

Abstract
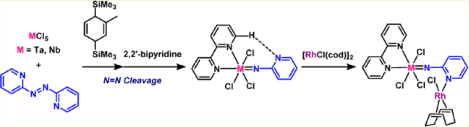
We report the syntheses of 2-pyridylimido complexes of tantalum and niobium by N=N bond cleavage of 2,2′-azopyridine. Reaction of MCl5 (M = Ta and Nb) with 2,2′-azopyridine in the presence of 0.5 equiv of 1-methyl-3,6-bis(trimethylsilyl)-1,4-cyclohexadiene (abbreviated Si-Me-CHD) afforded a dark red solution (for Ta) and a dark blue solution (for Nb) with some insoluble precipitates. After removing the solids, another 0.5 equiv of Si-Me-CHD was added to each solution, giving [M(=Npy)Cl3]n (1a: M = Ta; 1b: M = Nb) through reductive cleavage of the N=N bond of 2,2′-azopyridine. The initial products of the above reactions were determined to be 2,2′-azopyridine-bridged dinuclear complexes, [(MCl4)2(μ-pyNNpy)] (2a: M = Ta; 2b: M = Nb), which were isolated by treating MCl5 with 2,2′-azopyridine and Si-Me-CHD in a 2:1:1 molar ratio. In 2a and 2b, the N=N bond was reduced to a single bond via two-electron reduction. Further reduction of complexes 2a and 2b with 1 equiv of Si-Me-CHD afforded complexes 1a and 1b. An anionic doubly μ-imido-bridged ditantalum complex, [nBu4N][Ta2(μ-Npy)2Cl7] (3a), was generated upon addition of nBu4NCl to complex 1a, while addition of nBu4NCl to niobium complex 1b gave a polymeric terminal imido complex, [nBu4N]n/2[{Nb(=Npy)Cl3}2(μ-Cl)]n/2 (3b). Complexations of 1a and 1b with 1 equiv of 2,2′-bipyridine resulted in the formation of mononuclear 2-pyridylimido complexes, M(=Npy)Cl3(bipy) (4a: M = Ta; 4b: M = Nb), whose main structural feature is intramolecular hydrogen bonding between the ortho hydrogen atom of 2,2′-bipyridine and the nitrogen atom of the pyridyl group on the imido ligand. Isolated 2-pyridylimido complexes 4a and 4b reacted with [RhCl(cod)]2 to produce the corresponding early–late heterobimetallic complexes, (bipy)MCl3(μ-Npy)RhCl(cod) (5a: M = Ta; 5b: M = Nb).
Graphical Abstract
INTRODUCTION
Imido complexes of early transition metals have been intensively investigated in inorganic chemistry because of their versatile reactivity in stoichiometric and catalytic reactions,1 in which the imido groups can function either as spectator ligands for stabilizing the high-valent metal center1d,g,i or as key intermediates in catalytic reactions, such as cycloaddition,2,3 nitrene transfer,4 hydroamination,5 and metathesis reactions.6 Importantly, substituents on the imido nitrogen atom can control not only the electronic properties of the metal (through either σ + π donation or σ + 2π donation) but also the steric bulk around the metal center, regulating the formation of mononuclear, dinuclear, or cluster complexes by μ2- and μ3-bridging nitrogen atoms. Furthermore, the pyridylimido ligand has the unique capability of connecting to a second metal center via pyridyl nitrogen coordination to form homo- and heterometallic clusters, though only a few titanium, vanadium, and molybdenum complexes have been reported so far (Chart 1).7
Chart 1.
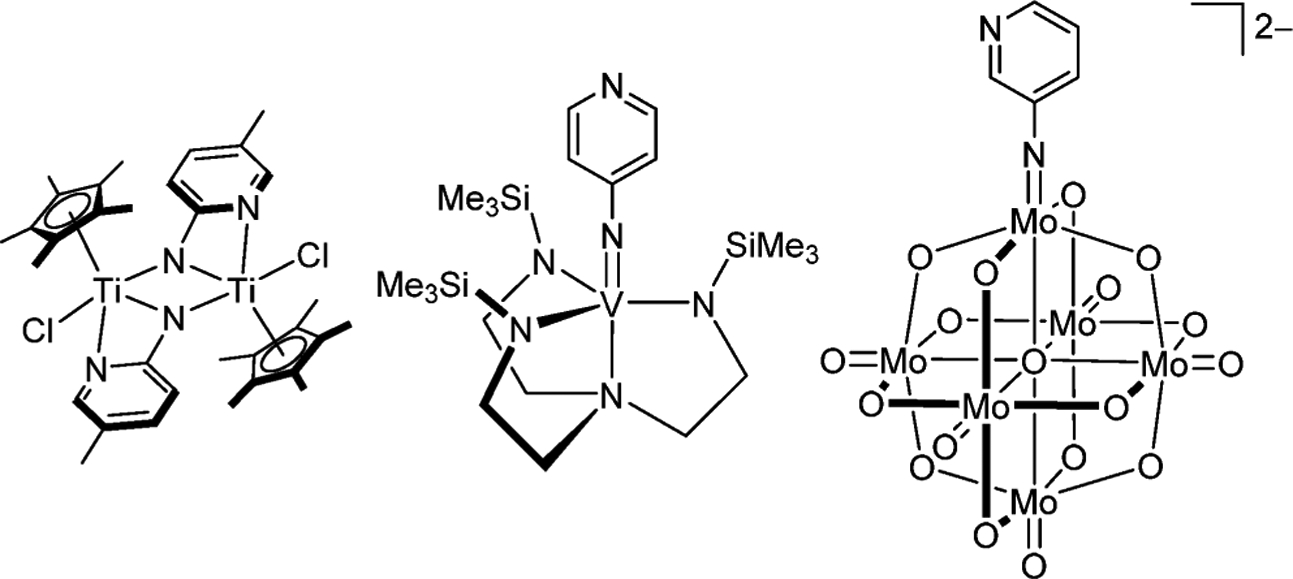
Examples of Pyridylimido Complexes of Early Transition Metals
With regard to the available synthetic methodologies for these pyridylimido complexes, there are three standard reactions: (1) salt-metathesis of a metal halide precursor with the lithium salt of pyridyl amide; (2) reaction of low-valent metal species with pyridyl azide; and (3) deprotonation of primary amine by metal oxo complexes. Each of these methods has limitations such as the following: (i) Lithium salt waste often hampers isolation of the desired complexes due to the formation of ate complexes. (ii) Special care is required to handle and treat potentially explosive organic azides. (iii) Additional promoting reagents are necessary to trap the water byproduct. Another promising synthetic route was recently developed by reductively cleaving the N=N bond of azo compounds using low-valent complexes of early transition metals,8 although reduction of the corresponding high-valent metal complexes required alkali metals or their derivatives as reducing reagents. Similar to the above salt-metathesis reaction, salt contamination has impeded the development of this method. In this context, we applied our methodology for preparing low-valent species of early transition metals in a salt-free manner using organosilicon-based reducing agents such as 1-methyl-3,6-bis(trimethylsilyl)-1,4-cyclohexadiene (abbreviated Si-Me-CHD) to prepare 2-pyridylimido complexes from 2,2′-azopyridine.9 We herein report the synthesis of 2-pyridylimido complexes of tantalum and niobium via reductive cleavage of the N=N bond of 2,2′-azopyridine by in situ generated MCl4 (M = Ta and Nb). In addition, we found that the newly prepared 2-pyridylimido complexes of tantalum and niobium served as unique metalloligands, coordinating to rhodium to form early late heterobimetallic complexes.
RESULTS AND DISCUSSION
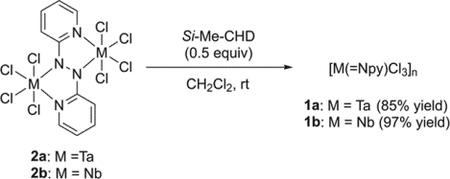
Treatment of MCl5 (M = Ta and Nb) with Si-Me-CHD (0.5 equiv) in the presence of 2,2′-azopyridine (0.5 equiv) gave a dark red solution (for Ta) and a dark blue solution (for Nb), respectively, together with small amounts of insoluble black precipitates. After removal of the solids, a second addition of Si-Me-CHD (0.5 equiv) to each solution induced the precipitation of (2-pyridylimido)tantalum complex 1a as a brown solid and (2-pyridylimido)niobium complex 1b as a pale-blue solid as in eq 1:
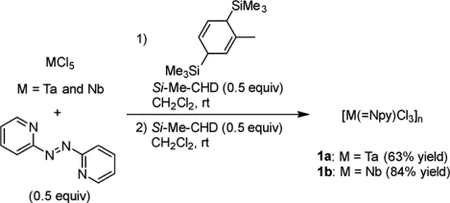 |
(1) |
The low solubility of 1a and 1b in noncoordinating solvents such as toluene, benzene, chloroform, and dichloromethane hampered their characterization by any spectroscopic methods; however, combustion analysis as well as their complexation with nBu4NCl and 2,2′-bipyridine revealed the formation of polymeric [M(=Npy)Cl3]n (1a: M = Ta; 1b: M = Nb) (vide infra).
To elucidate any complexes generated in each step in eq1, we examined the first reduction by mixing MCl5, Si-Me-CHD, and 2,2′-azopyridine in a 2:1:1 molar ratio, producing 2,2′-azopyridine-bridged dinuclear complexes of tantalum and niobium, [(MCl4)2(μ-pyNNpy)] (2a: M = Ta; 2b: M = Nb) as in eq 2:
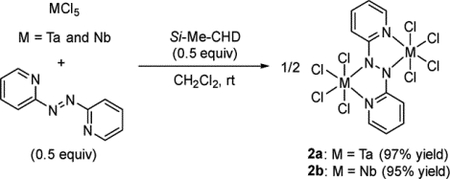 |
(2) |
It was assumed that in situ generated TaCl4 or NbCl4 reacted with 0.5 equiv of 2,2′-azopyridine to give 2a and 2b, respectively, as we already reported that the reaction of MCl5 with 0.5 equiv of Si-Me-CHD gave the corresponding MCl4.9a,f In fact, 2a and 2b were alternatively generated from reaction of the isolated MCl4 with 2,2′-azopyridine. The 1H NMR spectrum of complex 2a in C6D6 displayed one set of four resonances due to pyridyl ring protons at δ 5.64, 6.66, 7.64, and 8.42, while that of 2b showed almost the same pattern of four pyridyl protons at δ 5.62, 6.62, 7.77, and 8.34, suggesting a symmetric structure of 2a and 2b in solution. The dinuclear structures of 2a and 2b were determined by X-ray diffraction analyses (Figure 1 for 2a). Both complexes have two metal centers of MCl4 bridged by 2,2′-azopyridine, and each metal center adopts a pseudo-octahedral geometry with two nitrogen atoms of 2,2′-azopyridine and four chloride ligands. A notable structural feature is that the azo moiety was doubly reduced, resulting in a single N–N bonded azo-moiety (N1−N2 = 1.426(12) Å for 2a; N1−N2 = 1.4026(16) Å for 2b) and single M−N bonds (Ta1−N1 = 2.058(8) and Ta2−N2 = 2.077(8) Å for 2a; Nb1−N1 = 2.0731(12) and Nb2−N2 = 2.0829(12) Å for 2b). The doubly reduced 2,2′-azopyridine-bridged dinuclear structures of 2a and 2b are ascribed to the strong reducing ability of low-valent early transition metal centers and are significantly different from that of some 2,2′-azopyridine-bridged complexes of Cu, Re, and Ru. These complexes have N=N double bonded azo moieties as demonstrated by their bond distances of 1.248–1.372 Å due to the weak reducing ability of the late transition metal complexes as well as π-acceptor-coordinated metal complexes (Chart 2).10
Figure 1.
