

Abstract
The tumor suppressor p53 (TP53) protects the genome against cellular stress and is frequently mutated in cancer. Mutant p53 acquires gain-of-function oncogenic activities that are dependent on its enhanced stability. However, the mechanisms by which nuclear p53 is stabilized are poorly understood. Here, we demonstrate that stress-induced wild-type p53 and mutant p53 are regulated by the type I phosphatidylinositol phosphate kinase (PIPKIα or PIP5K1A) and its product phosphatidylinositol 4,5-bisphosphate (PI4,5P2). Nuclear PIPKIα directly binds to p53, resulting in the production and association of PI4,5P2 to p53. PI4,5P2 association promotes the interaction between p53 and small heat shock proteins (e.g. HSPB1 and HSPB5), which stabilize nuclear p53. Moreover, inhibition of PIPKIα or PI4,5P2-association results in p53 destabilization. Our results point to a previously unrecognized role of nuclear phosphoinositide signaling in regulating p53 stability and implicate this pathway as a promising therapeutic target in cancer.
Introduction
The lipid messenger phosphatidylinositol 4,5-bisphosphate (PI4,5P2) controls a broad range of normal cellular functions, while its dysregulated activity contributes to the pathogenesis of diverse diseases1. PI4,5P2 directly binds to protein targets known as PI4,5P2 effectors and regulates their function by modulating activity and localization2, 3. The majority of PI4,5P2 is generated by phosphorylation of PI4P and PI5P by type I and type II phosphatidylinositol phosphate kinases (PIPKs), respectively, and each type has α, β and γ isoforms in humans2, 3. Membrane PI4,5P2 rapidly diffuses in the lipid bilayer4 and thus PI4,5P2 generation is directly linked to its usage3. Often, PI4,5P2-generating enzymes are physically associated with PI4,5P2 effectors2, 3. PI4,5P2 is present in most membranes including the plasma membrane and membranes of the Golgi, endosome and endoplasmic reticulum2. PI4,5P2 is also found in the nucleus. Nuclear PI4,5P2 is distinct from the nuclear envelope and is found in non-membranous structures such as nuclear speckles2, 5–7. Although several nuclear PI4,5P2 effectors have recently been identified2, 6, the function of nuclear PI4,5P2 is poorly understood.
The tumor suppressor p53 protects the genome against multiple cellular stresses, including DNA damage, hypoxia, oxidative stress, metabolic stress, and oncogene activation8–10. In response to stress, wild-type p53 accumulates in the nucleus and controls transcription of target genes that regulate cell-cycle arrest, DNA repair, apoptosis, senescence, and metabolism9. The TP53 gene is frequently mutated in diverse cancers, and the majority of these alterations are missense mutations resulting in the expression of mutant p5311, 12. Often, the mutant p53 proteins accumulate in the nucleus and acquire oncogenic activities that promote tumor progression and chemoresistance. Although the enhanced stability of mutant p53 is critical for its oncogenic function, the molecular mechanisms regulating nuclear p53 stabilization are poorly understood. Here, we show that the stability of stress-activated wild-type p53 and mutant p53 is regulated by PI4,5P2. p53 associates with the PI4,5P2-generating enzyme, type Iα phosphatidylinositol-4-phosphate 5-kinase (PIPKIα) in the nucleus, and depletion or pharmacological inhibition of PIPKIα diminishes p53 stability. Moreover, PI4,5P2 generated by PIPKIα interacts to p53 via a polybasic motif in the C-terminal regulatory domain and in turn promotes binding of small heat shock proteins (sHSPs) HSP27 and αB-Crystallin. Both PI4,5P2 binding and recruitment of sHSP are required for stabilization of nuclear p53. Overall, these experiments point to a previously unanticipated role of nuclear phosphoinositide signaling in controlling p53 stability and implicate PIPKIα and the PIPKIα-p53-PI4,5P2-sHSP complex as promising therapeutic targets in cancer.
Results
PIPKIα controls mutant and stress-induced wild-type p53 stability
Although the function of PI4,5P2 at the plasma membrane and other membranes has been well characterized, PI4,5P2’s role in the nucleus remains largely enigmatic. Nevertheless, substantial quantities of PI4,5P2 and other phosphoinositides are present in the nucleus13, and nuclear-localized PIP kinases, PIPKIα, PIPKIγ, PIPKIIα, and PIPKIIβ, maintain the nuclear PI4,5P2 pool5. Based on a recent report that PIPKIIβ gene (PIP4K2B) amplification correlates with TP53 mutation in breast cancer and that PIPKIIα and PIPKIIβ promote the growth of mutant p53-expressing breast cancer cells14, we postulated that one or more PIP kinases regulate mutant p53 stability or function. To investigate this possibility, each of the nuclear-localized PIP kinases was deleted by the CRISPR-Cas9 system in MDA-MB-231 breast carcinoma cells expressing mutant p53 (R280K). Although deletion of PIPKIIα, PIPKIIβ or PIPKIγ had no impact on mutant p53 protein levels, PIPKIα deletion substantially reduced mutant p53 protein levels (Fig. 1a), but p53 mRNA levels were unaffected (Fig. 1b). Reduction of mutant p53 protein levels by PIPKIα deletion was not due to altered expression of components of the HSP90 chaperone or phosphoinositide 3-kinase (p110α) complexes (Fig. 1a) that have established roles in regulating p53 stability11. Transient knockdown of PIPKIα also reduced mutant p53 protein levels in cancer cell lines examined across a broad range of different TP53 missense mutations (Supplementary Fig. 1a, 1b).
Figure 1. PI4,5P2 generation by PIPKIα is required for stress-induced and mutant p53 stability.
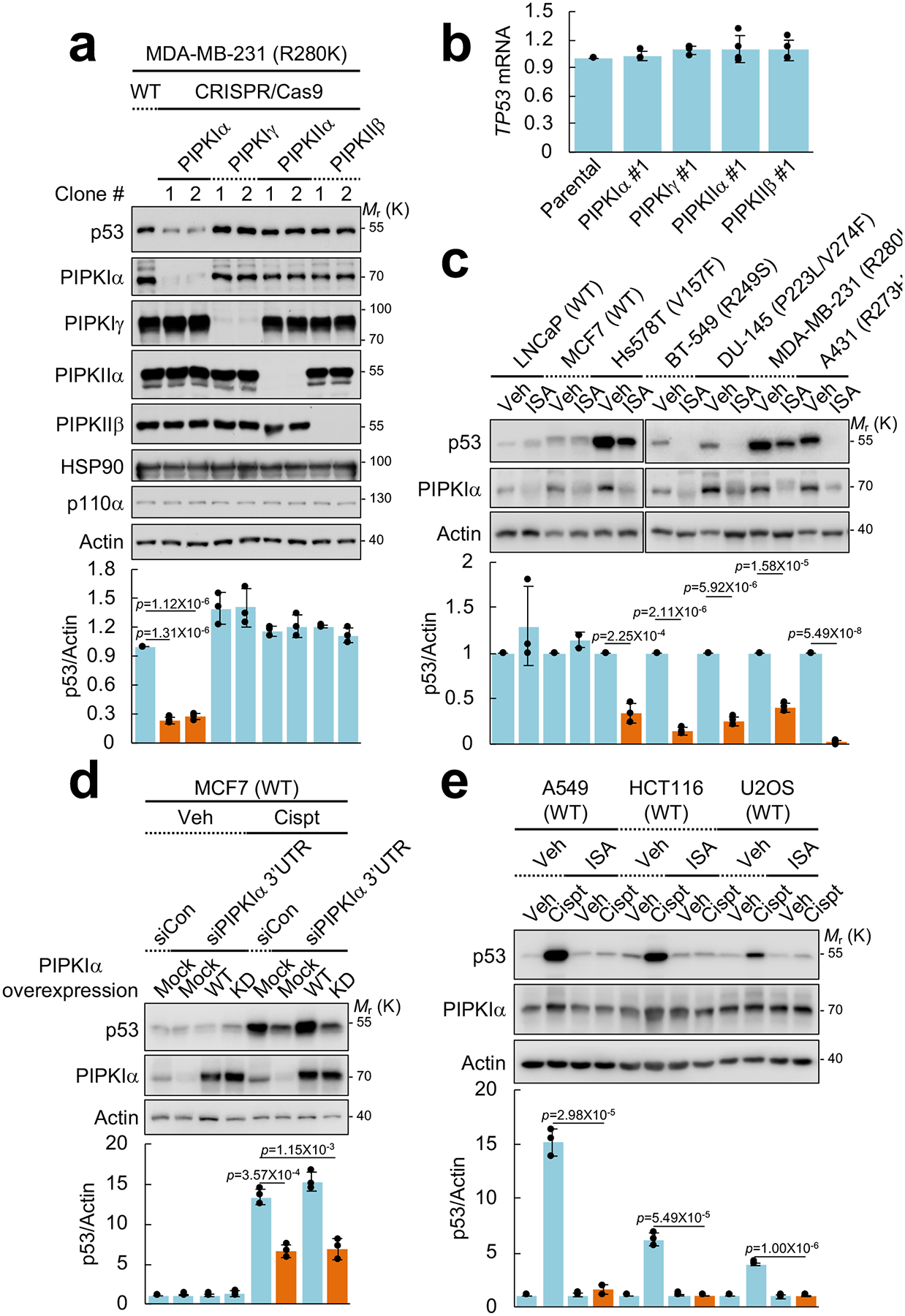
(A) Nuclear PIPKs were knocked out with CRISPR-Cas9 system in MDA-MB-231 cells. Expression of the indicated proteins was analyzed by immunoblotting (IB). Intensity of immunoblots was quantified and the graph is shown as mean ± SD n=3 independent experiments. One-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(B) RT-PCR analysis of TP53 mRNA. TP53 mRNA levels were normalized to GAPDH mRNA. The graph is shown as mean ± SD of n=4 independent experiments.
(C) The indicated cells were treated with 50 μM ISA-2011B (ISA) for 72 h. Protein expression was analyzed by IB, the intensity of p53 and actin bands was quantified, and the graph is shown as mean ± SD of n=3 independent experiments. An equivalent concentration of DMSO was used as a vehicle (Veh) control. One-sided paired Student t-tests were used for statistical analysis.
(D) MCF7 cells were transfected with the indicated siRNAs and DNA constructs. After 48 h transfection, cells were treated with 30 μM cisplatin (Cispt) for an additional 24 h. Expression of the indicated proteins was analyzed by IB, p53 immunoblots were quantified and the graph is shown as mean ± SD of n=3 independent experiments. WT, wild-type; KD, kinase-dead mutant; 3’UTR, 3’ untranslated region. One-sided paired Student t-tests were used for statistical analysis.
(E) Indicated cells expressing wild-type p53 were treated with 30 μM cisplatin and 50 μM of ISA-2011B for 24 h. Protein expression was analyzed by IB. p53 blots were quantified and the graph is shown as mean ± SD of n=3 independent experiments. One-sided paired Student t-tests were used for statistical analysis.
Inhibition of PIPKIα activity using a PIPKIα-selective inhibitor ISA-2011B15 decreased mutant p53 protein levels (Fig. 1c and Supplementary Fig. 1c, 1d). Under these conditions ISA-2011B treatment also reduced PIPKIα expression (Fig. 1c) as reported15. By contrast, PIPKIα knockdown or ISA-2011B treatment had no effect on wild-type p53 protein levels under unstressed conditions (Fig. 1c, 1d, 1e, and Supplementary Fig. 1a). These results indicate that inhibition of PIPKIα, but not other nuclear PIP kinases, reduces levels of mutant p53 protein encoded by several different TP53 missense mutations.
The stability of wild-type and mutant p53 has been reported to be regulated by distinct mechanisms11. Wild-type p53 protein levels are regulated by a set of E3 ubiquitin ligases including MDM2, MDMX, and CHIP8, 9. In response to stress including DNA damage, wild-type p53 is phosphorylated in the N-terminus, resulting in its dissociation from the E3 ubiquitin ligases and subsequent stabilization. In contrast, most mutant p53 proteins associate with the HSP90 chaperone complex, which is postulated to block access of the E3 ubiquitin ligases to mutant p53, promoting p53 stability11, 16, 17. However, recent reports demonstrate that inhibition of autophagy reduces both wild-type and mutant p53 levels18, 19, suggesting that the stability of wild-type and mutant p53 is regulated by at least partly common mechanisms.
We explored whether PIPKIα and PI4,5P2 regulate wild-type p53 stability in response to stress. Knockdown of PIPKIα blocked stabilization of wild-type p53 levels by the DNA damaging agent cisplatin (Fig. 1d). This phenotype was rescued by re-expression of wild-type PIPKIα but not by the kinase-dead (KD) mutant, indicating that PIPKIα-dependent PI4,5P2 production promotes stress-induced wild-type p53 stabilization. Furthermore, inhibition of PIPKIα by ISA-2011B suppressed cisplatin-induced wild-type p53 stabilization in the tested cell lines (Fig. 1e). It is important to note that the reduction of mutant p53 levels by ISA-2011B required longer incubation times (Supplementary Fig. 1c) as the turnover rate of mutant p53 is much slower than wild-type p5318, 20, 21. Moreover, prolonged treatment of cells with ISA-2011B reduced PIPKIα levels (Fig. 1c), while shorter ISA-2011B treatment did not alter PIPKIα levels (Fig. 1e). Taken together, these results indicate that inhibition of PIPKIα activity and PI4,5P2 generation is sufficient to suppress wild-type p53 stabilization by genotoxic stress.
