

Abstract
Genome-scale models (GEMs) of bacterial strains’ metabolism have been formulated and used over the past 20 years. Recently, with the number of genome sequences exponentially increasing, multi-strain GEMs have proved valuable to define the properties of a species. Here, through four major stages, we extend the original Protocol used to generate a GEM for a single strain to enable multi-strain GEMs: 1) Obtain or generate a high-quality model of a reference strain; 2) Compare the genome sequence between a reference strain and target strains to generate a homology matrix; 3) Generate draft strain-specific models from the homology matrix; 4) Manually curate draft models. These multi-strain GEMs can be used to study pan metabolic capabilities and strain-specific differences across a species, thus providing insights into its range of lifestyles. Unlike the original Protocol, this procedure is scalable and can be partly automated with the supplementary jupyter notebook tutorial. This Protocol Extension joins the ranks of other comparable methods for generating models such as CarveMe and KBase. This extension of the original Protocol takes in the order of weeks to multiple months to complete depending on the availability of a suitable reference model.
Keywords: Genome scale reconstruction, multi-strain model, multi-strain, Genome-scale model, GEM, metabolism, bacterial metabolism, metabolic network, metabolic network reconstruction, mathematical modeling, mathematical model, metabolic modeling
EDITORIAL SUMMARY
In this Protocol Extension, the Authors extend their original Protocol used to generate a genome-scale metabolic model for a single strain to enable multi-strain models to be made, which can be used to study pan metabolic capabilities and strain-specific differences across a species.
Introduction
In recent years, the exponential increase in the number of genome sequences has enabled us to investigate the variability across strains within the same species. As more genome sequences become available, significant differences in genomic content and functions across strains have been identified1. Therefore, researchers started to explore strain-specific variations using approaches such as pan-genome analyses2. These analyses showed that some species have a vast diversity of genes among its strains, resulting in remarkably different divergent phenotypes across strains3. However, despite the utility of pan-genome analysis based on gene lists, it does not provide mechanistic insight into phenotypic potential based on genetic and genomic variability within a species.
Over the past decade, genome-scale models (GEMs) of metabolism have proven to be valuable in understanding mechanistic links between genotype and phenotype4. GEMs are mathematical models of metabolic network reconstructions5. They allow computation of the systems-level metabolic functions from genome sequences and extend the power of pan-genome analyses towards sequence-based evaluation of the phenotypic variation of a species. So far, the majority of studies based on metabolic network reconstructions, and GEMs derived from them, have been focused on a single strain of a species. This includes a large number of studies based on our previously published metabolic network reconstruction Protocol6.
A strain-specific GEM can be expanded into models for multiple strains of the same species. Rapid mapping of the gene content in a GEM from a reference strain onto multiple strains’ genome sequences of interest is now possible. This process allows one to utilize highly curated knowledge bases assembled over many decades, upon which a metabolic reconstruction is based, to quickly study a freshly sequenced isolate. Using this process, recent studies have successfully identified strain-specific metabolic differences and their association with lifestyle of the strains for multiple species7–12. These studies lead to an understanding of strain diversity, for species with both large and small pangenomes. Using GEMs to characterize pan-genomes is thus likely to be a widely used method as thousands of strain sequences will become available for species across the microbial phylogenetic tree.
It is worth noting that other methods for the generation of GEMs from existing reconstructions are available, namely CarveMe 13 and functions within KBase14 (Supplementary Table 1). CarveMe relies on the use of a universal model that is then filtered to a specific model by solving a mixed integer linear program. KBase executes a proteome comparison and utilizes that information to infer reactions to keep within a new model. The reliance on the universal model within CarveMe may limit the achievable specificity particularly in regard to biomass equations. CarveMe also possesses the unique functionality to produce ensemble models and microbial community models. KBase benefits from ease-of-use and potential integration with other KBase functions, however the implementation is restricted to the KBase interface and limited in customizability.
In this Protocol, we extend our original metabolic reconstruction Protocol6 to instruct users on building multi-strain GEMs from an existing reference model. We will provide guidance for the reconstruction and application of strain-specific models and show how a reference strain is mapped to other strains within the same species. Furthermore, we provide a detailed tutorial (Supplementary Tutorial) along with the step-by-step instructions to guide readers through the Protocol and its efficient implementation. The application of the workflow is rapid, and it can be partly automated.
Applications
A highly curated reference reconstruction represents a highly organized and structured assembly of organism information. This accumulated knowledge can be efficiently extended to generate strain-specific models by combining comparative genomics and genome-scale metabolic modeling (Figure 1). By analyzing multiple strains, it becomes possible to investigate the range of evolutionary outcomes for a species. GEMs allow for the prediction of growth capabilities and auxotrophies across a bacterial species. These predictions have provided insight into the lifestyle and diversity of the members of a species. For example, metabolic capabilities predicted using multi-strain GEMs have been used to build classification schema capable of organizing strains into nutrient niche7, serovars9, and pathogenicity12. Multi-strain GEMs provide a platform with which to begin to combat limitations identified with reconstruction efforts13 regarding completeness and the coverage of the reactome.
Figure 1: Applications of multi-strain GEMs:
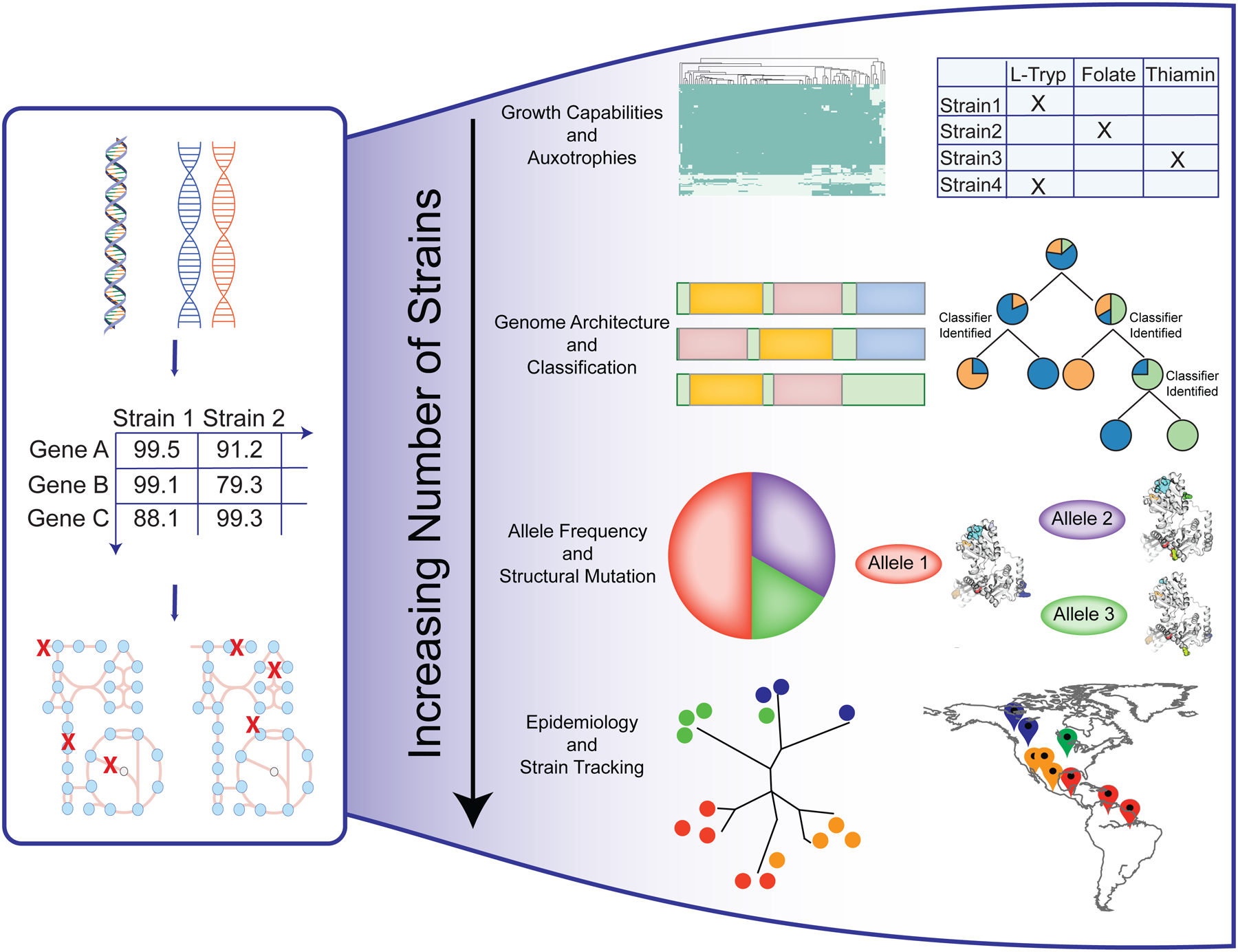
The workflow of genome comparison to generate a homology matrix of percentage identity values (PIDs), which is in turn used to generate strain-specific models of the target strains. The number of strains considered in this fashion enables various types of analyses including: 1) comparison of strain nutrient utilization and identification of strain-specific auxotrophies; 2) interrogation of genome architecture and classification of strains by niche or by pathotype; 3) investigation of allele frequencies among strains and mapping to protein structural information; and, 4) linking to epidemiology and tracking of strains/infections.
