

Abstract
CDK8 is a transcription-regulating kinase that controls TGF-β/BMP-responsive SMAD transcriptional activation and turnover through YAP1 recruitment. However, how the CDK8/YAP1 pathway influences SMAD1 response in cancer remains unclear. Here we report that SMAD1-driven epithelial-to-mesenchymal transition (EMT) is critically dependent on matrix rigidity and YAP1 in a wide spectrum of cancer models. We find that both genetic and pharmacological inhibition of CDK8 and its homologous twin kinase CDK19 leads to abrogation of BMP-induced EMT. Notably, selectively blocking CDK8/19 specifically abrogates tumor cell invasion, changes in EMT-associated transcription factors, E-cadherin expression and YAP nuclear localization both in vitro and in vivo in a murine syngeneic EMT model. Furthermore, RNA-seq meta-analysis reveals a direct correlation between CDK8 and EMT-associated transcription factors in patients. Our findings demonstrate that CDK8, an emerging therapeutic target, coordinates growth factor and mechanical cues during EMT and invasion.
Introduction
Bone morphogenetic proteins (BMPs) are members of the TGF-β family and much like TGF-β are pleiotropic cytokines regulating variety of cellular processes including differentiation, cell proliferation, and epithelial-to mesenchymal transition (EMT) during embryonic development and in cancer [1–4]. The precise context of BMP functions in cancer, however, are less clear due in part to both tumor suppressive and tumor promoting functions for BMPs [5–11], with alterations in the expression of several BMP family members observed in cancer [7, 12–14].
BMPs exert their responses primarily via serine–threonine kinase receptors [15]. Activation of BMP signaling is initiated by C-terminal phosphorylation and translocation of SMAD1/5/8 with co-SMAD (SMAD4) to the nucleus to regulate target genes [16–18]. In the nucleus, phosphorylation of residues in the linker region of SMAD1 by cyclin-dependent kinase 8 (CDK8) in response to BMP has been shown to be important for transcription and transcription-coupled turnover [19]. CDK8 and its isoform CDK19, together with their binding partner Cyclin C and MED12 and MED13 proteins form a regulatory module of the transcriptional Mediator complex connecting transcription-initiating factors with RNA polymerase II (Pol II). Of note, CDK8/19 kinase inhibition does not affect the phosphorylation of Pol II associated with actively transcribed genes. CDK8/19 has been shown to be responsible for Pol II CTD phosphorylation of silent genes that are activated by specific signals [4, 20–22].
CDK8-mediated SMAD1 linker phosphorylation, which occurs in the nucleus, creates a binding site for and recruitment of Yes-associated protein 1 (YAP1/YAP/ YAP65) to the C-terminal phosphorylated/BMP-activated SMAD1 [19]. Although YAP1 is a well-established mechanosensitive protein [23, 24] with oncogenic functions in cancer, the contextual link between YAP1 and BMP responses remain to be established. We recently demonstrated that BMP-induced EMT alters cancer cell stiffness and response to external mechanical force [25]. Thus, we predict that one such context is changes in the cellular extracellular matrix (ECM) environment that impacts matrix rigidity and cell–ECM interactions. As BMPs have established roles in highly-mechanical bone tissue and during developmental stages where tensional homeostasis is associated with tissue pathologies [26, 27], we predict that these alterations in cancer cell stiffness and mechanical force response, under BMP-induced EMT, may be, in part, owing to changes in ECM rigidity and cell–ECM interactions [26, 27]. The previously established relationship between CDK8 and YAP1 via SMADs suggests a possible mechanism linking mechanical control and alterations in the ECM to SMAD1 transcriptional changes in cancer. This relationship is clinically relevant due to the identification and emergence of CDK8 as an oncogene and as a therapeutic target in multiple cancers [28–33].
The present work examines the relationship between matrix cues and CDK8 in the context of BMP-induced EMT in cancer. We find here that substrate rigidity directly impacts BMP-induced EMT, suggesting that matrix mechanics is key in regulating BMP responses. Our findings point to a direct role for the mechanosensitive YAP1 in mediating BMP4-induced EMT on rigid substrates, which is abrogated on compliant substrates. Notably, using a highly selective small-molecule kinase inhibitor of CDK8/19, which has recently entered clinical trials, and shRNA knockdown of CDK8 and its isoform CDK19 we demonstrate that CDK8/19 loss and inhibition suppress BMP-induced EMT. We find that inhibition of CDK8/19 specifically abrogates invasion in vivo in a murine syngeneic EMT model of invasion. CDK8/19 expression also correlates with EMT-associated genes in cancer patients. These findings together suggest that matrix mechanics plays a key role in regulating the functional effects of BMP4 functions, which are mediated by the therapeutically targetable CDK8/19, and thus integrates both growth factor and mechanical signals in cancer.
Results
Matrix rigidity regulates BMP-induced EMT
To test the effect of mechanical tension on BMP-induced EMT, we confirmed and established cancer cell lines as models for BMP-induced EMT. Two human cancer cell lines Panc1 (human pancreatic) and OvCa429 (human ovarian) have previously been reported to undergo BMP4induced EMT [5]. Consistently, we find that BMP4 robustly alters the actin cytoskeleton, increases the mRNA levels of EMT-associated transcription factors SNAI1, SNAI2 (Fig. 1a, b), and increases transwell Matrigel invasion in response to BMP4 (Fig. 1c). We report here a third model, murine mammary epithelial cell Py2T, previously reported to undergo TGF-β induced EMT [34], as being responsive to BMP4 as well (Fig. 1). All three cell lines manifested these changes in response to BMP4 with SNAI1/2 being induced by BMP4 within 12–48 h, depending on the cell line. Additional EMT-associated transcription factors such as ZEB1, were also examined. However, we determined that although ZEB1 mRNA levels were increased in response to BMP4 in human Panc1 and murine Py2T cells (Supp Fig. 1A), no significant increase was observed in OvCa429 cells. Based on these results, transcription factors SNAI1 and SNAI2 were evaluated throughout the study as markers of EMT. E-cadherin protein expression, another commonly used EMT marker, was also significantly downregulated in response to BMP (BMP2/BMP4) requiring longer BMP treatment times of 3–4 days (Fig. 1d, e), which is consistent with previously reported TGF-β-mediated downregulation of E-cadherin in Py2T cells, which was also reported to occur after several days [34].
Fig. 1.

