

Abstract
Background:
Several reports have identified different patterns of Parkinson’s disease progression in individuals carrying missense variants in GBA or LRRK2 genes. The overall contribution of genetic factors to the severity and progression of Parkinson’s disease, however, has not been well studied.
Objectives:
To test the association between genetic variants and the clinical features of Parkinson’s disease on a genomewide scale.
Methods:
We accumulated individual data from 12 longitudinal cohorts in a total of 4093 patients with 22,307 observations for a median of 3.81 years. Genomewide associations were evaluated for 25 cross-sectional and longitudinal phenotypes. Specific variants of interest, including 90 recently identified disease-risk variants, were also investigated post hoc for candidate associations with these phenotypes.
Results:
Two variants were genomewide significant. Rs382940(T>A), within the intron of SLC44A1, was associated with reaching Hoehn and Yahr stage 3 or higher faster (hazard ratio 2.04 [1.58–2.62]; P value = 3.46E-8). Rs61863020(G>A), an intergenic variant and expression quantitative trait loci for α−2A adrenergic receptor, was associated with a lower prevalence of insomnia at baseline (odds ratio 0.63 [0.52–0.75]; P value = 4.74E-8). In the targeted analysis, we found 9 associations between known Parkinson’s risk variants and more severe motor/cognitive symptoms. Also, we replicated previous reports of GBA coding variants (rs2230288: p.E365K; rs75548401: p.T408M) being associated with greater motor and cognitive decline over time, and an APOE E4 tagging variant (rs429358) being associated with greater cognitive deficits in patients.
Conclusions:
We identified novel genetic factors associated with heterogeneity of Parkinson’s disease. The results can be used for validation or hypothesis tests regarding Parkinson’s disease. © 2019 International Parkinson and Movement Disorder Society
Keywords: Apolipoprotein E, GBA, genomewide association study, Parkinson’s disease
Parkinson’s disease (PD) is clinically defined by its motor features of rigidity, bradykinesia, gait disturbance, and tremor. Although these prominent features are important for diagnosis, patients with PD also suffer from many nonmotor features, such as constipation, urinary incontinence, orthostatic hypotension, rapid eye movement sleep behavior disorder (RBD), apathy, hyposmia, and cognitive impairment.1 Moreover, patients develop motor complications, including wearing off and dyskinesia, as side effects of medication. The onset, intensity, and progression of these different PD clinical features vary among individuals, and the mechanisms underlying this heterogeneity are not well understood.
Recent genomewide studies have identified 90 common variants associated with the risk of PD, with an overall heritability estimated to be between 22% to 27%.2,3 Although previous studies have indicated the importance of genetic contributions to disease risk, the contribution of genetic factors to PD progression and heterogeneity has not been well studied. Investigating genetic factors associated with disease progression and heterogeneity in disease presentation is an important step in elucidating the underlying molecular mechanisms and identifying better patient stratification in clinical trials.4
Longitudinal patient cohorts are powerful resources that can be used to explore the impact of genetics on the trajectory of PD-related phenotypes; the inherent precision of repeated measurements over time provides more power to detect these associations. However, the available number of participants in each study is usually not enough to conduct a genomewide association study (GWAS). In this study, we accumulated 22,307 follow-up visits from 4093 patients across 12 cohorts (Table 1) and performed meta-analyses of longitudinal GWAS on the progression markers of PD. Using the results from this meta-analysis, we evaluated how known risk variants, including the 90 recently identified variants for PD,3 GBA protein coding mutations, and APOE tagging variants were associated with the progression of phenotypes. To maximize the utility of this work to other researchers, we have made all results from this study publicly searchable and available for download (https://pdgenetics.shinyapps.io/pdprogmetagwasbrowser/)
TABLE 1.
Summary characteristics of 13 datasets (12 cohorts)
| Dataset | DATATOP | DIGPD_chip | DIGPD_neuroX | HBS | NET-PD_LS1 | OSLO | PARKFIT | PARKWEST | PDBP | PICNICS | PPMI | PRECEPT/POSTCEPT | PROPARK |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | 428 | 108 | 235 | 503 | 340 | 318 | 332 | 147 | 515 | 117 | 441 | 318 | 291 |
| Total number of visits | 3303 | 268 | 811 | 1,206 | 2209 | 2086 | 664 | 294 | 2360 | 358 | 2436 | 4983 | 1599 |
| Number of visits per participants, median [1Q,3Q] | 7 [6,8] | 2 [1, 3.25] | 4 [3, 4.5] | 3 [2,3] | 7 [6,8] | 6 [3, 10] | 2 [2,2] | 2 [2,2] | 5 [1,7] | 3 [2,4] | 6 [5,7] | 16 [14, 17] | 6 [6,6] |
| Follow-up in years | 1.22 (0.41) | 1.52 (1.60) | 2.54 (1.30) | 1.53 (0.84) | 4.47 (1.48) | 12.08 (6.91) | 1.97 (0.00) | 3.04 (0.09) | 1.97 (1.73) | 3.03 (1.62) | 4.88 (2.05) | 6.78 (0.95) | 4.64 (1.13) |
| Age at diagnosis | 58.73 (9.12) | 59.04 (10.14) | 60.44 (9.43) | 62.27 (10.40) | 60.84 (9.07) | 54.43 (10.14) | 60.85 (8.62) | 67.16 (9.29) | 58.59 (10.18) | 68.95 (9.38) | 61.05 (9.82) | 59.45 (9.18) | 53.11 (10.58) |
