

Summary
Hungateiclostridium thermocellum DSM 1313 has biotechnological potential as a whole‐cell biocatalyst for ethanol production using lignocellulosic renewable sources. The full exploitation of H. thermocellum has been hampered due to the lack of simple and high‐throughput genome engineering tools. Recently in our research group, a thermophilic bacterial CRISPR–Cas9‐based system has been developed as a transcriptional suppression tool for regulation of gene expression. We applied ThermoCas9‐based CRISPR interference (CRISPRi) to repress the H. thermocellum central metabolic lactate dehydrogenase (ldh) and phosphotransacetylase (pta) genes. The effects of repression on target genes were studied based on transcriptional expression and product formation. Single‐guide RNA (sgRNA) under the control of native intergenic 16S/23S rRNA promoter from H. thermocellum directing the ThermodCas9 to the promoter region of both pta and ldh silencing transformants reduced expression up to 67% and 62% respectively. This resulted in 24% and 17% decrease in lactate and acetate production, correspondingly. Hence, the CRISPRi approach for H. thermocellum to downregulate metabolic genes can be used for remodelling of metabolic pathways without the requisite for genome engineering. These data established for the first time the feasibility of employing CRISPRi‐mediated gene repression of metabolic genes in H. thermocellum DSM 1313.
This work describes the development of CRISPRi for the thermophilic Hungateiclostridium thermocellum and its application to silence two genes in central metabolism. These data established for the first time the feasibility of employing CRISPRi‐mediated gene repression in H. thermocellum DSM 1313.
![]()
Introduction
Microbial conversion of lignocellulosic renewable feedstock to fuels, chemicals and other bio‐based products significantly reduces the dependence on non‐sustainable fossil energy‐based processes. This contributes to the transition towards a bio‐based economy. Non‐model production microbes with native ability of solubilizing lignocellulose have been improved for robust industrial production processes (Akinosho et al., 2014; Lynd et al., 2016; Kumar and Sharma, 2017). The anaerobic thermophilic bacterium Hungateiclostridium thermocellum is proficient at solubilizing cellulose as it possesses a hydrolytic multienzyme complex called the cellulosome. This enzyme complex solubilizes cellulose to cellodextrins that are further metabolized to produce ethanol, acetate, l‐lactate, formate, hydrogen gas and carbon dioxide (McBee, 1954). Cellulose solubilization has the potential to improve product formation but not cost‐effective for the conversion of lignocellulose to ethanol. Consolidated bioprocessing (CBP) combines enzyme production, cellulose hydrolysis and fermentation into a single step, offering an economical advantage to the current multistep process (Jiang et al., 2017). In CBP no exogenous hydrolytic enzymes are added, thereby reducing the costs. Moreover, pretreatment, hydrolysis and fermentation steps are combined in one vessel, reducing the number of unit operations (Bhalla et al., 2013). Besides, fermentation efficiency is improved as it also lowers the cost of the production of biofuels and biochemicals. To this end, the efficient genetic engineering of H. thermocellum would render it a highly valuable ‘microbial cell factory’ for biotechnological exploitation.
Metabolic engineering of H. thermocellum is necessary to improve the yield of ethanol or alternative preferred products from mixed‐acid fermentation. However, the current genetic toolbox for H. thermocellum is still very limited. A plasmid‐based homologous recombination gene disruption method, including hpt and pyrF‐based counter selection, has been developed (Tripathi et al., 2010). Using hpt and pyrF as a counter‐selection marker few genes such as pta, ldh, adhE, cipA, cel48S, spooA and recA were deleted to interrogate the genetic base for specific phenotypes and to create modified strains for different functions (Olson et al., 2010; Argyros et al., 2011; Brown et al., 2011; Olson et al., 2013; van der Veen et al., 2013; Groom et al., 2018; Lo et al., 2019). This tool has not been used extensively, as it is a laborious and time‐consuming process. Subsequently, a Targetron system derived from the thermophilic cyanobacterium Synechococcus elongatus has been developed into a genome engineering tool for gene disruption in thermophiles, specifically H. thermocellum (Mohr et al., 2013). Targetron is a genome editing technology derived from mobile group II introns, which efficiently creates site‐specific integrations for gene knockout and knock‐in clostridia. The main limitations for this system include the inability to generate clean knockouts, reduced target specificity due to lack of intron‐encoded protein (IEP) recognition sites for small genes, the variation in the intron‐integration efficiency and the presence of ectopic intron insertion events (Song et al., 2015; Bruder et al., 2016; Joseph et al., 2018).
The development of simple, and preferably high‐throughput, genome engineering tools is crucial for efficient metabolic engineering, as well as for a full exploitation of this thermophile. The CRISPR–Cas9 technology has provided a precise technique for genome editing, paving the way for a new era in molecular biology. In addition, CRISPRi has commenced as an effective technique for gene downregulation and can be used to modulate gene expression. This approach uses a nuclease‐deficient Cas9 (dCas9) in conjunction with spacer region of a single‐guide RNA to repress specific genes targeting at the promoter and start of the gene. CRISPRi systems have been established in mesophilic clostridial species, including Clostridium acetobutylicum and Clostridium beijerinckii (Li et al., 2016; Wang et al., 2016; Wen et al., 2017). Recently, a thermophilic Cas9, ThermoCas9, has been isolated from the thermophilic bacterium Geobacillus thermodenitrificans T12 (Daas et al., 2018). It is a type‐IIC Cas9 that is active over a broad temperature range. A dCas9 variant has been generated and used for CRISPRi silencing of the lactate dehydrogenase activity in the thermophilic Bacillus smithii (Mougiakos et al., 2017).
High repression of gene expression has been reported when the dCas9‐sgRNA ribonucleoprotein complex targets the non‐coding DNA strand (5’–3’) of a gene, specifically close to the ATG start codon, whereas the effect is less pronounced if the coding strand (3’–5’) is targeted. Furthermore, effective gene silencing is observed when targeting the promoter region, with the strongest effect at the −35 sequence, being independent of the DNA strand (Qi et al., 2013). It has been hypothesized that the inhibition of the transcription is due to a physical collision between the RNA polymerase and the dCas9‐sgRNA complex. During elongation, the RNA polymerase encounters the dCas9‐sgRNA, which is bound to the non‐template DNA, pausing the transcription elongation. Alternatively, if dCas9‐sgRNA complex is bound to the template strand, slight repressive effect can be observed as the RNA polymerase could still read through the complex. The sgRNA faces the RNA polymerase, which could unzip the complex by helicase activity upon targeting the template strand (Qi et al., 2013). Moreover, gene knockdowns are less likely to be lethal and frequently permit analysing surviving mutants where drastic knockout techniques fail (Qi et al., 2013; Pyne et al., 2014; Peters et al., 2016).
In this study, ThermodCas9 was adapted for use in H. thermocellum as a tool to enable silencing of gene expression. The CRISPRi system was implemented to effectively knockdown metabolic genes such as lactate dehydrogenase (ldh) and phosphotransacetylase (pta) via appropriate sgRNA design. Selective repression of the ldh and pta genes decreased lactate and acetate production respectively. Hence, demonstrating the feasibility of employing CRISPRi for the metabolic engineering of H. thermocellum and production of bio‐derived chemicals.
