

Abstract
Objective:
Atherosclerosis is a leading cause of death in developed countries. MicroRNAs act as fine-tuners of gene expression and have been shown to have important roles in the pathophysiology and progression of atherosclerosis. We, and others, previously demonstrated that microRNA-144 (miR-144) functions to post-transcriptionally regulate ATP Binding Cassette Transporter A1 (ABCA1) and plasma high-density lipoprotein (HDL) cholesterol levels. Here, we explore how miR-144 inhibition may protect against atherosclerosis.
Approach and Results:
We demonstrate that miR-144 silencing reduced atherosclerosis in male, but not female low-density lipoprotein receptor null (Ldlr–/–) mice. MiR-144 antagonism increased circulating HDL cholesterol levels, remodeled the HDL particle, and enhanced reverse cholesterol transport (RCT). Notably, the effects on HDL and RCT were more pronounced in male mice suggesting sex-specific differences may contribute to the effects of silencing miR-144 on atherosclerosis. As a molecular mechanism, we identify the oxysterol metabolizing enzyme CYP7B1 as a miR-144 regulated gene in male, but not female mice. Consistent with miR-144-dependent changes in CYP7B1 activity, we show decreased levels of 27-hydroxycholesterol, a known pro-atherogenic sterol and the endogenous substrate for CYP7B1 in male, but not female mice.
Conclusions:
Our data demonstrate silencing miR-144 has sex-specific effects and that treatment with antisense oligonucleotides to target miR-144 might result in enhancements in reverse cholesterol transport and oxysterol metabolism in patients with cardiovascular disease.
Keywords: microRNA, cholesterol, atherosclerosis, oxysterol
Subject codes: Animal Models of Human Disease, Basic Science Research, Lipids and Cholesterol, Mechanisms, Atherosclerosis
Graphical Abstract
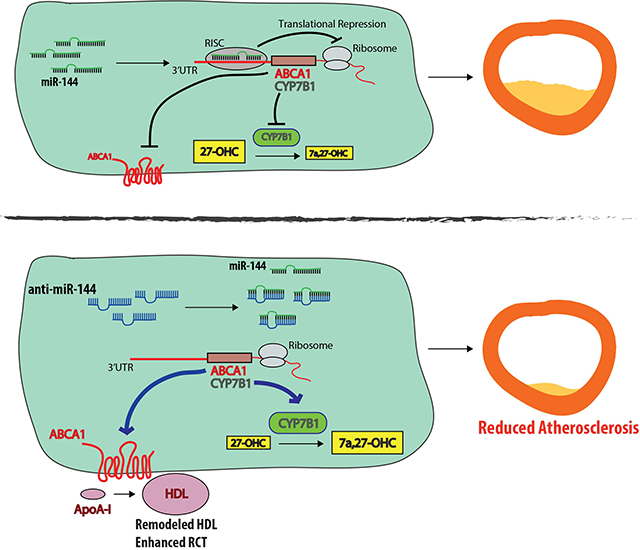
Introduction
MicroRNAs (miRs) are small noncoding RNAs, approximately 20–22 nucleotides in length, that target sets of genes in complimentary pathways to regulate gene expression. Generally, miRNAs regulate gene expression post-transcriptionally by base-pair binding to target mRNAs. Typically, when miRNAs bind in near-perfect complementarity to their target mRNAs, they trigger mRNA degradation by Ago2 cleavage1, 2. When miRNAs bind with imperfect or partial complementarity they impair mRNA stability and inhibit protein translation1, 2. Several miRNAs have been described to regulate lipid metabolism and cholesterol homeostasis3–9.
We, and others, previously demonstrated that microRNA-144 (miR-144) functions to post-transcriptionally target ATP-Binding Cassette Transporter A1 (ABCA1). We identified miR-144 as an FXR-induced microRNA in hepatocytes that reduced ABCA1 protein levels and activity, reduced plasma HDL cholesterol levels and in vitro, decreased cholesterol efflux from primary mouse hepatocytes to ApoAI5. In a parallel submission, Ramirez et al. identified miR-144 as an LXR-induced microRNA in macrophages, and demonstrated that overexpression or silencing of miR-144 decreased or increased cholesterol efflux, respectively, to Apo-AI in vitro in Huh7 and J774 cells10. It is well appreciated that ABCA1 plays a key role both in the hepatic generation of pre-beta HDL, the precursor of HDL, and in the efflux of cholesterol from sterol-loaded macrophages11–13. Macrophages constitutively express scavenger receptors that facilitate the uptake of oxidized/modifed cholesterol-rich LDL. Subsequent efflux of cellular cholesterol to prevent the formation of cholesterol-loaded foam cell formation is dependent upon the two sterol transporters ABCA1 and ABCG113. In a previous study Hu et al. used an agomir to overexpress miR-144 and demonstrated that increased levels of miR-144 decreased both macrophage ABCA1 levels and cholesterol efflux in vitro14. Consistent with these results, agomir treatment of Apoe–/– mice was shown to result in a decrease in both plasma HDL levels and reverse cholesterol transport and, in addition, accelerated progression of atherosclerosis14.
Based on these prior observations, we speculated that elevated levels of miR-144 in macrophage foam cells due to increased concentrations of LXR agonists might limit macrophage cholesterol efflux capacity by restricting ABCA1 activity. We also hypothesized that long-term silencing of miR-144 would result in an increase in hepatic expression of ABCA1 protein, increased plasma HDL cholesterol levels and in enhanced reverse cholesterol transport (RCT). We further predicted that such changes might accelerate the regression of pre-existing atherosclerotic lesions and attenuate the development of disease. Importantly no studies using antagomirs to silence miR-144 have been performed in vivo in the context of atherosclerosis. We therefore set out to determine whether silencing miR-144 in vivo would lead to a reduction in atherosclerosis.
In the current study we demonstrate that silencing miR-144 reduced atherosclerotic lesion area in male, but not female Ldlr–/– mice. Antagonism of miR-144 increased circulating plasma HDL cholesterol levels, remodeled the HDL particle, and enhanced RCT. Notably, the effects of anti-miR-144 treatment on HDL and RCT were more pronounced in male mice suggesting sex-specific differences may contribute to the effects of silencing miR-144 on atherosclerosis development. Indeed, we show that silencing miR-144 induced hepatic levels of the oxysterol metabolizing enzyme CYP7B1 in male mice. One consequence was a decrease in the levels of 27-hydroxycholesterol, the endogenous substrate for CYP7B1 and a known pro-atherogenic sterol. In contrast, compared to male mice, endogenous hepatic Cyp7b1 mRNA levels are 4-fold lower in female mice and these levels are unaffected by silencing miR-144. These data demonstrate that there are different effects of silencing miR-144 in male and female mice and that silencing miR-144 may be a possible therapeutic intervention for cardiovascular disease.
Methods
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Mice, Diets and Treatments.
Male and female, 12-week-old Ldlr–/– mice (The Jackson Laboratory) on a C57BL6/J background were maintained on a 12 hour/12 hour light/dark cycle with unlimited access to food and water. All atherosclerosis studies adhered to the American Heart Association guidelines for experimental atherosclerosis studies15. For chronic miR-144 silencing atherosclerosis studies, male Ldlr–/– mice were maintained on a western diet containing 21% fat and 0.2% cholesterol (R esearch Diets D12079B) for 16 weeks. Mice were injected intraperitoneally with 10 mg/kg control (n=8–11 mice/group), or an equivalent volume of vehicle/PBS, or 10 mg/kg anti-miR-144 2’fluoro/methoxyethyl-modified (2’F/MOE-modified) phosphorothioate backbone antisense oligonucleotides (Regulus Therapeutics; ASO). For acute miR-144 silencing atherosclerosis studies, male Ldlr–/– mice were maintained on a western diet (as described above) for 16 weeks. After 16 weeks of western diet-feeding, mice were randomized into 4 groups. One group was sacrificed immediately, as Baseline (n=23). The remaining three groups of mice were switched to normal laboratory diet for 4 weeks (NIH31 modified mouse/rat diet catalog number 7013, Harlan Teklad), and further randomized into three treatment groups; vehicle/no treatment (PBS; n=19–21 mice/group), 2’F/MOE control anti-miR oligonucleotide, or 2’F/MOE anti-miR-144 oligonucleotide. Mice received 2 injections of 10 mg/kg anti-miR or an equivalent volume of PBS the first week, spaced two days apart, and then once a week thereafter for 4 weeks. Atherosclerosis studies in female Ldlr–/– mice were carried out as described above as in male mice. For all studies, mice were fasted overnight before euthanization. Aortic roots were embedded in OCT medium and frozen immediately, and liver tissue was snap-frozen under liquid nitrogen and stored at −80C.
Plasma Lipid Analysis.
Plasma total and HDL cholesterol were determined as previously described16. Plasma sterol analysis was performed at the UCSD Lipidomics Core17.
LC-ESI-MS/MS analysis.
HDL (10 μg protein) isolated as previously described18, and tryptic digests of HDL (1 μg protein) were injected onto a C18 trap column (Paradigm Platinum Peptide Nanotrap, 0.15 × 50 mm; Michrom BioResources, Inc.), as previously described18. ESI was performed usinga CaptiveSpray source (Michrom BioResources, Inc.) at a 10 mL/min flow rate and 1.4 kV setting. HDL digests were introduced into the gas phase by ESI and positive ion mass spectra were acquired with an orbitrap mass spectrometer (Fusion, Thermo Electron Corp.) using data-development acquisition, as previously described18.
Protein identification and quantification.
MS/MS spectra were matched using the Comet search engine against a mouse UniProt database as previously described18. Proteins were quantified using peptide spectra matches (PSMs): the total number of MS/MS spectra detected for a protein as previously described18. PSMs for each protein, normalized to total PSMs for peptides from each sample, were used to calculate a normalized PSM to compare the relative protein composition of HDL particles from individual mice.
RNA isolation and quantitative PCR.
Liver tissue and aorta (comprising the ascending aorta, aortic arch, and 2mm of thoracic aorta) were homogenized and total RNA extracted using QIAzol reagent (Invitrogen Life Technologies). For miRNA measurements, 100 ng total RNA was reverse transcribed using the High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems), and miR-144 was detected by Taqman RT-qPCR using specific primers to hsa-miR-144–3p and normalized to SnoRNA 202 (Applied Biosystems). For all other gene expression analysis, 500 ng total RNA was reverse transcribed as above, and gene expression determined using a Lightcycler480 Real-time qPCR machine and Lightcycler480 mastermix (Roche). Relative gene expression was determined using an efficiency corrected method and efficiency was determined from a 3-log serial dilutions standard curve made from cDNA pooled from all samples. Primers were designed across exon-exon boundaries and are available on request. Results were normalized to 36b4 mRNA. Primer sequences are available upon request.
Western blotting.