| Year from diagnosis | 1.15 (1.16) | 2.78 (1.56) | 2.52 (1.57) | 4.02 (4.62) | 1.48 (0.97) | 1.76 (4.69) | 5.14 (4.40) | 0.14 (0.12) | 6.31 (5.44) | 0.23 (0.48) | 0.55 (0.54) | 0.80 (0.84) | 6.61 (4.67) |
| Female (%) | 142 (33.2) | 44 (40.7) | 91 (38.7) | 174 (34.6) | 122 (35.9) | 106 (33.3) | 109 (32.8) | 55 (37.4) | 202 (39.2) | 43 (36.8) | 159 (36.1) | 105 (33.0) | 105 (36.1) |
| Having Hoehn & Yahr scale 2 or larger at baseline (%) | 197 (46.0) | 68 (65.4) | 151 (64.3) | 420 (84.5) | - | 22 (100.0) | 314 (94.6) | 85 (57.8) | 426 (83.0) | 62 (53.0) | 242 (54.9) | 198 (62.5) | 271 (96.1) |
| Years of education | 14.19 (3.29) | 12.27 (2.92) | 12.28 (3.17) | 15.14 (1.72) | 15.46 (2.23) | - | - | 11.31 (3.24) | 15.24 (4.56) | 12.17 (2.89) | 15.58 (2.98) | 15.93 (3.15) | 11.95 (4.10) |
| Use of levodopa (%) | 0 (0.0) | 64 (59.8) | 161 (68.5) | 353 (70.2) | 171 (50.6) | - | - | 0 (0.0) | 409 (83.1) | 35 (29.9) | 3 (0.9) | 0 (0.0) | 199 (68.4) |
| Use of agonist (%) | 0 (0.0) | 88 (81.5) | 163 (69.4) | 198 (39.4) | 231 (68.3) | - | - | 0 (0.0) | 277 (56.3) | 22 (18.8) | 0 (0.0) | 1 (0.3) | 218 (74.9) |
| Hyposmia (%) | 0 (0.0) | 33 (30.8) | 66 (28.4) | - | - | - | - | 53 (36.1) | 295 (63.6) | - | 185 (44.6) | - | 170 (63.7) |
| Cognitive impairment (%) | 24 (5.6) | 1 (0.9) | 2 (0.9) | 59 (11.9) | 27 (7.9) | - | 55 (16.6) | 27 (18.4) | 119 (23.2) | 11 (9.4) | 35 (7.9) | 3 (0.9) | 76 (27.1) |
| Motor fluctuations (%) | - | 14 (13.1) | 32 (13.6) | 198 (39.9) | 87 (25.7) | - | - | 4 (2.7) | 176 (37.3) | 0 (0.0) | 0 (0.0) | - | 92 (32.3) |
| Dyskinesias (%) | 3 (0.7) | 4 (3.7) | 13 (5.5) | 168 (33.8) | 5(1.5) | - | - | 2(1.4) | 118 (25.0) | 0 (0.0) | 0 (0.0) | - | 80 (27.8) |
| Depression (%) | 11 (2.6) | 29 (27.1) | 77 (33.3) | 28 (10.3) | 31 (9.2) | - | - | 20 (13.6) | 59 (12.5) | 26 (22.2) | 141 (33.8) | 72 (22.6) | 48 (16.6) |
| Restless legs syndrome (%) | - | 16 (15.7) | 34 (14.5) | 30 (10.4) | - | - | - | - | 107 (24.4) | - | 27 (0.65) | - | - |
| Constipation (%) | 9 (2.1) | 17 (16.0) | 48 (20.7) | - | - | - | - | 17 (11.6) | 255 (54.0) | 27 (23.1) | 149 (33.8) | - | 137 (47.1) |
| Rapid eye movement sleep behavior disorder (%) | - | - | - | - | - | - | - | - | 217 (49.4) | - | 104 (24.9) | - | - |
| Daytime sleepiness (%) | 3 (0.7) | 51 (48.6) | 104 (44.3) | - | - | - | - | 23 (15.6) | 178 (37.7) | 24 (20.5) | 61 (14.7) | - | 125 (43.0) |
| Insomnia (%) | 11 (2.6) | 31 (30.4) | 86 (36.6) | 170 (33.9) | - | - | - | 45 (30.6) | 332 (70.3) | 59 (50.4) | 109 (24.7) | - | 83 (28.5) |
| Hoehn & Yahr scale 3 or greater (%) | 0 (0.0) | 2(1.9) | 3(1.3) | 59 (11.9) | 10 (2.9) | (0.0) | 17(5.1) | 11 (7.5) | 81 (15.8) | 12 (10.3) | 2 (0.5) | (0.0) | 115 (40.8) |
| SEADL 70 or less (%) | 3 (0.7) | 30 (30.0) | 12(5.1) | - | 2 (0.6) | - | - | 5 (3.4) | 75 (15.9) | - | 2 (0.5) | 1 (0.3) | - |
| Hoehn & Yahr scale | 1.61 (0.53) | 1.76 (0.56) | 1.77 (0.54) | 2.13 (0.63) | - | 3.27 (0.55) | 2.08 (0.33) | 1.87 (0.58) | 2.03 (0.73) | 1.63 (0.66) | 1.50 (0.50) | 1.75 (0.48) | 2.51 (0.79) |
| UPDRS_scaled | −0.07 (0.98) | 0.08 (1.00) | −0.03 (0.96) | −0.00 (1.00) | 0.00 (1.01) | - | −0.06 (0.96) | −0.02 (1.01) | −0.07 (1.01) | −0.01 (0.98) | - | 0.02 (1.00) | - |
| UPDRS1_scaled | - | −0.10 (0.99) | −0.02 (0.90) | 0.01 (1.01) | 0.01 (1.06) | - | - | −0.03 (1.00) | 0.03 (1.01) | - | 0.00 (4.31) | −0.03 (0.96) | - |
| UPDRS2 scaled | - | 0.13 (1.01) | −0.06 (0.98) | 0.00 (1.00) | 0.01 (0.98) | - | - | −0.03 (1.00) | −0.00 (1.01) | - | 0.00 (4.19) | −0.01 (1.00) | - |
| UPDRS3_scaled | - | 0.09 (0.99) | −0.02 (0.96) | −0.01 (1.00) | 0.00 (1.00) | 0.61 (1.21) | - | −0.01 (1.00) | −0.09 (1.00) | - | −0.00 (8.87) | 0.04 (0.99) | - |
| UPDRS4_scaled | - | −0.09 (1.55) | −0.17 (0.81) | −0.20 (0.86) | −0.33 (0.71) | - | - | −0.25 (0.86) | −0.12 (0.94) | - | - | - | - |
| MMSE | 29.01 (1.32) | 28.59 (1.66) | 28.21 (1.75) | 28.39 (2.17) | - | - | 28.09 (1.61) | 27.86 (2.29) | - | 28.70 (1.44) | - | 29.28 (1.08) | 27.05 (2.50) |
| SEADL | 91.56 (6.49) | 64.96 (41.02) | 88.54 (14.94) | - | 91.88 (5.84) | - | - | 89.39 (7.42) | 84.58 (14.84) | - | 93.79 (6.11) | 92.78 (5.26) | - |
Continuous variables were summarized as mean (standard deviation). 1Q, first quantile; 3Q, third quantile. DATATOP, Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism; DIGPD_chip, Drug Interaction With Genes in Parkinson’s Disease genotyped by lllumina Multi-Ethnic Genotyping Array; DIGPD_neuroX: Drug Interaction With Genes in Parkinson’s Disease genotyped by NeuroX; HBS, Harvard Biomarkers Study; NET-PD_LS1, National Institutes of Health Exploratory Trials in Parkinson’s Disease Large Simple Study 1; OSLO, the Oslo Parkinson’s Disease study; PARKFIT, the ParkFit study; PARKWEST, Norwegian ParkWest Study; PDBP, Parkinson’s Disease Biomarker Program; PICNICS, Parkinsonism Incidence and Cognitive and Non-motor heterogeneity In Cambridgeshire; PPM I, Parkinson’s Progression Markers Initiative; PreCEPT/PostCEPT, Parkinson Research Examination of CEP-1347 Trial study with its subsequent prospective study; PROPARK, Profiling Parkinson’s Disease Study; MMSE, Mini Mental State Examination; SEADL, Schwab and England Activities of Daily Living Scale; UPDRS, Unified Parkinson Disease Rating Scale; UPDRS1, UPDRS part 1; UPDRS2, part 2; UPDRS3, part 3; UPDRS4, part 4.