PIPKIα directly interacts with nuclear p53
As PIP kinases often bind to proteins whose function they control, these interacting proteins are often PI4,5P2 effectors2, 3, 22, 23. Thus, the potential interaction between PIPKIα and p53 was explored. Under unstressed conditions, mutant p53 coimmunoprecipitated with PIPKIα (Fig. 2a) and DNA damage stress modestly increased the interaction (Supplementary Fig. 2a). Mutant p53 is known to interact with heat shock protein (HSP) 70 and 90 chaperones that control its stability24, and these HSPs also coimmunoprecipitated with PIPKIα (Fig. 2a). The association of PIPKIα with wild-type p53 was not detected in unstressed cells (Fig. 2a), but was dramatically increased by DNA damaging agents (cisplatin and etoposide) and oxidative stress by tert-butylhydroquinone (tBHQ) (Fig. 2b and Supplementary Fig. 2b). MDM2 is known to interact with wild-type p5316, 21 and coimmunoprecipitated with PIPKIα in unstressed cells, but this interaction was diminished by cisplatin treatment and was not detected in cells expressing mutant p53 (Supplementary Fig. 2a, 2b). Collectively, these data indicate that PIPKIα forms a stress-induced complex with wild-type and mutant p53 and selective proteins in the p53-interacting complexes in vivo.
Figure 2. PIPKIα associates with p53 in the nucleus.
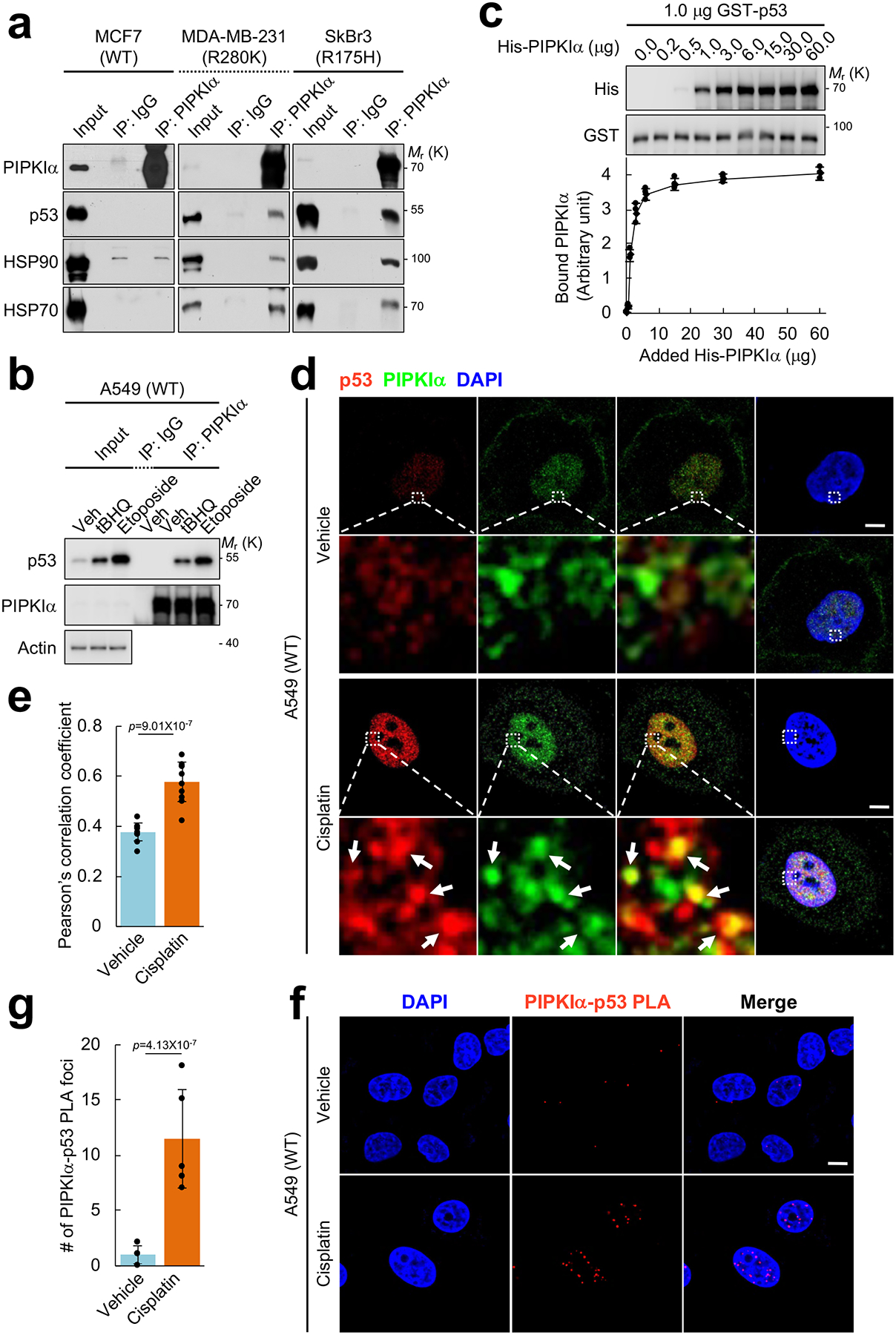
(A) Endogenous PIPKIα was immunoprecipitated (IP’ed) from the indicated breast cancer cells and the associated proteins were analyzed by IB. Normal immunoglobulin (IgG) was used as a negative control. Representative data of n=3 independent experiments were shown.
(B) A549 cells were treated with 100 μM tBHQ, 100 μM etoposide, or a vehicle control (DMSO) for 24 h. Endogenous PIPKIα was IP’ed and the associated proteins were analyzed by IB. Representative data of n=3 independent experiments were shown.
(C) 1.0 μg recombinant GST-p53 immobilized on glutathione beads was incubated with the indicated amount of His-PIPKIα in vitro. The complex was pulled down and p53-bound PIPKIα was analyzed by IB with an anti-His antibody. The graph is shown as mean ± SD of n=3 independent experiments.
(D and E) A549 cells were treated with 30 μM cisplatin for 24 h before processed for immunofluorescence (IF) staining against PIPKIα and p53. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope and processed by ImageJ. White arrows indicate the colocalized signals. The experiments were repeated three times and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm.
(F and G) A549 cells were treated with 30 μM cisplatin for 24 h before processed for proximity ligation assay (PLA) between PIPKIα and p53. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope. The red PLA signal was quantified by LAS X (Leica) and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
The potential for a direct interaction of PIPKIα with p53 was explored. Recombinant p53 was incubated with an increasing amount of recombinant PIPKIα and p53-bound PIPKIα was analyzed (Fig. 2c top). The binding curve showed saturable binding (Fig. 2c bottom), indicating that PIPKIα directly and specifically binds to p53.
To explore this interaction in cells, the subcellular localization of the PIPKIα-p53 complex was examined by immunofluorescence. Cisplatin treatment increased the nuclear PIPKIα signal (Fig. 2d and Supplementary Fig. 2c, 2d), indicating that PIPKIα locates to the nucleus in response to genotoxic stress. Under these conditions, PIPKIα partially colocalized with wild-type p53 in the nucleus (Fig. 2d, 2e). The interaction between PIPKIα and p53 was quantified using a proximity ligation assay (PLA)25 that detects two closely located epitopes in a complex (less than 40 nm apart)26. DNA damage stress dramatically increased the nuclear PIPKIα-p53 PLA signal (Fig. 2f, 2g), supportive of a regulated association. The nuclear PIPKIα-p53 PLA foci localized to compartments where DAPI staining was low (Supplementary Fig. 2e, 2f), suggesting that PIPKIα and p53 coexist in subnuclear compartments that are discrete from chromosome domains, consistent with other reports5, 7.
p53 associates with PI4,5P2 in the nucleus
The PI4,5P2 generation is transient and PI4,5P2-generating enzymes such as PIPKIα often associate with PI4,5P2 effectors2. To test if p53 is a PI4,5P2 effector, PI4,5P2 binding was quantified using a liposome sedimentation assay27 (Fig. 3a). Liposomes were made with 10% phosphoinositide of either synthetic or natural source. Synthetic lipids (PI, PI3P, PI4P, PI5P, PI3,4P2, PI3,5P2, PI4,5P2, and PI3,4,5P3) uniformly have dioctanoyl acyl chains. Natural bovine brain purified lipids (PI, PI4P, and PI4,5P2) have variable acyl chain composition but are largely arachidonate and palmitoleate1. All synthetic lipids exhibited background binding to recombinant p53. Significantly, p53 binding to native brain PI4,5P2 was robust, signifying that the acyl chain composition of PI4,5P228, 29 is important for p53 binding (Fig. 3a). The binding of p53 to PI4,5P2 was analogous to that of the pleckstrin homology (PH) domain of phospholipase C δ1 (PLC δ1-PH) (Fig. 3b and Supplementary Fig. 3a, 3b) that is a well-established PI4,5P2 binding protein30, 31, indicating that p53 is a genuine PI4,5P2-binding protein. The PI4,5P2-binding domains are often mediated by a cluster of positively charged amino acids known as a polybasic motif (PBM)2, 23, 27, 32 and a putative PBM is present in the C-terminal regulatory domain (CTD) of p53. To determine if this PBM is responsible for PI4,5P2 binding, 5 lysines and/or 1 arginine in the CTD were mutated to neutral glutamine creating 6Q or R379Q mutants (Supplementary Fig. 3a). These mutations dramatically reduced PI4,5P2 binding in vitro (Fig. 3b and Supplementary Fig. 3b). Collectively, these results establish that p53 specifically binds to PI4,5P2 via a PBM in the CTD of p53.
Figure 3. PI4,5P2 specifically binds to p53 and the PI4,5P2-associated p53 localizes in subnuclear compartments.
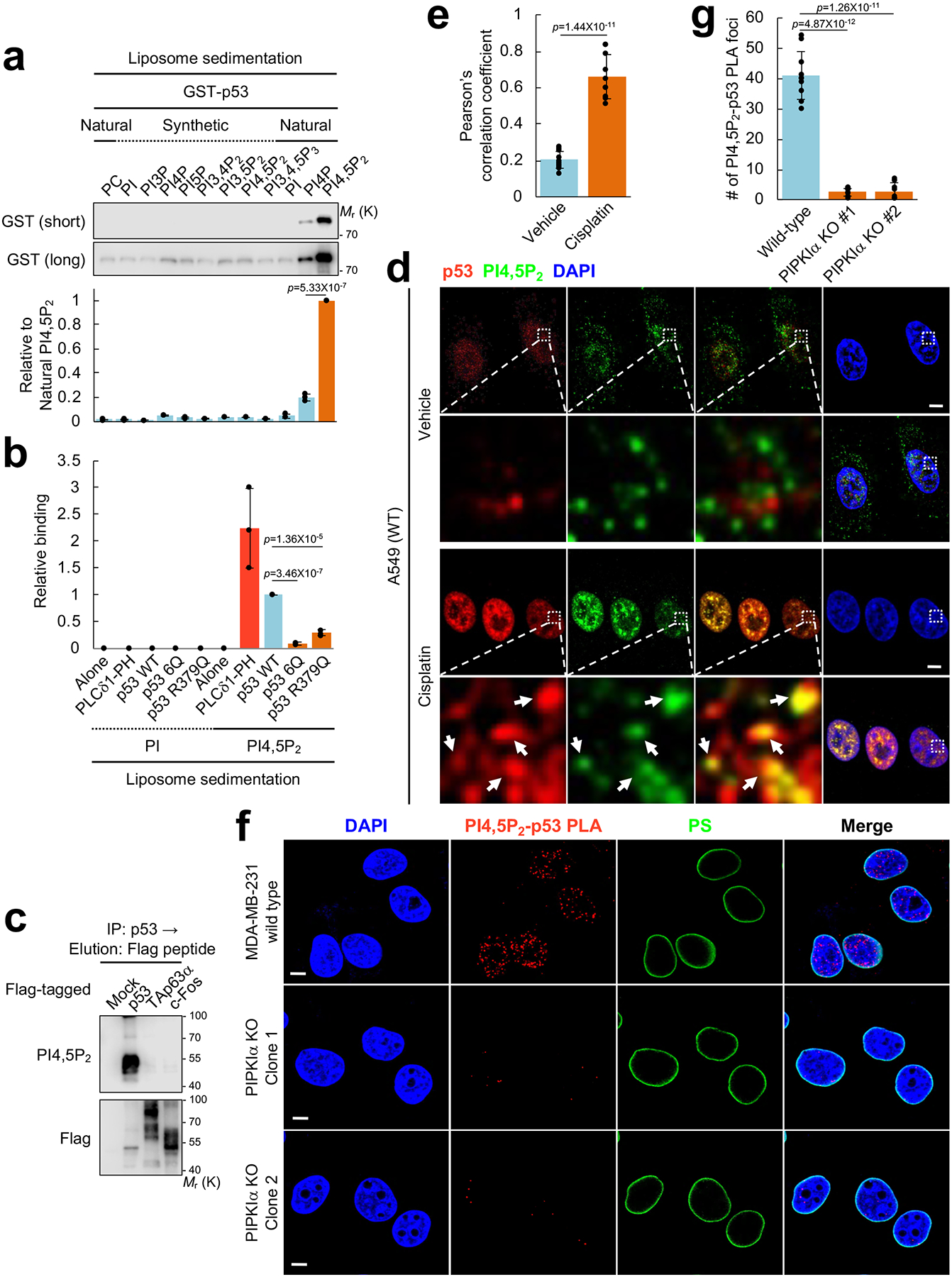
(A) 0.5 μM GST-p53 was incubated with 5.0 μM liposomes containing the indicated phosphoinositides. Liposomes were sedimented and the associated p53 was analyzed by IB with an anti-GST antibody. The GST immunoblot intensity was quantified and the graph is shown as mean ± SD of n=3 indent experiments.
