

Abstract
Background
Some authors have suggested that loxapine is more effective than typical antipsychotics in reducing the negative symptoms of schizophrenia, that extrapyramidal adverse effects are not usually seen at clinically effective antipsychotic doses and that it should therefore be classed as atypical.
Objectives
To determine the effects of loxapine compared with placebo, typical and other atypical antipsychotic drugs for schizophrenia and related psychoses.
Search methods
For this 2007 update, we searched the Cochrane Schizophrenia Group's Register (January 2007).
Selection criteria
We included all randomised controlled clinical trials relevant to the care of schizophrenia that compared loxapine to other treatments.
Data collection and analysis
We independently inspected abstracts ordered papers, re‐inspected and quality assessed these. For dichotomous data we calculated relative risks (RR) and their 95% confidence intervals (CI) on an intention‐to‐treat basis based on a fixed effects model. We calculated numbers needed to treat/harm (NNT/NNH) where appropriate. For continuous data, we calculated weighted mean differences (WMD) again based on a fixed effects model.
Main results
We were able to include 41 studies in this review. Compared with placebo, loxapine has an antipsychotic effect (Global effect ‐ not improved at six weeks: n=78, 2 RCTs, RR 0.30 CI 0.1 to 0.6 NNT 3 CI 3 to 5). It is as effective as typical drugs in the short term (4 ‐12 weeks) (Global effect: n=580, 13 RCTs, RR 0.86 CI 0.7 to 1.1; mental state: n=915, 6 RCTs, RR 0.89 CI 0.8 to 1.1). Very limited heterogeneous data suggest that, given intramuscularly (IM), loxapine may be at least as sedating as IM haloperidol and thiothixene. Loxapine is also as effective as atypicals (risperidone, quetiapine) (n=468, 6 RCTs, RR mental state not improved 1.07 CI 0.8 to 1.5). Adverse effect profile is similar to typicals but loxapine may cause more extrapyramidal adverse effects when compared with atypicals (n=340, 4 RCTs, RR 2.18 CI 1.6 to 3.1).
Authors' conclusions
Loxapine is an antipsychotic which is not clearly distinct from typical or atypical drugs in terms of its effects on global or mental state. Loxapines profile of adverse effects is similar to that of the older generation of antipsychotic drugs.
Keywords: Humans, Antipsychotic Agents, Antipsychotic Agents/adverse effects, Antipsychotic Agents/therapeutic use, Dopamine Antagonists, Dopamine Antagonists/adverse effects, Dopamine Antagonists/therapeutic use, Loxapine, Loxapine/adverse effects, Loxapine/therapeutic use, Randomized Controlled Trials as Topic, Schizophrenia, Schizophrenia/drug therapy
Plain language summary
Loxapine for schizophrenia
Schizophrenia is a chronic and relapsing serious mental illness with a lifetime prevalence of about 1% worldwide.
Typical and atypical antipsychotics provide a treatment for people with schizophrenia, with either a reduction in the episodes of psychosis or a reduction in the severity of the symptoms. However, a proportion of people still do not respond adequately to antipsychotic medication. Typical and atypical antipsychotics are associated with serious adverse effects which are not only uncomfortable for patients, but can also be associated with subsequent reduced compliance with treatment and therefore relapse in the illness.
Loxapine is an antipsychotic drug available in Belgium, Canada, Denmark, France, Germany, Iceland, Ireland, Netherlands, New Zealand, Spain, the UK and the USA. We systematically evaluated the effects of this antipsychotic and were able to include 41 randomised trials following the updates in 2005 and 2007.
Loxapine may be effective for the treatment of schizophrenia but does not differ greatly from the older typical antipsychotics (chlorpromazine, trifluperazine, perphenazine) or other atypicals (risperidone, quetipine) in respect of treatment efficacy. Loxapine, however, may cause more extrapyramidal adverse effects compared with atypical drugs.
Background
For schizophrenia, choosing the most appropriate, effective and tolerable antipsychotic drug is key to maximising the usefulness of medication. According to recent treatment guidelines, both conventional/typical and atypical antipsychotic drugs may be reasonable choices in the treatment of schizophrenia (APA 1994).
Typical antipsychotic drugs, such as haloperidol, chlorpromazine, and trifluoperazine are widely used as the first line treatment for people with schizophrenia, in the acute as well as in chronic forms of the illness (APA 1994, Silverstone 1995). However, the atypical class of antipsychotic drugs, most of which have been formulated relatively recently, have made important inroads into this traditional approach (Wood MacKenzie 1998, Adams 1999).
Atypical is a term widely used to describe some antipsychotics which have a low propensity to produce movement disorders, sedation and raised serum prolactin (Kerwin 1994). The pharmacokinetic profiles of the atypical drugs are either clozapine‐like or risperidone‐like (Kerwin 1996). There is some suggestion that the different adverse effect profiles of the atypical antipsychotic group make them more acceptable to people with schizophrenia (Casey 1997). Certainly, the adverse effects of the typical drugs, such as movement disorders and sedation, are problematic and can result in poor compliance with treatment (CWG 1998).
The positive symptoms (delusions, hallucinations, and disordered thinking) of schizophrenia seem more responsive to the typical antipsychotic drugs than the negative symptoms. This latter cluster of symptoms (poverty of speech, lack of motivation, apathy and inability to express emotions (Carpenter 1994)), is very disabling and may respond better to the atypical antipsychotic drugs (APA 1994, Silverstone 1995), although this too has not been adequately established (Kane 1996).
Atypical antipsychotics are more expensive than conventional drugs (Wood MacKenzie 1998), but it has been suggested that if they do indeed reduce a person's need for inpatient services their use would result in a net reduction of costs (Buckley 1997, Glazer 1997). Loxapine is an old, inexpensive D2/D3 receptor antagonist with a higher affinity for D3 than D2. It also has histamine (H1), serotonin (5‐HT2) and adrenergic (alpha 1)‐blocking activities. Structurally it is related to clozapine. Some authors have suggested that loxapine is more effective than other 'typical' antipsychotics in reducing the negative symptoms of schizophrenia (Vanelle 1994), that extrapyramidal side‐effects are not usually seen at clinically effective antipsychotic doses (Thomas 1998) and that it should therefore be classed as an atypical antipsychotic.
Technical background Former designation: oxilapine (Figure 1). Dopamine D2/D3 receptors antagonist with higher affinity for D3 than for D2. Also 5‐HT2 antagonist. Structurally related to clozapine. Used in the treatment of schizophrenic disorders. Dosage 50 to 200 mg/day. First French reports in 1965. Plasma half‐life after oral administration to healthy people three to four hours; terminal half‐life of a major 8‐hydroxy‐metabolite about eight hours. Loxapine as been known as oxilapine, LW 3170, SUM 3170, CL 71.563 (succinate), CL 62362 and S 805. It is sold as Desconex (Spain); Loxapac (Belgium, Canada, Denmark, France, Germany, Iceland, Ireland, Netherlands, New Zealand, UK); Loxitane (USA).
1.
Loxapine ‐ structure
Objectives
To determine the clinical effects and safety of loxapine compared with placebo, typical and atypical antipsychotics, for treating schizophrenia and related psychoses.
As secondary objectives, we proposed to investigate: i. whether people with schizophrenia described as 'treatment resistant' differed in their response from those whose illnesses were not designated as such; ii. whether people having predominantly positive or negative symptoms of schizophrenia were more responsive to loxapine than those without this designation; and iii. whether people experiencing their first episode of schizophrenia differed in their response from those at a later stage of their illness.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials. Where a trial was described as 'double‐blind', but it was only implied that the study was randomised, we included these trials in a sensitivity analysis. If there was no substantive difference within primary outcomes (see types of outcome measures) when these 'implied randomisation' studies were added, then we included these in the final analysis. If there was a substantive difference, we only used clearly randomised trials and described the results of the sensitivity analysis in the text. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week.
Types of participants
We included people with schizophrenia and other types of schizophrenia‐like psychosis (e.g. schizophreniform and schizoaffective disorders), irrespective of the diagnostic criteria used. There is no clear evidence that the schizophrenia‐like psychoses are caused by fundamentally different disease processes or require different treatment approaches (Carpenter 1994). If possible, people with dementing illnesses, depression and primary problems associated with substance misuse were excluded.
Types of interventions
1. Loxapine: any dose. 2. Placebo. 3. Any other antipsychotic agent, divided into the atypical antipsychotics (amisulpiride, aripiprazole, clozapine, olanzapine, risperidone, quetiapine, sertindole, zotepine) and the typical antipsychotics (chlorpromazine, flupenthixol, fluphenazine, haloperidol, pericyazine, sulpiride, thioridazine, trifluperazine, zuclopenthixol).
Types of outcome measures
1. Death ‐ suicide or natural causes
2. Leaving the study early
3. Clinical response 3.1 Clinically significant response in global state ‐ as defined by each of the studies 3.2 Average score/endpoint or change in global state 3.3 Clinically significant response in mental state ‐ as defined by each of the studies 3.4 Average score/endpoint or change in mental state 3.5 Clinically significant response on positive symptoms ‐ as defined by each of the studies 3.6 Average score/endpoint or change in positive symptoms 3.7 Clinically significant response on negative symptoms‐ as defined by each of the studies 3.8 Average score/endpoint or change in negative symptoms
4. Extrapyramidal adverse effects 4.1 Incidence of use of antiparkinson drugs 4.2 Clinically significant extrapyramidal adverse effects‐ as defined by each of the studies 4.3 Average score/change in extrapyramidal adverse effects
5. Other adverse effects, general and specific
6. Service utilization outcomes 6.1 Hospital admission 6.2 Days in hospital 6.3 Change in hospital status
7. Economic outcomes
8. Quality of life/satisfaction with care for either recipients or carers by directly asking participants 8.1. Significant change as defined by each of the studies 8.2 Average score/change in quality of life/ satisfaction.
We grouped outcomes into the short term (up to six weeks), medium term (seven to 26 weeks) and long term (over 26 weeks).
Search methods for identification of studies
1. Searches for the 2005 and 2007 update
1.1 Electronic searching We searched the Cochrane Schizophrenia Group Trials Register (February 2005, February 2007, and September 12, 2013) using the phrase:
[(*loxapin* or *oxilapin* or *loxpac* or *loxitane* or *desconex* or *cloxazepin* or *amoxapin* or *cl‐71*) in REFERENCE and (*loxapin* or *oxilapin* or *loxpac* or *loxitane* or *desconex* or *cloxazepin* or *amoxapin*) in STUDY]
This register is compiled by systematic searches of major databases, hand searches and conference proceedings (see Group Module).
1.2 Reference searching We inspected the references of all identified studies, included or excluded, for more studies.
2. Searches for past versions of this review (Murphy 2000). 2.1 Electronic searches
2.1.1 We searched Biological Abstracts (January 1980 ‐ February 1999) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or Loxpac or Loxitane or Desconex or oxilapine)]
2.1.2 We searched the Cochrane Library (Issue 1, 1999) using the phrase:
[loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or loxpac or loxitane or desconex or oxilapine]
2.1.3 We searched the Cochrane Schizophrenia Group's Register (January 1999) using the phrase:
[loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or Loxpac or Loxitane or Desconex or oxilapine or #42 =149 or #42=692]
(#42 is the field within this register that contains the intervention code and 149 and 692 is loxapine).
2.1.4 We searched CINAHL (1982 ‐ 1999) using the phrase:
[(explode "Psychotic‐Disorders"/all topical subheadings/all age subheadings or (schizo* in ti,ab) or (psychoti* in ti,ab) or (psychosis in ti,ab) or (psychoses in ti,ab) or ((chronic* or sever*) with mental* with (ill or illness* or disorder*))) and (loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or Loxpac or Loxitane or Desconex or oxilapine)]
2.1.5 We searched EMBASE (BIDS) (January 1980 ‐ February 1999) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or Loxpac or Loxitane or Desconex or oxilapine or explode "LOXAPINE"/ all subheadings)]
2.1.6 We searched MEDLINE (January 1966 ‐ February 1999) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or Loxpac or loxitane or desconex or oxilapine]
2.1.7 We searched PsycLIT (January 1974 ‐ February 1999) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or loxpac or loxitane or desconex or oxilapine)]
2.1.8 We searched LILACS (January 1982 ‐ February 1999) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or loxpac or loxitane or desconex or oxilapine)]
2.1.9 We searched PSYNDEX (January 1977 ‐ February 1999) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and (loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or loxpac or loxitane or desconex or oxilapine)]
2.1.10 We performed specific searches for randomised trials of loxapine in the following databases:
Pharmaceutical databases available on the Dialog Corporation Datastar service (July 1999): ADIS Inpharma; ADIS LMS drug alerts; IDIS Drug File; PharmLine; Pharma Marketing Service.
Pharmaceutical databases available on the Dialog Corporation Dialog service (July 1999): CAB Health (international coverage of health journal and non‐journal material); Conference Papers Index; Derwent Drug File; Dissertation Abstracts; Extramed; Federal Research in Progress (US federally funded research); International Pharmaceutical Abstracts; JICST‐EPLus (Japanese Science and Technology); Mental Health Abstracts; NTIS (national Technical Information Service); Pascal; Pharmaprojects; USP‐DI ‐ Drug information for the health care professional
using the following schizophrenia search terms: schizo$ psychotic$ psychoses psychosis ((chronic$ or sever$) near2 mental$) near2 (ill$ or disorder$)
combined with the following search terms: trial$ random$ (singl$ or doubl$ or trebl$ or tripl$) near (blind$ or mask$) placebo or standard adj treatment study or studies rct$ crossover$ control or controlled or controls
and the phrase:
[and (loxapine or LW‐3170 or SUM‐3170 or CL‐71.563 or loxpac or loxitane or desconex)]
2.1.11 We performed citation searches on key authors.
2.2 Reference lists We searched all references of selected articles for further relevant trials.
2.3 Authors of studies We contacted the first authors of included studies when necessary to clarify data, and asked for additional studies.
2.4 Pharmaceutical company We contacted Wyeth Pharmaceuticals to obtain data on unpublished trials.
Data collection and analysis
1. Selection of trials We independently inspected citations identified in the search. We identified potentially relevant abstracts and ordered full papers to be reassessed for inclusion and methodological quality. We discussed and reported any disagreement. For the update in 2005 and 2007, we (AC and VP) were involved in inspecting the citations from the additional search.
2. Assessment of methodological quality We assessed the methodological quality of included studies using the criteria described in The Cochrane Handbook (Higgins 2005), which is based on the degree of allocation concealment. Poor concealment has been associated with overestimation of treatment effect (Schulz 1995). Category A includes studies in which allocation has been randomised and concealment is explicit. Category B studies are those which have randomised allocation but in which concealment is not explicit. Category C studies are those in which allocation has neither been randomised nor concealed. Only trials that are stated to be randomised (categories A or B of the handbook) were included in this review. The categories are defined below:
A. Low risk of bias (adequate allocation concealment) B. Moderate risk of bias (some doubt about the results) C. High risk of bias (inadequate allocation concealment).
3. Data management 3.1 Data extraction We (MF, BM, JW) independently extracted data from selected trials. When disputes arose we attempted to resolve these by discussion. When this was not possible and further information was necessary to resolve the dilemma, we did not enter data and added the trial to the list of those awaiting assessment. Data from French studies were extracted by PC. For the update in 2005 and 2007, AC and VP extracted data, and (WW and JX) extracted data from Chinese papers, JW data extracted French papers, and EC Brazilian papers.
4. Data analysis 4.1 Binary data For binary outcomes we calculated the relative risk (RR) and its 95% confidence interval (CI) based on the fixed effects model. Relative Risk is more intuitive (Boissel 1999) than odds ratios and odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect. When the overall results were significant we calculated the number needed to treat (NNT) and the number‐ needed‐ to‐ harm (NNH). Where people were lost to follow up at the end of the study, we assumed that they had had a poor outcome, except for the event of death, and once they were randomised they were included in the analysis (intention‐to‐treat /ITT analysis).
Where possible, we made efforts to convert outcome measures to binary data. This can be done by identifying cut off points on rating scales and dividing participants accordingly into "clinically improved" or "not clinically improved". It was generally assumed that if there had been a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005a, Leucht 2005b). It was recognised that for many people, especially those with chronic or severe illness, a less rigorous definition of important improvement (e.g. 25% on the BPRS) would be equally valid. If individual patient data were available, the 50% cut‐off was used for the definition in the case of non‐chronically ill people and 25% for those with chronic illness. If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
4.2 Continuous data 4.2.1 Skewed data: Continuous data on outcomes in trials relevant to mental health issues are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data we applied the following standards to continuous final value endpoint data before inclusion: (a) standard deviations and means were reported in the paper or were obtainable from the authors; (b) when a scale started from zero, the standard deviation, when multiplied by two, should be less than the mean (otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution ‐ Altman 1996); In cases with data that are greater than the mean we entered these into 'Other data' table as skewed data. If a scale starts from a positive value (such as PANSS, which can have values from 30 to 210) the calculation described above in (b) should be modified to take the scale starting point into account. In these cases skewness is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score. We reported non‐normally distributed data (skewed) in the 'other data types' tables.
For change data (mean change from baseline on a rating scale) it is impossible to tell whether data are non‐normally distributed (skewed) or not, unless individual patient data are available. After consulting the ALLSTAT electronic statistics mailing list, we entered change data in RevMan analyses and reported the finding in the text to summarise available information. In doing this, we assumed either that data were not skewed or that the analysis could cope with the unknown degree of skew.
4.2.2 Summary statistic: For continuous outcomes we estimated a weighted mean difference (WMD) between groups based on a fixed effects model.
4.2.3 Valid scales: A wide range of instruments are available to measure mental health outcomes. These instruments vary in quality and many are not valid, and are known to be subject to bias in trials of treatments for schizophrenia (Marshall 2000). Therefore we only included continuous data from rating scales if the measuring instrument had been described in a peer‐reviewed journal.
4.2.4 Endpoint versus change data Where both final endpoint data and change data were available for the same outcome category, we only presented final endpoint data. We acknowledge that by doing this much of the published change data may be excluded, but argue that endpoint data is more clinically relevant and that if change data were to be presented along with endpoint data, it would be given undeserved equal prominence. Authors of studies reporting only change data are being contacted for endpoint figures.
4.3 Cluster trials Studies increasingly employ cluster randomisation (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a unit‐of‐analysis error (Divine 1992) whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes Type I errors (Bland 1997, Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation co‐efficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a design effect. This is calculated using the mean number of participants per cluster (m) and the intraclass correlation co‐efficient (ICC) [Design effect=1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999). If cluster studies had been appropriately analysed taking into account intra‐class correlation coefficients and relevant data documented in the report, we synthesised these with other studies using the generic inverse variance technique.
5. Test for heterogeneity Firstly, we considered all the included studies within any comparison to judge for clinical heterogeneity. Then we visually inspected graphs to investigate the possibility of statistical heterogeneity. We supplemented this by using primarily the I‐squared statistic. This provides an estimate of the percentage of variability due to heterogeneity rather than chance alone. Where the I‐squared estimate was greater than or equal to 50%, we interpreted this as indicating the presence of considerable levels of heterogeneity (Higgins 2003). Where heterogeneity was present, reasons for this were investigated. If it substantially altered the results, we did not summate data, but presented the data separately and investigated reasons for heterogeneity.
6. Addressing publication bias We entered data from all identified and selected trials into a funnel graph (trial effect versus trial size) in an attempt to investigate the likelihood of overt publication bias (Egger 1997).
7. Sensitivity analyses We analysed the effect of including studies with high attrition rates in a sensitivity analysis, and we also investigated, where possible, whether there were differences between: i. people with schizophrenia described as 'treatment‐resistant' and those whose illness was not designated as such; ii. people having predominantly positive or negative symptoms of schizophrenia and those without this designation; and iii. people experiencing their first episode of schizophrenia and those at a later stage of their illness.
8. General Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for loxapine.
Results
Description of studies
For substantive descriptions of studies please see Included and Excluded Studies table.
1. Excluded studies We excluded twenty nine studies, mainly because they were not randomised controlled trials (RCTs), or controlled clinical trials (CCTs).
We excluded two studies, Jones 2006 and Lewis 2006 because they randomised first generation antipsychotics against second generation antipsychotics rather than loxapine versus another treatment. One excluded study, Leone 1982, randomised people who had borderline personality problems and two trials, Lourido 1979 and Versiani 1980, included those who had both a psychosis and an organic illness. Finally, one trial, Serafetinides 1971, reported only electroencephalogram (EEG) readings as the main outcome and another, Maes 1996, only drug plasma levels. No clinically relevant outcomes could be gleaned from these studies. We were unable to extract any usable outcome measures in Bueno 1979 and Rainaut 1975 and they were added to the list of excluded studies. 2. Awaiting assessment No studies await assessment.
3. Ongoing studies We identified no ongoing studies from the search.
4. Included studies We included a total of 41 studies. One included study (Moyano 1975) did not state if they used random allocation but did report using "double blind" methodology. All data within this study suggested that randomisation did occur so it is included. All except two trials were both randomised and double blind. Dubin 1996 and Tuason 1986 used "modified" blinding. Staff administering the injection knew what treatment they were giving, whilst staff undertaking outcome rating were blind to treatment. Three studies seemed to be independent of assistance from the two main companies involved, Cynamid (India) and Lederle (US) (Shopsin 1972, Schiele 1975, Simpson 1976). All other trials either received assistance from industry, either in the form of study drugs, grants, or by, at least, study authorship. Thirteen studies were conducted in China.
4.1 Length of trials Most rapid tranquillisation studies were of very short duration, lasting between 72 hours and 10 days. The remainder were between 21 days and 12 weeks for the typical comparison. One study (Vyas 1980) evaluated participants for six months (Vyas 1980).
4.2 Participants Six studies employed operationalised diagnoses of schizophrenia and six others confirmed diagnoses by having had two psychiatrists work independently. Eight trials classed participants as chronically ill, with a minimum duration of illness being two years but three specifically randomised those who had been admitted for psychiatric emergencies (Dubin 1996, Fruensgaard 1977, Tuason 1986). Studies comparing loxapine in rapid tranquillisation all operationalised their definition of disturbed as '...characterized by symptoms such as agitation, excitement, aggressiveness, delusions and hallucinations'. Dubin 1996 required a Brief Psychiatric Rating Scale (BPRS) inclusion score of six to seven, and Tuason 1984 a score of eight, on the BPRS symptom categories of anxiety, conceptual disorganisation, tension, hostility, suspiciousness, hallucinatory behaviour, uncooperativeness, unusual thought content and excitement. Malik 1980 included participants whose ages ranged between 14 and 19 years but all other studies included people over the age of 18.
4.3 Setting All but one study, Xue 2004, involved people in, or newly admitted to hospital.
4.4 Study size There were three larger studies involving more than 200 patients in each study (Tu 2004, Xue 2004, Huang 1997), otherwise most trials included between 50 and 100 people.
4.5 Interventions Seven studies used a placebo control group (Clark 1972, Clark 1975, Clark 1977, Charalampous 1974, Pool 1976, Selman 1976, Van Der Velde 1975). Nine studies used trifluperazine (Bagadia 1980, Bishop 1970, Clark 1975, Gallant 1971, Kiloh 1976, Malik 1980, Moyano 1975, Seth 1979, Simpson 1976), 14 studies used chlorpromazine, four used thiothixene, three used haloperidol, four used risperidone, two used perphenazine, two clozapine, one quetiapine, one sulpride, and one used thioridazine. Loxapine was given as two relatively low fixed doses in Clark 1977 (100 mg and 50 mg). In trials from India, low doses of loxapine were also compared with low doses of comparator drugs (Dube 1976, Malik 1980, Seth 1979, Vyas 1980). In other studies, loxapine was given in mean doses of about 100 mg/day (range 20 to 150 mg/day). Chlorpromazine was given in doses ranging from 200‐1500 mg/day, trifluoperazine from 5‐60 mg/day, and oral thiothixene 20‐60 mg/day. In trials from China, five used chlorpromazine as a comparator, four used risperidone, two used clozapine, one used quetiapine and another sulpiride and perphenazine.