Another inherent strength of multi-strain reconstruction is scalability. The number of strains considered may be increased with ease. Scalability, in turn, enables new applications. On the order of hundreds of strains, it becomes possible to use multi-strain GEMs to investigate allele frequencies of genes within a network context14. The reconstructed networks provide insight into potential evolutionary hotspots that become linked to calculated phenotypes through the use of GEMs14. Additionally, the higher number of strains considered allows for applications with a wider perspective. For example, studying the global epidemiology of infection and the tracking of strains by their indicated abilities and classifications become possible. It is worth noting that the number of strains considered may also potentially influence the complexity and time for downstream analysis. Preliminary results become rapidly available through this approach, however if additional strains are candidates for extensive curation this increases the time required for future analysis.
Advantages and limitations
Multi-strain GEMs provide us with a comprehensive and high-resolution knowledge base of metabolic diversity across strains of a species of interest. The models enable accurate and rapid computational prediction of auxotrophies and nutrient utilization capability across strains from only genome sequences without the need for experiments. The results then allow us to calculate correlations between strain-specific metabolic variations and attributes of the strain’s lifestyle (such as host specificity) or health outcomes such as strain-specific implications in inflammatory bowel disease7,8,12,15. The reconstruction of multi-strain GEMs is much faster than reconstructing a reference model from scratch, yet still highly informative.
However, the user should also keep in mind the limitations before starting the multi-strain GEM reconstruction. First, it can be time-consuming to build multi-strain GEMs for species lacking a reference model, as approximately six months to a year is needed to build a GEM de novo. Second, this Protocol Extension works best with well-annotated species, since a lack of information may result in an incomplete model and inaccurate predictions. Nevertheless, strain-specific GEMs will also enable the discovery of knowledge gaps for less well-studied species. Third, multi-strain GEMs will be most valuable for species with significant differences in genomic content across strains. If strains within the species have limited genetic variability, the strain-specific GEM will be very similar and provide limited new information. Such similarity can be quickly evaluated by examining the openness of the pan-genome for the strains of interest. Finally, basic coding skills are required for this Protocol Extension. Previous experience with bioinformatics analysis, coding languages (especially python), and usage of GEMs will accelerate the process significantly.
Experimental Design
This Protocol Extension consists of four major stages to utilize the output of a high-quality genome-scale metabolic reconstruction6 to create multiple strain-specific models derived from the reference organism (Figure 2). These stages are described further in the following sections. These stages are also summarized within a pseudocode format (Supplementary Methods) Following the steps delineated here will result in draft strain-specific models based on genetic similarity to the original strain that can be used as a starting point to feed directly into Stage 2 of the original Protocol6 for further refinement and evaluation, or for immediate comparative investigation. The time-consuming nature of the base reconstruction approach of the original Protocol6 results in limited scalability; this approach of generating models for multiple strains through homology relationships represents a means of more rapidly extrapolating the knowledge contained within the highly-curated reconstruction. One caveat to consider when applying this approach is the metabolic diversity inherent to the species of interest. If the species is not particularly genetically diverse, then the resulting models will likewise be highly similar.
Figure 2:
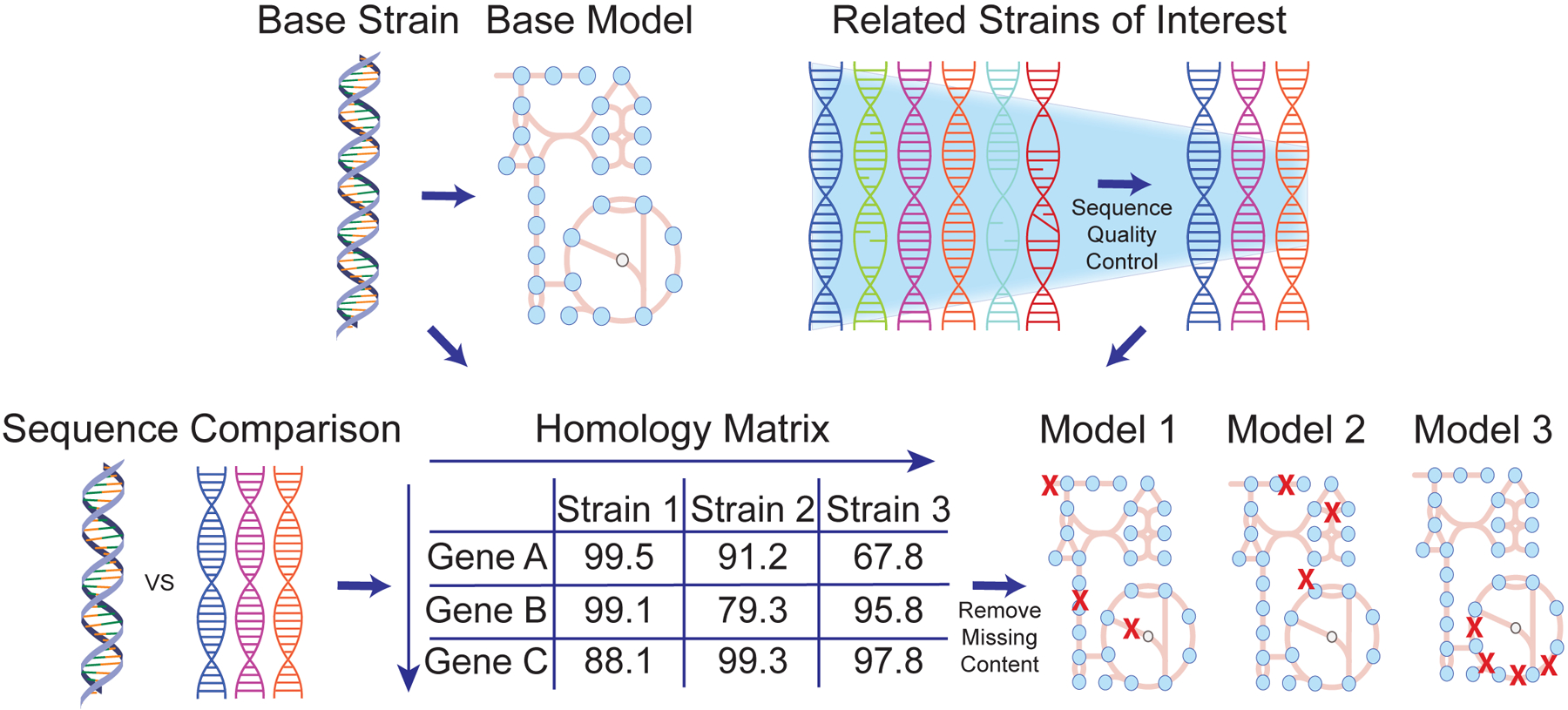
Overall workflow for multi-strain GEMs generation.