Methods
Cohorts
A total of 12 longitudinal cohorts of PD patients recruited across North America, Europe, and Australia were included in our study. The following observational studies were included: the Drug Interaction with Genes in Parkinson’s Disease (DIGPD), the Harvard Biomarkers Study, the Oslo Parkinson’s Disease study (partly including retrospective data) (OSLO), the Norwegian ParkWest study (PARKWEST), the Parkinson’s Disease Biomarker Program, the Parkinsonism Incidence and Cognitive and Non-Motor Heterogeneity in Cambridgeshire, the Parkinson’s Progression Markers Initiative (PPMI), and the Profiling Parkinson’s Disease Study. The 4 cohorts included were randomized clinical trials: the Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism (DATATOP), the National Institutes of Health Exploratory Trials in Parkinson’s Disease Large Simple Study 1, the ParkFit study (PARKFIT), and the Parkinson Research Examination of CEP-1347 Trial study with its subsequent prospective study (PreCEPT/PostCEPT). More details of these cohorts are described in Appendix. Participants’ information and genetic samples were obtained under appropriate written consent and with local institutional and ethical approvals.
Phenotyping
Each cohort had a different set of recorded biomarkers and phenotypes associated with PD. We selected continuous and binomial biomarkers based on their clinical importance and availability. For continuous outcomes, we collected the scores of Hoehn and Yahr staging scale (HY),5 total and sub-scores of the Unified Parkinson’s Disease Rating Scale (UPDRS) or the Movement Disorder Society–revised version (MDS-UPDRS),6 Mini-Mental State Examination, Montreal Cognitive Assessment (MoCA),7 and the modified Schwab and England Activities of Daily Living Scale (SEADL). With the exception of the subscores of UPDRS/MDS-UPDRS part 4, the total scores and subscores of the UPDRS and MDS-UPDRS were normalized to the population-baseline mean and standard deviation and converted to z values. The subscores of the UPDRS/MDS-UPDRS part 4, measuring complication of treatment, were normalized to the mean and standard deviation of all observations because the score was 0 at the baseline for the de novo PD cohorts. We also determined whether the subjects were recorded as presenting the following binomial outcomes during participant visits: constipation, cognitive impairment, depression, daytime sleepiness, HY stage of 3 or worse (HY3), hyposmia, insomnia, motor fluctuation, RBD, restless legs syndrome, and a SEADL of 70 or less (SEADL70). Because study-specific criteria for these binomial outcomes were not consistent among the studies, we tried to use the common criteria for these binomial outcomes if we had access to the raw data from the studies. The details of the definitions of binomial outcomes are provided in Supplemental Table 1.
Genetics Data
The genotyping was conducted with NeuroX, a targeted chip for neurodegenerative disease,8 for National Institutes of Health Exploratory Trials in Parkinson’s Disease Large Simple Study 1, a part of the DIGPD (DIGPD_neuroX), Harvard Biomarkers Study, Parkinson’s Disease Biomarker Program, and PRE-CEPT. The rest of DIGPD (DIGPD_chip) were genotyped using Illumina Multi-Ethnic Genotyping Array (Illumina, Inc. San Diego, CA). Participants in DATATOP, OSLO, PARKFIT, PARKWEST, the Parkinsonism Incidence and Cognitive and Non-Motor Heterogeneity in Cambridgeshire, and the Profiling Parkinson’s Disease Study were genotyped using Illumina Infinium OmniExpress array (Illumina, Inc. San Diego, CA). Whole-genome sequencing data was used for PPMI, with the detailed methods for genome sequencing provided on the PPMI website (https://www.ppmi-info.org/).
Variant inclusion criteria consisted of call rate >0.95, minor allele frequency (MAF) > 0.01, and Hardy-Weinberg equilibrium test statistic >1E-4. Participants were excluded with the following criteria: high missingness (>5% for genotyped variants), sex discordance, extreme heterozygosity (F statistics >0.15), non-European ancestry confirmed by joint analysis with HapMap 3 data using principal component (outside of mean 6 standard deviation in the first principle component [PC1] or the second principle component [PC2] for European reference samples),9 and excessive relatedness (pairwise kinships >0.125). We used PLINK version 1.9 for the above filtering.10
For all samples and variants passing quality control, imputation was conducted for chromosomes 1 to 22 using Minimac3 using the Haplotype Reference Consortium panel (r1.1) and Eagle version 2.3 for phasing at the Michigan Imputation Server,11 with the exception of the whole-genome sequenced PPMI dataset. Single nucleotide polymorphisms with an imputation quality of R2 less than 0.3 and MAF <1% were excluded. After quality control, the number of variants were approximately 2.6 to 2.9 million in the National Institutes of Health Exploratory Trials in Parkinson’s Disease Large Simple Study 1, DIGPD_neuroX, Harvard Biomarkers Study, Parkinson’s Disease Biomarker Program, and PRECEPT; 7.7 to 7.8 million in the Parkinsonism Incidence and Cognitive and Non-Motor Heterogeneity in Cambridgeshire study, the Profiling Parkinson’s Disease Study, PARKWEST, DATATOP, PARKFIT, DIGPD, and OSLO; and 8.6 million in PPMI. Note that the cohorts genotyped by NeuroX had relatively less genome coverage than the others.
Cohort-Level Analyses
We conducted a separate GWAS for each cohort per phenotype of interest. In addition, DIGPD cohorts were analyzed separately according to the genotyping array used (DIGPD_neuroX cohort and DIGPD_chip cohort). Each outcome was analyzed by an additive model with covariates. For the binomial outcomes at baseline visit, when the outcomes were positive for more than 5% of participants and >20 counts, logistic regression analyses were conducted. Those without the binomial outcome at baseline were followed-up until either censored or the development of the outcome. If more than 20 events were observed during follow-ups, the outcome was analyzed using Cox proportional hazard models with time-varying covariates. For the analysis of continuous traits, linear mixed models were used to evaluate the variants’ associations for the outcome. Age at diagnosis, year from diagnosis to the observation, sex and the first three principle components (PC1–3) were adjusted for in all analyses. In addition, the following covariates were associated with the outcome of interest in a backward stepwise manner: quadratic age, quadratic years from diagnosis, years of education, medication status (levodopa usage, dopamine agonist usage, using either dopamine agonist or levodopa), and a HY score of 2 or more at the first observation (except for the models regressing for HY score itself or UPDRS motor score). These covariates were selected per study using Akaike’ information criteria for logistic models and Cox survival models, and conditional Akaike information criteria (cAIC) for linear mixed effect models. The cohort level analyses were conducted with R (version 3.5.0; https://www.r-project.org/) and rvtests.12 R package “cAIC4” was used to calculate cAIC.13
Meta-Analyses
The results from the cohort-level analyses were combined using an inverse variance weighted fixed effect model. If the study-specific genomic inflation factor was more than 1.2, the study was excluded from the meta-analysis. Of the 204 GWAS, 5 were excluded based on these criteria. For the other cohorts, the overall α error was corrected using the genomic inflation factor before the meta-analysis. Meta-analyses were carried out with METAL.14 From the meta-analysis results, we only evaluated variants with MAF > 0.05 because of statistical power constraints. We also excluded variants with MAF variability greater than 15% across cohorts. Further exclusions at the meta-analysis level include variants with Cochran’s Q-test for heterogeneity <0.05 and a total participant N < 1000. The null hypothesis was tested with a significance level of 5E-8 on a 2-sided test. For genomewide signals, additional visualization and functional analyses were conducted using LocusZoom,15 FUMA (version 1.3.3d; http://fuma.ctglab.nl/snp2gene/).16 FUMA is a web-based annotation tool using Multi-marker Analysis of GenoMic Annotation (MAGMA) to conduct gene-based tests, a gene-set analysis, and a tissue expression analysis. We applied a default setting. Also, we explored the eQTLGen database (http://www.eqtlgen.org/)17 and meta-analyzed expression data in the brain accessible from the study by Qi and colleagues.18 Associations with the variants of interest, including the recently identified 90 risk variants for PD, known LRRK2 and GBA variants, and APOE, were extracted from the meta-analysis results. We exploratory evaluated the associations of these variants and clinical features based on the significance level of 0.05, applying the Bonferroni adjustment of a maximum of 25 tests per variant (raw P value <0.002).