4.6 Outcomes 4.6.1 Improvement: Definition of improvement consisted of an analysis of variance from the Brief Psychiatric Rating Scale (BPRS), Clinical Global Impression (CGI) and Nurse's Observation Scale for In‐patient Evaluation (NOSIE) in 14 studies. Discharge rates were used in Shopsin 1972; Tuason 1986 gave BPRS endpoint scores. Many studies used BPRS but data were either impossible to extract from graphs or so limited as to render them unusable. Nine studies reported binary results for the CGI. Again many trials employed this simple scale but data were often reported inadequately making outcomes unusable. Only Shopsin 1972 and Simpson 1976 reported discharge from hospital. Information collected on adverse events and adverse effects were not standardised. For example, one study (Selman 1976) reported five separate forms of muscle cramp, yet others ignored this outcome. Satisfaction with care came from a single study (Malik 1980). Of the 13 Chinese studies, common outcome measures were BPRS and CGI scales.
4.6.2 Missing outcomes: As most participants were hospitalised there are no data relating to outcomes such as admission. No study reported usable behaviour data as rated by the commonly used NOSIE scale. Such short trials are also not focusing on clinically relevant outcomes such as quality of daily functioning, 'employed', 'trouble with the police', satisfaction with care. Unfortunately no cost data were reported. No study reassured the reader as regards mortality.
4.6.3 Continuous data: Details of the scales that supplied usable data for this review are shown below. Reasons for exclusion of data from other instruments are given under 'Outcomes' in the 'Included studies' table.
4.6.3.1Global state 4.6.3.1.1 Clinical Global Impression Scale ‐ CGI Scale (Guy 1976) This is used to assess both severity of illness and clinical improvement, by comparing the conditions of the person standardised against other people with the same diagnosis. A seven‐point scoring system is usually used with low scores showing decreased severity and/or overall improvement. Bishop 1970, Clark 1972, Clark 1975, Dube 1976, Fruensgaard 1978, Kiloh 1976, Malik 1980, Moore 1975, Moyano 1975, Pool 1976, Schiele 1975 and Vyas 1980 reported data from this scale.
4.6.3.2 Mental state 4.6.3.2.1 Brief Psychiatric Rating Scale ‐ BPRS (Overall 1962) This is used to assess the severity of abnormal mental state. The original scale has 16 items, but a revised 18‐item scale is commonly used. Each item is defined on a seven‐point scale varying from 'not present' to 'extremely severe', scoring from 0‐6 or 1‐7. Scores can range from 0 to 126, with high scores indicating more severe symptoms. Du 2003, Tuason 1986, Wang 1996, Huang 1997, Xue 2004 reported data from this scale.
4.6.3.2.2 Positive and Negative Syndrome Scale ‐ PANSS (Kay 1986) This schizophrenia scale has 30 items, each of which can be defined on a seven‐point scoring system varying from 1 ‐ absent to 7 ‐ extreme. This scale can be divided into three sub‐scales for measuring the severity of general psychopathology, positive symptoms (PANSS‐P), and negative symptoms (PANSS‐N). A low score indicates lesser severity. Li 2004, Li 2005a, Li 2005b, Liu 2005, Lu 2003, Zhang 2005 reported data from this scale.
4.6.3.3 Adverse effects: general 4.6.3.3.1 Dosage Record and Treatment Emergent Symptoms Scale ‐ DOTES (Guy 1976) This side effect tool seems less of a scale, where the degree and severity of a symptom is recorded, and more of a checklist. The DOTES seems to record the presence or absence of a list of adverse effects. These dichotomous outcomes are then easily and usefully employed within a systematic review.
4.6.3.3.2 Treatment Emergent Symptom Scale ‐TESS (Guy 1976) This checklist assesses a variety of characteristics for each adverse event, including severity, relationship to the drug, temporal characteristics (timing after a dose, duration and pattern during the day), contributing factors, course, and action taken to counteract the effect. Symptoms can be listed a priori or can be recorded as observed by the investigator. Du 2003, Liu 2005, Xue 2004Wang 1996 reported data from this scale.
Risk of bias in included studies
1. Randomisation Two studies, Clark 1975 and Dubin 1996 fall into quality category A (adequate concealment of allocation). The others were category B as it was not clear exactly how randomisation had been conducted. The reader, therefore, is not assured that the introduction of bias was minimised at this crucial stage. It has been shown that poor reporting of randomisation increases the risk of presenting 'significant' outcomes (Chalmers 1983, Schulz 1995).
2. Blinding to interventions and outcomes Only Bishop 1970, and Shopsin 1972 did not clearly describe adequate precautions for blinding of treatment. Two studies in the rapid tranquillisation comparison did reassure the reader that they used staff independent of the study to evaluate the outcomes, although those giving the interventions were not blind (Dubin 1996, Tuason 1986). No included trial tested the adequacy of the blindness of those rating outcomes.
3. Follow‐up In placebo studies 25% of people left the studies early, in comparison to trials where a typical antipsychotic was the control. The latter lost 32% of people before completion. In the rapid tranquillisation studies, 29% left early and in the high dose versus 8% in the low dose group (Clark 1977). In Tuason 1984 and Tuason 1986 participants were discharged if better and not followed up. This added significantly to drop out, and contributed to Tuason 1984 being excluded from analysis within this review other than for the outcome 'leaving the study early'. Kramer 1978 also reported attrition of over 50%, so only data for the outcome of 'leaving the study early' were entered into the review. Tuason 1986 had exactly 50% dropout, and as the review protocol described how to manage studies above and below that threshold, but not exactly 50%, we decided to include it. Many studies actively excluded participants from analysis. Reporting of the explicit reasons for study attrition or exclusion from analyses was poor. Of the 13 Chinese studies, six reported participants dropping out of the study early in both loxapine and control groups.
The barely adequate reporting of randomisation, possible lack of double blindness for these outcomes and unclear reasons for loss to follow up would suggest that all estimates of effect of the experimental intervention may be exaggerated (Moher 1998).
Effects of interventions
1. The search The original search resulted in the inclusion of 28 studies. Following the updates in 2005 and 2007, we have been able to include an additional 13 studies. The total of trials in this review now stands at 41. Of the additional 13 studies in the update, all were from China. Following the update, an additional four studies were placed in the exclusion table, therefore giving a total of 29 excluded studies.
2. COMPARISON 1. LOXAPINE versus PLACEBO
2.1 Leaving the study early/removal from analysis The numbers of people leaving the study early by six weeks were equivocal (n=82, 2 RCTs, RR 0.43 CI 0.2 to 1.1), although, significantly fewer people left early in medium term studies (n=96, 2 RCTs, RR 0.52 CI 0.3 to 1.0, NNT 5 CI 4 to 117).
A few people were removed from analyses, mostly because they did not complete the first two weeks of the trial, but there were no apparent differences between loxapine and placebo (n=151, 3 RCTs, RR 0.74 CI 0.3 to 2.2).
2.2 Global effect ‐ Not improved We found short term data significantly favoured loxapine, with fewer participants not responding to treatment (n=78, 2 RCTs, RR 0.30 CI 0.1 to 0.6, NNT 3 CI 3 to 5) compared with placebo. Medium term data also significantly favoured loxapine (n=71, 2 RCTs, RR 0.54 CI 0.4 to 0.8, NNT 3 CI 3 to 8) with more participants in the placebo group not improving.
No clear differences were demonstrated for the outcome of needing additional antipsychotic/sedative medication both short and medium term data.
2.3 Mental state We found only limited mental state data. For the outcome of increased anxiety/tension, no significant differences were found between loxapine and placebo.
2.4 Adverse effects Those taking loxapine (short term) were more likely to experience an adverse event than those allocated to placebo (n=67, 2 RCTs, RR 1.51 CI 1.0 to 2.2, NNH 4 CI 3 to 48). Medium term data also revealed loxapine users had a worse outcome (n=38, 1 RCT, RR 2.60 CI 1.1 to 6.0, NNH 3 CI 2 to 33).
2.4.1 Anticholinergic effects We found reports of blurred vision were equivocal. We found participants taking placebo were more likely to suffer constipation (n=54, 1 RCT, RR 0.27 CI 0.1 to 0.9, NNH 3 CI 2 to 11). Dry mouth (short term) data were not significantly different.
2.4.2 Cardiovascular problems ECG abnormalities and hypotension are reported in selected small studies and neither outcome showed clear differences between groups. We found the loxapine group were significantly more likely to experience tachycardia compared with placebo (Clark 1972 n=36, RR 9.00 CI 1.3 to 63.9, NNH 3 CI 2 to 68).
2.4.3 Gastrointestinal problems Frequency of abdominal pain favoured loxapine compared with placebo (Van Der Velde 1975, n=54, RR 0.33 CI 0.1 to 0.9), although the high dropout from the placebo group heavily influences this outcome. Similarly, fewer people taking loxapine experienced nausea/vomiting than those on placebo, but the high dropout from the placebo group adds 75% weighting to this outcome (nausea/vomiting: n=119, 3 RCTs, RR 0.41 CI 0.2 to 1.0, NNH 4 CI 2 to 8).
2.4.4 Movement disorders Generally data were very limited but showed that there were no statistically significant difference between loxapine and placebo in terms of akathisia, akinesia, bradykinesia, drooling, dyskinesia, dystonia, muscle cramp, oculogyric crisis, thick speech and tremor. Loxapine resulted in significantly more participants experiencing extrapyramidal symptoms compared with placebo (n=89, RR 9.68 CI 3.2 to 29.6, NNH 2 CI 2 to 7). We found the loxapine group needed significantly more antiparkinsonian medication than the placebo group (n=67, 2 RCTs, RR 4.11 CI 1.6 to 10.8, NNH 3 CI 2 to 15) during short term evaluation. However, 12 weeks data (Clark 1977) were not significantly different (loxapine 8/25, placebo 0/13), although a clear trend is present, favouring placebo. Short term data for rigidity were not significantly different, but 12 week evaluation showed those given placebo had significantly fewer incidences of rigidity (n=74, 2 RCTs, RR 9.78 CI 1.3 to 75.6, NNH 4 CI 2 to 124).
2.4.5 Neurological problems There are limited data on ataxia, dizziness and seizures. Data tend to favour loxapine but, are heavily influenced by high dropout in the placebo. For the outcome weakness we found data significantly favoured loxapine (Van Der Velde 1975, n=54, RR 0.33 CI 0.1 to 0.9), although this is again heavily influenced by high drop out in the placebo group.
2.4.6 Sleep problems We found drowsiness (short term, n=67, 2 RCTs, RR 2.46 CI 1.1 to 5.6, NNH 4 CI 2 to 60) and medium term data (n=74, 2 RCTs, RR 6.75 CI 1.6 to 27.9, NNH 3 CI 2 to 25) significantly favoured placebo. No significant differences were found for insomnia.
2.4.7 Weight changes From limited short and medium term data participants given loxapine did not gain more weight than those allocated to placebo (n=102, 3 RCTs, RR 2.18 CI 0.7 to 6.6), although weight loss did occur more frequently in the loxapine group (n=74, 2 RCTs, RR 0.24 CI 0.1 to 0.9, NNH 5 CI 4 to 27) at the end of 12 weeks evaluation.
2.4.8 Other problems Most results in this category are influenced by the dropout rate of the placebo group in Van Der Velde 1975, and as a consequence results tend to favour loxapine.
3. COMPARISON 2. LOXAPINE versus TYPICAL ANTIPSYCHOTICS
3.1 Leaving the study early / removed from analysis Sixteen percent (100/638) of those taking loxapine and 14% (91/667) allocated typical drugs left before the completion of the trials (short and medium term data) that contribute to this review (n=1305, 16 RCTs, RR 1.11 CI 0.9 to 1.4). Small numbers were removed from the analysis, usually because they did not complete the first few days of the study, but there were no differences between loxapine and other typical drugs (n=793, 11 RCTs, RR 0.99 CI 0.5 to 1.8).
3.2 Global effect We found all global outcomes (not improved, not ready for discharge, needing additional antipsychotic/sedative medication) were not significantly different between loxapine and typical antipsychotics. Both short term (6 RCTs) and medium term (7 RCTs) data showed that in terms of CGI 'not improved' loxapine does not differ from typicals (n=580, RR 0.86 CI 0.7 to 1.1). We found participant's global impression of their illness were also equivocal, and showed no clear differences between loxapine and trifluoperazine.
3.3 Mental state As for the loxapine versus typical antipsychotic comparison, we found dichotomous mental state data measured by BPRS or PANSS, from six studies revealed no significant difference between loxapine and typical antipsychotics (n=915, RR 0.89, CI 0.8 to 1.1). We found BPRS endpoint data favoured loxapine (n=465, 3 RCTs, WMD ‐1.80 CI ‐2.9 to ‐0.7) compared with those given typical antipsychotics. However, data from a single study measuring mental state PANSS scores did not support this finding (Liu 2005, n=80, WMD ‐1.75 CI ‐8.6 to 5.1). Average change scores from the BPRS scale significantly favoured loxapine (n=465, 3 RCTs, WMD ‐1.38 CI ‐2.6 to ‐0.2). Specific mental state outcomes, anxiety, behaviour changes, depression, excitement, restlessness, and violence/aggression came from small studies and no significant differences were found.
3.4 Adverse effects
3.4.1 TESS We found side effects from treatments were significantly lower in the loxapine group, but data are heterogeneous (I squared=76%).
3.4.2 Any adverse effect We found both short and medium term data to be equivocal between loxapine and typical antipsychotics ‐ 'any adverse event'. When analysed together (n=627, 14 RCTs) we found the risk of such an event to be identical in each group (RR 0.96 CI 0.9 to 1.1).
3.4.3 Anticholinergic effects We found no significant differences for the outcomes, blurred vision, constipation, dry mouth and unspecified anticholinergic effects between loxapine and the other active drugs.
3.4.4 Cardiovascular problems Similarly for hypertension, ECG abnormalities, hypotension, syncope, and tachycardia, we found no significant differences between loxapine and the typical antipsychotic groups.
3.4.5 Gastrointestinal We found no significant differences between loxapine and typical antipsychotic drugs for outcomes such as abdominal pain, loss of appetite, diarrhoea, constipation, stomach trouble and nausea and vomiting.
3.4.6 Movement disorders Several symptoms including, agitation, akathisia, dyskinesia, dystonia, extrapyramidal, excess salivation, fixed stare, heavy muscles, muscle cramp, oculogyric crisis, rigidity, thick speech, tremor, akinesia, twisting movement are reported, but we did not find any significant differences between loxapine and typical antipsychotic drugs. About half the people on either loxapine or a typical antipsychotic drug experienced an extrapyramidal effect and 40% needed supplementary anticholinergic medication. For the outcome 'needing antiparkinsonian medication' we found no difference between loxapine and those given typical antipsychotics (n=302, 7 RCTs, RR 1.04 CI 0.8 to 1.3).
3.4.7 Neurological When outcomes such as confusion, ataxia, clumsiness, dizziness and seizures are reported, we found no significant differences between loxapine and typical antipsychotics.
3.4.8 Sleep problems We found some suggestion that loxapine is more sedating than typical drugs (medium term) (n=408, 6 RCTs, RR 1.38 CI 1.0 to 1.9, NNH 13 CI 6 to 244). Short term data, however, did not reveal any significant differences (n=279, 6 RCTs, RR 1.14 CI 0.8 to 1.7). Overall, data on fatigue (n=54, 1 RCT, RR 0.65 CI 0.2 to 2.4) and lethargy (n=108, 2 RCTs, RR 1.69 CI 0.8 to 3.8) revealed no significant differences. For insomnia, we found medium term data favoured the loxapine group (n=189, 2 RCTs, RR 0.30 CI 0.1 to 0.7) compared with those given typical antipsychotics (NNH 6 CI 5 to 13). However, short term outcomes (n=137, 2 RCTs) were equivocal.
3.4.9 Weight changes We found no significant differences between loxapine and typical drugs for outcomes related to weight change.
3.4.10 Others None of the abnormal blood results were serious and there we found no differences in their rate of occurrence between groups. Other limited data for anxiety, headache, ringing in ears and libidinal decrease were equivocal. Only medium term data on rash were significantly different (n=184, 4 RCTs, RR 0.20 CI 0.1 to 0.8, NNH 11 CI 9 to 37). Four week data for rash were not significantly different (n=95, 2 RCTs, RR 0.34 CI 0.1 to 1.3) between loxapine and typical antipsychotics.
4. COMPARISON 3. LOXAPINE IM versus TYPICAL ANTIPSYCHOTICS IM FOR RAPID TRANQUILLISATION
4.1 Withdrawn from analysis or leaving the study early by 72 hours More people allocated to the intramuscular (IM) haloperidol or thiothixene left or were withdrawn early than those given IM loxapine, although the data were not statistically significant (n=115, 2 RCTs, RR 0.58 CI 0.3 to 1.1).
4.2 General effect ‐ tranquillisation When rapid tranquillisation is necessary, data favoured IM loxapine when comparing with IM haloperidol and thiothixene at one hour (not sedated n=145, 3 RCTs, RR 0.62 CI 0.5 to 0.8, but data are heterogeneous I‐squared 77%); homogeneous data at six to 24 hours (n=54, 1 RCT, RR 0.39 CI 0.04 to 3.5), and requiring further sedation (n=115, 2 RCTs, RR 1.21 CI 0.6 to 2.4) did not reveal any significant differences.
4.3 Mental state ‐ BPRS Tuason 1986 reported mean BPRS endpoint scores and the standard deviation of these means. We felt that these standard deviations were in fact standard errors and converted them to deviations. In any event, we found no suggestion of a significant difference between IM loxapine and IM haloperidol.
4.4 Adverse effects No differences were found between IM loxapine and comparator drugs for outcomes such as 'any adverse event' and various movement disorders, for example dyskinesia, dystonia, oculogyric crisis, rigidity tremor, and dizziness. Other physiological effects showed no significant difference between groups.
5. COMPARISON 4. LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE One small (n=25) study compared a higher fixed dose to a lower fixed dose and placebo (Clark 1977). Low dose was considered 50 mg/day, and high, 100 mg/day. Unsurprisingly, because of the limited power of a study of 25 participants no differences are seen in global effect, leaving the study early and a variety of adverse effects. The high dose group did not need any additional sedating drugs than those on the low dose (RR 1.30 CI 0.5 to 3.2) and were no more likely to leave the study early (RR 0.22 CI 0.01 to 4.1) or experience any adverse event (RR 1.55 CI 0.9 to 2.7).
6. COMPARISON 5. LOXAPINE versus ATYPICAL ANTIPSYCHOTICS
6.1 Leaving the study early / removed from analysis Three percent (3/110) of those taking loxapine and 2% (2/108) allocated atypical drugs left before the completion of the trials that contribute to this review (n=218, 3 RCTs, RR 1.26 CI 0.3 to 5.0). Small numbers were removed from the analysis, because they did not complete the first week of the study, but there were no differences between loxapine and atypical antipsychotic drugs (n=68, 1 RCT, RR 5.00 CI 0.3 to 100.4).
6.2 Mental state As for the loxapine versus atypical antipsychotic comparison, mental state data, from BPRS and PANSS scales, revealed no significant difference: (PANSS, 'not improved' n=468, 6 RCTs, RR 1.07 CI 0.8 to 1.5) and (BPRS, 'endpoint score' n=60, 1 RCT, WMD 1.38 CI ‐5.8 to 8.6). Also, we found PANSS endpoint scores to be equivocal (n=408, 5 RCTs, WMD ‐1.13 CI ‐4.1 to 1.8).
6.3 Adverse effects Seven studies (n=528) examined the difference in adverse effects between loxapine and atypical antipsychotics and adverse effects.
6.3.1 TESS We found no significant difference between loxapine and atypical antipsychotics (Du 2003, n=60, WMD 0.05 CI ‐0.04 to 0.1) when treatment emergent side effects were measured.
6.3.2 Movement disorder There were no statistically significant differences between loxapine and atypical antipsychotics for symptoms of tremor (n=123, 2 RCTs, RR 1.08 CI 0.8 to 1.4), increased activity (n=68, 1 RCT, RR 0.11 CI 0.01 to 2.0), agitation (n=87, 1 RCT, RR 0.11 CI 0.01 to 2.0) and akathisia (n=63, 1 RCT, RR 6.79 CI 0.4 to 126.2). However, four studies reported extrapyramidal adverse effects, and we found this occurred more often in the loxapine group compared with atypicals (n=340, RR 2.18 CI 1.6 to 3.1, NNH 5 CI 3 to 9).
6.3.3 Cardiovascular Li 2005b and Li 2005a reported data on ECG abnormalities and we found no significant difference between groups (n=165, 2 RCTs, RR 1.80 CI 0.7 to 4.6).
6.3.4 Sleep Li 2005b and Li 2005a also reported on insomnia, and we found that those given the atypical risperidone were significantly more likely to experience insomnia compared with the loxapine group (n=165, RR 0.18 CI 0.04 to 0.8, NNH 9 CI 8 to 33). Also, we found the risperidone group had significantly greater 'sleep disturbance' compared with the loxapine group (Wang 2005b, n=63, RR 0.19 CI 0.1 to 0.8, NNH 4 CI 4 to 17).
6.3.5 Other adverse effects We found data from Wang 2005b showed no significant differences between the loxapine and atypical (risperidone) group regarding anxiety (n=63, RR 0.97, CI 0.3 to 3.5), dermatitis (n=63, RR 0.32 CI 0.01 to 7.7), amenorrhea (n=63, RR 0.32 CI 0.01 to 7.7) and enuresis (n=63, RR 0.32 CI 0.01 to 7.7). Both Lu 2003 and Wang 2005b reported on leucopoenia and we found that the atypical group (clozapine) had a statistically higher proportion of leucopoenia compared with the loxapine group (n=185, RR 0.12 CI 0.02 to 1.0, NNH 15 CI 14 to 264).
7. Sensitivity analysis We were unable to undertake the proposed sensitivity analysis for people with schizophrenia described as 'treatment resistant', people having predominantly positive or negative symptoms of schizophrenia and people experiencing their first episode of schizophrenia.
8. Funnel plot for publication bias With only two studies in the placebo comparison, three in the rapid tranquillisation studies and one in the high versus low dose comparison, it was impossible to undertake the proposed funnel plot for publication bias. Within the loxapine versus typical antipsychotics, for the outcomes of 'Global effect: 1. Not improved' and 'Leaving the study early' funnel plots were possible and there is no suggestion of asymmetry.
Discussion
1. Generalisability Eighteen of the included studies were set in the USA and randomised people mostly without operationally diagnosed disorders uncomplicated by co‐morbidity or ill health. Four studies were from India (Dube 1976, Malik 1980, Seth 1979, Vyas 1980) and used relatively low doses of drugs. Dube 1976 reported no adverse effects what so ever, whilst Malik 1980 recorded that every participant (all adolescents) has experienced adverse effects. Following the update in 2005 and 2007, 13 studies were added from China, all of which used the Chinese Classification of Mental Disorders (CCMD) as a means of diagnosing disorders. With limited operationalisation of diagnosis, varied groups of participants, and intervention regimens, the results of this review should be more generalisable than if very strict definitions and care regimens had been employed (Elwood 1982).
2. Limited data The collection and quality of reporting of data were disappointing. Most trials report only three to 12 week outcomes, with the exception of Vyas 1980, a six month study. No studies reported on economic outcomes or quality of life and only one gave data on satisfaction with care (Malik 1980). Only two of the included studies reported on discharge from hospital (Shopsin 1972, Simpson 1976). Many included trials reported 'efficacy' as an analysis of variance from the BPRS, CGI and NOSIE endpoint scores, or a maximum percentage improvement, and therefore much valuable data were unusable. Frequently mean scores were reported without a standard deviation or standard error, rendering data useless for a quantitative review such as this. The removal of people from analysis within a trial can rarely be justified (Hollis 1999). We have reported the rate of attrition and the rate of withdrawal from the analyses where this has happened. It is easy to criticise studies from the 1970s, by 2007 standards and knowledge, but this review is one where poor reporting of outcomes does seem to do a disservice to all concerned.
3. COMPARISON 1. LOXAPINE versus PLACEBO
3.1 Leaving the study early That fewer people in the loxapine group (medium term) left the study early than those on placebo may suggest that loxapine had a favourable effect on mental state and behaviour. Alternatively, it is possible that the sedation that loxapine affords may have made study attrition less likely. Even in the control group only about 30% of people left the studies early, which is considerably less than many more recent trials.
3.2 Global improvement People taking loxapine were more likely to have a global improvement than those allocated placebo (NNT 3); about 60% of people taking loxapine improved. It is surprising, however, that the need for additional antipsychotic/sedative medication did not mirror this result. It may be that those within these studies were not severe enough to be likely to ever need additional sedation.
3.3 Mental state Mental state data relating to loxapine versus placebo is almost non‐existent. The outcome of increased anxiety/tension is reported and data is in favour of those allocated to loxapine but, especially in a drug introduced into use over 20 years ago, more data would have been expected.