Results
Selection of promoters
To establish the ThermodCas9‐based CRISPRi tool in H. thermocellum, functional promoters are required for efficient expression of the ThermodCas9 and the sgRNA. The pThermoCas9i plasmid (Mougiakos et al., 2017) encodes the catalytically inactive variant of ThermoCas9 (ThermodCas9) and the non‐targeting sgRNA‐expressing module under the control of the constitutive xylL promoter from B. smithii and pta promoter from Bacillus coagulans respectively. This plasmid was introduced in H. thermocellum DSM 1313 via electroporation as described by Olson et al. (Olson and Lynd, 2012). To validate expression of both genes, RNA was isolated from H. thermocellum transformants containing the non‐targeting pThermoCas9i plasmid that were grown in CP medium. RT‐PCR on cDNA showed that the ThermodCas9 gene was expressed, but the sgRNA was not expressed (Fig. 1). From this, we concluded that the xylL promoter from B. smithii is functional in H. thermocellum, but the pta promoter from B. coagulans is not.
Figure 1.
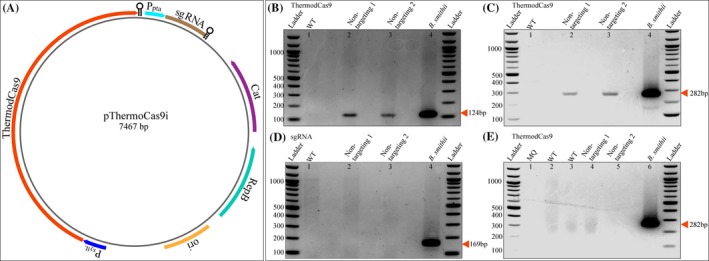
RT‐PCR of viable H. thermocellum transformants, containing the pThermoCas9i plasmid. A. Schematic illustration of plasmid pThermoCas9i_NT (Mougiakos et al., 2017). Thermodcas9 gene under control of the B. smithii xylL promoter; sgRNA‐expressing module under control of the B. coagulans pta promoter; and pNW33n backbone. B, C and D. Non‐targeting 1 and 2, independent pThermoCas9i_NT (Mougiakos et al., 2017) transformants of H. thermocellum (lanes 2 and 3) showing the RT‐PCR products of 124 bp (B) and 282 bp (C) from thermodcas9 cDNA and the absence of the RT‐PCR product of 169 bp (D) from the sgRNA; WT, RT‐PCR with H. thermocellum DSM 1313 wild‐type cDNA; B. smithii, RT‐PCR with B. smithii harbouring pThermoCas9i_NT (Mougiakos et al., 2017) cDNA. E) MQ, RT‐PCR with Milli‐Q water (lane 1); WT, RT‐PCR with H. thermocellum DSM 1313 wild‐type DNA (lane 2); WT, RT‐PCR with H. thermocellum DSM 1313 wild‐type RNA (lane 3); Non‐targeting 1 and 2, RT‐PCR with H. thermocellum transformants 1 and 2 RNA, respectively, (lane 4 and 5); and B. smithii, RT‐PCR with B. smithii harbouring pThermoCas9i_NT (Mougiakos et al., 2017) cDNA (lane 6).
Promoter screening for the establishment of CRISPRi system in H . thermocellum DSM 1313
Based on the previous results, it was necessary to replace the B. coagulans pta promoter with an alternative one to efficiently express the sgRNA. To develop an efficient system, we decided to assess the use of native promoter of small ribosomal intergenic 16S/23S rRNA from H. thermocellum. Due to the limited number of characterized promoters from this anaerobic thermophile, we selected approximately 250 bp long upstream sequence of the 16S and 23S rRNA genes anticipating the presence of promoters (Appendix S1). Ribosomal promoters are preferred for efficient sgRNA expression (Li et al., 2013; Xu et al., 2014). We replaced the pta promoter of the initial constructed plasmids with the predicted intergenic 16S/23S rRNA promoters and tested sgRNA expression by RT‐PCR (Fig. 2). The intergenic 16S/23S rRNA promoter drove sgRNA gene expression. Therefore, we chose the xylL promoter to drive ThermodCas9 gene expression and the 16S/23S rRNA promoter to drive sgRNA expression.
Figure 2.
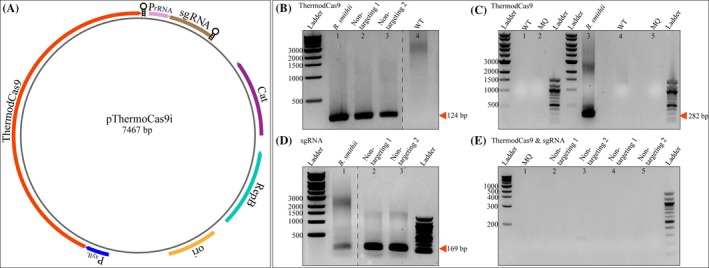
RT‐PCR of viable H. thermocellum transformants, containing the pThermoCas9i plasmid with rRNA promoters. A. Schematic illustration of plasmid pThermoCas9i_NT (Mougiakos et al., 2017). The thermodcas9 gene under control of the B. smithii xylL promoter; sgRNA‐expressing module under control of the native rRNA promoter; and pNW33n backbone. B. Non‐targeting 1 and 2, independent pThermoCas9i_NT (Mougiakos et al., 2017) transformants of H. thermocellum (lanes 2 and 3) showing the RT‐PCR product of 124 bp from thermodcas9 cDNA; WT, RT‐PCR with H. thermocellum DSM 1313 wild‐type cDNA; B. smithii, RT‐PCR with B. smithii harbouring pThermoCas9i_NT (Mougiakos et al., 2017) cDNA. C. WT, RT‐PCR with H. thermocellum DSM 1313 wild‐type DNA (lane 1 and 4); MQ, RT‐PCR with Milli‐Q water (lane 2 and 5); and B. smithii, RT‐PCR with B. smithii harbouring pThermoCas9i_NT cDNA (lane 3). D. Non‐targeting 1 and 2, independent pThermoCas9i_NT (Mougiakos et al., 2017) transformants of H. thermocellum (lanes 2 and 3) showing the RT‐PCR product of 169 bp from the sgRNA. E) RT‐PCR with the RNA from non‐targeting 1 and 2, independent pThermoCas9i_NT (Mougiakos et al., 2017) transformants of H. thermocellum.