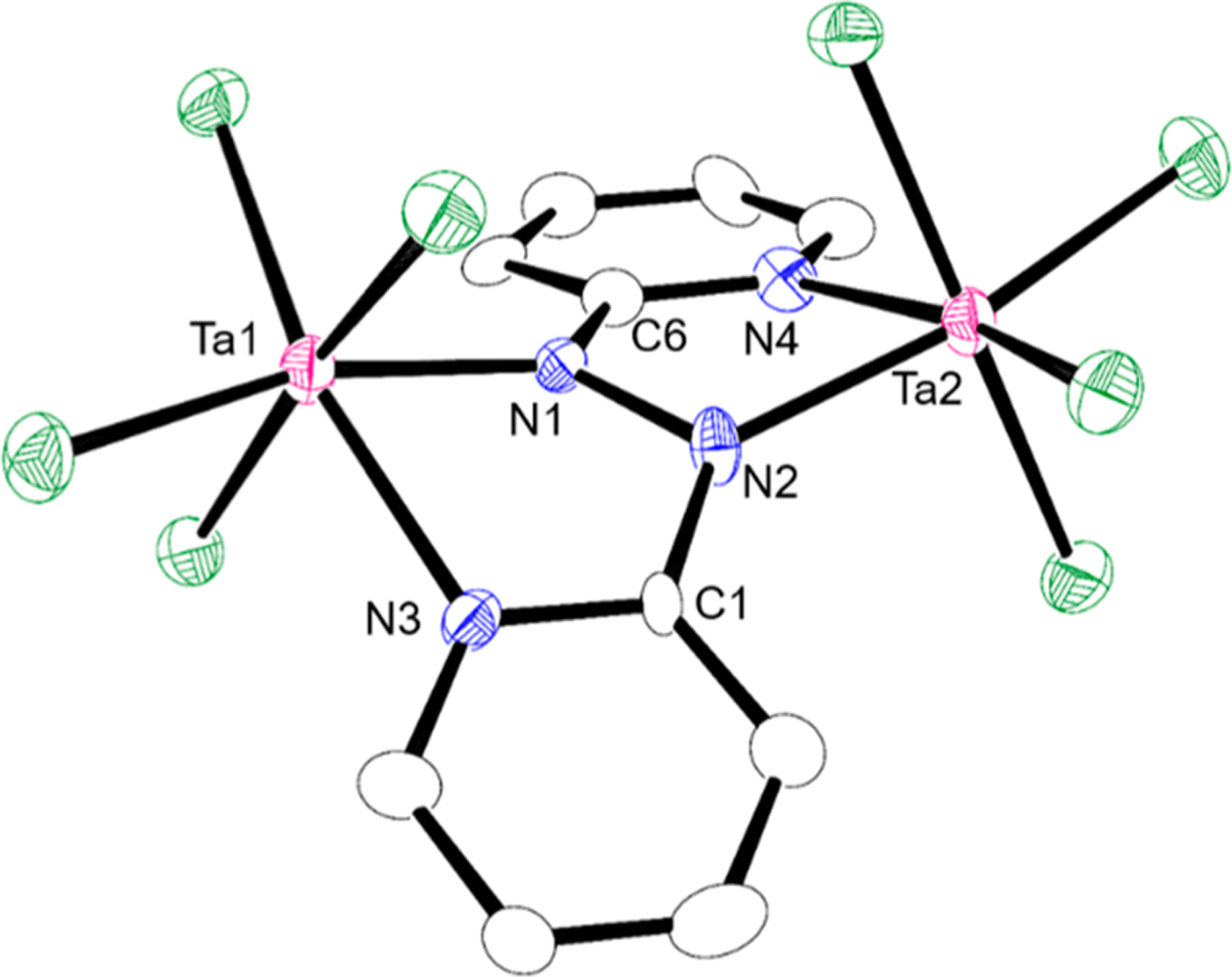
Molecular structure of 2a and with 50% thermal ellipsoids. All hydrogen atoms and solvent molecules are omitted for clarity. Selected bond distances (Å) and angle (deg) for 2a: Ta1–N1, 2.058(8); Ta1–N3, 2.213(9); Ta2–N2, 2.077(8); Ta2–N4, 2.198(8); N1–N2, 1.426(12); N2–C1, 1.387(14); N1–C6, 1.380(13); C1–N2–N1–C6, 33.10(3). The structure of 2b is given in the Supporting Information because 2b is isostructural to 2a. Selected bond distances (Å) and angle (deg) for 2b: Nb1–N1, 2.0731(12); Nb1–N3, 2.2255(13); Nb2–N2, 2.0829(12); Nb2–N4, 2.2164(12); N1–N2, 1.4026(16); N2–C1, 1.3910(19); N1–C6, 1.3935(18); C1–N2–N1–C6, 33.02(9).
Chart 2.
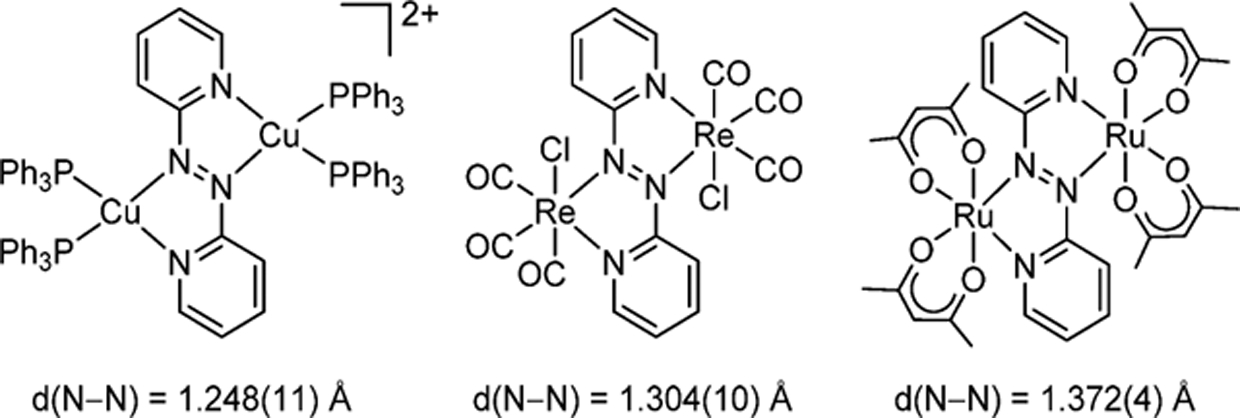
Examples of 2,2′-Azopyridine-Bridged Dinuclear Complexes
We next conducted the reduction of 2a and 2b corresponding to the second reduction step in eq1: First, 0.5 equiv of Si-Me-CHD was added to each solution of 2a and 2b in dichloromethane at room temperature to spontaneously precipitate 1a and 1b, along with the elimination of 2 equiv of Me3SiCl as in eq 3:
 |
(3) |
Such a two-electron reduction process was consistent with their electrochemical behaviors: Cyclic voltammetry of 2a and 2b in dichloromethane containing 0.1 M [nBu4N][BArF4] (ArF = 3,5-(CF3)2C6H3) with a scan rate of 100 mV/s exhibited one irreversible two-electron reduction wave ([2a]0/[2a]2−: E = −0.658 V; [2b]0/[2b]2−: E = −0.299 V vs Cp2Fe+/0) corresponding to reductive cleavage of the single N−N bond induced by two-electron transfer from the two metal center to the bridging N−N bond (see the Supporting Information). Because of the general tendency for the stability of the high-oxidation state third-row transition metals, the reduction potential of 2a was negative compared with that of 2b.
The formula of the precipitated compound [Ta(=Npy)Cl3]n (1a) was deduced by its complexations with nBu4NCl and 2,2′-bipyridine (Scheme 1). Complex 1a reacted with 0.5 equiv of nBu4NCl in dichloromethane to afford a clear brown solution, from which an anionic doubly bridged μ-imido dinuclear tantalum complex (3a) was isolated. Complex 3a was characterized by NMR spectroscopy and X-ray analysis. The 1H NMR spectrum of 3a in CD2Cl2 displayed four resonances due to the pyridine ring protons at δ 7.21, 7.35, 8.05, and 8.87. Figure 2 shows the dinuclear molecular structure of 3a. The N−N bond in 3a is fully cleaved (N1⋯N2 distance, 2.558 Å), which provides evidence that parent 1a also likely has a fully cleaved N–N bond prior to complexation with chloride. The two tantalum atoms of 3a are bridged by two 2-pyridylimido ligands in a dissymmetric fashion, where one of two tantalum centers possesses two κ2-pyridyl coordination with a seven-coordinate, distorted pentagonal bipyramidal geometry, and the other tantalum atom adopts a typical octahedral geometry of four chloride atoms and two bridging nitrogen atoms. The Ta1⋯Ta2 distance (3.1669(9) Å) shows no tantalum–tantalum bond. The Ta2–N1 (1.918(8) Å) and Ta2–N2 (1.944(10) Å) bonds are shorter than the Ta1–N1 (2.164(10) Å) and Ta1–N2 (2.122(9) Å) bond due to the pπ–dπ interaction of the μ-N atoms to the Ta2 center. A different coordination number of two metal centers for doubly μ-imido dinuclear metal complexes was also reported for [Ti2(μ-Npy)2Cl4(thf)3] and [Zr2(μ-NR)2Cl4(thf)3].7c,11
Scheme 1.
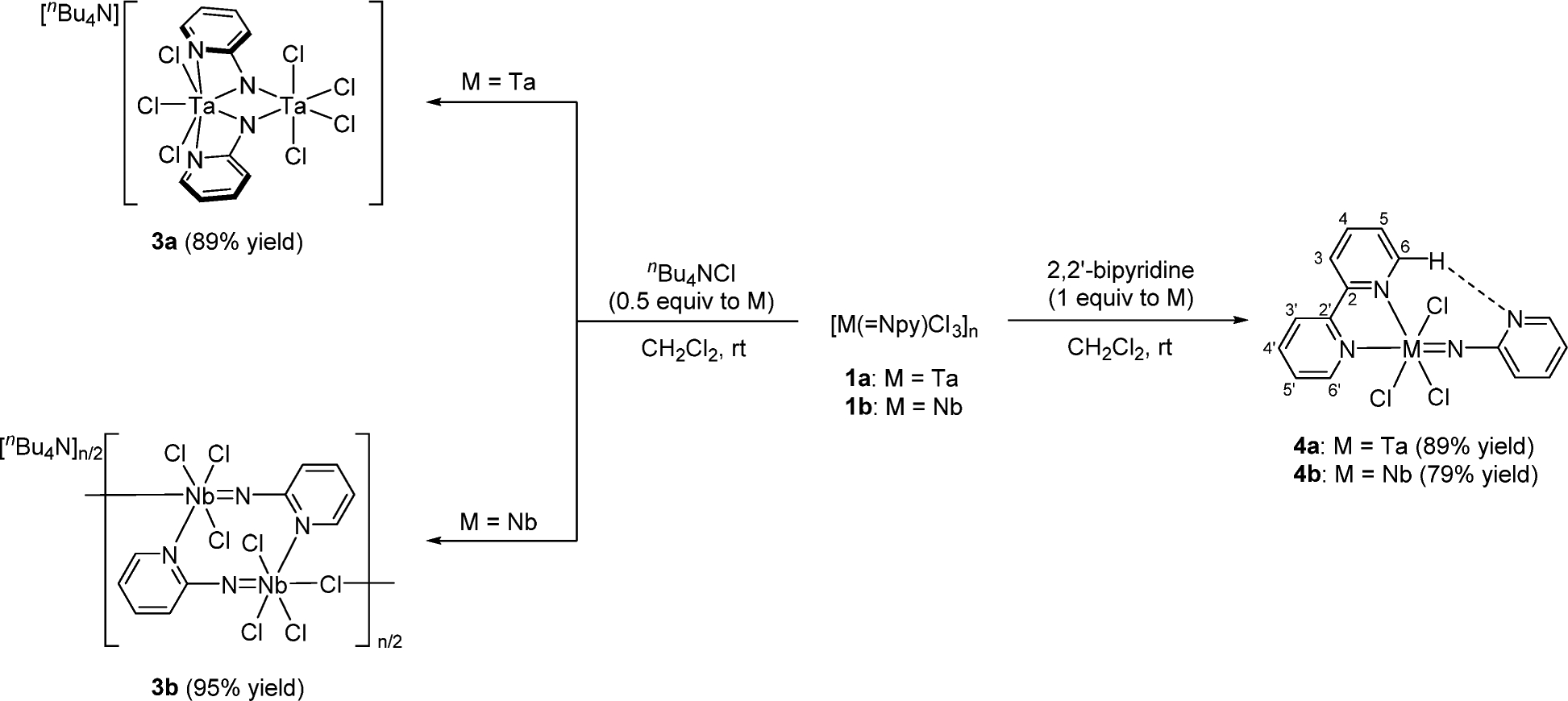
Complexation of 1a and 1b with nBu4NCl and 2,2′-Bipyridine
Figure 2.
