate and butyrate are the three dominant SCFAs which composed more than 85% of total short‐chain fatty acids (Petra and Flint, 2009) or even more. So in the current study, the concentration of total SCFAs was symbolized with the summarized concentrations of these three main SCFAs. The concentration of acetate, propionate and butyrate in each faecal sample was separated and quantified using a gas chromatograph (GC; GC‐14B, Shimadzu, Japan; capillary column 30 m × 0.32 mm × 0.25 μm film thickness/VARIAN CP‐3800, Varian, Palo Alto, CA, USA) as previously described by Franklin et al. (2002). Faecal samples (1 g) were thawed and suspended in 2‐ml of distilled water in a screw‐capped tube. After being vortexed, each sample was centrifuged (12 000g) at 4°C for 10 min. The supernatant (1‐ml) was transferred into a 2‐ml centrifuge tube and mixed with 0.2‐ml metaphosphoric acid and kept at 4°C for 30 min. The mixtures were then centrifuged (12 000g) again at 4°C for 10 min. Aliquots of the supernatant (1‐μl) were analysed by GC as stated above. Briefly, a flame ionization detector was used with an oven temperature at 100–150°C. The polyethylene glycol column was operated with a highly purified N2 as the carrier gas at 1.8 ml min−1. The minimal detectable limit for each SCFA was 0.01 mmol l−1.
DNA extraction and sequencing
The genomic DNA for each faecal sample was extracted using the QIAamp DNA stool Mini Kit (Qiagen, Hilden, Germany). The extracted DNA was stored at −80°C until used for PCR amplification and DNA sequencing. The ITS1 regions were amplified using specific primers ITS5‐1737F (GGAAGTAAAAGTCGTAACAAGG) and ITS2‐2043R (GCTGCGTTCTTCATCGATGC) with barcodes. The 30‐µl PCR reaction included 15 µl of Phusion® High‐Fidelity PCR Master Mix (New England Biolabs), each of 0.2‐µM forward and reverse primer, and 10‐ng template DNA. The procedure of PCR amplification was initiated with a denaturation at 98°C for 1 min, followed by 30 cycles of a denaturation at 98°C for 10 s, an annealing at 50°C for 30 s, and an extension at 72°C for 30 s and finally ended by an extension at 72°C for 5 min. The PCR products were detected with electrophoresis using 2% agarose gel, samples with bright main strip between 400 and 450 bp were chosen for further experiments. PCR products for each sample were mixed in equidensity ratios, and the mixture was purified with Qiagen Gel Extraction Kit (Qiagen, Germany) and quantified with spectrophotometer Nanodrop 2000 (Thermo Scientific, United States). Sequencing libraries were generated using TruSeq® DNA PCR‐Free Sample Preparation Kit (Illumina, USA) following manufacturer's recommendations and index codes were added. The quality of the library was assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. Finally, the library was sequenced on an Illumina HiSeq 2500 platform and 250 bp paired‐end reads were generated. All reads were deposited at the National Center for Biotechnology Information (NCBI) and can be accessed in the Short Read Archive (SRA) under accession number PRJNA492811.
Bioinformatics analysis
Paired‐end reads were merged using FLASH (Fast Length Adjustment of Short reads), a very fast and accurate analysis tool, and the splicing sequences were called raw tags (Tanja and Salzberg, 2011). Quality filtering on the raw tags was performed under specific filtering conditions to obtain the high‐quality clean tags according to the QIIME (V1.9.1) quality controlled process (Caporaso et al., 2010). The tags were compared with the reference database (Unite Database) using UCHIME algorithm to detect chimera sequences, and then the chimera sequences were removed to obtain the effective tags (Edgar, 2013). A category with the highest score from the annotation results under the set conditions was selected and displayed for each OTU using BLAST method. Finally, the thirty faecal samples yielded a total of 2 578 478 sequence reads, and 2 354 182 sequences (91.3%) representing the high‐quality reads were used for the analysis of OTUs. The average length of sequences for each sample was from 182 to 213 base pairs. The quality distribution of all samples ranged between Q20 and Q30, in which most of the samples showed close to Q20. The percent error rate for each sample was below 1% (Table S3).