3.4 Adverse effects Those allocated loxapine were more likely to experience 'any adverse event' than those randomised to placebo (NNH 3). In any event, it is difficult to know how to interpret 'any adverse event' but the suggestion is that loxapine is an active compound. Limited data (Clark 1972, n=36) suggests that loxapine may cause tachycardia (NNH 3), although ECG data did not indicate loxapine disrupted cardio‐function over a 12 week period. Because of high dropout from the placebo group on one dominant study (Van Der Velde 1975), it is difficult to comment on the results favouring loxapine over placebo. It is possible that loxapine may have caused movement disorders and these outcomes would have been masked by the increased use of antiparkinsonian medication in the loxapine group.
4. COMPARISON 2. LOXAPINE versus TYPICAL ANTIPSYCHOTICS These results were characterised by the similarity in outcomes between the various loxapine doses and comparison drugs.
4.1 Leaving the study early / removal from analysis The loxapine group were no different to typical drugs for causing study attrition. About 16% of people left these studies early and again, this is considerably better than would be expected in more recent studies. Most trials were conducted in the 1970s and 1980s. Three, however, also with very low attrition, were undertaken between 2004 and 2005. It is possible that those designing studies could learn from these trials. 4.2 Global effect Whilst the CGI data, a measure of global impairment, were used within all studies, none reported it as a continuous outcome. Nine included studies reported a binary result based on the CGI and we found no suggestion of a difference between loxapine and the drugs of comparison. Global effects were also reported as readiness for discharge (Shopsin 1972, Simpson 1976), needing additional antipsychotic or sedative medication (Clark 1975, Moore 1975) and participant's impression of change (Malik 1980). Although data for these latter outcomes are based on very small numbers they are consistent with the dichotomised CGI. Loxapine is not clearly less or more effective than typical antipsychotics. Overall, in the short to medium term, over 70% of people given loxapine do experience global improvement.
4.3 Mental state Four studies (Huang 1997, Tu 2004, Wang 1996, Xue 2004) using dichotomous mental state data measured by BPRS, showed no statistically significant difference between loxapine and typical antipsychotics. Of these four studies, three also used continuous data measured by BPRS, in addition to reporting dichotomous data (Huang 1997, Wang 1996, Xue 2004). The continuous data measured by Huang 1997 and Tu 2004 revealed no statistically significant difference between loxapine and typical antipsychotics. However, the dichotomous and continuous mental state data measured by Xue 2004 showed contradictory results, with the dichotomous data showing no statistical significance and the continuous data favouring loxapine in a statistically significant manner. This was, perhaps, a chance result. Again, as for the global outcomes, loxapine seems little different to the typical group of antipsychotic drugs.
4.4 Adverse effects Comment must be made about what appears to be fairly unsystematic approach to collecting data on adverse effects, with only five studies reporting on the tools used for collecting data (Kramer 1978, Liu 2005, Shopsin 1972, Wang 1996, Xue 2004). Other studies are suspected to have used a treatment emergent symptoms checklist, but they do not make this clear. Fourteen studies provided data on the outcome of 'any adverse event'. Although it is difficult to know what this outcome means in clinical terms, it is reassuring to know that loxapine seemed no different to the other typical drugs; about two thirds of both groups experience an adverse event. The limited and patchy adverse event data are, nevertheless, consistent. Loxapine is not clearly different from other drugs in its adverse effect profile. It does not clearly cause less movement disorders or need for anticholinergic medication than the typical drugs (˜50%). Sedation revealed no differences in short term evaluation, but medium term data did suggest that loxapine is more sedating (NNH 13); more data is needed however to support this finding, as this was heavily influenced by one study (Zhang 2005). Two studies (Kiloh 1976, Zhang 2005) indicate that loxapine causes less insomnia compared with typicals. Measures of weight change came mostly from small studies conducted up to no more than 12 weeks, and we found no significant differences between loxapine and typical antipsychotics. Other adverse effects also proved equivocal with the exception of skin rash which occurred significantly more in the loxapine group. However, the data (4 RCTs) were influenced by the Clark 1972 study, otherwise no difference in rates of rash would have occurred, and this is possibly a spurious finding.
4.5 Missing outcomes In such short term explanatory studies data on service utilisation, economic outcomes, quality of life and satisfaction with care are commonly not recorded. The long term effects of loxapine are currently unknown. This is no different than other more recently formulated drugs but loxapine has been used for nearly a quarter of a century with minimal long term data.
5. COMPARISON 3. LOXAPINE versus TYPICAL ANTIPSYCHOTICS FOR RAPID TRANQUILLISATION All three studies in this comparison started with a rapid tranquillisation phase followed by an oral extension stage. Dubin 1996 did not report any data other than tranquillisation and adverse effects, Fruensgaard 1977 did not report on the extension phase, and Tuason 1986 had 50% loss to follow up.
5.1 Withdrawn from analysis or leaving the study early by 72 hours Even if there is no clear difference in levels of sedation and mental state outcomes, fewer participants given loxapine were withdrawn from the study than those given typical antipsychotics, which may suggest that loxapine may be more acceptable to clinicians, if not patients, than either haloperidol or thiothixine.
5.2 General effect ‐ tranquillisation / sedation Data are poor and it is unclear why they are heterogeneous. In any effect there is no clear differences demonstrated between IM loxapine and haloperidol or thiothixene.
5.3 Mental state We are waiting confirmation that the trialists (Tuason 1986) did erroneously report standard errors as deviations but, in any event, there is no difference between IM loxapine and IM haloperidol. This probably is to be expected at such a short follow up period of 72 hours, where sedation/ tranquillisation would have been expected but not necessarily an antipsychotic effect. The other two studies used the BPRS but did not report data.
5.4 Adverse effects From the small studies no statistically significant difference were found between IM loxapine and haloperidol or thiothixene regarding adverse effects. Perhaps, had there been larger trials conducted over a longer period differences may have become apparent.
6. COMPARISON 4. LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE Only 25 people have been randomised to high or low dose loxapine (Clark 1977). Such a small study would be likely to miss real differences so all data should be considered hypothesis‐generating. There were no differences at all for global effect, study attrition and adverse effects. Perhaps these data do support generation of the hypothesis that there is no real difference in terms of effect between high and low dose loxapine.
7. COMPARISON 5. LOXAPINE versus ATYPICAL ANTIPSYCHOTICS Four studies used risperidone, two clozapine, and one used quetiapine as comparator.
7.1 Leaving the study early / removed from analysis There were no differences between loxapine and atypical drugs. However, very few people left these recent three Chinese studies (˜3%). This may not simply reflect good trial design, but also a difference in health care culture between China and the rest of the world. However, 1.3 billion people live in this large country.
7.2 Mental state There were no statistical differences found between loxapine and atypical antipsychotics in each of the seven studies all from China. Four hundred and sixty eight people have entered this comparison and still there is no suggestion of a difference between this inexpensive drug and risperidone, clozapine, or quetiapine.
7.3 Adverse effects Seven studies (n=528) examined the difference in adverse effects between loxapine and atypical antipsychotics.
There were no statistically significant differences between loxapine and atypical antipsychotics for symptoms of tremor, increased activity, and akathisia. However, we did find that extrapyramidal adverse effects in four studies (Li 2005a, Li 2005b, Lu 2003, Wang 2005b) were more frequent and of statistical significance, in the loxapine treatment group compared with the atypicals risperidone and clozapine. This would fit with the impression that loxapine is a valuable antipsychotic but with an adverse effect profile not much different from the older generation of antipsychotic drugs.
ECG abnormalities were more common in the loxapine group, but not significantly different compared with atypical risperidone. Also, we found in the studies by Li 2005a and Li 2005b that those given risperidone were more likely to experience insomnia compared with the loxapine, while in the Wang 2005b study the risperidone group were significantly associated with 'sleep disturbance' compared with those given loxapine. There were no significant differences found between loxapine and quetiapine regarding anxiety, dermatitis, amenorrhea and enuresis. Two studies (Lu 2003; Wang 2005b) reported incidences of leucopoenia in the atypical clozapine group, and we found this to be statistically significant compared with loxapine.
7.4 Missing outcomes In such short term explanatory studies data on service utilisation, economic outcomes, quality of life and satisfaction with care are commonly not recorded. The medium or long term effects of loxapine are currently unknown. This is no different than other more recently formulated drugs but loxapine has been used for nearly a quarter of a century with minimal long term data.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia Loxapine is an antipsychotic drug. For those whose illness results in situations where rapid tranquillisation is needed, loxapine is an option. When loxapine is compared with typical and atypical drugs, most data relate to six to 12 weeks, with very little being known beyond that period. Limited data suggest that in terms of global outcomes or mental state ratings, loxapine is as effective as these drugs. Loxapine does appear to produce a similar degree of adverse effects compared with typicals and atypical drugs but loxapine may cause more extrapyramidal adverse effects than some atypicals.
2. For clinicians Very limited trial data suggest that loxapine has very similar effects to typical and atypical drugs and its intramuscular preparation may be as acutely sedating as IM haloperidol or thiothixene. It may be under‐used for the care of those with schizophrenia and be a real option when idiosyncratic intolerance to other compounds has occurred.
3. For managers or policy makers There are almost no data on service utilisation for loxapine. This is probably a function of the studies having been undertaken when there was less emphasis on these outcomes.
Implications for research.
1. General All except one study considerably preceded the first CONSORT statement (Begg 1996), so quality of data reporting might be expected to be lower than at present. However, clear and full reporting of all outcomes that were in fact measured would have resulted in this review being more informative. Denominator data were not always clearly presented. Authors should present raw data rather than in graphical format. If p‐values are used, the exact value and test should be reported.
2. Specific Loxapine seems to be a potent antipsychotic drug. In the context of all other research priorities it is hard to know if further trials of loxapine are possible. Certainly, the use of loxapine in the acute situation may be worth investigating within the context of a well planned, conducted and reported randomised controlled trial. This should only take place in areas where loxapine is used in this way and, at present, we know of no places where this happens. A reasonable comparison intervention would be haloperidol plus promethazine (Table 1).
1. Suggested design for future study.
| Methods | Participants | Interventions | Outcomes | Notes |
| Allocation: randomised, block, fully explicit description.. Duration: 2 weeks. | Diagnosis: not prestipulated, acute agression thought to be due to serious mental illness. N=300.* Age: adults. Sex: both. | 1. Loxapine: dose 25‐50 mg/6‐12 hours IM. N=150. 2. Haloperidol + promethazine: dose 5‐10mg, 25‐50 mg IM stat. N=150. | Tranquil by 20, 40, 60, 90, 180 mins. Need for further medication. Need to call doctor again. Time in restraints. General functioning. Behaviour. Symptoms. Adverse events. Satisfaction with care. Economic outcomes. | * powered to be able to identify a difference of ˜20% between groups for primary outcome with adequate degree of certainty. |
Feedback
Included studies
Summary
Category: Included studies This review does not include Clark 1972 which is included in the Cochrane Library Chlorpromazine review. In addition to a chlorpromazine and placebo arm, this study has a loxapine arm.
Reply
The reviewers would like to thank the commentator for drawing our attention to the omission of the study, which had been identified in the original electronic search. The study will be included in the next update of the review and placed in the 'awaiting assessment' section in the interim.
Contributors
Comment received from Paul Waraich, University of British Columbia,Canada, November 2000. Reply by Mark Fenton, Scarborough, UK, November 2000.
What's new
| Date | Event | Description |
|---|---|---|
| 14 October 2015 | Amended | The contributions of a previous review author, Jo Wood, have been moved to Acknowledgements. |
History
Protocol first published: Issue 3, 1999 Review first published: Issue 1, 2000
| Date | Event | Description |
|---|---|---|
| 2 December 2013 | Amended | 32 new references from search (September 12, 2013) were added to 'Classification pending references' section of the review. |
| 26 April 2008 | Amended | Converted to new review format. |
| 6 August 2007 | New citation required and conclusions have changed | Substantive amendment |
Notes
Cochrane Schizophrenia Group internal peer review complete (see Module). External peer review scheduled.
Acknowledgements
Thanks to Julie Glanvile from the Centre for Reviews and Dissemination, University of York who undertook all electronic searches of the original search in 1999 and to Judy Wright from the Cochrane Schizophrenia group who undertook electronic searches for the 2007 update. Thanks to Evandro Coutinho who provided translation for a Brazilian study and Tessa Grant who provided translation for a French study. Thanks to Professor Clive Adams and Ajit Kumar for their support.
Jo Wood helped select studies, extract data, summate data and produce the original report in 2008. She has not been involved in the writing of subsequent versions. We would like to thank her for these previous contributions.
Data and analyses
Comparison 1. LOXAPINE versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early ‐ any reason | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 short term ‐ up to 6 weeks | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.16, 1.13] |
| 1.2 medium term ‐ by 12 weeks | 2 | 96 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.28, 0.98] |
| 2 Removed from analysis | 3 | 151 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.26, 2.17] |
| 3 Global effect: 1. Not improved | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 short term ‐ up to 4 weeks | 2 | 78 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.14, 0.63] |
| 3.2 medium term ‐ up to 12 weeks | 2 | 71 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.36, 0.82] |
| 4 Global effect: 2. Needing additional antipsychotic/sedative drugs | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 short term ‐ up to 6 weeks | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.60, 3.20] |
| 4.2 medium term‐ 7 ‐ 26 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.36, 1.13] |
| 5 Mental state: Specific symptoms ‐ anxiety/tension | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 short term ‐ up to 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.14, 1.08] |
| 6 Adverse effects: 1. Any adverse event | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 up to 6 weeks | 2 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.51 [1.04, 2.17] |
| 6.2 by 12 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.6 [1.12, 6.01] |
| 7 Adverse effects: 2. Anticholinergic effects ‐ specific symptoms | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 blurred vision ‐ 12 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.77 [0.21, 67.89] |
| 7.2 constipation ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.09, 0.85] |
| 7.3 constipation ‐ 12 weeks | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.4 dry mouth ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.14, 1.08] |
| 8 Adverse effects: 3. Cardiovascular problems | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 ECG abnormalites ‐ 6 weeks | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.73 [0.54, 5.59] |
| 8.2 ECG abnormalities ‐ 12 weeks | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.46, 2.32] |
| 8.3 blood pressure ‐ hypertension ‐ 12 weeks | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 69.09] |
| 8.4 blood pressure ‐ hypotension, 12 weeks | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.77 [0.21, 67.89] |
| 8.5 tachycardia ‐ 12 weeks | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [1.27, 63.89] |
| 9 Adverse effects: 4. Gastrointestinal problems | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 abdominal pain ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.12, 0.89] |
| 9.2 nausea or vomiting ‐ short term, up to 6 weeks | 3 | 119 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.41 [0.18, 0.97] |
| 10 Adverse effects: 5. Movement disorders | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 akathisia ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.31, 1.50] |
| 10.2 akathisia ‐ 12 weeks | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.65 [0.57, 38.23] |
| 10.3 akinesia ‐ 12 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.77 [0.21, 67.89] |
| 10.4 bradykinesia ‐ 12 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.85 [0.28, 83.66] |
| 10.5 drooling ‐ 12 weeks | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.43 [0.41, 29.03] |
| 10.6 dyskinesia ‐ 4 weeks | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.47 [0.44, 27.24] |
| 10.7 dyskinesia ‐ 12 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.77 [0.21, 67.89] |
| 10.8 dystonia ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.20, 1.22] |
| 10.9 dystonia ‐ 12 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.77 [0.21, 67.89] |
| 10.10 extrapyramidal symptoms ‐ 4 weeks, unspecified | 2 | 89 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.68 [3.17, 29.55] |
| 10.11 muscle cramp ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.14, 1.08] |
| 10.12 needing additional anticholinergic medication ‐ 4 weeks | 2 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.11 [1.56, 10.83] |
| 10.13 needing additional anticholinergic medication ‐ 12 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.15 [0.57, 147.14] |
| 10.14 oculogyric crisis ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.20, 1.22] |
| 10.15 rigidity ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.53, 1.84] |
| 10.16 rigidity ‐ 12 weeks | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.78 [1.27, 75.55] |
| 10.17 thick speech ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.25, 1.36] |
| 10.18 tremor ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.25, 1.36] |
| 10.19 tremor ‐ 12 weeks | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.0 [0.39, 126.48] |
| 11 Adverse effects: 6. Neurological | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 ataxia ‐ up to 6 weeks | 2 | 93 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.14, 1.03] |
| 11.2 dizziness ‐ up to 6 weeks | 2 | 93 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.21, 1.00] |
| 11.3 weakness ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.12, 0.89] |
| 11.4 seizures ‐ 12 weeks | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.68] |
| 12 Adverse effects: 7. Sleep problems | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 drowsiness ‐ 4 weeks | 2 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.46 [1.09, 5.56] |
| 12.2 drowsiness ‐ 12 weeks | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.75 [1.63, 27.89] |
| 12.3 insomnia ‐ up to 6 weeks | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.25, 1.39] |
| 13 Adverse effects: 8. Weight changes | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 weight increase ‐ 4 weeks | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.11, 2.94] |
| 13.2 weight increase ‐ 12 weeks | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.65 [0.91, 48.77] |
| 13.3 weight loss ‐ 4 weeks | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.11, 2.94] |
| 13.4 weight loss ‐ 12 weeks | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.07, 0.87] |
| 14 Adverse effects: 9.Others | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 abnormal blood results ‐ up to 6 weeks | 2 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.32, 1.14] |
| 14.2 abnormal blood results ‐ 12 weeks | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.53 [0.76, 3.07] |
| 14.3 eye pigments ‐ 12 weeks | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 14.79] |
| 14.4 headache ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.11, 0.75] |
| 14.5 lactation ‐ 12 weeks | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.6 libido ‐ decrease ‐ 4 weeks | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.7 ringing in ears ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.14, 1.08] |
| 14.8 skin problems ‐ rash, up to 6 weeks | 2 | 93 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.09, 0.85] |
| 14.9 skin problems ‐ rash, 12 weeks | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
1.1. Analysis.
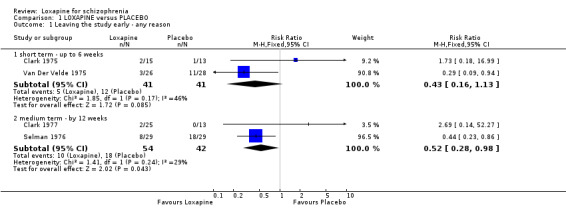
Comparison 1 LOXAPINE versus PLACEBO, Outcome 1 Leaving the study early ‐ any reason.
1.2. Analysis.
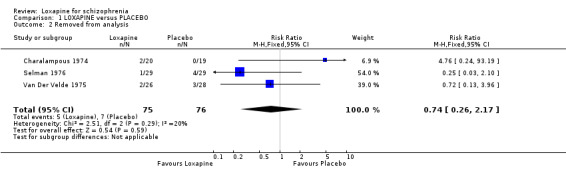
Comparison 1 LOXAPINE versus PLACEBO, Outcome 2 Removed from analysis.
1.3. Analysis.
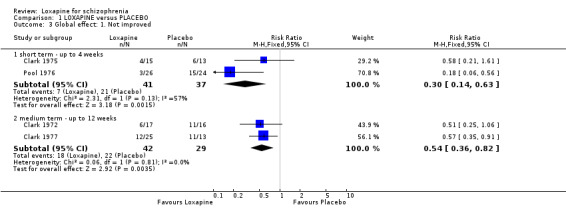
Comparison 1 LOXAPINE versus PLACEBO, Outcome 3 Global effect: 1. Not improved.
1.4. Analysis.
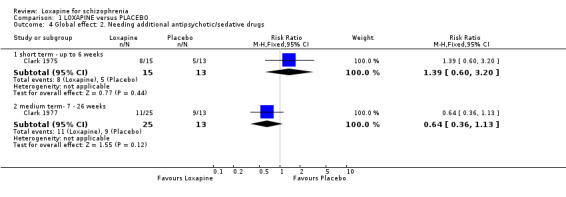
Comparison 1 LOXAPINE versus PLACEBO, Outcome 4 Global effect: 2. Needing additional antipsychotic/sedative drugs.
1.5. Analysis.
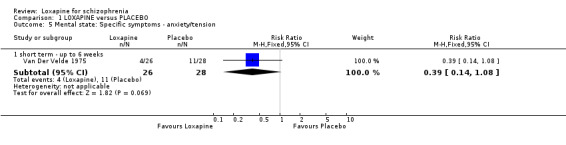
Comparison 1 LOXAPINE versus PLACEBO, Outcome 5 Mental state: Specific symptoms ‐ anxiety/tension.
1.6. Analysis.
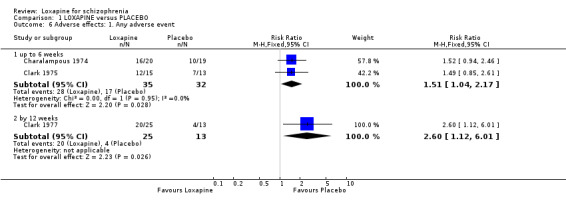
Comparison 1 LOXAPINE versus PLACEBO, Outcome 6 Adverse effects: 1. Any adverse event.
1.7. Analysis.
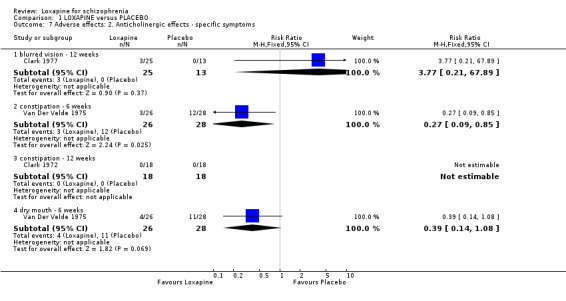
Comparison 1 LOXAPINE versus PLACEBO, Outcome 7 Adverse effects: 2. Anticholinergic effects ‐ specific symptoms.
1.8. Analysis.
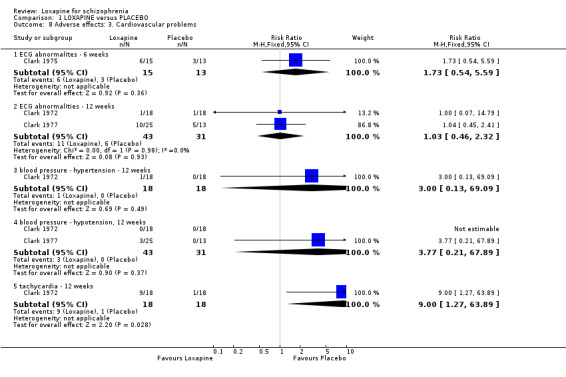
Comparison 1 LOXAPINE versus PLACEBO, Outcome 8 Adverse effects: 3. Cardiovascular problems.
1.9. Analysis.
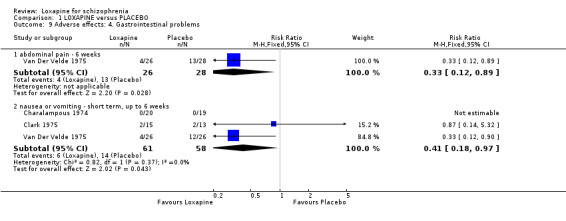
Comparison 1 LOXAPINE versus PLACEBO, Outcome 9 Adverse effects: 4. Gastrointestinal problems.
1.10. Analysis.
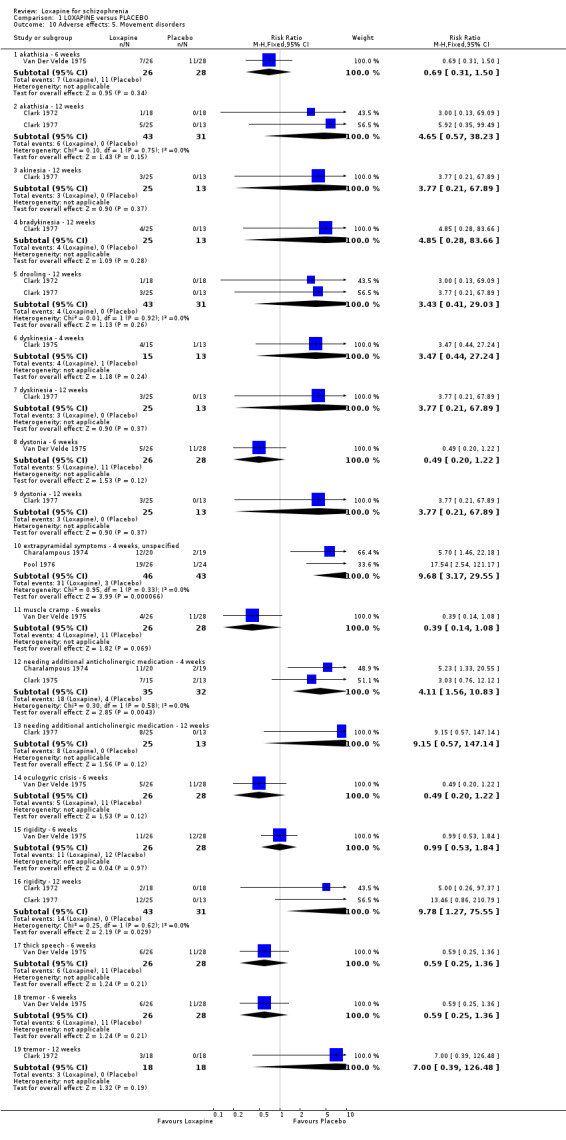
Comparison 1 LOXAPINE versus PLACEBO, Outcome 10 Adverse effects: 5. Movement disorders.