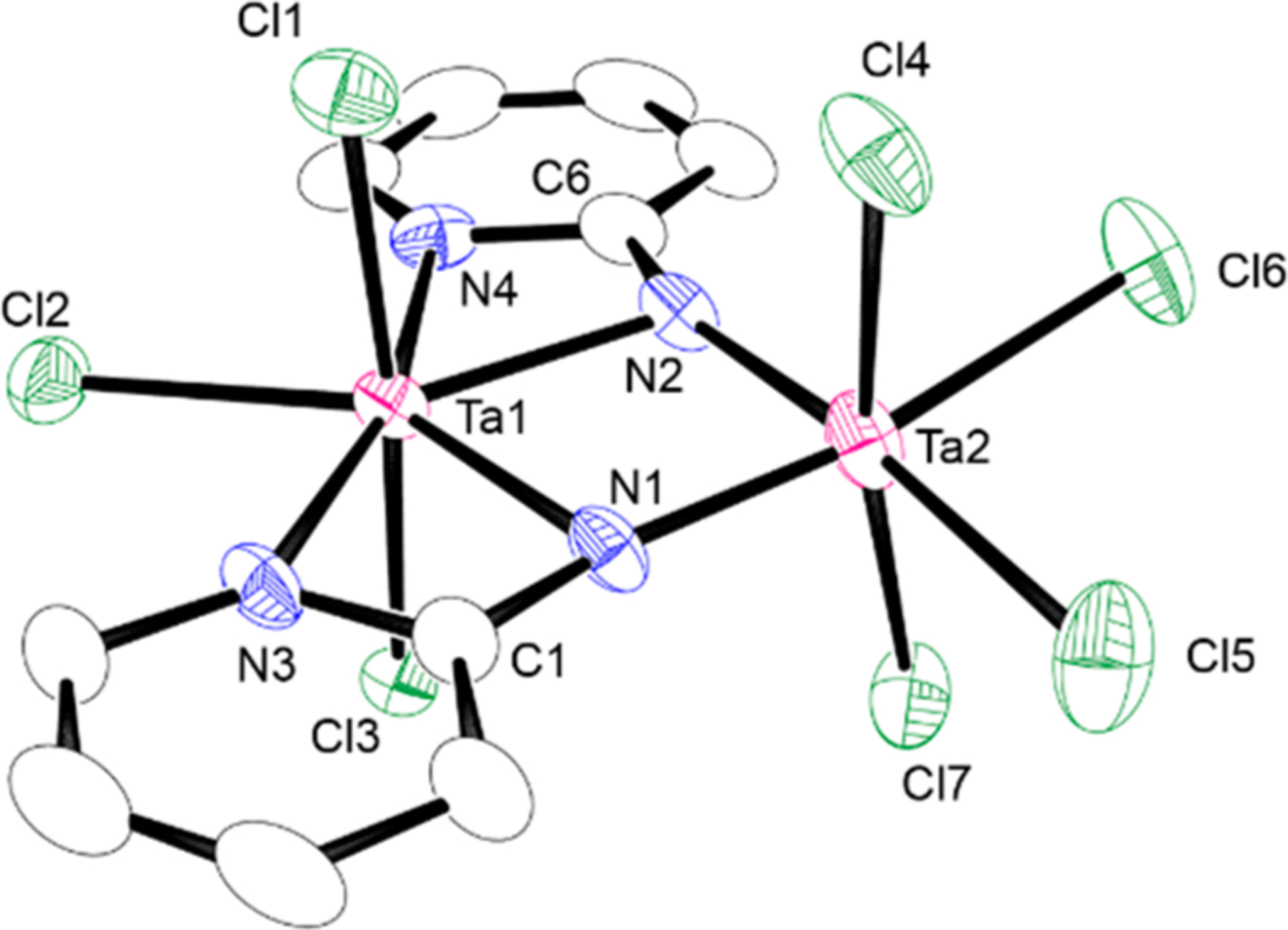
Molecular structure of anionic part of 3a with 30% thermal ellipsoids. All hydrogen atoms and countercation are omitted for clarity. Selected bond distances (Å) and angles (deg): N1⋯N2, 2.558; Ta1⋯Ta2, 3.1669; Ta1–N1, 2.164(10); Ta1–N2, 2.122(9); Ta1–N3, 2.222(9); Ta1–N4, 2.225(10); Ta2–N1, 1.918(8); Ta2–N2, 1.944(10); Ta1–N1–Ta2, 101.6(4); Ta1–N1–C1, 96.0(7); Ta2–N1–C1, 160.7(8); Ta1–N2–Ta2, 102.2(4); Ta1–N2–C6, 98.4(7); Ta2–N2–C6, 158.0(8).
In contrast to the formation of the anionic dinuclear complex of 3a, anionic polymeric compound 3b was formed upon adding 0.5 equiv of nBu4NCl to 1b in dichloromethane (Scheme 1). During the reaction, green-colored microcrystals were grown, although the solution color did not change. The overall molecular structure of 3b was revealed by X-ray diffraction analysis, though the quality of the diffraction data was low because it could not be recrystallized, and only the connectivity of the molecular structure was clarified. The monomeric unit comprises a dimer of terminal imido species [Nb(=Npy)Cl3]2, where the pyridine nitrogen atom of the 2-pyridylimido ligand bound to a NbCl3 moiety coordinates to the neighboring niobium atom of the other Nb(=Npy)Cl3 unit, forming an eight-membered cyclic ring, and an additional chloride atom links to the dimer unit. Each niobium atom adopts a six-coordinate octahedral geometry where the bridging chloride ligand occupies a position trans to the 2-pyridylimido ligand.
The addition of 2,2′-bipyridine to a suspension of 1a in dichloromethane gave a clear solution, from which mononuclear imido complex 4a was isolated in 89% yield (Scheme 1). Complex 4a was fully characterized by NMR spectroscopy as well as X-ray diffraction analysis. The 1H NMR spectrum of complex 4a in CD2Cl2 showed one set of four resonances at δ 6.93, 7.09, 7.81, and 8.48 assignable to the pyridyl ring protons bound to the imido nitrogen atom, along with the other set of signals attributed to 2,2′-bipyridine at δ 7.81, 8.26, 8.34, 9.63, and 10.45, the latter two of which were assigned to H6 (δ 10.45 with 3J = 5.5 Hz) and H6′ (δ 9.63 with 3J = 5.4 Hz). The significantly lower field-shifted resonance for H6 indicated an intramolecular hydrogen bond between the H atom bound to C6 of 2,2′-bipyridine and the nitrogen atom of the pyridyl group of the 2-pyridylimido ligand. Further evidence was provided by its 13C NMR spectrum, where two resonances due to C6 and C6′ of 2,2′-bipyridine were observed as magnetically nonequivalent resonances at δ156.9 (1JC–H = 191 Hz) and 150.6 (1JC–H = 186 Hz). A similar downfield shift and large 1JC–H value were reported for 2-(1-vinyl-1H-pyrrol-2-yl)-pyridine, which has an intramolecular hydrogen bond between the hydrogen atom bound to the vinyl group and the nitrogen atom of the pyridine moiety.12 Figure 3 shows the mononuclear structure of 4a, in which the tantalum atom possesses a six-coordinate pseudo-octahedral geometry with the 2-pyridylimido ligand and one of two nitrogen atoms of the 2,2′-bipyridine ligand at the axial positions. Additionally, the nitrogen atom, N2, of the pyridyl ring points toward H6 bound to C6 with the N2⋯C6 distance (3.289 Å), which lies in the range of weak hydrogen bond distances.12 Although the bond distance of Ta–N1 (1.782(9) Å) and the angle of Ta–N1–C1 (173.9(7)°) are normal for typical 6e-donating imido ligands,1a the 2-pyridylimido ligand is slightly bent toward the C6 of 2,2′-bipyridine. Similarly, niobium complex 4b was prepared in 79% yield by treating 1b with 2,2′-bipyridine and characterized by spectral and X-ray diffraction methods. The 1H NMR spectrum of niobium complex 4b in CD2Cl2 displayed almost the same pattern with the characteristic two doublet signals at δ 9.58 (3J = 5.0 Hz) and 10.13 (3J = 5.2 Hz) for H6′ and H6 of the 2,2′-bipyridine bound to the niobium center due to an intramolecular hydrogen bond between the H atom bound to C6 of 2,2′-bipyridine and the nitrogen atom of the 2-pyridyl group. The crystal structure of 4b is isostructural to that of 4a, and its drawing is provided in the Supporting Information.
Figure 3.
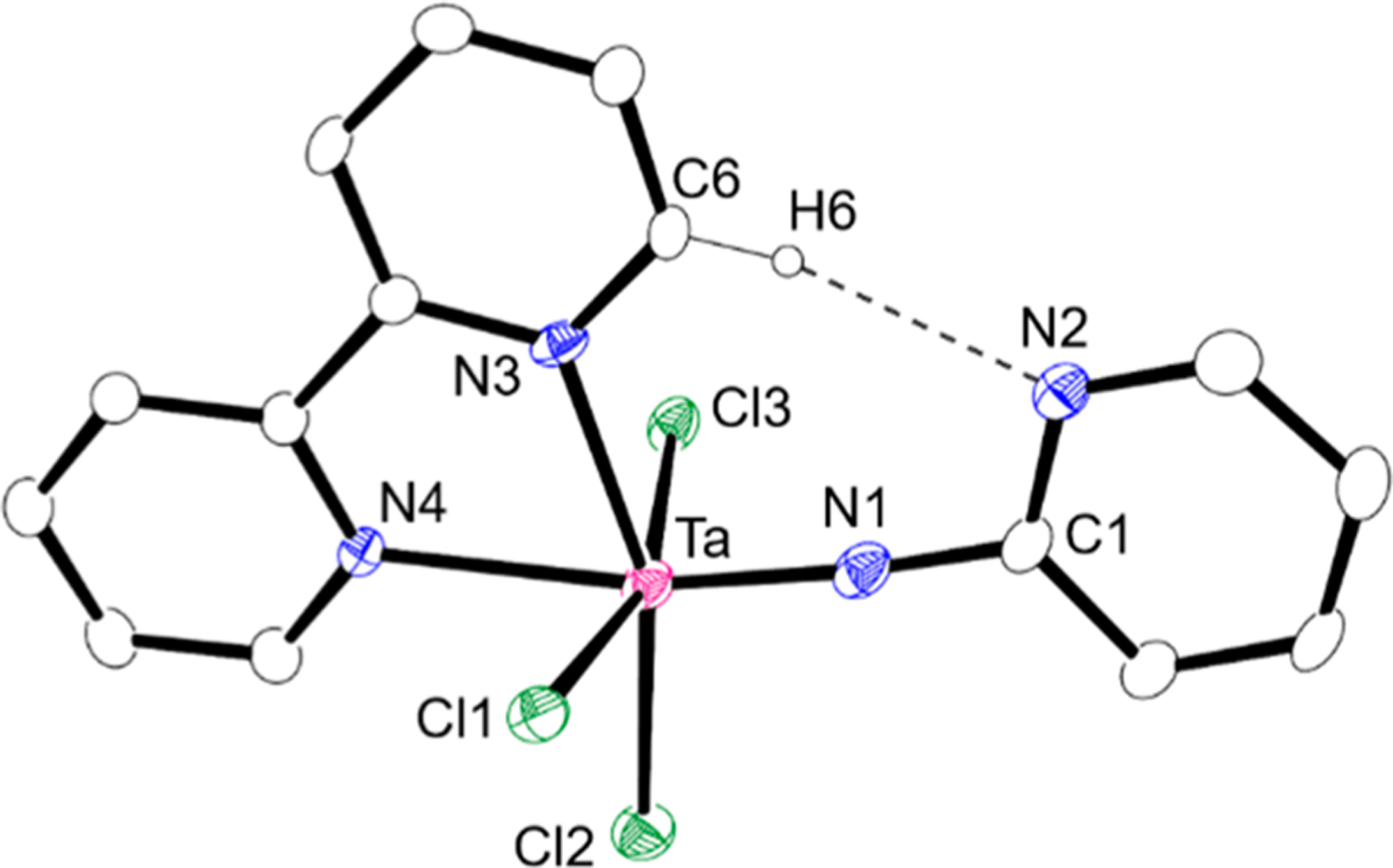
Molecular structure of 4a with 50% thermal ellipsoids. All hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angle (deg) for 4a: Ta–N1, 1.782(9); Ta–N3, 2.232(8); Ta–N4, 2.368(3); N2⋯C6, 3.289; Ta–N1–C1, 173.9(7). The structure of 4b is given in the Supporting Information. Selected bond distances (Å) and angle (deg) for 4b: Nb–N1, 1.774(2); Nb–N3, 2.259(2); Nb–N4, 2.370(2); N2⋯C6, 3.296; Nb–N1–C1, 174.2(2).
We investigated the coordination of the 2-pyridylimido ligand of 1a and 1b to some transition metal chlorides, since it was reported that the pyridyl moiety of the (4-pyridylimido)-vanadium complex shown in Chart 1 coordinates to [RhCl-(CO)2]2 and [W(=NEt)Cl4]2 to form the corresponding heterometallic complexes.7e We thus conducted reactions of 1a and 1b with several early and late transition metal complexes; however, no reaction proceeded, probably due to the strong coordination of the 2-pyridylimido moiety of 1a and 1b to the Lewis acidic tantalum and niobium centers. In contrast, the free 2-pyridyl moiety in complexes 4a and 4b was capable of coordinating to other transition metals because the 2-pyridyl nitrogen atom has only a weak intramolecular hydrogen bond (vide supra). Treatment of 4a and 4b with 0.5 equiv of [RhCl(cod)]2 in dichloromethane led to the formation of the corresponding early late heterometallic complexes 5a and 5b as microcrystalline solids as in eq 4:
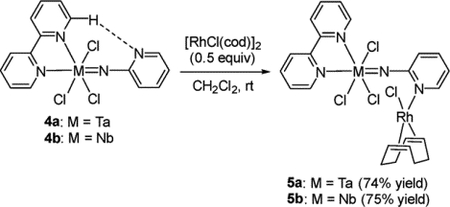 |
(4) |
In the 1H NMR spectrum, a doublet signal (δ 10.24) for H6 of the 2,2′-bipyridine ligand of 5a in CD2Cl2 shifted to a field magnetically higher than that of 4a (δ 10.45), probably due to the loss of the intramolecular hydrogen bonding. We observed an equilibrium between 5a and a mixture of 4a and [RhCl(cod)]2 in a 65:35 ratio by the 1H NMR measurement at 303 K. Similar equilibrium was observed for niobium complex 5b with a 55:45 ratio for 5b and a mixture of 4b and [RhCl(cod)]2 at 303 K. The thermodynamic parameters (ΔH = −5.3(2) kcal mol−1, ΔS = −9.5(8) e.u., ΔG303K = −2.4(5) kcal mol−1 for 5a; ΔH = −7.2(4) kcal mol−1, ΔS = −17(2) e.u. ΔG303K = −2.1(9) kcal mol−1 for 5b) for the equilibria were evaluated by van’t Hoff plots (see the Supporting Information Such negative ΔS values are consistent with the dominance of 5a and 5b as the temperature decreased.
The early late heterobimetallic structures of complexes 5a and 5b were determined by X-ray diffraction, and the ORTEP drawing of 5a is shown in Figure 4. The molecular structure of Nb–Rh complex 5b is included in Supporting Information. The 2-pyridyl moiety on the imido ligand is bound to the rhodium center to form tantalum–rhodium heterobimetallic structure. The Ta–N1 bond distance (1.787(3) Å) is similar to that of 4a, while the Ta–N1–C1 bond angle (166.7(22)°) is slightly smaller than that of 4a. This lower linearity of the imido ligand in 5a probably arises from the steric repulsion between coordinated rhodium complex and chloride ligands on the tantalum center.