Liver tissue was homogenized and protein was extracted using RIPA buffer, with protease inhibitor cocktail mix (1 Complete MINI EDTA-free protease inhibitor tablet (Roche); 25 μg/mL calpain inhibitor (Sigma); 10 μg/mL leupeptin (Sigma); 2 μg/mL aprotinin (Sigma); 200 μM PMSF (Sigma)). 25 μg protein was separated on an SDS-PAGE gel (BioRad) and transferred to nitrocellulose. Membranes were incubated overnight with antibodies to ABCA1 (1:1000; a gift from Dr. John Parks, Wake Forest University)19, PDI (1:1,000; Cell Signaling) and CYP7B1 (1:1000; a gift from Dr. David Russell, UT Southwestern)20. Proteins were detected with HRP-conjugated secondary antibodies (1:10,000; GE Healthcare) and visualized using an AI600 Imager (GE Healthcare).
In vivo RCT Assay.
Resident peritoneal macrophages were prepared from C57BL/6J mice as previously described3, 21. For RCT assays, macrophages were incubated with 37.5 μg/mL acetylated LDL (acLDL) and 5 μCi/mL 3H-cholesterol for 24 hours, as previously described3, 21. Cells were resuspended in ice-cold PBS, and 1 × 106 cells were injected intraperitoneally into individually housed mice fed a western diet and treated with either PBS, control anti-miR or anti-miR-144 for 4 weeks, as described above. Prior to injection, an aliquot of cells was counted using a liquid scintillation counter to determine baseline radioactivity. Blood was obtained by retro-orbital bleeding at 6 hr and 24 hr after macrophage injection, and after 48 hr at sacrifice. An aliquot of plasma was used for liquid scintillation counting immediately at each time point. Feces were collected for 48 hr after macrophage injection and homogenized overnight in 50% NaOH, after which an aliquot was removed for liquid scintillation counting. At sacrifice, liver samples were collected and incubated with hexane/isopropanol (3:2) and dried overnight. Lipids were solubilized and radioactivity determined by liquid scintillation counting. RCT to plasma, liver and feces was calculated as a percentage of total radioactivity injected at baseline.
Atherosclerotic Lesion Analysis.
Atherosclerotic lesion analysis was performed according to the procedure outlined in the American Heart Association statement15. Hearts embedded in OCT were serially sectioned through the aortic root (10 μM) from the ascending aortic segment to the aortic sinus. Slides were stained with Oil Red O for lesion quantification or used for immunohistochemical analysis as previously described22, 23. Lesion size was measured with ImageProPlus software by averaging 6 sections of each aorta. Quantification of the lesion size/coverage in the entire descending aorta by en face analysis was performed as described22, 23. Briefly, each entire aorta (from the ascending aorta to the iliac bifurcation) was harvested and fixed by 4% paraformaldehyde overnight. The aorta was opened longitudinally and was stained with Sudan IV and three low power photomicrographs taken that correspond to the aortic arch (from the aortic root to the first intercostal), the thoracic segment (from the first intercostal to the mesenteric artery), and the abdominal segment ending at the iliac artery bifurcation. The total area of the aorta and the area corresponding to the lesion (staining positive with Sudan IV) were determined using ImageProPlus software in each of the three sections and the values routinely added together to determine the percent lesion coverage.
Analysis of Human Carotid Plaques.
Human atherosclerotic carotid artery lesion samples were obtained from patients who underwent carotid endarterectomy surgery (CEA) for high-grade (>50% NASCET)24 stenosis at the Department of Vascular Surgery, Karolinska University Hospital, Stockholm, Sweden, and were enrolled in the Biobank of Karolisnka Endarterectomies (BiKE)25, or patients enrolled in the Munich Vascular Biobank26. Symptoms of plaque instability were defined as transitory ischemic attack (TIA), minor stroke (MS), and amaurosis fugax (retinal TIA) in the BiKE cohort. In the Munich Vascular Biobank, retrieved plaque was divided into 3–6 segments (3–4mm) which were snap frozen and stored at −80C or fixed in formalin (4%) overnight, decalcified with ethylenediaminetetraacetic acid (Sigma), embedded in paraffin and sectioned. Sections were stained with Hematoxylin and Eosin as well as Elastica van Gleeson27. Plaque vulnerability was assessed according to the Rothewell & Regrave criteria28, as well as the American Heart Association classification after Stary29. For BiKE samples, patients without qualifying symptoms within 6 months prior to surgery were categorized as asymptomatic and indication for CEA based on results from the Asymptomatic Carotid Surgery Trial (ACST)30. In total, n=10 plaques from symptomatic (Ruptured), n=10 plaques from asymptomatic (Stable) patients, and n=10 plaques from healthy controls were used in this study. For Munich Vascular Biobank samples, RNA was extracted from 10 paques (5 male, 5 female patients) with stable fibrous caps and 10 plaques with unstable/ruptured fibrous caps (5 male, 5 female patients). Whole carotid artery plaque and RNA isolation were performed as described25. The Taqman High Capacity cDNA Transcription Kit (Thermo Fisher Scientific) was used for cDNA synthesis, and primer assays for human miR-144, U6, GAPDH, and CYP7B1 (Thermo Fisher Scientific) were used to detect changes in expression levels.
Statistics.
Statistical analysis was performed using Prism Graphpad software (V7.0). All results are mean ±SEM or ±SD, as stated in the figure legends. Normality was determined using D’Agostino-Pearson and/or Shapiro-Wilk normality testing. P values for normally distributed data were calculated using either a 2-tailed Student’s t-test or a one-way ANOVA with either Tukey’s or Sidak’s post hoc analysis as indicated in the figure legends. P values for nonnormally distributed data were calculated using the Mann-Whitney rank sum test of the Kruskal-Wallis test with Dunn’s multiple comparisons testing. A P < 0.05 was considered significant and statistical significance is shown as described in the figure legends.
Study Approval.
All animal experiments were approved by the Office of Animal Research Oversight (OARO) and the Institutional Animal Care and Use Committee (IACUC) at the University of California Los Angeles. All patients provided written consent according to the Declaration of Helsinki. Experiments using human samples were approved by the local ethics committee in Munich and Stockholm, and the University of California Los Angeles Institutional Review Board.
Results
Chronic long-term silencing of miR-144 attenuates progression of atherosclerosis.
To test the hypothesis that long-term silencing of miR-144 would protect against atherosclerosis, we used a 2’-F/MOE anti-miR-144 compound to silence miR-144. We previously showed that this anti-miR-144 compound to effectively silences miR-144 in vivo in C57BL6/J mice5. Here, we fed Ldlr–/– mice (n=10 mice/group) a western diet (WD) for 16 weeks to promote atherosclerotic disease and concurrently treated mice with either vehicle (PBS), control anti-miR, or anti-miR-144 (Figure 1A). Anti-miR-144 treatment did not significantly affect body weight gain or total plasma cholesterol levels (Supplemental Figure I A–B). RNA analysis of the livers of these mice demonstrated that miR-144 was effectively silenced in the animals treated with the anti-miR-144 compound (Figure 1B). As we previously reported, mRNA levels of the miR-144 target Abca1 were unchanged, but mRNA levels of miR-144 target genes Nfe2l2 and Rarb were significantly increased in mice treated with anti-miR-144 (Supplemental Figure I C). Chronic treatment with anti-miR-144 did not cause any toxicity as measured by plasma AST and ALT enzyme levels (Supplemental Figure I D–E). ABCA1 protein levels were significantly increased (Figure 1C), and consistent with increased ABCA1 levels and activity, plasma HDL cholesterol levels were increased 20% with anti-miR-144 treatment (Figure 1D). En face analysis of Sudan IV-stained aortae revealed a significant 35% decrease in lesion area in mice receiving anti-miR-144 (Figure 1E–F). Oil red O staining of aortic root sections also demonstrated reduced lipid accumulation and lesion area in animals treated with anti-miR-144 (Figure 1G–H). Thus, our data demonstrates that blocking miR-144 partially protects mice from the development of atherosclerosis.
Figure 1. Anti-miR-144 Treatment Attenuates Atherosclerosis Progression.
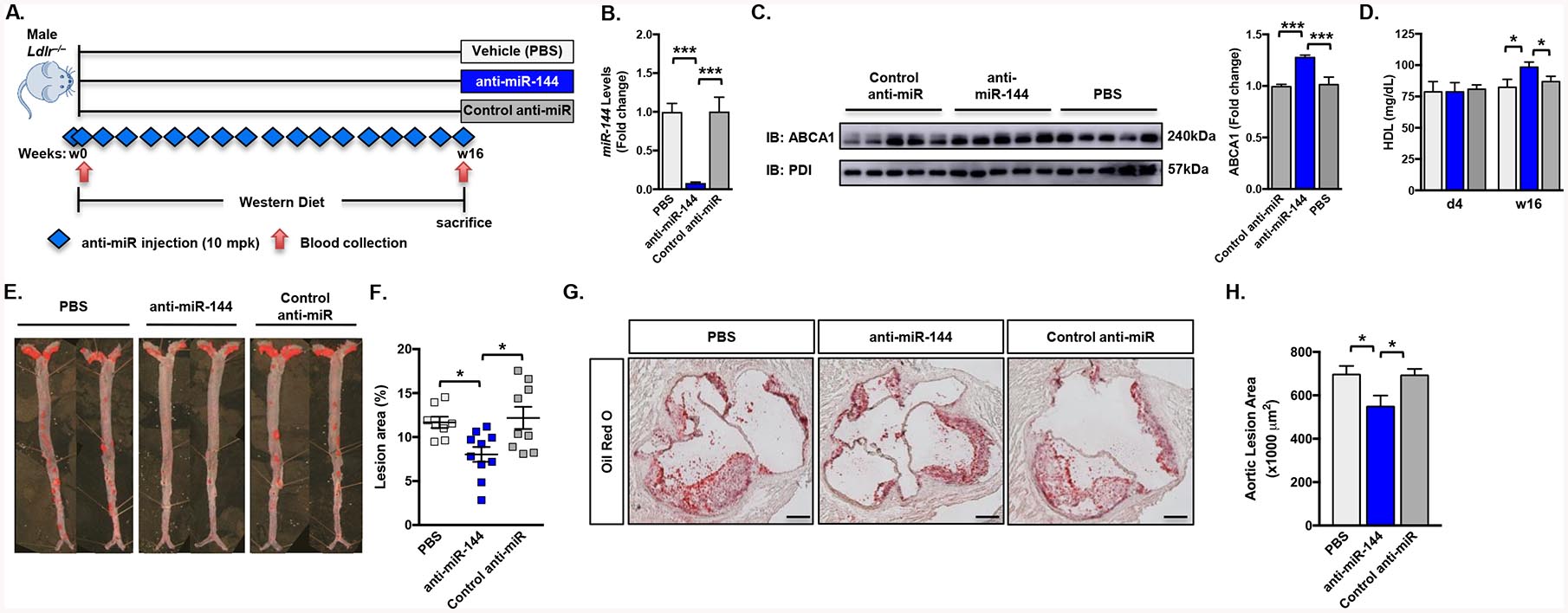
(A) Male Ldlr–/– mice were fed a Western diet for 16 weeks and treated with either vehicle (PBS), control anti-miR or anti-miR-144 2’F/MOE oligonucleotides at a dose of 10 mg/kg (n=10 mice/group). Blue diamonds indicate injection frequency. Red arrows indicate blood collection. (B) Hepatic expression of miR-144 quantified by Taqman® RT-qPCR. (C) Western blot (representative n=5 mice/group) and quantification (n=10 mice/group) of hepatic ABCA1 protein. (D) Plasma HDL cholesterol levels at the start of the study (d4; day 4) and after 16 weeks (w16) of Western diet feeding. (E) Representative Sudan IV-stained en face aortas. (F) Quantification of en face lesion area of mice (n=8–10 mice/group) after 16 weeks of Western diet feeding. Horizontal bars indicate the mean, and individual symbols indicate individual mice. (G) Aortic root lesions from mice described in (A) were stained with Oil Red O. Original magnification x50. Scale bar, 200. (H) Quantification of Oil Red O-stained aortic root sections (20 sections/mouse, n=4 mice/group). Data are presented as mean ±SEM. Statistical significance was determined by one-way ANOVA. * p<0.05, ** p<0.01, *** p<0.001.