BMP induces EMT in a spectrum of cancer cell lines. Human ovarian cancer cell line OvCA429, human pancreatic cancer cell line PANC1, and murine mammary cancer cell line Py2T were treated with 10 nM BMP4 for 4 days and a immunostained for phalloidin actin. b mRNA expression levels of SNAI1 and SNAI2 from indicated cells in the presence or absence of 10 nM BMP4 for 24 h analyzed by qRT-PCR. Values are normalized to their untreated controls. c Matrigel transwell invasion in response to BMP4 after 18 h of invasion. Images are of cells on the underside of the filter taken at × 10 and are representative of two independent biological replicates done in triplicate. *p < 0.05, **p < 0.01, ***p < 0.001. Graphs are representative of two independent biological trials done in triplicate for each cell line. (d Indicated cell lines treated with BMP2/4 as in a for indicated numbers of days and analyzed for E-cadherin protein levels by western blots or e by immunofluorescence
To test the impact of matrix rigidity in these cell lines we utilized fibronectin-conjugated polyacrylamide hydrogel gels with an elastic modulus of 0.5 kPa (soft) or 8 kPa (stiff) (Methods). We found that cells on the different substrates exhibited differences in morphology with cells on 0.5 kPa substrate appearing qualitatively less spread under regular growth conditions (Fig. 2a). BMP4 treatment of cells on the stiffer 8 kPa substrate resulted in a further change in morphology including elongation and spreading compared with untreated cells (Fig. 2a). BMP4 treatment did not change the morphology of cells on the 0.5kPa (soft) substrate (Fig. 2a). This observation was recapitulated in all three EMT cell lines tested. Consistent with the changes in gross morphological appearances, BMP4 treatment of cells on 8 kPa substrate resulted in accumulation of actin stress fibers and de-localization of E-cadherin from cell–cell junctions when compared with untreated cells (Fig. 2b) and reduction of E-cadherin at the mRNA level (Supp Fig. 1B). BMP4-induced alterations on 8 kPa were similar to alterations on glass (Fig. 1a). On the 0.5 kPa substrate E-cadherin in BMP4-treated cells remained tightly localized around cell–cell junctions (Fig. 2b), with no significant changes at the mRNA level (Supp Fig. 1B). Examination of EMT-associated transcription factors indicate that both the human and murine cell lines increased SNAI1 and SNAI2 mRNA levels in response to BMP4, significantly more robustly in cells on the 8 kPa substrate as compared with the 0.5 kPa substrate (Fig. 2c). Collectively, these data indicate that mechanical tension and matrix rigidity play an important role in modulating BMP4-induced EMT.
Fig. 2.

BMP4-induced EMT is suppressed on compliant substrates. a Phase contrast images of cells plated on soft (0.5 kPa) and rigid (8 kPa) fibronectin-coated hydrogels and treated with 10 nM BMP4 for up to 5 days to monitor any changes in morphology (Methods). b Immunofluorescence images of BMP4-treated OvCa429 and Panc1 cells plated on soft (0.5 kPa) and rigid (8 kPa) fibronectin-coated hydrogels as described in Methods. Cells were then immunostained for E-cadherin and phalloidin/actin as indicated. Scale bars = 20 μm. c Realtime quantitative PCR of SNAI1 and SNAI2 in OvCa429, Panc1, and Py2T cells plated on soft (0.5 kPa) and rigid (8 kPa) hydrogels followed by treatment with 10 nM BMP4 (Methods). Values are normalized to their untreated control. Graphs are representative of two independent biological trials done in triplicate. *p < 0.05, **p < 0.01, ***p < 0.001
We next investigated the requirement of SMAD1 for cancer EMT in response to BMP4 using shRNA to knockdown SMAD1 (Fig. 3a) or inhibited BMP-induced SMAD1 C-terminus phosphorylation using the small-molecule inhibitor Dorsomorphin that inhibits the kinase activity of the BMP Type I receptors [8, 35] in human cell lines (Supp Fig. 1C). We found that specifically reducing SMAD1 levels (Fig. 3a and Supp Fig. 1D) or the use of Dorsomorphin significantly reduced BMP4-induced increase in SNAI1 and SNAI2 transcripts (Fig. 3b, Supp Fig. 1e) and suppressed BMP4-induced cell invasion compared to control cells (Fig. 3c). Although these data do not rule out the possible contributions of non-SMAD pathways [36], they confirm SMAD1’s significant role in mediating BMP4-induced EMT in the different cancer cell line models. We next tested whether matrix rigidity dependent changes in BMP4-induced EMT were caused by changes in SMAD1 and SMAD4 nuclear translocation in response to BMP4 on compliant substrates. As prior studies have indicated that cell density can regulate cell functions in response to TGF-β [37, 38], all experiments/conditions on the different substrates were conducted at 50–70% confluence to ensure that BMP-induced EMT was not suppressed on soft substrates owing to cell density. BMP4induced SMAD1 and SMAD4 translocation to the nucleus occurred robustly on rigid substrates (8 kPa) within 1 h of BMP4 treatment (Fig. 3d for SMAD1 and Supp Fig. 1F for SMAD4). However, both SMAD1 and SMAD4 were excluded from the nucleus in cells plated on compliant gels (0.5 kPa) (Fig. 3d and Supp Fig. 1F). These findings together suggest that failure to mount a nuclear SMAD1 response upon BMP4 treatment on substrates of 0.5 kPa rigidity likely leads to a failure to mount an EMT response as well.
Fig. 3.

BMP4-induced EMT is SMAD1 dependent. qRT-PCR of mRNA transcript levels for a SMAD1 and b SNAI1 and SNAI2 in OvCa429 and Panc1 cells transiently infected as described in methods with shRNA to SMAD1 or Scr controls and treated with 10 nM BMP4 for 24 h. Values are normalized to their respective untreated control (black horizontal line in b). c Images of shSMAD1- or control shScr–OvCa429 and Panc1 cells untreated or treated with BMP4 (10 nM), on Matrigel coated transwells after invasion for 18 h. Images are representative of two independent biological trials done in triplicate. Right graph represents the number of invading cells from four independent fields and two independent trials. d Immunofluorescence images of OvCa429, Panc1, and Py2T cells plated on soft (0.5 kPa) and rigid (8 kPa) fibronectin-coated hydrogels with or without 10 nM BMP4 treatment for 1 h followed by immunostaining with antiSMAD1. Overlay images are shown with the nuclear stain 4’6-diamidino-2-phenylindole. Scale bar = 20 μm. Quantitation of nuclear to cytoplasmic fluorescence intensity ratio is presented for each cell line (right graph). Error bars represent the standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001
Mechanoresponsive YAP1 is required for BMP-induced EMT
To define the mechanistic impact of substrate rigidity on BMP4-induced EMT, we hypothesized a role for YAP1, a protein capable of relaying mechanical signals exerted by ECM rigidity to the nucleus [23]. To test a direct role for YAP1, we used two shRNAs to YAP1 in the two human cell lines, Panc1, and OvCa429, (Fig. 4a). We find that BMP4-induced increase in EMT transcription factors SNAI1 and SNAI2 was suppressed 2.5- and ninefold in OvCa429 and Panc1 shYAP1 cells, respectively, compared with control plko.1 cells (Fig. 4b, and Supp Fig. 2A for second shRNA to YAP1). shRNA to YAP1 also suppressed BMP4induced OvCa429 and Panc1 cell invasion by 10 and 14fold, respectively, compared with control cells (Fig. 4c, Supp Fig. 2B). These data implicate YAP1 in mediating BMP4-dependent EMT responses. As YAP1 is active in the nucleus under conditions of high matrix rigidity [23] and is required for BMP4-induced EMT (Fig. 4a–c), we asked whether BMP4 could induce nuclear localization of YAP1 to facilitate EMT. We find that BMP4 increases the nuclear localization of YAP1 on rigid substrates (8kPa), compared with untreated cells (Fig. 4d). Consistent with the impact of BMP4 on increased nuclear localization of YAP1 on rigid substrates and previous studies demonstrating reduced phosphorylation of nuclear YAP1, we find reduction in phosphorylated levels of YAP1 on rigid substrates in response to BMP4 as well (Supp Fig. 2C). Notably, BMP4 failed to increase nuclear localization of YAP1 on soft substrates (0.5 kPa, Fig. 4d) consistent with a lack of BMP4-induced EMT on 0.5 KPa substrates (Fig. 3). These YAP1 shRNA findings, together with reduced EMT in shSMAD1 cells (Fig. 2), suggest a role for both nuclear YAP1 and SMAD1 in BMP4 mediated EMT responses in a rigidity dependent manner.
Fig. 4.