Figure 4.
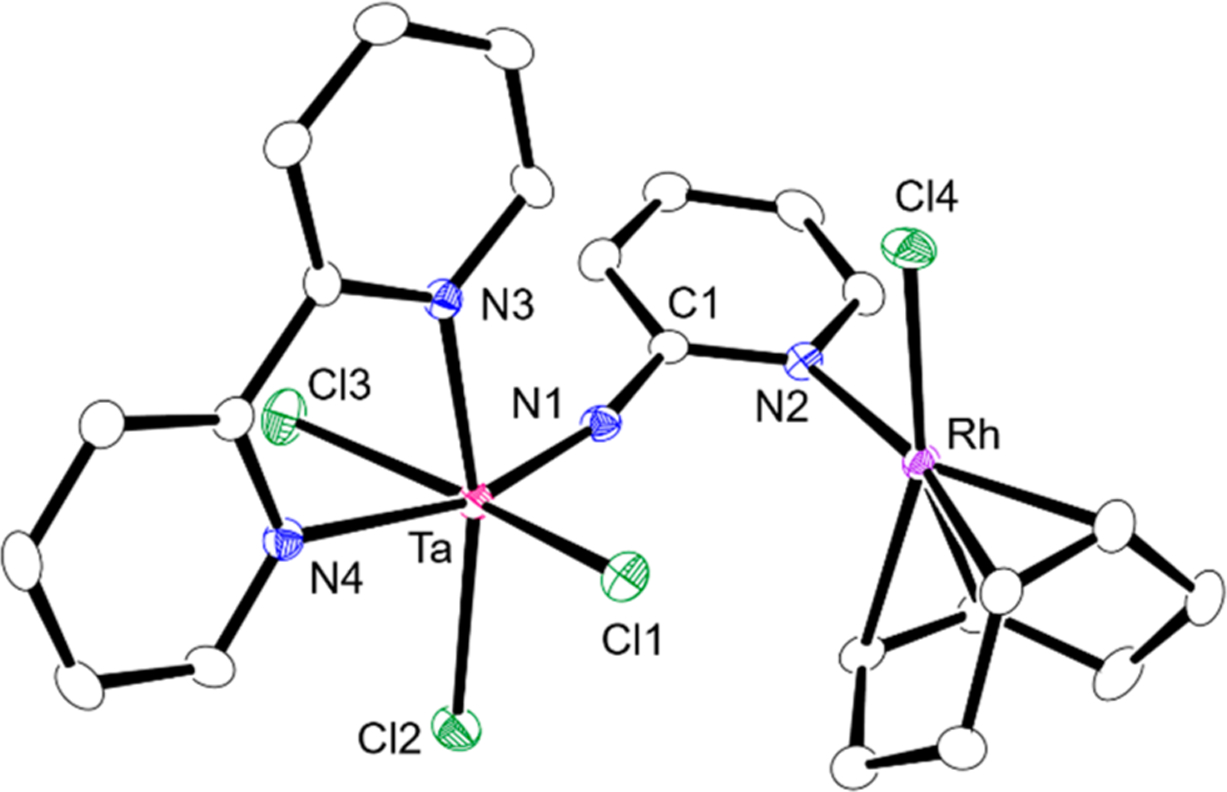
Molecular structure of 5a with 50% thermal ellipsoids. All hydrogen atoms and solvent molecules are omitted for clarity. Selected bond distances (Å) and angles (deg) for 5a: Ta–N1, 1.787(3); N1–C1, 1.376(4); Ta–N3, 2.240(2); Ta–N4, 2.348(3); Ta–Cl1, 2.3818(8); Ta–Cl2, 2.3640(8); Ta–Cl3, 2.3927(9); Rh–N2, 2.100(3); Rh–Cl4, 2.3956(9); N1–Ta–N4, 163.67(10); Ta–N1–C1, 166.7(22). The structure of 5b is given in the Supporting information. Selected bond distances (Å) and angle (deg) for 5b: Nb–N1, 1.7804(14); N1–C1, 1.383(2); Nb–N3, 2.2531(14); Nb–N4, 2.3545(14); Nb–Cl1, 2.3876(5); Nb–Cl2, 2.3710(5); Nb–Cl3, 2.4054(5); Rh–N2, 2.0999(15); Rh–Cl4, 2.3909(5); N1–Nb–N4, 163.29(5); Nb–N1–C1, 165.92(12).
CONCLUSION
We found that the N=N bond of 2,2′-azopyridine was reductively cleaved upon treating 2,2′-azopyridine with low-valent metal chlorides derived from MCl5 (M = Ta; Nb) in the presence of a salt-free reducing agent, Si-Me-CHD, giving 2-pyridylimido complexes [M(=Npy)Cl3]n (1a: M = Ta, 1b: M = Nb). The reaction was stepwise; prior to the N=N cleavage process, two-electron-reduced 2,2′-azopyridine-bridged dinuclear complexes 2a and 2b were formed as a consequence of the reaction of 2,2′-azopyridine with in situ generated MCl4. The addition of chloride to 1a induced the formation of the doubly μ-imido dinuclear tantalum complex, [nBu4N][Ta2(μ-Npy)2Cl7] (3a), while the reaction of 1b with chloride afforded an anionic polymeric terminal imido niobium complex, [nBu4N]n/2[{Nb(=Npy)Cl3}2(μ-Cl)]n/2 (3b). In contrast, mononuclear 2-pyridylimido complexes, M(=Npy)-Cl3(bipy) (4a, M = Ta; 4b, M = Nb), were obtained by complexation of 2,2′-bipyridine with 1a and 1b. We demonstrated that complexes 4a and 4b acted as metalloligands whose pyridyl nitrogen atom coordinated to the rhodium center of [RhCl(cod)]2, resulting in the formation of early late heterobimetallic complexes 5a and 5b. Further studies of the reaction of the M=Npy moiety with the additional metal on the 2-pyridyl nitrogen atom are ongoing in our laboratories.
EXPERIMENTAL SECTION
General Remarks.
All manipulations involving air- and moisture-sensitive organometallic compounds were carried out under argon atmosphere using standard Schlenk techniques or in an argon-filled glovebox. 2,2′-Azopyridine13 and 1-methyl-3,6-bis(trimethylsilyl)-1,4-cyclohexadiene (Si-Me-CHD)14 were prepared according to the literature. C6D6 and CD2Cl2 were purchased and purified by distillation over CaH2. All other reagents were purchased from commercial resources and used as received. Anhydrous dichloromethane, hexane, and toluene were purchased from Kanto Chemical and further purified by passage through activated alumina under positive argon pressure as described by Grubbs et al.15 1H NMR (400 MHz), 13C{1H} NMR (100 MHz), 2D 1H–1H COSY, 2D 1H–13C HMQC, and 1H–13C HMBC spectra were measured on a BRUKER AVANCE III 400 MHz spectrometer. Chemical shifts were reported in parts per million (ppm) and referenced to the residual proton signal of the solvent (1H: δ = 7.15 and 5.32 ppm for C6D6 and CD2Cl2, respectively) or the solvent itself (13C{1H}: δ = 128.06 and 53.84 ppm for C6D6 and CD2Cl2, respectively). The elemental analysis was recorded by using PerkinElmer 2400 at the Faculty of Engineering Science, Osaka University. Melting point was measured in sealed tubes under an argon atmosphere (BUCHI Melting Point M-565).
Synthesis of [Ta(=Npy)Cl3]n (1a).
Synthesis from TaCl5.
To a suspension of TaCl5 (897 mg, 2.50 mmol) in dichloromethane (30 mL) at room temperature was added a solution of Si-Me-CHD (299 mg, 1.25 mmol) in dichloromethane (12 mL). The color of the reaction mixture changed to gray. After the mixture was stirred for 10 min, a solution of 2,2-azopyridine (231 mg, 1.26 mmol) in dichloromethane (12 mL) was added. The reaction mixture was stirred for 2 h, and the precipitate was removed by filtration. To the dark red filtrate was added at room temperature a solution of Si-Me-CHD (301 mg, 1.26 mmol) in dichloromethane (12 mL). After the mixture was stirred for 18 h, the supernatant was decanted. The resulting solids were washed with dichloromethane (10 mL) and then dried under vacuum to give 1a as brown powder in 63% yield (597 mg, 1.57 mmol), 355 °C (dec). Anal. Calcd. for C4H4Cl3N2Ta: C, 15.83; H, 1.06; N, 7.38. Found: C, 16.10; H, 1.02; N, 7.48. Hydrolysis of the solid of 1a afforded 2-aminopyridine, revealing that 1a possesses the 2-pyridylimido ligand.
Synthesis from 2a.
To a solution of 2a (963 mg, 1.16 mmol) in dichloromethane (20 mL) at room temperature was added a solution of Si-Me-CHD (278 mg, 1.17 mmol) in dichloromethane (10 mL). A brown powder slowly precipitated. After the reaction mixture was stirred for 24 h at room temperature, the liquid was decanted and the brown precipitates were washed with dichloromethane (15 mL × 3), and then dried under vacuum to give 1a as a brown powder in 85% yield (744 mg, 1.96 mmol).
Synthesis of [Nb(=Npy)Cl3]n (1b).
Synthesis from NbCl5.
To a suspension of NbCl5 (392 mg, 1.45 mmol) in dichloromethane (15 mL) at room temperature was added a solution of Si-Me-CHD (166 mg, 0.696 mmol) in dichloromethane (7 mL). The color of the reaction mixture changed to brown. After the mixture was stirred for 10 min, a solution of 2,2′-azopyridine (137 mg, 0.741 mmol) in dichloromethane (7 mL) was added. The reaction mixture was stirred for 2 h, and the precipitate was removed by filtration. To the dark blue filtrate was added at room temperature a solution of Si-Me-CHD (175 mg, 0.734 mmol) in dichloromethane (7 mL). After the mixture was stirred for 23 h, the supernatant was decanted. The resulting solids were washed with dichloromethane (20 mL × 2) and then dried under vacuum to give 1b as pale blue powder in 84% yield (357 mg, 0.817 mmol), 260–261 °C (dec). Anal. Calcd for C5H4Cl3N2Nb: C, 20.61; H, 1.38; N, 9.61. Found: C, 20.39; H, 1.49; N, 9.31. Hydrolysis of the solid of 1b afforded 2-aminopyridine, revealing that 1b possesses the 2-pyridylimido ligand.
Synthesis from 2b.
To solution of 2b (838 mg, 1.28 mmol) in dichloromethane (15 mL) at room temperature was added a solution of Si-Me-CHD (322 mg, 1.35 mmol) in dichloromethane (10 mL). A pale-blue powder slowly precipitated. After the reaction mixture was stirred for 17 h at room temperature, the liquid was decanted, and the pale blue precipitates were washed with dichloromethane (5 mL × 2), and then dried under vacuum to give 1b as a pale blue powder in 97% yield (727 mg, 2.50 mmol).
Synthesis of (TaCl4)2(μ-pyNNpy) (2a).
To a suspension of TaCl5 (500 mg, 1.40 mmol) in dichloromethane (15 mL) at room temperature was added a solution of Si-Me-CHD (166 mg, 0.696 mmol) in dichloromethane (7 mL). The color of the reaction mixture changed to gray. After the mixture was stirred for 10 min, a solution of 2,2′-azopyridine (131 mg, 0.711 mmol) in dichloromethane (7 mL) was added. The reaction mixture was stirred for 3 h, and then the precipitate was removed by filtration. All volatiles were removed under vacuum. The resulting solid was washed with hexane (10 mL) and then dried under vacuum to give 2a as a black powder in 97% yield (561 mg, 0.676 mmol), mp 209–210 °C (dec). Single crystals suitable for the X-ray diffraction were obtained from the saturated benzene solution. 1H NMR (400 MHz, C6D6, 303 K): δ 5.64 (ddd, 3J = 6.6 Hz, 3J = 6.7 Hz, 4J =1.0 Hz, 2H, H5), 6.66 (ddd, 3J = 8.1 Hz, 3J = 8.0 Hz, 4J =1.6 Hz, 2H, H4), 7.64 (dd, 3J = 8.8 Hz, 2H, H3), 8.42 (ddd, 3J = 6.1 Hz, 4J = 1.6 Hz, 2H, H6). 13C{1H} NMR (100 MHz, C6D6, 303 K): δ 118.3 (C5), 120.4 (C3), 141.3 (C4), 146.9 (C6) 162.3 (C2). Anal. Calcd. for C10H8Cl8N4Ta2: C, 14.48; H, 0.97; N, 6.75. Found: C, 14.89; H, 0.66; N, 6.34.
Synthesis of (NbCl4)2(μ-pyNNpy) (2b).