(B) The indicated GST-tagged 0.5 μM recombinant proteins were incubated with 5.0 μM liposomes containing PI or PI4,5P2. Liposomes were sedimented and the associated proteins were analyzed by IB with an anti-GST antibody (Supplementary Fig. 3b). The GST immunoblot intensity was quantified and the graph is shown as mean ± SD of n=3 independent experiments.
(C) The indicated Flag-tagged transcription factors were transiently transfected in H1299 cells for 48 h. Proteins were IP’ed with an anti-Flag gel and eluted with a Flag peptide. Protein complexes were analyzed by IB with an anti-PI4,5P2 KT10 antibody. Representative data of n=3 independent experiments were shown.
(D and E) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining against PI4,5P2 and p53. An anti-PI4,5P2 antibody (Echelon Biosciences) was used for the immunostaining study. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope and processed by ImageJ. White arrows indicate the colocalized signals. The experiments were repeated 3 times and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm.
(F and G) MDA-MB-231 cells with wild-type PIPKIα or CRISPR-Cas9 knockout (KO) of PIPKIα (two independent clones) were treated with 30 μM cisplatin for 24 h before processed for PLA between PI4,5P2 and p53. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope. The red PLA signal was quantified by LAS X (Leica) and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
As p53 binds to DNA (also negatively charged), the specificity of the p53 interaction with PI4,5P2 was compared to other DNA binding proteins (Fig. 3c). p53, the p53 homolog p63 (TAp63α)33, or c-Fos34 was expressed in cells and the associated complex was analyzed by an anti-PI4,5P2 antibody KT1035. p53, but not p63 nor c-Fos, retained PI4,5P2 interaction, indicating that the p53-PI4,5P2 interaction is a specific event. Furthermore, the endogenous interaction between p53 and PI4,5P2 was examined in non-transformed and cancer cell lines. In the tested cells, stress-induced wild-type and mutant p53 robustly associated with PI4,5P2 (Supplementary Fig. 3c).
The colocalization of p53 and PI4,5P2 in vivo was assessed by immunofluorescence. DNA damage stress induced by cisplatin stimulated the nuclear accumulation of PI4,5P2 and PI3,4,5P3 (Supplementary Fig. 3d–3i) consistent with a recent report36. PI3P nuclear staining was also increased while PI4P and PI3,4P2 remained unchanged. Importantly, a substantial fraction of nuclear PI4,5P2 colocalized with DNA damage-induced wild-type p53 (Fig. 3d, 3e). The PI4,5P2-p53 association was also examined using PLA. The PI4,5P2-mutant p53 PLA signal was largely detected in the nucleoplasm and was ~95% reduced by PIPKIα deletion (Fig. 3f, 3g), supporting that PI4,5P2-generated by PIPKIα is responsible for p53 association. In unstressed conditions, minimal nuclear PI4,5P2-wild-type p53 PLA signal was detected. DNA damage robustly induced the nuclear PI4,5P2-p53 PLA signal but not the PLA signal for p53 with other phosphoinositides tested (Supplementary Fig. 3j–3n). Taken together, these data demonstrate that PI4,5P2 generated by PIPKIα associates with p53 in the nucleus.
PI4,5P2-associated p53 is found in distinct subnuclear areas
The nuclear PI4,5P2 pool is distinct from the nuclear membrane5, 37 and consistently the p53-PI4,5P2 association was observed largely in the nucleoplasm (Fig. 4a and Supplementary Fig. 4a, 4b). A closer examination via p53-PI4,5P2 PLA showed that the vast majority of the PLA signal was located in the nucleus at sites distinct from the nuclear membrane (Fig. 4b and Supplementary Fig. 4c, 4d). To more precisely map where p53 and PI4,5P2 interact in the nucleus, a nuclear speckle marker SC-35 was used. Previously, PIPKIα and PI4,5P2 was reported to localize partly in nuclear speckles6 and consistently PI4,5P2 and p53 partially colocalized with SC-35-positive nuclear speckles (Supplementary Fig. 4e). In addition, a recent report indicated that nuclear PI4,5P2 levels increase in response to DNA damage36, and our results further demonstrate that PIPKIα, PI4,5P2, and p53 co-localize to DNA damage sites (γH2A.X-positive foci) in the nucleus upon cisplatin treatment (Fig. 4c and Supplementary Fig. 4f–4k). Importantly, knockdown of PIPKIα reduced the colocalization of PI4,5P2 and p53 at the DNA damage sites (Fig. 4d, 4e, and Supplementary Fig. 5a–5f). Taken together, these findings signify that p53 interacts with PI4,5P2 in distinct regions of the nucleus, and suggest that the interaction of p53 with PIPKIα and PI4,5P2 targets p53 to distinct subnuclear areas.
Figure 4. PI4,5P2-bound p53 localizes in subnuclear compartments distinct from the nuclear membrane.
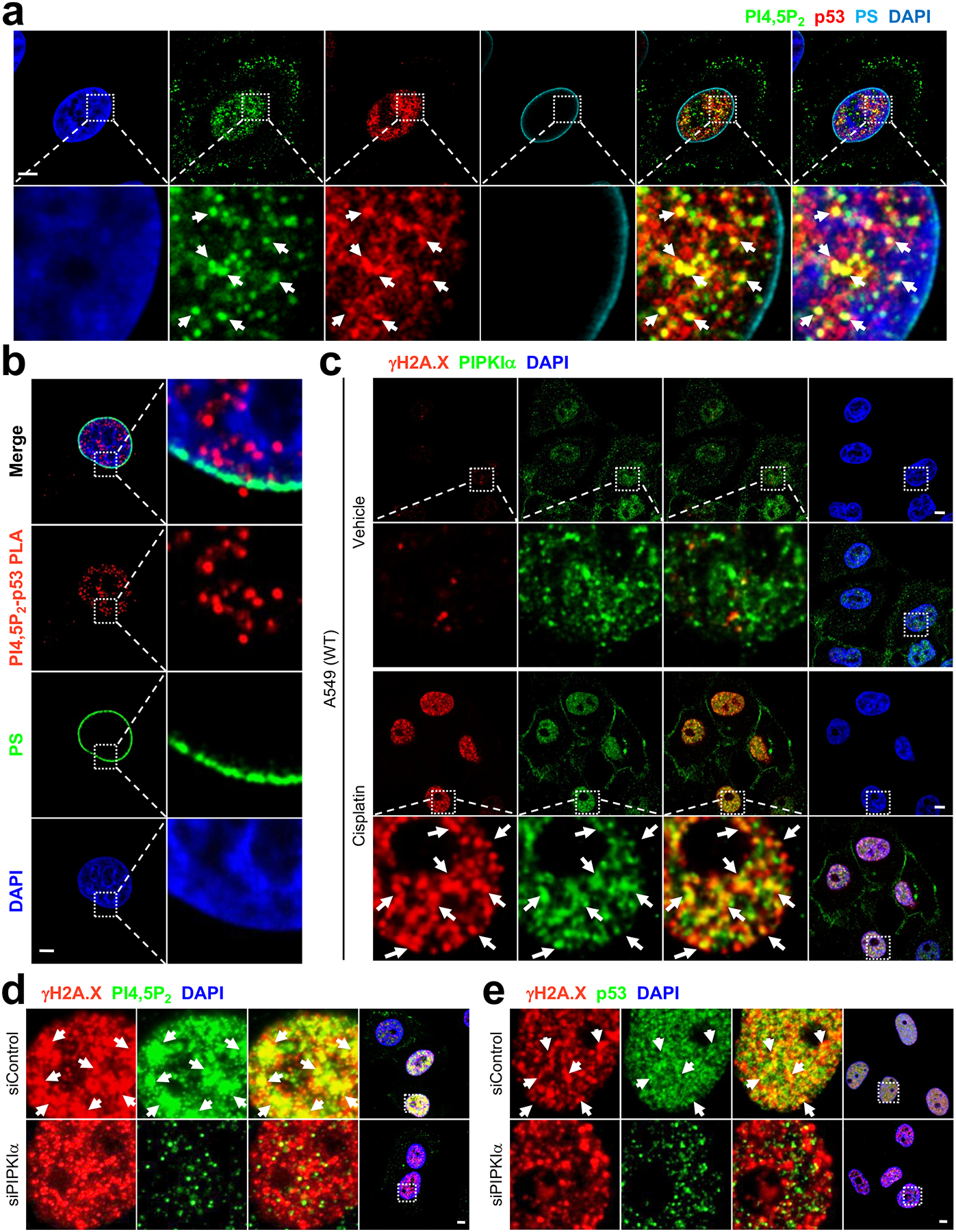
(A and C) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated molecules. An anti-phosphatidylserine (PS) antibody was used to stain the nuclear membrane66. An anti-gH2A.X antibody67 was used as a marker for DNA damage. DNA stain DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope. Scale bar, 5 μm. The experiments were repeated 3 times.
(B) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between PI4,5P2 and p53. DAPI was used to stain nucleic acids. The images were taken by Leica SP8 confocal microscope. Scale bar, 5 μm. The experiments were repeated 3 times.
(D and E) A549 cells were transfected with the indicated siRNAs for 48 and then treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated molecules. The images were taken with a Leica SP8 confocal microscope. Scale bar, 5 μm. The experiments were repeated 3 times.
PI4,5P2-binding controls stability of stress-induced wild-type and mutant p53
We showed that the PI4,5P2-binding region is on the CTD of p53 (Fig. 3). The CTD of p53 is a regulatory region that controls its function and stability21. Initially, we assessed whether PI4,5P2 binding regulated mutant p53 stability. The 6Q mutation was introduced into cDNAs encoding the two most frequent TP53 missense mutations found in cancer (R175H and R248Q)11,17, and the effect on mutant p53 stability was tested. When stably expressed in p53-null PC3 cells, protein levels of each p53 6Q mutant were significantly diminished compared to their wild-type CTD counterparts (Fig. 5a). However, mRNA levels were not altered (Fig. 5b), indicating that the 6Q mutation reduced the stability of mutant p53 protein. The diminished stability of mutant p53 was not due to defective binding of 6Q mutants to PIPKIα (Supplementary Fig. 5g). These results suggest that mutant p53 is stabilized by PIPKIα-generated PI4,5P2 binding to its CTD. To further test if PI4,5P2 is required for p53 stability, a PI4,5P2-specific phosphatase inositol polyphosphate-5-phosphatase E (INPP5E)38, 39 was expressed and its impact on wild-type p53 stabilization by cisplatin was measured. Overexpression of wild-type INPP5E significantly reduced wild-type p53 accumulation in response to genotoxic stress but the phosphatase-dead mutant had no effect (Fig. 5c). Additionally, specific phosphoinositide binding domains30, 40 were expressed and their impact on p53 stability was tested. Sequestration of either PI3P by expression of the FYVE domain of Hrs (Hrs-FYVE) or PI3,4,5P3 by the PH domain of Akt1 (Akt1-PH) did not affect wild-type p53 stabilization by cisplatin (Fig. 5d). In contrast, sequestration of PI4,5P2 by PLCδ1-PH significantly reduced p53 levels, further supporting that PI4,5P2 is critical for p53 stability. Sequestration of PI4P by Osh2-PH also reduced stress-induced wild-type p53 levels, suggesting that conversion of PI4P to PI4,5P2 (by PIPKIα) is required to maintain p53 stability. Of note, we previously demonstrated that these phosphoinositide binding domains are localized in the nucleus41.
Figure 5. PI4,5P2 binding controls the stability of stress-induced wild-type p53 and mutant p53.