Operational taxonomic units (OTUs) were picked using the de novo OTU picking protocol with a 97% similarity threshold. Alpha diversity was estimated based on observed species (observed OTUs), ACE, Chao1 (estimated number of OTUs), Goods_coverage and Shannon’s diversity index. Beta diversity was used to evaluate the differences of samples in species complexity. Alpha and beta diversity were determined with QIIME (version 1.9.1) and displayed with R software (version 2.15.3). Weighted and unweighted Unifrac were based on a phylogenetic tree that was generated with an approximate‐maximum‐likelihood model in FastTree (Linux 64‐bit). Principal coordinate analysis (PCoA) was performed and visualize from complex and multidimensional data. The plot was constructed based on the weighted Unifrac distance. Non‐MetricMulti‐Dimensional Scaling (NMDS) was employed to visualize the relationships between samples by two‐dimensional ordination plotting. The differences among intra‐group samples and between groups were compared using multi‐response permutation procedure (MRPP), and with 1000 times of permutations. The separation was tested using R in Anosim.
Statistical analysis
For the data on the concentration of SCFAs and the relative abundance of certain fungal species, descriptive statistics were performed to evaluate whether the data were normal distributed using the statistical software SPSS 20.0 (SPSS Inc., Chicago, IL, USA). For those data with non‐normal distribution, log transformation was then used to assess the availability of one‐way ANOVA test. If the converted data were still not normal distributed (e.g. the concentration of acetate and butyrate), a non‐parametric Kruskal–Wallis (K‐W) test was then performed to compare the difference of the data among groups, followed by pairwise Mann–Whitney U tests which was used to evaluate the difference within the two groups. One‐way ANOVA test and the Fisher’s Least Significant Difference Test (LSD) procedure were used to compare the difference of the normal distributed data among groups. All data were presented as means ± standard deviation (SD) if normally distributed or median ± interquartile range (IQR) if skewed. The confidence interval was 95%, and the differences were considered to be significant when α = 0.05 (P < 0.05).
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals.
Supporting information
Table S1. The α‐diversity indexes of each sample.
Table S2 . The P‐value of Spearman analysis.
Table S3 . The number of assembled reads and parameters for quality control of fecal sample from each pig.
Table S4 . The level of main nutrients in the diets of three breeds of pigs.
Table S5 . The fecal score for each pig.
Fig. S1 . The rank abundance curve for each sample.
Fig. S2 .The unweighted Unifrac clustering of all samples.
Fig. S3 . Non‐metric multi‐dimensional scaling (NMDS) profile of fungal community structure in the fecal samples of all pigs.
Fig. S4 . The Venn diagrams for samples from the three different breeds of pigs.
Fig. S5 . Fungal composition at genus level in the fecal samples from the three different breeds of pigs.
Fig. S6 . Histogram of the linear discriminant analysis (LDA) coupled with effect size (LEfSe) using the default parameters.
Fig. S7 . The network among major fungal genera in the feces of pigs in the three different groups.
Fig. S8 . Thenetwork analysis of correlation among the major fungal genera in CH pigs.
Fig. S9 . The network analysis of correlation among the major fungal genera in YS pigs.
Fig. S10 . The network analysis of correlation among the major fungal genera in TB pigs.
Fig. S11 . Map of China and Sichuan Province.
Acknowledgements
We would like to thank Dr. Isaac Cann from University of Illinois at Urbana‐Champaign and Dr. Weihuan Fang from Zhejiang University for their help in language and writing.
Microbial Biotechnology (2020) 13(2), 509–521
Funding Information
We would like to acknowledge the following funding sources: The work presented in this manuscript was supported by the National Natural Science Foundation of China (NSFC, grant number 31872369, 31672436 and 31730091).
Data availability statement
Sequences supporting the results of this article are available in the GenBank Database (https://www.ncbi.nlm.nih.gov/genbank/) under the accession number: PRJNA492811.