Acute inhibition of miR-144 reduces atherosclerosis.
Cardiovascular disease (CVD) is the number one cause of death and patients typically present with established atherosclerotic disease. As such strategies that can reverse disease, and prevent further development are clinically attractive. Therefore we turned to a more clinically relevant model of atherosclerosis intervention. Male Ldlr–/– mice (n=12–14 mice/group) were fed a western diet for 16 weeks to establish atherosclerotic lesions, after which one group of animals were sacrificed (Figure 2A; baseline). All remaining animals were then switched to a chow diet to attenuate further development and progression of atherosclerosis and divided into three groups treated with either vehicle, control anti-miR, or anti-miR-144 for 4 weeks (Figure 2A). Treatment with anti-miR-144 for 4 weeks effectively reduced miR-144 levels (Figure 2B) but did not significantly effect body weight, total plasma cholesterol, hepatic Abca1 mRNA levels, or plasma AST/ALT enzyme levels (Supplemental Figure II A–E). ABCA1 protein levels and plasma HDL cholesterol levels were increased (45% and 15%, respectively) in mice treated with anti-miR-144 (Figure 2C–D), confirming effective miR-144 silencing.
Figure 2. Anti-miR-144 Treatment Accelerates Regression of Atherosclerosis.
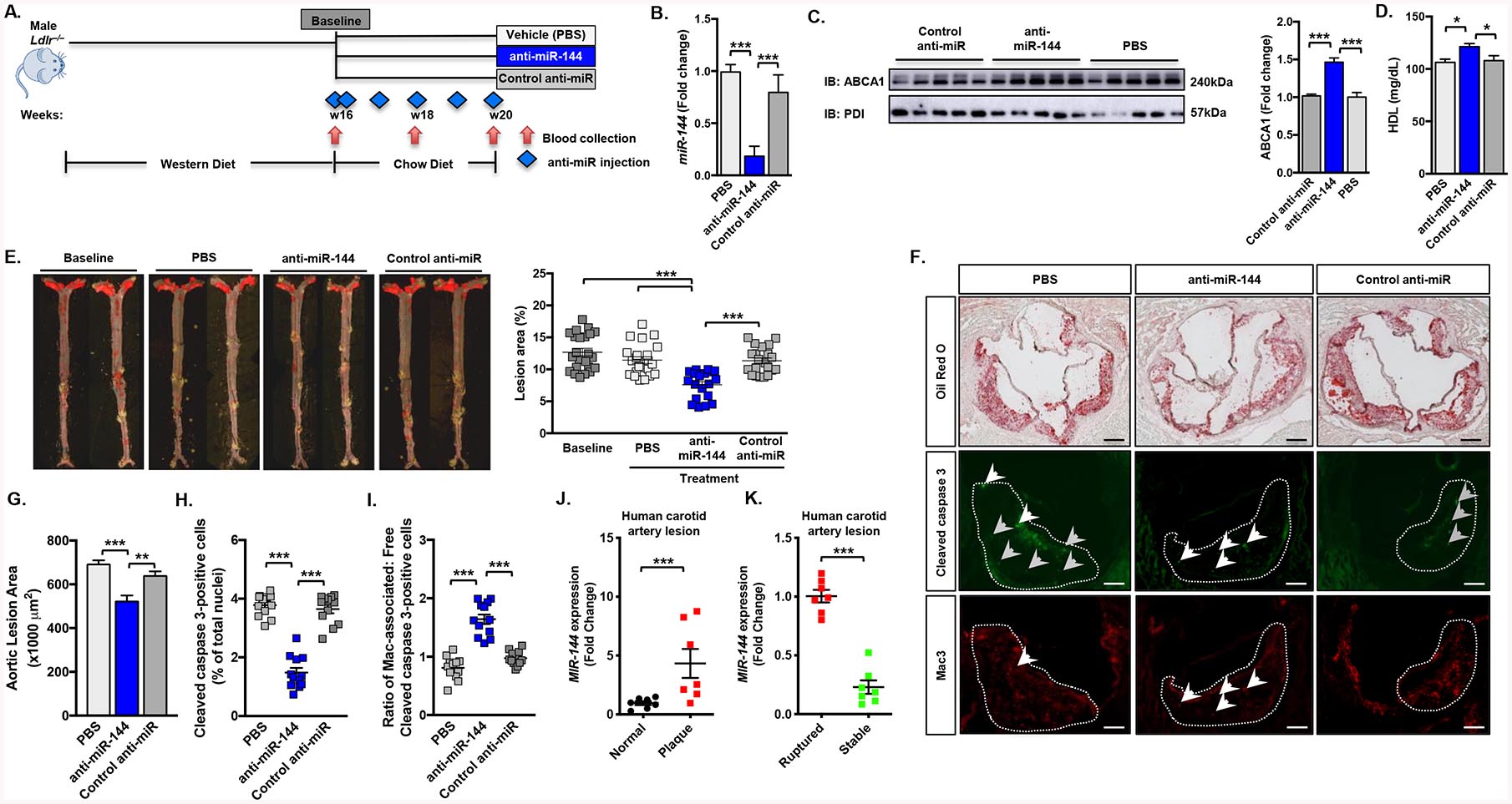
(A) Male Ldlr–/– mice were fed a Western diet for 16 weeks. They were then switched to a regular chow diet and treated for 4 weeks with either vehicle (PBS), control anti-miR or anti-miR-144 2’F/MOE oligonucleotides at a dose of 10 mg/kg (n=18–20 mice/group). Blue diamonds indicate injection frequency. Red arrows indicate blood collection. (B) Hepatic expression of miR-144 from mice quantified by Taqman® RT-qPCR. (C) Western blot (representative n=5 mice/group) and quantification (n=10 mice/group) of hepatic ABCA1 protein. (D) Plasma HDL cholesterol levels after 4 weeks of treatment. (E) Representative en face aortae and quantification of en face lesion area of mice (n=18–20 mice/group) at baseline (16 weeks of Western diet) and after 4 weeks of the indicated treatment. Horizontal bars indicate the mean, and individual symbols indicate individual mice. (F) Aortic root lesions were stained with Oil Red O and antibodies for Cleaved caspase 3 (a marker for apoptosis) and Mac-3 (a marker for macrophages). The white dotted line demarcates aortic lesion area. Grey arrowheads indicate “free” apoptotic cells and white arrowheads indicate macrophage-associated apoptotic cells. Original magnification x50. Scale bar, 200 mm (Oil Red O), 125 mm (immunohistochemistry). (G) Quantification of Oil Red O-stained aortic root sections (20 sections/mouse, n=4 mice/group). (H) Aortic root sections were stained for Cleaved caspase 3 and DAPI. The total number of Cleaved caspase 3-positive cells were 36 ±3.2, 32 ±1.4, 12 ±1.1 for PBS, control anti-miR, and anti-miR-144-treated mice, respectively. Data are presented as the percentage of positive cells of total nuclei. (I) Aortic root sections were co-stained for Mac-3 and Cleaved caspase 3. Each cleaved caspase 3-positive cell was determined to be either associated or not associated with a macrophage, and the data are presented as the ratio of macrophage-associated (Mac-associated) apoptotic cells to free apoptotic cells. The total number of Mac-3-positive cells were 142 ±4.3, 157 ±3.9, and 127 ±2.8 for PBS, control anti-miR, and anti-miR-144-treated mice, respectively. Horizontal bars indicate the mean, and individual symbols indicate individual mice. (J) Relative expression levels (fold change) of MIR-144 in healthy arteries (Normal; n=7) or carotid arteries from patients with atherosclerosis (Plaque; n=7) from patients enrolled in the Biobank of Karolinska Endarterectomies (BiKE). (K) Relative expression levels (fold change) of MIR-144 in human atherosclerotic carotid artery lesions from asymptomatic (Stable; n=7) vs. symptomatic (Ruptured; n=7) patients enrolled in the Biobank of Karolinska Endarterectomies (BiKE). Data are presented as mean ±SEM. Statistical significance was determined by one-way ANOVA (C-F, I-K) or Students t-test (L-M). * p<0.05, ** p<0.01, *** p<0. 001.
Treatment with anti-miR-144 significantly reduced atherosclersosis, as anti-miR-144-treated mice had 35% smaller lesions as compared to animals treated with vehicle or the control anti-miR (Figure 2E). As expected, there was no further increase in atherosclerosis in saline/PBS or control anti-miR-treated mice after 4 weeks on the chow diet (Figure 2E). Analysis of Oil red O-stained aortic root lesions demonstrated that animals treated with anti-miR-144 also had reduced lesion area (Figure 2F–G).
Advanced atherosclerotic lesions have significant amounts of cell death, and a major cause of necrotic core formation is defective phagocytic clearance or efferocytosis of apoptotic cells. We assayed apoptotic cells by measuring the percentage of lesion cells with caspase 3 activation by staining for cleaved caspase 3. The lesions of mice treated with anti-miR-144 had fewer apoptotic cells compared with control mice (Figure 2F and 2H). To determine if this decrease in apoptotic cells was associated with changes in efferocytosis we performed a standard assay where lesions are co-stained for apoptosis (cleaved caspase 3) and macrophages (Mac-3) and quantified for apoptotic cells (cleaved caspase 3-positive) that are either associated with a macrophage (indicative of an efferocytic event) or not associated with a macrophage (free). Data are presented as the ratio of associated/free apoptotic cells, with higher values representing increased efferocytosis. We found increased efferocytosis in lesions of mice treated with anti-miR-144 (Figure 2I), suggesting one mechanism for the smaller lesion area and decreased numbers of apoptotic cells is improved efferocytosis. Macrophage infiltration, as assessed by quantifying CD68 transcript abundance in the aortas, was greater in anti-miR-144-treated animals as compared to controls (Supplemental Figure II F). Steady state mRNA levels for the M2 macrophage markers Arg1, Cd206, and Il10 were significantly elevated (Supplemental Figure II G) and levels of the M1 macrophage markers Il1b, Il6, and Tnfa (Supplemental Figure II H) were significantly decreased, suggesting that these infiltrating macrophages are M2 macrophages.