To a suspension of NbCl5 (399 mg, 1.48 mmol) in dichloromethane (15 mL) at room temperature was added a solution of Si-Me-CHD (177 mg, 0.740 mmol) in dichloromethane (7 mL). The color of the reaction mixture changed to brown. After the mixture was stirred for 10 min, a solution of 2,2′-azopyridine (139 mg, 0.754 mmol) in dichloromethane (7 mL) was added. The reaction mixture was stirred for 2 h, and then the precipitate was removed by filtration. All volatiles were removed under vacuum. The resulting solid was washed with hexane (20 mL) and then dried under vacuum to give 2b as a blackish-blue powder in 95% yield (458 mg, 0.701 mmol), mp 190–191 °C (dec). Single crystals suitable for X-ray diffraction were obtained from the saturated benzene solution. 1H NMR (400 MHz, C6D6, 303 K): δ 5.62 (ddd, 3J = 7.2 Hz, 3J = 6.0 Hz, 4J =1.1 Hz, 2H, H5), 6.62 (ddd, 3J = 8.8 Hz, 3J = 7.2 Hz, 4J =1.7 Hz, 2H, H4), 7.77 (ddd, 3J = 8.7 Hz, 2H, H3), 8.34 (ddd, 3J = 6.0 Hz, 4J = 1.7 Hz, 5J = 0.7 Hz, 2H, H6). 13C NMR (100 MHz, C6D6, 303 K): δ 119.2 (C5), 122.4 (C3), 141.4 (C4), 148.2 (C6), 161.7 (C3). Anal. Calcd. for C10H8Cl8N4Nb2(C6H6)0.1: C, 19.25; H, 1.31; N, 8.47. Found: C, 19.46; H, 1.28; N, 8.21. Inclusion of benzene is due to the remaining of a small amount of benzene in the recrystallized sample even after evacuation.
Synthesis of [nBu4N][Ta2(μ-Npy)2Cl7] (3a).
To a suspension of la (200 mg, 0.527 mmol) in dichloromethane (7 mL) at room temperature was added a solution of nBu4NCl (74.5 mg, 0.268 mmol) in dichloromethane (5 mL). The color of the mixture changed to brown. After the reaction mixture was stirred for 12 h, the volatiles were removed under vacuum. The residue was washed with hexane (5 mL) and then dried to afford 3a as a brown powder in 89% yield (243 mg, 0.234 mmol), mp 85–87 °C. Single crystals suitable for the X-ray diffraction were obtained from the saturated dichloromethane solution. 1H NMR (400 MHz, CD2Cl2 303 K): δ 1.01 (t, 12H, 3J = 7.4 Hz, NCH2CH2CH2CH3), 1.45 (m, 8H, NCH2CH2CH2CH3), 1.65 (m, 8H, NCH2CH2CH2CH3), 3.20 (m, 8H, NCH2CH2CH2CH3), 7.21 (ddd, 2H, 3J = 7.5 Hz, 3J = 5.5 Hz, 4J = 1.0 Hz, H5), 7.35 (br d, 2H, H3), 8.05 (ddd, 2H, 3J = 8.3 Hz, 3J = 7.6 Hz, 4J =1.8 Hz, H4), 8.87 (ddd, 2H, 3J = 4.9 Hz, 3J =1.8 Hz, 4J = 0.9 Hz, H6). 13c{1H} NMR (100 MHz, CD2Cl2, 303 K): δ 13.9 (NCH2CH2CH2 CH3), 20.2 (NCH2CH2CH2CH3), 24.5 (NCH2CH2CH2CH3), 59.5 (NCH2CH2CH2CH3), 118.2 (C3), 119.5 (C5), 140.9 (C6), 144.9 (C4), 172.3 (C2). Anal. Calcd. for C26H44Cl7N5Ta2: C, 30.12; H, 4.28; N, 6.76. Found: C, 29.94; H, 3.99; N, 6.49.
Synthesis of [nBu4N]n/2[{Nb(=Npy)Cl3}2(μ-Cl)]n/2 (3b).
To a suspension of 1b (104 mg, 0.356 mmol) in dichloromethane (1 mL) at room temperature was added a solution of nBu4NCl (49.5 mg, 0.178 mmol) in dichloromethane (2 mL). The color of the mixture changed to green. After the reaction mixture was stirred for 19 h, the volatiles were removed under vacuum. The resulting solids were washed with dichloromethane (1 mL × 4) and then dried to give 3b as a green powder in 95% yield (145 mg, 0.168 mmol), mp 207–209 °C (dec). Single crystals suitable for the X-ray diffraction were obtained from the saturated dichloromethane solution. Anal. Calcd for C26H44Cl7N5Nb2: C, 36.29; H, 5.15; N, 8.14. Found: C, 35.66; H, 5.00; N, 8.11. Deviation of the carbon value in elemental analysis is probably due to a small amount of contamination of insoluble 1b.
Synthesis of Ta(=Npy)Cl3(bipy) (4a).
A solution of 2,2′-bipyridine (47.4 mg, 0.303 mmol) in dichloromethane (5 mL) was added to a suspension of 1a (111 mg, 0.293 mmol) in dichloromethane (5 mL) at room temperature. After stirring for 37 h, all volatiles were removed under reduced pressure. The resulting solids were washed with hexane (5 mL × 6) and then dried to give 4a as a pale yellow powder in 89% yield (140 mg, 0.261 mmol), mp 163 °C (dec). Single crystals suitable for the X-ray diffraction were obtained from the saturated dichloromethane solution. 1H NMR (400 MHz, CD2Cl2, 303 K): δ 6.93 (ddd, 3J = 7.4 Hz, 3J = 5.0 Hz, 4J =1.0 Hz, 1H, H4 of py), 7.09 (d, 3J = 8.0 Hz, 1H, H3 of py), 7.80–7.83 (m, 3H, H5 and H5′ of bipy and H5 of py), 8.26 (tdd, 3J = 8.0 Hz, 3J = 4.9 Hz, 4J = 1.6 Hz, 2H, H4 and H4′ of bipy), 8.34 (d, 3J = 8.1 Hz, 2H, H3 and H3′ of bipy), 8.48 (ddd, 3J = 5.0 Hz, 4J = 1.9 Hz, 5J = 0.8 Hz, 1H, H4 of py), 9.63 (d, 3J = 5.4 Hz, 1H, H6′ of bipy), 10.45 (d, 3J = 5.5 Hz, 1H, H6 of bipy). 13C{1H} NMR (100 MHz, CD2Cl2 303 K): δ 120.7 (C4 of py), 121.5 (C6 of py), 123.4 (C3 of bipy), 123.6 (C3′ of bipy), 127.8 (C5 of bipy), 128.2 (C5′ of bipy), 137.8 (C5 of py), 141.2 (C4′ of bipy), 142, 3 (C4 of bipy), 148.0 (C3 of py), 150.6 (C6′ of bipy), 152.0 (C2’ of bipy), 152.4 (C2 of bipy), 156.9 (C6 of bipy), 163.9 (C2 of py). Anal. Calcd. for C15H12Cl3N4Ta: C, 33.64; H, 2.26; N, 10.46. Found: C, 33.08; H, 2.05; N, 10.02. The small deviation of the carbon value in elemental analysis is probably due to a small amount of contamination of insoluble 1a.
Synthesis of Nb(=Npy)Cl3(bipy) (4b).
A solution of 2,2′-bipyridine (218 mg, 1.40 mmol) in dichloromethane (20 mL) was added to a suspension of 1b (406 mg, 1.39 mmol) in dichloromethane (20 mL) at room temperature. After stirring for 21 h, all volatiles were removed under reduced pressure. The resulting solids were washed with toluene (10 mL × 4) and hexane (5 mL × 2) and then dried to give 4b as a blue-gray powder in 79% yield (486 mg, 1.08 mmol), mp 236 °C (dec). Single crystals suitable for the X-ray diffraction were obtained from the saturated dichloromethane solution. 1H NMR (400 MHz, CD2Cl2, 303 K): δ 7.11 (ddd, 3J = 7.4 Hz, 3J = 4.9 Hz, 4J = 0.9 Hz, 1H, H4 of py), 7.36 (d, 3J = 8.0 Hz, 1H, H3 of py), 7.72 (ddd, 3J = 7.3 Hz, 3J = 5.8 Hz, 4J = 1.1 Hz, 1H, H5 of py), 7.76–7.81 (m, 3H, H5 and H5′ of bipy and H5 of py), 8.18–8.24 (m, 3H, H5 and H5′ of bipy and H6 of py), 8.30 (d, 3J = 4.2 Hz, 1H, H3 of bipy), 8.32 (d, 3J = 4.4 Hz, 1H, H3′ of bipy), 8.48 (dd, 3J = 4.8 Hz, 4J = 0.9 Hz, 1H, H4 of py), 9.59 (d, 3J = 4.8 Hz, 1H, H6′ of bipy), 10.12 (d, 3J = 5.4 Hz, 1H, H6 of bipy). 13C{1H} NMR (100 MHz, CD2Cl2, 303 K): δ 120.1 (C4 of py), 122.1 (C6 of py), 123.1 (C3 of bipy), 123.3 (C3′ of bipy), 127.4 (C5 of bipy), 127.6 (C5′ of bipy), 138.2 (C5 of py), 141.0 (C4′ of bipy), 141.9 (C4 of bipy), 149.0 (C3 of py), 150.4 (C6′ of bipy), 151.6 (C2′ of bipy), 152.0 (C2 of bipy), 155.8 (C6 of bipy), 163.3 (C2 of py). Anal. Calcd for C15H12Cl3N4Nb: C, 40.26; H, 2.70; N, 12.52. Found: C, 39.84; H, 2.46; N, 12.23.
Synthesis of (bipy)TaCl3(μ-Npy)RhCl(cod) (5a).
A solution of [RhCl(cod)]2 (108 mg, 0.219 mmol) in dichloromethane (3 mL) was added to a solution of 4a (234 mg, 0.437 mmol) in dichloromethane (3 mL). The color of the reaction mixture changed to brown, and an orange powder precipitated. After the reaction mixture was stirred for 2 h, the mixture was concentrated to 3 mL, and then the supernatant was removed. The powder was washed with cold dichloromethane (1 mL × 4) and dried under vacuum to give 5a as an orange powder in 74% yield (254 mg, 0.325 mmol), mp 200–201 °C (dec). Single crystals suitable for the X-ray diffraction were obtained from the saturated dichloromethane solution. 1H NMR (400 MHz, CD2Cl2, 303 K): δ 2.63 (br s, COD), 3.69 (br s, COD), 4.39 (br s, COD), 4.58 (br s, COD), 6.94 (t, 3J = 6.4 Hz, 1H, H4 of py), 7.49 (d, 3J = 8.2 Hz, 1H, H3 of py), 7.72 (t, 3J = 7.8 Hz, 1H, H5 of py), 7.78–7.87 (m, 2H, H5 and H5′ of bipy), 8.25 (t, 3J = 7.8 Hz, 1H, H4 or H4′ of bipy), 8.30 (t, 3J = 8.1 Hz, 1H, H4 or H4′ of bipy), 8.38 (d, 3J = 8.1 Hz, 2H, H3 and H3′ of bipy), 8.82 (d, 3J = 5.3 Hz, 1H, H4 of py), 9.69 (d, 3J = 5.1 Hz, 1H, H6′ of bipy), 10.23 (d, 3J = 5.4 Hz, 1H, H6 of bipy). 13C{1H} NMR (100 MHz, CD2Cl2, 303 K): δ 30.6 (cod), 30.9 (cod), 31.9 (cod), 84.3 (cod), 85.8 (cod), 86.0 (cod), 120.9 (C4 of py), 123.5 (C6 of py), 123.6 (C3 of bipy), 126.3 (C3′ of bipy), 127.8 (C5 of bipy), 128.6 (C5′ of bipy), 137.8 (C5 of py), 141.3 (C4′ of bipy), 142.6 (C4 of bipy), 148.6 (C3 of py), 150.7 (C6′ of bipy), 151.7 (C2′ of bipy), 152.4 (C2 of bipy), 157.2 (C6 of bipy), 163.9 (C2 of py). Anal. Calcd for C23H24Cl4N4TaRh(CH2Cl2)0.5: C, 34.23; H, 3.06; N, 6.79. Found: C, 34.15; H, 3.15; N, 6.89. Inclusion of dichloromethane is due to the remaining of CH2Cl2 in the lattice for the recrystallized sample even after evacuation.
Synthesis of (bipy)NbCl3(μ-Npy)RhCl(cod) (5b).
A solution of [RhCl(cod)]2 (110 mg, 0.223 mmol) in dichloromethane (3 mL) was added to a solution of 4b (200 mg, 0.488 mmol) in dichloromethane (3 mL). The color of the reaction mixture changed to dark green, and an orange microcrystalline powder precipitated. After the reaction mixture was stirred for 2 h, the mixture was concentrated to 3 mL, and then the supernatant was removed. The powder was washed with cold dichloromethane (1 mL × 6) and dried under vacuum to give 5b as an orange powder in 75% yield (235 g, 0.338 mmol), mp 211–212 °C (dec). Single crystals suitable for the X-ray diffraction were obtained from the saturated dichloromethane solution. 1H NMR (400 MHz, CD2Cl2, 303 K): δ 2.66 (br s, COD), 3.69 (br s, COD), 4.24 (br s, COD), 4.65 (br s, COD), 7.12 (t, 3J = 6.2 Hz, 1H, H4 of py), 7.69 (t, 3J = 8.2 Hz, 1H, H5 of py), 7.72–7.83 (m, 2H, H5 and H5′ of bipy), 7.91 (d, 3J = 8.1 Hz, 1H, H3 of py), 8.18 (t, 3J = 8.2 Hz, 1H, H4 or H4′ of bipy), 8.20–8.36 (m, 3H, H4 or H4′ of bipy, H3 and H3′ of bipy), 8.80 (d, 3J = 4.8 Hz, 1H, H4 of py), 9.66 (d, 3J = 4.9 Hz, 1H, H6′ of bipy), 9.98 (d, 3J = 5.0 Hz, 1H, H6 of bipy). 13C{1h} NMR (100 MHz, CD2Cl2, 303 K): δ 30.9 (cod), 31.9 (cod), 85.0 (cod), 85.1 (cod), 122.3 (C6 of py), 123.0 (C3 of bipy), 123.4 (C3′ of bipy), 125.6 (C4 of py), 128.1 (C5 of bipy), 138.2 (C5 of py), 141. One (C4′ of bipy), 142.2 (C4 of bipy), 149.2 (C3 of py), 150.6 (C6′ of bipy), 151.3 (C2′ of bipy), 152.1 (C2 of bipy), 156.3 (C6 of bipy), a signal of C2 of py was not observed due to the low sensitivity of NMR analysis. Anal. Calcd for C23H24Cl4N4NbRh(CH2Cl2)0.5: C, 38.32; H, 3.42; N, 7.61. Found: C, 37.86; H, 3.35; N, 7.51. Inclusion of dichloromethane is due to the remnant of CH2Cl2 in the lattice for the recrystallized sample even after evacuation.