Transcriptional repression on lactate dehydrogenase (ldh) and phosphotransacetylase (pta) genes
To investigate the utility of CRISPRi for gene repression in H. thermocellum, we aimed to target the non‐template strand of both lactate dehydrogenase (ldh) and phosphotransacetylase (pta) genes. We constructed pThermoCas9i vectors with the xylL promoter expressing the ThermodCas9 and the intergenic 16S/23S rRNA promoter controlling the sgRNA together with a non‐targeting spacer (pThermoCas9i_NT) or a spacer targeting the non‐template DNA strand overlapping either the promoter region or the start of the gene for both ldh (ldh_P; ldh_S) and pta (pta_P; pta_S) genes respectively. Targeting and non‐targeting plasmids were electroporated to H. thermocellum DSM 1313. A drop of two orders of magnitude in transformation efficiency was observed when using the targeting plasmid (102 CFU µg DNA−1) in comparison with the controls (104 CFU µg DNA−1). All the tested colonies for the targeting plasmid showed a positive band for the expression of the ThermodCas9 and sgRNA. These clones were grown in 50 ml CP medium containing 6 µg ml−1 thiamphenicol (TmR) and cultured to OD600 ~ 0.6 and > 1.0. Five millilitres of cells were sampled for RNA extraction and qRT‐PCR analysis to determine the downregulation of the silencing transformants. qRT‐PCR was performed using gyrA and recA as house‐keeping genes. Eventually, recA was used as a reference gene as it appeared more consistent in expression than gyrA (data not shown).
The qRT‐PCR analysis (Fig. 3) on RNA isolated from exponentially growing cells harvested at an OD600 ~ 0.6 showed 62 % and 58% reduction in ldh gene expression, using the expression in control transformant cells with the pThermoCas9i_NT (NT control) as the reference, when targeting the promoter (ldh_P) and start of the gene region (ldh_S) respectively. When harvested at the early stationary phase at an OD600 > 1.0, the repression effect was still retained 43% for the ldh_P, but the ldh_S transformant no longer showed repression and had the same expression level as the NT control. Similarly, at an OD600 ~ 0.6 for the pta‐targeting transformants the silencing effect was 67% and 62% when targeting the promoter (pta_P) and start of the gene (pta_S) respectively. When the OD600 > 1.0, the expression was 45% and 39% reduced for the pta_P and pta_S transformants respectively. These data indicate that CRISPRi suppressed the gene expression, and the silencing efficacy was high in comparison to the control.
Figure 3.
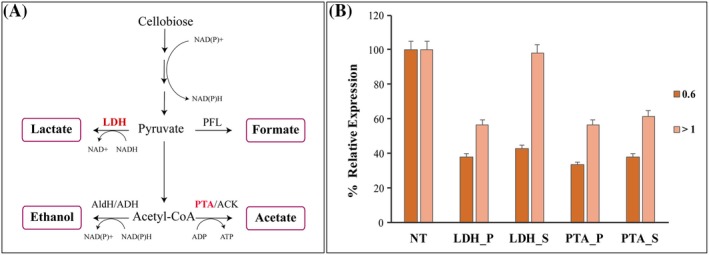
Effective suppression of ldh and pta genes by CRISPRi. A. Metabolic pathway representation of H. thermocellum DSM1313 for conversion of cellobiose to ethanol and major organic acids production. (ACK, acetate kinase; ADH, alcohol dehydrogenase; AldH, aldehyde dehydrogenase; LDH, lactate dehydrogenase; PFL, pyruvate‐formate lyase; and PTA, phosphotransacetylase). B. Relative ldh and pta gene expression was evaluated using qRT‐PCR in comparison with control non‐targeting (NT) at an OD600 – 0.6 and 1.0 respectively; positive silencing transformants for ldh gene referred as ldh_P – targeting the promoter, ldh _S – targeting the start of the gene; positive silencing transformants for pta gene referred as pta_P – targeting the promoter, pta_S – targeting the start of the gene. H. thermocellum DSM 1313 cells were transformed with both non‐targeting pThermoCas9i_NT and targeting pThermoCas9i_ldh/pta plasmids. The TmR colonies of both the non‐targeting and targeting plasmids were transferred to 50 ml CP (5 g l−1 yeast extract) medium containing 6 µg ml−1 TmR and cultured to OD600 ~ 0.6 and > 1.0, and 5 ml cells were sampled for qRT‐PCR analysis with technical triplicates. The expression levels were normalized to those in the NT control. Data represent the mean values of three biological replicates and the standard deviation (SD). The level of significance of the differences when results were compared was estimated by means of analysis of variance (ANOVA), with α = 0.05.
In addition, the impact of the silencing on the other respective genes involved in product formation was evaluated. At OD600 ~ 0.6, the ldh gene for pta silencing transformants targeting the promoter (ldh_E1) and start of the gene (ldh_E2) showed enhanced expression respectively compared with the NT control. For the early stationary phase cultures, the ldh_E1 expression was same as NT control but increased for the ldh_E2. Contrarily, the pta gene had reduced expression compared with the NT control at early exponential phase, for both the ldh silencing transformants (pta_E1, targeting the promoter and pta_E2, targeting the start of the gene). Hence, the non‐targeted metabolic pta and ldh genes responded differently to the silencing of ldh and pta transformants respectively (data not shown).
Impact of gene silencing on product formation using HPLC
To evaluate the effect of repression of ldh and pta gene on organic acids production, silencing transformants were cultured to an OD600 ~ 0.5–1.5, in anaerobic bottles containing 50 ml CP medium (1 g l−1 yeast extract) for 2 days. Samples were taken at two time points for HPLC analysis for the two transformants in comparison with the NT control. Moreover, the fermentation profile was observed to see the effect of knockdown of ldh and pta genes on other organic acids and ethanol production.
It was clearly noted that repression of ldh and pta genes at the promoter region led to a 24% and 17% reduction on lactate and acetate production in comparison with NT cells respectively (Fig. 4). At time point T1 (OD600 ~ 0.5–0.7), the acetate production in the pta silencing transformants (pta_P and pta_S) was suppressed. In contrast, the lactate production for these transformants was increased around 25% to the NT control. For the ldh silencing transformants (ldh_P and ldh_S), lactate was reduced drastically with slender decrease in acetate as well. Other products for both ldh and pta silencing transformants such as formate had slightly better yield in all silencing transformants whereas ethanol did not show any significant differences from the control. When the OD600 > 1.0 (T2), the yield (mol of lactate/mol of cellobiose consumed) for ldh_P was 0.36 and ldh_S was 0.55 whereas that of NT was 0.48. Thus, lactate production was still low for the ldh_P transformant. Contrarily, both the pta silencing transformants had similar levels of acetate, formate and ethanol production compared with the NT control (Appendix S2, Fig. S1).
Figure 4.