It has been previously demonstrated that systemically delivered 2’-F/MOE anti-microRNA compounds can reach tissues other than the liver, such as the aorta3. We therefore analyzed miR-144 in the aortae of mice treated as in Figure 2 and observed significant silencing of miR-144 in mice treated with anti-miR-144 (Supplemental Figure II I). Thus, our results suggests that anti-miR-144 is not only being targeted in liver, but also in macrophages within atherosclerotic plaques. Together these data suggest that treatment with anti-miR-144 resulted in more anti-inflammatory macrophages with enhanced efferocytic ability in the aortae, which likely contributes to the reduced atherosclerosis observed in mice treated with anti-miR-144.
To determine whether miR-144 levels are different in human atherosclerotic plaques, we measured the levels of miR-144 in human patients with carotid atherosclerosis and control arteries (Figure 2J). We found miR-144 levels to be elevated in human plaques consistent with increased macrophage foam cells and increased levels of LXR agonists. MiR-144 levels were also significantly reduced in plaques from asymptomatic vs. symptomatic human carotid plaques (Figure 2K), suggesting that reduced miR-144 expression levels are associated with differences in plaque remodeling.
Silencing miR-144 enhance reverse cholesterol transport and remodels the HDL particle in vivo.
The data of Figures 1–2 demonstrate that silencing miR-144 in hypercholesterolemic Ldlr–/– mice is atheroprotective. Hu et al. demonstrated that overexpression of miR-144 to supraphysiologic levels using an agomir decreased plasma HDL cholesterol levels by 25% and decreased RCT to plasma (15%), liver (25%), and feces (20%)14. However, there are no prior studies reporting the converse, that is treatment of mice with antagomir to reduce miR-144 levels and determining changes in RCT in vivo. To determine whether the increased HDL levels in hypercholesterolemic Ldlr–/– mice treated with anti-miR-144 leads to increased RCT, we performed an in vivo RCT assay (Figure 3A). Mice treated with anti-miR-144 had significantly increased RCT to plasma (30%; Figure 3B), liver (80%; Figure 3C), and feces (50%; Figure 3D). This assay follows the movement of 3H-cholesterol from wildtype macrophages that have been loaded with cholesterol in vitro and whose function has not been affected by prior treatment with anti-miR-144 oligonucleotides.
Figure 3. Anti-miR-144 Treatment Increases Reverse Cholesterol Transport and HDL Efflux Capacity.
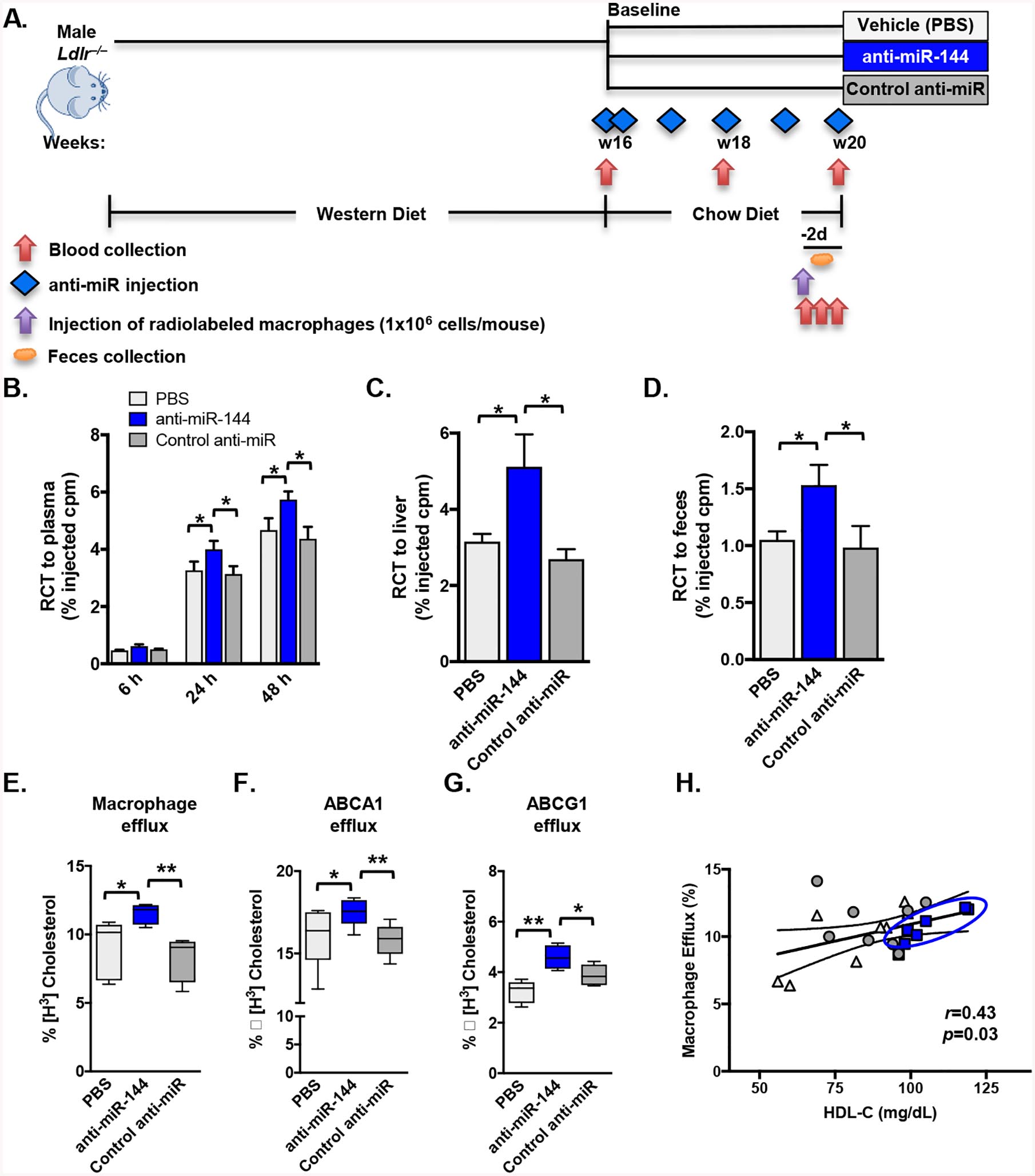
(A-D) Mice were treated as in Figure 2. Male Ldlr–/– mice (n=10 mice/group) treated with either vehicle (PBS), control anti-miR or anti-miR-144 were injected subcutaneously with 3H-cholesterol-labeled, acLDL-loaded resident peritoneal macrophages 2 days prior to sacrifice (A) and blood collected at 6, 24 and 48h post-injection of radiolabeled macrophages. (B-D) Data are expressed as the percentage of the 3H-cholesterol tracer relative to that of the total cpm tracer injected ±SEM. (B) Time course of 3H-cholesterol distribution in plasma. (C) Hepatic 3H-cholesterol tracer levels after 48 hours. (D) Fecal 3H-cholesterol tracer levels. Feces were collected continuously from 0 to 48 hours after injection. (E-G) Serum HDL was obtained by polyethylene glycol (PEG) precipitation of ApoB-containing lipoproteins from plasma-derived serum from mice treated as in Figure 3A. (E) Macrophage cholesterol efflux capacity (cAMP-stimulated J774 macrophages), (F) ABCA1-specific cholesterol efflux (mifepristone-stimulated BHK cells minus BHK cells incubated in medium alone) and (G) ABCG1-specific cholesterol efflux (mifepristone-stimulated BHK cells minus BHK cells incubated in medium alone) were measured after a 4 hour incubation with serum HDL (2.8% v/v) isolated from mice treated as in Figure 3A. (H) The relationship of plasma HDL cholesterol concentration and HDL efflux capacity was quantified by Pearson’s correlation. Data are presented as mean ±SEM. Horizontal bars indicate the mean (E-G). Statistical significance was determined by one-way ANOVA. * p<0.05, ** p<0.01.
To address whether HDL particles from mice treated with anti-miR-144 have enhanced cholesterol efflux capacity we performed in vitro HDL sterol efflux capacity functional assays using radiolabeled J774 macrophages (Figure 3E) or BHK cells overexpressing inducible forms of ABCA1 (Figure 3F) or ABCG1 (Figure 3G). We isolated HDL from mice treated with either anti-miR-144 or control anti-miR, and we show that silencing miR-144 significantly increased sterol efflux capacity (Figure 3, E–G), compared to HDL isolated from control mice. Additionally, the miR-144-dependent increase in efflux capacity was significantly correlated with plasma HDL cholesterol concentrations (Figure 3H; r=0.43, p=0.03), consistent with the observed increased HDL levels in mice treated with anti-miR-144.
To determine whether anti-miR144 treatment affected the protein composition of HDL particles, we performed unbiased shotgun mass spectrometric analysis of HDL isolated from mice treated with either control anti-miR or anti-miR-144 (Figure 4A). Spectral counting was used to identify differentially expressed proteins18. APOA1, the major constituent of HDL, was unaffected by miR-144 (Figure 4A–B). We identified 7 proteins as differentially expressed (p<0.05) on HDL isolated from mice treated with anti-miR-144 (Figure 4A–D). APOA2, MUP2, MUP6, MUP20, and SAA1 levels were increased (Figure 4B–D), whereas APOC4 and PF4 levels were decreased (Figure 4B and 4D). HDL-dependent cholesterol efflux capacity was significantly correlated with the levels of APOA2 (r=0.68, p=0.04; Figure 4E) and inversely correlated with levels of APOC4 (r=0.65, p0.04; Supplemental Figure III A), consistent with previously observed increases in cholesterol efflux to particles containing elevated APOA231–33. Despite increases in the acute phase response proteins SAA1 and SAA2 (Figure 4D), the cholesterol efflux capacity of the HDL particles did not correlate with levels of SAA1 (r=0.37, p=0.3) or SAA2 (r=0.26, p=0.46) (Supplemental Figure III B–C). Taken together, these data suggest that silencing miR-144 results in the production of an HDL particle that is more efficient at promoting cholesterol efflux from cells, a finding that may contribute to the increased flux of cholesterol through the RCT pathway in vivo (Figure 3).
Figure 4. Silencing miR-144 Remodels the HDL Particle.