References
- AOAC International (1990) Official Methods of Analysis of AOAC International, Vol. II, 16th. [Google Scholar]
- Arumugam, M. , Raes, J. , Pelletier, E. , Le, P.D. , Yamada, T. , Mende, D.R. , et al (2011) Enterotypes of the human gut microbiome. Nature 473: 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auchtung, T.A. , Fofanova, T.Y. , Stewart, C.J. , Nash, A.K. , Wong, M.C. , Gesell, J.R. , et al (2018) Investigating colonization of the healthy adult gastrointestinal tract by fungi. Msphere 3: e00092‐00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, A.K. , Kelly, S.A. , Legge, R. , Ma, F. , Low, S.J. , Kim, J. , et al (2010) Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA 107: 18933–18938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges, F.M. , De, T.P. , Sarmiento, M. , De, M.O. , Pereira, M. , Toledo, I.V. , et al (2018) Fungal diversity of human gut microbiota among eutrophic, overweight, and obese individuals based on aerobic culture‐dependent approach. Curr Microbiol 75: 726–735. [DOI] [PubMed] [Google Scholar]
- Bresinsky, A. , and Schötz, A. (2006) Behaviour in cultures and habitat requirements of species within the genera Loreleia and Rickenella (Agaricales). Acta Mycologica 41: 189–208. [Google Scholar]
- Caporaso, J.G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F.D. , Costello, E.K. , et al (2010) QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerfbensussan, N. , and Gaboriaurouthiau, V. (2010) The immune system and the gut microbiota: friends or foes? Nat Rev Immunol 10: 735–744. [DOI] [PubMed] [Google Scholar]
- Colhoun, J. (2008) Dictionary of the Fungi. 106: 507–508. [Google Scholar]
- Diao, H. , Yan, H.L. , Xiao, Y. , Yu, B. , Yu, J. , He, J. , et al (2016) Intestinal microbiota could transfer host Gut characteristics from pigs to mice. BMC Microbiol 16: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10: 996–998. [DOI] [PubMed] [Google Scholar]
- Filippo, C.D. , Cavalieri, D. , Paola, M.D. , Ramazzotti, M. , Poullet, J.B. , Massart, S. , et al (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA 107: 14691–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin, M.A. , Mathew, A.G. , Vickers, J.R. , and Clift, R.A. (2002) Characterization of microbial populations and volatile fatty acid concentrations in the jejunum, ileum, and cecum of pigs weaned at 17 vs 24 days of age. J Anim Sci 80: 2904–2910. [DOI] [PubMed] [Google Scholar]
- Frey‐klett, P. , Burlinson, P. , Deveau, A. , Barret, M. , Tarkka, M. , and Sarniguet, A. (2011) Bacterial‐fungal interactions: hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol Mol Biol Rev 75: 583–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallen‐Adams, H.E. , and Suhr, M.J. (2016) Fungi in the healthy human gastrointestinal tract. Virulence 8: 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallen‐Adams, H.E. , Kachman, S.D. , Kim, J. , Legge, R.M. , and Martínez, I. (2015) Fungi inhabiting the healthy human gastrointestinal tract: a diverse and dynamic community. Fungal Ecol 15: 9–17. [Google Scholar]
- Hamer, H.M. , Jonkers, D. , Venema, K. , Vanhoutvin, S. , Troost, F.J. , and Brummer, R.J. (2010) Review article: the role of butyrate on colonic function. Aliment Pharm Ther 27: 104–119. [DOI] [PubMed] [Google Scholar]
- Heisel, T. , Montassier, E. , Johnson, A. , Al‐Ghalith, G. , Lin, Y.W. , Wei, L.N. , et al (2017) High‐fat diet changes fungal microbiomes and interkingdom relationships in the murine gut. Msphere 2: e00351‐00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, C. , Dollive, S. , Grunberg, S. , Chen, J. , Li, H. , Wu, G.D. , et al (2013) Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS ONE 8: e66019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Nie, Y. , Chen, J. , Zhang, Y. , Wang, Z. , Fan, Q. , et al (2016) Gradual changes of gut microbiota in weaned miniature piglets. Front Microbiol 7: 1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, R. , Ji, J. , Yang, X. , Lv, Y. , Zhang, M. , and Ge, Y. , et al. (2016) Investigation on host susceptibility of Tibetan pig to infection of porcine reproductive and respiratory syndrome virus through viral challenge study. Vet Microbiol 183: 62–68. [DOI] [PubMed] [Google Scholar]