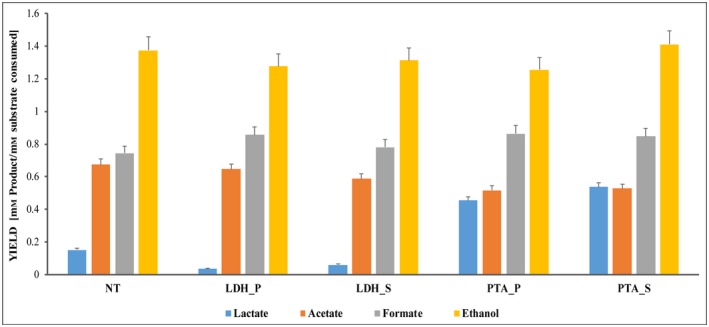
Effects of CRISPRi‐mediated suppression on lactate and acetate production and impact on the fermentation profile. For lactate and acetate production, positive transformants referred as ldh_P, ldh _S, pta_P, pta_S and control ldh_non‐targeting (NT) were analysed in HPLC at OD600 – 0.5–0.7 (T1). The silencing transformants for ldh gene referred as ldh_P – targeting the promoter, ldh _S – targeting the start of the gene; positive silencing transformants for pta gene referred as pta_P – targeting the promoter, pta_S – targeting the start of the gene. H. thermocellum DSM 1313 cells were transformed with both non‐targeting pThermoCas9i_NT and targeting pThermoCas9i_ldh and pta plasmids. The TmR colonies of both the non‐targeting and targeting plasmids were transferred to 50 ml CP medium (1 g l−1 yeast extract) containing 6 µg ml−1 TmR and cultured to OD600 ~ 0.5–0.7 (T1) and > 1.0 (T2), and 1 ml cells were sampled for 2 days for HPLC analysis. Data represent the mean values of three biological replicates and the standard deviation (SD). The level of significance of the differences when results were compared was estimated by means of analysis of variance (ANOVA), with α = 0.05.
In summary, HPLC showed decrease in acetate and lactate production at an exponential phase but in the late stationary phase the fermentation products come to same yields as the NT cells. This shows that despite that ThermodCas9 can silence specific metabolic genes, H. thermocellum can escape the silencing upon prolonged incubation.
Discussion
CRISPRi offers reduction in gene expression and can be used to interrogate gene function and modulate cellular activities. Recent studies have explored CRISPRi to control the metabolic pathways in E. coli and other non‐model organisms for improved production of numerous biotechnological products (Mougiakos et al., 2018). In this study, we applied the CRISPR‐ThermoCas9‐based transcriptional regulation tool for the first time an effective system in H. thermocellum DSM 1313. The existing methods for genome editing and transcriptional regulation purposes are laborious, regularly inefficient and technically complex in the anaerobic bacteria, which are known to be challenging for genetic manipulation. The recent discovery of a thermostable Cas9 coupled to its transformation into a powerful genetic tool has opened new possibilities for genome manipulation of the thermophilic bacteria (Mougiakos et al., 2017). The dCas9‐based CRISPRi allows the transcriptional regulation of the gene of interest without completely disrupting its function, resulting in less pleiotropic effects than gene knockouts (Dominguez et al., 2016; Peters et al., 2016).
To evaluate the functionality of the CRISPRi tool in thermophilic clostridia, we targeted two genes of the central metabolism; the lactate dehydrogenase (ldh) and phosphotransacetylase (pta) in H. thermocellum DSM 1313. It has been proven that the deletions of these genes are not detrimental for its survival and allows the increase in ethanol production (Argyros et al., 2011; Biswas et al., 2014; Papanek et al., 2015). We constructed two ThermodCas9 expressing vectors, with sgRNA genes targeting the promoter and start of the gene plus a control plasmid with a non‐targeting sgRNA. Based on previous studies, the target sites were located in the non‐template strand, near the promoter region and start of both ldh and pta genes. According to literature, the highest transcriptional downregulation effect was expected to occur where ThermodCas9 targets the promoter or beginning of the coding sequence, the lowest repression in the transformants where the middle of the gene was targeted (Larson et al., 2013; Qi et al., 2013; Yao et al., 2016).
The first steps towards the adaptation of ThermodCas9‐based CRISPRi tool showed the xylL promoter from Bacillus smithii allows the expression of ThermodCas9 protein in H. thermocellum. However, the pta promoter from Bacillus coagulans failed to transcribe the gene encoding the sgRNA. Bacilli and clostridia are closely related bacteria that belong to the phylum Firmicutes (Talukdar et al., 2015). The close‐relatedness among these two classes might explain the functionality of the xylL promoter from Bacillus smithii in H. thermocellum, specifically because both are thermophilic strains. Bacillus coagulans is also a facultative anaerobic thermophilic bacterium, but the reason for the non‐functioning of the pta promoter in H. thermocellum remains unclear.
To develop an efficient CRISPRi tool, we successfully managed to get the activity of putative native intergenic 16S/23S rRNA promoter for the expression of the chimeric sgRNA. The promoter region within 16S/23S intergenic spacer sequence has homology to the prokaryotic consensus promoter structure. In E. coli, it has been shown that this promoter has a role in the transcription of ribosomal RNAs (Mankin et al., 1987; Zacharias and Wagner, 1989). Now, we show that this is also likely to be the case for H. thermocellum.
This study shows the first attempt to exploit the CRISPRi tool in thermophilic clostridia to modulate gene expression using the central metabolism phosphotransacetylase (pta) and lactate dehydrogenase (ldh) genes as examples. The results showed downregulation levels of 67% and 62%, respectively, when the promoter and the start of pta gene were targeted. Consequently, the organic acids production changed in comparison with NT control: acetate production decreased between 17% and 27%; formate production increased between 5% and 12%; and ethanol had no significant change. As studied before, the pta knockout mutant generated using the Targetron system had 13.5% and 81.5% decreased acetate and lactate production, respectively, and 42% improvement in ethanol production (Mohr et al., 2013). Contrarily, we observed 30% of increased lactate production and this correlated well with the upsurge in ldh gene expression for pta silencing transformants. Argyros et al. (2011) have shown similar impact of improved lactate titre with pta knockout. In addition, no significant increase in ethanol production was noted in comparison with the NT control. The plausible explanation for this could be despite of the silencing, the residual expression still resulted in sufficient enzyme activity for the cell to still produce acetate and ATP instead of ethanol and NAD(P). Besides, further genetic engineering of pyruvate metabolism is required to increase ethanol production as discussed in previous studies (Dash et al., 2017; Tian et al., 2017).
The ldh silencing transformants ldh_P and ldh_S similarly had 62% and 58% downregulation levels with decrease in lactate production in comparison with NT control. Mohr et al. (2013) have shown with the ldh knockout mutant a 4% reduction in lactate production in comparison with wild type. In contrast, we showed 24% decrease in lactate production and 15% increase in formate; however, no increase in acetate or ethanol was detected. This was also noted with minor reduction in pta gene expression at exponential phase for both ldh_P and ldh_S. In another study, the ldh mutants of H. thermocellum were characterized, acetate titre was also not altered. However, no prominent difference was observed in gene expression and acetate production in the stationary phase. Thus, signifying lactate dehydrogenase deletion does not affect the flux through the pyruvate node if ethanol is produced (van der Veen et al., 2013). Moreover, for ethanol production, there were no significant differences observed when compared to the NT control. This result was comparable to earlier reports of H. thermocellum pta mutant (Tripathi et al., 2010). Future work in developing a H. thermocellum strain could involve pta and ldh multiplex silencing or knockout using dead or active thermophilic Cas9, respectively, to study if there is any impact on product formation. Nevertheless, the genetic system developed here demonstrates a step forward towards manipulating H. thermocellum strain with slight effects on other genes in the metabolic pathways.