X-ray Crystallographic Analysis.
The crystals were mounted on a CryoLoop (Hampton Research Corp) with a layer of light mineral oil and placed in a nitrogen stream at 113(1) K. All measurements were made on a Rigaku Xtalab P200 diffractometer using multilayer mirror monochromated Mo Kα (0.71076 Å) radiation. The structures were solved by SHELXS-201316 and refined on F2 by full-matrix least-squares method, using SHELXL-2013.17 Non-hydrogen atoms were anisotropically refined. H-atoms were included in the refinement on calculated positions riding on their carrier atoms. The function minimized was [Σw(Fo2 − Fc2)2] (w = 1/[σ2(Fo)2 + (aP)2 + bP]), where P = (Max(Fo2, 0) + 2Fc2)/3 with σ2(Fo2) from counting statistics. The functions R1 and wR2 were (Σ||Fo| − |Fc||)/Σ|Fo| and [Σw(Fo2 − Fc2)2/Σ(wFo4)]1/2, respectively. The ORTEP-3 program18 was used to draw the molecule.
Supplementary Material
ACKNOWLEDGMENTS
K.K. thanks the financial support by the JSPS Research Fellowships for Young Scientist. H.T. acknowledges the financial support by JSPS KAKENHI grant No. 15KK0186, a Fund for the Promotion of Joint International Research (Fostering Joint International Research), and Multidisciplinary Research Laboratory System of Graduate School of Engineering Science, Osaka University. K.M. acknowledges financial supports by JSPS KAKENHI Grant Nos. 15H05808 and 15K21707 in Precisely Designed Catalysts with Customized Scaffolding (No. 2702). Financial support was provided by the National Institutes of Health (1R35GM119457), and the Alfred P. Sloan Foundation (I.A.T. is a 2017 Sloan Fellow).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorg-chem.9b02043.
Summary of electrochemical analyses of 2a and 2b (PDF)
Accession Codes
CCDC 1939439–1939445 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).Representative books and reviews for imido complexes of early transition metals:; (a) Nugent WA; Mayer JM Metal-Ligand Multiple Bonds; Wiley-Interscience: New York, 1988. [Google Scholar]; (b) Wigley DE Organoimido Complexes of the Transition Metals. Prog. Inorg. Chem 2007, 42, 239–482. [Google Scholar]; (c) Gianetti TL; La Pierre HS; Arnold J Group 5 Imides and Bis(imide)s as Selective Hydrogenation Catalysts. Eur. J. Inorg. Chem 2013, 2013, 3771–3783. [Google Scholar]; (d) Schrock RR Recent Advances in High Oxidation State Mo and W Imido Alkylidene Chemistry. Chem. Rev 2009, 109, 3211–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zarubin DN; Ustynyuk NA Methods of synthesis of Group 4–9 Transition Metal Imido Complexes. Russ. Chem. Rev 2006, 75, 671–707. [Google Scholar]; (f) Hazari N; Mountford P Reactions and Applications of Titanium Imido Complexes. Acc. Chem. Res 2005, 38, 839–849. [DOI] [PubMed] [Google Scholar]; (g) Bolton PD; Mountford P Transition Metal Imido Compounds as Ziegler-Natta Olefin Polymerisation Catalysts. Adv. Synth. Catal 2005, 347, 355–366. [Google Scholar]; (h) Giesbrecht GR; Gordon JC Lanthanide Alkylidene and Imido Complexes. Dalton Trans 2004, 2387–2393. [DOI] [PubMed] [Google Scholar]; (i) Schrock RR Recent Advances in Olefin Metathesis by Molybdenum and Tungsten Imido Alkylidene Complexes. J. Mol. Catal. A: Chem 2004, 213, 21–30. [DOI] [PubMed] [Google Scholar]; (j) Antiñolo A; Fajardo M; Otero A; Prashar S Sandwich and Half-Sandwich (Imido)niobium Complexes. Eur. J. Inorg. Chem 2003, 2003, 17–28. [Google Scholar]; (k) Eikey RA; Abu-Omar MM Nitrido and Imido Transition Metal Complexes of Groups 6–8. Coord. Chem. Rev 2003, 243, 83–124. [Google Scholar]; (l) Duncan AP; Bergman RG Selective Transformations of Organic Compounds by Imidozircocene Complexes. Chem. Rec 2002, 2, 431–445. [DOI] [PubMed] [Google Scholar]; (m) Gade LH; Mountford P New Transition Metal Imido Chemistry with Diamido-donor Ligands. Coord. Chem. Rev 2001, 216–217, 65–97. [Google Scholar]; (n) Mountford P New Titanium Imido Chemistry. Chem. Commun 1997, 2127–2134. [Google Scholar]; (o) Nugent WA; Haymore BL Transition Metal Complexes Containing Organoimido (nr) and Related Ligands. Coord. Chem. Rev 1980, 31, 123–175. [Google Scholar]
- (2).Representative references for cycloaddition reaction by early transition metal imido complexes:; (a) Schwarz AD; Nova A; Clot E; Mountford P Titanium Alkoxyimido (Ti = N−OR) Complexes: Reductive N–O Bond Cleavage at the Boundary Between Hydrazide and Peroxide Ligands. Chem. Commun 2011, 47, 4926–4928. [DOI] [PubMed] [Google Scholar]; (b) Schofield AD; Nova A; Selby JD; Manley CD; Schwarz AD; Clot E; Mountford PM = Nα Cycloaddition and Nα−Nβ Insertion in the Reactions of Titanium Hydrazido Compounds with Alkynes: A Combined Experimental and Computational Study. J. Am. Chem. Soc 2010, 132, 10484–10497. [DOI] [PubMed] [Google Scholar]; (c) Bolton PD; Feliz M; Cowley AR; Clot E; Mountford P Ti = NR vs Ti–R’ Functional Group Selectivity in Titanium imido Alkyl Cations from an Experimental Perspective. Organometallics 2008, 27, 6096–6110. [Google Scholar]; (d) Bouwkamp MW; Batinas AA; Witte PT; Hubregtse T; Dam J; Meetsma A; Teuben JH; Hessen B Relative Reactivity of the Metal–Amido versus Metal–Imido Bond in Linked Cp-Amido and Half-Sandwich Complexes of Vanadium. Organometallics 2008, 27, 4071–4082. [Google Scholar]; (e) Vujkovic N; Ward BD; Maisse-François A; Wadepohl H; Mountford P; Gade LH Imido-Alkyne Coupling in Titanium Complexes: New Insights into the Alkyne Hydroamination Reaction. Organometallics 2007, 26, 5522–5534. [Google Scholar]; (f) Lokare KS; Ciszewski JT; Odom AL Group-6 Imido Activation by a Ring-Strained Alkyne. Organometallics 2004, 23, 5386–5388. [Google Scholar]; (g) Zuckerman RL; Bergman RG Mechanistic Investigation of Cycloreversion/Cycloaddition Reactions between Zirconocene Metallacycle Complexes and Unsaturated Orgnanic Substrates. Organometallics 2001, 20, 1792–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zuckerman RL; Bergman RG Structural Factors that Influence the Course of Overall [2 + 2] Cycloaddition Reactions between Imidozirconocene Complexes and Heterocumulenes. Organometallics 2000, 19, 4795–4809. [Google Scholar]; (i) Lee YS; Bergman RG Generation of Oxozirconocene Complexes from the Reaction of Cp2(THF)Zr = N-t-Bu with Organic and Metal Carbonyl Functionalities: Apparently Divergent Behavior of Transient [Cp2Zr = O]. J. Am. Chem. Soc 1996, 118, 6396–6406. [Google Scholar]; (j) Lee SY; Bergman RG Mechanism and Regiochemistry of Azametallacyclobutene Formation from Imidozirconocene Complexes and Alkynes. Tetrahedron 1995, 51, 4255–4276. [Google Scholar]; (k) Hanna TA; Baranger AM; Walsh PJ; Bergman RG Formation of α,β-Unsaturated Imines and Successful Trapping of Oxozirconocene in a [4 + 2] Azaoxametallacyclohexene Retrocycloaddition. J. Am. Chem. Soc 1995, 117, 3292–3293. [Google Scholar]; (l) Meyer KE; Walsh PJ; Bergman RG A Mechanistic Study of the Cycloaddition-Cycloreversion Reactions of the Zirconium-Imido Complex Cp2Zr(N-t-Bu)(THF) with Organic Imines and Azides. J. Am. Chem. Soc 1995, 117, 974–985. [Google Scholar]; (m) Walsh PJ; Hollander FJ; Bergman RG Monomeric and Dimeric Zirconocene imido Compounds: Synthesis, Structure, and Reactivity. Organometallics 1993, 12, 3705–3723. [Google Scholar]; (n) Baranger AM; Walsh PJ; Bergman RG Variable Regiochemistry in the Stoichiometric and Catalytic Hydroamination of Alkynes by Imidozirconium Complexes Caused by an Unusual Dependence of the Rate Law on Alkyne Structure and Temperature. J. Am. Chem. Soc 1993, 115, 2753–2763. [Google Scholar]; (o) Walsh PJ; Hollander FJ; Bergman RG Generation, Alkyne Cycloaddition, Arene C–H Activation, N–H Activation, and Dative ligand Trapping Reactions of the First Monomeric Imidozirconocene (Cp2Zr = NR) Complexes. J. Am. Chem. Soc 1988, 110, 8729–8731. [Google Scholar]
- (3).(a) Kawakita K; Beaumier EP; Kakiuchi Y; Tsurugi H; Tonks IA; Mashima K Bis(imido)vanadium(V)-Catalyzed [2 + 2+1] Coupling of Alkynes and Azobenzenes Giving Multisubstituted Pyrroles. J. Am. Chem. Soc 2019, 141, 4194–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chiu H-C; See XY; Tonks IA Dative Directing Group Effects in Ti-Catalyzed [2 + 2+1] Pyrrole Synthesis: Chemo- and Regioselective Alkyne Heterocoupling. ACS Catall. 2019, 9, 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Davis-Gilbert ZW; Kawakita K; Blechschmidt DR; Tsurugi H; Mashima K; Tonks IA In Situ Catalyst Generation and Benchtop-Compatible Entry Points for TiII/TiIV Redox Catalytic Reactions. Organometallics 2018, 37, 4439—4445. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Davis-Gilbert ZW; Wen X; Goodpaster JD; Tonks IA Mechanism of Ti-Catalyzed Oxidative Nitrene Transfer in [2 + 2+1] Pyrrole Synthesis from Alkynes and Azobenzene. J. Am. Chem. Soc 2018, 140, 7267–7281. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chiu H-C; Tonks IA Trimethylsilyl-Protected Alkynes as Selective Cross-Coupling Partners in Titanium-Catalyzed [2 + 2+1] Pyrrole Synthesis. Angew. Chem., Int. Ed 2018, 57, 6090–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Pearce AJ; See XY; Tonks IA Oxidative Nitrene Transfer from Azides to Alkynes via Ti(II)/Ti(IV) Redox Catalysis: Formal [2 + 2+1] Synthesis of Pyrroles. Chem. Commun 2018, 54, 6891–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Davis-Gilbert ZW; Yao LJ; Tonks IA Ti-Catalyzed Multicomponent Oxidative Carboamination of Alkynes with Alkenes and Diazenes. J. Am. Chem. Soc 2016, 138, 14570–14573. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Gilbert ZW; Hue RJ; Tonks IA Catalytic Formal [2 + 2+1] Synthesis of Pyrroles from Alkynes and Diazenes via TiII/TiIV Redox Catalysis. Nat. Chem 2016, 8, 63–68. [DOI] [PubMed] [Google Scholar]