Along with the step-by-step procedures in this Protocol Extension, we also provide a tutorial (Supplementary Tutorial) to generate strain-specific models for five E. coli strains from a reference model. The Supplementary Tutorial includes 3 jupyter notebooks that are focused on stages 2 and 3 (the genome sequence comparison and generation of homology matrix stage, and the creation of strain-specific draft models) to guide the steps that could be automated in this Protocol Extension.
Overview of the procedure
Stage 1: Steps 1–4 Obtain a high-quality genome-scale starting reference reconstruction.
To generate strain-specific reconstructions, a high-quality single-strain base reconstruction generated through the use of the original Protocol6 is a necessary starting point. Published reconstruction efforts usually include this output as a supplementary data file in either SBML or JSON file formats. Additionally, a number of reconstruction repositories exist, such as BiGG, BioModels, and MetaNetX16–18. If a reconstruction for a reference strain in the species of interest is not available, then the original Protocol can be executed to produce one6. The resulting output can then be used as the starting point to generate multi-strain models. It is possible that for certain organisms there could be multiple available models that have been independently reconstructed. This represents a potential opportunity to broaden the reference knowledgebase. In this case the user can either reconcile the base reconstructions for a single strain into a single reconstruction of highest confidence through careful manual curation of the content or run this Protocol Extension using each base reconstruction in turn and compare the resulting draft models of interest. After obtaining (or generating) a reference reconstruction, it is necessary to evaluate its quality to determine its suitability for use as a reference reconstruction. To evaluate the reference reconstruction, refer to Stage 4 of the original Protocol6. Recently, a testing suite called Memote has become available that evaluates a number of quality control/quality assurance features of a GEM in a drag and drop fashion19. Once a curated, quality reference reconstruction is either obtained or generated, it can be used in the following steps to generate strain-specific GEMs.
Stage 2: Steps 5–13 Genome Sequence Comparison and Generation of Homology Matrix
Stage 2 is to identify and acquire the sequenced genomes of different strains from the species of interest. Publicly available genome data is available in sources such as NCBI or PATRIC20,21. How many and which strains to include depends on the given research question posed. Criteria for genome selection could possibly include particular isolation location, existence of associated metadata, and phenotype or pathotype information. One should keep in mind the phylogenetic distance between reference strain and target strains as this will directly impact the utility of mapping the content of the original reconstruction. As a means of quality assurance, it is important to keep track of the identifiers of the publicly available genomes used. Within Notebook 1 (Supplementary Tutorial) we begin by acquiring a small set of E. coli genomes from NCBI. In the described workflow and corresponding tutorials, we assume that the user is starting with annotated GenBank files for the strains of interest (see Box 1).
Box 1: A commentary on genome annotation and assembly:
Genome annotation and assembly are both well documented and established techniques within the bioinformatics field42 If the research effort is using publicly available genomes, most will likely be annotated. However, when utilizing newly sequenced genomes or those lacking annotation, it is necessary to perform annotation. While a plethora of tools exist for executing genome annotation43,44, it is important to use a consistent tool to prevent potential errors/bias. One potentially useful annotation software package is Prokka45. If one is interested in following this Protocol Extension to generate models of newly sequenced strains it will also be necessary to perform genome assembly. This raises the question of the sequence quality required to generate multi-strain models. One means of assessing quality is through coverage. While the specific requirement may vary from species to species, we analyzed how varying coverage impacts the resulting assembly metrics of N50 and number of contigs (Supplementary Figure 2). For the purposes of using an assembled genome, it is important to have sufficient coverage that demonstrates saturation in these metrics. In the case of the E. coli strain discussed in the supplementary figure 2 we see this to be at around 70X coverage.
After identifying and obtaining the genomes for the target strains of interest, the next step is to identify the orthologous genes between each strain and the reference strain. This step is detailed within Notebook 1 (Supplementary Tutorial). While a plethora of techniques exist to perform this function, we recommend utilizing NCBI protein BLAST to identify bidirectional best hits as it is widely adopted by the community, scriptable, and reliable. This method is utilized within the provided scripts (Supplementary Tutorial). Following the identification of homologous genes in each of the target strains, the results can be unified into a single Pandas dataframe of the percentage identity values (PID). This dataframe is then filtered down to contain all the genes within the reference reconstruction. The output of these steps is the homology matrix consisting of N x M PIDs, where there are N rows of the genes within the reference reconstruction and M columns of the target strains (Figure 2). The penultimate step is to apply a threshold to binarize this matrix into a presence/absence matrix detailing which genes are absent within the target strain. We suggest utilizing a cutoff of 80% percentage sequence identity covering at least 25% of the query gene length or above to consider the gene present within the target strain. However, this threshold is an adjustable parameter and the effect of genes retained in draft strain-specific models is dependent on how genetically similar the target strains are to the reference strain (Supplementary Figure 1).
A supplementary final step is to execute a nucleotide BLAST. Many reference genomes have undergone extensive manual curation within the annotation, so there may be discrepancies with automatically annotated target strains. By executing a BLAST on raw nucleotide sequences there is a secondary comparison made to catch potentially unannotated open reading frames within a given target strain. In addition, for each open reading frame (ORF) identified to pass the nucleotide sequence similarity threshold but missing from the annotations, a quality check for premature stop codons within the sequence is performed as these ORFs likely result in a nonfunctional protein. This process is also detailed within Notebook 1 (Supplementary Tutorial). The nucleotide BLAST provides an added catch to avoid excluding genes from strain-specific models due to lack of annotation. The final binarized homology matrix can then be used in concert with COBRA methods22–29 to create and save strain-specific models of the target strains.