The main challenge for applying thermophilic CRISPR‐Cas system seems to be its robustness. In previous studies, this system has been successfully applied in B. smithii to reduce the highly expressed ldhL gene expression for about 90% resulting in 40% reduction in lactate production (Mougiakos et al., 2017). Similarly, we have repressive effect for both pta and ldh genes with reduction in acetate and lactate, respectively. However, at stationary phase the cells overcome the repressive effect with less impact on product formation. This CRISPRi loss‐of‐function effect has been shown in several growth studies where essential genes were targeted (Zhao et al., 2016; Liu et al., 2017), correlating it with the presence of suppressor mutations in the dead Cas9 coding sequence at stationary phase. Even a single mutation of the first seven nucleotides intensely decreased repression, suggesting the importance of the ‘seed region’ sequence for binding of both the type I and type II CRISPR systems (Jinek et al., 2012; Qi et al., 2013). Therefore, it is likely that the importance of these metabolic genes pose a strong selection pressure for the cells that have overcome this burden by suppressor mutations for survival affecting the repression at stationary phase. Further studies are required to demonstrate that this is actually the case.
To date, H. thermocellum strains have been genetically engineered, improving the production of ethanol using cellulose as a carbohydrate source. Two main strategies have been followed. On one hand, competing pathways for carbon flux are disrupted (Argyros et al., 2011; Biswas et al., 2014; Papanek et al., 2015; Kannuchamy et al., 2016). In another approach, competing pathways for electron flux are eliminated (Biswas et al., 2015; Rydzak et al., 2015; Lo et al., 2017). This is the first time a silencing tool has been applied to H. thermocellum DSM1313 to transcriptionally regulate the expression of genes involved in central metabolism. In this case, qRT‐PCR has shown clear effect on the repression of both the genes in the exponential growth phase which leads to efficient redistribution of the organic acids and ethanol in H. thermocellum DSM1313.
Experimental procedures
Bacterial strains and growth conditions
All bacterial strains used in this study are listed in Table S1, Supporting information. High efficiency NEB (New England Biolabs, CFU ~ 109 μg pUC19−1) chemically competent E. coli DH5α was used for cloning purposes. E. coli Acella (AcellaTM Chemically Competent cells, Edge Bio) was used for plasmid propagation and isolation when H. thermocellum DSM 1313 was transformed to ensure the transfer of Dam‐methylated plasmids. E. coli strains were cultured in Lysogeny‐Broth (LB) medium (1% tryptone, 1% NaCl, 0.5% yeast extract) at 37ºC and 200 rpm. When appropriate the medium was supplemented with 20 μg ml−1 chloramphenicol.
Hungateiclostridium thermocellum DSM 1313 was obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ). H. thermocellum DSM 1313 wild type and transformants were grown anaerobically in CFTÜD adapted from Olson and Lynd (2012) (0.13% ammonium sulphate, 0.15% potassium phosphate monobasic, 0.013% calcium chloride dehydrated, 0.05% l‐cysteine hydrochloride, 1.15% MOPS sodium salt, 0.0001% ferrous sulfate heptahydrate, 0.26% magnesium chloride hexahydrate, 0.45% BD BBL™ Yeast Extract, 0.5% Na‐resazurin solution 0.1% w/v and 0.3% sodium citrate tribasic dihydrate) broth and agar media at 50–60ºC range (Olson and Lynd, 2012). For plasmid selection after electrotransformation, 6 μg ml−1 thiamphenicol was added. For RNA isolation and HPLC analysis, the silencing transformants were grown in CP medium adapted from Plugge, 2005 (0.408 g l−1 potassium dihydrogen phosphate, 0.534 g l−1 sodium phosphate dibasic dihydrate, 0.3 g l−1 ammonium chloride, 0.3 g l−1 sodium chloride, 0.1 g l−1 magnesium chloride hexahydrate, 0.11 g l−1 calcium chloride dihydrate, 4.0 g l−1 sodium bicarbonate, 1.0 g l−1 l‐cysteine and 5.0 g l−1 (RNA isolation) or 1.0 g l−1 (HPLC) yeast extract (BD Bacto), 0.5 mg resazurin, 1 ml vitamin solution, 1 ml trace elements solution I and 1 ml trace elements solution II. The final volume of 50 ml medium was dispensed in serum bottles under 80:20 N2/CO2 atmosphere with ∼70 kPa overpressure and then autoclaved. A stock solution comprising sodium bicarbonate and l‐cysteine was autoclaved separately and added later. Likewise, vitamin stock solution was added to calcium chloride dihydrate solution after it was autoclaved. Cellobiose as a carbon source was also autoclaved separately and added later to a final concentration of 5.0 g l−1 (Plugge, 2005). The vitamin solution, 1000 × concentrated, contained per litre 20 mg biotin, 20 mg folic acid, 100 mg pyridoxine‐HCl, 50 mg thiamine‐HCl, 50 mg riboflavin, 50 mg nicotinic acid, 50 mg Ca‐d‐pantothenate, 1 mg vitamin B12, 50 mg 4‐aminobenzoid acid and 50 mg lipoic acid.Trace elements solution I, 1000 × concentrated, contained per litre 50 mM HCl, 61.8 mg H3BO4, 99.0 mg MnCl2·4H2O, 1.49 g FeCl2·4H2O, 119 mg CoCl2·6H2O, 23.8 mg NiCl2·6H2O, 68.2 mg ZnCl2 and 17.0 mg CuCl2·2H2O. Trace elements solution II, which was 1000 × concentrated, contained per litre 10 mM NaOH, 17.3 mg Na2SeO3, 33.0 mg Na2WO4·2H2O and 24.2 mg Na2MoO4·2H2O.
CRISPRi plasmids construction
Both the plasmids and primers used in this study are listed in Table S2, Supporting information. Plasmids were built using fragments with the appropriated overhangs via NEBuilder® HiFi DNA assembly cloning kit according to the manufacturer’s protocol. All PCR reactions for amplification of fragments were performed with the NEB Q5® High‐Fidelity DNA polymerase (M0491). PCR fragments were subjected to 1% w/v agarose gel electrophoresis and isolated using ZymocleanTM Gel DNA Recovery kit. Plasmid pThermoCas9i (Mougiakos et al., 2017) was used as a template for the construction of all the plasmids. Plasmids were designed for the knockdown of the lactate dehydrogenase (ldh: Clo1313_1160) and phosphotransacetylase (pta; Clo1313_1185) in the genome of H. thermocellum DSM 1313. Two different protospacers were selected for each targeted gene and a non‐targeting with random sequences was used as a control (Table S3, Supporting information). They differ on the position within the targeted gene. The target sites are the promoter, start of the gene and also the non‐targeting spacer with random sequences. To block transcription, the non‐template strand of the gene was targeted.Plasmids with alternative promoters to drive the sgRNA expression were based on the previously built ThermodCas9 plasmids by exchanging the B. coagulans pta promoter of the sgRNA expression module with 250 bp of the H. thermocellum intergenic region of the 16S and 23S rRNA genes (Table S1). All plasmids were introduced by heat shock into chemically competent E. coli DH5α cells (Sambrook et al., 1989). Plasmids were isolated from selected single transformants by using the GeneJET Plasmid Miniprep Kit® (Thermo Scientific). Plasmid sequences were confirmed by standard sequencing from Macrogen (MACROGEN Inc. DNA Sequencing Service; Amsterdam, The Netherlands) using the primers present in Table S4, Supporting information. After sequence confirmation, plasmids were electroporated into E. coli Acella (Sambrook et al., 1989) before being introduced in H. thermocellum by electroporation as described by Olson and Lynd (2012).