- (4).Representative references for nitrene transfer reaction by early transition metal imido complexes:; (a) Heins SP; Wolczanski PT; Cundari TR; MacMillan SN Redox Non-Innocence Permits Catalytic Nitrene Carbonylation by (dadi)Ti = NAd (Ad = adamantyl). Chem. Sci 2017, 8, 3410–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kriegel BM; Bergman RG; Arnold J Nitrene Metathesis and Catalytic Nitrene Trasfer Promoted by Niobium Bis(imido) Complexes. J. Am. Chem. Soc 2016, 138, 52–55. [DOI] [PubMed] [Google Scholar]; (c) Yousif M; Tjapkes DJ; Lord RL; Groysman S Catalytic Formation of Asymmetric Carbodiimides at Mononuclear Chromium(II/IV) Bis(alkoxide) Complexes. Organometallics 2015, 34, 5119–5128. [Google Scholar]; (d) Nguyen AI; Zarkesh RA; Lacy DC; Thorson MK; Heyduk AF Catalytic Nitrene Transfer by Zirconium(IV) Redox-Active Ligand Complex. Chem. Sci 2011, 2, 166–169. [Google Scholar]; (e) Heyduk AF; Zarkesh RA; Nguyen AI Designing Catalyst for Nitrene Transfer Using Early Transition Metals and Redox-Active Ligands. Inorg. Chem 2011, 50, 9849–9863. [DOI] [PubMed] [Google Scholar]; (f) Zarkesh RA; Ziller JW; Heyduk AF Four-Electron Oxidative Formation of Aryl Diazenes Using a Tantalum Redox-Active Ligand Complex. Angew. Chem., Int. Ed 2008, 47, 4715–4718. [DOI] [PubMed] [Google Scholar]
- (5).Representative reviews for hydroamination reaction by early transition metal imido complexes:; (a) Müller TE; Hultzsch KC; Yus M; Foubelo F; Tada M Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev 2008, 108, 3795–3892. [DOI] [PubMed] [Google Scholar]; (b) Severin R; Doye S The Catalytic Hydroamination of Alkynes. Chem. Soc. Rev 2007, 36, 1407–1420. [DOI] [PubMed] [Google Scholar]; (c) Pohlki F; Doye S The Catalytic Hydroamination of Alkynes. Chem. Soc. Rev 2003, 32, 104–114. [DOI] [PubMed] [Google Scholar]; (d) Müller TE; Beller M Metal-Initiated Amination of Alkenes and Alkynes. Chem. Rev 1998, 98, 675–703. [DOI] [PubMed] [Google Scholar]
- (6).Representative references for metathesis reaction by early transition metal imido complexes:; (a) Hamzaoui B; Pelletier JDA; Abou-Hamad E; Basset J-M Well-Defined Silica-Supported Zirconium-Imido Complexes Mediated Heterogeneous Imine Metathesis. Chem. Commun 2016, 52, 4617–4620. [DOI] [PubMed] [Google Scholar]; (b) Zhizhko PA; Zhizhin AA; Belyakova OA; Zubavichus YV; Kolyagin YG; Zarubin DN; Ustynyuk NA Oxo/Imido Heterometathesis Reactions Catalyzed by a Silica-Supported Tantalum Imido Complex. Organometallics 2013, 32, 3611–3617. [Google Scholar]; (c) Darwish W; Seikel E; Käsmarker R; Harms K; Sundermeyer J Synthesis and X-ray Crystal Structures of Imido and Ureato Derivatives of Titanium(IV) Phthalocyanine and Their Application in the Catalytic Formation of Carbodiimides by Metathesis from Isocyanates. Dalton Trans 2011, 40, 1787–1794. [DOI] [PubMed] [Google Scholar]; (d) Burland MC; Pontz TW; Meyer TY Role of Trace Amine in the Metathesis of Imines by CpTa(=NR)Cl2. Organometallics 2002, 21, 1933–1941. [Google Scholar]; (e) Zuckerman RL; Krska SW; Bergman RG Zirconium-Mediated Metathesis of Imines: A Study of the Scope, Longevity, and Mechanism of a Complicated Catalytic System. J. Am. Chem. Soc 2000, 122, 751–761. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Bruno JW; Li XJ Use of Niobium(III) and Niobium(V) Compounds in Catalytic Imine Metathesis under Mild Conditions. Organometallics 2000, 19, 4672–4674. [Google Scholar]; (g) Cantrell GK; Geib SJ; Meyer TY Ring-Opening of a Cyclic Imine: The First Step of Imine ROMP. Organometallics 1999, 18, 4250—4252. [Google Scholar]; (h) Wang W-D; Espenson JH Metathesis Reactions of Tris(adamantylimido)methylrhenium and Aldehydes and Imines. Organometallics 1999, 18, 5170–5175. [Google Scholar]; (i) Cantrell GK; Meyer TY Catalytic C = N Bond Formation by Metal-Imide-Mediated Imine Metathesis. J. Am. Chem. Soc 1998, 120, 8035–8042. [Google Scholar]; (j) Krska SW; Zuckerman RL; Bergman RG Use of Steric Hindrance and a Metallacyclobutene Resting State to Develop Robust and Kinetically Characterizable Zirconium-Based Imine Metathesis Catalysts. J. Am. Chem. Soc 1998, 120, 11828–11829. [Google Scholar]; (k) McInnes JM; Mountford P Transition Metal Imide/Organic Imine Metathesis Reactions: Unexpected Observations. Chem. Commun 1998, 1669–1670. [Google Scholar]; (l) Cantrell GK; Meyer TY Azaheteroalkene Metathesis: Reaction of Imines with Molybdenum(VI) Bis(imide) Complexes. Chem. Commun 1997, 1551–1552. [Google Scholar]; (m) Cantrell GK; Meyer TY Transition-Metal-Catalyzed Imine Metathesis. Organometallics 1997, 16, 5381–5383. [Google Scholar]; (n) Meyer KE; Walsh PJ; Bergman RG A Mechanistic Study of the Cycloaddition-Cycloreversion Reactions of the Zirconium-Imido Complex Cp2Zr(N-t-Bu)(THF) with Organic Imines and Azides. J. Am. Chem. Soc 1995, 117, 974–985. [Google Scholar]; (o) Meyer KE; Walsh PJ; Bergman RG Zirconium-mediated Imine Metathesis. Synthesis of 2,4-Diaza-1-zirconiacyclobutanes and the Mechanism of Their Reactions with Imines and Alkynes. J. Am. Chem. Soc 1994, 116, 2669–2670. [Google Scholar]; (p) Birdwhistell KR; Boucher T; Ensminger M; Harris S; Johnson M; Toporek S Catalysis of Phenyl Isocyanate Condensation to N,N’-Diphenylcarbodiimide via Vanadium Oxo and Imido Complexes. Organometallics 1993, 12, 1023–1025. [Google Scholar]
- (7).(a) Pedrosa S; Vidal F; Lee LM; Vargas-Baca I; Gómez-Sal P; Mosquera MEG Imido-Pyridine Ti(IV) Compounds; Synthesis of Unusual Imido-Amido Heterobimetallic Derivatives. Dalton Trans. 2015, 44, 11119–11128. [DOI] [PubMed] [Google Scholar]; (b) Lv C; Hu J; Khan RNN; Zhang J; Wei Y Postfunctionalization of Polyoxometalates: An Efficient Strategy to Construct Organic-Inorganic Zwitterions. Dalton Trans. 2015, 44, 16698–16702. [DOI] [PubMed] [Google Scholar]; (c) Duan X-E; Yuan S-F; Tong H-B; Bai S-D; Wei X-H; Liu D-S Metal (Mg, Fe, Co, Zr and Ti) Complexes Derived from Aminosilyl Substituted Aminopyridinato Ligand: Synthesis, Structures and Ethylene Polymerization Behaviors of the Group 4 Complexes. Dalton Trans. 2012, 41, 9460–9467. [DOI] [PubMed] [Google Scholar]; (d) Qian Y-L; Deng F-J; Li F-J; Liu Y; Wang C-X; Liu S-Y; Cao A-H; Qu S-S Novel Intramolecular Coordination Chemistry of Some New Metallocene Complexes. Chin. J. Chem 2001, 19, 1009–1022. [Google Scholar]; (e) Li Z; Huang J; Yao T; Qian Y; Leng M Synthesis of Titanium μ-Arylimido and μ-Pyridylimido Complexes Bearing (Un)substituted Cyclopentadienyl Ligand. J. Organomet. Chem 2000, 598, 339–347. [Google Scholar]; (f) Hill PL; Yap GPA; Rheingold AL; Maatta EA Organoimido Ligands with Remote Functionality: A p-Pyridylimido Complex of Vanadium(V) and its Use as a Metalloligand. J. Chem. Soc., Chem. Commun 1995, 737–738. [Google Scholar]
- (8).(a) Ikeda H; Nishi K; Tsurugi H; Mashima K Metathesis Cleavage of an N=N Bond in Benzo[c]cinnolines and Azobenzenes by Triply-Bonded Ditungsten Complexes. Chem. Commun 2018, 54, 3709–3711. [DOI] [PubMed] [Google Scholar]; (b) Wijeratne GB; Zolnhofer EM; Fortier S; Grant LN; Carroll PJ; Chen C-H; Meyer K; Krzystek J; Ozarowski A; Jackson TA; et al. Electronic Structure and Reactivity of a Well-Defined Mononuclear Complex of Ti(II). Inorg. Chem 2015, 54, 10380–10397. [DOI] [PubMed] [Google Scholar]; (c) Milsmann C; Turner ZR; Semproni SP; Chirik PJ Azo N=N Bond Cleavage with a Redox-Active Vanadium Compound Involving Metal-Ligand Cooperativity. Angew. Chem., Int. Ed 2012, 51, 5386–5390. [DOI] [PubMed] [Google Scholar]; (d) Kaleta K; Arndt P; Beweries T; Spannenberg A; Theilmann O; Rosenthal U Reactions of Group 4 Metallocene Alkyne Complexes with Azobenzene: Formation of Diazametallacyclopropenes and N=N Bond Activation. Organometallics 2010, 29, 2604–2609. [Google Scholar]; (e) Tsai Y-C; Wang P-Y; Lin K-M; Chen S-A; Chen J-M Synthesis and Reactions of β-Diketiminato Divanadium(I) Inverted-sandwich Complexes. Chem. Commun 2008, 205–207. [DOI] [PubMed] [Google Scholar]; (f) Monillas WH; Yap GPA; MacAdams LA; Theopold KH Binding and Activation of Small Molecules by Three-Coordinate Cr(I). J. Am. Chem. Soc 2007, 129, 8090–8091. [DOI] [PubMed] [Google Scholar]; (g) Kilgore UJ; Yang X; Tomaszewski J; Huffman JC; Mindiola DJ Activation of Atmospheric Nitrogen and Azobenzene N=N Bond Cleavage by Transient Nb(III) Complex. Inorg. Chem 2006, 45, 10712–10721. [DOI] [PubMed] [Google Scholar]; (h) Komuro T; Matsuo T; Kawaguchi H; Tatsumi K Synthesis of a Vanadium(III) Tris(arylthiolato) Complex and Its Reactions with Azide and Azo Compounds: Formation of a Sulfenamide Complex via Cleavage of an Azo N=N Bond. Inorg. Chem 2005, 44, 175–177. [DOI] [PubMed] [Google Scholar]; (i) Lentz MR; Vilardo JS; Lockwood MA; Fanwick PE; Rothwell IP Synthetic and Mechanistic Studies of the Four-Electron Reduction of Dioxygen, N=N, and N = O Double Bonds by Tungsten(II) Aryloxide Compounds. Organometallics 2004, 23, 329–343. [Google Scholar]; (j) Aubart MA; Bergman RG Tantalum-Mediated Cleavage of an N=N Bond in an Organic Diazene (Azoarene) to Produce an Imidometal (M = NR) Complex: An η2-Diazene Complex Is Not an Intermediate. Organometallics 1999, 18, 811–813. [Google Scholar]; (k) Gray SD; Thorman JL; Adamian VA; Kadish KM; Woo LK Synthesis, Electrochemistry, and Imido Transfer Reactions of (TTP)Ti(η2-PhN=NPh). Inorg. Chem 1998, 37, 1–4. [DOI] [PubMed] [Google Scholar]; (l) Barry JT; Chisholm MH; Folting K; Huffman JC; Streib WE Reactions of W2(H)(OR)7, W2(OR)6(py)2 and W4(OCH2cC4H7)12 Compounds (R = Pri, CH2But, cC5H9) with Azobenzene, 1,2-Diphenylhydrazine and 1,1-Dimethylhydrazine. Polyhedron 1997, 16, 2113–2133. [Google Scholar]; (m) Gray SD; Thorman JL; Berreau LM; Woo LK Alkoxido, Amido, and Imido Derivatives of Titanium(IV) Tetrato-lylporphyrin. Inorg. Chem 1997, 36, 278–283. [Google Scholar]; (n) Lockwood MA; Fanwick PE; Eisenstein O; Rothwell IP Mechanistic Studies of the Facile Four-Electron Reduction of Azobenzene at a Single Tungsten Metal Center. J. Am. Chem. Soc 1996, 118, 2762–2763. [Google Scholar]; (o) Duchateau R; Williams AJ; Gambarotta S; Chiang MY Carbon–Carbon Double-Bond Formation in the Intermolecular Acetonitrile Reductive Coupling Promotedn by a Mononuclear Titanium(II) Compound. Preparation and Characterization of Two Titanium(IV) Imido Derivatives. Inorg. Chem 1991, 30, 4863–4866. [Google Scholar]; (p) Hill JE; Profilet RD; Fanwick PE; Rothwell IP Synthesis, Structure, and Reactivity of Arylozo(imido)titanium Complexes. Angew. Chem., Int. Ed. Engl 1990, 29, 664–665. [Google Scholar]; (q) Canich JAM; Cotton FA; Duraj SA; Roth WJ The Preparation of Ta2Cl6(PhN)2(Me2S)2 by Reaction of Ta2Cl6(Me2S)3 with PhNNPh: Crystal Structure of the Product. Polyhedron 1986, 5, 895. [Google Scholar]; (r) Cotton FA; Duraj SA; Roth WJ A New Double Bond Metathesis Reaction: Conversion of An Niobium:niobium and An Nitrogen:nitrogen Bond into Two Niobium:niobium Bonds. J. Am. Chem. Soc 1984, 106, 4749–4751. [Google Scholar]; (s) Gambarotta S; Floriani C; Chiesi-Villa A; Guastini C Cyclopentadienyldichlorotitanium(III): A Free-radical-like Reagent for Reducing Azo(N:N) Multiple Bonds in Azo and Diazo Compounds. J. Am. Chem. Soc 1983, 105, 7295–7301. [Google Scholar]; (t) Gambarotta S; Floriani C; Chiesi-Villa A; Guastini C Nitrogen-Nitrogen Multiple Bond Cleavage and Reduction in Diphenyldiazomethane and Azobenzen by a Titanium(III) Complex. J. Chem. Soc., Chem. Commun 1982, 1015–1017. [Google Scholar]; (u) Wiberg N; Häring H-W; Schubert U Darstellung und Struktur des Komplexes [C5H5Cr(NSiMe3)2]2 (Zur Reaktion von Cyclopentadienylmetall-chloriden mit Bis(trimethylsilyl)diazen). Z. Natur/orsch., B: J. Chem. Sci 1978, 33, 1365–1369. [Google Scholar]