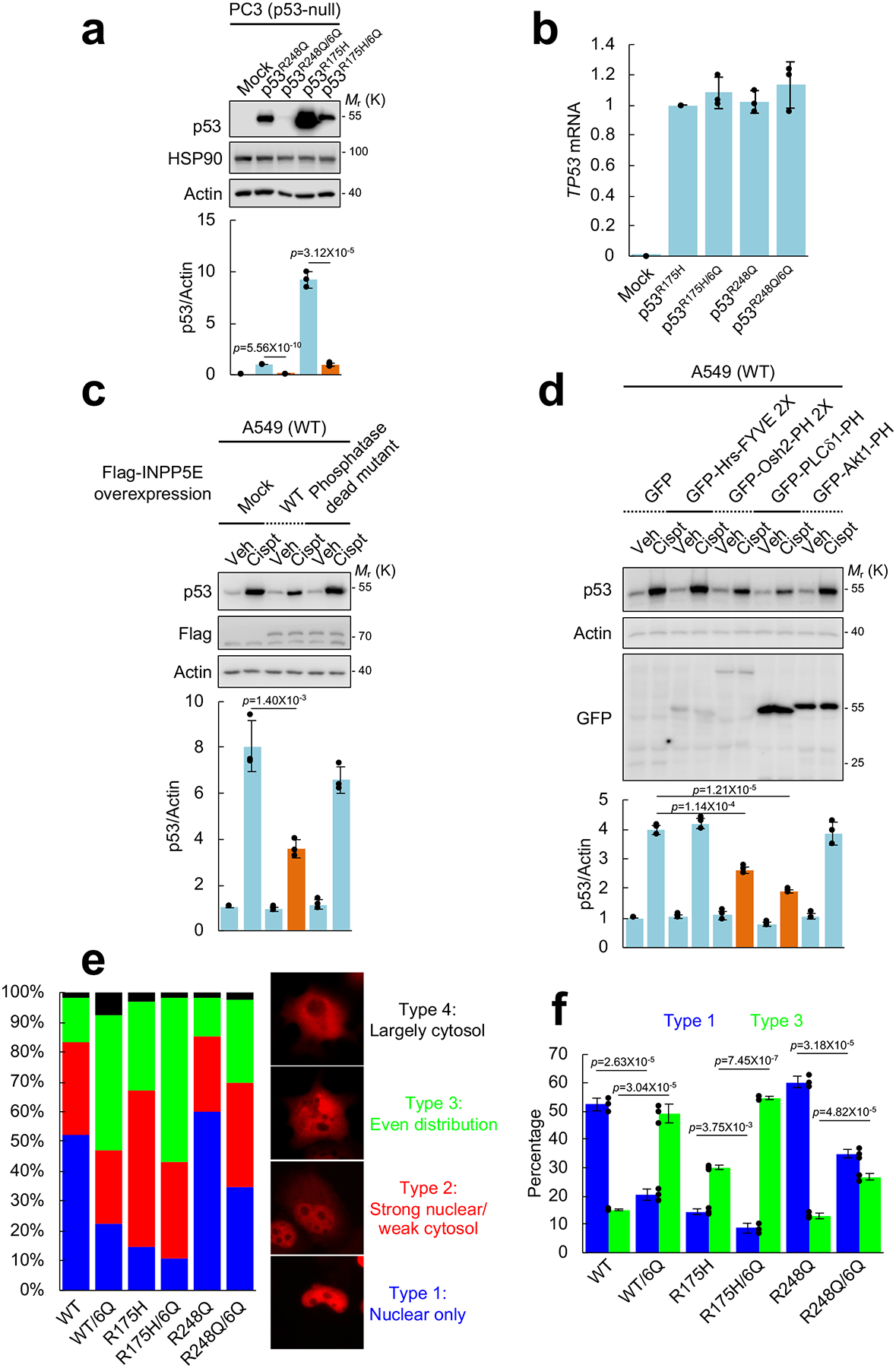
(A) The indicated p53 constructs were stably expressed in p53-null PC3 prostate cancer cells. Expression of the indicated proteins was analyzed by IB. Empty vector (Mock) was used as a negative control. The intensity of immunoblots was quantified and the graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(B) RT-PCR analysis of TP53 mRNA. TP53 mRNA levels were normalized to GAPDH mRNA. The graph is shown as mean ± SD of n=3 independent experiments.
(C) A549 cells were transiently transfected with the indicated INPP5E constructs for 60 h. Cells were treated with 30 μM cisplatin for 12 h. Expression of the indicated molecules was analyzed by IB. The intensity of immunoblots was quantified and the graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(D) The indicated GFP-tagged FYVE or PH domain constructs were transiently transtected in A549 cells for 60 h. Cells were treated with 30 μM cisplatin for 12 h. Expression of the indicated molecules was analyzed by IB. The intensity of immunoblots was quantified and the graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(E and F) The indicated p53 constructs were transiently transfected in p53-null H1299 cells for 24 h. Cells were fixed and analyzed by immunofluorescence for p53. DAPI was used to stain nucleic acids. Four different distribution patterns of p53 expression were observed as categorized. At least 100 cells were counted for each condition and the graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
To investigate whether PI4,5P2 binding regulates the subcellular localization of p53, p53 proteins were transiently expressed in p53-null H1299 cells and visualized by immunofluorescence. Different p53 proteins showed distinct distribution patterns as reported42. p53 6Q mutants were more frequently found in the cytoplasmic compartment compared to their wild-type CTD counterparts (Fig. 5e, 5f, and Supplementary Fig. 5h). These results indicate that PI4,5P2 binding to p53 may regulate its nuclear localization.
Nuclear p53 associates with small heat shock proteins
To investigate the mechanisms by which PIPKIα and PI4,5P2 regulate p53 stability, the immunoprecipitated p53 complex from cytosolic and nuclear fractions were analyzed for associated molecules (Fig. 6a, 6b, and Supplementary Fig. 6a–6c). Genotoxic stress dramatically enhanced the interaction between wild-type p53 and PI4,5P2 in the nucleus (Fig. 6a and Supplementary Fig. 6a). Mutant p53 was constitutively associated with PI4,5P2 in the nuclear fraction and cisplatin only modestly enhanced this interaction. Furthermore, knockdown of PIPKIα reduced nuclear wild-type and mutant p53 interaction with PI4,5P2 (Supplementary Fig. 6b, 6c), indicating that PI4,5P2 generated by PIPKIα is transferred to and interacts with p53 in the nucleus.
Figure 6. The sHSP HSP27 associates with nuclear p53.
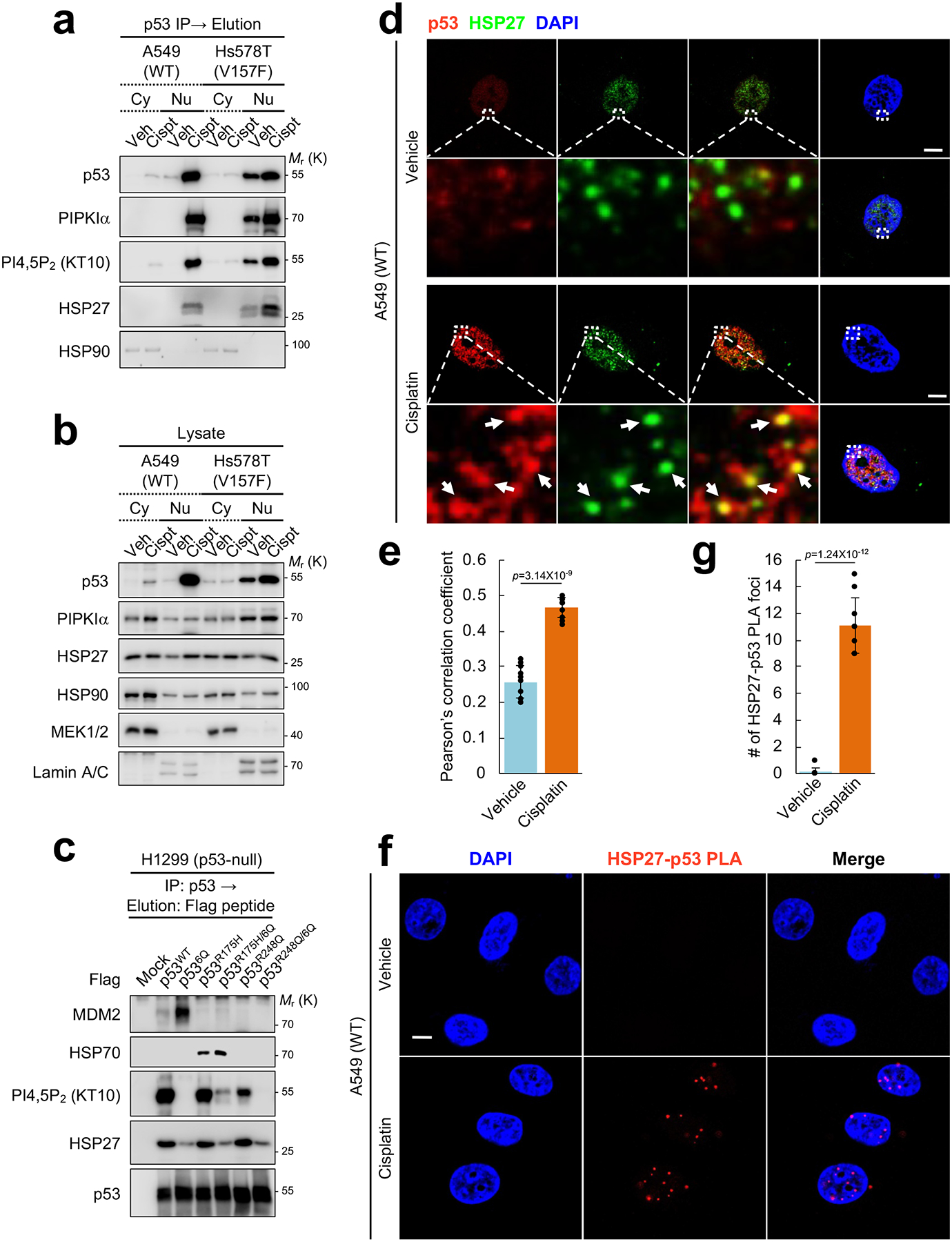
(A and B) A549 and Hs578T cells were treated with with 30 μM cisplatin for 24 h. Cells were fractionated into cytosolic and nuclear compartments using NE-PER™ reagent (Thermo Fisher Scientific). Endogenous p53 was IP’ed from equal amount of cytosolic and nuclear lysates and eluted with 0.2 M glycine (pH 2.5). The p53 associated complexes were analyzed by IB with the indicated antibodies (A). Expression of the indicated proteins was analyzed by IB with indicated antibodies (B). The quality of fractionation was validated by a cytosolic marker MEK1/2 and a nuclear marker Lamin A/C (B). Cy, cytosolic fraction; Nu, nuclear fraction. Representative data of n=3 independent experiments were shown.
(C) p53-null H1299 cells were transiently transfected with the indicated Flag-p53 constructs for 24 h. Flag-p53 proteins were immunoprecipitated with an anti-Flag antibody and eluted with Flag peptide. Associated molecules were analyzed by IB with the indicated antibodies. Representative data of n=3 independent experiments were shown.
(D and E) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining against HSP27 and p53. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope and processed by ImageJ. White arrows indicate the colocalized signals. The experiments were repeated three times and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm.
(F and G) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between HSP27 and p53. DAPI was used to stain nucleic acids. The images were taken by with a Leica SP8 confocal microscope. The red PLA signal was quantified by LAS X (Leica) and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated 3 times. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
It has been well-documented that wild-type p53 stability is controlled by a set of E3 ubiquitin ligases and MDM2 has a dominant role8, 20, 21. Mutant p53 stability has been reported to be regulated by unique protein interactions, including the HSP90-HSP70 chaperone complex12, 16, 24, 43. However, each of these known interacting proteins associated with p53 largely in the cytosolic fraction (Fig. 6a and Supplementary Fig. 6a), suggesting that the stability of nuclear p53 is regulated by distinct mechanisms. To identify the molecular components of the nuclear PIPKIα-p53-PI4,5P2 complex, wild-type and two mutant TP53 cDNAs (R175H and R248Q) and their corresponding PI4,5P2 binding-defective 6Q constructs were transiently expressed in p53-null H1299 cells. p53-interacting proteins were analyzed by mass spectrometry and immunoprecipitation. Ectopic expression of p53 6Q constructs resulted in similar protein levels as the other constructs (Fig. 6c). However, the 6Q p53 proteins had little or no PI4,5P2 association, confirming that the 6Q mutants abrogated PI4,5P2 interaction in vivo, consistent with their loss of PI4,5P2 binding in vitro (Fig. 3b). MDM2 interacted with wild-type p53, but not with mutant p53 (Fig. 6c) as previously described43, 44. Notably, MDM2 binding to wild-type p53 was markedly enhanced in the p53 6Q mutant (Fig 6c), suggesting that PI4,5P2 binding may promote wild-type p53 stability by blocking MDM2 binding and subsequent p53 degradation. Consistent with the prior reports of the HSP90-HSP70 chaperone complex regulating mutant p53 stability24, 44, the R175H mutant p53, but not the R248 mutant nor wild-type p53, associated with HSP70. Moreover, the R175H/6Q mutant did not disrupt binding of HSP70 (Fig. 6c), indicating that HSP70 is unlikely to contribute to the observed stabilization of mutant p53 by PI4,5P2 binding.
In contrast, one of the components of the p53 complex identified by mass spectrometry (Supplementary Fig. 6d), the small heat shock protein (sHSP) HSP27/HSPB1 interacted with both wild-type and mutant p53 in the nuclear fraction (Fig. 6a), while its interaction with the corresponding p53 6Q proteins was markedly reduced (Fig. 6c). HSP27/HSPB1 is one of the 10 human sHSP family members (HSPB1–10). These sHSPs form homo- or hetero-oligomeric complexes and the binding of sHSP complexes enhances target protein stability45. Further, HSP27 is reported to control p53 signaling46 and translocate to the nucleus upon stress47.