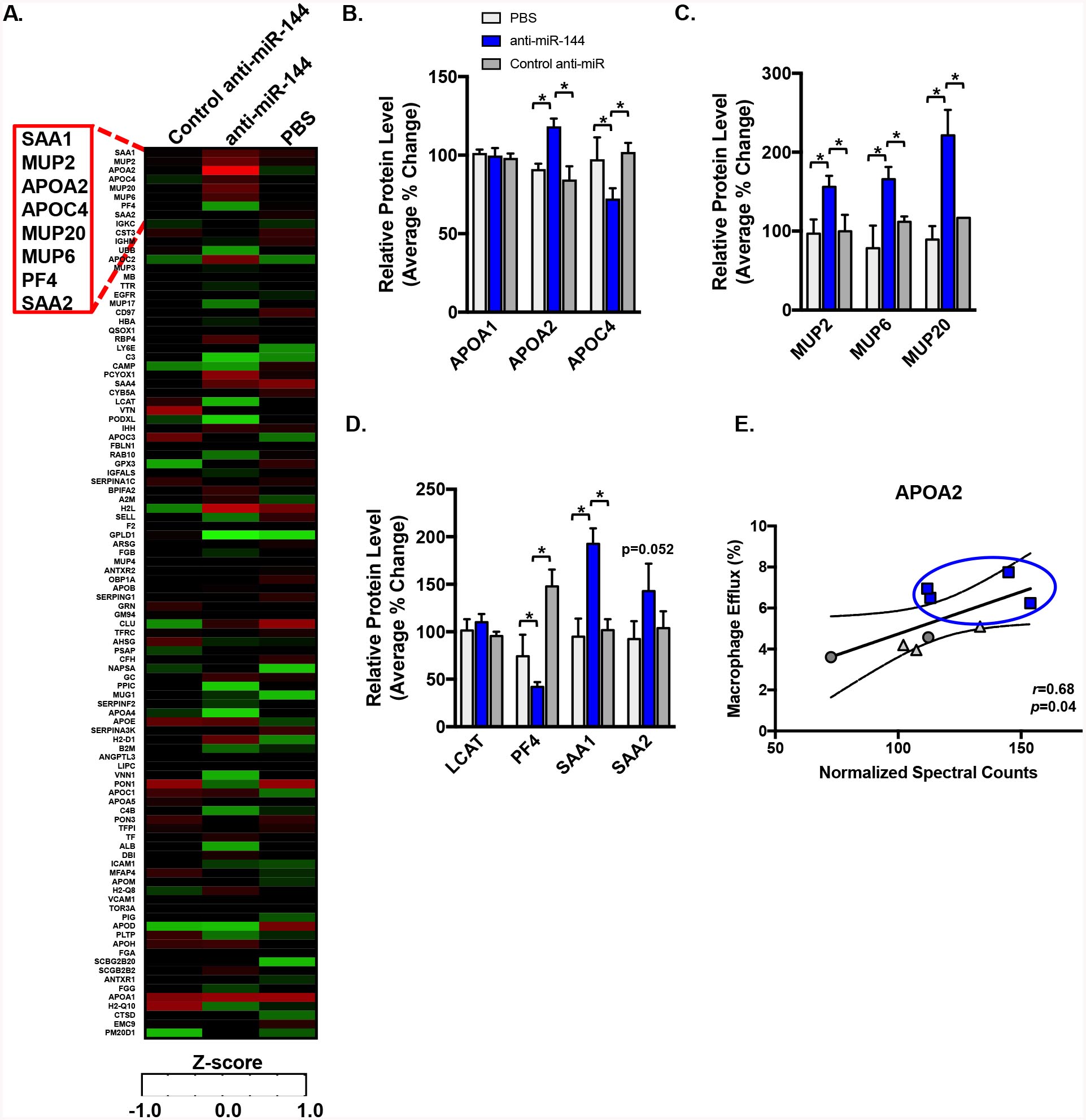
LC-ESI-MS/MS analysis of proteins on HDL isolated by ultracentrifugation from mice (n=5–6 mice/group) treated as in Figure 2. Proteins were quantified by spectral counting (total number of peptides identified for a given protein normalized to total spectral counts). (A) Heat map of differentially expressed proteins, with relative abundance calculated as Z-scores generated from adjusted spectral counts. (B-D) Quantification of representative HDL proteins. Data are presented as mean ±SEM. Statistical significance was determined by one-way ANOVA. * p<0.05. (E) Relationship between APOA2 levels on HDL and macrophage efflux capacity of HDL particles was quantified by Pearson’s correlation.
Identification of novel miR-144-regulated pathways.
To better understand the molecular targets of miR-144 in atherosclerosis regression, we performed global gene expression analysis from the livers of mice treated with control anti-miR and anti-miR-144 (Figure 5A). Pathway analysis of the most differentially regulated genes identified both significantly induced and repressed pathways (Figure 5B). One of the most highly induced genes was Cyp7b1 (blue dot/arrow; Figure 5A). Indeed, RT-qPCR analysis confirmed a miR-144-dependent induction in Cyp7b1 mRNA (Figure 5C) and Western Blotting analysis shows that CYP7B1 protein levels were alos elevated in the livers of animals treated with anti-miR-144, compared to control anti-miR-treated livers (Figure 5D). To determine whether Cyp7b1 is a direct target of miR-144, we cloned the 3’UTR downstream of a luciferase reporter gene, as we have done previously for Abca15. MiR-144 was able to reduce luciferase activity in a concentration-dependent manner, demonstrating that Cyp7b1 is a direct target of miR-144 (Figure 5E).
Figure 5. Identification of Novel Pathways Regulated by miR-144 in Atherosclerosis.
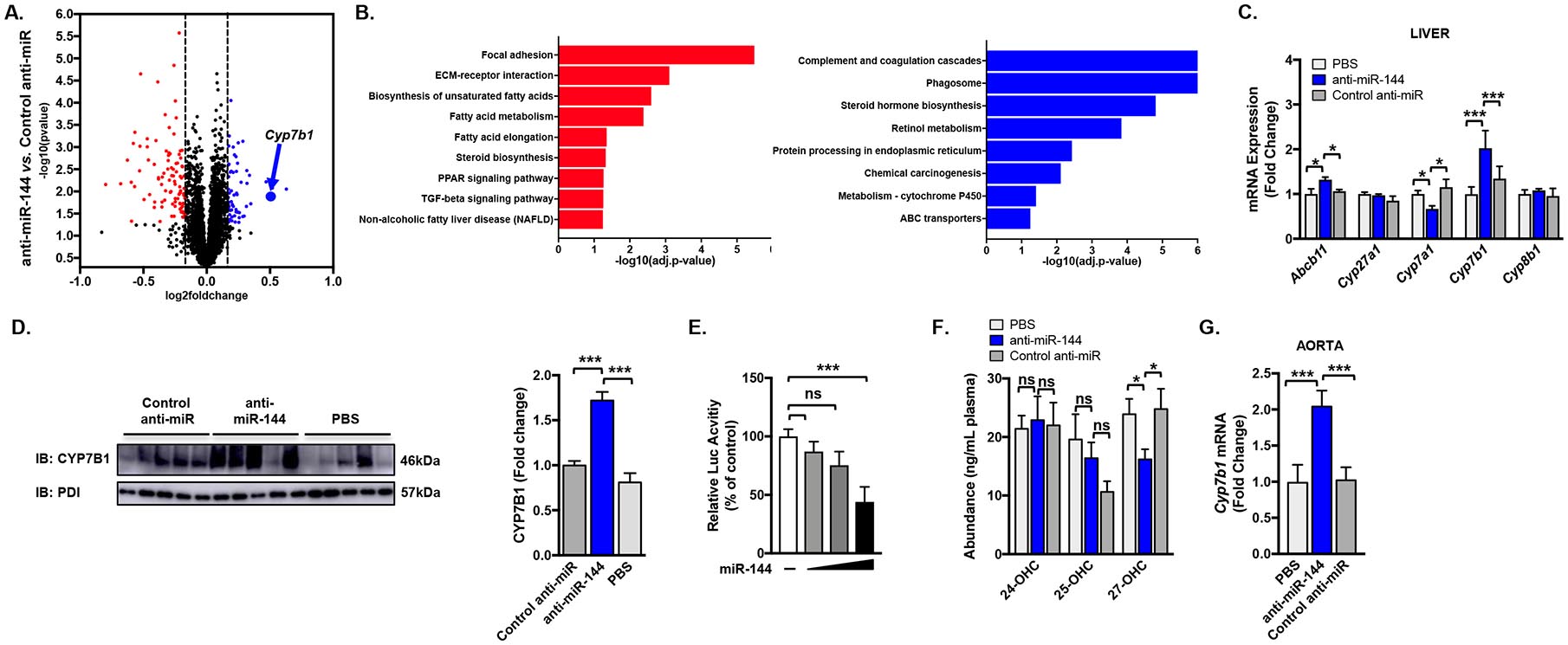
(A) RNA-Seq was performed on livers of mice treated with either control anti-miR or anti-miR-144 as in Figure 2. Gene expression profiling was performed in triplicate for each condition. Log2 fold changes indicates the change in expression after comparison of anti-miR-144 with control anti-miR treatment. Red = significantly down-regulated, blue = significantly up-regulated. (B) Differentially expressed genes were associated with biological functions using gene-set and KEGG pathway enrichment analysis and GO processes annotations. Red = significantly down-regulated pathways, blue = significantly up-regulated pathways. (C) Validation of hepatic mRNA expression of Abcb11, Cyp27a1, Cyp7a1, Cyp7b1, and Cyp8b1 as measured by RT-qPCR. (D) Western blot of hepatic CYP7B1 protein (representative n=5 mice/group). (E) Luciferase activity of the Cyp7b1 3’UTR reporter plasmid was determined 48 hours after transfection and normalized to b-galactosidase activity. The activity of the reporter plasmid in the absence of a miRNA overexpression plasmid was independently set to 100%. Results are representative of 3 independent experiments. (F) Plasma concentrations of 24-, 25-, and 27-hydroxycholesterol. (G) Expression of Cyp7b1 mRNA in the aortae of mice treated as in Figure 2. Data are presented as mean ±SEM. Statistical significance was determined by one-way ANOVA. * p<0.05, ** p<0.01, *** p<0.001.