1.11. Analysis.
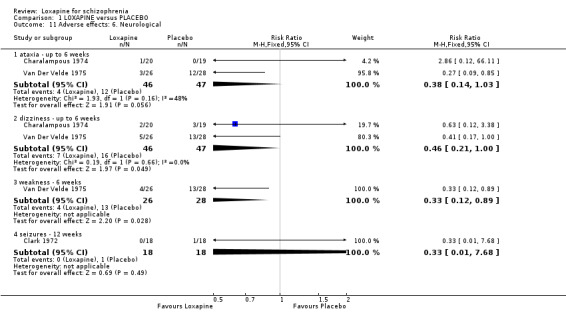
Comparison 1 LOXAPINE versus PLACEBO, Outcome 11 Adverse effects: 6. Neurological.
1.12. Analysis.
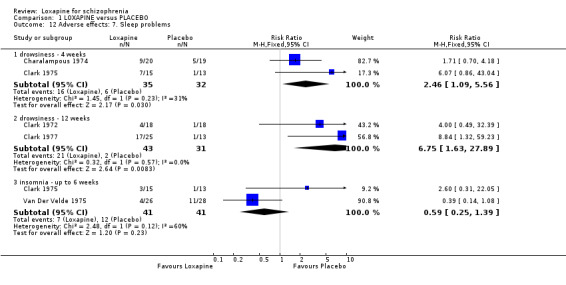
Comparison 1 LOXAPINE versus PLACEBO, Outcome 12 Adverse effects: 7. Sleep problems.
1.13. Analysis.
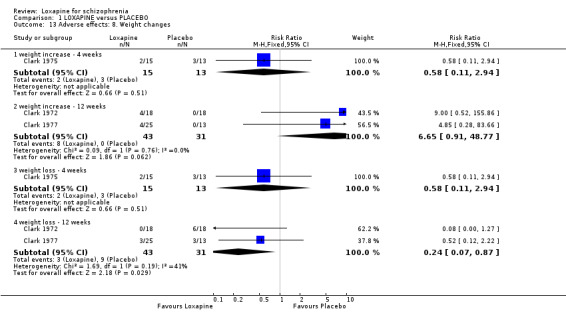
Comparison 1 LOXAPINE versus PLACEBO, Outcome 13 Adverse effects: 8. Weight changes.
1.14. Analysis.
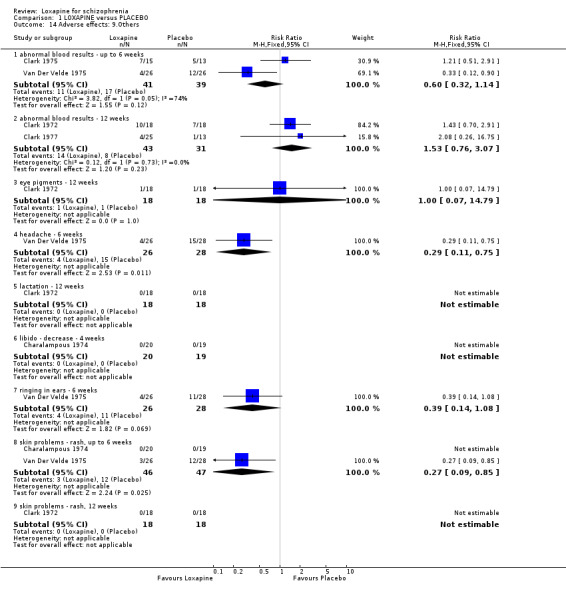
Comparison 1 LOXAPINE versus PLACEBO, Outcome 14 Adverse effects: 9.Others.
Comparison 2. LOXAPINE versus TYPICAL ANTIPSYCHOTICS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early ‐ any reason | 16 | 1305 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.89, 1.38] |
| 1.1 short term ‐ up to 6 weeks | 7 | 380 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.84, 1.34] |
| 1.2 medium term ‐ 7 ‐ 26 weeks | 9 | 925 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.76, 1.97] |
| 2 Removed from analysis | 11 | 793 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.54, 1.79] |
| 3 Global effect: 1. Not improved (CGI) | 13 | 580 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.68, 1.09] |
| 3.1 short term ‐ up to 6 weeks | 6 | 294 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.58, 1.21] |
| 3.2 medium term ‐ 7 ‐ 26 weeks | 7 | 286 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.65, 1.19] |
| 4 Global effect: 2. Not ready for discharge ‐ up to 4 weeks | 2 | 73 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.87, 1.60] |
| 5 Global effect: 3. Needing additional antipsychotic/sedative drugs ‐ up to 6 weeks | 2 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.62, 2.12] |
| 6 Global effect: 4. Participant rating of illness | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 did not feel better ‐ 4 weeks | 2 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.69, 2.21] |
| 6.2 much, or very much better ‐ 4 weeks | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.73, 1.37] |
| 6.3 worse ‐ 4 weeks | 2 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.04, 3.01] |
| 6.4 would not prefer to stay on medication ‐ 4 weeks | 2 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.78, 1.81] |
| 6.5 prefer another medication ‐ 4 weeks | 2 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.78, 1.81] |
| 7 Mental state: 1a. General ‐ not improved, by 8 weeks (BPRS/PANSS) | 6 | 915 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.76, 1.05] |
| 8 Mental state: 1b. General ‐ average endpoint score, by 8 weeks (BPRS, high score=worse) | 3 | 465 | Mean Difference (IV, Fixed, 95% CI) | ‐1.80 [‐2.92, ‐0.67] |
| 9 Mental state: 1c. General ‐ average endpoint score, by 8 weeks (PANSS, high score=worse) | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | ‐1.75 [‐8.60, 5.10] |
| 10 Mental state: 1d. General ‐ average change score (BPRS, high score=worse) | 3 | 465 | Mean Difference (IV, Fixed, 95% CI) | ‐1.38 [‐2.60, ‐0.16] |
| 11 Mental state: 2. Specific | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 anxiety ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.35, 5.81] |
| 11.2 anxiety ‐ 7 ‐ 26 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.58] |
| 11.3 behaviour changes (not specified), by 12 weeks | 2 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.59, 1.50] |
| 11.4 depression ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.05, 5.19] |
| 11.5 excitement ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.19, 20.77] |
| 11.6 restlessness ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.38, 4.16] |
| 11.7 restlessness ‐ 7 ‐ 26 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.58] |
| 11.8 violence or aggression ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.19, 20.77] |
| 12 Adverse effects: 1. Average change score, by 8 weeks (TESS, high score=worse) | 3 | 340 | Mean Difference (IV, Fixed, 95% CI) | ‐0.07 [‐0.11, ‐0.03] |
| 13 Adverse effects: 2. Any adverse event | 14 | 627 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.88, 1.06] |
| 13.1 up to 6 weeks | 7 | 318 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.91, 1.13] |
| 13.2 7 ‐ 26 weeks | 7 | 309 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.76, 1.05] |
| 14 Adverse effects: 3. Anticholinergic effects ‐ specific symptoms | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 blurred vision ‐ up to 6 weeks | 4 | 187 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.63, 1.39] |
| 14.2 blurred vision ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.52, 4.79] |
| 14.3 constipation ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.92, 1.40] |
| 14.4 constipation ‐ 7 ‐ 26 weeks | 3 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.48, 2.96] |
| 14.5 dry mouth ‐ up to 6 weeks | 3 | 151 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.76, 2.39] |
| 14.6 dry mouth ‐ 7 ‐ 26 weeks | 3 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.60, 2.26] |
| 14.7 nasal congestion ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.11, 3.32] |
| 15 Adverse effects: 4. Cardiovascular problems | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 hypertension ‐ 12 weeks | 1 | 37 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.05, 5.33] |
| 15.2 ECG abnormalites ‐ up to 4 weeks | 2 | 76 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.37, 1.90] |
| 15.3 ECG abnormalities ‐ up to 12 weeks | 4 | 456 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.11, 1.47] |
| 15.4 hypotension ‐ 7 ‐ 26 weeks | 5 | 280 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.46, 1.52] |
| 15.5 syncope ‐ 8 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.7 [0.30, 24.43] |
| 15.6 tachycardia ‐ 7 to 26 weeks | 6 | 365 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.78, 1.47] |
| 15.7 unspecified ‐ 12 weeks | 2 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.45, 1.54] |
| 16 Adverse effects: 5. Gastrointestinal problems | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 abdominal pain ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.35, 5.81] |
| 16.2 appetite loss ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.49, 1.70] |
| 16.3 constipation ‐ 8 weeks | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.25, 99.95] |
| 16.4 diarrhoea ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.05, 5.19] |
| 16.5 diarrhoea ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.01, 3.60] |
| 16.6 nausea or vomiting ‐ 4 weeks | 2 | 95 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.15, 3.60] |
| 16.7 nausea or vomiting ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.8 [0.17, 18.75] |
| 16.8 stomach trouble ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.12, 3.68] |
| 17 Adverse effects: 6. Movement disorders | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 agitation ‐ 8 weeks | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.01, 3.18] |
| 17.2 akathisia ‐ up to 6 weeks | 3 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.52, 2.19] |
| 17.3 akathisia ‐ up to 12 weeks | 3 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.80, 1.88] |
| 17.4 akinesia ‐ 4 weeks | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.51, 158.85] |
| 17.5 dyskinesia ‐ 4 weeks | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.29, 3.03] |
| 17.6 dystonia ‐ up to 6 weeks | 4 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 [0.91, 3.54] |
| 17.7 dystonia ‐ up to 12 weeks | 2 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.59, 2.42] |
| 17.8 extrapyramidial ‐ up to 4 weeks | 4 | 169 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.77, 1.23] |
| 17.9 extrapyramidal ‐ up to 12 weeks | 4 | 314 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.85, 1.38] |
| 17.10 excess salivation ‐ 4 weeks | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.87, 2.31] |
| 17.11 excess salivation ‐ 7 ‐ 26 weeks | 3 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.39, 2.19] |
| 17.12 fixed stare ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.20, 3.27] |
| 17.13 heavy muscles ‐ up to 6 weeks | 2 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.21, 2.32] |
| 17.14 muscle cramp ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.35, 5.81] |
| 17.15 muscle spasm ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.30, 3.87] |
| 17.16 muscle spasm ‐ 26 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.20, 19.78] |
| 17.17 needing additional anticholinergic medication ‐ up to 6 weeks | 7 | 302 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.81, 1.33] |
| 17.18 needing additional anticholinergic medication ‐ up to 12 weeks | 3 | 120 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.71, 1.72] |
| 17.19 oculogyric crisis ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.40, 4.48] |
| 17.20 rigidity ‐ up to 6 weeks | 4 | 212 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.96, 1.50] |
| 17.21 rigidity ‐ 7 ‐ 26 weeks | 3 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.51, 2.06] |
| 17.22 thick speech ‐ up to 6 weeks | 2 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.44, 3.39] |
| 17.23 tremor ‐ up to 6 weeks | 4 | 212 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.81, 1.51] |
| 17.24 tremor ‐ 7 to 26 weeks | 4 | 184 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.54, 1.32] |
| 17.25 twisting movement ‐ 8 weeks | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.19, 20.90] |
| 18 Adverse effects: 7. Neurological problems | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 18.1 ataxia ‐ 4 weeks | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.03, 2.15] |
| 18.2 clumsiness ‐ 26 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
| 18.3 confusion/cloudiness ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.20, 3.27] |
| 18.4 confusion/cloudiness ‐ 7 to 26 weeks | 2 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.85 [0.61, 13.24] |
| 18.5 dizziness, fainting, weakness ‐ up to 6 weeks | 2 | 95 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.37, 2.36] |
| 18.6 dizziness/fainting, weakness ‐ 7 ‐ 26 weeks | 3 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.59 [0.67, 3.75] |
| 18.7 giddiness ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.4 [0.08, 1.89] |
| 18.8 seizures ‐ up to 12 weeks | 3 | 302 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.94 [0.45, 34.72] |
| 18.9 unsteadiness ‐ 26 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.35, 25.68] |
| 19 Adverse effects: 8. Sleep problems | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 19.1 drowsiness / sedation ‐ up to 6 weeks | 6 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.79, 1.65] |
| 19.2 drowsiness/ sedation ‐ up to 12 weeks | 6 | 408 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.02, 1.86] |
| 19.3 fatigue ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.17, 2.44] |
| 19.4 insomnia ‐ up to 6 weeks | 3 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.56, 1.81] |
| 19.5 insomnia ‐ up to 12 weeks | 2 | 189 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.13, 0.69] |
| 19.6 lethargy ‐ up to 6 weeks | 2 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [0.77, 3.75] |
| 20 Adverse effects: 9. Weight changes | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 20.1 weight increase ‐ 6 weeks | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.10, 2.16] |
| 20.2 weight increase 12 weeks | 2 | 169 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.30, 1.10] |
| 20.3 weight loss ‐ 6 weeks | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.12, 3.19] |
| 20.4 weight loss ‐ 12 weeks | 2 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.58, 3.31] |
| 21 Adverse effects: 10. Others | 16 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 21.1 abnormal blood results ‐ up to 6 weeks | 5 | 235 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.60, 2.22] |
| 21.2 abnormal blood results ‐ up to 12 weeks | 5 | 506 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.76, 1.47] |
| 21.3 anxiety ‐ 8 weeks | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.07, 1.20] |
| 21.4 difficulty swallowing ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.20, 3.27] |
| 21.5 headache ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.35, 5.81] |
| 21.6 headache ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.24, 7.48] |
| 21.7 libido ‐ decrease ‐ 4 weeks | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.01, 4.11] |
| 21.9 opthalmic changes ‐ 12 weeks | 2 | 86 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.15, 6.81] |
| 21.10 ringing in ears ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.35, 5.81] |
| 21.11 skin problems ‐ rash ‐ 4 weeks | 2 | 95 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.09, 1.33] |
| 21.12 skin problems ‐ rash ‐ 7 ‐ 26 weeks | 4 | 184 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.05, 0.77] |
| 21.13 swelling of hands/face ‐ 4 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.28, 1.61] |
| 21.14 swelling of hands/face ‐ 26 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 96.13] |
| 21.15 tingling sensation ‐ 6 weeks | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.17, 2.44] |
| 21.16 lactation ‐ 12 weeks | 1 | 37 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.02, 8.09] |
| 21.17 sweating ‐ 72 hours | 1 | 47 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.09 [0.20, 21.48] |
| 21.18 sweating ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.01, 7.09] |
| 21.19 excitement ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.05 [0.96, 17.12] |
| 21.20 depression ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.2 [0.48, 3.02] |
| 21.21 lacrimation ‐12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.71 [0.12, 63.84] |
| 21.22 breathlessness ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.71 [0.12, 63.84] |
| 21.23 bulimia ‐ 12 weeks | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.71 [0.12, 63.84] |
| 21.24 hypersalivation ‐ 8 weeks | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.55, 7.27] |
2.1. Analysis.
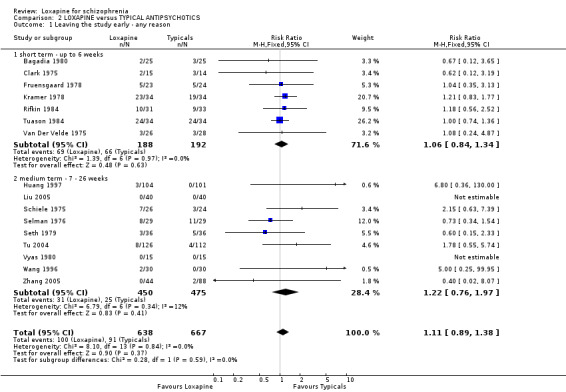
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 1 Leaving the study early ‐ any reason.
2.2. Analysis.
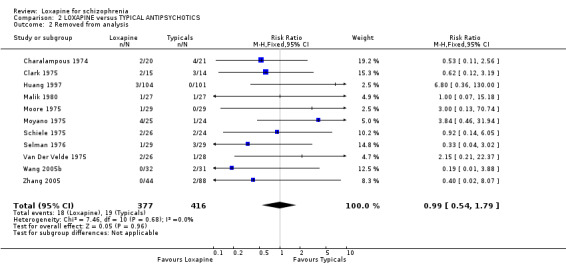
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 2 Removed from analysis.
2.3. Analysis.
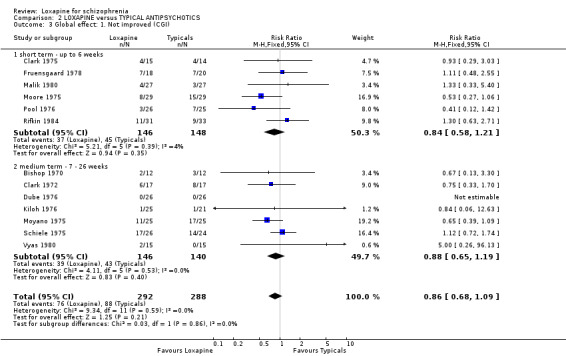
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 3 Global effect: 1. Not improved (CGI).
2.4. Analysis.
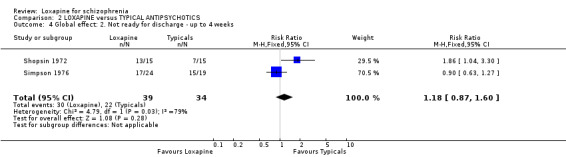
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 4 Global effect: 2. Not ready for discharge ‐ up to 4 weeks.
2.5. Analysis.
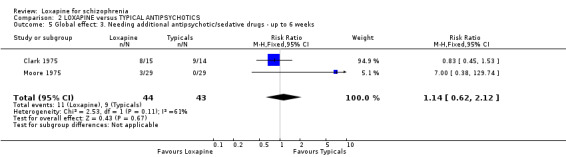
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 5 Global effect: 3. Needing additional antipsychotic/sedative drugs ‐ up to 6 weeks.
2.6. Analysis.
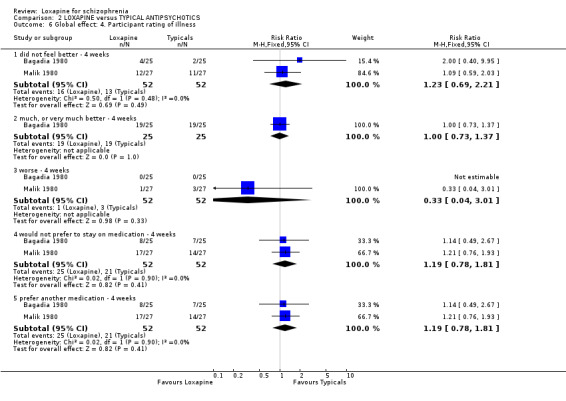
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 6 Global effect: 4. Participant rating of illness.
2.7. Analysis.
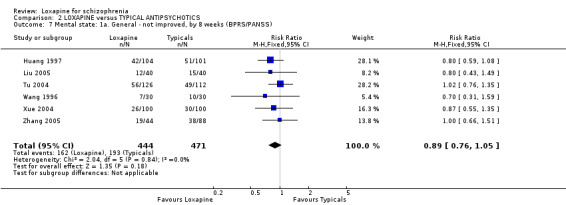
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 7 Mental state: 1a. General ‐ not improved, by 8 weeks (BPRS/PANSS).
2.8. Analysis.
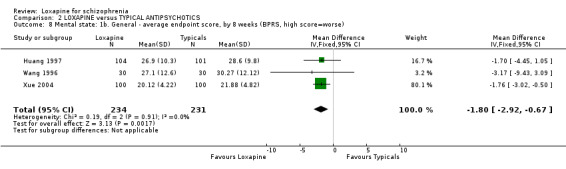
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 8 Mental state: 1b. General ‐ average endpoint score, by 8 weeks (BPRS, high score=worse).
2.9. Analysis.
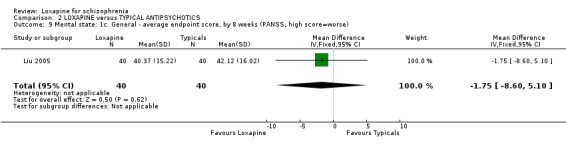
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 9 Mental state: 1c. General ‐ average endpoint score, by 8 weeks (PANSS, high score=worse).
2.10. Analysis.
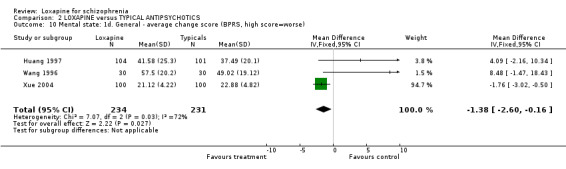
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 10 Mental state: 1d. General ‐ average change score (BPRS, high score=worse).
2.11. Analysis.
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 11 Mental state: 2. Specific.
2.12. Analysis.
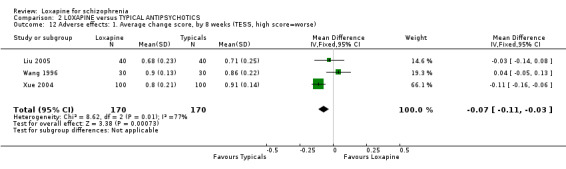
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 12 Adverse effects: 1. Average change score, by 8 weeks (TESS, high score=worse).
2.13. Analysis.
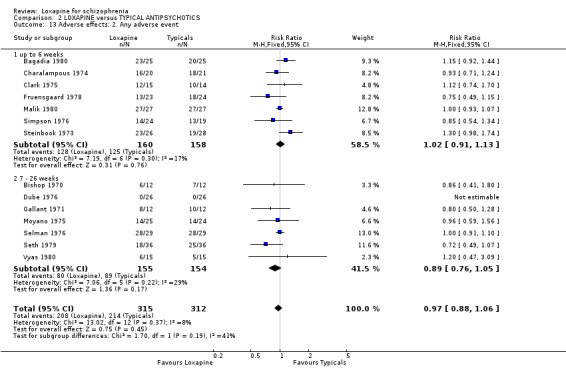
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 13 Adverse effects: 2. Any adverse event.
2.14. Analysis.
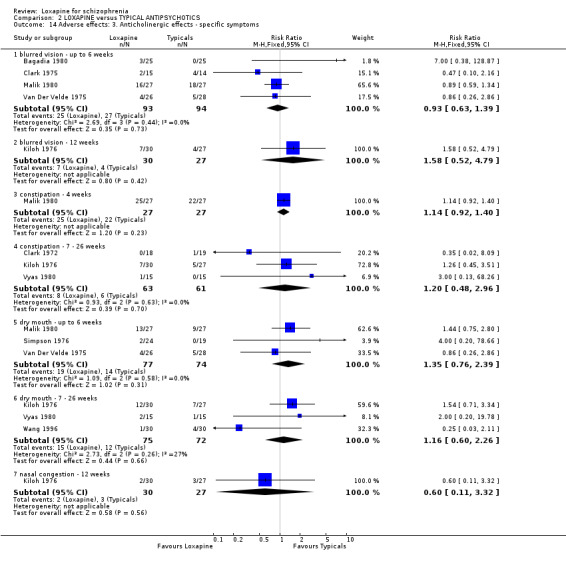
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 14 Adverse effects: 3. Anticholinergic effects ‐ specific symptoms.