RNA isolation
RNA isolation of H. thermocellum mutants, harbouring the pThermoCas9i plasmids, was performed using 5 ml of overnight cultures in the mid‐log phase (OD600 nm reached 0.6). In order to harvest the cells, the culture was brought inside the anaerobic chamber where 5 ml was transferred to a 50 ml Greiner tube. Cells were centrifuged aerobically at 4800 g and 4ºC for 15 min. The supernatant was carefully decanted, and the pellet was resuspended in 0.5 ml of ice cold TE buffer (pH 8.0). The pellet was placed on ice during the next steps of the protocol, unless otherwise specified. The cell suspension was divided in two 2 screw‐capped tubes containing 0.5 g of zirconium beads, 30 μl of 10% w/v SDS, 30 μl of 3 M sodium acetate (pH 5.2) and 500 μl of Roti® aqua Phenol (Carl Roth GmbH, Karlsruhe, Germany). Cells were disrupted in the Fastprep apparatus (MP Biomedicals, Solon, OH, USA) at speed 6 for 40 s, centrifuged at 10 000 rpm and 4ºC, for 5 min. Three hundred microlitres of aqueous phase was transferred to a sterile 1.5 ml Eppendorf tube, and 300 μl of chloroform‐isoamyl alcohol (Carl Roth GmbH, Karlsruhe, Germany) was added. The mixture was centrifuged at 14 000 rpm and 4ºC, for 3 min. Two hundred and fifty microlitres of water phase was transferred to a sterile 1.5 ml Eppendorf tube and mixed with 250 μl of the Lysis/Binding buffer of the Maxwell® 16 LEV Total RNA Cells Kit. Consequently, the rest of the steps were followed according to the RNA isolation Kit. The purified RNA was measured in the NanoDrop spectrophotometer to determine the quality and the concentration of the elution and stored at −80ºC.
First‐strand cDNA synthesis and RT‐PCR
The first‐strand cDNA synthesis was performed in a 20‐µl reaction, containing 2500 ng of RNA, 125 ng of random primers, 10 mM dNTP mix, 5× First‐Strand Buffer, 0.1 M DTT, the SuperScriptTM III RT and RNAse‐free water, following the manufacturer’s instruction of the SuperScriptTM III Reverse Transcriptase protocol (Invitrogen (Life Technologies Europe BV), The Netherlands). The cDNA as well as RNA was used in RT‐PCR to analyse the expression of both, the ThermodCas9 protein and the sgRNA. The RNA was used as a negative control. The primers BG11642 and BG11643 were used to amplify 169 bp of the sgRNA using the NEB Q5® High‐Fidelity DNA polymerase. The primers BG11636 and BG11637 were used to amplify 282 bp of the ThermodCas9 using the NEB Q5® High‐Fidelity DNA polymerase.
Quantitative real time PCR
To assess the functionality of ThermodCas9 based CRISPRi as a silencing tool, a quantitative real‐time PCR (RT‐qPCR) was performed using the cDNA synthesized from the RNA of H. thermocellum DSM 1313, expressing the ThermodCas9 and the sgRNA targeting the lactate dehydrogenase (ldh) and phosphate acetyltransferase (pta) genes. As controls, the cDNA was also synthesized from the RNA of the wild‐type H. thermocellum DSM 1313 strain and the transformant strains with ThermodCas9 and the non‐targeting sgRNA. It was used to measure the relative expression level of the ldh and pta genes of the transformant H. thermocellum, comparing it to the transformant H. thermocellum with the ThermodCas9 and the non‐targeting sgRNA.
RT‐qPCR was performed by using the iQTM SYBR® Green Supermix from Bio‐Rad. The final volume of the reaction was set to 20 μl; thus, all the components were scaled accordingly relevant to the manufacturer’s protocol. The cDNA samples were diluted in sterile Milli‐Q water. The amount of cDNA used as a template was relative to 50 ng of RNA. The house‐keeping gene used to measure the relative expression was the recombinase A (recA) of H. thermocellum DSM 1313. The primers used to amplify the ldh and pta genes of H. thermocellum DSM 1313 were BG14575, BG14580 and BG15853, BG15854 respectively (Table S6, Supporting information). The RT‐qPCR was run in a Bio‐Rad C1000 Thermal Cycler.
High‐pressure liquid chromatography
A high‐pressure liquid chromatography (HPLC) system ICS‐5000 was used for the organic acids and ethanol quantification. The system has Aminex HPX 87H column from Bio‐Rad Laboratories and is equipped with a UV1000 detector operating on 210 nm and a RI‐150 40 °C refractive index detector. The mobile phase consisted of 0.16 N H2SO4, and the column was operated at 0.8 ml min−1. All samples were diluted 4:1 with 10 mM DMSO in 0.01 N H2SO4. The H. thermocellum NT control, ldh and pta silencing transformant strains were grown in CP medium (1 g l−1 yeast extract) for 2 days and samples were taken at different time points from OD600 ~ 0.5–1.5 to analyse using HPLC. Sugars (cellobiose, glucose, ethanol and glycerol) and organic acids (acetic acid, lactic acid, succinic acid and formic acid) were used as standards with a concentration range between 1.25 and 25 mM.
Conflict of interest
None declared.
Supporting information
Fig. S1. For lactate and acetate production, positive transformants referred as ldh_P, ldh _S, pta_P, pta_S and control ldh_non‐targeting (NT) were analyzed in HPLC at OD600 > 1.0 (T2). The silencing transformants for ldh gene referred as ldh_P – targeting the promoter, ldh _S – targeting the start of the gene; positive silencing transformants for pta gene referred as pta_P – targeting the promoter, pta_S – targeting the start of the gene. H. thermocellum DSM 1313 cells were transformed with both non‐targeting pThermoCas9i_NT and targeting pThermoCas9i_ldh and pta plasmids. The TmR colonies of both the non‐targeting and targeting plasmids were transferred to 50 ml CP medium (1g l−1 yeast extract) containing 6 µg ml−1 TmR and cultured to OD600 ~ 0.5–0.7 (T1) and > 1.0 (T2), and 1 ml cells were sampled for 2 days for HPLC analysis. Data represent the mean values of three biological replicates and the standard deviation (SD). The level of significance of the differences when results were compared was estimated by means of analysis of variance (ANOVA), with α = 0.05.
Table S1. Bacterial strains used in the present study.
Table S2. Plasmids used in the present study.
Table S3. Spacers used in the present study.
Table S4. Primers used in the present study.
Appendix S1. Promoter intergenic 16S/23S rRNA_C. thermocellum DSM1313.
Appendix S2. Effects of CRISPRi‐mediated suppression on lactate and acetate production and impact on the fermentation profile.