Colocalization of HSP27 with nuclear p53 was examined by immunofluorescence. Upon genotoxic stress, the HSP27 nuclear signal was increased (Supplementary Fig. 6e, 6f), consistent with a previous report47. Under these conditions, the colocalization of HSP27 with wild-type p53 was increased in the nuclear regions (Fig. 6d, 6e). Notably, the HSP27-p53 PLA signal was dramatically increased by cisplatin treatment (Fig. 6f, 6g, and Supplementary Fig. 6g, 6h).
Small heat shock proteins stabilize p53
We next explored the functional role of sHSPs in regulating p53 stability. Knockdown of HSP27 or PIPKIα significantly attenuated wild-type p53 (Fig. 7a) and mutant p53 stability (Fig. 7b). Moreover, silencing PIPKIα or inhibiting PIPKIα with ISA-2011B significantly reduced the genotoxic stress-induced interaction between wild-type p53 and HSP27 (Fig. 7c). These data suggest that PIPKIα-mediated PI4,5P2 interaction with p53 stabilizes p53 by recruiting HSP27 to the complex.
Figure 7. PI4,5P2 binding to p53 promotes its interaction with small heat shock proteins in vivo.
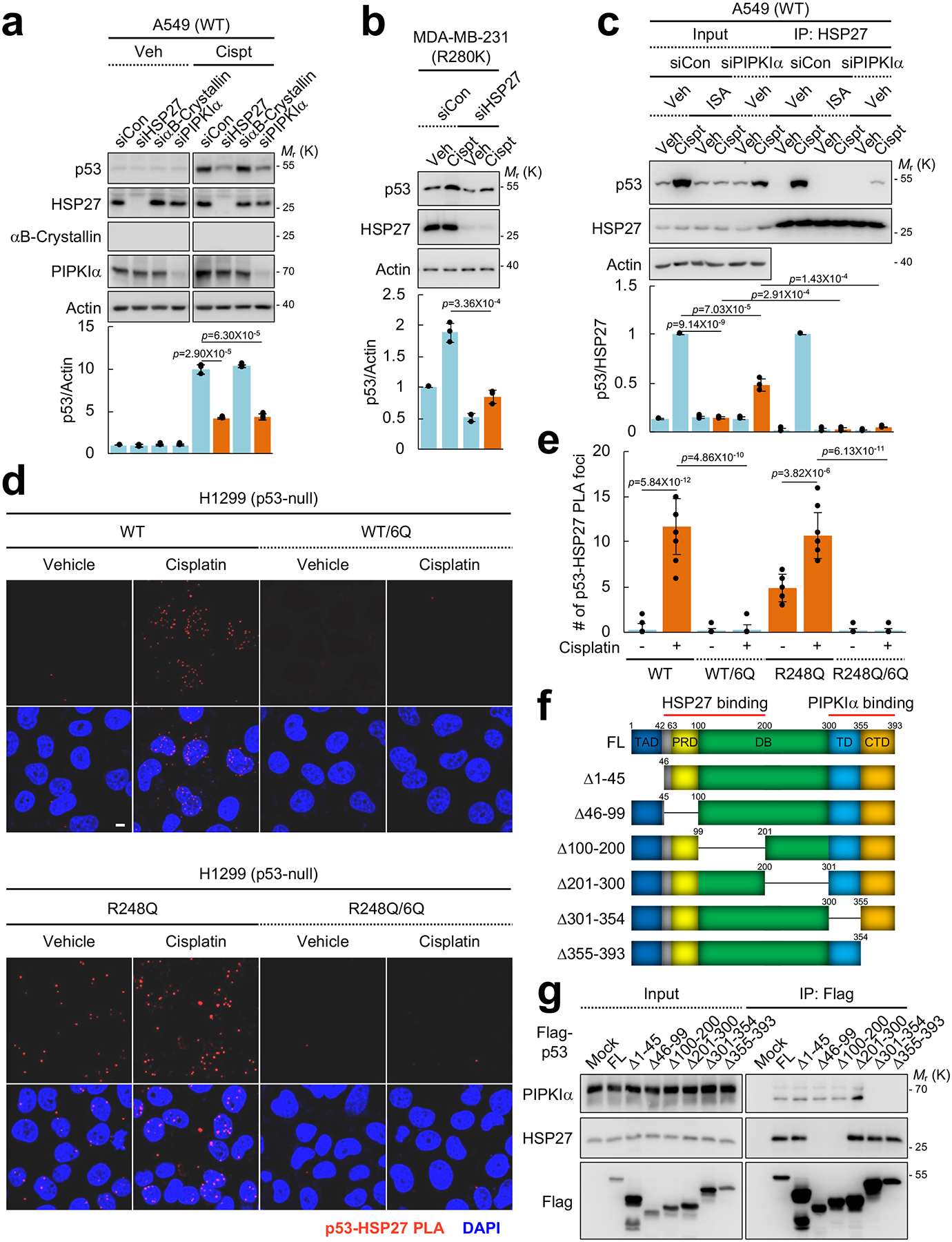
(A) The indicated proteins were transiently knocked down with siRNAs for 48 h in A549 cells. Cells were treated with 30 μM cisplatin for 24 h and protein expression was analyzed by IB. The graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(B) MDA-MB-231 cells were transfected with the indicated siRNAs for 48 h and then treated with 30 μM cisplatin for 24 h. Protein expression was analyzed by IB. The graph is shown as mean ± SD of n=3 independent experiments.
(C) A549 cells were transfected with PIPKIα siRNAs for 48 h and then treated with 30 μM cisplatin and 50 μM ISA-2011B for 24 h. Endogenous HSP27 was immunoprecipitated, the associated p53 was analyzed by IB, and the graph is shown as mean ± SD of n=3 independent experiments.
(D and E) p53-null H1299 cells were transiently transfected with the indicated p53 constructs for 24 and then treated with 30 μM cisplatin for 24 h. Cells were fixed and processed for PLA between exogenous p53 and endogenous HSP27. DAPI was used to stain nucleic acids. The red PLA signal was quantified by LAS X (Leica) and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated 3 times. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(F) A schematic representation of p53 truncation mutants used in the study.
(G) p53-null H1299 cells were transiently overexpressed with Flag-tagged p53 truncation mutants for 48 h. p53 proteins were IP’ed with an anti-Flag gel and the associated endogenous PIPKIα and HSP27 were detected by IB. The experiments were repeated three times.
To validate the functional role of HSP27 in PI4,5P2-mediated p53 stabilization, we expressed wild-type, R248Q mutant p53 and the 6Q counterparts in p53-null H1299 cells, and the association with endogenous HSP27 was tested by immunofluorescence and PLA (Fig. 7d, 7e, and Supplementary Fig. 7a, 7b). Genotoxic stress by cisplatin-induced wild-type p53 association with HSP27, while R249Q mutant p53 constitutively associated with HSP27 and the association was only modestly enhanced by genotoxic stress. Importantly, HSP27 association with WT/6Q and R248Q/6Q was dramatically reduced compared to wild-type and R248Q p53. These data support that the nuclear PI4,5P2 binding to p53 promotes the subsequent association of HSP27, and HSP27 binding is critical for both wild-type and mutant p53 stability (Fig. 7a, 7b).
In parallel studies, a yeast-2 hybrid screen identified a homolog of HSP27, the sHSP αB-Crystallin/HSPB5, as a putative PIPKIα-binding protein. αB-Crystallin was reported to interact with wild-type p53 and translocate to the nucleus in response to genotoxic stress47, 48. Both recombinant HSP27 and αB-Crystallin directly bound to PIPKIα (Supplementary Fig. 7c, 7d), validating the yeast 2-hybrid results. By immunoblotting, we could not detect αB-Crystallin expression in MCF7, A549, PC3, and H1299 cells. In clinical breast carcinomas, αB-Crystallin is predominantly expressed in aggressive (ER/PR/HER2)-triple-negative breast tumors49 and its expression correlates with mutant p53 protein stabilization50. Knockdown of αB-Crystallin or PIPKIα in cancer cells reduced stress-induced wild-type (Supplementary Fig. 7e, 7f) and mutant p53 levels (Supplementary Fig. 7g). Additionally, mutant p53 and PIPKIα coimmunoprecipitated with αB-Crystallin, and knockdown of PIPKIα reduced the p53-αB-Crystallin interaction (Supplementary Fig. 7h–7j). Collectively, these findings indicate that sHSPs including αB-Crystallin and HSP27 play an important functional role in stabilizing p53 by a PIPKIα-dependent mechanism.
To further explore the potential functional relationship between PIPKIα and sHSPs in stabilizing p53, the PIPKIα and HSP27 binding regions on p53 were mapped using a series of p53 truncation mutants (Fig. 7f). Deletion of the tetramerization domain (TD) and the C-terminal regulatory domain (CTD) of p53 eliminated the interaction with PIPKIα (Fig. 7g), indicating that PIPKIα binds to p53 via the TD and CTD regions. The HSP27 binding region was within the proline-rich domain (PRD) and the DNA-binding domain (DBD) of p53 that are distinct from the PIPKIα and PI4,5P2 binding regions (Fig. 3b, 7f, 7g, and Supplementary Fig. 3a, 3b). These data indicate that PIPKIα and PI4,5P2 bind to similar regions in the C-terminus of p53, while HSP27 binds to the PRD and DBD of p53.
Dual regulation of small heat shock protein recruitment to the p53 complex by PIPKIα and PI4,5P2
To directly evaluate the effects of PI4,5P2 on the p53-sHSP interaction, binding studies with recombinant proteins were performed. PI4,5P2 enhanced HSP27 and αB-Crystallin binding to wild-type p53, while sHSP binding was markedly diminished in the p53 6Q mutant even in the presence of PI4,5P2 (Fig. 8a, 8b). Phosphoinositides other than PI4,5P2 tested had little or no effect on the p53-sHSP interaction (Fig. 8c, 8d, and Supplementary Fig. 8a), consistent with the specific interaction of p53 with PI4,5P2 (Fig. 3 and Supplementary Fig. 3). The mutated five lysine residues (Supplementary Fig. 3a) in the 6Q mutants are also sites for post-translational modification including ubiquitination, acetylation, and methylation in p538. These modifications are reported to regulate nuclear localization, stability, and activity of p53. To distinguish between these potential contributions of the modifications on p53 stability, a p53 R379Q mutant was utilized because no modifications on the R379 residue have been reported8. The R379Q mutant significantly reduced PI4,5P2 association in vivo, although to a lesser extent than the 6Q mutant (Supplementary Fig. 8b). When stably expressed in p53-null PC3 cells, the protein levels of mutant p53 were reduced by the R379Q mutation (Supplementary Fig. 8c). The R379Q mutant exhibited reduced interaction with endogenous HSP27 in vivo (Supplementary Fig. 8d) and diminished interaction with purified HSP27 and αB-Crystallin in vitro (Fig. 8a, 8b).
Figure 8. PI4,5P2 binding to p53 controls small heat shock protein binding to p53 in vitro.