- (9).(a) Tsurugi H; Mashima K Salt-Free Reduction of Transition Metal Complexes by Bis(trimethylsilyl)cyclohexadiene, -dihydropyrazine, and −4,4′-bipyridinylidene Derivatives. Acc. Chem. Res 2019, 52, 769–779. [DOI] [PubMed] [Google Scholar]; (b) Tsurugi H; Mashima K A New Protocol to Generate Catalytically Active Species of Group 4–6 Metals by Organosilicon-Based Salt-Free Reductants. Chem. - Eur. J 2019, 25, 913. [DOI] [PubMed] [Google Scholar]; (c) Saito T; Nishiyama H; Kawakita K; Nechayev M; Kriegel B; Tsurugi H; Arnold J; Mashima K Reduction of (tBuN =)NbCl3(py)2 in a Salt-Free Manner for Generating Nb(IV) Dinuclear Complexes and Their Reactivity toward Benzo[c]cinnoline. Inorg. Chem 2015, 54, 6004–6009. [DOI] [PubMed] [Google Scholar]; (d) Saito T; Nishiyama H; Tanahashi H; Kawakita K; Tsurugi H; Mashima K 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes as Strong Salt-Free Reductants for Generating Low-Valent Early Transition Metals with Electron-Donating Ligands. J. Am. Chem. Soc 2014, 136, 5161. [DOI] [PubMed] [Google Scholar]; (e) Tsurugi H; Tanahashi H; Nishiyama H; Fegler W; Saito T; Sauer A; Okuda J; Mashima K Salt-Free Reducing Reagent of Bis(trimethylsilyl)cyclohexadiene Mediates Multielectron Reduction of Chloride Complexes of W(VI) and W(IV). J. Am. Chem. Soc 2013, 135, 5986–5989. [DOI] [PubMed] [Google Scholar]; (f) Tsurugi H; Saito T; Tanahashi H; Arnold J; Mashima K Carbon Radical Generation by d0 Tantalum Complexes with α-Diimine Ligands through Ligand-Centered Redox Processes. J. Am. Chem. Soc 2011, 133, 18673–18683. [DOI] [PubMed] [Google Scholar]; (g) Arteaga-Müller R; Tsurugi H; Saito T; Yanagawa M; Oda S; Mashima K New Tantalum Ligand-Free Catalyst System for Highly Selective Trimerization of Ethylene Affording 1-Hexene: New Evidence of a Metallacycle Mechanism. J. Am. Chem. Soc 2009, 131, 5370–5371. [DOI] [PubMed] [Google Scholar]
- (10).(a) Frantz S; Sieger M; Hartenbach I; Lissner F; Schleid T; Fiedler J; Duboc C; Kaim W Structure, Electrochemistry, Spectroscopy, and Magnetic Resonance, including High-Field EPR, of {(μ-abpy)[Re(CO)3X]2}0/•-, where abpy = 2,2′-Azobispyridine and X = F, Cl, Br, I. J. Organomet. Chem 2009, 694, 1122–1133. [Google Scholar]; (b) Sarkar B; Patra S; Fiedler J; Sunoj RB; Janardanan D; Lahiri GK; Kaim W Mixed-Valent Metals Bridged by a Radical Ligand: Fact or Fiction Based on Structure-Oxidation State Correlations. J. Am. Chem. Soc 2008, 130, 3532–3542. [DOI] [PubMed] [Google Scholar]; (c) Sarkar B; Patra S; Fiedler J; Sunoj RB; Janardanan D; Mobin SM; Niemeyer M; Lahiri GK; Kaim W Theoretical and Experimental Evidence for a New Kind of Spin-Coupled Singlet Species: Isomeric Mixed-Valent Complexes Bridged by a Radical Anion Ligand. Angew. Chem., Int. Ed 2005, 44, 5655–5658. [DOI] [PubMed] [Google Scholar]; (d) Frantz S; Fiedler J; Hartenbach I; Schleid T; Kaim W A Complete Series of Tricarbonylhalidorhenium(I) Complexes (abpy)Re(CO)3(Hal), Hal = F, Cl, Br, I; abpy = 2,2′-azobispyridine: Structures, Spectroelectrochemistry and EPR of Reduced Forms. J. Organomet. Chem 2004, 689, 3031–3039. [Google Scholar]; (e) Kaim W Complexes with 2,2 ‘-Azobispyridine and Related ‘S-frame’ Bridging Ligands Containing the Azo Function. Coord. Chem. Rev 2001, 219–221, 463–488. [Google Scholar]; (f) Hartmann H; Scheiring T; Fiedler J; Kaim W Structures and Spectroelectrochemistry (UV-vis, IR, EPR) of Complexes [(OC)3ClRe]n (abpy), n = 1, 2; abpy = 2,2’-azobispyridine. J. Organomet. Chem 2000, 604, 267–272. [Google Scholar]; (g) Doslik N; Sixt T; Kaim W The First Structural Characterization of an Azoaromatic Radical Anion Stabilized by Dicopper(I) Coordination. Angew. Chem., Int. Ed 1998, 37, 2403–2404. [DOI] [PubMed] [Google Scholar]; (h) Kaim W; Kohlmann S; Jordanov J; Fenske D Kantenverkmpfung zweier Metallchelat-Fünfringe: Strukturen und Magnetismus von Mo0-, CuI-und CoII- Komplexen mit dem “S-Frame”-Liganden Azo-2,2′ -pyridin. Z. Anorg. Allg. Chem 1991, 598–599, 217–234. [Google Scholar]; (i) Kaim W; Kohlmann S Four Bridging Bis Chelate Ligands with Very Low Lying π* Orbitals. MO Pertubation Calculatiions, Electrochemistry, and Spectroscopy of Mononuclear and Binuclear Group 6 Metal Tetracarbonyl Complexes. Inorg. Chem 1987, 26, 68–77. [Google Scholar]; (j) Kaim W; Kohlmann S Semireduced Bridging Ligands Containing −N=N-Multiple Bond Coordination Sites. ESR Study of Binuclear Group 6 Metal Carbonyl Complexes. Inorg. Chem 1986, 25, 3442–3448. [Google Scholar]; (k) Kohlmann S; Ernst S; Kaim W Extremely Long-Wavelength Charge-Transfer Absorptions of Binuclear Complexes with Azo-Modified 2,2′ -Bipyridyl Ligands. Angew. Chem., Int. Ed. Engl 1985, 24, 684–685. [Google Scholar]; (l) Baldwin DA; Lever ABP; Parish RV Complexes of 2,2′-Azopyridine with Iron(II), Cobalt(II), Nickel(II), Copper(I), and Copper(n). Inorg. Chem 1969, 8, 107–115. [Google Scholar]; (m) Grzeskowiak R; Beadle PJ Metal Complexes of Azopyridines. Inorg. Nucl. Chem. Lett 1967, 3, 245–248. [Google Scholar]
- (11).Dubberley SR; Evans S; Boyd CL; Mountford P New and versatile routes to zirconium imido dichloride compounds. Dalton Trans. 2005, 1448–1458. [DOI] [PubMed] [Google Scholar]
- (12).(a) Afonin AV; Pavlov DV; Albanov AI; Tarasova OA; Nedolya NA Experimental and Theoretical Study of the Intramolecular C–H⋯N and C–H⋯S Hydrogen Bonding Effects in the 1H and 13C NMR Spectra of the 2-(Alkylsulfanyl)-5-amino-1-vinylpyrroles: a Particular State of Amine Nitrogen. Magn. Reson. Chem 2013, 51, 414–423. [DOI] [PubMed] [Google Scholar]; (b) Afonin AV; Ushakov IA; Vashchenko AV; Simonenko DE; Ivanov AV; Vasil’tsov AM; Mikhaleva AI; Trofimov BA C–H⋯N and C–H⋯O Intramolecular Hydrogen Bonding Effects in the 1H, 13C and 15N NMR Spectra of the Configurational Isomers of 1-Vinylpyrrole-2-carbaldehyde Oxime Substantiated by DFT Calculations. Magn. Reson. Chem 2009, 47, 105–112. [DOI] [PubMed] [Google Scholar]; (c) Laungani AC; Keller M; Slattery JM; Krossing I; Breit B Cooperative Efect of a Classical and a Weak Hydrogen Bond for the Metal-Induced Construction of Self-Assembled β-Turn Mimic. Chem. - Eur. J 2009, 15, 10405–10422. [DOI] [PubMed] [Google Scholar]; (d) Arunima; Kurur ND Cross-Correlated Relaxation Evidence for the Intramolecular C–H⋯N Hydrogen Bond in Solution. Chem. Phys. Lett 2005, 401, 470–474. [Google Scholar]; (e) Cappelli A; Giorgi G; Anzini M; Vomero S; Ristori S; Rossi C; Donati A Characterization of Persistent Intramolecular C–H⋯X(N,O) Bonds in Solid State and Solution. Chem. - Eur. J 2004, 10, 3177–3183. [DOI] [PubMed] [Google Scholar]; (f) Alekseyeva ES; Batsanov AS; Boyd LA; Fox MA; Hibbert TG; Howard JAK; MacBride JAH; Mackinnon A; Wade K Intra- and Inter-Molecular Carboranyl C–H⋯N Hydrogen Bonds in Pyridyl-Containing ortho-Carboranes. Da/ton Trans 2003, 475–482. [Google Scholar]; (g) Afonin AV; Ushakov IA; Kuznetsova SY; Andriyankova LV Influence of the C–H⋯N Intramolecular Interaction on the Spatial Structures and 1H and 13C NMR Parameters of Heteroaryl Vinyl Ethers and Sulfides. Magn. Reson. Chem 2003, 41, 557–566. [Google Scholar]; (h) Afonin AV; Ushakov IA; Kuznetsova SY; Petrova OV; Schmidt EY; Mikhaleva AI C–H⋯X (X = N, O, S) Intramolecular Interaction in 1-Vinyl-2-(2′-heteroaryl)pyrroles as Monitored by 1H and 13C NMR Spectroscopy. Magn. Reson. Chem 2002, 40, 114–122. [Google Scholar]; (i) Afonin AV; Vashchenko AV; Fujiwara H Structural Effects in NMR spectroscopy of Vinylic Compound 1. Investigation of Intramolecular specific Interactions C–H⋯X in Hetaryl Vinyl Ethers by 1H, 13C, 15N, and 17O NMR Spectroscopies and Quantum-Chemical Calculations. Bull. Chem. Soc. Jpn 1996, 69, 933–945. [Google Scholar]; (j) Li C; Sammes MP Hydrogen Bonds Involving Polar CH Groups. Part 10. Intramolecular Hydrogen Bonds in 1-(ω-Substituted-alkyl) Bis(phenylsulphonyl)methanes. J. Chem. Soc., Perkin Trans 1 1983, 2193–2196. [Google Scholar]; (k) Bruce R St L; Cooper MK; Freeman HC; McGrath BG Evidence for an Intramolecular C–H⋯X Hydrogen Bond in (E)-5-Methylpyridine-2-carboxaldehyde-2′-pyridylhydrazonetetracarbonylmolybdenum(0) from Its Crystal Structure and Proton Magnetic Resonance Spectrum. Inorg. Chem 1974, 13, 1032–1037. [Google Scholar]
- (13).Fang W; Liu X; Lu Z; Tu T Photoresponsive Metallo-Hydrogels Based on Visual Discrimination of the Positional Isomers through Selective Thixotropic Gel Collapse. Chem. Commun 2014, 50, 3313–3316. [DOI] [PubMed] [Google Scholar]
- (14).Laguerre M; Dunogues J; Calas R; Duffaut N Silylation D’hydrocarbures Mono-Aromatiques Mono- ou Dissubstitues. J. Organomet. Chem 1976, 112, 49–59. [Google Scholar]
- (15).Pangborn AB; Giardello MA; Grubbs RH; Rosen RK; Timmers FJ Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar]
- (16).(a) Sheldrick GM A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]; (b) Altomare A; Cascarano G; Giacovazzo C; Guagliardi A; Burla MC; Polidori G; Camalli M SIR92 - A Program for Automatic Solution of Crystal Structures by Direct Methods. J. Appl. Crystallogr 1994, 27, 435–436. [Google Scholar]
- (17).Sheldrick GM Crystal Structure of Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Farrugia LJ WinGX Suite for Small-Molecule SingleCrystal Crystallography. J. Appl. Crystallogr 1999, 32, 837–838. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.