Acknowledgements
The authors thank Ioannis Mougiakos for providing the ThermodCas9 plasmids and technical assistance with plasmid design. We are grateful to Daniel Olson for his advice on the transformation protocol of H. thermocellum DSM 1313.
Microbial Biotechnology (2020) 13(2), 339–349
Funding Information
This work was supported by Corbion BV, The Netherlands; The European Union Marie Skłodowska‐Curie Innovative Training Networks (ITN) [contract number 642068].
References
- Akinosho, H. , Yee, K. , Close, D. , and Ragauskas, A. (2014) The emergence of Clostridium thermocellum as a high utility candidate for consolidated bioprocessing applications. Front Chem 2: 66–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyros, D. A. , Tripathi, S. A. , Barrett, T. F. , Rogers, S. R. , Feinberg, L. F. , Olson, D. G. , et al (2011) High ethanol titers from cellulose by using metabolically engineered thermophilic, anaerobic microbes. Appl Environ Microbiol 77: 8288–8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla, A. , Bansal, N. , Kumar, S. , Bischoff, K.M. , and Sani, R.K. (2013) Improved lignocellulose conversion to biofuels with thermophilic bacteria and thermostable enzymes. Biores Technol 128: 751–759. [DOI] [PubMed] [Google Scholar]
- Biswas, R. , Prabhu, S. , Lynd, L.R. , and Guss, A. M. (2014) Increase in ethanol yield via elimination of lactate production in an ethanol‐tolerant mutant of Clostridium thermocellum . PLoS ONE One 9: e86389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas, R. , Zheng, T. , Olson, D.G. , Lynd, L.R. , and Guss, A.M. (2015) Elimination of hydrogenase active site assembly blocks H2 production and increases ethanol yield in Clostridium thermocellum . Biotechnol Biofuels 8: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, S.D. , Guss, A.M. , Karpinets, T.V. , Parks, J.M. , Smolin, N. , Yang, S. , et al (2011) Mutant alcohol dehydrogenase leads to improved ethanol tolerance in Clostridium thermocellum . Proc Natl Acad Sci USA 108: 13752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruder, M.R. , Pyne, M.E. , Moo‐Young, M. , Chung, D.A. , and Chou, C.P. (2016) Extending CRISPR‐Cas9 technology from genome editing to transcriptional engineering in the genus Clostridium . Appl Environ Microbiol 82: 6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daas, M.J.A. , Vriesendorp, B. , van de Weijer, A.H. P. , van der Oost, J. , and van Kranenburg, R. (2018) Complete genome sequence of Geobacillus thermodenitrificans T12, a potential host for biotechnological applications. Curr Microbiol 75: 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash, S. , Khodayari, A. , Zhou, J. , Holwerda, E.K. , Olson, D.G. , Lynd, L.R. , and Maranas, C.D. (2017) Development of a core Clostridium thermocellum kinetic metabolic model consistent with multiple genetic perturbations. Biotechnol Biofuels 10: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez, A.A. , Lim, W.A. , and Qi, L.S. (2016) Beyond editing: repurposing CRISPR‐Cas9 for precision genome regulation and interrogation, Nature reviews . Mol Cell Biol 17: 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom, J. , Chung, D. , Kim, S.K. , Guss, A. , and Westpheling, J. (2018) Deletion of the Clostridium thermocellum recA gene reveals that it is required for thermophilic plasmid replication but not plasmid integration at homologous DNA sequences. J Ind Microbiol Biotechnol 45: 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y. , Xin, F. , Lu, J. , Dong, W. , Zhang, W. , Zhang, M. , et al (2017) State of the art review of biofuels production from lignocellulose by thermophilic bacteria. Biores Technol 245: 1498–1506. [DOI] [PubMed] [Google Scholar]
- Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J. A. , and Charpentier, E. (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science (New York, N.Y.) 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph, R.C. , Kim, N.M. , and Sandoval, N.R. (2018) Recent developments of the synthetic biology toolkit for Clostridium . Front Microbiol 9: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannuchamy, S. , Mukund, N. , and Saleena, L. M. (2016) Genetic engineering of Clostridium thermocellum DSM1313 for enhanced ethanol production. BMC Biotechnol 16(Suppl 1): 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A.K. , and Sharma, S. (2017) Recent updates on different methods of pretreatment of lignocellulosic feedstocks: a review. Bioresour Bioprocess 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, M.H. , Gilbert, L.A. , Wang, X. , Lim, W.A. , Weissman, J.S. , and Qi, L.S. (2013) CRISPR interference (CRISPRi) for sequence‐specific control of gene expression. Nat Protoc 8: 2180–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J.‐F. , Norville, J.E. , Aach, J. , McCormack, M. , Zhang, D. , Bush, J. , et al (2013) Multiplex and homologous recombination–mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31: 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Jendresen, C.B. , Grunberger, A. , Ronda, C. , Jensen, S.I. , Noack, S. , and Nielsen, A.T. (2016) Enhanced protein and biochemical production using CRISPRi‐based growth switches. Metab Eng 38: 274–284. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Gallay, C. , Kjos, M. , Domenech, A. , Slager, J. , van Kessel, S.P. , et al (2017) High‐throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae . Mol Syst Biol 13: 931–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, J. , Olson, D.G. , Murphy, S.J.‐L. , Tian, L. , Hon, S. , Lanahan, A. , et al (2017) Engineering electron metabolism to increase ethanol production in Clostridium thermocellum . Metab Eng 39: 71–79. [DOI] [PubMed] [Google Scholar]
- Lo, J. , Zheng, T. , Hon, S. , Olson, D.G. , and Lynd, L.R. (2019) ,,Correction for Lo et al.,Correction for Lo Clostridium thermocellum and Thermoanaerobacterium saccharolyticum . J Bacteriol 201: e00405‐00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynd, L.R. , Guss, A.M. , Himmel, M.E. , Beri, D. , Herring, C. , Holwerda, E.K. , et al (2016) Advances in consolidated bioprocessing using Clostridium thermocellum and Thermoanaerobacter saccharolyticum . Indust Biotechnol 10: 365–394. [Google Scholar]
- Mankin, A.S. , Skripkin, E.A. , and Kagramanova, V.K. (1987) A putative internal promoter in the 16 S/23 S intergenic spacer of the rRNA operon of archaebacteria and eubacteria. FEBS Lett 219: 269–273. [DOI] [PubMed] [Google Scholar]