Stage 3: Steps 14–23 Creation of Strain-Specific Draft Models
The genome comparison executed in Stage 2 provides information on which genes within the base reconstruction are lacking a homologous gene within each target strain genome. By utilizing the “remove_genes” function from the “cobra.manipulation.delete” module of COBRApy, the appropriate genes can be removed from a model. Notebook 2 (Supplementary Tutorial) demonstrates how to properly implement this technique. For every target strain of interest, a copy of the base reconstruction is created and appropriate genes, as per the homology matrix, are deleted from each model, creating a draft strain-specific model. This process is repeated for each strain of interest. Additionally, the genes retained in each strain-specific model are updated at this stage to reflect the locus_tags in the target strain’s annotation. This process is executed using the “rename_genes” function from the “cobra.manipulation.modify” module according to another generated matrix of all the gene names, mapping the gene identifiers constructed within Stage 2. Depending on the annotation platform it may be worthwhile to add additional locus tag information to stratify multiple namespaces. For example, if the genomes used were re-annotated with Prokka it could be useful to add NCBI locus tags to the gene objects within the model. Additional information can be stored within the “notes” field of a gene object. The updated draft models are then ready for further evaluation.
The next step is functional evaluation of the draft strain-specific models and this begins by determining which of them are able to be optimized through linear programming for biomass objective flux, i.e., in silico growth. At this point, a combination of automated gap-filling methods and manual curation are used to determine which nutrients need to be supplemented to the in silico media to achieve positive biomass yield. Gap-filling methods have been well documented30–33, and the results generated can be used to enable growth in strain-specific models found to have auxotrophies.
This step is executed in an iterative fashion across all target strains and reflects a critical step in any reconstruction effort. It is important to keep in mind the differences between the two model types to be gap-filled: 1) models of true auxotrophs that require only a supplementation of extracellular nutrient to enable biomass production and 2) models in which metabolic reaction gap-filling is necessary and thus offers a potential for discovery of alternative pathways. Ideally, gap-filling should always be supported by literature information and/or validated experimentally. In this context, we refer to the gap-filling required to obtain a functional network that can produce biomass. It is also worth noting that in some cases where there are known biomass composition variants, instead of gap-filling the model to enable growth, the biomass reactions should be modified. Alternate biomass formulations may substantially affect model predictions and have been shown to be variable across species and conditions34. The base biomass reaction may also be highly variable across strains in certain species. For example, O antigen structures are highly variable across Gram-negative strains and the corresponding biosynthetic pathways vary extensively, requiring a separate pan-species reconstruction effort.9 Therefore, instead of directly taking the biomass reaction from the base strain, we recommend that the users customize biomass reaction for strains of interest by generating or collecting strain-specific experimental data, when available. A recently developed workflow can also help users generate the biomass reactions in a data-driven and unbiased fashion34.
Stage 4: Steps 24–28 Curation of Strain-Specific Models
At this juncture, a group of functional models for the identified target strains has been produced, and may be used in their current form to generate preliminary predictions and direct future studies. Any known strain capabilities present an opportunity to perform a validation step to inspect whether the strain-specific models can still accurately predict known phenotypes. Additionally, all, or select models depending on interest and/or time constraints, can now be extensively manually curated as per the original Protocol6 to produce a high-quality reconstruction. In this case, the models produced would be used as input to the original Protocol6 at Stage 2: Reconstruction Refinement. This would refine these models from derivative draft strain-specific models to curated reconstructions of specific strains. This effort will involve adding strain-specific metabolism not present in the original reconstruction. One useful technique here would be to annotate the pangenome to potentially catch genes with divergent nucleotide sequence but similar functional machinery which may have appeared due to horizontal gene transfer events. While additional manual curation of the generated strain-specific models would yield more accurate predictions, it is worth noting that the group of draft models represents a valuable resource.
Various analyses can be conducted such as determining differing growth capabilities across nutrient environments. An example of this for carbon source utilization is demonstrated in Notebook 3 (Supplementary Tutorial). In this analysis, growth in different nutrient conditions can quickly be predicted. Starting from a minimal media condition, the current growth-supporting nutrients for carbon, nitrogen, phosphorous, or sulfur can be removed, and an appropriate list of nutrients looped through to determine whether alternative sources of carbon, nitrogen, phosphorus, and sulfur support growth. This process is repeated for each strain in the group of strain-specific models. Experimental validation of the multi-strain predictions is ideal. The resulting in silico predicted growth capabilities can then be used to examine which strains are similar in terms of metabolic phenotype. This approach has proven fruitful in providing an additional level of discrimination in numerous past studies and represents one of the immediate benefits of extending a reconstruction to construct strain-specific models.
Stage 5: Applications of Multi-Strain GEMs
Once a collection of functional models of the identified target strains has been generated, they can be used in a variety of ways (see Applications). This fifth stage includes a range of techniques to select from, determined by the research to be conducted. Given the breadth of the potential applications, they are not addressed in this Protocol Extension.
Materials
Annotated genome sequences of interest
Annotated sequences of interest can either be downloaded from public databases or generated by the user through sequencing. In this Protocol Extension, we start with annotated GenBank files that contain the annotation and sequence sections that can be directly downloaded from NCBI. Several other guides document how to assemble and annotate genomes of interest34,35.
Reference GEM
Reference GEMs have already been reconstructed for many well studied organisms (see Supplementary Table 1). The available GEMs can mostly be found and downloaded from publications or public databases such as BiGG Models16. Reference models can be in various formats such as SBML, MAT and JSON. If the Reference model has not been built for the species of interest, please refer to the original Protocol6 for details of building a detailed, reference reconstruction.
- Equipment Standard personal computer with the following software/packages properly installed:
- BLAST (v 2.9.0 tested)
- Python (v 3.5.2 tested)
Software
- Python Packages: pandas (v0.23.0 tested), seaborn (v0.8.1 tested), biopython (1.71 tested), jupyter notebook (v5.2.3 tested)
- All python packages can be installed directly with pip command. If the users are more comfortable with anaconda, all packages are available in anaconda installation as well.
- CobraPy: the installation steps and tutorial can be found on https://cobrapy.readthedocs.io/en/latest/. To ensure the performance of the scripts in the Supplementary Tutorial, use version 0.13.0
Procedure
Stage 1: Reconstruction of base model
Timing: 6 months - 1 year
-
1
Obtain reference model. Download a reference model from BiGG Models (http://bigg.ucsd.edu/), publications or other databases (see Supplementary Table 1). The resulting draft strain-specific models will reflect the namespace of the base reconstruction. CRITICAL STEP: Models in the BiGG database16 have been pre-checked for quality, so it is a recommended resource if your organism of interest is available. While BiGG is recommended, any consistent reconstruction where the gene product rules are linked to a genome annotation, producing a model that can be loaded to COBRApy will work within this Protocol Extension.
-
2
Build reference model if not available. If the reference model is not available, reconstruct a model from scratch following the original reconstruction Protocol6 or start from draft models reconstructed in previous studies36,37 (see Supplementary Table 2) and follow the original Protocol6.
-
3
Quality control. Regardless of the source, perform quality control analysis on the base model by uploading the model to Memote (https://memote.io/)19 for quality checking. Once the report is available, check the following two important measures: 1. All metrics in the consistency section 2. Uniform Metabolite Identifier Namespace. These metrics ensure that the model is properly standardized. In addition, check if the model is functional by performing growth simulations to ensure firstly that there is no growth when exchange reactions are closed. Refer to the computational method developed by Fritzmeier et al. 38 to identify and remove erroneous energy-generating cycles. And secondly that the growth prediction is consistent with experimental observations including nutrient utilization and metabolite secretion (if data available).