YAP1 enhances BMP4-induced EMT. a Real-time quantitative PCR of YAP1 mRNA in control and knockdown cell lines as indicated. b Relative change in transcription factors determined by qRT-PCR of SNAI1 and SNAI2 in OvCa429 and Panc1 cells expressing shYAP1 or shPlko.1 after being treated with BMP4 (10 nM) as described in Methods. Values are normalized to their untreated control (black horizontal line in b) c OvCa429 and Panc1 cells expressing shYAP1 or shPlko.1 were plated in transwell chambers coated with Matrigel in the absence or presence of BMP4 (10 nM). Cell invasion imaged after 18 h. (Right) Graphical representation of number of invaded cells from four independent fields. Error bars represent the standard error of the mean. d Immunofluorescence images of OvCa429, Panc1, and Py2T cells plated on soft (0.5 kPa) and rigid (8 kPa) fibronectin-coated PA hydrogels and then treated with BMP4 (10 nM) for 1 h. Cells were then immunostained with anti-YAP1. Scale bar = 20μm. Quantitation of nuclear to cytoplasmic fluorescence intensity ratio is presented for each cell line (right graphs). *p < 0.05, **p < 0.01, ***p < 0.001
Kinase activity of CDK8 is required for BMP4-induced EMT
YAP1-SMAD1 interaction and YAP1’s ability to mediate SMAD1 activity has been shown to be dependent on the activity of CDK8 that facilitates recruitment of YAP1 to C- terminal phosphorylated SMAD1 in the nucleus [19]. To test a role for the kinase activity of CDK8 and its paralog CDK19 in BMP4-induced EMT, we used a selective small-molecule ATP competitive inhibitor of CDK8/19, Senex in B, an early-stage clinical drug candidate [39]. Treatment of both human cell lines OvCa429 and Panc1 and murine Py2T cells resulted in reduction in BMP4-induced SMAD1 linker phosphorylation at Ser206 [19] (Fig. 5a). Senex in B treatment prevented BMP4-induced increases in the induction of the mRNA of SNAI1 and SNAI2 (Fig. 5b) and also reduced BMP4-dependent E-cadherin repression in the cancer EMT models (Supp Fig. 2D). Senexin B treatment also led to a significant suppression of BMP4-induced invasion in both the human and murine cell lines occurring in a dose-dependent manner (Fig. 5c, Supp Fig. 2E 250 nM–5 μM). Senexin B alone did not have a significant effect on in vitro invasion (Fig. 5c, Supp Fig. 2E). As our findings using substrates with different rigidity suggested a role for nuclear YAP1 in BMP4-induced EMT (Fig. 4), we examined whether CDK8/19 kinase inhibition impacted BMP4-induced YAP1 nuclear localization. We found that Senexin B treatment reduced YAP1 nuclear localization only in response to BMP4, as compared with cells treated with BMP4 alone in the absence of Senexin B (Fig. 5d). These results demonstrate that the kinase activity of CDK8/19, which was previously reported to be required for YAP1 interaction with SMAD1 in the nucleus [19], is necessary for BMP4-induced EMT.
Fig. 5.

CDK8/19 kinase activity is required for BMP4-induced EMT. a Effect of Senexin B on BMP4 (30 min treatment) induced SMAD1 phosphorylation at Ser206 in indicated cell lines in the absence or presence of Senexin B as indicated. Quantitation normalized to total SMAD1 for pSer206 from two independent biological trials for each cell line using LI-COR Biosciences linear range quantitation tool are presented (right graph). b Real-time quantitative PCR showing fold change in mRNA levels of SNAI1, SNAI2 transcription factors in indicated cells pre-treated with Senexin B for 30 min, then treated with BMP4 (10 nM) for 24 h. Graphs are representative of two independent biological trials done in triplicate. Error bars represent the standard error of the mean. c OvCa429 and Panc1 cells were pre-treated with Senexin B for 30 mins and plated in transwell chambers coated with Matrigel in the absence or presence of BMP4 (10 nM). Cells were allowed to invade for 18 h. (Graph) Graphical representation (right graph) of invading cells from four independent fields representing three independent biological trials. Error bars represent the standard error of the mean. d Immunofluorescence images of OvCa429, Panc1, and Py2T cells pre-treated for 30 mins with Senexin B (5 μM), then treated with BMP4 (10 nM) for 1 h followed by immunostaining with YAP1 Scale bars = 20μm. Quantitation of nuclear to cytoplasmic fluorescence intensity ratio is presented for each cell line (right graphs) *p < 0.05, **p < 0.01, ***p < 0.001
CDK8 /CDK19 are required for BMP4-induced EMT
As Senexin B inhibits the kinase activity of both twin kinases, CDK8 and CDK19, we used lentiviral vector-based shRNA against either CDK8 or CDK19 in human cell lines Panc1 and OvCa429 (Fig. 6a) as an independent test for the role of CDK8/19. Knockdown of CDK8 or CDK19 diminished CDK8 and CDK19 expression respectively relative to plko.1. control cells (Fig. 6a). Reducing the expression of CDK8 and CDK19 individually reduced BMP4-induced SMAD1 linker phosphorylation compared with control cells (Fig. 6b).
Fig. 6.

Knockdown of CDK8/19 suppresses BMP4-induced EMT. a Real-time quantitative PCR of CDK8 and CDK19 in OvCa429 and Panc1 cells upon transduction of shRNA to either CDK8 or CDK19 compared with control cells (either Scramble or vector plko.1 as indicated). b Stable CDK8 or CDK19 knockdown OvCa429 and Panc1 cells were treated with BMP4 (10 nM) for 1 h and then analyzed, via immunoblotting, with the indicated antibodies. c Immunofluorescence images of OvCa429 and Panc1 cells expressing shScr, shplko.1, shCDK8, or shCDK19, treated with BMP4 for 1 h and then immunostained with anti-YAP1. Scale bar = 20 μm. Quantitation of nuclear to cytoplasmic fluorescence intensity ratio is presented for each cell line (right graphs) *p < 0.05, **p < 0.01, ***p < 0.001. d Real-time quantitative PCR showing the mRNA levels for transcription factors SNAI1 and SNAI2 in Panc1 shPlko.1, shCDK8/19, or shCDK19 and treated with BMP4 (10 nM) for 24 h. Graphs are representative of two independent biological trials experiments done in triplicate. e OvCa429 and Panc1 shCDk8, shCDk19 or control derivatives as in a were allowed to invade for 18 h through transwell chambers coated with Matrigel in the absence or presence of BMP4 (10 nM). Images are cells on the underside of the chamber taken at × 10 and are representative of two independent biological trials done in triplicate. Graphical representation of invading cells from four independent fields is presented (Right graph). Error bars represent the standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001
We next examined the effects of shRNA to CDK8 or CDK19 on YAP1 nuclear localization in response to BMP4. We find that only in OvCa429 cells, shRNA to CDK8 or CDK19 had a modest effect on lowering YAP1 nuclear localization. However, in both human cell lines OvCa429 and Panc1, BMP4-induced YAP1 nuclear localization was robustly suppressed by shRNA to CDK8 and CDK19 compared with control cells (Fig. 6c) similar to the effects of Senexin B on reduced nuclear localization of YAP1 in response to BMP4 (Fig. 5d).
shRNA to CDK8 or CDK19 also inhibited BMP4-induced increases in SNAI1 and SNAI2 mRNA when compared with control cells (Fig. 6d) albeit to different extents by shRNA to CDK8 or shRNA to CDK19 in different cell lines. Transwell Matrigel invasion induced by BMP4 in both human cancer EMT models was significantly suppressed in shCDK8 and shCDK19 cells to similar extents when compared to control cells (Fig. 6e). The findings were recapitulated using a second independent shRNA to CDK8 and CDK19 as well (Supp Fig. 2F). These data indicate that the twin kinases CDK8/CDK19 are required for BMP4-induced cancer EMT.