The summary of analytical processes is shown in Figure 1.
FIG. 1.
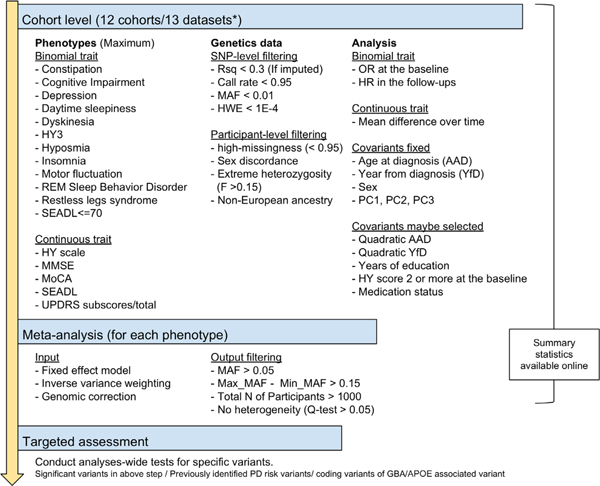
Graphical overview of the analysis strategy. *Drug Interaction with Genes in Parkinson’s Disease cohort was analyzed separately depending on the genotyping system. AAD, age at diagnosis; HR, hazard ratio; HWE, Hardy-Weinberg equilibrium test; HY, Hoehn and Yahr scale; HY3, Hoehn and Yahr score; MAF, minor allele frequency; MMSE, Mini-Mental State Examination; MoCA, Montreal Cognitive Assessment; OR, odds ratio; PC, principal components; PD, Parkinson’s disease; REM, rapid eye movement; Rsq, R square; SEADL, Schwab and England Activities of Daily Living Scale; UPDRS, Unified Parkinson’s Disease Rating Scale; YfD, years from diagnosis to observation. [Color figure can be viewed at wileyonlinelibrary.com]
Data Availability
The summary statistics of the meta-analysis results, including the ones that were not evaluated in this article, are publicly available for convenient browsing and downloading (https://pdgenetics.shinyapps.io/pdprogmetagwasbrowser/).
Results
Novel GWAS Associations With PD Progression Markers
The cohort characteristics are provided in Table 1. Overall, we analyzed 4093 participants with 22,307 longitudinal data points for a median of 3.81 years. These cohorts varied in the years between enrollment and diagnosis as well as follow-up durations. DATATOP, PARKWEST, PPMI, and PreCEPT/PostCEPT enrolled untreated PD patients, whereas others enrolled both treated and untreated patients. Considering the difference in design and recruitment strategies in the cohorts (Supporting Information Appendix), it is important to adjust for baseline characteristics as well as the follow-up lengths per cohort level. All cohort-specific models for analysis are listed in Supplemental Table 2.
In total, 204 GWAS were conducted and combined into 33 meta-analyses. A total of 8 meta-analyses were not evaluated because of the small number of total participants in the analyses (N total <1000). Those excluded were baseline analyses for RBD, restless legs syndrome, and SEADL70 and longitudinal analyses for constipation, daytime sleepiness, hyposmia, RBD, and restless legs syndrome. Therefore, we investigated 9 binomial traits at baseline, 7 binomial traits for survival, and 9 continuous traits over the follow-ups. The genomic inflation factor was the mean value of 0.993, standard deviation of 0.023, and the range was 0.951–1.031 across meta-analyses. The study-specific genomic inflation factors are provided in Supplemental Table 3.
One association with the progression of PD was of genomewide significance (P value <5.00E-08). The minor allele of rs382940 (chr9:108058562T>A), an intronic variant of SLC44A1, was associated with a higher hazard ratio (HR) of reaching HY stage 3.0 or greater (HR 2.04 [1.58–2.62], P value = 3.46E-8; estimates in a random effect model, 1.97 [1.38–2.81], P value = 1.96E-4). When considering the baseline observations, the minor allele of rs61863020 (chr10:112956055G>A), an intergenic variant, was significantly associated with the lower baseline odds ratio (OR) of having insomnia (OR 0.63 [0.52–0.75], P value = 4.74E-8; the same estimates and P value in a random effect model). Locus plots and forest plots for these 2 associations are shown in Figure 2. Cochran’ Q statistics, I2, and forest plots all showed no evidence of heterogeneity for these associations (Fig. 2).
FIG. 2.
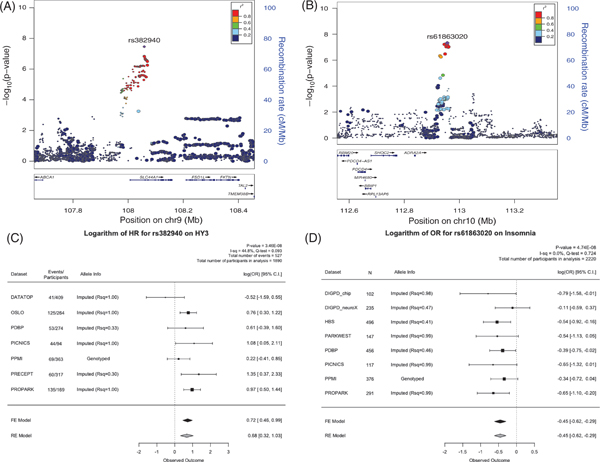
LocusZoom plots and forest plots of the 2 genome-wide significant hits. (A) The locus plot for rs382940, which is associated with HY3. (B) The locus plot for rs61863020, which is associated with insomnia. (C) The forest plot for rs382940. (D) The forest plot for rs61863020. ADRA2A, α−2A adrenergic receptor; C.I., confidence interval; DATATOP, Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism; FE, Fixed effect mode; HBS, Harvard Biomarkers Study; HR, hazard ratio; HY3, Hoehn and Yahr score; OR, odds ratio; PARKWEST, Norwegian ParkWest Study; PDBP, Parkinson’s Disease Biomarker Program; PICNICS, Parkinsonism Incidence and Cognitive and Non-motor heterogeneity In Cambridgeshire; PPMI, Parkinson’s Progression Markers Initiative; PreCEPT, Parkinson Research Examination of CEP-1347 Trial study; PROPARK, Profiling Parkinson’s Disease Study; Rsq, R square RE, Random effect model.