CRITICAL STEP: The quality of the multi-strain models generated from this Protocol Extension will be highly dependent on the reference model. So, it is especially important to start with a high-quality reconstruction and experimentally validated model.
-
4
Obtain base strain genome annotation. Download the reference strain genome annotation. Retrieve the GenBank file that contains the genome sequence and annotation, which were originally used to reconstruct the reference GEM and extract the modeled coding DNA sequences and corresponding unique locus tags. CRITICAL STEP: This is important because the creation of draft multi-strain model is depended on the sequence annotation of the base strain.
Stage 2: Sequence comparison & generation of homology matrix
Timing: days - weeks
-
5
Download annotated genomes for different strains of interest in GenBank format. Genomes of interest can be downloaded from various public databases such as National Center for Biotechnology Information (NCBI) and Pathosystems Resource Integration Center (PATRIC)20. Instructions to download annotated genome sequences from NCBI can be found here: https://www.ncbi.nlm.nih.gov/guide/howto/dwn-genome/, and instructions to download genome sequences from PATRIC can be found here: https://docs.patricbrc.org/user_guides/data/index.html#download-data. Or users can follow the Supplementary Tutorial to download the GenBank files using jupyter notebooks.
!CAUTION: We recommend downloading GenBank files that contain both sequence and annotation information. If annotation is not available for the target strains, see Box 1 for our recommendations and tips on genome annotation. To ensure consistency, the annotation pipeline used for target strains should be the same as the pipeline used for reference strain.
-
6
Quality control of the genome sequences. Calculate and check the coverage (if available), N50 score and number of contigs of the genome sequences. To determine the threshold for the above quality metrics, consider performing similar analysis shown in Supplementary Figure 1. Discard genome sequences that do not pass the quality test.
CRITICAL STEP: More reliable results can be obtained from genome sequences with coverage > 70x. Adjust the threshold for quality metrics such as N50 score and number of contigs based on your organism of interest, as they are highly dependent on the organism. If time permits, use sensitivity analysis 38 to find the most appropriate threshold.
-
7
Generate Fasta files from GenBank files. Use the Genbank files to generate fasta files for both protein and nucleotide sequences (see Notebook 1 in Supplementary Tutorial). Protein fasta files are then used as input for the following BLAST operation in Step 9 to identify homologous proteins across strains.
-
8
Identify candidate metabolic functions. The previous genome annotation (Step 4) should provide E.C. numbers for genes involved in metabolic function. Extract genes with E.C. numbers from annotations and the following steps are focused on these metabolic genes only.
-
9
BLAST the genomes of interest against the reference strain. Perform bidirectional protein BLAST39 to identify the sequence similarity of metabolic proteins in strains of interest compared to the reference strain. Use BLASTp (output format 6) to record both query/subject ID and percentage identity matches (PID).
CRITICAL STEP: Bidirectional BLAST uses both the reference strain or the other strain as reference BLAST database and selects the best bidirectional hits (BBH) based on BLAST result in both directions to identify orthologs. Note that we recommend filtering mapping results based on coverage of alignment length. (see Notebook1 in Supplementary Tutorial)
-
10
Filter the BLAST result for only proteins in the base model. Identify the list of proteins included in the base model and keep only the BLAST results for these protein genes for the following analysis.
-
11
Create a homology matrix summarizing the results for all strains of interest. Identify the BBHs of all proteins between reference strain and strains of interest. Compile the PID of all BBHs in the base model for all strains into a homology matrix, where the columns represent the strains, and the rows represent the protein.
-
12
Create binarized homology matrix for genes in the model. Select a threshold for PID to determine the presence/absence of proteins in all strains. The matrix is binary with 1 representing presence, and 0 representing absence. Similar to the homology matrix, it should have M strains * N proteins.
CRITICAL STEP: Adjust the threshold for PID accordingly depending on your data and purpose (see Supplementary Figure 1 for how PID threshold affects the number of genes retained in strain-specific models). The threshold of 80% used in the Supplementary Tutorial is quite stringent as some tools use the sequence identity cutoff of 50% to identify gene orthologs40.
-
13
Nucleotide BLAST to check unannotated open reading frames. To ensure that we do not miss any genes in the target strains due to lack of annotation during BLASTp, we perform nucleotide BLAST between the reference strain and nucleotide sequences of the target strains (fna files containing contigs). In addition, we also look for premature stop codons in genes of interest to exclude non-functional proteins. Record any inconsistencies observed in gene absence/present results generated by BLASTp and BLASTn, as they are potential candidates for manual curation.
Stage 3: Creation of draft multi-strain models
Timing: days - weeks
-
14
Identify missing reactions. Based on the presence/absence matrix from Step 12 and the gene-protein-reaction (GPR) established in the reference strain reconstruction, identify the genes missing in each strain and reactions encoded by the missing genes.
-
15
Remove missing genes/reactions. For each strain of interest, start with the reference strain. Remove the identified missing gene/reaction for each strain from the starting base strain using COBRApy function remove_genes. Save the modified model as the draft strain-specific model.
!CAUTION: Multiple functions in COBRApy allow the user to delete reactions, but make sure to use function remove_genes with the the parameter “remove_reactions=True” to remove both the missing genes and reactions.
-
16
Update the GPR in the draft models. Using the query/subject ID obtained in step 9, match the genes in the base model with genes in the strains of interest to update the gene names in the strain-specific model. Optionally, to ensure that all possible encoding genes of a metabolic reaction are included in strain-specific models, one can refer to the full BLAST result from Step 9 and identify cases of additional pertinent homologs (potential paralogs) that are not BBH but also pass the PID threshold, and update the GPR accordingly (e.g., “Gene A” -> “Gene A or Gene B”).
-
17
Check biomass reaction. Make sure that the metabolites are general to all strains of interest. Remove the metabolites which are specific to the reference strain or its unique microenvironment. If strain-specific experimental omics data-sets are available, coefficients of metabolites in the biomass reaction could also be adjusted accordingly using the BOFdat workflow41.
-
18
Simulate growth. For each strain-specific model, simulate for growth under the same medium condition as the base model using COBRApy function model.optimize(). A minimal medium condition is preferred if the recipe is available to identify potential auxotrophies. To modify the medium composition, change the constraint on the exchange reactions (see original Protocol Step 37 for more details6). If the simulated growth rate is less than 0.001 and the objective status is “optimal”, skip Steps 19 to 23 and proceed to stage 4 directly. Otherwise continue with Sstep 19. ?Troubleshooting
!CAUTION: Adjust the lower bound of the exchange reactions to allow uptake of extracellular nutrients. Ensure the exchange reactions of the metabolites missing from the medium are closed (lower bound set to 0).
-
19
Identify strain-specific auxotrophies. Simulate biomass yield in a rich medium (set all nutrient exchanges to −5 mmol/gDW/hr). If the yield obtained is less than 0.001 go to Step 22. Otherwise, find the minimal number of nutrient supplementations needed to support in silico growth using the find_nutrient_supplementation function. Review the literature for reports of an experimentally validated auxotrophic phenotype for your strain. If possible, acquire the strain and validate the auxotrophic phenotype experimentally.