CDK8/19 expression correlates with EMT genes in human cancer and promotes EMT-associated invasion in vivo
Our studies indicate that CDK8/19 expression and kinase activity is required for BMP4-induced EMT in cancer models including ovarian cancer. Therefore, we next assessed whether the expression of these genes correlated with common EMT markers in human high-grade serous ovarian tumors. We acquired RNAseq data for 283 high-grade serous ovarian tumors (Fig. 7a, b) from the Cancer Genome Atlas (TCGA) project [40] and first confirmed that positive correlations (Pearson) existed between upregulated EMT markers. SNAI1, SNAI2, TWIST1, TWIST2, ZEB1, and ZEB2 each had a significant positive correlation, ranging from 0.38 to 0.78, with the expression of every other EMT marker (Fig. 7a, b). We further confirmed the significant negative correlation between CDH1, expression of which is lost during EMT, and SNAI2, TWIST1 and ZEB1; SNAI1, ZEB2, and TWIST2 were negatively correlated but only trended towards significance (Fig. 7a, b). As illustrated in Fig. 7a, consistent with our in vitro results, expression of neither CDK8 nor CDK19 was negatively correlated with CDH1. However, CDK8 expression had a significant positive correlation with SNAI1, SNAI2, ZEB1, TWIST1, and TWIST2 (Pearson correlation: 0.14–0.24 p = 5.3E-04, 5.2E-04, 0.0051, 0.0173, 5.1E-05, respectively; Fig. 7a), whereas CDK19 had a significant positive correlation with ZEB1 and TWIST1 (Pearson correlation: 0.16–0.22, p = 2.0E-04, 0.0089, respectively, Fig. 7a). Coupled with our in vitro data, these results suggest that CDK8, and to a lesser extent CDK19, expression is significantly associated with EMT.
Fig. 7.
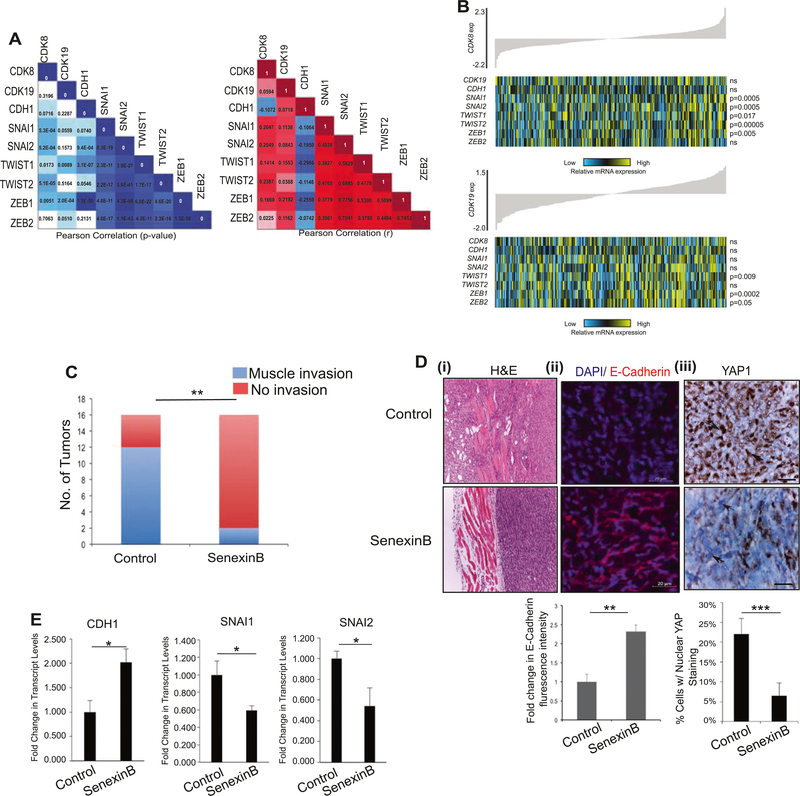
CDK8/19 correlates with markers of EMT in patients and inhibits EMT-associated invasion in vivo. a Pearson correlation demonstrating association between CDK8 and CDK19 with EMT markers in a panel of 283 high-grade serous ovarian tumors from TCGA. The left panel shows resulting p values while the right indicates the r-value. b Heat map of expression of CDK8 (upper panel) correlated with the expression of EMT markers SNAI1, SNAI2, TWIST1, TWIST2, and ZEB1 in high-grade serous ovarian tumors (n = 283). CDK19 expression (lower panel) corresponds with TWIST1 and ZEB1 mRNA levels. In each panel, tumor samples are arranged based on CDK8 or CDK19 expression, respectively, and relative expression of each gene is shown in the associated heat map; blue indicates low expression, whereas yellow corresponds to high mRNA levels. c–e 1 × 106 Py2T cells were injected into right and left mammary fat pads of FVB mice (n = 8/group, 16 tumors). After the appearance of measurable tumors on day 6, mice were randomized and fed either Senexin B medicated diet (Methods) or a normal diet. Mice were euthanized 32 days after implantation. c Tumors were analyzed at necropsy. Graph represents the number of tumors in either group with notable invasion into the muscle **p < 0.005 Χ2-test. d (i–iii) Representative images of tumor sections from c (i) H&E staining of invasion into muscle from tumors in c, (ii) confocal fluorescence images after immunostaining of tumor sections for E- cadherin and DAPI as indicated and described in Methods. Graph below represents fluorescence intensity quantitation from average of 10 random areas per sample from two independent tumors per group normalized to control tumor sections. (iii) Immunohistochemical staining of tumor sections labeled using anti-YAP1 as described in Methods. Arrows indicate YAP stained/unstained nuclei. Scale bar = 20μm. Graph below represents quantitation of percentage of tumor cells with nuclear YAP staining calculated in five random areas per sample from two independent tumors per group (Methods). e Real-time quantitative PCR of RNA extracted from tumors in c showing the mRNA levels for E-Cadherin (CDH1), and transcription factors SNAI1 and SNAI2 (n = 3 tumors/group) normalized to control tumors
We next tested the therapeutic relevance of CDK8/19 inhibition to EMT-associated invasion in vivo by using the transplantable murine Py2T cells. Py2T cells were chosen because they recapitulate cancer EMT in vivo that may serve as the initial step in metastasis [41, 42]. These cells form tumors in a syngeneic model, undergo EMT-like changes during invasion in vivo [34, 43], and also undergo BMP-induced EMT as determined here (Fig. 1). Py2T cells were transplanted into the mammary fat pads of syngeneic female FVB mice [34]. Mice were then randomized to receive either a normal diet or diet with Senexin B for a period of 32 days. Py2T tumors are rapidly growing tumors but do not give rise to distant metastasis as demonstrated previously [34] and as observed by us here as well. Tumor volume was measured every week and tumors were harvested and analyzed at day 32 for local invasion. Although Senexin B-treated mice had larger tumors (Supp Fig. 3A) necropsy analysis and analysis of Hematoxylin & Eosin (H&E) staining of histological sections revealed a much lower degree of invasion in the Senexin B-treated group (Fig. 7c, d). Specifically, 12 out of 16 tumors in the control group (Fig. 7c)—invaded the muscle and exhibited regions of streaming invading cells (Fig. 7d, H&E). In contrast, Py2T tumors from mice receiving Senexin B, were characterized by clear noninvasive fronts (Fig. 7d i, H&E). Only 2 out of 16 Senexin B-treated mice exhibited some muscle invasion with fewer regions of passive invasion/mesenchymal areas compared to control tumors. Analysis of the amount and localization of E-cadherin, a marker lost in EMT, revealed that Py2T control tumors were largely negative for E-cadherin, with limited amounts detectable at cell–cell junctions (Fig. 7d ii) consistent with prior studies [34]. Senexin B-treated tumors in contrast appeared more differentiated and strongly expressed E-cadherin (Fig. 7d ii 2.5-fold increase in Senexin B-treated tumors). As our in vitro studies implicate nuclear YAP1 in EMT-associated invasion (Fig. 4), with CDK8/19 inhibition by Senexin B leading to reduced EMT-associated nuclear YAP1 (Fig. 5), we tested if Senexin B treatment in vivo affected YAP1 nuclear localization. We find a striking reduction in the percentage of tumor cells with YAP1 in the nuclei in tumors from Senexin B-treated mice as compared with tumors from the control group (Fig. 7d iii, threefold reduction in Senexin B-treated tumors). Further analysis of RNA extracted from tumors from three mice in each group revealed an increase in E-cadherin mRNA in Senexin B-treated mice (Fig. 7e) in agreement with immunohistochemistry of E-cadherin in the tumors (Fig. 7d ii) and a parallel decrease in EMT markers SNAI1 and SNAI2 in tumors from Senexin B-treated mice (Fig. 7e). Put together, these in vivo findings suggest that CDK8/19 inhibition is sufficient at maintaining an epithelial and noninvasive phenotype and in suppressing local invasion in vivo, recapitulating the in vitro findings.