- Koh, A. Y. (2013) Gastrointestinal colonization of fungi. Curr Fungal Infec Rep 7: 144–151. [Google Scholar]
- Leonel, A.J. , and Alvarez‐Leite, J. I. (2012) Butyrate: implications for intestinal function. Curr Opin Clin Nutr Metab Care 15: 474–479. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Li, X. , Sun, W.K. , Cheng, C. , Chen, Y.H. , Zeng, K. , et al (2016) Comparison of liver microRNA transcriptomes of Tibetan and Yorkshire pigs by deep sequencing. Gene 577: 244–250. [DOI] [PubMed] [Google Scholar]
- Li, J. , Chen, D. , Yu, B. , He, J. , Zheng, P. , Mao, X. , et al (2017) Fungi in gastrointestinal tracts of human and mice: from community to functions. Microb Ecol 75: 821–829. [DOI] [PubMed] [Google Scholar]
- Luo, Y.H. , Yong, S. , Wright, A.‐D.G. , Zhang, L.L. , Hauke, S. , and Zhu, W.Y. (2012) Lean breed landrace pigs harbor fecal methanogens at higher diversity and density than obese breed Erhualian pigs. Archaea 2012: 605289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejewska, D. , Skonieczna‐Zydecka, K. , Lukomska, A. , Gutowska, I. , Dec, K. , Kupnicka, P. , et al (2018) The short chain fatty acids and lipopolysaccharides status in Sprague‐Dawley rats fed with high‐fat and high‐cholesterol diet. J Physiol Pharmacol 69: 205–210. [DOI] [PubMed] [Google Scholar]
- Mayu, K. , Sae, H. , Takero, H. , Atsuhiko, I. , and Ikuo, K. (2015) Dietary gut microbial metabolites, short‐chain fatty acids, and host metabolic regulation. Nutrients 7: 2839–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcorist, A.L. , Miller, R.B. , Bird, A.R. , Keogh, J.B. , Noakes, M. , Topping, D.L. , et al (2011) Fecal butyrate levels vary widely among individuals but are usually increased by a diet high in resistant starch. J Nutr 141: 883–889. [DOI] [PubMed] [Google Scholar]
- Oro, L. , Ciani, M. , and Comitini, F. (2014) Antimicrobial activity of Metschnikowia pulcherrima on wine yeasts. J Appl Microbiol 116: 1209–1217. [DOI] [PubMed] [Google Scholar]
- Pajarillo, E.A. , Chae, J.P. , Balolong, M.P. , Kim, H.B. , Seo, K.S. , and Kang, D.K. (2014) Pyrosequencing‐based analysis of fecal microbial communities in three purebred pig lines. J Microbiol 52: 646–651. [DOI] [PubMed] [Google Scholar]
- Parfrey, L.W. , Walters, W.A. , Lauber, C.L. , Clemente, J.C. , Berglyons, D. , Teiling, C. , et al (2014) Communities of microbial eukaryotes in the mammalian gut within the context of environmental eukaryotic diversity. Front Microbiol 5: 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petra, L. , and Flint, H.J. (2009) Diversity, metabolism and microbial ecology of butyrate‐producing bacteria from the human large intestine. Fems Microbiol Lett 294: 1–8. [DOI] [PubMed] [Google Scholar]
- Qin, J. , Li, R. , Raes, J. , Arumugam, M. , Burgdorf, K.S. , Manichanh, C. , et al (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, X. , Zhang, F. , Yang, X. , Wu, N. , Jiang, W. , Li, X. , et al (2015) Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium‐induced colitis. Sci Rep 5: 10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redhead, S.A. , Moncalvo, J.M. , Vilgalys, R. , and Lutzoni, F. (2002) Phylogeny of agarics: partial systematics solutions for bryophilous omphalinoid agarics outside of the Agaricales (Euagarics). Mycotaxon 82: 151–168. [Google Scholar]
- Scupham, A.J. , Presley, L.L. , Wei, B. , Bent, E. , Griffith, N. , Mcpherson, M. , et al (2006) Abundant and diverse fungal microbiota in the murine intestine. Appl Environ Microb 72: 793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sian, K.C. , Rokiah, I. , Zhenli, K. , Mei, Y.S. , Koon, Y.S. , Ling, C.C. , et al (2016) Isolation and characterization of an atypical Metschnikowiasp. strain from the skin scraping of a dermatitis patient. PLoS ONE 11: e0156119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisti, M. , and Savini, V. (2014) Antifungal properties of the human Metschnikowia strain IHEM 25107. Folia Microbiol 59: 263–266. [DOI] [PubMed] [Google Scholar]