2.15. Analysis.
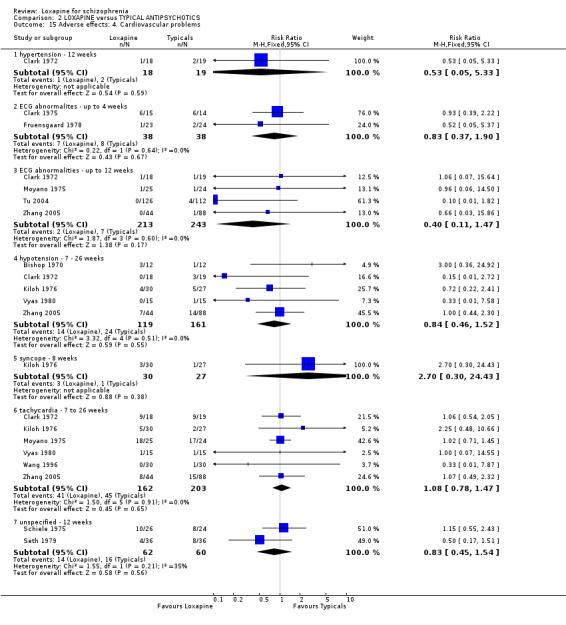
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 15 Adverse effects: 4. Cardiovascular problems.
2.16. Analysis.
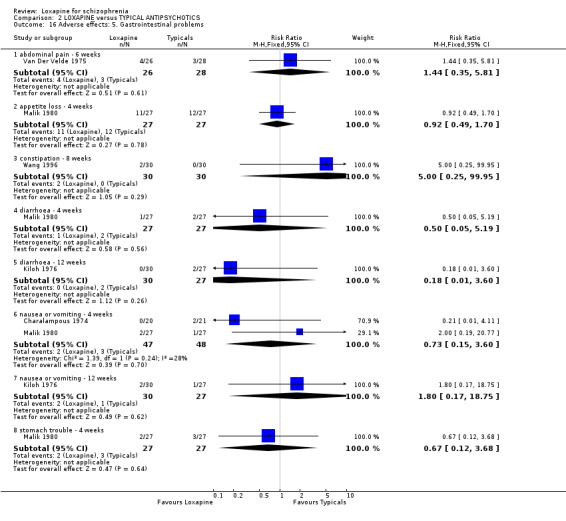
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 16 Adverse effects: 5. Gastrointestinal problems.
2.17. Analysis.
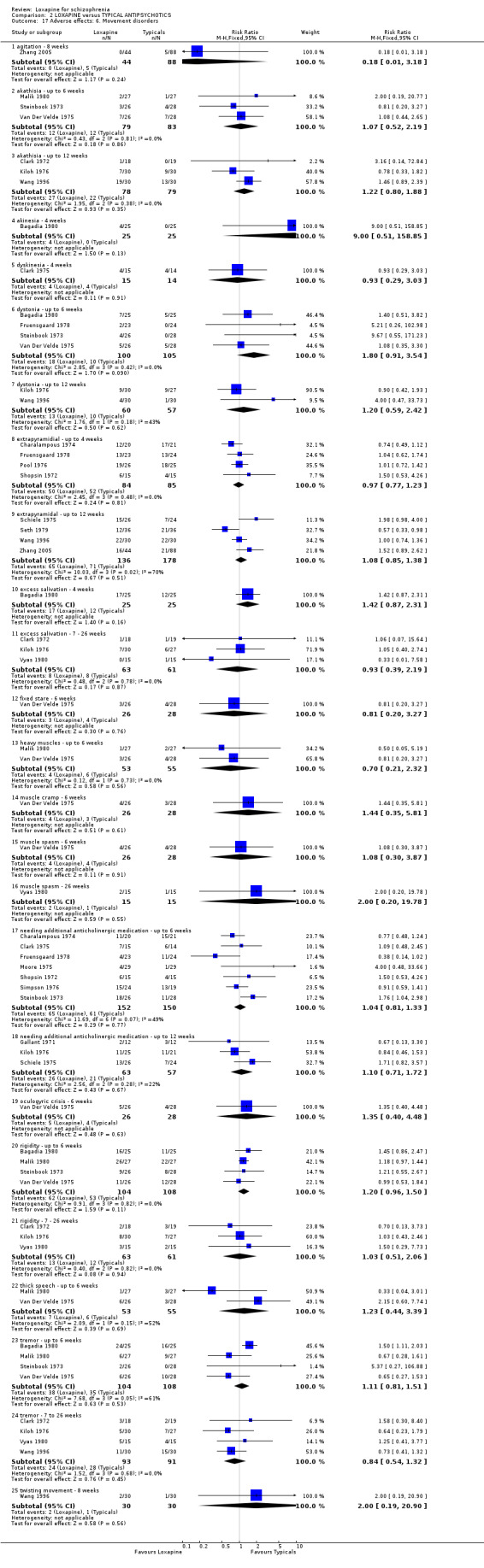
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 17 Adverse effects: 6. Movement disorders.
2.18. Analysis.
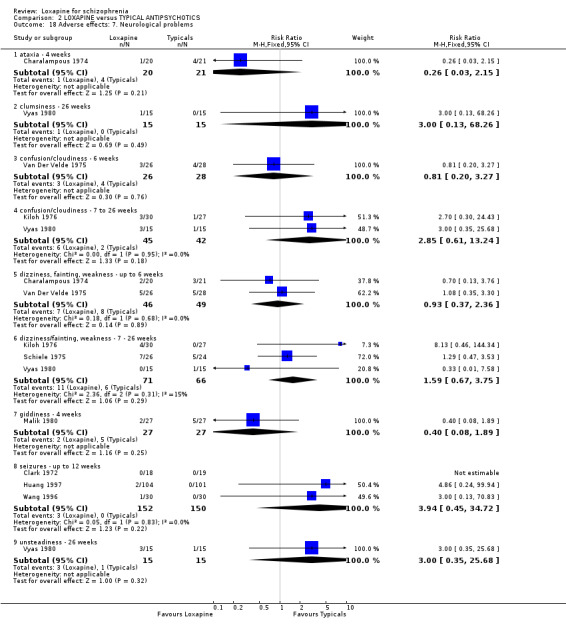
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 18 Adverse effects: 7. Neurological problems.
2.19. Analysis.
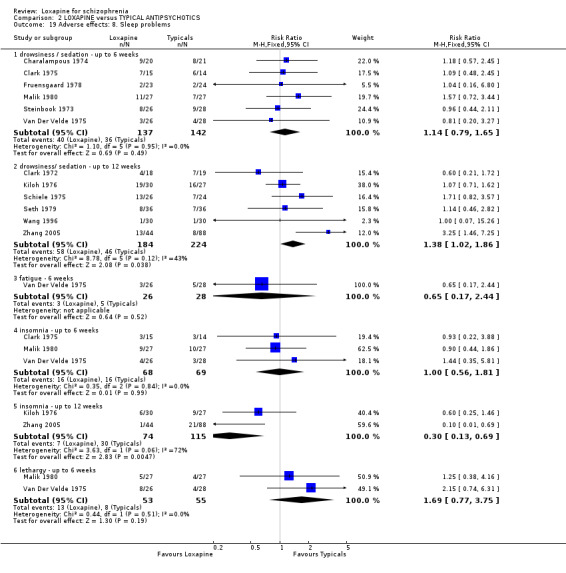
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 19 Adverse effects: 8. Sleep problems.
2.20. Analysis.
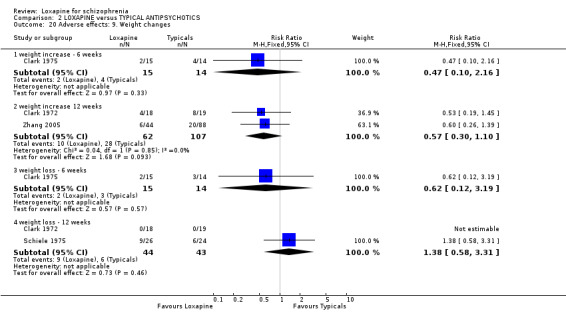
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 20 Adverse effects: 9. Weight changes.
2.21. Analysis.
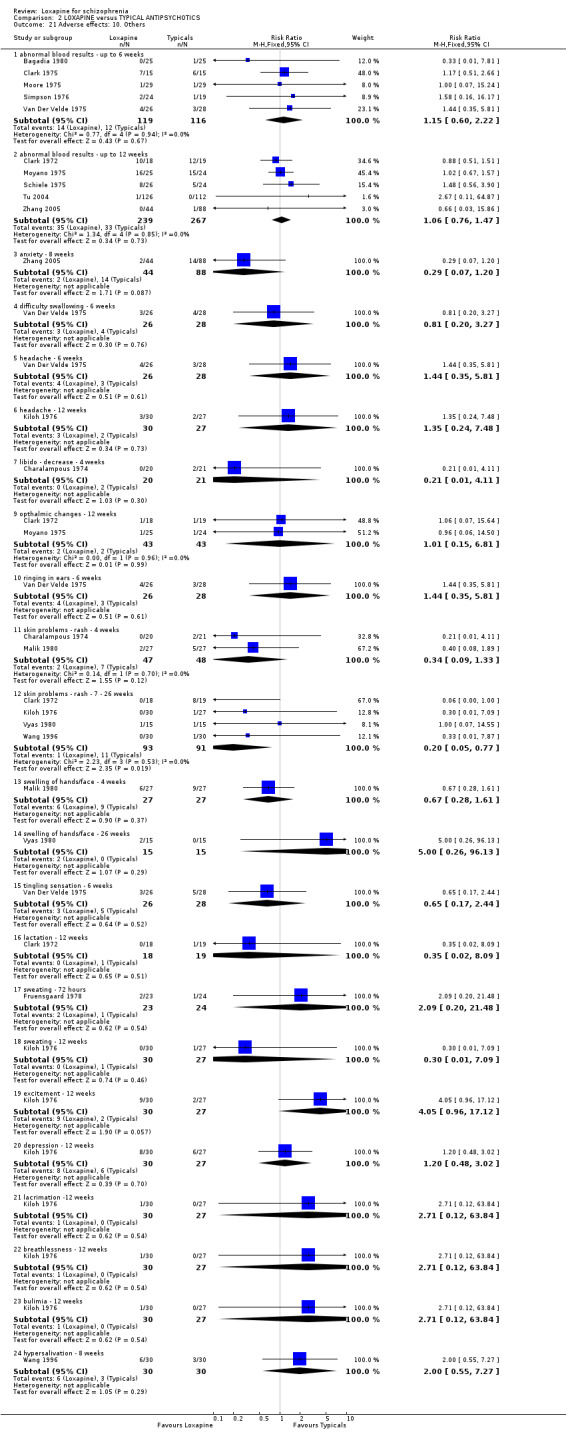
Comparison 2 LOXAPINE versus TYPICAL ANTIPSYCHOTICS, Outcome 21 Adverse effects: 10. Others.
Comparison 3. LOXAPINE IM versus TYPICAL ANTIPSYCHOTIC IM FOR RAPID TRANQUILISATION.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Withdrawn from or leaving the study early ‐ by 72 hours | 2 | 115 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.31, 1.08] |
| 2 General effect: Not tranquilised | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 not sedated / still hostile ‐ at 1hour | 3 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.47, 0.83] |
| 2.2 not sedated / still hostile ‐ at 6‐24 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.04, 3.49] |
| 2.3 requiring further sedation ‐ up to 6 days | 2 | 115 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.21 [0.62, 2.38] |
| 3 Mental state: Average endpoint score ‐ at 72 hours (BPRS, high score=worse) | 1 | 47 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐7.01, 7.01] |
| 4 Adverse effects: 1. Any event ‐ 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.44, 1.45] |
| 5 Adverse effects: 2. Movement ‐ specific symptoms | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 akathisia ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.51, 2.64] |
| 5.2 drooling ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.14, 4.26] |
| 5.3 dyskinesia ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.06, 6.02] |
| 5.4 dystonia ‐ 72 hours | 2 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.42, 1.99] |
| 5.5 oculogyric crisis ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.01, 3.04] |
| 5.6 rigidity ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.12, 2.90] |
| 5.7 tremor ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.01, 3.04] |
| 5.8 thick tongue ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.06, 6.02] |
| 6 Adverse effects: 3. Other | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 anticholinergic ‐ 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.48, 5.76] |
| 6.2 blood test abnormalities ‐ 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
| 6.3 dizziness ‐ 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.0 [0.82, 44.00] |
| 6.4 drowsiness/fatigue ‐ 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.36, 4.97] |
| 6.5 increased blood pressure ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.18, 7.64] |
| 6.6 increased pulse rate ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.06, 6.02] |
| 6.7 nervousness ‐ 72 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.06, 6.02] |
| 6.8 pain at injection site ‐ 72 hours | 2 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.5 [0.29, 7.73] |
| 6.9 palpatations ‐ 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
3.1. Analysis.
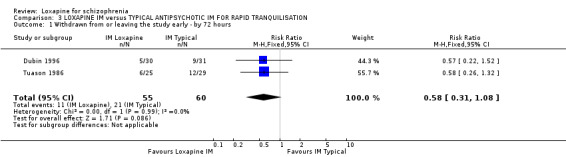
Comparison 3 LOXAPINE IM versus TYPICAL ANTIPSYCHOTIC IM FOR RAPID TRANQUILISATION, Outcome 1 Withdrawn from or leaving the study early ‐ by 72 hours.
3.2. Analysis.
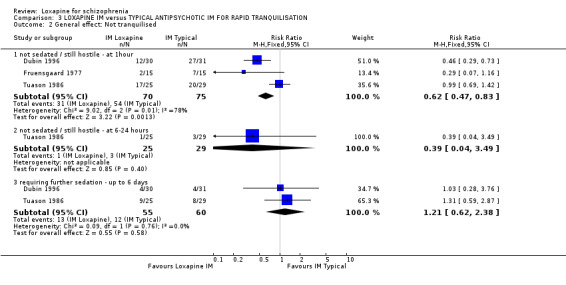
Comparison 3 LOXAPINE IM versus TYPICAL ANTIPSYCHOTIC IM FOR RAPID TRANQUILISATION, Outcome 2 General effect: Not tranquilised.
3.3. Analysis.
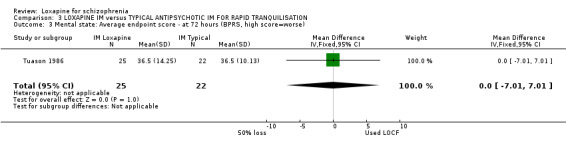
Comparison 3 LOXAPINE IM versus TYPICAL ANTIPSYCHOTIC IM FOR RAPID TRANQUILISATION, Outcome 3 Mental state: Average endpoint score ‐ at 72 hours (BPRS, high score=worse).
3.4. Analysis.
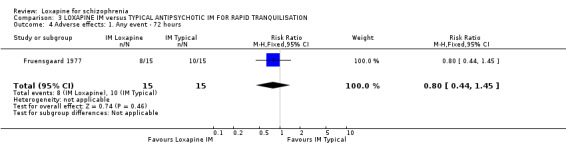
Comparison 3 LOXAPINE IM versus TYPICAL ANTIPSYCHOTIC IM FOR RAPID TRANQUILISATION, Outcome 4 Adverse effects: 1. Any event ‐ 72 hours.
3.5. Analysis.
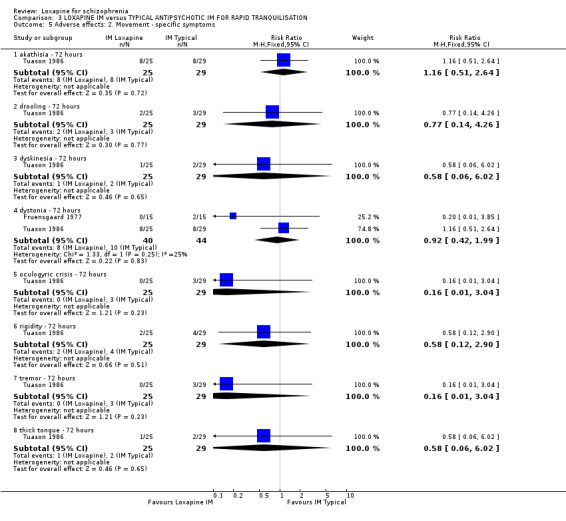
Comparison 3 LOXAPINE IM versus TYPICAL ANTIPSYCHOTIC IM FOR RAPID TRANQUILISATION, Outcome 5 Adverse effects: 2. Movement ‐ specific symptoms.
3.6. Analysis.
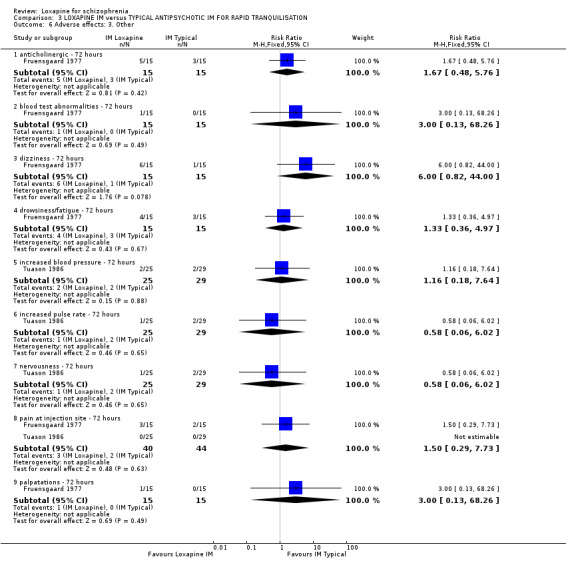
Comparison 3 LOXAPINE IM versus TYPICAL ANTIPSYCHOTIC IM FOR RAPID TRANQUILISATION, Outcome 6 Adverse effects: 3. Other.
Comparison 4. LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early ‐ any reason ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.01, 4.08] |
| 2 Global effect: 1. Not improved ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.22, 1.34] |
| 3 Global effect: 2. Needing additional antipsychotic/sedative drugs ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.3 [0.53, 3.17] |
| 4 Adverse effects: 1. Any event ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.88, 2.72] |
| 5 Adverse effects: 2. Anticholinergic effects ‐ specific symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 blurred vision ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.01, 2.70] |
| 6 Adverse effects: 3. Cardiovascular problems | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 blood pressure ‐ hypotension ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.01, 2.70] |
| 6.2 ECG abnormalites ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.63 [0.60, 4.38] |
| 7 Adverse effects: 4. Movement disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 akathisia ‐ 12 weeks | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.29, 3.03] |
| 7.2 akinesia ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.06, 5.24] |
| 7.3 bradykinesia ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.04, 3.02] |
| 7.4 drooling ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.01, 2.70] |
| 7.5 dyskinesia ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.06, 5.24] |
| 7.6 dystonia ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.06, 5.24] |
| 7.7 needing additional anticholinergic medication ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.35, 3.40] |
| 7.8 rigidity ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.33, 1.79] |
| 7.9 tremor ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.37, 2.20] |
| 8 Adverse effects: 5. Sleep problems | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 drowsiness ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.71, 2.09] |
| 9 Adverse effects: 6. Weight changes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 weight increase ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.18, 6.53] |
| 9.2 weight loss ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.06, 5.24] |
| 10 Adverse effects: 7. Others ‐ abnormal blood results ‐ 12 weeks | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.04, 3.02] |
4.1. Analysis.
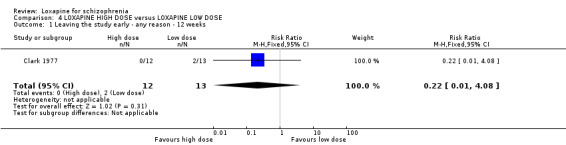
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 1 Leaving the study early ‐ any reason ‐ 12 weeks.
4.2. Analysis.
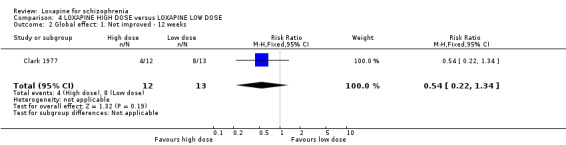
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 2 Global effect: 1. Not improved ‐ 12 weeks.
4.3. Analysis.
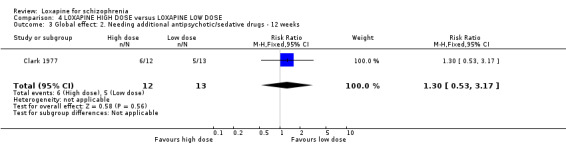
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 3 Global effect: 2. Needing additional antipsychotic/sedative drugs ‐ 12 weeks.
4.4. Analysis.
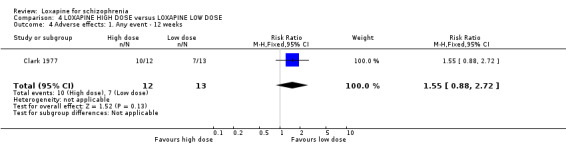
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 4 Adverse effects: 1. Any event ‐ 12 weeks.
4.5. Analysis.
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 5 Adverse effects: 2. Anticholinergic effects ‐ specific symptoms.
4.6. Analysis.
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 6 Adverse effects: 3. Cardiovascular problems.
4.7. Analysis.
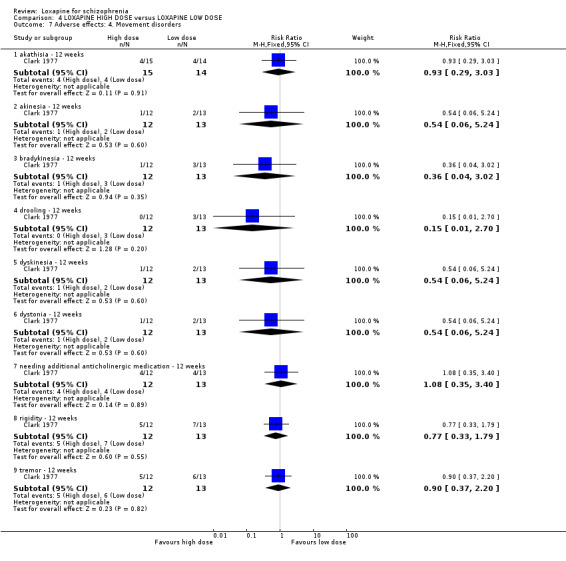
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 7 Adverse effects: 4. Movement disorders.
4.8. Analysis.
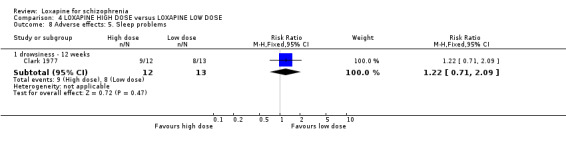
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 8 Adverse effects: 5. Sleep problems.
4.9. Analysis.
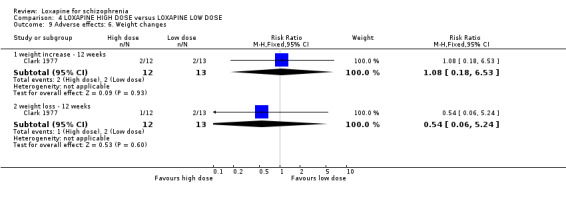
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 9 Adverse effects: 6. Weight changes.
4.10. Analysis.
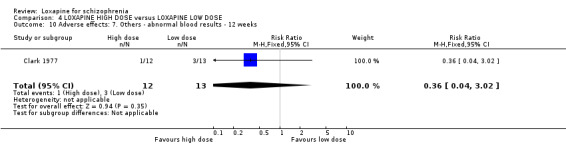
Comparison 4 LOXAPINE HIGH DOSE versus LOXAPINE LOW DOSE, Outcome 10 Adverse effects: 7. Others ‐ abnormal blood results ‐ 12 weeks.