Discussion
Our findings here suggest the significance of therapeutically targeting CDK8/19 for the inhibition of EMT-associated transcriptional changes and invasion that is dependent on integration of growth factor signaling (BMP/SMAD1) and mechanical (YAP1) cues in cancer. We find here that reducing matrix rigidity inhibits BMP-induced EMT, which occurs in a SMAD1 and YAP1-dependent manner. Previous studies have shown that BMP is able to signal through SMAD1 and SMAD2/3 [36]. Although our findings do not rule out possible contributions of SMAD2/3 and other non-SMAD mechanisms, we find that SMAD1 is sufficient for BMP4-induced EMT in cancer models. Matrix rigidity can drive TGF-β-induced EMT with reduced rigidity leading to elevated TGF-β induced apoptosis [44]. Interestingly, YAP1 has independently been shown to be required for both TGF-β induced EMT and apoptosis [45]. In contrast to TGF-β, we find that although BMP-induced EMT was inhibited on softer substrates, changes in apoptosis on softer substrates were not significant in response to BMP (data not shown). The apoptotic effect of TGF-β on softer substrates was reported to be mediated via non-SMAD mechanisms [44] and possible differences in BMP and TGF-β-induced non-SMAD mechanisms may explain the absence of apoptosis on softer substrates observed here. Moreover, as BMP4-induced EMT seems to be primarily driven by SMAD1, the absence of nuclear translocation of SMAD1/SMAD4 explains a lack of EMT on softer substrates. YAP1 has a well-documented role in mediating mechanical responses, with decreased rigidity resulting in YAP1 exclusion from the nucleus [23]. We find that BMP4 promotes nuclear accumulation of YAP1 on rigid substrates, consistent with a recent study in astrocytes on BMP2induced YAP activation (nuclear localization) [46], suggesting broad relevance of our findings. Using knockdown approaches we find a critical role for YAP1 in mediating BMP4-induced EMT. BMP4 was unable to override the absence of mechanical control on YAP1 translocation on softer substrates. As SMAD nucleocytoplasmic trafficking is primarily regulated at the level of nuclear retention [47–49], we speculate that absence of mechanical tension leading to reduced YAP1 in the nucleus impacts SMAD¼ nuclear retention. A similar model has also been recently proposed for TAZ in mediating SMAD2/3 retention downstream of TGF-β during embryonic stem cell self-renewal [50] and is likely to be relevant for BMP signaling as well.
The interaction between SMAD1 and YAP1 in the nucleus was previously demonstrated to depend on CDK8 [19]. Based on the requirement of YAP1 in SMAD1dependent EMT, we tested the role of CDK8/19, an emerging oncogene and therapeutic target. Blocking the kinase activity of CDK8/19 with Senexin B and shRNA knockdown of CDK8 or CDK19 inhibited BMP4-induced SNAI1, SNAI2, invasion, YAP1 nuclear localization, and SMAD1 linker phosphorylation. These findings are the first to implicate CDK19 along with CDK8 as a potential kinase for SMAD1. On the other hand, we also find that CDK8/19 are required for the increased expression of the EMT-associated transcription factors along with the increase in cell invasion induced by BMP signaling. No consistent significant differences were noted in changes in Matrigel invasion in vitro and SNAI1 and SNAI2 across cell lines produced by Senexin B alone in the absence of BMP induction, suggesting that the kinase activity of CDK8/19 cooperates with secondary signals (in this case, BMP) rather than regulates the expression of pertinent genes by itself, as previously found for the effect of CDK8/19 on estrogen receptor [51] and NFkB [4]. Notably, systemic treatment of mice with Senexin B in an in vivo model of EMT [34], significantly suppressed local tumor invasion into the muscle layer with inhibition of lowering of E-cadherin, and reduction in transcription factors SNAI1/2 in the tumors. Most strikingly, reduction in nuclear YAP1 in the tumors of Senexin B-treated mice was noted, further demonstrating the impact of CDK8/19 inhibition on nuclear YAP1 in a complete tumor environment. Based on our in vivo and in vitro studies, we propose that suppression of cancer cell invasion is a key outcome of CDK8/19 inhibition. These findings and the correlation between CDK8 and EMT transcription factors in patients and its emerging role as a therapeutic target warrant exploring clinical use.
Materials and Methods
Cell lines and culture conditions and reagents
OvCa429 cells were obtained from Duke Gynecology/Oncology Bank (Durham, NC), Panc1 cells were from ATCC (Manassas, VA) and Py2T cells were a kind gift from Gerhard Christofori [34]. Panc1 and Py2T cells were cultured in DMEM containing L-glutamine, 10% fetal bovine serum (FBS) and 100 U of penicillin–streptomycin. OvCa429 cells were cultured in RPMI containing L-glutamine, 10% FBS, and 100 U of penicillin–streptomycin. All cells used for experiments were confirmed mycoplasma-free (MycoAlert PLUS mycoplasma detection kit, Lonza, Basel, Switzerland). Antibodies: E-cadherin came from BD Biosciences (#610181) (San Jose, CA, USA) and β-actin came from Sigma (#A2228) (St. Louis, MO, USA), YAP1 (#sc101199) from Santa Cruz Biotechnology (San Cruz, CA, USA), pYAP (#4711), YAP1 for IHC (#14074), pSMAD1 Ser206 (#5753), pSMAD1/5/8 (#9511), SMAD1 (#6944) and SMAD4 (#9515), pSTAT727 (#9177) antibodies were from Cell Signaling (Danvers, MA, USA). Senexin B was provided by Senex Biotechnology (Columbia, SC, USA). For rigidity experiments, cells were plated on 0.5 kPa (soft) or 8 kPa (rigid) substrates obtained from Matrigen Life Technologies (Brea, CA, USA), (Softslip 12-well and Softwell 6-well) that were coated with 10 μg/mL fibronectin (Cultrex® #3420–001-01) in 1 × PBS for 1 h. All cells were allowed to sit on substrates for at least 12 h before BMP4 treatment. Cell-plating densities were carefully determined so as to obtain qualitatively equal confluence between substrates.