-
20
Check and report the genetic basis for the auxotrophy: Retrieve the missing genes identified by “find_nutrient_supplementation” as the genetic basis for the nutrient requirement and run BLASTn as a final quality check to ensure that no matches are found. If no matches are found, supplement the in silico medium for that strain-specific model with one of the sets of nutrients returned by “find_nutrient_supplementation”. If the genes are found, add the reactions back into the strain-specific model, and adjust of the PID threshold used in Step 12 if needed. If positive yield is achieved skip Steps 21–23 and proceed to stage 4.
-
21
Check biomass metabolite synthesis. For strain-specific models which cannot simulate nonzero positive yield, simulate the production of each metabolite in the biomass reaction. To do so, create demand reactions which consume metabolites included in the biomass reaction. A demand reaction is a pseudo-reaction with a lower bound of 0 and upper bound of 1000 which allows for a metabolite to leave the cell. Instead of the biomass reaction, iteratively set one demand reaction as the objective to optimize for the production of each biomass precursor. If the flux through the demand reaction is less than 0.0001, model simulations suggest that this biomass precursor cannot be produced. ?Troubleshooting
-
22
Identify missing essential reaction using gap filling. Use the gapfill function in COBRApy to identify the minimum number of reactions that need to be added to the strain-specific model to enable the production of those biomass precursors which cannot be synthesized. Use the original reference model as the reaction repository to draw reactions from in the gap-filling step. Once the genetic basis for the simulated phenotype is identified, the curator should decide whether to exclude the precursor from the biomass reaction or add the gap filling reactions back. ?Troubleshooting
-
23
Identify genetic evidence for missing essential reactions. For the reactions identified in the previous step, look for evidence in the genome and identify why they were deleted in the previous steps. Adjust sequence similarity threshold if needed and repeat the analysis from Step 12. If no genetic evidence is found, proceed with stage 4 to identify potential strain-specific alternative pathways.
Stage 4: Curation of strain-specific models
Timing: days - weeks
-
24
Identify strain-specific genes absent from reference model. Inspect the genes with E.C. number from strains of interest that are not present in the reference strain. Cross referencing models of related organisms may be helpful in this step.
-
25
Identify novel metabolic reactions. Identify metabolic reactions corresponding to the strain-specific genes identified in Step 23 using public databases including Uniprot (https://www.uniprot.org/), ModelSEED (http://modelseed.org/), KEGG (https://www.genome.jp/kegg/) and BIOCYC (https://biocyc.org/). Add the metabolic reaction to the model using COBRApy (see details in original Protocol6 Steps 6–11). If the reaction is already present in the model, update the GPR of the reaction to include the strain-specific gene.
!CAUTION: Make sure that the metabolite naming scheme for the novel reactions is consistent with the model standard to enable flux simulation through the newly-added reaction.
-
26
Repeat growth simulation. Ensure that draft models which were originally are able to simulate growth can still do so. Check if the models which failed to grow before can now simulate growth with newly-added reactions. If not, add back the missing essential reaction to enable follow-up analysis as it may be due to unknown alternative pathways. Ensure that the model does not have futile cycles after adding new reactions.
CRITICAL STEP: Growth simulation results could have been altered after adding novel strain-specific reactions. So even if the model was predicted to grow in stage 3, double-check here to ensure growth.
-
27
Quality check the models. Following the instructions in the original Protocol6, perform quality check on the models generated including their mass/charge balance, dead-end metabolites/reactions and blocked reactions, etc.?Troubleshooting
-
28
Validate strain-specific models. Perform experiments or collect experimental data from the literature on the metabolic capabilities of the strains of interest. Data useful for validation include known secretion products, growth on different nutrient sources, auxotrophy and knock-out phenotypes (see original Protocol Steps 81 and 82 for details of model validation against experimental observations6). As with all GEMs, better experimental characterization of the strains of interest will improve the in silico results. Thus, increasing the accuracy of the biochemical composition of the biomass function for strains of interest is of value.
!CAUTION: In order to maximize the accuracy of model prediction, ensure the simulation condition (constraint, strain, media) is consistent with the experimental condition.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1.
A troubleshooting table that describes the step, problem, possible reason and solution.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 18 | Strains of interest cannot simulate growth |
|
|
| 22 | Gap-filling failure | Gap-filling algorithm of choice does not produce reasonable or reliable results. | Utilize the gap-filling algorithm provided in Supplementary Tutorial Notebook 3 to identify which missing reactions will enable growth. This approach is designed to work in concert with this Protocol Extension and provides a starting point for manual curation. |
| 22 | No genetic evidence was found for gap-filled reactions |
|
|
| 27 | Experimental results do not match model predictions |
|
|
Timing
The timing of the entire process is estimated under the assumption that the user has basic coding experience and is working with prokaryotes. The timing also depends on multiple factors: 1) Availability of the base model: the timing will be significantly reduced if the user starts with an available and high-quality base model. 2) Number of strains. While a good portion of the workflow can be automated (see Supplementary Tutorial), manual curation is still necessary for each strain-specific model, resulting in longer time needed with increased number of strains. 3) Experience with coding/GEMs. If the user has worked with GEMs and is comfortable with coding (especially python), the timing will be greatly reduced with the help of the Supplementary Tutorial. 4) Computational resources. This factor will only come into play in the BLAST step if the user is working with large genomes and many strains. Otherwise a personal computer should be sufficient.
Stage 1 (Steps 1–4) (base model reconstruction if model not available): 6 months - 1 year depending on the size of the genome, annotation quality and availability of the metabolic knowledge
Stage 2 (Steps 5–13): days to weeks depending on the number of strains and availability of computational resources
Stage 3 (Steps 14–23): days to weeks depending on the number of strains
Stage 4 (Steps 24–28): days to weeks depending on the number of strains
Anticipated results
This Protocol will result in multi-strain genome-scale metabolic models that not only can serve as a comprehensive knowledge base for the species of interest but will also allow computation of metabolic capabilities for different strains from just their genome sequences. Compared to single-strain-based GEMs, multi-strain GEMs can also be queried for strain-specific metabolic genes/reactions. Multi-strain GEMs will also allow various simulations including growth on different nutrient sources and gene knockouts to allow us to obtain a high-resolution understanding of the metabolic phenotypes displayed by different strains.
Supplementary Material
Supplementary Figure 1: Genes Retained Per Strain at Incrementing PID Thresholds. The number of genes retained in each strain-specific model is dependent on the threshold utilized for binarization of the homology matrix. The effect of the threshold will also be dependent on how closely related the target strains are to the reference strain. For example, within the strains in the Supplementary Tutorial notebooks we see that CU651637.1 and CP002167.1 are more dissimilar to reference model iML1515 as the drop off in retained genes occurs in a steeper fashion. We suggest using a threshold of 80 when comparing strains of the same species to ensure a sufficient similarity metric to include a gene in the draft models.
Supplementary Figure 2: Resulting Assembly Statistics at Various Coverage. To investigate the effect of coverage on overall assembly statistics of N50 and Number of Contigs, we randomly sampled reads of the BOP27 strain, which has been sequenced to extremely high coverage (400X), enabling this analysis. Analyzing the resulting assemblies at coverages ranging from 10X to 100X, we see from comparing the metrics that at 70X the assembly quality mostly saturates and as such we recommend included genomes have at least this much coverage.