To evaluate the potential molecular mechanism for the two genomewide signals, we explored eQTL datasets in the blood and brain17,18 and functional annotation of the GWAS summary statistics using FUMA. Although it is in a regulatory region of SLC44A1, rs382940 itself was not reported to be an eQTL in the blood or brain. Gene-based tests using the GWAS summary statistics for reaching HY3 showed that SLC44A1 was significant gene-wise (P value = 5.8E-07 < Bonferroni correction threshold = 2.7E-6; Supplemental Fig. 1). Rs61863020 was a significant eQTL for α−2A adrenergic receptor (ADRA2A; P value = 7.2E-4, the Bonferroni corrected P value = 6.5E-3, up-regulation for A allele) in the brain.
In the meta-analysis results from the other clinical outcomes, rs382940 was associated with higher scores in the UPDRS part 2 and part 3 (UPDRS2_scaled: 0.36 [0.15–0.57], P value = 8.21E-04; UPDRS3_scaled: 0.29 [0.14–0.45], P value = 2.18E-04). These findings are consistent with the primary association of rs382940 and reaching HY3, which is a significant motor milestone (bilateral signs on clinical examination and the emergence of postural instability). Except for the association with having insomnia at baseline, rs61863020 was not significantly associated with other clinical variables in this analysis after adjusting for 25 tests. Of note, the variant was not associated with the development of insomnia in the survival analysis. This could be the result of low power of the analysis (N = 1112) or the variant may be important for the development of insomnia in an earlier phase of the disease.
Targeted Assessment for the PD Risk Variants
Of the 90 risk variants from the recently published PD GWAS, rs34637584 (LRRK2 p.G2019S) and rs76763715 (GBA p.N370S) were not available in the meta-analyses because of their MAF < 0.01. The remaining 88 PD GWAS risk SNPs were assessed in our 25 GWAS summary sets, resulting in evaluations of 2022 candidate associations. A total of 112 associations between known genetic risk variants and clinical markers had raw P values less than 0.05. After Bonferroni correction for all evaluated candidate associations, 9 surpassed the threshold of the analyses-wide significance for the maximum of 25 analyses per variant (raw P value <0.002). The directions of these associations generally indicated that having the higher risk allele was associated with more severe deficits in both the cognitive and motor domains of PD, but not for sleeping problems. Having the risk allele (A) of rs1293298 (intron variant of CTSB) was associated with a lower risk of developing insomnia (HR 0.79 [0.69-0.91], P value = 1.2E-3), and the risk allele (A) of rs6500328 (intron variant of NOD2) and (A) of rs76116224 (intergenic variant close to 3- end of KCNS3) were associated with a lower prevalence of daytime sleepiness at baseline (OR 0.76 [0.64-0.90], P value = 1.4E-3; OR 0.47 [0.32-0.68], P value = 8.4E-5; respectively). Among the 9 associations with analysis-wide significance, 3 were significant after adjusting for 88 variants (raw P value <5.68E-4), and 1 among them had testwide significance (raw P value <2.47E-5). Figure 3 shows the strength of the associations for the selected variants with associations of analyses-wide significance in at least 1 analysis. This figure suggests that some risk variants were associated with specific clinical features. For example, rs35749011 was associated with both the HR of cognitive impairment at testwide significance (HR 2.45 [1.64-3.65] for the minor allele, P value = 1.1E-5) and lower MoCA score over time at analyses-wide significance (−1.16 [−1.89 to −0.43], P value = 0.0018). Although it is an intergenic variant whose closest gene is KRTCAP2, the variant is in high linkage disequilibrium (r2 = 0.78) with rs2230288 (GBA p.E365K)19,20 and has a similar spectrum of phenotype associations as rs2230288. Other notable variants with variant-wide significance were rs76904798, the intergenic variant close to the 5’ end of LRRK2, for reaching HY3 (HR 1.32 [1.14-1.54] with the minor allele of T, P value = 3.0E-4), and rs76116224 and the baseline OR of having daytime sleepiness mentioned previously. The detailed information for all of the test results is provided as supplemental material (Supplemental Table 4 and Supplemental Fig. 2).
FIG. 3.
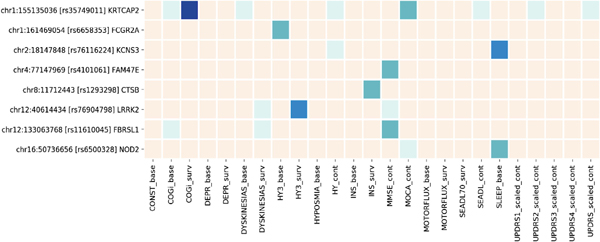
Heatmap of the Parkinson’s disease genome-wide association study (GWAS) loci associated with progression markers. Cream, P value >0.05; light green, P value <0.05; green, P value <0.002; blue, P value <5.68E-4; dark blue, P value <2.47E-5. Suffix of “base” indicates the logistic regression model at baseline, “surv” for the survival analysis during the course, and “cont” for the linear mixed effect model for continuous outcome analyzed by linear mixed model. CONST, constipation; COGi, cognitive impairment; DEPR, depression; HY3, Hoehn and Yahr score; INS, insomnia; MMSE, Mini-Mental State Examination; MOCA, Montreal Cognitive Assessment; SEADL, the modified Schwab and England Activities of Daily Living Scale; SEADL70, the modified Schwab and England Activities of Daily Living Scale; SLEEP, daytime sleepiness; UPDRS, Unified Parkinson’s Disease Rating Scale or the Movement Disorder Society–revised UPDRS, scaled at the baseline (UPDRS1–3) or during the course.
GBA Protein Coding Variants and APOE Tagging Variants
In the focused analyses for GBA coding variants, rs75548401, GBA p.T408M, was associated with the faster development of HY3 (HR 2.35 [1.58–3.49], P value = 2.5E-5). rs2230288, GBA p.E365K, was associated with the higher odds of having cognitive impairment at baseline (OR 2.05 [1.33–3.18], P value = 1.3E-3), faster development of cognitive impairment (HR 2.58 [1.71–3.89], P value = 5.5E-6), and lower MoCA score at the analysis-wide significance (β −1.23 [−1.97 to −0.50], P value = 1.0E-3) (Fig. 4). We previously reported these associations,21 and we were able to confirm them in our updated analysis with more stringent multiple-testing correction (false discovery rate vs. Bonferroni).
FIG. 4.