CYP7B1/oxysterol 7α-hydroxylase metabolizes 27-hydroxycholesterol to 7α, 27-dihydroxycholesterol34, and 27-hydroxycholesterol has been shown to potently exacerbate atherosclerosis progression35, 36. We hypothesized that increases in hepatic CYP7B1 enzyme activity in mice treated with anti-miR-144 would result in reduced levels of the pro-atherogenic oxysterol, 27-hydrocholesterol. Consistent with this hypothesis, we show that plasma 27-hydroxycholesterol levels were decreased 34% in mice treated with anti-miR-144, compared to those injected with PBS or control anti-miR (Figure 5F). These changes were specific to 27-hydroxycholesterol, as other oxysterols did not change with anti-miR-144 treatment (Figure 5F and Supplemental Figure IV A). Macrophages also express Cyp7b1 and produce 27-hydroxycholesterol so we analyzed Cyp7b1 mRNA levels in the aortae of mice treated with anti-miR-144. We observed significantly increased Cyp7b1 expression in aortae isolated from mice treated with anti-miR-144, compared to mice treated with the control anti-miR (Figure 5G), suggesting that local effects of miR-144 on macrophages may also contribute to the decreased circulating levels of 27-hydroxycholesterol (Figure 5F). Hepatic CYP7B1 can also function in the alternative pathway for the synthesis of bile acids20, 34, 37. However, mice treated with anti-miR-144 had no significant differences in total biliary bile acid levels (Supplemental Figure IV B). Taken together, our data suggest that silencing miR-144 also induces Cyp7b1 mRNA resulting in increased CYP7B1 activity, thereby decreasing circulating levels of the pro-atherogenic oxysterol, 27-hydroxycholesterol.
Antagonism of miR-144 alters oxysterol metabolism in a sexually dimorphic manner to protect against atherosclerosis.
Cyp7b1 is known to be differentially expressed in male and female mice20, 34, which prompted us to investigate whether there may be sex-specific effects of the regulation of Cyp7b1 by miR-144. These previous studies20, 34 have demonstrated that hepatic Cyp7b1 expression is 2- to 3-fold higher in male mice compared to female mice, and loss of Cyp7b1 led to a greater increase in 27-hydroxycholesterol levels, suggesting that there are also differences in the generation of 27-hydroxycholesterol in male and female mice. Indeed, comprehensive analysis of gene expression in somatic tissues between sexes has previously identified cytochrome P450 enzymes as among the most highly sexually dimorphic gene families in mouse liver38 and analysis of the livers of over 100 strains of genetically unique in-bred mice, demonstrates that Cyp7b1 is among the most upregulated genes in the livers of male mice compared to female mice (Supplemental Figure V C).
It is well appreciated that sex differences can have dramatic effects on disease and that females are often protected, an effect that is attributed to the protective effects of estrogen. To determine potential sex differences in the role of miR-144 and atherosclerosis, we fed female Ldlr–/– a western diet for 16 weeks to establish atherosclerosis, as we did with male Ldlr–/– mice in Figure 2A. As in Figure 2, one group of animals were sacrificed after 16 weeks of western diet (baseline), and the remaining animals were switched to a chow diet to attenuate further development and progression of atherosclerosis and treated with either saline, control anti-miR, or anti-miR-144 for 4 weeks. Basal levels of miR-144 were 50% elevated in female mice, compared to males, suggesting miR-144 itself may be differentially regulated in males and females (Figure 6A). Silencing miR-144 was effective in female mice with 75% reduction in miR-144 levels, and comparable to that seen in male mice (80%), however residual miR-144 levels were almost 2-fold higher in female mice (0.37 vs. 0.19; Figure 6A). Surprisingly, anti-miR-144 treatment had no significant effect on atherosclerotic lesion burden in female mice (Figure 6B). Silencing miR-144 increased plasma HDL cholesterol by 13% (Figure 6C), comparable to that seen in male mice (15%; Figure 2D), with no change in total cholesterol levels (Supplemental Figure V A).
Figure 6. Silencing miR-144 does not protect against atherosclerosis in female mice.
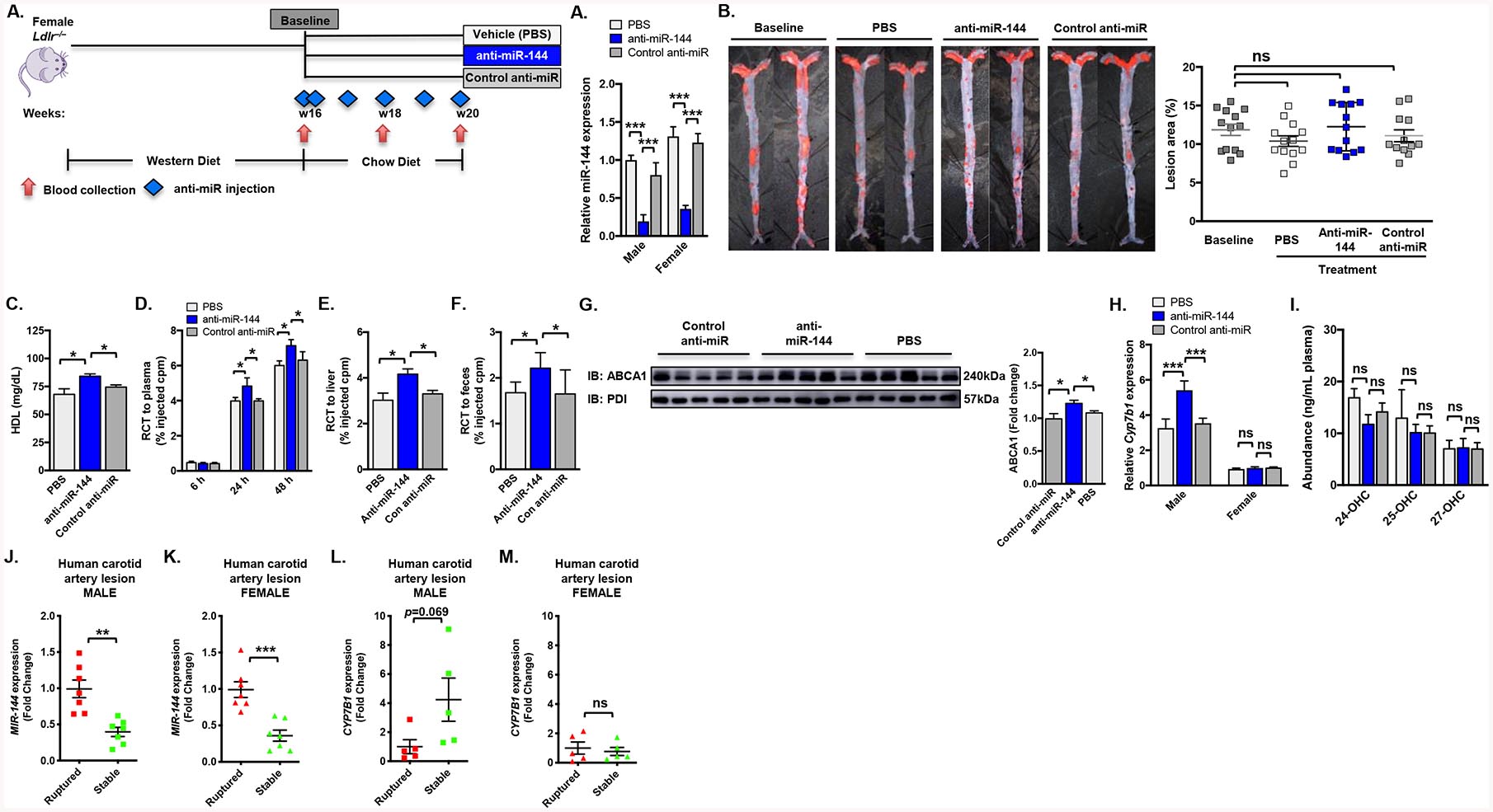
Female Ldlr–/– mice were fed a Western diet for 16 weeks and then subsequently treated for 4 weeks with either vehicle (PBS), control anti-miR or anti-miR-144 2’F/MOE oligonucleotides at a dose of 10 mg/kg (n=13–18 mice/group). (A) Hepatic expression of miR-144 in male mice from Figure 2 and female mice, quantified by Taqman® RT-qPCR. (B) Representative Sudan IV-stained en face aortas and quantification of en face lesion area of mice (n=13–18 mice/group) at baseline (16 weeks of Western diet) and after 4 weeks of the indicated treatment. Horizontal bars indicate the mean, and individual symbols indicate individual mice. (D-F) Female Ldlr–/– mice (n=10 mice/group) treated with either vehicle (PBS), control anti-miR or anti-miR-144 were injected subcutaneously with 3H-cholesterol-labeled, acLDL-loaded resident peritoneal macrophages 2 days prior to sacrifice and blood collected at 6, 24 and 48h post-injection of radiolabeled macrophages. Data are expressed as the percentage of the 3H-cholesterol tracer relative to that of the total cpm tracer injected ±SEM. (D) Time course of 3H-cholesterol distribution in plasma. (E) Hepatic 3H-cholesterol tracer levels after 48 hours. (F) Fecal 3H-cholesterol tracer levels. Feces were collected continuously from 0 to 48 hours after injection. (G) Western blot of hepatic ABCA1 protein from mice treated as in Figure 2 (representative n=5 mice/group). (H) Hepatic expression of Cyp7b1 in male mice from Figure 2 and female mice, quantified by RT-qPCR. (I) Plasma concentrations of 24-, 25-, and 27-hydroxycholesterol. (J-M) Relative MIR144 (J-K) and CYP7B1 (L-M) expression levels (fold change) in human atherosclerotic artery lesions from patients from the Munich Vascular Biobank. Data are presented as mean ±SEM. Statistical significance was determined by one-way ANOVA. * p<0.05, *** p<0.001.
Because we observed significant increases in plasma HDL cholesterol levels, but no change in atherosclerotic lesion area, we repeated our in vivo RCT analysis, but this time in female Ldlr–/– mice (treated as in Figure 2 with vehicle (PBS), control anti-miR, or anti-miR-144). In female mice we observed increased that silencing miR-144 increased RCT to plasma (20%; Figure 6D), liver (33%; Figure 6E), and feces (31%; Figure 6F). Notably, these changes were significantly less robust that those observed in male mice (30%, 80%, and 50%, respectively). Further, when we analyzed the livers of female mice treated with anti-miR-144, we observed a more modest 20% increase ABCA1 protein levels (Figure 6G), which was significantly less than we observed in male mice (50% Figure 2C vs. 20% Figure 6G). Together, these data suggest that reduced levels of flux through the RCT pathway could contribute to the absence of changes in lesions in female mice treated with anti-miR-144.
In agreement with previous studies, hepatic Cyp7b1 expression was 3-fold higher in the livers of PBS-treated male mice compared to female mice (Figure 6H). In contrast to the induction of Cyp7b1 mRNA observed in anti-miR-144-treated male mice, hepatic and aorta Cyp7b1 mRNA levels were unchanged in female mice treated with anti-miR-144 (Figure 6H and Supplemental Figure V D). Consistent with the absence of any changes in Cyp7b1 expression, there was no effect on plasma 27-hydroxycholesterol levels (Figure 6I), other oxysterols (Supplemental Figure V B), or bile acid levels (Supplemental Figure V E). To determine whether silencing miR-144 differentially regulates other sexually dimorphic expressed hepatic genes, we overlayed our miR-144 differentially expressed genes (from Figure 5) with all sexually dimorphic genes between male and female livers (Supplemental Figure V C). Several other sexually dimorphic genes were also significantly differentially regulated by silencing miR-144 (including Elovl3 and Slco1a1; Supplemental Figure V C, blue and black boxed genes), suggesting miR-144 is not only regulated differently in males and females, but may also target different genes in either sex. To determine whether these differences in Cyp7b1 in atherosclerosis are also observed in human patients, we obtained stable and ruptured plaque samples from male and female patients undergoing carotid endarterectomy from the Munich Vascular Biobank26. Consistent with the human plaque data in Figure 2K, expression of MIR-144 was significantly reduced in stable plaques in both male and female patients (Figure 6J–K). CYP7B1 levels were increased in stable plaques in male patients (Figure 6L), but not in female patients (Figure 6M), suggesting that levels of CYP7B1 may be important in plaque composition and stability in humans. These data, together with the data in male and female mice, suggest that the sex differences in miR-144 regulation of cholesterol efflux and oxysterol pathways is responsible in part for the effects on atherosclerosis observed with silencing miR-144 in male versus female mice.