Supplementary Table 1: Comparison with other Model Generation Tools: We compared our Protocol Extension with existing model generation tools Kbase and CarveMe in terms of their functions, user interfaces, advantages, limitations, and applications in the generation of multi-strain models.
Supplementary Table 2: Relevant Data Sources for Multi-Strain Approach
Supplementary Methods: Pseudo-code for generation of multi-strain GEMs. This pseudo-code provides succinct instructions to produce multi-strain GEMs to facilitate automation and understanding of this Protocol Extension.
Supplementary Tutorial: Three jupyter notebooks detailing the entire Protocol Extension as laid out within the Procedure. The first details executing sequence comparison to generate homology matrix, the second details generation of multi-strain models from the homology matrix, and then the third details the beginning investigation of strain-specific capabilities using the draft models.
Figure 3: Workflow to generate base model from reference genome sequence.
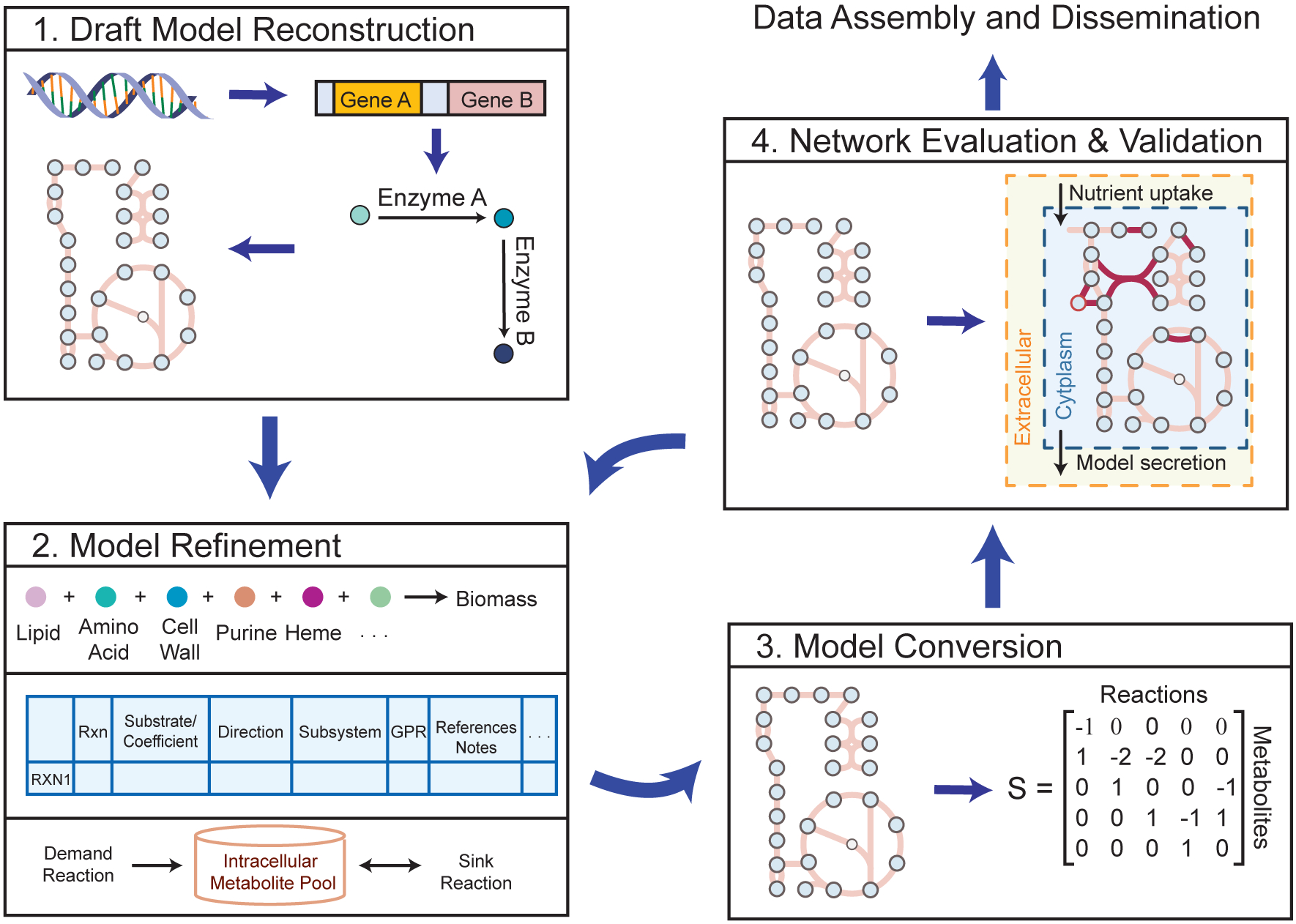
This workflow presents the four major steps involved in reconstructing a reference model as outlined originally by Thiele et al.6.
Acknowledgements:
This research was supported by NIH Grant: 1-U01-AI124316.
Footnotes
Competing interests:
The authors declare no competing interests.
References
- 1.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev 2005; 15: 589–594. [DOI] [PubMed] [Google Scholar]
- 2.Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Current Opinion in Microbiology. 2008; 11: 472–477. [DOI] [PubMed] [Google Scholar]
- 3.Rasko DA, Rosovitz MJ, Myers GSA, Mongodin EF, Fricke WF, Gajer P et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 2008; 190: 6881–6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bordbar A, Monk JM, King ZA, Palsson BO. Constraint-based models predict metabolic and associated cellular functions. Nat Rev Genet 2014; 15: 107–120. [DOI] [PubMed] [Google Scholar]
- 5.O’Brien EJ, Monk JM, Palsson BO. Using Genome-scale Models to Predict Biological Capabilities. Cell 2015; 161: 971–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiele I, Palsson BØ. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc 2010; 5: 93–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monk JM, Charusanti P, Aziz RK, Lerman JA, Premyodhin N, Orth JD et al. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc Natl Acad Sci U S A 2013; 110: 20338–20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang X, Monk JM, Mih N, Du B, Sastry AV, Kavvas E et al. Escherichia coli B2 strains prevalent in inflammatory bowel disease patients have distinct metabolic capabilities that enable colonization of intestinal mucosa. BMC Syst Biol 2018; 12: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seif Y, Kavvas E, Lachance J-C, Yurkovich JT, Nuccio S-P, Fang X et al. Genome-scale metabolic reconstructions of multiple Salmonella strains reveal serovar-specific metabolic traits. Nat Commun 2018; 9: 3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Norsigian CJ, Kavvas E, Seif Y, Palsson BO, Monk JM. iCN718, an Updated and Improved Genome-Scale Metabolic Network Reconstruction of Acinetobacter baumannii AYE. Front Genet 2018; 9: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fouts DE, Matthias MA, Adhikarla H, Adler B, Amorim-Santos L, Berg DE et al. What Makes a Bacterial Species Pathogenic?:Comparative Genomic Analysis of the Genus Leptospira. PLOS Neglected Tropical Diseases. 2016; 10: e0004403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosi E, Monk JM, Aziz RK, Fondi M, Nizet V, Palsson BØ. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc Natl Acad Sci U S A 2016; 113: E3801–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monk J, Nogales J, Palsson BO. Optimizing genome-scale network reconstructions. Nat Biotechnol 2014; 32: 447–452. [DOI] [PubMed] [Google Scholar]
- 14.Monk JM, Lloyd CJ, Brunk E, Mih N, Sastry A, King Z et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat Biotechnol 2017; 35: 904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang X, Monk JM, Nurk S, Akseshina M, Zhu Q, Gemmell C et al. Metagenomics-Based, Strain-Level Analysis of Escherichia coli From a Time-Series of Microbiome Samples From a Crohn’s Disease Patient. Front Microbiol 2018; 9: 2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King ZA, Lu J, Dräger A, Miller P, Federowicz S, Lerman JA et al. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res 2016; 44: D515–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganter M, Bernard T, Moretti S, Stelling J, Pagni M. MetaNetX.org: a website and repository for accessing, analysing and manipulating metabolic networks. Bioinformatics 2013; 29: 815–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Novere N, Bornstein B, Broicher A, Courtot M, Donizelli M, Dharuri H et al. BioModels Database: a free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res 2006; 34: D689–D691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lieven C, Beber ME, Olivier BG, Bergmann FT, Chauhan S, Correia K et al. Memote: A community driven effort towards a standardized genome-scale metabolic model test suite. bioRxiv. 2018; 350991. [Google Scholar]
- 20.Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res 2014; 42: D581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2012; 40: D13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis NE, Nagarajan H, Palsson BO. Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nat Rev Microbiol 2012; 10: 291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ebrahim A, Lerman JA, Palsson BO, Hyduke DR. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst Biol 2013; 7: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heirendt L, Arreckx S, Pfau T, Mendoza SN, Richelle A, Heinken A et al. Creation and analysis of biochemical constraint-based models: the COBRA Toolbox v3.0. arXiv [qbio.QM]. 2017http://arxiv.org/abs/1710.04038. [DOI] [PMC free article] [PubMed]
- 25.Palsson BØ. Systems Biology: Constraint-based Reconstruction and Analysis. Cambridge University Press, 2015. [Google Scholar]