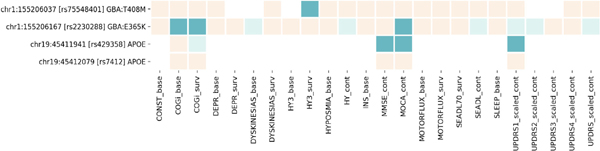
Heatmap of the GBA and APOE variants associated with progression markers. Cream, P value >0.05; light green, P value <0.05; green, Bonferroni-corrected P value <0.05. Suffix of “base” indicates the logistic regression model at baseline, “surv” for the survival analysis during the course, and “cont” for the linear mixed effect model for continuous outcome analyzed by linear mixed model. CONST, constipation; COGi, cognitive impairment; DEPR, depression; HY3, Hoehn and Yahr score; INS, insomnia; MMSE, Mini-Mental State Examination; MOCA, Montreal Cognitive Assessment; SEADL, the modified Schwab and England Activities of Daily Living Scale; SEADL70, the modified Schwab and England Activities of Daily Living Scale; SLEEP, daytime sleepiness; UPDRS, Unified Parkinson’s Disease Rating Scale or the Movement Disorder Society–revised UPDRS, scaled at the baseline (UPDRS1–3) or during the course.
The C allele of rs429358, the tagging variant for the APOE E4 allele, was associated with lower MiniMental State Examination (β −0.20 [−0.33 to −0.07], P value = 2.8E-3) and lower MoCA (β−0.52 [−0.86 to −0.17], P value = 3.4E-3) as expected (Fig. 4). Moreover, it was associated with higher UPDRS part 1 scores (β 0.12 [0.04–0.20] in z score, P value = 4.5E-3). We did not have enough evidence to conclude that the APOE E4 allele was associated with the prevalence of cognitive impairment at baseline (P value = 0.4) or its development during follow-ups (P value = 0.034). The T allele of rs7412, tagging APOE E2, showed no association with these measurements as predicted.
Discussion
We conducted GWAS using longitudinal data from multiple PD cohorts to investigate markers of PD progression and heterogeneity. Of the 25 meta-analyses that we evaluated, we identified 2 variant–phenotype associations with genomewide significance.
We also evaluated the summary statistics to assess clinical value of the variants of interest.
One of our genomewide hits, rs382940, in the intron of SLC44A1, was associated with a faster rate of progression to reach HY3. SLC44A1, soluble carrier 44A1, is also referred to as choline transporter-like protein 1. The gene is ubiquitously expressed in the brain, colon, thyroid, and other organs and is involved in choline transport. No associations with PD and this variant or the gene itself have been reported so far although it has been studied in several vitro and vivo studies.22–25 Further investigation is warranted. The search of the Brain eQTL database suggested that another GWAS signal, rs61863020, was associated with ADRA2A expression, a gene reported to be associated with the arousal/sleep state.26 ADRA2A is consistently expressed in locus coeruleus as well as nigral dopamine neurons and pyramidal neurons of the human brain (http://www.humanbraincode.org/).27
The ADRA2A-encoded alpha2 adrenoreceptor modulates norepinephrine levels. In addition, norepinephrine28 and its receptors29,30 have been linked to PD in multiple model systems. Interestingly, neither of the variants were reported to be associated with the incidence of PD in the recent case-control analysis of PD.3 A case-control study cannot address some mechanisms that contribute to the heterogeneity of PD such as genetic effects only relevant to cases or interactions with PD treatments. The discrepancy between the case-control GWAS and our study may reflect this point.
In the targeted assessments, we confirmed the previous results of the associations between GBA risk variants and motor and cognitive aspects of PD.31–35 In contrast with GBA variants, association studies of APOE and cognitive function in PD have yielded mixed results previously.36–40 Our data supported the association of APOE and cognitive function on the following 2 measurements: Mini-Mental State Examination and MoCA.
The strength of the current study is the hypothesis-free approach of GWAS, which can be powerful in identifying new associations and expanding our biological knowledge base. Although the associations here should be replicated and further investigated with vivo/vitro experiments, these findings suggest the prioritization of the 2 variants and loci for future validations. We have reported all of the summary results on our publicly accessible site to benefit researchers so that they may conduct/replicate the analysis of variants of interest in their own research.
The major limitation of this study is the heterogeneity of the cohorts, which is apparent in several ways: baseline characteristics, definitions of binomial outcomes, patterns for clinical care during the course of follow-up, the platforms for genotyping/sequencing, and sample acquisition/enrollment practices. By meta-analyzing at the dataset-level and exercising careful quality control throughout, we tried to extract the most generalizable and reliable results across cohorts.
Another limitation is the power of the study. Although we have aggregated the largest collection of longitudinal data in PD genetics so far, more data would be needed to identify relatively small differences expected within PD patients when compared with the case-versus-control setting. From the meta-analysis results, we estimated that if we had 30% more participants in the same setting, we would have had at least 1 variant of the genomewide significance (5E-8) in 21 of 25 phenotypes (Supplemental Table 5). In addition, our study results can be a valuable resource for validation and hypothesis testing as we have shown in our targeted analysis. The study website aims to provide other researchers with a tool to explore variants of their interest easily for all included phenotypes (https://pdgenetics.shinyapps.io/pdprogmetagwasbrowser/).
In our survival analysis, we did not explicitly check the proportional hazard assumption. If a variant effect changed over time, the result would be interpreted as the average HR over time. Also, the result would be biased when 1 of the covariates violated the proportional hazard assumption in our model and it was also associated with the variant dosage. Replication is important in this regard as well.
Finally, the study participants were restricted to individuals with European ancestry. We are now striving to collect more data, including from populations that are underrepresented in this study, to improve our understanding of this topic in future studies.