- Sokol, H. , Leducq, V. , Aschard, H. , Pham, H.P. , Jegou, S. , Landman, C. , et al (2016) Fungal microbiota dysbiosis in IBD. Gut 66: 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudipta, G. , Mehla, R.K. , Sirohi, S.K. , and Biswajit, R. (2010) The effect of dietary garlic supplementation on body weight gain, feed intake, feed conversion efficiency, faecal score, faecal coliform count and feeding cost in crossbred dairy calves. Trop Anim Health Pro 42: 961–968. [DOI] [PubMed] [Google Scholar]
- Suhr, M.J. , and Hallen‐Adams, H.E. (2015) The human gut mycobiome: pitfalls and potentials−a mycologist's perspective. Mycologia 107: 1057–1073. [DOI] [PubMed] [Google Scholar]
- Summers, K.L. , Frey, J.F. , Ramsay, T.G. , and Arfken, A.M. (2019) The piglet mycobiome during the weaning transition: a pilot study. J Anim Sci 97: 2855–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanja, M. , and Salzberg, S.L. (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27: 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauktornisielo, S.M. , Vieira, J.M. , Cecília, M. , Carneiro, V.S. , and Govone, J.S. (2007) Fatty acid production by four strains of Mucor hiemalis grown in plant oil and soluble carbohydrates. Afr J Biotechnol 6: 1840–1847. [Google Scholar]
- Tremaroli, V. , and Bäckhed, F. (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489: 242–249. [DOI] [PubMed] [Google Scholar]
- Yang, S.L. , Wang, Z.G. , Liu, B. , Zhang, G.X. , Zhao, S.H. , Yu, M. , et al (2003) Genetic variation and relationships of eighteen Chinese indigenous pig breeds. Genet Sel Evol 35: 657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S. , Zhang, H. , Mao, H. , Yan, D. , Lu, S. , Lian, L. , et al (2011) The local origin of the tibetan pig and additional insights into the origin of Asian Pigs. PLoS ONE 6: e28215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W. , Meng, F. , Peng, J. , Han, P. , Fang, F. , Ma, L. , et al (2014) Isolation and identification of a cellulolytic bacterium from the Tibetan pig's intestine and investigation of its cellulase production. Electron J Biotechnol 17: 262–267. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The α‐diversity indexes of each sample.
Table S2 . The P‐value of Spearman analysis.
Table S3 . The number of assembled reads and parameters for quality control of fecal sample from each pig.
Table S4 . The level of main nutrients in the diets of three breeds of pigs.
Table S5 . The fecal score for each pig.
Fig. S1 . The rank abundance curve for each sample.
Fig. S2 .The unweighted Unifrac clustering of all samples.
Fig. S3 . Non‐metric multi‐dimensional scaling (NMDS) profile of fungal community structure in the fecal samples of all pigs.
Fig. S4 . The Venn diagrams for samples from the three different breeds of pigs.
Fig. S5 . Fungal composition at genus level in the fecal samples from the three different breeds of pigs.
Fig. S6 . Histogram of the linear discriminant analysis (LDA) coupled with effect size (LEfSe) using the default parameters.
Fig. S7 . The network among major fungal genera in the feces of pigs in the three different groups.
Fig. S8 . Thenetwork analysis of correlation among the major fungal genera in CH pigs.
Fig. S9 . The network analysis of correlation among the major fungal genera in YS pigs.
Fig. S10 . The network analysis of correlation among the major fungal genera in TB pigs.
Fig. S11 . Map of China and Sichuan Province.
Data Availability Statement
Sequences supporting the results of this article are available in the GenBank Database (https://www.ncbi.nlm.nih.gov/genbank/) under the accession number: PRJNA492811.