- McBee, R.H. (1954) The characteristics of Clostridium thermocellum . J Bacteriol 67: 505–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr, G. , Hong, W. , Zhang, J. , Cui, G.Z. , Yang, Y. , Cui, Q. , et al (2013) A targetron system for gene targeting in thermophiles and its application in Clostridium thermocellum . PLoS ONE One 8: e69032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougiakos, I. , Mohanraju, P. , Bosma, E.F. , Vrouwe, V. , Finger Bou, M. , Naduthodi, M.I.S. , et al (2017) Characterizing a thermostable Cas9 for bacterial genome editing and silencing. Nat Commun 8: 1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougiakos, I. , Bosma, E.F. , Ganguly, J. , van der Oost, J. , and van Kranenburg, R. (2018) Hijacking CRISPR‐Cas for high‐throughput bacterial metabolic engineering: advances and prospects. Curr Opin Biotechnol 50: 146–157. [DOI] [PubMed] [Google Scholar]
- Olson, D.G. , and Lynd, L.R. (2012) Transformation of Clostridium thermocellum by electroporation. Methods Enzymol 510: 317–330. [DOI] [PubMed] [Google Scholar]
- Olson, D.G. , Tripathi, S.A. , Giannone, R.J. , Lo, J. , Caiazza, N.C. , Hogsett, D.A. , et al (2010) Deletion of the Cel48S cellulase from Clostridium thermocellum . Proc Natl Acad Sci USA 107: 17727–17732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, D.G. , Giannone, R.J. , Hettich, R.L. , and Lynd, L.R. (2013) Role of the CipA scaffoldin protein in cellulose solubilization, as determined by targeted gene deletion and complementation in Clostridium thermocellum . J Bacteriol 195: 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanek, B. , Biswas, R. , Rydzak, T. , and Guss, A.M. (2015) Elimination of metabolic pathways to all traditional fermentation products increases ethanol yields in Clostridium thermocellum . Metab Eng 32: 49–54. [DOI] [PubMed] [Google Scholar]
- Peters, J.M. , Colavin, A. , Shi, H. , Czarny, T.L. , Larson, M.H. , Wong, S. , et al (2016) A Comprehensive, CRISPR‐based functional analysis of essential genes in bacteria. Cell 165: 1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plugge, C.M. (2005) Anoxic media design, preparation, and considerations. Methods Enzymol 397: 3–16. [DOI] [PubMed] [Google Scholar]
- Pyne, M.E. , Moo‐Young, M. , Chung, D.A. , and Chou, C.P. (2014) Expansion of the genetic toolkit for metabolic engineering of Clostridium pasteurianum: chromosomal gene disruption of the endogenous CpaAI restriction enzyme. Biotechnol Biofuels 7: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, L.S. , Larson, M.H. , Gilbert, L.A. , Doudna, J.A. , Weissman, J.S. , Arkin, A.P. , and Lim, W.A. (2013) Repurposing CRISPR as an RNA‐guided platform for sequence‐specific control of gene expression. Cell 152: 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydzak, T. , Lynd, L.R. , and Guss, A.M. (2015) Elimination of formate production in Clostridium thermocellum . J Ind Microbiol Biotechnol 42: 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. , Fritsch, E. F. , and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, xxxviii + 1546 pp. [Google Scholar]
- Song, C.W. , Lee, J. , and Lee, S.Y. (2015) Genome engineering and gene expression control for bacterial strain development. Biotechnol J 10: 56–68. [DOI] [PubMed] [Google Scholar]
- Talukdar, P.K. , Olguin‐Araneda, V. , Alnoman, M. , Paredes‐Sabja, D. , and Sarker, M.R. (2015) Updates on the sporulation process in Clostridium species. Res Microbiol 166: 225–235. [DOI] [PubMed] [Google Scholar]
- Tian, L. , Perot, S.J. , Hon, S. , Zhou, J. , Liang, X. , Bouvier, J.T. , et al (2017) Enhanced ethanol formation by Clostridium thermocellum via pyruvate decarboxylase. Microb Cell Fact 16: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi, S.A. , Olson, D.G. , Argyros, D.A. , Miller, B.B. , Barrett, T.F. , Murphy, D.M. , et al (2010) Development of pyrF‐based genetic system for targeted gene deletion in Clostridium thermocellum and creation of a pta mutant. Appl Environ Microbiol 76: 6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen, D. , Lo, J. , Brown, S. D. , Johnson, C.M. , Tschaplinski, T.J. , Martin, M. , et al (2013) Characterization of Clostridium thermocellum strains with disrupted fermentation end‐product pathways. J Ind Microbiol Biotechnol 40: 725–734. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Zhang, Z.T. , Seo, S.O. , Lynn, P. , Lu, T. , Jin, Y.S. , and Blaschek, H.P. (2016) Bacterial genome editing with CRISPR‐Cas9: deletion, integration, single nucleotide modification, and desirable "clean" mutant selection in Clostridium beijerinckii as an example. ACS Synthet Biol 5: 721–732. [DOI] [PubMed] [Google Scholar]
- Wen, Z. , Minton, N.P. , Zhang, Y. , Li, Q. , Liu, J. , Jiang, Y. , and Yang, S. (2017) Enhanced solvent production by metabolic engineering of a twin‐clostridial consortium. Metab Eng 39: 38–48. [DOI] [PubMed] [Google Scholar]
- Xu, T. , Li, Y. , Van Nostrand, J.D. , He, Z. , and Zhou, J. (2014) Cas9‐based tools for targeted genome editing and transcriptional control. Appl Environ Microbiol 80: 1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, L. , Cengic, I. , Anfelt, J. , and Hudson, E.P. (2016) Multiple gene repression in cyanobacteria using CRISPRi. ACS Synthet Biol 5: 207–212. [DOI] [PubMed] [Google Scholar]
- Zacharias, M. , and Wagner, R. (1989) Functional characterization of a putative internal promoter sequence between the 16S and the 23S RNA genes within the Escherichia coli rrnB operon. Mol Microbiol 3: 405–410. [DOI] [PubMed] [Google Scholar]
- Zhao, H. , Sun, Y. , Peters, J.M. , Gross, C.A. , Garner, E.C. , and Helmann, J.D. (2016) Depletion of undecaprenyl pyrophosphate phosphatases disrupts cell envelope biogenesis in Bacillus subtilis . J Bacteriol 198: 2925–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. For lactate and acetate production, positive transformants referred as ldh_P, ldh _S, pta_P, pta_S and control ldh_non‐targeting (NT) were analyzed in HPLC at OD600 > 1.0 (T2). The silencing transformants for ldh gene referred as ldh_P – targeting the promoter, ldh _S – targeting the start of the gene; positive silencing transformants for pta gene referred as pta_P – targeting the promoter, pta_S – targeting the start of the gene. H. thermocellum DSM 1313 cells were transformed with both non‐targeting pThermoCas9i_NT and targeting pThermoCas9i_ldh and pta plasmids. The TmR colonies of both the non‐targeting and targeting plasmids were transferred to 50 ml CP medium (1g l−1 yeast extract) containing 6 µg ml−1 TmR and cultured to OD600 ~ 0.5–0.7 (T1) and > 1.0 (T2), and 1 ml cells were sampled for 2 days for HPLC analysis. Data represent the mean values of three biological replicates and the standard deviation (SD). The level of significance of the differences when results were compared was estimated by means of analysis of variance (ANOVA), with α = 0.05.
Table S1. Bacterial strains used in the present study.
Table S2. Plasmids used in the present study.
Table S3. Spacers used in the present study.
Table S4. Primers used in the present study.
Appendix S1. Promoter intergenic 16S/23S rRNA_C. thermocellum DSM1313.
Appendix S2. Effects of CRISPRi‐mediated suppression on lactate and acetate production and impact on the fermentation profile.