Conclusion
With 4093 participants and 22,307 longitudinal data points over a median of 3.81 years, we performed 25 GWAS meta-analyses. We found 2 genomewide significant signals: the rate to reach HY3 during the disease course and rs382940, and the prevalence of insomnia at baseline and rs61863020. We also conducted targeted assessments of previously published variants of interest using the GWAS results. These results provide valuable insights into how genetic factors contribute to the heterogeneity of PD and disease progression.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments:
We thank all of the study participants and their families and the investigators and members of the following studies: Parkinson Study Group: Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism (DATATOP); Drug Interaction with Genes in Parkinson’s Disease (DIGPD); Harvard Biomarkers Study (HBS); NET-PD_LS1, National Institutes of Health Exploratory Trials in Parkinson’s Disease Large Simple Study 1; Oslo PD study; ParkFit Study; The Norwegian ParkWest Study (PARKWEST); Parkinson’s Disease Biomarker Program (PDBP); Parkinsonism Incidence and Cognitive and Non-motor heterogeneity In Cambridgeshire (PICNICS); Parkinson’s Progression Markers Initiative (PPMI); Parkinson Study Group: Parkinson Research Examination of CEP-1347 Trial (PreCEPT) and its following study (PostCEPT); Profiling Parkinson’s Disease Study (PROPARK); and Morris K. Udall Centers for Parkinson’s Research (Udall). We also thank the several grants and financial supporters of the previous studies. DATATOP was supported by a Public Health Service grant (NS24778) from the National Institute of Neurological Disorders and Stroke (NINDS) and by grants from the General Clinical Research Centers Program of the National Institutes of Health at Columbia University (RR00645), the University of Virginia (RR00847), the University of Pennsylvania (RR00040), the University of Iowa (RR00059), Ohio State University (RR00034), Massachusetts General Hospital (RR01066), the University of Rochester (RR00044), Brown University (RR02038), Oregon Health Sciences University (RR00334), Baylor College of Medicine (RR00350), the University of California (RR00827), Johns Hopkins University (RR00035), the University of Michigan (RR00042), and Washington University (RR00036), the Parkinson’s Disease Foundation at Columbia-Presbyterian Medical Center, the National Parkinson Foundation, the Parkinson Foundation of Canada, the United Parkinson Foundation, Chicago, the American Parkinson’s Disease Association, New York, and the University of Rochester. DIGPD is supported by Assistance Publique Hôpitaux de Paris and funded by a grant from the French Ministry of Health (PHRC 2008, AOM08010) and a grant from the Agence Nationale pour la Sécurité des Médicaments (ANSM 2013). HBS is supported by the Harvard NeuroDiscovery Center, Michael J. Fox Foundation, NINDS U01NS082157, U01NS100603, and the Massachusetts Alzheimer’ Disease Research Center NIA P50AG005134; National Institutes of Health Exploratory Trials in Parkinson’s Disease Large Simple Study 1 was supported by NINDS Grant U01NS043128. OSLO is supported by the Research Council of Norway and South-Eastern Norway Regional Health Authority. ParkFit is supported by ZonMw (the Netherlands Organization for Health Research and Development [75020012]) and the Michael J. Fox Foundation for Parkinson’s Research, VGZ (health insurance company), GlaxoSmithKline, and the National Parkinson Foundation. ParkWest is supported by the Research Council of Norway, the Western Norway Regional Health Authority, Stavanger University Hospital Research Funds, and the Norwegian Parkinson’s Disease Association. PDBP is a consortium with NINDS initiative. PICNICS has received funding from the Cure Parkinson’s Trust and the Van Geest Foundation and is supported by the National Institute of Health Research Cambridge Biomedical Research Centre. PPMI is supported by the Michael J. Fox Foundation for Parkinson’s Research. PreCEPT and PostCEPT were funded by NINDS 5U01NS050095-05 and the Department of Defense Neurotoxin Exposure Treatment Parkinson’s Research Program, Grant W23RRYX7022N606, the Michael J. Fox Foundation for Parkinson’s Research, Parkinson’s Disease Foundation, Lundbeck Pharmaceuticals, Cephalon Inc, Lundbeck Inc, John Blume Foundation, Smart Family Foundation, RJG Foundation, Kinetics Foundation, National Parkinson Foundation, Amarin Neuroscience LTD, CHDI Foundation Inc, National Institutes of Health (NHGRI, NINDS), and the Columbia Parkinson’s Disease Research Center. PROPARK is funded by the Alkemade-Keuls Foundation, Stichting Parkinson Fonds, Parkinson Vereniging, and The Netherlands Organisation for Health Research and Development. Udall is supported by NINDS.
Relevant conflicts of interests/financial disclosures: Hirotaka Iwaki: grants, Michael J. Fox Foundation, Jodi Maple-Grødem: grants, Norwegian Parkinson’s Disease Association, Norwegian Health Association. Jean-Christophe Corvol: advisory boards, Biogen, Air Liquide, BrainEver, Theranexus, BMS, Zambon, Pfizer, Ipsen, Abbvie; grants, Michael J. Fox Foundation, Actelion, Ipsen. Lasse Pihlstrøm: grants, Norwegian Health Association, South-East Norway Regional Health Authority, Norwegian Parkinson Research Fund, Michael J. Fox Foundation. Khanh-Dung H. Nguyen: stock ownership in medically related fields, Biotech/Pharmaceutical Industry. Kirsten M. Scott: grants, Wellcome Trust PhD Fellowship. Vivianna M. Van Deerlin: grants, National Institutes of Health NS-053488. Aaron G. Day-Williams: stock ownership in medically related fields, Biogen, Merck. Alexis Brice: advisory boards, FWO, ERC; grants, JPND, ANR, Eranet Neuron, Association France Parkinson. Alastair J. Noyce: honoraria, Britannia Pharmaceuticals; grants, Parkinson’s UK (G-1606). Jonathan R. Evans: advisory boards, AbbVie, Global Kinetics, Allergan; honoraria, UCB, Allergan, AbbVie. David P. Breen: grants, Wellcome Clinical Research Career Development Fellowship. Karol Estrada: stock ownership in medically related fields, Biogen. David K. Simon: consultancies, Lysosomal Therapeutics, Inc.; advisory boards, Weston Brain Institute; honoraria, Parkinson Study Group, Harvard Medical School, Michael J. Fox Foundation, Biogen; grants, National Institutes of Health, Weston Brain Institute, Mission Therapeutics, Inc., and BioElectron Technologies. Bernard Ravina: stock ownership in medically related fields, Voyager Therapeutics; consultancies, Michael J. Fox Foundation. Mathias Toft: grants, Research Council of Norway, South-Eastern Norway Regional Health Authority, Michael J. Fox Foundation, Norwegian Parkinson Research Fund. Bastiaan R. Bloem: consultancies, Abbvie, Zambon; advisory boards, Michael J. Fox Foundation; honoraria, speaker fees from Abbvie, Zambon, Bial; grants, Netherlands Organization for Scientific Research, the Michael J. Fox Foundation, UCB, Abbvie, the Stichting Parkinson Fonds, the Hersenstichting Nederland, the Parkinson’s Foundation, Verily Life Sciences, the Topsector Life Sciences and Health, and the Parkinson Vereniging. Daniel Weintraub: consultancies, Acadia, Alkahest, Anavex Life Sciences, BlackThorn Therapeutics, Bracket, Clintrex LLC, Sunovion, Theravance Biopharma, and the CHDI Foundation. Roger A. Barker: consultancies, FCDI, LCT, BlueRock, Nova Nordisk, Cellino, Locate-Bio; royalties, Springer; Wiley; grants, EU, NIHR, PUK, CPT, Rosetrees Trust, MRC, Wellcome Trust, Evelyn Trust. Caroline H. Williams-Gray: grants, MRC Clinician Scientist fellowship, the NIHR Cambridge Biomedical Research Centre and grants from the Michael J. Fox Foundation, the Rosetrees Trust, the Evelyn Trust, and Addenbrookes Charitable Trust. Bart P. van de Warrenburg: advisory boards, member of medical advisory boards patient organizations; royalties, Reed Elsevier (for chapter in Dutch Neurology textbook); grants, Radboud University Medical Centre, ZonMW, Hersenstichting, Bioblast Pharma. Jacobus J. Van Hilten: grants, The Alkemade-Keuls Foundation, Stichting Parkinson Fonds, Parkinson Vereniging, The Netherlands Organisation for Health Research and Development. Clemens R. Scherzer: grants, National Institutes of Health Grants U01NS082157, U01NS095736, U01NS100603; consultancies, Sanofi. Mike A. Nalls: consultancies, Lysosomal Therapies Inc., Vivid Genomics Inc., Kleiner Perkins Caufield & Byers, Neuron23, Inc., Michael J. Fox Foundation.