Generation of stable and transient knockdown cells using either adenovirus or lentiviral shRNAs
For SMAD1 knockdown, OvCa429 and Panc1 cells were infected with 100 MOI of two SMAD1 shRNA adenovirus constructs or scrambled (Scr) controls as described previously [52, 53] (sequence in Table 1). Lentiviral shRNA plasmids were obtained from Sigma and are also listed in Table 1. All lentiviral plasmids were amplified in Stbl3 bacteria and extracted using the QIAprep® Miniprep kit (Qiagen, Hilden, Germany). Lentiviral production was carried out using pCMV-dR8.91 and pCMV-VSV-G packaging constructs in 293FT cells. The lentiviral plasmid, pCMV-dR8.91 and pCMV-VSV-G were mixed at 5:4:1 ratio and co-transfected into 293FT cells using lipofectamine 3000 Reagent (Thermo Fisher Scientific, Waltham, MA, USA). Lentivirus-containing supernatants were harvested 48 h after transfection, filtered using a 0.45 μm filter, and diluted with fresh culture media to transduce target cells in the presence of 8 μg/ml PolyBrene (hex-adimethrinebromide). Transduced cells were selected with puromycin. For YAP1 knockdown, OvCa429, and Panc1 cells were infected at a 1:8 ratio of the YAP1 shRNA lentiviral construct. For CDK8/19 and CDK19 knockdown, OvCa429 and Panc1 cells were infected at a 1:4 ratio of the CDK8/19 and CDK19 shRNA lentiviral construct. OvCa429 and Panc1 cells were selected in puromycin at 3 μg/ml and 5 μg/ml, respectively. All shRNA lentiviral constructs were provided by COBRE Functional Genomics Core (University of South Carolina, Columbia, SC, USA). The knockdown was confirmed by qPCR (sequences in Table 2).
Table 1.
shRNA sequence
| YAP1 | CCGGCCCAGTTAAATGTTCACCAATCTCGAGATTGGTGAACATTTAACTGGGTTTTTG (TRCN0000107265) |
| CCGGGACCAATAGCTCAGATCCTTTCTCGAGAAAGGATCTGAGCTATTGGTCTTTTTG (TRCN0000107268) | |
| CDK8 | CCTCTGGCATATAATCAAGTT (TRCN0000000491 for the 5’UTR and CDS regions) |
| GAACCTGGTATGGGCCATGAG (TRCN0000199779 Sigma for the 3’UTR region) | |
| CDK19 | CGTTCGTATTTATCTAGTTTC (TRCN0000195069 Sigma for the 3’UTR); |
| GCTTGTAGAGAGATTGCACTT | |
| (TRCN0000003140 Open Biosystems CDS region) | |
| SMAD1 | CCTGTCATTATTGCTTACT as used previously [52, 53] |
| CACACACCTTGGTAACATA as used previously [52, 53] | |
| Scr | TTCTCCGAACGTGTCACGT as used previously [52, 53] |
Table 2.
qPCR Primers
| 1 | RPL13A F human: AGA TGG CGG AGG TGC AG |
| RPL13A R human: GGC CCA GCA GTA CCT GTT TA | |
| 2 | E-cadherin F human: GAT AAT CCT CCG ATC TTC AAT CCC |
| E-cadherin R human: CAA TAT GGT GTA TAC AGC CTC CC | |
| 3 | Snail F human: AAG ATG CAC ATC CGA AGC CA |
| Snail R human: CAG TGG GAG CAG GAG AAT GG | |
| 4 | SNAI1–2 F human: TCG GAA GCC TAA CTA CAG CGA |
| SNAI1–2 R human: AGA TGA GCA TTG GCA GCG AG | |
| 5 | Slug F human: CGA ACC CAC ACA TTG CCT TG |
| Slug R human: GTG AGG GCA AGA GAA AGG CT | |
| 6 | Zeb1 F human: CTG CTC CCT GTG CAG TTA CA |
| Zeb1 R human: GTG CAC TTG AAC TTG CGG TT | |
| 7 | Yap1 F human: TGA CCC TCG TTT TGC CAT GA |
| Yap1 R human: GTT GCT GCT GGT TGG AGT TG | |
| 8 | Cdk8 F human: AAG TTG GCC GAG GCA CTT AT |
| Cdk8 R human: ATG CCG ACA TAG AGA TCC CA | |
| 9 | Cdk19 F human: GTT TCA CCG TGC ATC AAA AGC |
| Cdk19 R human: ACC CAA TTT GCA TGG AGG TAA TG | |
| 10 | Smad1 F human: AAT TCC GGG GGT ATT GGC AG |
| Smad1 R human: AAG TAA CCC AGT CAG CAC CG | |
| 11 | RPL13A F mouse: CAA GGT TGT TCG GCT GAA GC |
| RPL13A R mouse: GCT GTC ACT GCC TGG TAC TT | |
| 12 | E-cadherin F mouse: CAA CGA TCC TGA CCA GCA GT |
| E-cadherin R mouse: TGT ATT GCT GCT TGG CCT CA | |
| 13 | Snail F mouse: AAG ATG CAC ATC CGA AGC CA |
| Snail R mouse: CAG TGG GAG CAG GAG ATT GG | |
| 14 | Slug F mouse: CGAACCCACACATTGCCTTG |
| Slug R mouse: GTGAGGGCAAGAGAAAGGCT | |
| 15 | Zeb1 F mouse: CTG CTC CCT GTG CAG TTA CA |
| Zeb1 R mouse: GTC CAC TTG AAC TTG CGG TT | |
| 16 | SNAI1–2 F mouse: AAG ATG CAC ATC CGA AGC CA |
| SNAI1–2 R mouse: CAG TGG GAG CAG GAG AAT GG |
Quantitative polymerase chain reaction (qPCR)
For all qRT-PCR reactions, Panc1 cells were treated with BMP4 for 12 h, OvCa429 for 24 h, and Py2T cells for 48 h unless otherwise indicated. In experiments with Senexin B, cells were pre-treated with Senexin B for 30 min prior to BMP4 treatments unless otherwise indicated. Total RNA was isolated from cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA). 1μg of RNA was reverse transcribed to cDNA using 5 × iScript Reverse Transcription Supermix (#1708840) (Bio-Rad, Hercules, CA, USA) and the Advanced Universal SYBR Green Supermix (Bio-Rad #1725271). qRT-PCR primer sequences are listed in Table 2.
Matrigel invasion assays
Invasion assays were performed using 24-well transwells (Greiner Bio-One, Kremsmünster, Austria; ThinCerts™, 24-well 8.0 um) coated with 400 μg/ml Matrigel (BD Biosciences #3248404). Cells (50 000) in serum free media were plated in the upper chamber, and allowed to invade for 18 h toward serum media in the lower chamber. Filters were stained with Three Step Stain (Richard-Allan Scientific, San Diego, CA, USA). Filters were removed and mounted onto glass slides. Cells on the filter were counted using an Olympus DP21 microscope at × 10 magnification.