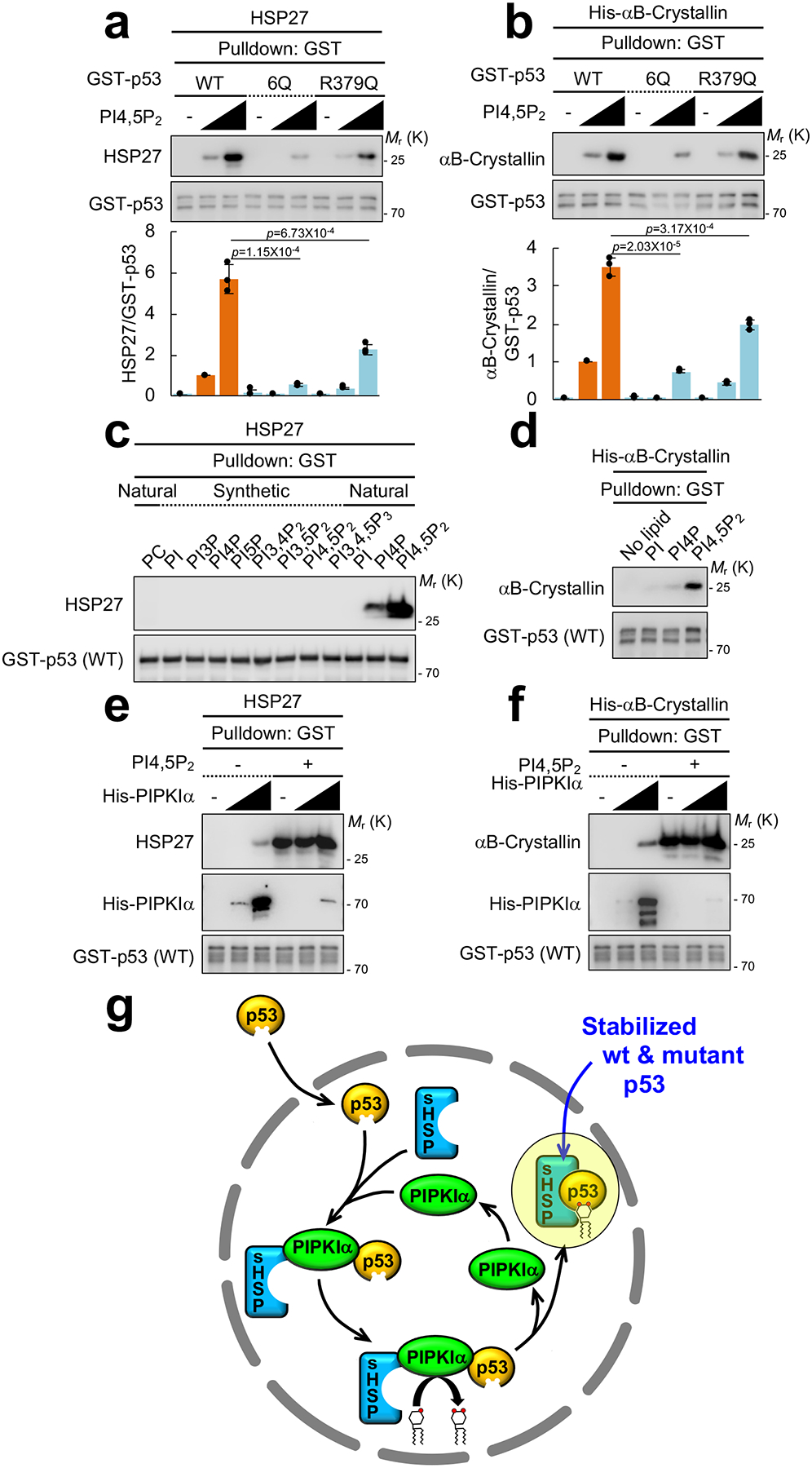
(A and B) Recombinant 0.1 μM GST-p53 and 0.5 μM untagged HSP27 or His-αB-Crystallin were incubated with 0, 1, or 10 μM PI4,5P2. GST-p53 proteins were pulled down and the associated HSP27 or His-αB-Crystallin was analyzed with an anti-HSP27 or an anti-αB-Crystallin antibody. The graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(C) 0.1 μM GST-p53 and 0.5 μM untagged HSP27 were incubated with 2.0 μM liposomes containing the indicated phosphoinositides. GST-p53 was pulled down and the associated HSP27 was analyzed by IB. Representative data of n=3 independent experiments were shown.
(D) 0.1 μM GST-p53 and 0.5 μM His-αB-Crystallin were incubated with 1 μM PI, PI4P, or PI4,5P2. GST-p53 was pulled down and the associated His-αB-Crystallin was analyzed by IB. Representative data of n=3 independent experiments were shown.
(E and F) 0.1 μM GST-p53, 0.5 μM untagged HSP27 or His-αB-Crystallin and 0, 0.01 and 0.1 μM His-PIPKIα were incubated with 1 μM PI4,5P2. GST-p53 was pulled down and the associated HSP27 or His-αB-Crystallin and His-PIPKIα was analyzed by IB. Representative data of n=3 independent experiments were shown.
(G) A schematic model of how p53 stability is regulated by PIPKIα, PI4,5P2 and small heat shock proteins (sHSPs).
PIPKIα directly interacted with both p53 and the sHSPs (Fig. 2c, 7f, 7g, and Supplementary Fig. 7c, 7d). This suggests that PIPKIα regulates the association between sHSPs and p53 by first binding independently to p53 and sHSPs and then promoting the PI4,5P2-dependent interaction between p53 and sHSPs. To explore this possibility, recombinant sHSPs where added to wild-type p53 protein with increasing amounts of PIPKIα protein in the absence or presence of PI4,5P2 (Fig. 8e, 8f). In the absence of PI4,5P2, sHSPs bound weakly to wild-type p53, but in the presence of PI4,5P2, the binding of sHSPs to p53 was markedly enhanced, while PIPKIα binding to p53 was virtually abolished. These data support a model whereby the stability of p53 is regulated by nuclear PIPKIα and its product PI4,5P2; nuclear PIPKIα binds to both p53 and sHSPs, and its product PI4,5P2 subsequently binds to the p53 CTD, promoting the p53-sHSP interaction and releasing PIPKIα from the complex (Fig. 8g).
Discussion
We have discovered a signaling pathway in which the lipid kinase PIPKIα and its PI4,5P2 product regulate the stability of stress-induced wild-type and mutant p53 in the nucleus by a step-wise mechanism that confers exquisite temporal and spatial specificity (Fig. 8g). In this pathway, PIPKIα binds directly to both wild-type and mutant p53 as well as the sHSP chaperones αB-Crystallin and HSP27. The binding of PIPKIα to wild-type p53 is dependent on genotoxic and oxidative stress, while PIPKIα interacts constitutively with mutant p53. These results are consistent with observations suggesting that mutant p53 is constitutively activated in cancer cell lines, perhaps by stress pathways that are intrinsic to cancer cells51. The interaction of PIPKIα with p53 results in the generation of PI4,5P2 that binds to p53 in its CTD, a domain previously implicated in regulating p53 stability8, 9, 11, 21. The bound PI4,5P2 stimulates the recruitment of sHSPs that stabilize both stress-induced wild-type and mutant p53. Additionally, PI4,5P2 stimulates the dissociation of PIPKIα from the p53-sHSP complex, enabling the dissociated PIPKIα to initiate assembly of new PIPKIα-PI4,5P2-p53-sHSP complexes. This model suggests a catalytic cycling mechanism of PIPKIα in p53 stabilization (Fig. 8g).
It has been well-documented that the stability of stress-induced wild-type p53 is controlled by a set of E3 ubiquitin ligases including MDM2, MDMX, and CHIP8, 9. We observed that MDM2 interacted with wild-type p53 in the cytoplasm consistent with a report demonstrating that MDM2 is predominantly localized in the cytoplasm where it targets wild-type p53 for ubiquitin-mediated proteasomal degradation52. Moreover, genotoxic stress stimulated the interaction between PIPKIα and wild-type p53 in the nucleus, while displacing MDM2 from this complex. In contrast, the stability of mutant p53 is reported to be regulated by the HSP90 chaperone complex24, 44. As we and others42, 43 report, mutant p53 is largely localized in the nucleus, whereas the HSP90 chaperone complex is primarily found in the cytoplasm53. Additionally, we observed that HSP70 bound a subset of mutant p53 proteins and its interaction with mutant p53 was not altered by the 6Q CTD mutation that disrupted PI4,5P2 binding (Fig. 6c), distinguishing it from the sHSPs. These results suggest that the stability of nuclear p53 cannot be fully explained by the canonical MDM2 and HSP90 pathways. The discovery that nuclear p53 stability is regulated by a novel PIPKIα-p53-PI4,5P2-sHSP protein-lipid complex adds a new stratum of regulation for nuclear p53.
Based on our data and the existing literature, we hypothesize that the interaction between PI4,5P2 and the CTD of p53 induces a conformational change and this gives the sHSPs access to the DBD for binding (Fig. 7f, 7g). In turn, the association of sHSPs with p53 blocks the interaction of p53 with its degradation machinery including MDM2. Consistent with this model, the PI4,5P2-binding defective mutant 6Q interacts with MDM2 more robustly than wild-type p53 (Fig. 6c). The CTD of p53 is a natively unstructured region and it is well-established that post-translational modifications in the CTD affect functions in other domains including the DBD8, 54, 55. Not only mutant p53 but also wild-type p53 have an intrinsic proclivity to form homo- and hetero-aggregates and the DBD is responsible for self aggregation42, 56, 57. sHSPs do not have ATPase activity, but under stress conditions sHSPs form large oligomeric complexes that bind to unfolded clients58. This chaperone-like activity of sHSPs protects client proteins from aggregation and degradation47, 58–60 and may also support specific functions. Importantly, αB-Crystallin directly binds to the DBD of p5348 and HSP27 also associated with p53 via the DBD (Fig. 7f, 7g). Furthermore, sHSPs including αB-Crystallin and HSP27 translocate to the nucleus upon stress47, 59, consistent with our data (Fig. 6d, 6e). Indeed, sHSPs have been reported to be present in nuclear speckles61 consistent with our results that p53 and PI4,5P2 colocalize with the nuclear speckle marker SC-35 (Supplementary Fig. 4e). Also, HSP27 targets to γH2A.X-positive DNA damage sites and has been previously implicated in DNA double-stranded break repair62 consistent with our results localizing PIPKIα, PI4,5P2 and p53 to DNA damage sites (Fig. 4c–4e, and Supplementary Fig. 5a–5f). Collectively, these findings point to a model whereby sHSPs may stabilize nuclear p53 by preventing protein aggregation and inhibiting recruitment of components of the p53-degradation machinery. In addition, the sHSPs and PI4,5P2 may facilitate assembly of functional p53 complexes that control both wild-type and mutant p53 functions.
This nuclear PI4,5P2 pathway for controlling p53 stability has significant implications for cancer. Although PIPKIα, mutant p53, and sHSPs have independently been implicated in tumor progression12, 15, 58, 60, our results indicate that these proteins form a carefully orchestrated molecular complex that may contribute to these effects. Intriguingly, p53 mutation was recently reported to lead to an increase in phosphoinositides63 that are favored substrates for PIPKIα64. This observation suggests a positive feedback regulation of the PIPKIα-mutant p53 axis in cancer: mutant p53 promotes PI4,5P2 generation by PIPKIα by increasing substrate availability, and PIPKIα stabilizes mutant p53 levels to promote tumor progression and chemoresistance. Moreover, αB-Crystallin expression correlates with p53 protein stabilization in clinically aggressive triple-negative breast cancers50 and the PIPKIα gene is commonly amplified in breast cancer65, suggesting that this complex may contribute to the pathogenesis of these and perhaps other cancers. Given the critical role of mutant p53 stability in its oncogenic activity11, 12, 24, our results point to PIPKIα and the PIPKIα-p53-PI4,5P2-sHSP complex as therapeutic targets for cancer.
Supplementary Material
Supplementary Figure 1. PIPKIα depletion or pharmacological inhibition reduces mutant p53 protein levels. (A) siRNAs against PIPKIα were transiently transfected in the indicated cells for 72 h. Protein expression was analyzed by IB, p53 blots were quantified and the graph is shown as mean ± SD of n=3 independent experiments. Scrambled siRNAs (siCon) were used as a negative control. One-sided paired Student t-tests were used for statistical analysis here and elsewhere (*, p<0.05; **, p<0.01).
(B) SkBr3 cells stably expressing shRNA against a scrambled sequence or PIPKIα were fixed and immunostained with PIPKIα and p53 antibody. Representative data of n=3 independent experiments were shown. Scale bar, 30 μm.
(C) MDA-MB-231 cells were treated with vehicle control or 50 μM ISA-2011B for the indicated time. Cell lysates were analyzed by IB with the indicated antibodies. Representative data of n=3 independent experiments were shown.
(D) A431 cells were treated with vehicle control or 50 μM ISA-2011B for 72 h. Cells were fixed and immunostained with PIPKIα and p53 antibody. DAPI was used to stain nucleic acids. Representative data of n=3 independent experiments were shown. Scale bar, 30 μm.
Supplementary Figure 2. PIPKIα associates with the p53 complex in subnuclear compartments. (A and B) The indicated cells were treated with 30 μM cisplatin for 24 h. Endogenous PIPKIα was IP’ed and the associated proteins were analyzed by IB. Representative data of n=3 independent experiments were shown.
(C and D) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of PIPKIα. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope. The mean intensity of the green channels in the nuclear region (DAPI positive) was quantified by LAS X (Leica). The green signal from cisplatin-treated group was normalized to the untreated control. The graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times.
(E and F) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between PIPKIα and p53. DAPI was used to stain nucleic acids. The z-stack images were taken with a Leica SP8 confocal microscope with each frame over a 0.2 μm thickness. The 3D view was generated by ImageJ. Scale bar, 5 μm. Representative data of n=3 independent experiments were shown.
Supplementary Figure 3. PI4,5P2 accumulates in the nucleus in response to genotoxic stress and associates with p53. (A) Schematic representation of p53 domains (top). 1 or 6 basic residues in the CTD were mutated to glutamine (Q) to generated PI4,5P2-binding defective mutant R379Q or 6Q. TAD, transactivation domain; PRD, proline-rich domain; DBD, DNA-binding domain; TD, tetramerization domain; CTD, C-terminal regulatory domain.
(B) The indicated GST-tagged 0.5 μM recombinant proteins were incubated with 5.0 μM liposomes containing PI or PI4,5P2. Liposomes were sedimented and the associated proteins were analyzed by IB with an anti-GST antibody. The experiments were repeated 3 times.
(C) The indicated cells were treated with 30 μM cisplatin for 24 h. Endogenous p53 was IP’ed and IP samples were eluted with 0.2 M glycine (pH 2.5). Elution was analyzed by IB with an anti-PI4,5P2 KT10 antibody. Representative data of n=3 independent experiments were shown.
(D to I) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated phosphoinositides. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope. The mean intensity of the green channels in the nuclear region (DAPI positive) was quantified by LAS X (Leica). The green signal from cisplatin-treated group was normalized to its untreated control. The graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times.