Discussion
In the current manuscript we have demonstrated that silencing miR-144 in Ldlr–/– male mice significantly inhibits the progression and promotes regression of atherosclerosis. Previous studies with antagomirs of miR-144 focused on in vitro cholesterol efflux studies with Huh7, J774, and THP-1 cells10, and did not analyze effects on cholesterol efflux in vivo. We show for the first time that silencing miR-144 enhances RCT in vivo, resulting in a 50% increase in fecal 3H-cholesterol. These data are in agreement with studies using an agomir to overexpress miR-144, where Hu et al. observed the converse decrease in RCT in Apoe–/– mice treated with miR-14414. Additionally, we observed significant changes in Abcb11/Bsep and Cyp7a1 expression in the liver that are consistent with enhance biliary secretion and cholesterol excretion from the body. Taken together, these data suggest that one mechanism by which silencing miR-144 improves RCT is by increasing flux of cholesterol through the reverse cholesterol transport pathway to be excreted in the feces.
We also report for the first time that anti-miR-144 treatment had significant effects on the properties and composition of plasma HDL. Using proteomics we identified numerous proteins that were differentially regulated in HDL isolated from mice treated with anti-miR-144. Of the significantly regulated proteins, only Saa1 and Saa2 mRNA levels were significantly induced in the livers of mice treated with anti-miR-144. Although increased levels of SAA1 and SAA2 in plasma have been associated with increased inflammation39, HDL particles containing SAA1 and/or SAA2 have been demonstrated to be cleared more rapidly from the circulation40, 41, suggesting that the HDL in mice treated with anti-miR-144 may be preferentially cleared by the liver and may explain, in part, the increased RCT observed in mice treated with anti-miR-144. APOA2 was increased in HDL particles isolated from mice treated with anti-miR-144 and APOA1 levels were unchanged by anti-miR-144 treatment. The protective effects of APOA1 are well established42–44. However, the effects of APOA2 are less well understood and conflicting results have been observed with some groups reporting reduced cholesterol efflux capacity of HDL particles containing APOA2 and increased atherosclerosis45, 46 and others reporting that HDL with APOA2 are actually smaller particles and have better efflux capacity32, 33. Taken together, our data on HDL protein composition and cholesterol efflux in vitro and in vivo suggest that one mechanism by which miR-144 silencing improves RCT is by generating an HDL particle that is more efficient at cholesterol efflux and preferentially removed from the circulation.
We also showed that antagonizing miR-144 promotes regression of atherosclerosis in male mice, but not in female mice. We were surprised and intrigued to observed that silencing miR-144 did not have an effect on atherosclerotic lesions in female Ldlr–/– mice. We initially profiled miR-144 levels to determine whether the anti-miR-144 was effective at silencing miR-144 in female mice. We observed a significant reduction in the levels of miR-144, that was comparable to that seen in male mice treated with anti-miR-144 (75% reduction in female mice vs. 80% reduction in male mice). Interestingly, relative miR-144 levels were 1.3-fold higher in control female livers compared to male controls, and after miR-144 silencing there was 2-fold more residual miR-144 in female livers compared to male livers. We also determined the effect of miR-144 silencing on ABCA1 protein levels in the livers of female mice, and observed a significant increase in hepatic ABCA1 protein in female mice treated with anti-miR-144, however this effect was smaller in female mice compared to male mice (50% increase in male mice vs. 20% increase in female mice). This difference in the regulation of ABCA1 may be due to the increased residual miR-144 in female livers after treatment with anti-miR-144. The observed smaller increase in ABCA1 protein levels also translated into a smaller increase in RCT to plasma (20% in females vs. 30% in males), liver (33% in females vs. 50% in males), and feces (31% in females vs. 80% in males) in female mice treated with anti-miR-144.
MicroRNAs can regulate hundreds, if not thousands of genes47. Often thought of as rheostats that fine-tune gene expression, microRNAs frequently regulate sets of genes in common pathways or processes. To better understand the changes in gene expression following silencing of miR-144 we performed global gene expression analysis and show that silencing miR-144 resulted in significant changes in hepatic gene expression (a total of 152 differentially regulated genes), and likely also genes in other tissues, including the artery wall (as shown in Supplemental Figures II and V). These changes could be direct or indirect effects of silencing miR-144. As we have mentioned above, a given microRNA can be predicted to target several hundred genes (miR-144 has over 1,400 predicted mRNA targets), and at least 70% of mRNAs have at least one, if not several, predicted microRNA binding sites in their 3’UTR48–50. At least 4 microRNAs have been reported to regulate ABCA1, including miR-144, in multiple tissues and cell types4–6, 8, 10. The specific contributions of each individual microRNA in regulating ABCA1 will be determined by the abundance of each specific microRNA and the abundance of ABCA1 itself in different tissues. Additionally, we, and others, have demonstrated that miR-144 is regulated by different stimuli under different cellular contexts5, 10, adding a further layer of regulation to this system of fine-tuning gene expression. Similar to Abca1, the 3’UTR of Cyp7b1 is also large and is therefore likely subject to extensive post-transcriptional regulation. Indeed, the Cyp7b1 3’UTR harbors binding sites for miR-33, miR-758, and miR-26, microRNAs that have been shown to regulate Abca1 and lipid metabolism. The regulation of Abca1 and/or Cyp7b1 by miR-144 will likely also be influenced by 1) the number of binding sites for a specific microRNA within a given 3’UTR, and 2) the relative expression of other miR-144 targets that can compete for the microRNA binding in their 3’UTR. This may also be further complicated by competing endogenous RNAs, transcripts that chare the microRNA response element with the microRNA target genes and can influence each other by competing for microRNA binding51. Therefore, it will require extensive further studies to determine if any of these additional mechanisms also contribute to the reduction in atherosclerosis observed with anti-miR-144 treatment. Additionally, the differences in atheroprotection observed in male and female mice and the data from human plaques may have important implications for silencing miR-144 as a potential therapeutic in humans. Nonetheless, when taken together, our data suggest that treatment with antisense oligonucleotides to target miR-144 might result in enhancements in reverse cholesterol transport and oxysterol metabolism in patients with cardiovascular disease.
Supplementary Material
Highlights.
Atherosclerosis is a leading cause of death in developed countries.
MicroRNAs are fine-tuners of gene expression shown to have important roles in pathophysiology of atherosclerosis.
We demonstrate that silencing miR-144 reduces atherosclerosis in male, but not female, mice.
We identify the oxysterol metabolizing enzyme CYP7B1 as a miR-14-regulated target gene in male, but not female, mice.
Our data demonstrate that targeting miR-144 might result in enhancements in reverse cholesterol transport and oxysterol metabolism in patients with cardiovascular disease.
Acknowledgements
a) Acknowledgements: EJT and TQDAV conceived and designed the research. JC, AC, BLC, XW, NP, LM, EJT, and TQDAV conducted experiments. XW and TS provided assistance in conducting histological analysis of en face atherosclerotic lesions. UH and LM provided access to human samples from the Biobank of Karolinska Endarterectomies and the Munich Vascular Biobank, and LM performed qPCR analysis of human carotid artery plaques. NP performed proteomic and in vitro analyses of HDL particles. JC, AC, BLC, NP, LM, EJT and TQDAV performed data analysis. EJT and TQDAV wrote and edited the manuscript. All authors reviewed the manuscript.
b) Sources of Funding: EJT is supported by NIH grants HL118161 and HL136543; TS is supported by NIH grant HL139549; and TQDAV is supported by NIH grants HL122677 and DK112119 and in part by HL028481 and DK102559. TQDAV was also supported by the UCLA Clinical and Translational Science Institute (UL1TR000124); and the American Heart Association (SDG18440015). EJT and TQDAV were also supported by the UCLA/UCSD Diabetes Research Center (DK063491). We thank Dr. Peter Edwards for his guidance and input on this manuscript. We also thank Dr. Peter Tontonoz and members of the UCLA Atherosclerosis Research Unit for feedback and suggestions. We thank Drs. Alan Fogleman and Mohamad Navab for Ldlr−/− mice used in Figure 1. We thank Tieyan Han for technical assistance with histologic analysis of en face lesions. We thank Dr. Jake Lusis for access to the hybrid mouse diversty panel and Calvin Pan for assisting with HMDP analyses. We thank Regulus Therapeutics for anti-miR compounds. We thank Tomas Vaisar and Chongren Tang for the gift of BHK cells overexpressing ABCA1 and ABCG1.
Non-standard Abbreviation and Acronyms
- 2’F/MOE
2’fluoro/methoxyethyl
- ABCA1
ATP Binding Cassette Transporter A1
- ABCG1
ATP Binding Cassette Transporter G1
- AcLDL
Acetylated LDL
- ASO
Anti-sense oligonucleotide
- APO
Apolipoprotein
- CYP7B1
Cytochrome P450 enzyme 7B1
- CVD
Cardiovascular disease
- HDL
High-density Lipoprotein
- microRNA-144
miR-144
- MUP
Major urinary protein
- Ldlr
Low-density lipoprotein receptor
- PSM
Peptide spectra matches
- PF
Platelet factor
- RCT
Reverse cholesterol transport
- SAA
Serum amyloid A
Footnotes
Disclosures: The authors have declared that no conflict of interest exists.