Immunofluorescence
Standard Immunofluorescence methods were used as described previously [54]. Imaging was performed using either an Olympus IX81 motorized inverted microscope or Zeiss LSM700 confocal microscope (COBRE Microscopy and Flow Cytometry Core, University of South Carolina, Columbia, SC, USA). For immunofluorescence of E-cadherin on hydrogels, OvCa429, Panc1, and Py2T cells were treated with 10 nM BMP4 for 5, 3, and 4 days, respectively, based on analysis of E-cadherin levels in Supplementary Figure 1.
In vivo studies
Study approval
All animal experimental protocols were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of South Carolina under an approved Protocol (AUP 2329–101161-121916).
Female FVB mice (aged 6–7 weeks) were obtained from Taconic Biosciences (Hudson, NY, USA). Animals were housed at the animal research facility (DLAR) at the University of South Carolina under BL2 conditions. All mouse procedures were approved by the IACUC at USC. Single-cell suspensions of Py2T cells (1 × 106 cells in phosphate-buffered saline) were injected into the right and left mammary fat pads (n = 8 mice/group, 16 total tumors per group). After the appearance of measurable tumors on day 6, mice were randomized into either receiving normal chow or CDK8/19 inhibitor Senexin B medicated diet (1350 mg Senexin B-dimaleate per kilogram of control diet, Research Diets (New Brunswick, NJ, USA), D12450B). Tumor volumes were then measured twice a week after the first week by measuring perpendicular tumor diameters (length and width, L > W) with calipers. Tumor volumes (V) were calculated using the formula V = L × W × W/2. At the end-point of study, mice were euthanized; tumor weights were recorded and, during necropsy, invasion into muscle and other tissues quantitatively assessed.
Histology and immunohistochemical staining
The formalin-fixed tissues were processed, paraffin-embedded, sectioned at 10 μm and stained with hematoxylin and eosin.
For all immunohistochemical staining, formalin-fixed, paraffin-embedded serial sections of Py2T xenografts at 5 μm were used. For YAP labeling, de-paraffinized and rehydrated sections were incubated overnight at 4 °C with antibodies against YAP1 (rabbit monoclonal, cat# 14074, diluted 1:250). Following incubation with primary antibodies, sections were processed with EnVision + System-HRP kits (DakoCytomation, Carpinteria, CA) according to the kit protocol. The chromogen was diaminobenzidene and sections were counter stained with 1% methyl green. The negative control was carried out without primary antibody incubation. Intensity and degree of staining for YAP1 were evaluated independently by two blinded investigators (AC, ZM) as described previously [55]. For each tissue section the percentage of cells with nuclear YAP1 staining was calculated in five randomly chosen areas per sample by each of two independent researchers (AC, ZM). The average of 10 calculations per sample is reported. For E-cadherin immunofluorescence, sections were de-paraffinized and rehydrated by washing in an ethanol gradient. Antigen was retrieved by boiling sections in 10 mM sodium citrate buffer (pH 6.0) for 10 min. Tissues were blocked in 10% goat serum and incubated with primary antibody targeting E-cadherin (1:125) and secondary Alexa594 antibody (1:500). Nuclei were stained with DAPI and tissues were mounted for imaging. The negative control was carried out without primary antibody incubation. Images were collected using a Zeiss LSM 510 inverted confocal microscope using a × 20 Plan-Apochromat objective. Images were acquired by Ergonomic system microscope Leica DM1000 LED and Leica Application Suite EZ. Integrated intensity of fluorescence images was quantified using Fiji for Image J. Average of 10 areas per sample from two independent tumors per group is reported.
Patient data analysis
Upper quartile normalized RSEM values for 283 high-grade serous ovarian tumors were acquired from the TCGA data portal. Data were log transformed and median centered. All genes present in fewer than 80% of samples were excluded from our analyses. Missing values were set to NaN. Expression data for CDK8, CDK19, and specific EMT markers were extracted from the data and a Pearson correlation was performed using the ‘corr’ function in MATLAB. The Pearson correlation and associate p values are reported in Supplementary Figure 3.
Statistical analysis
All in vitro experiments were conducted at least three times and in triplicate when applicable or as indicated in figure legends. In vitro experiments were analyzed using parametric statistics (analysis of variance global test with Bonferroni-corrected two-tailed Student’s t-tests as post hoc tests) and presented as mean ± SEM. In cases where data were normalized to control, one-sample Student’s t-test was used with an expected value of 1 or 100% in order to decrease the likelihood of a type I error. All clinical and xenograft data were analyzed using nonparametric statistics. PS Power and sample size calculations were used to confirm the minimum number of animals/tumors required for rejecting the null hypothesis that the experimental and control groups are equal with the probability (power) 0.950. The Type I error probability associated with this test of the null hypothesis is 0.05. Outcome assessments for in vivo studies was performed in a blinded manner
Supplementary Material
Acknowledgements
We thank the COBRE Functional Genomics Core (USC Center for Targeted Therapeutics) for lentiviral preparations, COBRE Microscopy and Flow cytometry Core (USC Center for Targeted Therapeutics) for assistance with imaging, Qi Zhang, Zach Mack, Nick Lenze, Priyanka Singh, and Pratik Patel for technical assistance. Grant Support: NIH P20GM109091 (E.V.B., K.M., I.B.R.), Marsha Rivkin Foundation (K.M.), Ovarian Cancer Research Fund (K. M.), University of South Carolina startup funds (K.M.), University of South Carolina SPARC Graduate Student Research, and Predoctoral NIH GM122379–01 (L.M.J.).
Footnotes
Equal first co-authorship: Anne Serrao and Laura M. Jenkins
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41388–018-0316-y) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest I.B.R. is the founder and president and J.L., M.C., H.K., K.M, and E.V.B. are consultants of Senex Biotechnology, Inc. Other authors declare no conflict of interest.