(J to N) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between p53 and the indicated phosphoinositides. DAPI was used to stain nucleic acids. The images were taken by Leica SP8 confocal microscope. The red PLA signal was quantified by LAS X (Leica) and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
Supplementary Figure 4. PI4,5P2 and p53 colocalize in nuclear speckles and DNA damage sites. (A and B) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of PI4,5P2 and p53. An anti-PS antibody was used to stain the nuclear membrane. The z-stack images were taken with a Leica SP8 confocal microscope with each frame over a 0.2 μm thickness. The 3D view was generated by ImageJ. Scale bar, 5 μm. The experiments were repeated 3 times.
(C and D) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between PI4,5P2 and p53. An anti-PS antibody was used to stain the nuclear membrane. DAPI was used to stain nucleic acids. The z-stack images were taken with a Leica SP8 confocal microscope with each frame over a 0.2 μm thickness. The 3D view was generated by ImageJ. Scale bar, 5 μm. The experiments were repeated 3 times.
(E) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated molecules. The images were taken with a Leica SP8 confocal microscope. Scale bar, 5 μm. The experiments were repeated 3 times.
(F to K) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated molecules. The images were taken with a Leica SP8 confocal microscope and processed by ImageJ. White arrows indicate the colocalized signals. The experiments were repeated three times and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
Supplementary Figure 5. PI4,5P2 binding controls the nuclear localization of p53. (A to F) A549 cells were transfected with the indicated siRNAs for 48 h and then treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated molecules. The images were taken with a Leica SP8 confocal microscope and processed by ImageJ. White arrows indicate the colocalized signals. The experiments were repeated 3 times and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(G) Flag-p53 constructs were co-transfected with HA-PIPKIα in HEK293 cells. Flag-p53 proteins were immunoprecipitated and the associated PIPKIα was analyzed by IB with an anti-HA antibody. Representative data of n=3 independent experiments were shown.
(H) The indicated p53 constructs were transiently transfected in p53-null H1299 cells for 24 h. Cells were fixed and analyzed for p53 immunofluorescence. Cells were photographed under 400X magnification. Scale bar, 30 μm. Representative data of n=3 independent experiments were shown.
Supplementary Figure 6. HSP27 is a novel binding partner of nuclear p53. (A) Cells were treated with 30 μM cisplatin for 24 h, and cytoplasmic (Cy) and nuclear (Nu) proteins were fractionated. p53 was IP’ed and IP samples were eluted with 0.2 M glycine (pH 2.5). Elution was analyzed by IB with the indicated molecules and the graph is shown as mean ± SD of 3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(B and C) Cells were transfected with the indicated siRNAs. After 48 h, cells were treated with 30 μM cisplatin for an additional 24 h. Cytoplasmic and nuclear proteins were fractionated and analyzed by IB. MEK1/2 and Lamin A/C were used as cytoplasmic and nuclear markers, respectively (top). p53 was immunoprecipitated from fractionated cell lysates and eluted. The elution was resolved by SDS-PAGE. Protein blots were analyzed by IB with an anti-PI4,5P2 antibody (KT10 clone) or an anti-p53 antibody (DO1 clone). Representative data of n=3 independent experiments were shown.
(D) The indicated p53 constructs were transiently transfected in p53-null H1299 cells for 48 h. p53 was IP’ed and IP samples were eluted with 0.2 M glycine (pH 2.5). Elution was analyzed by mass spectrometry. The table shows a selected list of detected proteins. The experiments were repeated twice.
(E and F) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of HSP27. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope. The mean intensity of the green channels in the nuclear region (DAPI positive) was quantified by LAS X (Leica). The green signal from cisplatin-treated group was normalized to the untreated control. The graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times.
(G and H) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between HSP27 and p53. DAPI was used to stain nucleic acids. The z-stack images were taken with a Leica SP8 confocal microscope with each frame over a 0.2 μm thickness. The 3D view was generated by ImageJ. Scale bar, 5 μm. Representative data of n=3 independent experiments were shown.
Supplementary Figure 7. Small heat shock proteins are required for mutant p53 stability. (A and B) p53-null H1299 cells were transiently transfected with the indicated p53 constructs for 24 and then treated with 30 μM cisplatin for 24 h. Cells were fixed and processed for IF staining against HSP27 and p53. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope and processed by ImageJ. White arrows indicate the colocalized signals. The experiments were repeated three times and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(C and D) Recombinant His-PIPKIα was incubated with untagged HSP27 or His-αB-Crystallin. HSP27 and His-αB-Crystallin were pulled down with an anti-HSP27 and an anti-αB-Crystallin antibody, respectively. Associated His-PIPKIα was detected with an anti-PIPKIα antibody. Representative data of n=3 independent experiments were shown.
(E) MCF7 cells were transfected with PIPKIα siRNA for 48 h. Cells were treated with 10 μM cisplatin for an additional 16 h. Protein expression was analyzed by IB, p53 blots were quantified and the graph is shown as mean ± SD of 3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(F) LNCaP cells were transfected with the indicated siRNAs for 48 h. Cells were treated with 10 μM cisplatin for an additional 16 h. Protein expression was analyzed by IB, p53 blots were quantified and the graph is shown as mean ± SD of 3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(G) In the indicated breast cancer cells, PIPKIα or αB-Crystallin was knocked down with siRNAs for 72 h. Protein expression was analyzed by IB. Representative data of n=3 independent experiments were shown.
(H) αB-Crystallin was IP’ed in MDA-MB-468 cells and the associated proteins analyzed by IB.
(I and J) PIPKIα was knocked down for 72 h in MDA-MB-468 cells. αB-Crystallin was IP’ed and the associated mutant p53 was analyzed by IB. The graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
Supplementary Figure 8. A point mutant of p53 that diminishes PI4,5P2 binding reduces p53 stability. (A) 0.1 μM GST-p53 and 0.5 μM untagged HSP27 were incubated with 1 μM PI, PI4P, or PI4,5P2. GST-p53 was pulled down and the associated HSP27 was analyzed by IB.
(B) The indicated p53 constructs were transiently expressed in p53-null H1299 cells for 24 h. p53 was immunoprecipitated and eluted with 0.1 M glycine (pH 2.5) and the associated molecules were analyzed by IB. The graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(C) In PC3 cells (p53-null), the indicated mutant p53 constructs were stably expressed and analyzed by IB. The graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(D) The indicated p53 constructs were transfected in p53-null H1299 cells for 24 h. Endogenous HSP27 was immunoprecipitated and the associated p53 was analyzed by IB. The graph is shown as mean ± SD of 3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
Acknowledgments
We would like to thank Addgene and Dr. Jiandong Chen (H. Lee Moffitt Cancer Center and Research Institute) for the p53 constructs. We are also grateful to Dr. Jenny L. Persson (Lund University) and Kiyoko Fukami (Tokyo University of Pharmacy and Life Sciences) for generously sharing ISA-2011B and the KT10 antibody, respectively. We would like to thank members of the R.A.A. and V.L.C. laboratories for helpful discussions. This work was supported in part by a National Institutes of Health grant GM114386 (R.A.A.), Department of Defense Breast Cancer Research Program grants W81XWH-17-1-0258 (R.A.A.) and W81XWH-17-1-0259 (V.L.C.), and a grant from the Breast Cancer Research Foundation (V.L.C.).
References
- 1.Balla T Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93, 1019–1137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi S, Thapa N, Tan X, Hedman AC & Anderson RA PIP kinases define PI4,5P(2)signaling specificity by association with effectors. Biochimica et biophysica acta 1851, 711–723 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson RA, Boronenkov IV, Doughman SD, Kunz J & Loijens JC Phosphatidylinositol phosphate kinases, a multifaceted family of signaling enzymes. J Biol Chem 274, 9907–9910 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Golebiewska U, Nyako M, Woturski W, Zaitseva I & McLaughlin S Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol Biol Cell 19, 1663–1669 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barlow CA, Laishram RS & Anderson RA Nuclear phosphoinositides: a signaling enigma wrapped in a compartmental conundrum. Trends Cell Biol 20, 25–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellman DL et al. A PtdIns4,5P2-regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature 451, 1013–1017 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Boronenkov IV, Loijens JC, Umeda M & Anderson RA Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing premRNA processing factors. Mol Biol Cell 9, 3547–3560 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai C & Gu W p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med 16, 528–536 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bieging KT, Mello SS & Attardi LD Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 14, 359–370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kruiswijk F, Labuschagne CF & Vousden KH p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 16, 393–405 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Freed-Pastor WA & Prives C Mutant p53: one name, many proteins. Genes Dev 26, 1268–1286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller PA & Vousden KH Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25, 304–317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barlow CA, Laishram RS & Anderson RA Nuclear phosphoinositides: a signaling enigma wrapped in a compartmental conundrum. Trends Cell Biol 2010, 25–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emerling BM et al. Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell 155, 844–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Semenas J et al. The role of PI3K/AKT-related PIP5K1alpha and the discovery of its selective inhibitor for treatment of advanced prostate cancer. Proc Natl Acad Sci U S A 111, E3689–3698 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiech M et al. Molecular mechanism of mutant p53 stabilization: the role of HSP70 and MDM2. PLoS One 7, e51426 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller PA & Vousden KH p53 mutations in cancer. Nat Cell Biol 15, 2–8 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Vakifahmetoglu-Norberg H et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev 27, 1718–1730 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147, 223–234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks CL & Gu W p53 regulation by ubiquitin. FEBS Lett 585, 2803–2809 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kruse JP & Gu W Modes of p53 regulation. Cell 137, 609–622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heck JN et al. A conspicuous connection: structure defines function for the phosphatidylinositol-phosphate kinase family. Crit Rev Biochem Mol Biol 42, 15–39 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Choi S, Houdek X & Anderson RA Phosphoinositide 3-kinase pathways and autophagy require phosphatidylinositol phosphate kinases. Adv Biol Regul (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexandrova EM et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 523, 352–356 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fredriksson S et al. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol 20, 473–477 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Lam F, Cladiere D, Guillaume C, Wassmann K & Bolte S Super-resolution for everybody: An image processing workflow to obtain high-resolution images with a standard confocal microscope. Methods 115, 17–27 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Choi S et al. IQGAP1 is a novel phosphatidylinositol 4,5 bisphosphate effector in regulation of directional cell migration. The EMBO journal 32, 2617–2630 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein DE, Lee A, Frank DW, Marks MS & Lemmon MA The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J Biol Chem 273, 27725–27733 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Rohacs T, Chen J, Prestwich GD & Logothetis DE Distinct specificities of inwardly rectifying K(+) channels for phosphoinositides. J Biol Chem 274, 36065–36072 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Balla T & Varnai P Visualization of cellular phosphoinositide pools with GFP-fused protein-domains. Curr Protoc Cell Biol Chapter 24, Unit 24 24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varnai P & Balla T Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol 143, 501–510 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis AE et al. Identification of nuclear phosphatidylinositol 4,5-bisphosphate-interacting proteins by neomycin extraction. Mol Cell Proteomics 10, M110 003376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatterjee A et al. U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of DeltaNp63alpha. Cell Cycle 7, 1231–1237 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy LO, Smith S, Chen RH, Fingar DC & Blenis J Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol 4, 556–564 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Fukami K et al. Antibody to phosphatidylinositol 4,5-bisphosphate inhibits oncogene-induced mitogenesis. Proc Natl Acad Sci U S A 85, 9057–9061 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang YH et al. DNA damage causes rapid accumulation of phosphoinositides for ATR signaling. Nat Commun 8, 2118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shah ZH et al. Nuclear phosphoinositides and their impact on nuclear functions. FEBS J 280, 6295–6310 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Xu Q et al. Phosphatidylinositol phosphate kinase PIPKIgamma and phosphatase INPP5E coordinate initiation of ciliogenesis. Nat Commun 7, 10777 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humbert MC et al. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci U S A 109, 19691–19696 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balla T & Varnai P Visualizing cellular phosphoinositide pools with GFP-fused protein-modules. Sci STKE 2002, pl3 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Choi S et al. Agonist-stimulated phosphatidylinositol-3,4,5-trisphosphate generation by scaffolded phosphoinositide kinases. Nat Cell Biol 18, 1324–1335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu J et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 7, 285–295 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Peng Y, Chen L, Li C, Lu W & Chen J Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J Biol Chem 276, 40583–40590 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Li D et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res 9, 577–588 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arrigo AP et al. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett 581, 3665–3674 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Venkatakrishnan CD et al. HSP27 regulates p53 transcriptional activity in doxorubicin-treated fibroblasts and cardiac H9c2 cells: p21 upregulation and G2/M phase cell cycle arrest. Am J Physiol Heart Circ Physiol 294, H1736–1744 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Adhikari AS, Sridhar Rao K, Rangaraj N, Parnaik VK & Mohan Rao C Heat stress-induced localization of small heat shock proteins in mouse myoblasts: intranuclear lamin A/C speckles as target for alphaB-crystallin and Hsp25. Exp Cell Res 299, 393–403 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Watanabe G et al. alphaB-crystallin: a novel p53-target gene required for p53-dependent apoptosis. Cancer Sci 100, 2368–2375 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moyano JV et al. AlphaB-crystallin is a novel oncoprotein that predicts poor clinical outcome in breast cancer. J Clin Invest 116, 261–270 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koletsa T et al. alphaB-crystallin is a marker of aggressive breast cancer behavior but does not independently predict for patient outcome: a combined analysis of two randomized studies. BMC Clin Pathol 14, 28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Marchenko ND et al. Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ 17, 255–267 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Passinen S, Valkila J, Manninen T, Syvala H & Ylikomi T The C-terminal half of Hsp90 is responsible for its cytoplasmic localization. Eur J Biochem 268, 5337–5342 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Friedler A, Veprintsev DB, Freund SM, von Glos KI & Fersht AR Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure 13, 629–636 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Laptenko O et al. The p53 C terminus controls site-specific DNA binding and promotes structural changes within the central DNA binding domain. Mol Cell 57, 1034–1046 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cino EA, Soares IN, Pedrote MM, de Oliveira GA & Silva JL Aggregation tendencies in the p53 family are modulated by backbone hydrogen bonds. Sci Rep 6, 32535 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ano Bom AP et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J Biol Chem 287, 28152–28162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malin D, Petrovic V, Strekalova E, Sharma B & Cryns VL alphaB-crystallin: Portrait of a malignant chaperone as a cancer therapeutic target. Pharmacol Ther 160, 1–10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bakthisaran R, Tangirala R & Rao Ch M Small heat shock proteins: Role in cellular functions and pathology. Biochimica et biophysica acta 1854, 291–319 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Arrigo AP & Gibert B HspB1, HspB5 and HspB4 in Human Cancers: Potent Oncogenic Role of Some of Their Client Proteins. Cancers (Basel) 6, 333–365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van den IP, Wheelock R, Prescott A, Russell P & Quinlan RA Nuclear speckle localisation of the small heat shock protein alpha B-crystallin and its inhibition by the R120G cardiomyopathy-linked mutation. Exp Cell Res 287, 249–261 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Sottile ML & Nadin SB Heat shock proteins and DNA repair mechanisms: an updated overview. Cell Stress Chaperones 23, 303–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Naguib A et al. p53 mutations change phosphatidylinositol acyl chain composition. Cell Rep 10, 8–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shulga YV, Anderson RA, Topham MK & Epand RM Phosphatidylinositol-4-phosphate 5-kinase isoforms exhibit acyl chain selectivity for both substrate and lipid activator. J Biol Chem 287, 35953–35963 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waugh MG Amplification of Chromosome 1q Genes Encoding the Phosphoinositide Signalling Enzymes PI4KB, AKT3, PIP5K1A and PI3KC2B in Breast Cancer. J Cancer 5, 790–796 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prudovsky I, Vary CP, Markaki Y, Olins AL & Olins DE Phosphatidylserine colocalizes with epichromatin in interphase nuclei and mitotic chromosomes. Nucleus 3, 200–210 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma A, Singh K & Almasan A Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol 920, 613–626 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. PIPKIα depletion or pharmacological inhibition reduces mutant p53 protein levels. (A) siRNAs against PIPKIα were transiently transfected in the indicated cells for 72 h. Protein expression was analyzed by IB, p53 blots were quantified and the graph is shown as mean ± SD of n=3 independent experiments. Scrambled siRNAs (siCon) were used as a negative control. One-sided paired Student t-tests were used for statistical analysis here and elsewhere (*, p<0.05; **, p<0.01).