Funding agencies: The Intramural Research Program the National Institute on Aging (Z01-AG000949-02), Biogen Idec, and the Michael J. Fox Foundation for Parkinson’s Research.
Footnotes
Full financial disclosures and author roles may be found in the online version of this article.
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 2015;30:1591–1601. [DOI] [PubMed] [Google Scholar]
- 2.Keller MF, Saad M, Bras J, et al. Using genome-wide complex trait analysis to quantify “missing heritability” in Parkinson’s disease. Hum Mol Genet 2012;21:4996–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nalls MA, Blauwendraat C, Vallerga CL, et al. Expanding Parkinson’s disease genetics: novel risk loci, genomic context, causal insights and heritable risk [published online ahead of print 2019]. bioRxiv. 10.1101/388165 [DOI] [Google Scholar]
- 4.Leonard H, Blauwendraat C, Krohn L, et al. Genetic variability and potential effects on clinical trial outcomes: perspectives in Parkinson’s disease [published online ahead of print 2018]. bioRxiv. 10.1101/427385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goetz CG, Poewe W, Rascol O, et al. Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: status and recommendations The Movement Disorder Society Task Force on rating scales for Parkinson’s disease. Mov Disord 2004;19:1020–1028. [DOI] [PubMed] [Google Scholar]
- 6.Goetz CG, Fahn S, Martinez-Martin P, et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): process, format, and clinimetric testing plan. Mov Disord 2007;22:41–47. [DOI] [PubMed] [Google Scholar]
- 7.Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699. [DOI] [PubMed] [Google Scholar]
- 8.Nalls MA, Bras J, Hernandez DG, et al. NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging 2015;36:1605.e7–1605.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.International HapMap 3 Consortium, Altshuler DM, Gibbs RA, et al. Integrating common and rare genetic variation in diverse human populations. Nature, 2010;467:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das S, Forer L, Schönherr S, et al. Next-generation genotype imputation service and methods. Nat Genet 2016;48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data: Table 1. Bioinformatics 2016;32:1423–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Säfken B, Rügamer D, Kneib T, Greven S. Conditional model selection in mixed-effects models with cAIC4 [published online ahead of print March 15, 2018]. arXiv:1803.05664. [Google Scholar]
- 14.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26: 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010; 26:2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Võsa U, Claringbould P, Westra H-J, et al. Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis [published online ahead of print October 19, 2018]. bioRxiv. 10.1101/447367 [DOI] [Google Scholar]
- 18.Qi T, Wu Y, Zeng J, et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun 2018;9:2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berge-Seidl V, Pihlstrøm L, Maple-Grødem J, et al. The GBA variant E326K is associated with Parkinson’s disease and explains a genome-wide association signal. Neurosci Lett 2017;658:48–52. [DOI] [PubMed] [Google Scholar]
- 20.Blauwendraat C, Bras JM, Nalls MA, Lewis PA, Hernandez DG, Singleton AB. Coding variation in GBA explains the majority of the SYT11-GBA Parkinson’s disease GWAS locus. Mov Disord 2018; 33:1821–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwaki H, Blauwendraat C, Leonard HL, et al. Genetic risk of Parkinson disease and progression: Neurol Genet 2019;5:e348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Machová E, O’Regan S, Newcombe J, et al. Detection of choline transporter-like 1 protein CTL1 in neuroblastoma × glioma cells and in the CNS, and its role in choline uptake. J Neurochem 2009; 110:1297–1309. [DOI] [PubMed] [Google Scholar]
- 23.Schenkel LC, Singh RK, Michel V, et al. Mechanism of choline deficiency and membrane alteration in postural orthostatic tachycardia syndrome primary skin fibroblasts. FASEB J 2015;29:1663–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heffernan C, Jain MR, Liu T, et al. Nectin-like 4 complexes with choline transporter-like protein-1 and regulates schwann cell choline homeostasis and lipid biogenesis in vitro. J Biol Chem 2017;292:4484–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao P, He M, Zhang C, Geng C. Integrated analysis of gene expression signatures associated with colon cancer from three datasets. Gene 2018;654:95–102. [DOI] [PubMed] [Google Scholar]
- 26.Gelegen C, Gent TC, Ferretti V, et al. Staying awake—a genetic region that hinders α 2 adrenergic receptor agonist-induced sleep. Eur J Neurosci 2014;40:2311–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong X, Liao Z, Gritsch D, et al. Enhancers active in dopamine neurons are a primary link between genetic variation and neuropsychiatric disease. Nat Neurosci 2018;21:1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tong J, Hornykiewicz O, Kish SJ. Inverse relationship between brain noradrenaline level and dopamine loss in Parkinson disease. Arch Neurol 2006;63:1724. [DOI] [PubMed] [Google Scholar]
- 29.Srinivasan J, Schmidt WJ. Treatment with α2-adrenoceptor antagonist, 2-methoxy idazoxan, protects 6-hydroxydopamine-induced Parkinsonian symptoms in rats: neurochemical and behavioral evidence. Behav Brain Res 2004;154:353–363. [DOI] [PubMed] [Google Scholar]
- 30.Mittal S, Bjørnevik K, Im DS, et al. β2-adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017;357:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winder-Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013;136:392–399. [DOI] [PubMed] [Google Scholar]
- 32.Brockmann K, Srulijes K, Pflederer S, et al. GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015;30:407–411. [DOI] [PubMed] [Google Scholar]
- 33.Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol Aging 2016;37:209.e1–209.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis MY, Johnson CO, Leverenz JB, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 2016;73:1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher’ mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 2016;80:674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang X, Chen P, Kaufer DI, Tröster AI, Poole C. Apolipoprotein E and dementia in Parkinson disease. Arch Neurol 2006;63:189. [DOI] [PubMed] [Google Scholar]
- 37.Kurz MW, Dekomien G, Nilsen OB, Larsen JP, Aarsland D, Alves G. APOE alleles in Parkinson disease and their relationship to cognitive decline: a population-based, longitudinal study. J Geriatr Psychiatry Neurol 2009;22:166–170. [DOI] [PubMed] [Google Scholar]
- 38.Federoff M, Jimenez-Rolando B, Nalls MA, Singleton AB. A large study reveals no association between APOE and Parkinson’s disease. Neurobiol Dis 2012;46:389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mata IF, Leverenz JB, Weintraub D, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol 2014;71:1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paul KC, Rausch R, Creek MM, et al. APOE, MAPT, and COMT and Parkinson’s disease susceptibility and cognitive symptom progression. J Parkinsons Dis 2016;6:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The summary statistics of the meta-analysis results, including the ones that were not evaluated in this article, are publicly available for convenient browsing and downloading (https://pdgenetics.shinyapps.io/pdprogmetagwasbrowser/).