Comparison 5. LOXAPINE versus ATYPICAL ANTIPSYCHOTICS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early‐any reason ‐ 8 weeks | 3 | 218 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.32, 5.00] |
| 2 Removed from analysis ‐ 8 weeks | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.25, 100.43] |
| 3 Mental state: 1. Not Improved, up to 8 weeks (PANSS) | 6 | 468 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.75, 1.53] |
| 4 Mental state: 2a. Average endpoint score, by 8 weeks (BPRS, high score=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 1.38 [‐5.81, 8.57] |
| 5 Mental state: 2b. Average endpoint score, by 8 weeks (PANSS, high score=worse) | 5 | 408 | Mean Difference (IV, Fixed, 95% CI) | ‐1.13 [‐4.08, 1.81] |
| 6 Adverse effects: 1. Average change score, by 8 weeks (TESS, high score=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.05 [‐0.04, 0.14] |
| 7 Adverse effects: 2. Movement disorders ‐ 8 weeks | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 agitation | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.96] |
| 7.2 akathsia | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.79 [0.36, 126.24] |
| 7.3 extraparyamidal | 4 | 340 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.18 [1.55, 3.06] |
| 7.4 increased activity | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.99] |
| 7.5 tremor | 2 | 123 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.82, 1.42] |
| 8 Adverse effects: 3. Cardiovascular ‐ 8 weeks | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 ECG abnormal | 2 | 155 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 [0.70, 4.63] |
| 9 Adverse effects: 4. Sleep problems ‐ 8 weeks | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 insomnia | 2 | 155 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.04, 0.78] |
| 9.2 sleep disturbance | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.05, 0.81] |
| 10 Adverse effects: 5. Others ‐ 8 weeks | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 amenhorrhea | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.65] |
| 10.2 anxiety | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.27, 3.54] |
| 10.3 dermatitis | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.65] |
| 10.4 enuresis | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.65] |
| 10.5 haematological ‐ leucopenia | 2 | 185 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.02, 0.95] |
5.1. Analysis.
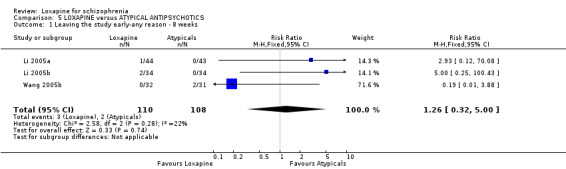
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 1 Leaving the study early‐any reason ‐ 8 weeks.
5.2. Analysis.
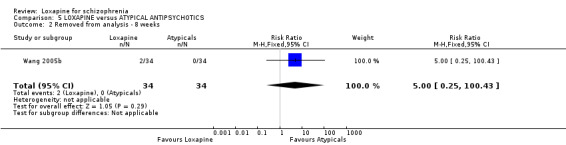
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 2 Removed from analysis ‐ 8 weeks.
5.3. Analysis.
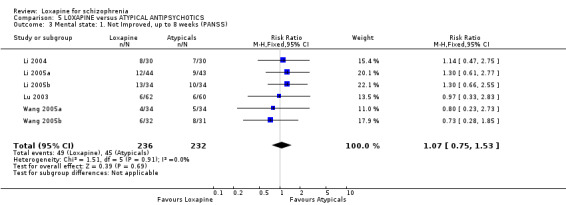
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 3 Mental state: 1. Not Improved, up to 8 weeks (PANSS).
5.4. Analysis.
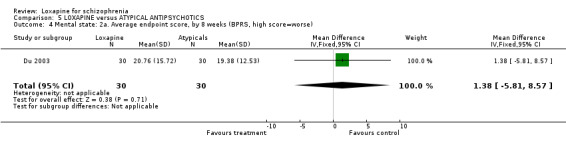
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 4 Mental state: 2a. Average endpoint score, by 8 weeks (BPRS, high score=worse).
5.5. Analysis.
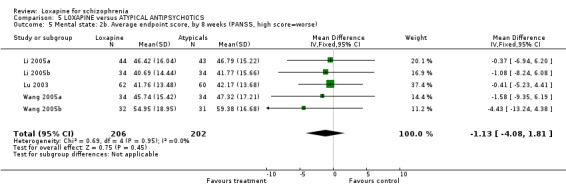
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 5 Mental state: 2b. Average endpoint score, by 8 weeks (PANSS, high score=worse).
5.6. Analysis.
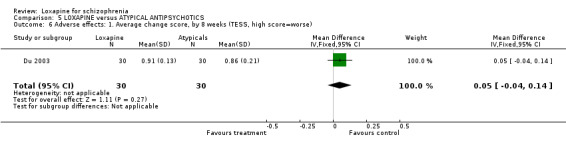
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 6 Adverse effects: 1. Average change score, by 8 weeks (TESS, high score=worse).
5.7. Analysis.
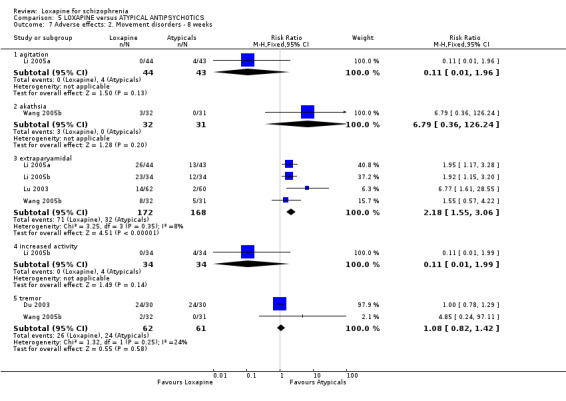
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 7 Adverse effects: 2. Movement disorders ‐ 8 weeks.
5.8. Analysis.
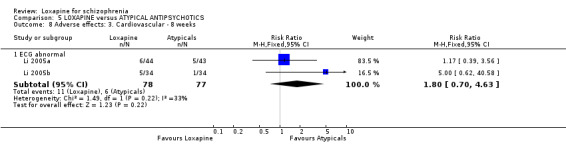
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 8 Adverse effects: 3. Cardiovascular ‐ 8 weeks.
5.9. Analysis.
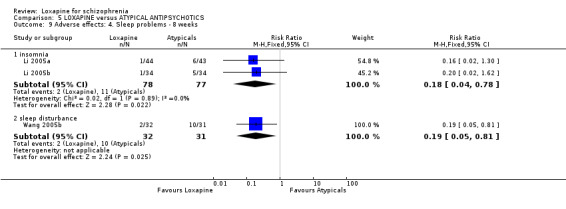
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 9 Adverse effects: 4. Sleep problems ‐ 8 weeks.
5.10. Analysis.
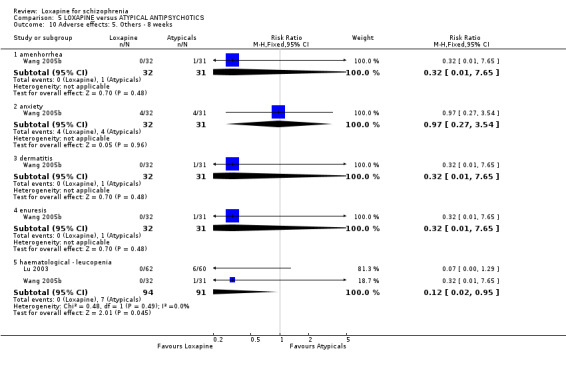
Comparison 5 LOXAPINE versus ATYPICAL ANTIPSYCHOTICS, Outcome 10 Adverse effects: 5. Others ‐ 8 weeks.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bagadia 1980.
| Methods | Allocation: randomised ‐ no further details. Blinding: double blind ‐ identical capsules. Duration: 4 weeks. | |
| Participants | Diagnosis: schizophrenia with onset between 13‐19 yrs (ICD‐10). N=55. Age: mean ˜17 years, range 14‐24. Sex: male and female. History: outpatients. | |
| Interventions | 1. Loxapine: dose 10 mg/day increased to 120 mg/day. N=25. 2. Trifluperazine: dose 2.5 mg/day increased up to 25 mg/day max. N=30. | |
| Outcomes | Leaving the study early.
Drug preference.
Patients self evaluation.
Adverse effects. Unable to use ‐ Global effect: CGI (no SD). Mental state: BPRS (no SD). Behaviour: NOSIE (no SD). Laboratory tests: Blood tests, ECG, opthmalogical, physiological (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Bishop 1970.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ no further information. Duration: 8 weeks ‐ preceded by 4 weeks washout + 2 week assessment period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, chronic ‐ no further details. N=24. Age: mean ˜ 44 years, range 30‐55. Sex: 12 M, 12 F. History: inpatients ˜ 17 years, range 5‐29 years. | |
| Interventions | 1. Loxapine: dose 20 mg/day, increased to 120 mg/day maximum. N=12. 2. Trifluoperazine: dose 10 mg/day, increased to 60 mg/day maximum. N=12. | |
| Outcomes | Global effect: CGI.
Adverse effects. Unable to use ‐ Mental state: BPRS (no usable data). Behaviour: NOSIE (no usable data). Laboratory tests: ECG (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Charalampous 1974.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical capsules. Duration: 4 weeks ‐ preceded by 1 week washout. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia (by 2 psychiatrists). N=60*. Age: mean ˜ 26 years, range 18‐53. Sex: 58 M, 2 F. History: inpatients, ill < 6 years, healthy, mean length ill ˜ 2.5 years. | |
| Interventions | 1. Loxapine: dose range 50 mg‐150 mg/day, mean 147.5 mg. N=20.
2. Thiothixene: dose range 20 mg‐60 mg/day, mean 51.9 mg. N=20.
3. Placebo. N=19. Chloral hydrate and trihexyphenidyl as required. |
|
| Outcomes | Excluded from analysis.
Adverse effects (TESS, use of anticholinergic drugs). Unable to use ‐ Global effect: CGI (no data). Mental state: BPRS (no SD). Behaviour: NOSIE (no data). Leaving the study early (not reported). Laboratory tests: (no data). Physiological measures: BP, ECG, weight (no data). |
|
| Notes | *One participant not accounted for. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Clark 1972.
| Methods | Allocation: random, stratified age and sex. Blinding: double blind‐identical capsules. Duration: 12 weeks, with 12 weeks washout. | |
| Participants | Diagnosis: schizophrenia, confirmed by project psychiatrist. N=55. Age: 21‐60 years. Sex: 31 M, 24 F. History: inpatients, ill for at least 2 years. | |
| Interventions | 1. Loxapine: dose 10 mg/day increased to 100 mg/day in 25 days. N=18
2. Chlorpromazine: dose 100 mg/day increased to 1gm/day . N=19.
3. Placebo. N=18. Antiparkinsonian medication allowed as required. |
|
| Outcomes | Global effect: CGI (improvement).
Adverse effects(physical examination, lab results, ECG and eye examination). Unable to use ‐ Global effect: CGI (severity) (no SD). Mental state: BPRS (no SD). Behaviour: NOSIE (no SD). |
|
| Notes | Weight increase or decrease ‐ 10 lbs. All blood test abnormalities combined. For CGI (improvement) reading by psychiatrist 2 used (randomly chosen by lots). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Clark 1975.
| Methods | Allocation: random ‐ pre‐randomised list, blocks of 3, provided by drug company. Blinding: double ‐ identical capsules. Duration: 4 weeks. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, confirmed by research psychiatrists. N=42. Age: range 21‐57 years. Sex: 21 M, 16 F, 6 unreported. History: ill > 2 years, healthy, not able to bear children. | |
| Interventions | 1. Loxapine: dose 100mg/day, mean 71mg/day. N=15. 2. Trifluoperazine: dose 50 mg/day, mean 36 mg/day. N=14. 3. Placebo. N=13. Short acting sedatives and antiparkinsonian medication as required. | |
| Outcomes | Global effect (CGI‐I, CGI‐S, use of additional sedation).
Adverse effects.
Leaving the study early.
Laboratory tests.
Physiological measures (ECG, weight). Unable to use ‐ Efficacy: (analysis of covariance ‐ no usable data). Mental state: BPRS (no SD). Behaviour: NOSIE (no usable data). Physiological measures: BP, pulse (no data). |
|
| Notes | 6 people removed from analysis but original group of allocation reported so ITT analysis possible. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Clark 1977.
| Methods | Allocation: random ‐ pre‐randomised list, blocks of 3, provided by drug company. Blinding: double ‐ identical capsules. Duration: 12 weeks ‐ "..effects of previous treatment allowed to dissipate over a period of 12 weeks" before trial. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia (DSM‐II). N=38. Age: range 21‐57 years. Sex: 11 M, 27 F. History: > 2 years ill & institutionalizations without remission, healthy, not pregnant, inpatients. | |
| Interventions | 1. Loxapine: dose 100 mg/day. N=12.
2. Loxapine: dose 50 mg/day. N=13.
3. Placebo. N=13. Short acting sedatives and antiparkinsonian medication as required. |
|
| Outcomes | Global effect (CGI‐I, CGI‐S, use of additional sedation).
Adverse effects.
Leaving the study early.
Laboratory tests.
Physiological measures (ECG, weight). Unable to use ‐ Efficacy: (analysis of covariance ‐ no usable data). Mental state: BPRS (no SD). Behaviour: NOSIE (no usable data). Physiological measures: BP, pulse (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Du 2003.
| Methods | Allocation: randomised. Blinding: none. Duration: 8 weeks. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3). N=60. Loxapine grp: 18 M, 12 F; average age 29. Risperidone grp: 17 M, 13 F; average age 26. History: hospitalised patients. | |
| Interventions | 1. Loxapine: dose range: 68‐305 mg. N=30. 2. Risperidone: dose range: 1‐6 mgs. N=30. | |
| Outcomes | Adverse effects: TESS.
Laboratory tests.
Physiological measures: ECG, EEG. Unable to use ‐ Global effect: CGI (no data). Mental State: BPRS (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Dube 1976.
| Methods | Allocation: randomised ‐ no further information. Blinding: double ‐ identical capsules. Duration: 12 weeks. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, no further information. N=52. Age: mean ˜ 32 years, range 18‐55. Sex: 52 M. History: < 2 years ill, healthy, no co‐existing mental illnesses. | |
| Interventions | 1. Loxapine: dose 20‐80 mg/day, mean 34.3 mg/day. N=26. 2. Chlorpromazine: dose 200‐800 mg/day, mean 320 mg/day. N=26. | |
| Outcomes | Global effect: CGI.
Adverse effects.
Leaving the study early. Unable to use ‐ Efficacy (analysis of covariance ‐ no usable data). Mental state: BPRS (no SD). Laboratory tests: (no data). Physiological measures: BP, ECG, pulse, ophthalmic change (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Dubin 1996.
| Methods | Allocation: randomised ‐ randomisation table. Blinding: double ‐ identical ampules, staff administering medication not blinded, assessors blind. Duration: 6 days ‐ preceded by 24h washout (only data from first 72 hours used). Setting: single centre. | |
| Participants | Diagnosis: schizophrenia (52), bipolar manic (9) (DSM‐III). N=61. Age: mean ˜ 35 years, range 18‐65. Sex: ˜27 M, ˜31 F. Inclusion : BPRS score of 6/7 in >2 prespecified symptom categories. History: admitted as psychiatric emergency, healthy, drug sensitivity, not pregnant or lactating, no co‐existing mental illness. | |
| Interventions | 1. Loxapine: dose mean 75.5 mg/day IM, range 25‐175 mg/day IM. N=30.
2. Thiothixene: dose mean 31 mg/day IM, range 20‐60 mg/day IM, N=31. IM for first 24 hours, then oral. IM injections every 30 minutes as needed, until BPRS reduced or sedation occurred. Chloral hydrate, trihexyphenidyl/benztropine as required. |
|
| Outcomes | Global effect (sedation, requiring further injections).
Dropped from study. Unable to use ‐ Global effect: CGI (no data). Mental state: BPRS (no SD). Side effects: (only data for 5 day oral phase available). Physiological measures: BP, pulse (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Fruensgaard 1977.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical ampules. Duration: 72 hours ‐ preceded by 12 h washout (study continued for 4 weeks but not reported). Setting: single centre. | |
| Participants | Diagnosis: acute schizophrenia (12), psychogenic psychosis (18). N=30. Age: mean ˜ 40 years, range 19‐65. Sex: 7 M, 23 F. Inclusion: healthy, not pregnant, no coexisting mental illness, mania or treatment with ECT in last 8 weeks. History: duration of present episode: <1week (14), 1 week‐1 month (13), >1month (3). | |
| Interventions | 1. Loxapine: dose 25‐50 mg/6‐12 hours IM, mean 130 mg/day IM. N=15.
2. Thiothixene: dose 2.5‐5 mg/6‐12 hours IM, mean 12 mg/day IM. N=15. Both given with biperiden. IM for first 24 hours, then oral. IM injections every 30 minutes as needed, until BPRS reduced or sedation occurred. |
|
| Outcomes | Global effect: CGI, sedation.
Adverse effects: pain at injection site. Unable to use ‐ Mental state: BPRS (no SD), continued aggression (no usable data). Physiological measures: BP, pulse (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Fruensgaard 1978.
| Methods | Allocation: randomised ‐ no further details. Blinding: double blind. Duration: group one ‐3 weeks, group two ‐ 12 weeks. Setting: multicenter. | |
| Participants | Diagnosis: acute schizophrenia (7, acute schizophreniform psychotic episodes or acute exacerbations of a chronic schizophrenic process), psychogenic (reactive) psychosis (15), chronic schizophrenia (25). N=47. Age: range 16‐67 years. Sex: 32 M, 15F. | |
| Interventions | 1. Loxapine: dose 10 mg bid increased to 150 mg/day. N=23.
2. Perphenazine: dose 8 mg bid increased to 120 mg/day. N=24. Chloralodolol used for insomnia and antiparkinsonian medications used as required. |
|
| Outcomes | Global effect: CGI.
Adverse effects.
Physiological effects, laboratory tests, ECG. Unable to use ‐ Mental state: BPRS (no data). Behaviour: NOSIE (no data). |
|
| Notes | Data of adverse effects and CGI were extracted after clubbing both acute and chronic patients. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Gallant 1971.
| Methods | Allocation: randomised by random numbers. Blinding: double blind. Duration: 8 weeks, with 4 weeks washout. | |
| Participants | Diagnosis: chronic schizophrenia. N=24. Age: range 30‐55. Sex: 11 M, 13F. Design: phase 2 trial. | |
| Interventions | 1. Loxapine: dose 20 mg/day increased to 120 mg/day. N=12.
2. Trifluperazine: dose 10 mg/day increased up to 60 mg/day. N=12. Antiparkinsonian medication given as required. |
|
| Outcomes | Adverse events. Unable to use ‐ Global effect: CGI (no data). Mental state: BPRS (no data). Behaviour: NOSIE (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Huang 1997.
| Methods | Allocation: randomised ‐ no further details Blinding: double blind. Duration: 8 weeks. Setting: multicenter. | |
| Participants | Diagnosis: schizophrenia. N=205. Age: mean ˜35 years, range 18‐60. Sex: 123 M, 82 F. History: hospitalised. | |
| Interventions | 1. Loxapine: dose range: 50‐300 mg. N=104. 2. Chlorpromazine: dose range: 75‐600 mg. N=101. | |
| Outcomes | Mental state: BPRS.
Global effect: CGI.
Adverse effects: TESS.
Physiological effects: EEG. Unable to use ‐ Laboratory tests (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Kiloh 1976.
| Methods | Allocation: randomised ‐ by a prearranged system. Blinding: double blind, no more details. Duration: 12 weeks with a 2 weeks washout. | |
| Participants | Diagnosis: schizophrenia (Slater and Roth criteria). N=57 Age: less than 69. Sex: no details available. History: inpatients, duration ill ‐ acute < 2 years, chronic > 2 years. | |
| Interventions | 1. Loxapine: dose 10 mg /day increased progressively ‐ acute group mean ˜37 mg/day (SD ˜22), chronic group 56 mg/day (SD 20). N=30.
2. Trifluperazine: dose 5mg/day increased progressively ‐ acute group mean ˜24 mg/day (SD 14.5), chronic group 31 mg/day (SD 11.7). N=27. Diazepam, barbiturates and benztropine for adverse effects as required. |
|
| Outcomes | Global effect: CGI.
Adverse effects ‐ physical examination, ophthalmic examination, laboratory tests, ECG. Unable to use ‐ Mental state: BPRS (no data). Behaviour: NOSIE (no data). |
|
| Notes | Data extracted clubbing both acute and chronic patients. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Kramer 1978.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical ampules. Duration: 4 weeks ‐ preceded by 2 week drug free period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, acute (DSM‐II). N=69. Age: mean ˜31 years, range >18‐57. Sex: 21 M, 35 F, 13 not reported. Exclusion: ill health, < 1 week of study medications. | |
| Interventions | 1. Loxapine: dose mean 74 mg/day. N=34.
2. Thioridazine: dose mean 442 mg/day. N=35. Doses individually titrated, antiparkinsonian medication as required. |
|
| Outcomes | Leaving the study early.
Dropped from analysis. Unable to use ‐ Global effect: CGI (>50% attrition). Mental state: BPRS (>50% attrition). Behaviour: NOSIE (>50% attrition). Side effects: DOTES (>50% attrition). Efficacy: (analysis of covariance). Laboratory tests (no data). Physiological measures: ECG, hand writing (>50% attrition). |
|
| Notes | Loss to follow up 60%. Only data from the outcome of 'leaving the study early' used. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Li 2004.
| Methods | Allocation: randomised ‐ no further details. Blinding: non blind. Duration: 8 weeks. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3). N=60. Age: mean ˜30, range 18‐60. Sex: 38M 22F. History: hospitalised. | |
| Interventions | 1. Loxapine: mean dose 208 mg/day (max 306 mg/day). N=30. 2. Clozapine: mean dose 415 mgs/day (max 600 mg/day). N=30. | |
| Outcomes | Mental state: PANSS. Unable to use ‐ Physiological measures: EEG (no data). Laboratory tests: bloods, urine (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Li 2005a.
| Methods | Allocation: randomised ‐ no further details. Blinding: not mentioned. Duration: 8 weeks. | |
| Participants | Diagnosis: schizophrenia (CCMD3). N=87. Age: mean ˜33. Sex: not reported. History: duration ill ˜ 5 years. | |
| Interventions | 1. Loxapine: dose, no average dose, max dose 272 mgs/day. N=44 2. Risperidone: dose, no average dose, max dose 6 mgs/day. N=43. | |
| Outcomes | Mental state: PANSS
Adverse effects: EPSE, abnormal ECG, agitation, insomnia. Unable to use ‐ Adverse effects: TESS (no data) |
|
| Notes | 1 dropout from the loxapine group after 1 week due to difficulty swallowing and hypermyotonia. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Li 2005b.
| Methods | Allocation: randomised ‐ no further details. Blinding: not mentioned. Duration: 8 weeks. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3). N=68. Age: mean ˜25, range 16‐50. Sex: 40 M, 28 F. History: hospitalised. | |
| Interventions | 1. Loxapine: dose range 34‐340 mg. N=34. 2. Risperidone: dose range 1‐7mg. N=34. | |
| Outcomes | Mental state: PANSS.
Adverse effects: TESS.
Physiological measures: EEG, ECG. Unable to use ‐ Haematological tests (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Liu 2005.
| Methods | Allocation: randomised ‐ no further details. Blinding: not mentioned. Duration 8 weeks. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3). N=80. Age: mean ˜28 years. Sex: 43 M, 37 F. History: mean duration ill ˜ 22 months. | |
| Interventions | 1. Loxapine: dose 68‐204 mgs/day. N=40. 2. Chlorpromazine: dose 250‐600 mgs/day. N=40. | |
| Outcomes | Mental state: PANSS. Adverse effects: TESS. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Lu 2003.
| Methods | Allocation: randomised ‐ no further details. Blinding: non‐blind. Duration: 6 weeks. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3). N=122. Age: mean ˜34 years, range 16‐56. Sex: 81M, 41F. History: hospitalised. | |
| Interventions | 1. Loxapine: dose range 34‐272 mgs. N=62. 2. Clozapine: dose range 25mgs‐600 mg. N=60. | |
| Outcomes | Mental state: PANSS. Adverse effects: TESS. Laboratory tests: Bloods, Urine, EEG. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Malik 1980.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical capsules. Duration: 28 days. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, no further details. N=54. Age: mean ˜17 years, range: >14‐19. Sex: 25 M, 27 F, 2 not reported. Exclusion: sensitivity to study drugs, ECT in last 8 weeks, co‐existing mental illness, ill health. | |
| Interventions | 1. Loxapine: dose mean 91.5 mg/day. N=27.
2. Trifluoperazine: dose mean 23.57 mg/day. N=27. Antiparkinsonian medication as required. |
|
| Outcomes | Global effect: CGI.
Adverse effects.
Drug preference.
Dropped from analysis. Unable to use ‐ Efficacy: analysis of covariance (no usable data). Mental state: BPRS (no SD). Behaviour: NOSIE (no usable data). Adverse effects: Use of antiparkinsonian drugs (no data). Physiological measures: BP, pulse (no data). Laboratory tests: (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Moore 1975.
| Methods | Allocation: no details. Blinding: double ‐ identical capsules. Duration: 6 weeks ‐ preceded by 2 week washout. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, acute or exacerbations of chronic. N=54*. Age: mean ˜ 37 years. Sex: 25 M, 27 F, 2 not reported. History: hospitalised. | |
| Interventions | 1. Loxapine: dose 20 mg‐120 mg/day. N=29.
2. Chlorpromazine: dose 200 mg‐1200 mg/day. N=29. Antiparkinsonian or sedative medication as required. |
|
| Outcomes | Global effect: CGI, use of additional sedation.