(B) SkBr3 cells stably expressing shRNA against a scrambled sequence or PIPKIα were fixed and immunostained with PIPKIα and p53 antibody. Representative data of n=3 independent experiments were shown. Scale bar, 30 μm.
(C) MDA-MB-231 cells were treated with vehicle control or 50 μM ISA-2011B for the indicated time. Cell lysates were analyzed by IB with the indicated antibodies. Representative data of n=3 independent experiments were shown.
(D) A431 cells were treated with vehicle control or 50 μM ISA-2011B for 72 h. Cells were fixed and immunostained with PIPKIα and p53 antibody. DAPI was used to stain nucleic acids. Representative data of n=3 independent experiments were shown. Scale bar, 30 μm.
Supplementary Figure 2. PIPKIα associates with the p53 complex in subnuclear compartments. (A and B) The indicated cells were treated with 30 μM cisplatin for 24 h. Endogenous PIPKIα was IP’ed and the associated proteins were analyzed by IB. Representative data of n=3 independent experiments were shown.
(C and D) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of PIPKIα. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope. The mean intensity of the green channels in the nuclear region (DAPI positive) was quantified by LAS X (Leica). The green signal from cisplatin-treated group was normalized to the untreated control. The graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times.
(E and F) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between PIPKIα and p53. DAPI was used to stain nucleic acids. The z-stack images were taken with a Leica SP8 confocal microscope with each frame over a 0.2 μm thickness. The 3D view was generated by ImageJ. Scale bar, 5 μm. Representative data of n=3 independent experiments were shown.
Supplementary Figure 3. PI4,5P2 accumulates in the nucleus in response to genotoxic stress and associates with p53. (A) Schematic representation of p53 domains (top). 1 or 6 basic residues in the CTD were mutated to glutamine (Q) to generated PI4,5P2-binding defective mutant R379Q or 6Q. TAD, transactivation domain; PRD, proline-rich domain; DBD, DNA-binding domain; TD, tetramerization domain; CTD, C-terminal regulatory domain.
(B) The indicated GST-tagged 0.5 μM recombinant proteins were incubated with 5.0 μM liposomes containing PI or PI4,5P2. Liposomes were sedimented and the associated proteins were analyzed by IB with an anti-GST antibody. The experiments were repeated 3 times.
(C) The indicated cells were treated with 30 μM cisplatin for 24 h. Endogenous p53 was IP’ed and IP samples were eluted with 0.2 M glycine (pH 2.5). Elution was analyzed by IB with an anti-PI4,5P2 KT10 antibody. Representative data of n=3 independent experiments were shown.
(D to I) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated phosphoinositides. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope. The mean intensity of the green channels in the nuclear region (DAPI positive) was quantified by LAS X (Leica). The green signal from cisplatin-treated group was normalized to its untreated control. The graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times.
(J to N) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between p53 and the indicated phosphoinositides. DAPI was used to stain nucleic acids. The images were taken by Leica SP8 confocal microscope. The red PLA signal was quantified by LAS X (Leica) and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
Supplementary Figure 4. PI4,5P2 and p53 colocalize in nuclear speckles and DNA damage sites. (A and B) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of PI4,5P2 and p53. An anti-PS antibody was used to stain the nuclear membrane. The z-stack images were taken with a Leica SP8 confocal microscope with each frame over a 0.2 μm thickness. The 3D view was generated by ImageJ. Scale bar, 5 μm. The experiments were repeated 3 times.
(C and D) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between PI4,5P2 and p53. An anti-PS antibody was used to stain the nuclear membrane. DAPI was used to stain nucleic acids. The z-stack images were taken with a Leica SP8 confocal microscope with each frame over a 0.2 μm thickness. The 3D view was generated by ImageJ. Scale bar, 5 μm. The experiments were repeated 3 times.
(E) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated molecules. The images were taken with a Leica SP8 confocal microscope. Scale bar, 5 μm. The experiments were repeated 3 times.
(F to K) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated molecules. The images were taken with a Leica SP8 confocal microscope and processed by ImageJ. White arrows indicate the colocalized signals. The experiments were repeated three times and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
Supplementary Figure 5. PI4,5P2 binding controls the nuclear localization of p53. (A to F) A549 cells were transfected with the indicated siRNAs for 48 h and then treated with 30 μM cisplatin for 24 h before processed for IF staining of the indicated molecules. The images were taken with a Leica SP8 confocal microscope and processed by ImageJ. White arrows indicate the colocalized signals. The experiments were repeated 3 times and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(G) Flag-p53 constructs were co-transfected with HA-PIPKIα in HEK293 cells. Flag-p53 proteins were immunoprecipitated and the associated PIPKIα was analyzed by IB with an anti-HA antibody. Representative data of n=3 independent experiments were shown.
(H) The indicated p53 constructs were transiently transfected in p53-null H1299 cells for 24 h. Cells were fixed and analyzed for p53 immunofluorescence. Cells were photographed under 400X magnification. Scale bar, 30 μm. Representative data of n=3 independent experiments were shown.
Supplementary Figure 6. HSP27 is a novel binding partner of nuclear p53. (A) Cells were treated with 30 μM cisplatin for 24 h, and cytoplasmic (Cy) and nuclear (Nu) proteins were fractionated. p53 was IP’ed and IP samples were eluted with 0.2 M glycine (pH 2.5). Elution was analyzed by IB with the indicated molecules and the graph is shown as mean ± SD of 3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(B and C) Cells were transfected with the indicated siRNAs. After 48 h, cells were treated with 30 μM cisplatin for an additional 24 h. Cytoplasmic and nuclear proteins were fractionated and analyzed by IB. MEK1/2 and Lamin A/C were used as cytoplasmic and nuclear markers, respectively (top). p53 was immunoprecipitated from fractionated cell lysates and eluted. The elution was resolved by SDS-PAGE. Protein blots were analyzed by IB with an anti-PI4,5P2 antibody (KT10 clone) or an anti-p53 antibody (DO1 clone). Representative data of n=3 independent experiments were shown.
(D) The indicated p53 constructs were transiently transfected in p53-null H1299 cells for 48 h. p53 was IP’ed and IP samples were eluted with 0.2 M glycine (pH 2.5). Elution was analyzed by mass spectrometry. The table shows a selected list of detected proteins. The experiments were repeated twice.
(E and F) A549 cells were treated with 30 μM cisplatin for 24 h before processed for IF staining of HSP27. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope. The mean intensity of the green channels in the nuclear region (DAPI positive) was quantified by LAS X (Leica). The green signal from cisplatin-treated group was normalized to the untreated control. The graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. The experiments were repeated at least 3 times.
(G and H) A549 cells were treated with 30 μM cisplatin for 24 h before processed for PLA between HSP27 and p53. DAPI was used to stain nucleic acids. The z-stack images were taken with a Leica SP8 confocal microscope with each frame over a 0.2 μm thickness. The 3D view was generated by ImageJ. Scale bar, 5 μm. Representative data of n=3 independent experiments were shown.
Supplementary Figure 7. Small heat shock proteins are required for mutant p53 stability. (A and B) p53-null H1299 cells were transiently transfected with the indicated p53 constructs for 24 and then treated with 30 μM cisplatin for 24 h. Cells were fixed and processed for IF staining against HSP27 and p53. DAPI was used to stain nucleic acids. The images were taken with a Leica SP8 confocal microscope and processed by ImageJ. White arrows indicate the colocalized signals. The experiments were repeated three times and the graph is shown as mean ± SD of n=10 cells. Scale bar, 5 μm. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(C and D) Recombinant His-PIPKIα was incubated with untagged HSP27 or His-αB-Crystallin. HSP27 and His-αB-Crystallin were pulled down with an anti-HSP27 and an anti-αB-Crystallin antibody, respectively. Associated His-PIPKIα was detected with an anti-PIPKIα antibody. Representative data of n=3 independent experiments were shown.
(E) MCF7 cells were transfected with PIPKIα siRNA for 48 h. Cells were treated with 10 μM cisplatin for an additional 16 h. Protein expression was analyzed by IB, p53 blots were quantified and the graph is shown as mean ± SD of 3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(F) LNCaP cells were transfected with the indicated siRNAs for 48 h. Cells were treated with 10 μM cisplatin for an additional 16 h. Protein expression was analyzed by IB, p53 blots were quantified and the graph is shown as mean ± SD of 3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(G) In the indicated breast cancer cells, PIPKIα or αB-Crystallin was knocked down with siRNAs for 72 h. Protein expression was analyzed by IB. Representative data of n=3 independent experiments were shown.
(H) αB-Crystallin was IP’ed in MDA-MB-468 cells and the associated proteins analyzed by IB.
(I and J) PIPKIα was knocked down for 72 h in MDA-MB-468 cells. αB-Crystallin was IP’ed and the associated mutant p53 was analyzed by IB. The graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
Supplementary Figure 8. A point mutant of p53 that diminishes PI4,5P2 binding reduces p53 stability. (A) 0.1 μM GST-p53 and 0.5 μM untagged HSP27 were incubated with 1 μM PI, PI4P, or PI4,5P2. GST-p53 was pulled down and the associated HSP27 was analyzed by IB.
(B) The indicated p53 constructs were transiently expressed in p53-null H1299 cells for 24 h. p53 was immunoprecipitated and eluted with 0.1 M glycine (pH 2.5) and the associated molecules were analyzed by IB. The graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(C) In PC3 cells (p53-null), the indicated mutant p53 constructs were stably expressed and analyzed by IB. The graph is shown as mean ± SD of n=3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).
(D) The indicated p53 constructs were transfected in p53-null H1299 cells for 24 h. Endogenous HSP27 was immunoprecipitated and the associated p53 was analyzed by IB. The graph is shown as mean ± SD of 3 independent experiments. Two-sided paired Student t-tests were used for statistical analysis (*, p<0.05; **, p<0.01).