References
- 1.Hogan BL. Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 1996;10: 1580–1594. [DOI] [PubMed] [Google Scholar]
- 2.Richter A, Valdimarsdottir L, Hrafnkelsdottir HE, Runarsson JF, Omarsdottir AR, Ward-van Oostwaard D, et al. BMP4 promotes EMT and mesodermal commitment in human embryonic stem cells via SLUG and MSX2. Stem Cells. 2014;32:636–648. [DOI] [PubMed] [Google Scholar]
- 3.Padgett RW, Wozney JM, Gelbart WM. Human BMP sequencescan confer normal dorsal-ventral patterning in the Drosophila embryo. Proc Natl Acad Sci USA. 1993;90:2905–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen M, Liang J, Ji H, Yang Z, Altilia S, Hu B, et al. CDK8/19 Mediator kinases potentiate induction of transcription by NFkappaB. Proc Natl Acad Sci USA. 2017;114:10208–10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon KJ, Kirkbride KC, How T, Blobe GC. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis. 2009;30:238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pickup MW, Owens P, Moses HL. TGF-beta, Bone Morphogenetic Protein, and Activin Signaling and the Tumor Microenvironment. Cold Spring Harbor perspectives in biology 2017; 9: a022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theriault BL, Shepherd TG, Mujoomdar ML, Nachtigal MW.BMP4 induces EMT and Rho GTPase activation in human ovarian cancer cells. Carcinogenesis. 2007;28:1153–1162. [DOI] [PubMed] [Google Scholar]
- 8.Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ampuja M, Alarmo EL, Owens P, Havunen R, Gorska AE, Moses HL, et al. The impact of bone morphogenetic protein 4 (BMP4) on breast cancer metastasis in a mouse xenograft model. Cancer Lett. 2016;375:238–244. [DOI] [PubMed] [Google Scholar]
- 10.Hover LD, Owens P, Munden AL, Wang J, Chambless LB, Hopkins CR, et al. Bone morphogenetic protein signaling promotes tumorigenesis in a murine model of high-grade glioma. Neuro Oncol. 2016;18:928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Owens P, Pickup MW, Novitskiy SV, Giltnane JM, Gorska AE, Hopkins CR, et al. Inhibition of BMP signaling suppresses metastasis in mammary cancer. Oncogene. 2015;34:2437–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Page C, Puiffe ML, Meunier L, Zietarska M, de Ladurantaye M, Tonin PN, et al. BMP-2 signaling in ovarian cancer and its association with poor prognosis. J Ovarian Res. 2009;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanco Calvo M, Bolos Fernandez V, Medina Villaamil V, Aparicio Gallego G, Diaz Prado S, Grande Pulido E. Biology of BMP signalling and cancer. Clin Transl Oncol. 2009;11:126–137. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama Y, Watanabe T, Tamura Y, Hashizume Y, Miyazono K, Ehata S. Autocrine BMP-4 signaling is a therapeutic target in colorectal cancer. Cancer Res. 2017;77:4026–4038. [DOI] [PubMed] [Google Scholar]
- 15.Yamashita H, Ten Dijke P, Heldin CH, Miyazono K. Bone morphogenetic protein receptors. Bone. 1996;19:569–574. [DOI] [PubMed] [Google Scholar]
- 16.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. [DOI] [PubMed] [Google Scholar]
- 18.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. [DOI] [PubMed] [Google Scholar]
- 19.Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139:757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donner AJ, Ebmeier CC, Taatjes DJ, Espinosa JM. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol. 2010;17:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galbraith MD, Donner AJ, Espinosa JM. CDK8: a positive regulator of transcription. Transcription. 2010;1:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galbraith MD, Allen MA, Bensard CL, Wang X, Schwinn MK, Qin B, et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell. 2013;153: 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. [DOI] [PubMed] [Google Scholar]
- 24.Wrighton KH. Mechanotransduction: YAP and TAZ feel the force. Nat Rev Mol Cell Biol. 2011;12:404. [DOI] [PubMed] [Google Scholar]
- 25.Osborne LD, Li GZ, How T, O’Brien ET 3rd, Blobe GC, Superfine R et al. TGF-beta regulates LARG and GEF-H1 during EMT to impact stiffening response to force and cell invasion. Mol Biol Cell 2014;25:3528–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. [DOI] [PubMed] [Google Scholar]
- 27.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. [DOI] [PubMed] [Google Scholar]
- 28.Broude EV, Gyorffy B, Chumanevich AA, Chen M, McDermott MS, Shtutman M, et al. Expression of CDK8 and CDK8-interacting genes as potential biomarkers in breast cancer. Curr Cancer Drug Targets. 2015;15:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crown J CDK8: a new breast cancer target. Oncotarget. 2017;8:14269–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDermott MS, Chumanevich AA, Lim CU, Liang J, Chen M, Altilia S, et al. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget. 2017;8: 12558–12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rzymski T, Mikula M, Wiklik K, Brzozka K. CDK8 kinase—An emerging target in targeted cancer therapy. Biochim Biophys Acta. 2015;1854:1617–1629. [DOI] [PubMed] [Google Scholar]
- 33.Xu W, Wang Z, Zhang W, Qian K, Li H, Kong D, et al. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/beta-catenin signaling pathway. Cancer Lett. 2015;356:613–627. [DOI] [PubMed] [Google Scholar]
- 34.Waldmeier L, Meyer-Schaller N, Diepenbruck M, Christofori G.Py2T murine breast cancer cells, a versatile model of TGFbeta-induced EMT in vitro and in vivo. PLoS ONE. 2012;7:e48651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. [DOI] [PubMed] [Google Scholar]
- 36.Holtzhausen A, Golzio C, How T, Lee YH, Schiemann WP, Katsanis N, et al. Novel bone morphogenetic protein signaling through Smad2 and Smad3 to regulate cancer progression and development. FASEB J. 2013;28:1248–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petridou S, Maltseva O, Spanakis S, Masur SK. TGF-beta receptor expression and smad2 localization are cell density dependent in fibroblasts. Invest Ophthalmol Vis Sci. 2000;41:89–95. [PubMed] [Google Scholar]
- 38.Nallet-Staub F, Yin X, Gilbert C, Marsaud V, Ben Mimoun S, Javelaud D, et al. Cell density sensing alters TGF-beta signaling in a cell-type-specific manner, independent from Hippo pathway activation. Dev Cell. 2015;32:640–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roninson IB, Porter DC, Wentland MP. CDK8-CDK19 selective inhibitors and their use in anti-metastatic and chemopreventative methods for cancer. US patent 9,321,737 (2016).
- 40.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Acloque H, Thiery JP, Nieto MA. The physiology and pathology of the EMT. Meeting on the epithelial-mesenchymal transition. EMBO Rep. 2008;9:322–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. [DOI] [PubMed] [Google Scholar]
- 43.Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. 2013;23:768–783. [DOI] [PubMed] [Google Scholar]
- 44.Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-beta1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23: 781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, He K, Hu Y, Guo X, Wang D, Shi W, et al. YAP modulates TGF-beta1-induced simultaneous apoptosis and EMT through upregulation of the EGF receptor. Sci Rep. 2017;7:45523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Z, Hu J, Pan J, Wang Y, Hu G, Zhou J, et al. YAP stabilizes SMAD1 and promotes BMP2-induced neocortical astrocytic differentiation. Development. 2016;143:2398–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inman GJ, Nicolas FJ, Hill CS. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-beta receptor activity. Mol Cell. 2002;10:283–294. [DOI] [PubMed] [Google Scholar]
- 48.Schmierer B, Hill CS. Kinetic analysis of Smad nucleocytoplasmic shuttling reveals a mechanism for transforming growth factor beta-dependent nuclear accumulation of Smads. Mol Cell Biol. 2005;25:9845–9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen HB, Rud JG, Lin K, Xu L. Nuclear targeting of transforming growth factor-beta-activated Smad complexes. J Biol Chem. 2005;280:21329–21336. [DOI] [PubMed] [Google Scholar]
- 50.Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol. 2008;10:837–848. [DOI] [PubMed] [Google Scholar]
- 51.Martina SJ, McDermott AAC, Chang-uk Lim, Jiaxin Liang, Mengqian Chen, Serena Altilia, David Oliver, Rae James M., Michael Shtutman, Hippokratis Kiaris, Balázs Győrffy, Roninson Igor B., Broude Eugenia V.. Inhibition of CDK8 Mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget. 2017;8:12558–12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varadaraj A, Patel P, Serrao A, Bandyopadhay T, Lee NY, Jazaeri AA, et al. Epigenetic regulation of GDF2 suppresses anoikis in ovarian and breast epithelia. Neoplasia. 2015;17:826–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pannu J, Nakerakanti S, Smith E, ten Dijke P, Trojanowska M. Transforming growth factor-beta receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J Biol Chem. 2007;282:10405–10413. [DOI] [PubMed] [Google Scholar]
- 54.Swaminathan V, Mythreye K, O’Brien ET, Berchuck A, Blobe GC, Superfine R. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res. 2011;71:5075–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chumanevich AA, Chaparala A, Witalison EE, Tashkandi H, Hofseth AB, Lane C, et al. Looking for the best anti-colitis medicine: A comparative analysis of current and prospective compounds. Oncotarget. 2017;8:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.