Adverse effects: TESS, use of antiparkinsonian drugs.
Dropped from analysis.
Laboratory tests. Unable to use ‐ Mental state: BPRS (no usable data). Behaviour: NOSIE (no usable data). Physiological tests: BP, ECG, ophthalmic tests, pulse (no data). |
|
| Notes | *Four participants not accounted for. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Moyano 1975.
| Methods | Allocation: no details. Blinding: double ‐ identical capsules. Duration: 12 weeks ‐ preceded by 4 week washout. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, chronic (no further details). N=49. Age: mean ˜ 47 years, all >21. Sex: 30 M, 19 F. History: prolonged drug treatment, hospitalised patients. Exclusion: co‐existing mental illness, ill health, < 4 weeks of study medication. | |
| Interventions | 1. Loxapine: dose 20 mg‐120 mg/day. N=25.
2. Trifluoperazine: dose 20 mg‐40 mg/day. N=24. Antiparkinsonian or sedative medication as required. |
|
| Outcomes | Global effect.
Adverse effects: TESS.
Dropped from analysis.
Physiological measures. (ophthalmic tests).
Laboratory tests. Unable to use ‐ Global effect: CGI, use of sedation (no data). Mental state: BPRS (no usable data). Behaviour: NOSIE (no usable data). Side effects: use of antiparkinsonian drugs (no data). Physiological measures: BP, ECG, EEG, pulse (no usable data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Pool 1976.
| Methods | Allocation: random (pre arranged procedure). Blinding: double blind, identical capsules in bottles which were numbered only with the person's study number. Duration: 4 weeks, with 5 day washout. | |
| Participants | Diagnosis: schizophrenia confirmed by two psychiatrists (no other details). N=75. Age range: 13‐18 years. Sex: 43 M, 32 F. History: inpatients. | |
| Interventions | 1. Loxapine: dose 10 mg/day increased to 200 mg/day, mean 87.5 mg/day. N=25.
2. Haloperidol: dose 2mg/day increased to 16 mg/day, mean 9.8 mg/day. N=25.
3. Placebo. N=25. Antiparkinsonian medications, sodium amobarbital used as required. |
|
| Outcomes | Global effect: CGI. Unable to use ‐ Behaviour: NOSIE (no data). Physical examinations (no data). Laboratory tests including ECG (no data). |
|
| Notes | Values of CGI are given in percentages. These were rounded to the nearest whole number. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Rifkin 1984.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical capsules, evaluation by psychiatrist blind to drug taken. Duration: 4 weeks. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, paranoid (RDC). N=64. Age: 18‐60 years. Sex: 41 M, 23 F. History: hospitalised. Exclusion: pregnant or risk of, co‐existing mental illnesses, ill health, recent amphetamine abuse, hospitalised patients. | |
| Interventions | 1. Loxapine: dose mean 128.6 mg/day (SD 38). N=31.
2. Chlorpromazine: dose mean 1288 mg/day (SD 358). N=33. Antiparkinsonian or benzodiazepine as required. |
|
| Outcomes | Global effect: CGI.
Leaving the study early. Unable to use ‐ Mental state: BPRS, use of additional sedation (no data). Behaviour: IMPS, NOSIE, Prerbid Asociality Adjustment Scale (no data). Side effects: use of antiparkinsonian drugs (no data). Laboratory tests: (no data). Physiological measures: BP, pulse (no usable data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Schiele 1975.
| Methods | Allocation: random ‐ no further details. Blinding: double ‐ identical opaque capsules. Duration: 12 weeks ‐ preceded by 1 week placebo period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, chronic, no further details. N=64*. Age: mean ˜45 years, range 25‐74. Sex: 50 M, 14 F. History: long term hospitalisation. Exclusion: ill health. | |
| Interventions | 1. Loxapine: dose mean 110 mg/day. N=26.
2. Chlorpromazine: dose mean 1100 mg/day. N=24. Antiparkinsonian or benzodiazepine as required. |
|
| Outcomes | Global effect: CGI.
Adverse effects (use of antiparkinsonian drugs).
Leaving the study early.
Laboratory tests. Unable to use ‐ Mental state: BPRS (no usable data). Behaviour: NOSIE (no usable data). Physiological measures: BP, ECG, ophthalmic tests, pulse (no usable data). |
|
| Notes | *3 people withdrawn from analysis ‐ original group uncertain. 4 people not included in BPRS and CGI analyses ‐ original group clear. Other data is reported for original total. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Selman 1976.
| Methods | Allocation: random ‐ no further details. Blinding: double ‐ identical capsules. Duration: 12 weeks ‐ preceded by 2 week placebo period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, acute or exacerbations of chronic (by 2 psychiatrists). N=87. Age: mean ˜ 32 years. Sex: 69 M, 18 F. History: long term hospitalisation. Exclusion: ill health, < 4 weeks of study medication. | |
| Interventions | 1. Loxapine: dose 50‐150 mg/day. N=29.
2. Haloperidol: dose 4‐12 mg/day. N=29.
3. Placebo. N=29. Antiparkinsonian, chloral hydrate or paraldehyde as required. |
|
| Outcomes | Global effect: CGI.
Adverse effects.
Leaving the study early.
Dropped from analysis. Unable to use ‐ Global effect: BPRS, CGI, NOSIE (analysis of covariance ‐ no usable data). Mental state: BPRS (no usable data). Behaviour: NOSIE (no usable data). Adverse effects: Use of antiparkinsonian drugs (no data). Physiological measures: ECG, ophthalmic tests (no data). Laboratory tests: (no data). |
|
| Notes | 5 categories of "muscle spasms" ‐ impossible to combine data for these outcomes. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Seth 1979.
| Methods | Allocation: random ‐ no further details. Blinding: double ‐ identical capsules. Duration: 12 weeks ‐ preceded by 4 week washout period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, chronic (by 2 psychiatrists). N=72. Age: mean ˜ 30 years, range <20‐49. Sex: 28 M, 36 F, 8 not reported. History: hospitalised patients. Exclusion: ill health, pregnant or risk of, substance abuse. | |
| Interventions | 1. Loxapine: dose 20‐90 mg/day. N=36. 2. Trifluoperazine: dose 5‐45 mg/day. N=36. | |
| Outcomes | Leaving the study early.
Dropped from analysis.
Adverse effects. Unable to use ‐ Global effect: CGI (no usable data). Mental state: BPRS (no usable data). Behaviour: NOSIE (no usable data). Physiological measures: BP, ECG, ophthalmic tests, pulse, temperature, weight (no data). Laboratory tests: (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Shopsin 1972.
| Methods | Allocation: random ‐ no further details. Blinding: double ‐ identical capsules, rated by independant psychologists. Duration: 3 weeks ‐ preceded by 7 day placebo washout period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, acute (SPS), undertaken by 2 psychiatrists. N=30. Age range: 21‐62 years. Sex: male and female (some participants not reported on)**. History: newly hospitalised. Exclusion: ill health, pregnant or risk of, substance abuse, unmanageable behaviour, refusal to take oral medication, spontaneous remission during placebo phase. Inclusion: demonstrating disturbance of affect and association. | |
| Interventions | 1. Loxapine: dose 30‐120 mg/day. N=15.
2. Chlorpromazine: dose 300‐1200 mg/day. N=15. Antiparkinsonian drugs, chlorayl hydrate and paraldehyde as required. |
|
| Outcomes | Global effect (discharge).
Leaving the study early.
Adverse effects: TESS. Unable to use ‐ Global effect: CGI (no usable data). Mental state: BPRS (no usable data), SSRS (no data). Behaviour: NOSIE, IMPS (no usable data). Physiological measures: ECG (no data). Laboratory tests: (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Simpson 1976.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical capsules. Duration: 4 weeks ‐ preceded by 3 day drug free period, evaluation carried out by the same physician, study lasted over 3 years. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, acute (no further details). N=43. Age: mean 32 years, range 16‐61. Sex: 27 M, 16 F. Exclusion: ill health. History: newly hospitalised. | |
| Interventions | 1. Loxapine: dose mean 74 mg/day, range 30‐120 mg/day. N=24.
2. Trifluoperazine: dose mean 35 mg/day, range 20‐50 mg/day. N=19. Antiparkinsonian drugs and chlorayl hydrate as required. |
|
| Outcomes | Global effect (discharge).
Leaving the study early.
Adverse effects (Neurological Rating Scale, Unwanted Effects Checklist). Unable to use ‐ Global effect: CGI (no usable data). Mental state: BPRS (no usable data). Physiological measures: BP, ECG, ophthalmic tests, pulse (no data). Laboratory tests (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Steinbook 1973.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical capsules. Duration: 6 weeks ‐ preceded by 3 day drug free period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, acute (no further details). N=54. Age: mean ˜34 years, range 21‐65. Sex: 16 M, 38 F. Exclusion: ill health, hospitalisation in last 6 months. History: newly admitted. | |
| Interventions | 1. Loxapine: dose range 30‐150 mg/day. N=26.
2. Chlorpromazine: dose range 10‐1200 mg/day. N=28. Antiparkinsonian medication as required. |
|
| Outcomes | Adverse effects (use of antiparkinsonian drugs). Unable to use ‐ Global effect: CGI (no usable data). Mental state: BPRS (no usable data). Behaviour: NOSIE (no usable data). Physiological measures: BP, ECG, ophthalmic tests, pulse (no data). Laboratory tests (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Tu 2004.
| Methods | Allocation: randomised. Blinding: not mentioned. Duration: 8 weeks. Setting: multicenter. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3). N=238. Age: adults, mean ˜32 years, range 21‐43. Sex: 130 M, 108 F. History: hospitalised. | |
| Interventions | 1. Loxapine. dose mean 113 mgs/day. N=126. 2. Chlorpromazine. dose mean 428 mgs/day. N=112. | |
| Outcomes | Global effect: CGI.
Mental state: BPRS.
Laboratory tests: ECG, bloods. Unable to use ‐ Laboratory tests: ECG & haematology (no data) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Tuason 1984.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical capsules. Duration: 4 weeks ‐ preceded by 8 hr drug free period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, paranoid (RDC). N=68. Age: mean ˜ 35 years, range 19‐61 years. Sex: 32 M, 36 F. Exclusion: pregnancy or the risk of, ill health, recent amphetamine abuse. History: mainly people with acute exacerbation of chronic illness, ill <1 week ‐ >6 months ‐ 20 years. | |
| Interventions | 1. Loxapine: dose range 30‐150 mg/day. N=34.
2. Chlorpromazine: dose range 300‐1500 mg/day. N=34. Doses individually titrated, antiparkinsonian and sedative medication as required. |
|
| Outcomes | Leaving the study early. Unable to use ‐ Global effect: CGI, use of additional sedative medication (>50% attrition). Mental state: BPRS (>50% attrition). Behaviour: NOSIE, IMPS (>50% attrition). Side effects: use of antiparkinsonian medication (>50% attrition). Physiological measures: BP, ECG, pulse (>50% attrition). Laboratory tests (>50% attrition). |
|
| Notes | Loss to follow up 70%. Only data from the outcome 'leaving the study early' included. People who had improved were discharged and not followed up ‐ adds to dropout over the 50% cut off point. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Tuason 1986.
| Methods | Allocation: randomised ‐ no further details. Blinding: "modified double" ‐ staff administering drugs not blinded, assessments blind. Duration: 24‐72 hours (IM phase) ‐ oral phase data not included. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, acutely psychotic (DSM‐III used beyond 3 days). N=54. Age: mean ˜35 years (SD ˜ 10), range 18‐65 years. Sex: 33 M, 19 F, 2 not reported. History: newly admitted. Inclusion criteria: > 7 on BPRS hostility & uncooperativeness, behaviour = hostile/aggressive/uncooperative/unmanageable. Exclusion: ill health, co‐existing mental illness condition. | |
| Interventions | 1. Loxapine: dose 25 mg IM, then 12.5‐25 mg/hour IM, max. 250 mg/day. N=25.
2. Haloperidol: dose 5mg IM, then 2.5‐5mg/hour IM, max. 100 mg/day. N=29. Antiparkinsonian drugs and chlorayl hydrate as required. |
|
| Outcomes | General effect (requiring extended period of medication > 24 hours).
Mental state: BPRS.*
Side effects (sedation ‐ ESBE, use of antiparkinsonian drugs).
Dropped from analysis.
Leaving the study early. Unable to use ‐ Global effect: CGI (no data). |
|
| Notes | 2 people withdrawn from analysis ‐ original group unclear. * BPRS scores reported with SD. Data thought not to be SD but standard error and reviewers have converted to SD. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Van Der Velde 1975.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical capsules. Duration: 6 weeks ‐ preceded by 14 day drug free period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, acute or acute exacerbation (by 2 psychiatrists & principal investigator). N=82. Age: mean ˜ 27 years. Sex: 43 M, 33 F, 6 not reported. Exclusion: not completing 2 weeks of study medication. History: 18/82 first episode, rest onset in last 6 years. | |
| Interventions | 1. Loxapine: dose range 50‐150 mg/day. N=26.
2. Thiothixene: dose range 20‐60 mg/day. N=28.
3. Placebo. N=28. Antiparkinsonian drugs and chlorayl hydrate as required. |
|
| Outcomes | Dropped from analysis.
Leaving the study early.
Adverse effects.
Laboratory tests. Unable to use ‐ Efficacy: (variance analyses ‐ no usable data). Global effect: CGI (no usable data). Mental state: BPRS (no usable data). Behaviour: NOSIE (no usable data). Adverse effects: Use of antiparkinsonian drugs (no data). Physiological measures: BP, EEG, pulse, temperature, weight (no data). |
|
| Notes | 6 people withdrawn from analyses ‐ original group clear so ITT analysis possible. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Vyas 1980.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ identical capsules. Duration: 6 months ‐ preceded by 15 day antipsychotic free period. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia, chronic ‐ no further details. N=30. Age: mean ˜ 32 years (SD ˜ 9), all >21. Sex: 17 M, 13 F. Exclusion: pregnant women, ill health, substance abuse. History: inpatients, hospitalisation duration <2 weeks‐>2 years. | |
| Interventions | 1. Loxapine: dose mean 44 mg/day, range 30‐90 mg/day. N=15.
2. Chlorpromazine: dose mean 453 mg/day, range 300‐900 mg/day. N=15. Antiparkinsonian drugs as required. |
|
| Outcomes | Global effect: CGI.
Leaving the study early.
Adverse effects. Unable to use ‐ Efficacy: (analyses of covariance ‐ no usable data). Mental state: BPRS (no usable data). Behaviour: NOSIE (no usable data). Adverse effects: Use of antiparkinsonian drugs (no usable data). Physiological measures: BP, ECG. pulse, ophthalmic tests, temperature, weight (no data). Laboratory tests: (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Wang 1996.
| Methods | Allocation: randomised ‐ no further details. Blinding: double blind. Duration: 8 weeks. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia (CCMD‐2). N=60. Age: mean ˜36 years, range 18‐60. Sex: M & F ‐ no further details. History: hospitalised. | |
| Interventions | 1. Loxapine: dose range 50‐300 mg/day. N=30. 2. Chlorpromazine: dose range 75 mg‐600 mg/day. N=30. | |
| Outcomes | Global effect: CGI.
Mental state: BPRS.
Adverse effects: TESS.
Physiological measures: Temperature, BP, Weight. Unable to use ‐ Physiological measures: temperature, BP, weight (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Wang 2005a.
| Methods | Allocation: randomised ‐ no further details. Blinding: not mentioned. Duration: 8 weeks Setting: single centre. | |
| Participants | Diagnosis: schizophrenia. N=68. Age: mean ˜33 years. Sex: 32 M, 36 F. History: duration ill 3‐12 months. | |
| Interventions | 1. Loxapine: dose mean 267 mg/day. N=34. 2. Quetiapine: dose mean 426 mg/day. N=34. | |
| Outcomes | Mental state: PANSS. Unable to use ‐ Adverse effects: TESS (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Wang 2005b.
| Methods | Allocation: randomised ‐ no further details. Blinding: not mentioned. Duration: 8 weeks. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3). N=63. Age: mean ˜29 years. Sex: 36 M, 30 F. History: hospitalised. | |
| Interventions | 1. Loxapine: dose mean 86 mgs/day. N=32. 2. Risperidone: dose mean 4 mgs/day. N=31. | |
| Outcomes | Mental state: PANSS.
Adverse effects: TESS.
Laboratory tests: ECG. Unable to use ‐ Haematology (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Xue 2004.
| Methods | Allocation: randomised ‐ no further details. Blinding: double blind. Duration: 8 weeks. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3). N=200. Age: mean ˜32 years. Sex: 90 M, 110 F. History: in community. | |
| Interventions | 1. Loxapine: dose range 34‐136 mg/day. N=100. 2. Chlorpromazine: dose range 250‐500 mg/day. N=100. | |
| Outcomes | Mental state: BPRS.
Adverse effects: TESS. Unable to use ‐ Laboratory tests: Bloods, ECG, EEG (no data) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Zhang 2005.
| Methods | Allocation: randomised ‐ no further details. Blinding: not mentioned. Duration: 8 weeks. Setting: single centre. | |
| Participants | Diagnosis: schizophrenia (CCMD3). N=134. Age: mean ˜30, range 18‐60. Sex: not reported. History: hospitalised, mean duration ill ˜ 4 years. | |
| Interventions | 1. Loxapine: dose range: 34‐68 mgs/day. N=44. 2. Perphenazine: dose range: 6‐12mgs/day. N=46. 3. Sulpride: dose range 300‐400 mgs/day. N=44. | |
| Outcomes | Mental state: PANSS. Unable to use ‐ Adverse effects: TESS (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
General BP ‐ blood pressure IM ‐ intramuscular ECG‐ electro cardiogram ITT ‐ intention‐to‐treat
Diagnostic tools RDC ‐ Research Diagnostic Criteria ICD‐ International classification of diseases
Global state scale CGI ‐ Clinical Global Impression ( CGI‐I ‐ Improvement , CGI‐S ‐ Severity)
Mental state scales BPRS ‐ Brief Psychiatric Rating Scale PANSS ‐ Postive and Negative Symptom Score SPS ‐ Symptom profile for schizophrenia SSRS ‐ Self rating Symptom Scale
Behaviour scale NOSIE ‐ Nurse's Observation Scale for In‐patient Evaluation IMPS ‐ Inpatient Multidimensional Psychiatric Scale
Side effect scales ESBE ‐ Evaluation of Sedative and Behavioral Effects of Parental Treatment TESS ‐ Treatment Emergent Symptom Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ananth 1980 | Allocation: not randomised, review article. |
| Bishop 1977 | Allocation: not randomised, review article. |
| Branchey 1981 | Allocation: randomised. Participants: people with chronic schizophrenia. Intervention: continuing or discontinuing loxapine, not initiation of loxapine as per protocol. |
| Brown 2003 | Allocation: randomised. Participants: people with any mental illness and with cocaine and amphetamine dependence. Intervention: continuing typical antipsychotic versus discontinuing typical antipsychotic and starting quetiapine, not starting loxapine as per protocol. |
| Bueno 1979 | Allocation: randomised. Participants: people with schizophrenia. Intervention: loxapine versus haloperidol. Outcomes no usable data. |
| Burdock 1974 | Allocation: not randomised, methodology paper. |
| Cottereau 1979 | Allocation: not randomised, case series. |
| Delteil 1980 | Allocation: not randomised, case series. |
| Jones 2006 | Allocation: random. Participants: people with schizophrenia. Interventions: 1st generation antipsychotics vs 2nd generation antipsychotics (rather than loxapine vs another treatment). |
| Leone 1982 | Allocation: random. Participants: people with borderline personality disorder, not schizophrenia. |
| Lewis 2006 | Allocation: random. Participants: people with schizophrenia. Interventions: 1st generation antipsychotics vs 2nd generation antipsychotics (rather than loxapine vs another treatment). |
| Lourido 1979 | Allocation: random. Participants: people with chronic psychoses associated with either organic brain syndrome or mental retardation, not clearly schizophrenia. |
| Maes 1996 | Allocation: not clear. Participants: either healthy people or those with schizophrenia. Interventions: loxapine versus placebo. Outcomes: plasma levels, no clinical outcomes. |
| Mahmoud 2004 | Allocation: random but open label. |
| Martin 1982 | Allocation: not randomised, case series. |
| Mattke 1975 | Allocation: not randomised, patients matched. |
| Nair 1976 | Allocation: not randomised, case series. |
| Paprocki 1976 | Allocation: not randomised, review article. |
| Paprocki 1977 | Allocation: randomised by consecutive admission, quality rating 'C'. |
| Rainaut 1975 | Allocation: randomised. Participants: people with schizophrenia. Interventions: loxapine versus thioridazine. Outcomes no usable data. |
| Serafetinides 1971 | Allocation: random. Participants: people with schizophrenia. Interventions: loxapine versus chlorpromazine. Outcomes: EEG readings, no clinical outcomes. |
| Serafetinides 1973 | Pooled data from 4 randomised control trials: Study 1 Allocation: no details, double blind. Participants: people with chronic schizophrenia. Intervention: chlorpromazine. Study 2. Allocation: no details, double blind. Participants: people with chronic schizophrenia. Intervention: haloperidol versus clopenthixol versus chlorpromazine. Study 3. Allocation: no details, double blind. Participants: people with chronic schizophrenia. Intervention: chlorpromazine versus molindone. Study 4. Allocation: no details, double blind. Participants: people with chronic schizophrenia. Intervention: chlorpromazine versus loxapine. Outcomes: EEG readings, no clinical outcomes available. |
| Simpson 1976b | Allocation: not randomised, case series. |
| Simpson 1978 | Allocation: not randomised. Participants: people with chronic schizophrenia. Intervention: oral loxapine versus parenteral loxapine, not loxapine versus placebo or any other intervention. |
| Ucer 1979 | Allocation: not randomised, case series. |
| Versiani 1978 | Allocation: by consecutive admission, quality 'C'. |
| Versiani 1980 | Allocation: random. Participants: chronic psychosis associated with organic brain syndrome or mental retardation, not clearly schizophrenia (two studies). |
| Vianna Filho 1975 | Allocation: by consecutive admission, quality rating 'C'. |
| Yu 2004 | Allocation: not randomised. |
Contributions of authors
Contributions: Mark Fenton ‐ prepared protocol, selected and acquired studies, extracted data, summated data, produced report.
Brendan Murphy ‐protocol writing, selected studies, extracted data, summated data.
Anne‐Marie Bagnall ‐ prepared protocol, undertook searches, selected and acquired studies, extracted data, summated data, produced report..
Pierre Chue ‐ data extraction from French studies, report writing.
Maria Leitner ‐ finding funding, inital input into the protocol and searches.
Sources of support
Internal sources
Leeds Social Services, UK.
Jansen Cilag, UK.
Centre for Reviews and Dissemination, University of York, UK.
Cochrane Schizophrenia Group, UK.
External sources
Centre for Reviews and Dissemination, University of York., UK.
Declarations of interest
Mark Fenton has lead Janssen, Lilly and Zeneca sponsored workshops for clinicians.
Jo Wood is Medical Information Officer for Janssen Cilag UK, and worked on this review to increase her knowledge of the process of systematic reviewing.
The Cochrane Schizophrenia Group has received general support funding from Eli Lilly during the years 1996‐9 (see Group Module).