- 26.Becker SA, Feist AM, Mo ML, Hannum G, Palsson BØ, Herrgard MJ. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nature protocols. 2007. March;2(3):727. [DOI] [PubMed] [Google Scholar]
- 27.Orth JD, Thiele I, Palsson BØ. What is flux balance analysis?. Nature biotechnology. 2010. March;28(3):245. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gelius-Dietrich G, Desouki AA, Fritzemeier CJ, Lercher MJ. Sybil–efficient constraint-based modelling in R. BMC systems biology. 2013. December;7(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gelius-Dietrich G, Desouki AA, Fritzemeier CJ, Lercher MJ. Sybil–efficient constraint-based modelling in R. BMC systems biology. 2013. December;7(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]; Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2. 0. Nature protocols. 2011. September;6(9):1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2. 0. Nature protocols. 2011. September;6(9):1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orth JD, Palsson BØ. Systematizing the generation of missing metabolic knowledge. Biotechnol Bioeng 2010; 107: 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan S, Reed JL. Advances in gap-filling genome-scale metabolic models and model-driven experiments lead to novel metabolic discoveries. Curr Opin Biotechnol 2018; 51: 103–108. [DOI] [PubMed] [Google Scholar]
- 32.Karp PD, Weaver D, Latendresse M. How accurate is automated gap filling of metabolic models? BMC Syst Biol 2018; 12: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reed JL, Patel TR, Chen KH, Joyce AR, Applebee MK, Herring CD et al. Systems approach to refining genome annotation. Proc Natl Acad Sci U S A 2006; 103: 17480–17484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ekblom R, Wolf JBW. A field guide to whole-genome sequencing, assembly and annotation. Evolutionary Applications. 2014; 7: 1026–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Angel VDD, Del Angel VD, Hjerde E, Sterck L, Capella-Gutierrez S, Notredame C et al. Ten steps to get started in Genome Assembly and Annotation. F1000Research. 2018; 7: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Magnúsdóttir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, Noronha A et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol 2017; 35: 81–89. [DOI] [PubMed] [Google Scholar]
- 37.Feist AM, Herrgård MJ, Thiele I, Reed JL, Palsson BØ. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol 2009; 7: 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christopher Frey H, Patil SR. Identification and Review of Sensitivity Analysis Methods. Risk Anal 2002; 22: 553–578. [PubMed] [Google Scholar]
- 39.Schmelling N Reciprocal Best Hit BLAST v1 (protocols.io.grnbv5e). protocols.io. doi: 10.17504/protocols.io.grnbv5e. [DOI]
- 40.Hulsen T, Huynen MA, de Vlieg J, Groenen PMA. Benchmarking ortholog identification methods using functional genomics data. Genome Biol 2006; 7: R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lachance J-C, Lloyd CJ, Monk JM, Yang L, Sastry AV, Seif Y et al. BOFdat: Generating biomass objective functions for genome-scale metabolic models from experimental data. PLOS Computational Biology. 2019; 15: e1006971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edwards DJ, Holt KE. Beginner’s guide to comparative bacterial genome analysis using next-generation sequence data. Microb Inform Exp 2013; 3: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Domselaar GH, Stothard P, Shrivastava S, Cruz JA, Guo A, Dong X et al. BASys: a web server for automated bacterial genome annotation. Nucleic Acids Res 2005; 33: W455–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanizawa Y, Fujisawa T, Nakamura Y. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 2018; 34: 1037–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seemann T Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014; 30: 2068–2069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Genes Retained Per Strain at Incrementing PID Thresholds. The number of genes retained in each strain-specific model is dependent on the threshold utilized for binarization of the homology matrix. The effect of the threshold will also be dependent on how closely related the target strains are to the reference strain. For example, within the strains in the Supplementary Tutorial notebooks we see that CU651637.1 and CP002167.1 are more dissimilar to reference model iML1515 as the drop off in retained genes occurs in a steeper fashion. We suggest using a threshold of 80 when comparing strains of the same species to ensure a sufficient similarity metric to include a gene in the draft models.
Supplementary Figure 2: Resulting Assembly Statistics at Various Coverage. To investigate the effect of coverage on overall assembly statistics of N50 and Number of Contigs, we randomly sampled reads of the BOP27 strain, which has been sequenced to extremely high coverage (400X), enabling this analysis. Analyzing the resulting assemblies at coverages ranging from 10X to 100X, we see from comparing the metrics that at 70X the assembly quality mostly saturates and as such we recommend included genomes have at least this much coverage.
Supplementary Table 1: Comparison with other Model Generation Tools: We compared our Protocol Extension with existing model generation tools Kbase and CarveMe in terms of their functions, user interfaces, advantages, limitations, and applications in the generation of multi-strain models.
Supplementary Table 2: Relevant Data Sources for Multi-Strain Approach
Supplementary Methods: Pseudo-code for generation of multi-strain GEMs. This pseudo-code provides succinct instructions to produce multi-strain GEMs to facilitate automation and understanding of this Protocol Extension.
Supplementary Tutorial: Three jupyter notebooks detailing the entire Protocol Extension as laid out within the Procedure. The first details executing sequence comparison to generate homology matrix, the second details generation of multi-strain models from the homology matrix, and then the third details the beginning investigation of strain-specific capabilities using the draft models.