Edited (no change to conclusions)
References
References to studies included in this review
Bagadia 1980 {published data only}
- Bagadia VN, Shah LP, Abhyankar RR. A double‐blind controlled trial of loxapine and trifluoperazine in adolescent schizophrenia. Current Therapeutic Research, Clinical and Experimental 1980;27(6):886‐96. [MEDLINE: ; PMID 361345] 361345 [Google Scholar]
Bishop 1970 {published data only}
- Bishop MP, Gallant DM. Loxapine: a controlled evaluation in chronic schizophrenic patients. Current Therapeutic Research ‐ Clinical and Experimental 1970;12(9):594‐7. [PubMed] [Google Scholar]
Charalampous 1974 {published data only}
- Charalampous KD, Freemesser GF, Malev J, Ford K. Loxapine succinate: A controlled double blind study in schizophrenia. Current Therapeutic Research 1974;6(8):829‐37. [PubMed] [Google Scholar]
Clark 1972 {published data only}
- Clark ML, Huber WK, Sullivan J, Wood F, Costiloe JP. Evaluation of loxapine succinate in chronic schizophrenia. Diseases of the Nervous System 1972;33:783‐91. [PubMed] [Google Scholar]
Clark 1975 {published data only}
- Clark ML, Paredes A, Costiloe JP, Wood F, Barrett A. Loxapine in newly admitted chronic schizophrenic patients. Journal of Clinical Pharmacology 1975;15(4):286‐94. [DOI] [PubMed] [Google Scholar]
Clark 1977 {published data only}
- Clark ML, Paredes A, Costiloe JP, Fulkerson FG, Wood F. Evaluation of two dose levels of loxapine succinate in chronic schizophrenia. Diseases of the Nervous System 1977;38(1):7‐10. [PubMed] [Google Scholar]
Du 2003 {published data only}
- Du B‐G, Fang R‐L. Control studies on curative effects of loxapine succinate and risperidone in schizophrenics. Journal of Clinical Psychosomatic Diseases 2003;9(1):16‐9. [CAJ] [Google Scholar]
Dube 1976 {published data only}
- Dube KC, Kumar N. Loxapine succinate: a comparative study with chlorpromazine. Current Therapeutic Research ‐ Clinical and Experimental 1976;19(6):653‐60. [PubMed] [Google Scholar]
Dubin 1996 {published data only}
- Dubin WR, Weiss KJ. Rapid tranquilization: a comparison of thiothixene with loxapine. Journal of Clinical Psychiatry 1986;47(6):294‐7. [PubMed] [Google Scholar]
Fruensgaard 1977 {published data only}
- Fruensgaard K, Korsgaard S, Jorgensen H, Jensen K. Loxapine versus haloperidol parenterally in acute psychosis with agitation. A double‐blind study. Acta Psychiatrica Scandinavica 1977;56(4):256‐64. [DOI] [PubMed] [Google Scholar]
Fruensgaard 1978 {published data only}
- Fruensgaard K, Wollenberg J, Hansen KM, Fensbo C, Sihm F. Loxapine versus perphenazine in psychotic patients. A double‐blind, randomized, multicentre trial. Current Medical Research and Opinion 1978;5(8):601‐7. [MEDLINE: ; PMID 361345] [DOI] [PubMed] [Google Scholar]
Gallant 1971 {published data only}
- Gallant, Bishop. Oxilapine versus Trifluoperazine. Psychopharmacology Bulletin 1971;7(1):40‐3. [08024] [Google Scholar]
Huang 1997 {published data only}
- Huang S, Qin Y, Wang L. Study on the efficacy of loxapine in treatment of schizo phrenia in comparison with chlorpromazine. Chinese New Drugs Journal 1997;6(3):161‐4. [MEDI9706] [Google Scholar]
Kiloh 1976 {published data only}
- Kiloh LG, Williams SE, Grant DA, Whetton PS. A double‐blind comparative trial of loxapine and trifluoperazine in acute and chronic schizophrenic patients. Journal of International Medical Research 1976;4(6):441‐8. [MEDLINE: ; PMID 800384] [DOI] [PubMed] [Google Scholar]
Kramer 1978 {published data only}
- Kramer MR, Thomas SPJ, Zorick FJ, Blackwell B. Relative efficacy and safety of loxapine succinate (Loxitane) and thioridazine hydrochloride (Mellaril) in the treatment of acute schizophrenia. Current Therapeutic Research 1978;23(5):619‐31. [Google Scholar]
Li 2004 {published data only}
- Li Y, Shang J, Li C. Control study of loxapine succinate and clozapine on curative effects in treatment of schizophrenics. Heath Psychology Journal 2004;12(3):202. [115139] [Google Scholar]
Li 2005a {published data only}
- Li L‐X, Li C‐Y. A comparative study of loxapine succinate and risperidone in the treatment of schizophrenia. Shandong Archives of Psychiatry 2005;18(4):246‐7. [CAJ MEDI0510] [Google Scholar]
Li 2005b {published data only}
- Li Zhong‐yi, HE Rui‐fang. Comparative study between loxapine and risperedone in treatment of schizophrenia. Journal of clinical psychological medicine 2005;15(1):11‐12. [MEDLINE: ] [Google Scholar]
Liu 2005 {published data only}
- Liu. Control study of loxapine and chlorpromazine in the treatment of schizophrenia. Medical Journal of Chinese People's Health 2005;17(9):503‐4. [Google Scholar]
Lu 2003 {published data only}
- Lu X‐Q, Zhu G‐L. Control studies of loxapine succinate and clozapine in treatment of schizophrenia. Journal of Clinical Psychosomatic Diseases 2003;9(3):157‐8. [115139] [Google Scholar]
Malik 1980 {published data only}
- Malik SC, Kumar K. Loxapine in adolescent schizophrenia ‐ a comparative study with trifluoperazine. Current Therapeutic Research ‐ Clinical and Experimental 1980;28(3):432‐46. [Google Scholar]
Moore 1975 {published data only}
- Moore DF. Treatment of acute schizophrenia with loxapine succinate (Loxitane) in a controlled study with chlorpromazine. Current Therapeutic Research 1975;18(1):172‐80. [PubMed] [Google Scholar]
Moyano 1975 {published data only}
- Moyano CZ. A double‐blind comparison of loxitane‐‐loxapine succinate and trifluoperazine hydrochloride in chronic schizophrenic patients. Diseases of the Nervous System 1975;36(6):301‐4. [PubMed] [Google Scholar]
Pool 1976 {published data only}
- Pool D, et al. A controlled evaluation of loxitane in seventy‐five adolescent schizophrenic patients. Current Therapeutic Research 1976;19(1):99‐104. [PsycINFO 1976‐10411‐001] [PubMed] [Google Scholar]
Rifkin 1984 {published data only}
- Rifkin A, Rieder E, Sarantakos S, Saraf K, Kane J. Is loxapine more effective than chlorpromazine in paranoid schizophrenia?. American Journal of Psychiatry 1984;141(11):1411‐3. [DOI] [PubMed] [Google Scholar]
Schiele 1975 {published data only}
- Schiele BC. Loxapine succinate: a controlled double‐blind study in chronic schizophrenia. Diseases of the Nervous System 1975;36(7):361‐4. [PubMed] [Google Scholar]
Selman 1976 {published data only}
- Selman FB, McClure RF, Helwig DH. Loxapine succinate ‐ A double‐blind comparison with haloperidol and placebo in acute schizophrenics [Loxapine succinate ‐ essai comparatif en double aveugle avec l'haloperidol et un placebo dans les psychoses aigues]. Psychologie Medicale 1979;11(7):1533‐40. [Google Scholar]
- Selman FB, McClure RF, Helwig H. Loxapine succinate: A double‐blind comparison with haloperidol and placebo in acute schizophrenics. Current Therapeutic Research 1976;19(6):645‐52. [PubMed] [Google Scholar]
Seth 1979 {published data only}
- Seth S, Mahal AS, Kumar KA. A double‐blind comparative trial of loxapine and trifluperazine in chronic schizophrenic patients. Current Therapeutic Research 1979;25(2):320‐9. [Google Scholar]
Shopsin 1972 {published data only}
- Gershon S. Loxapine vs chlorpromazine. Early clinical drug evaluation units report 1972;9:67‐70. [Google Scholar]
- Shopsin B, Pearson E, Gershon S, Collins P. A controlled double‐blind comparison between loxapine succinate and chlorpromazine in acute newly hospitalized schizophrenic patients. Current Therapeutic Research ‐ Clinical and Experimental 1972;14(11):739‐48. [PubMed] [Google Scholar]
Simpson 1976 {published data only}
- Simpson GM, Cuculic Z. A double‐blind comparison of loxapine succinate and trifluoperazine in newly admitted schizophrenic patients. Journal of Clinical Pharmacology 1976;16(1):60‐5. [DOI] [PubMed] [Google Scholar]
Steinbook 1973 {published data only}
- Steinbook R, Goldstein BJ, Brauzer B, Moreno SS, Jacobson AF. Loxapine: A double blind comparison with chlorpromazine in acute schizophrenic patients. Current Therapeutic Research ‐ Clinical and Experimental 1973;15(1):1‐7. [PubMed] [Google Scholar]
Tu 2004 {published data only}
- Tu Z‐M, Gao S‐R, Ye S‐N. Controlled study of loxapine and chlorpromazine in the treatment of schizophrenia. Medical Journal of Chinese People Health 2004;16(7):415‐7. [MED10505] [Google Scholar]
Tuason 1984 {published data only}
- Tuason VB, Escobar JI, Garvey M, Schiele B. Loxapine versus chlorpromazine in paranoid schizophrenia: A double blind study. Journal of Clinical Psychiatry 1984;45(4):158‐63. [PubMed] [Google Scholar]
Tuason 1986 {published data only}
- Tuason VB. A comparison of parenteral loxapine and haloperidol in hostile and aggressive acutely schizophrenic patients. Journal of Clinical Psychiatry 1986;47(3):126‐9. [PubMed] [Google Scholar]
Van Der Velde 1975 {published data only}
- Velde CD, Kiltie H. Effectiveness of loxapine succinate in acute schizophrenia: A comparative study with thiothixene. Current Therapeutic Research 1975;17(1):1‐12. [PubMed] [Google Scholar]
Vyas 1980 {published data only}
- Vyas BK, Kalla V. A six‐month double‐blind comparison of loxapine succinate and chlorpromazine in chronic schizophrenic patients. Current Therapeutic Research ‐ Clinical and Experimental 1980;28(1):16‐30. [Google Scholar]
Wang 1996 {published data only}
- Wang L, Wang Z, Wang Y. Using loxapine succinate and chlorpromazine for treating schizophrenia. Tianjing Medical Journal 1996;24(11):672‐5. [115139] [Google Scholar]
Wang 2005a {published data only}
- Wang. Control study of Loxapine and Quetiapine treating first onset schizophrenia. Chinese journal of behavioural medical science 2005;14(8):752. [Google Scholar]
Wang 2005b {published data only}
- Wang H‐Y, Wang G‐P, Pei G‐X. A comparative study of loxapine and risperidone in the treatment of schizophrenic patients. Shandong Archives of Psychiatry 2005;18(1):28‐9. [CAJ] [Google Scholar]
Xue 2004 {published data only}
- Xue S‐J, Huang J‐M, Yang M‐Z. A comparison study on loxapine and chlorpromazine in the treatment of schizophrenia. Medical Journal of Chinese People Health 2004;16(10):593‐4. [115139] [Google Scholar]
Zhang 2005 {published data only}
- Zhang. Control study of Loxapine treatment in schizophrenia. Sichuan Mental Health 2005;18(4):222‐4. [Google Scholar]
References to studies excluded from this review
Ananth 1980 {published data only}
- Ananth, JV, Steen N. The place of loxapine ‐ a new neuroleptic drug. Psychiatric Journal of the University of Ottawa 1980;5(4):279‐82. [Google Scholar]
Bishop 1977 {published data only}
- Bishop MP, Simpson GM, Dunnett CW, Kiltie H. Efficacy of loxapine in the treatment of paranoid schizophrenia. Psychopharmacology 1977;51:107‐15. [DOI] [PubMed] [Google Scholar]
Branchey 1981 {published data only}
- Branchey MH, Branchey LB, Richardson MA. Effects of neuroleptic adjustment on clinical condition and tardive dyskinesia in schizophrenic patients. American Journal of Psychiatry 1981;138(5):608‐12. [MEDLINE: ; PMID 7015884] [DOI] [PubMed] [Google Scholar]
- Branchey MH, Branehey LB, Richardson MA. Effects of gradual decrease and discontinuation of neuroleptics on clinical condition and tardive dyskinesia. Psychopharmacology Bulletin 1981;17(1):118‐20. [MEDLINE: ; PMID 7232639] [PubMed] [Google Scholar]
Brown 2003 {published data only}
- Brown ES, Nejtek VA, Perantie DC, Thomas NR, Rush AJ. Cocaine and amphetamine use in patients with psychiatric: Illness a randomized trial of typical antipsychotic continuation or discontinuation. Journal of Clinical Psychopharmacology 2003;23(4):384‐8. [EMBASE 2003319642] [DOI] [PubMed] [Google Scholar]
Bueno 1979 {published data only}
- Bueno JR. A double‐blind comparative clinical trial with loxapine succinate and haloperidol in the treatment of Schizophrenia [Ensaio clinico comparativo e duplo‐cego com succina de loxapina e haloperidol no tratamento da esquizofrenia]. Folha Medica 1979;78(1):47‐52. [CENTRAL 79169917; CENTRAL CN‐00316048] [Google Scholar]
Burdock 1974 {published data only}
- Burdock EI, Gershon S, Hardesty AS, Linden KJ. Problems and profits of cross‐national collaborative studies. Diseases of the Nervous System 1974;35(7, Sect 2):31‐2. [PubMed] [Google Scholar]
Cottereau 1979 {published data only}
- Cottereau MJ, Poirier MF, Loo H, Deniker P. The loxapine succinate: A new major tranquillizer [Le succinate de loxapine: un nouveau neuroleptique]. Encephale 1979;5:251‐67. [PubMed] [Google Scholar]
Delteil 1980 {published data only}
- Delteil P. Clinical trial of a new neuroleptic, loxapine [Essai clinique d'un nouveau neuroleptique la loxapine]. Psychiatrique 1980;56:915‐6. [Google Scholar]
Jones 2006 {published data only}
- Jones PB, Barnes TRE, Davies L, Dunn G, Lloyd H, Hayhurst KP, Murray RM, Markwick A, Lewis SW. Randomized controlled trial of the effect on quality of life of second‐ vs first‐generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1). Archives of General Psychiatry 2006;63(10):1079‐87. [MEDLINE: ; EMBASE 2006486244] [DOI] [PubMed] [Google Scholar]
Leone 1982 {published data only}
- Leone NF. Response of borderline patients to loxapine and chlorpromazine. Journal of Clinical Psychiatry 1982;43(4):148‐50. [PubMed] [Google Scholar]
Lewis 2006 {published data only}
- Lewis SW, Davies L, Jones PB, Barnes TRE, Murray RM, Kerwin R, Taylor D, Hayhurst KP, Markwick A, Lloyd H, Dunn G. Randomised controlled trials of conventional antipsychotic versus new atypical drugs, and new atypical drugs versus clozapine, in people with schizophrenia responding poorly to, or intolerant of, current drug treatment. Health Technology Assessment 2006;10(17):iii‐xi, 1‐182. [MEDLINE: ; EMBASE 2006300640; CINAHL 2009206784] [DOI] [PubMed] [Google Scholar]
Lourido 1979 {published data only}
- Lourido E, Santa Cruz A. Comparative treatment of chronic psychoses with loxapine and thioridazine: a double‐blind clinical study. Current Therapeutic Research 1979;25(5):681‐9. [Google Scholar]
Maes 1996 {published data only}
- Maes M, Bosmans E, Ranjan R, Vandoolaeghe E, et al. Lower plasma CC16, a natural anti‐inflammatory protein, and increased plasma interleukin‐l receptor antagonist in schizophrenia: Effects of antipsychotic drugs. Schizophrenia Research 1996;21(1):39‐50. [DOI] [PubMed] [Google Scholar]
Mahmoud 2004 {published data only}
- Mahmoud RA, Engelhart LM, Janagap CC, Oster G, Ollendorf D. Risperidone versus conventional antipsychotics for schizophrenia and schizoaffective disorder. Clinical Drug Investigation 2004;24(5):275‐86. [DOI] [PubMed] [Google Scholar]
Martin 1982 {published data only}
- Martin A. A multi‐center study of the effect of loxapine on 403 hospitalized, chronically psychotic patients [Efficacite et tolerance de la loxapine a propos d'un essai multicentrique sur 403 psychotiques hospitalises]. Psychologie Medicale 1982;14(9):1439‐52. [Google Scholar]
Mattke 1975 {published data only}
- Mattke DJ, Mombour W, Glotzner FL. The assesment of neuroleptogenic extrapyramidial syndroms in psychopharmalogical research [Erfassung neuroleptisch bedingter extrapymidaler syndrome in einem psychopharmakologischen forschungsprojekt]. Pharmakopsychiat 1975;1:36‐44. [DOI] [PubMed] [Google Scholar]
Nair 1976 {published data only}
- Nair NP, Decker BL, Schwartz G. Loxapine succinate in the treatment of chronic schizophrenia. Current Therapeutic Research 1976;20(6):802‐9. [PubMed] [Google Scholar]
Paprocki 1976 {published data only}
- Paprocki J, Barcala Peixoto MP, Andrade NM. A controlled double‐blind comparison between loxapine and haloperidol in acute newly hospitalized schizophrenic patients. Psychopharmacoly Bulletin 1976;12(2):32‐4. [PubMed] [Google Scholar]
Paprocki 1977 {published data only}
- Paprocki J, Versiani M. A double‐blind comparison between loxapine and haloperidol by parenteral route in acute schizophrenia. Current Therapeutic Research 1977;21(1):80‐100. [PubMed] [Google Scholar]
Rainaut 1975 {published data only}
- Rainaut J. Statistical analysis of the activity of loxapine and thioridazine in a double‐blind study [Analyse statistique de l'etude comparative double insu de l'activite de la loxapine et de la thioridazine]. Encephale 1975;1(2):147‐60. [MEDLINE: ; PMID 1100361] [PubMed] [Google Scholar]
Serafetinides 1971 {published data only}
- Serafetinides EA, Willis D, Clark ML. The EEG effects of dibenzoxazepines (loxapine succinate) as compared to CPZ: EEG changes as drug side effects. International Pharmacopsychiatry 1971;6(1):38‐44. [DOI] [PubMed] [Google Scholar]
Serafetinides 1973 {published data only}
- Serafetinides EA. Consistency and similarity of drug EEG responses in chronic schizophrenic patients. International Pharmacopsychiatry 1973;8(4):214‐6. [MEDLINE: ; PsycINFO 52‐05954; PMID 4589640] [DOI] [PubMed] [Google Scholar]
Simpson 1976b {published data only}
- Branchey MH, Branchey LB, Richardson MA. Effects of neuroleptic adjustment on clinical condition and tardive dyskinesia in schizophrenic patients. American Journal of Psychiatry 1981;138(5):608‐12. [DOI] [PubMed] [Google Scholar]
- Simpson GM, Branchey MH, Lee JH, Varga E. Two year trial of loxapine succinate in chronic psychotic patients. Diseases of the Nervour System 1976;37(5):305‐7. [PubMed] [Google Scholar]
Simpson 1978 {published data only}
- Simpson GM, Cooper TB, Lee JH, Young MA. Clinical and plasma level characteristics of intramuscular and oral loxapine. Psychopharmacology 1978;56(2):225‐32. [MEDLINE: ; PMID 417377] [DOI] [PubMed] [Google Scholar]
Ucer 1979 {published data only}
- Ucer E, Casey P. Effect of loxapine succinate in acutely ill schizophrenic outpatients. Current Therapeutic Research 1979;25(1):144‐9. [Google Scholar]
Versiani 1978 {published data only}
- Versiani M, Silva JA, Frota LH, Mundim FD. Double‐blind comparison between loxapine and haloperidol in the treatment of adolescent schizophrenic patients. Current Therapeutic Research 1978;24(5):559‐66. [Google Scholar]
Versiani 1980 {published data only}
- Versiani M, Silva JAR, Mundim FD. Loxapine versus thioridazine in the treatment of organic psychosis. Journal of International Medical Research 1980;8(1):22‐30. [DOI] [PubMed] [Google Scholar]
Vianna Filho 1975 {published data only}
- Vianna Filho U, Versiani Caldeira MV, Bueno J. Preliminary report on a double blind study comparing loxapine succinate with thiothixene in chronic schizophrenic patients. Jornal Brasileiro de Psiquiatria 1975;24(1):5‐27. [Google Scholar]
- Vianna‐Filho U, Versiani Caldeira V, Romildo Bueno J. The efficacy and safety of loxapine succinate in the treatment of schizophrenia: a comparative study with thiothixene. Current Therapeutic Research ‐ Clinical and Experimental 1975;18(3):476‐90. [PubMed] [Google Scholar]
Yu 2004 {published data only}
- Yu H, Luo J‐Y, Xia S‐Y. Treatment of 44 cases of refractory schizophrenia with loxapine. Herald of Medicine 2004;23(10):723‐5. [MEDLINE: ] [Google Scholar]
Additional references
Adams 1999
- Adams CE, Gray R, Daniels J, Thornley B, Philpott H, Wahlbeck K. A survey of UK consultant psychiatrists first and second choice drug treatments for schizophrenia. Schizophrenia Trials Meeting, Stratford‐upon‐Avon. 5‐9th May, 1999.
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [LOX020600] [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). 4th Edition. Washington DC: American Psychiatric Association, 1994. [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, Pitkin R, Rennie D, Schulz KF, Simel D, Stroup DF. Improving the quality of randomized controlled trials. The CONSORT statement. JAMA 1996;276:637‐9. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, Boutitie F, Nony P, Haugh M, Mignot G. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use. Therapie 1999;54(4):405‐11. [PubMed] [Google Scholar]
Buckley 1997
- Buckley PF. New dimensions in the pharmacologic treatment of schizophrenia and related psychoses. Journal of Clinical Pharmacology 1997;37:363‐78. [DOI] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT Jr, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Casey 1997
- Casey DE. The relationship of pharmacology to side effects. Journal of Clinical Psychiatry 1997;58(10):55‐62. [PubMed] [Google Scholar]
Chalmers 1983
- Chalmers TC, Celano P, Sacks HS, Smith H Jr. Bias in treatment assignment in controlled clinical trials. New England Journal of Medicine 1983;309:1358‐61. [DOI] [PubMed] [Google Scholar]
CWG 1998
- Collaborative Working Group on Clinical Trial Evaluations. Adverse effects of the atypical antipsychotics. Journal of Clinical Psychiatry 1998;59:17‐22. [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Abstracts of 8th International Cochrane Colloquium; 2000 Oct 25‐28th; Cape Town, South Africa. 2000.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elwood 1982
- Elwood PC. Randomised controlled trials: sampling. British Journal of Clinical Pharmacology 1982;13:631‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Glazer 1997
- Glazer WM, Johnstone BM. Pharmacoeconomic evaluation of antipsychotic therapy for schizophrenia. Journal of Clinical Psychiatry 1997;58(10):50‐4. [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83 876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. Early clinical drug evaluation (ECDEU) assessment manual for psychopharmacology: National Institute of Mental Health Publication No.76‐338. Washington DC. National Institute of Mental Health Publication 1976; Vol. 76‐338:217‐22.
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]. The Cochrane Library. Chichester, UK: John Wiley & Sons, Ltd., 2005. [Google Scholar]
Hollis 1999
- Hollis S, Campbell F. What is meant by intention to treat analysis? Survey of published randomised controlled trials. BMJ 1999;319:670‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kane 1996
- Kane JM. Factors which can make patients difficult to treat. British Journal of Psychiatry 1996;169(31):10‐4. [PubMed] [Google Scholar]
Kay 1986
- Kay SR. Positive and negative symptom (PANNS) scale manual. Schizophrenia Bulletin 1987;13:261‐78. [DOI] [PubMed] [Google Scholar]
Kerwin 1994
- Kerwin RW. The new atypical antipsychotics. A lack of extrapyramidal side‐effects and new routes in schizophrenia research. British Journal of Psychiatry 1994;164:141‐8. [DOI] [PubMed] [Google Scholar]
Kerwin 1996
- Kerwin R, Taylor D. New antipsychotics. A review of their current status and clinical potential. CNS Drugs 1996;6:71‐82. [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale Scores. British Journal of Psychiatry 2005;187:366‐71. [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. What does the PANSS mean?. Schizophrenia Research 2005;79:231‐8. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, Tugwell P, Klassen TP. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352:609‐13. [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Silverstone 1995
- Silverstone T, Turner P. Drug treatment in psychiatry.. TJ Press, 1995. [Google Scholar]
Thomas 1998
- Thomas CS, Lewis C. Which atypical antipsychotic?. British Journal of Psychiatry 1998;172:106‐9. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Vanelle 1994
- Vanelle JM, Olie JP, Levy‐Soussan P. New antipsychotics in schizophrenia: the French Experience. Acta Psychiatrica Scandinavica, Supplementum 1994;380:59‐63. [DOI] [PubMed] [Google Scholar]
Wood MacKenzie 1998
- Wood MacKenzie. Review of the Global Schizophrenia Market. PharmaForum 1998;39:9. [Google Scholar]
References to other published versions of this review
Murphy 2000
- Murphy BE, Bagnall A, Chue P, Fenton M, Leitner M, Wood J. Loxapine for schizophrenia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD001943] [DOI] [PubMed] [Google Scholar]