References
- 1.Iwakawa HO, Tomari Y. The functions of micrornas: Mrna decay and translational repression. Trends Cell Biol. 2015;25:651–665 [DOI] [PubMed] [Google Scholar]
- 2.Hutvágner G, Zamore PD. A microrna in a multiple-turnover rnai enzyme complex. Science. 2002;297:2056–2060 [DOI] [PubMed] [Google Scholar]
- 3.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of mir-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramirez CM, Dávalos A, Goedeke L, Salerno AG, Warrier N, Cirera-Salinas D, Suárez Y, Fernández-Hernando C. Microrna-758 regulates cholesterol efflux through posttranscriptional repression of atp-binding cassette transporter a1. Arterioscler Thromb Vasc Biol. 2011;31:2707–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Aguiar Vallim TQ, Tarling EJ, Kim T, Civelek M, Baldán Á, Esau C, Edwards PA. Microrna-144 regulates hepatic atp binding cassette transporter a1 and plasma high-density lipoprotein after activation of the nuclear receptor farnesoid x receptor. Circ Res. 2013;112:1602–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Novák J, Olejníčková V, Tkáčová N, Santulli G. Mechanistic role of micrornas in coupling lipid metabolism and atherosclerosis. Adv Exp Med Biol. 2015;887:79–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouimet M, Hennessy EJ, van Solingen C, Koelwyn GJ, Hussein MA, Ramkhelawon B, Rayner KJ, Temel RE, Perisic L, Hedin U, Maegdefessel L, Garabedian MJ, Holdt LM, Teupser D, Moore KJ. Mirna targeting of oxysterol-binding protein-like 6 regulates cholesterol trafficking and efflux. Arterioscler Thromb Vasc Biol. 2016;36:942–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rayner KJ, Suárez Y, Dávalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernández-Hernando C. Mir-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marquart TJ, Allen RM, Ory DS, Baldán A. Mir-33 links srebp-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A. 2010;107:12228–12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramírez CM, Rotllan N, Vlassov AV, Dávalos A, Li M, Goedeke L, Aranda JF, Cirera-Salinas D, Araldi E, Salerno A, Wanschel A, Zavadil J, Castrillo A, Kim J, Suárez Y, Fernández-Hernando C. Control of cholesterol metabolism and plasma high-density lipoprotein levels by microrna-144. Circ Res. 2013;112:1592–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor lxr alpha. Proc Natl Acad Sci U S A. 2000;97:12097–12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oram JF, Lawn RM, Garvin MR, Wade DP. Abca1 is the camp-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem. 2000;275:34508–34511 [DOI] [PubMed] [Google Scholar]
- 13.Tarling EJ, Edwards PA. Dancing with the sterols: Critical roles for abcg1, abca1, mirnas, and nuclear and cell surface receptors in controlling cellular sterol homeostasis. Biochim Biophys Acta. 2012;1821:386–395 [DOI] [PubMed] [Google Scholar]
- 14.Hu YW, Hu YR, Zhao JY, Li SF, Ma X, Wu SG, Lu JB, Qiu YR, Sha YH, Wang YC, Gao JJ, Zheng L, Wang Q. An agomir of mir-144–3p accelerates plaque formation through impairing reverse cholesterol transport and promoting pro-inflammatory cytokine production. PLoS One. 2014;9:e94997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, García-Cardeña G, Lusis AJ, Owens AP, Rosenfeld ME, Virmani R, American Heart Association Council on Arteriosclerosis TraVB, Sciences aCoBC. Recommendation on design, execution, and reporting of animal atherosclerosis studies: A scientific statement from the american heart association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157 [DOI] [PubMed] [Google Scholar]
- 16.Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. Abcg1 has a critical role in mediating cholesterol efflux to hdl and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131 [DOI] [PubMed] [Google Scholar]
- 17.Quehenberger O, Armando AM, Brown AH, Milne SB, Myers DS, Merrill AH, Bandyopadhyay S, Jones KN, Kelly S, Shaner RL, Sullards CM, Wang E, Murphy RC, Barkley RM, Leiker TJ, Raetz CR, Guan Z, Laird GM, Six DA, Russell DW, McDonald JG, Subramaniam S, Fahy E, Dennis EA. Lipidomics reveals a remarkable diversity of lipids in human plasma. J Lipid Res. 2010;51:3299–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pamir N, Pan C, Plubell DL, Hutchins PM, Tang C, Wimberger J, Irwin A, Vallim TQA, Heinecke JW, Lusis AJ. Genetic control of the mouse hdl proteome defines hdl traits, function, and heterogeneity. J Lipid Res. 2019;60:594–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JY, Timmins JM, Mulya A, Smith TL, Zhu Y, Rubin EM, Chisholm JW, Colvin PL, Parks JS. Hdls in apoa-i transgenic abca1 knockout mice are remodeled normally in plasma but are hypercatabolized by the kidney. J Lipid Res. 2005;46:2233–2245 [DOI] [PubMed] [Google Scholar]
- 20.Li-Hawkins J, Lund EG, Turley SD, Russell DW. Disruption of the oxysterol 7alpha-hydroxylase gene in mice. J Biol Chem. 2000;275:16536–16542 [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ. Hepatic expression of scavenger receptor class b type i (sr-bi) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest. 2005;115:2870–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldan A, Pei L, Lee R, Tarr P, Tangirala RK, Weinstein MM, Frank J, Li AC, Tontonoz P, Edwards PA. Impaired development of atherosclerosis in hyperlipidemic ldlr−/− and apoe−/− mice transplanted with abcg1−/− bone marrow. Arterioscler. Thromb. Vasc. Biol 2006;26:2301–2307 [DOI] [PubMed] [Google Scholar]
- 23.Tarling EJ, Bojanic DD, Tangirala RK, Wang X, Lovgren-Sandblom A, Lusis AJ, Bjorkhem I, Edwards PA. Impaired development of atherosclerosis in abcg1−/−apoe−/− mice: Identification of specific oxysterols that both accumulate in abcg1−/−apoe−/− tissues and induce apoptosis. Arterioscler. Thromb. Vasc. Biol 2010;30:1174–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naylor AR, Rothwell PM, Bell PR. Overview of the principal results and secondary analyses from the european and north american randomised trials of endarterectomy for symptomatic carotid stenosis. Eur J Vasc Endovasc Surg. 2003;26:115–129 [DOI] [PubMed] [Google Scholar]
- 25.Perisic L, Aldi S, Sun Y, Folkersen L, Razuvaev A, Roy J, Lengquist M, Akesson S, Wheelock CE, Maegdefessel L, Gabrielsen A, Odeberg J, Hansson GK, Paulsson-Berne G, Hedin U. Gene expression signatures, pathways and networks in carotid atherosclerosis. J Intern Med. 2016;279:293–308 [DOI] [PubMed] [Google Scholar]
- 26.Pelisek J, Hegenloh R, Bauer S, Metschl S, Pauli J, Glukha N, Busch A, Reutersberg B, Kallmayer M, Trenner M, Wendorff H, Tsantilas P, Schmid S, Knappich C, Schaeffer C, Stadlbauer T, Biro G, Wertern U, Meisner F, Stoklasa K, Menges AL, Radu O, Dallmann-Sieber S, Karlas A, Knipfer E, Reeps C, Zimmermann A, Maegdefessel L, Eckstein HH. Biobanking: Objectives, requirements, and future challenges-experiences from the munich vascular biobank. J Clin Med. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin H, Li DY, Chernogubova E, Sun C, Busch A, Eken SM, Saliba-Gustafsson P, Winter H, Winski G, Raaz U, Schellinger IN, Simon N, Hegenloh R, Matic LP, Jagodic M, Ehrenborg E, Pelisek J, Eckstein HH, Hedin U, Backlund A, Maegdefessel L. Local delivery of mir-21 stabilizes fibrous caps in vulnerable atherosclerotic lesions. Mol Ther. 2018;26:1040–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Redgrave JN, Gallagher P, Lovett JK, Rothwell PM. Critical cap thickness and rupture in symptomatic carotid plaques: The oxford plaque study. Stroke. 2008;39:1722–1729 [DOI] [PubMed] [Google Scholar]
- 29.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, american heart association. Circulation. 1995;92:1355–1374 [DOI] [PubMed] [Google Scholar]
- 30.Halliday A, Mansfield A, Marro J, Peto C, Peto R, Potter J, Thomas D, Group MRCACSTC. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: Randomised controlled trial. Lancet. 2004;363:1491–1502 [DOI] [PubMed] [Google Scholar]
- 31.Smith LE, Segrest JP, Davidson WS. Helical domains that mediate lipid solubilization and abca1-specific cholesterol efflux in apolipoproteins c-i and a-ii. J Lipid Res. 2013;54:1939–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Niimi M, Nishijima K, Waqar AB, Yu Y, Koike T, Kitajima S, Liu E, Inoue T, Kohashi M, Keyamura Y, Yoshikawa T, Zhang J, Ma L, Zha X, Watanabe T, Asada Y, Chen YE, Fan J. Human apolipoprotein a-ii protects against diet-induced atherosclerosis in transgenic rabbits. Arterioscler Thromb Vasc Biol. 2013;33:224–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melchior JT, Street SE, Andraski AB, Furtado JD, Sacks FM, Shute RL, Greve EI, Swertfeger DK, Li H, Shah AS, Lu LJ, Davidson WS. Apolipoprotein a-ii alters the proteome of human lipoproteins and enhances cholesterol efflux from abca1. J Lipid Res. 2017;58:1374–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stiles AR, McDonald JG, Bauman DR, Russell DW. Cyp7b1: One cytochrome p450, two human genetic diseases, and multiple physiological functions. J Biol Chem. 2009;284:28485–28489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Umetani M, Ghosh P, Ishikawa T, Umetani J, Ahmed M, Mineo C, Shaul PW. The cholesterol metabolite 27-hydroxycholesterol promotes atherosclerosis via proinflammatory processes mediated by estrogen receptor alpha. Cell Metab. 2014;20:172–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, Korach KS, Shaul PW, Mangelsdorf DJ. 27-hydroxycholesterol is an endogenous serm that inhibits the cardiovascular effects of estrogen. Nat Med. 2007;13:1185–1192 [DOI] [PubMed] [Google Scholar]
- 37.Schwarz M, Lund EG, Russell DW. Two 7 alpha-hydroxylase enzymes in bile acid biosynthesis. Curr Opin Lipidol. 1998;9:113–118 [DOI] [PubMed] [Google Scholar]
- 38.Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, Drake TA, Lusis AJ. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006;16:995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uhlar CM, Whitehead AS. Serum amyloid a, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–523 [DOI] [PubMed] [Google Scholar]
- 40.van der Westhuyzen DR, Cai L, de Beer MC, de Beer FC. Serum amyloid a promotes cholesterol efflux mediated by scavenger receptor b-i. J Biol Chem. 2005;280:35890–35895 [DOI] [PubMed] [Google Scholar]
- 41.Kim MH, de Beer MC, Wroblewski JM, Charnigo RJ, Ji A, Webb NR, de Beer FC, van der Westhuyzen DR. Impact of individual acute phase serum amyloid a isoforms on hdl metabolism in mice. J Lipid Res. 2016;57:969–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein ai. Nature. 1991;353:265–267 [DOI] [PubMed] [Google Scholar]
- 43.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant apoa-i milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA. 2003;290:2292–2300 [DOI] [PubMed] [Google Scholar]
- 44.Morgantini C, Imaizumi S, Grijalva V, Navab M, Fogelman AM, Reddy ST. Apolipoprotein a-i mimetic peptides prevent atherosclerosis development and reduce plaque inflammation in a murine model of diabetes. Diabetes. 2010;59:3223–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson WJ, Kilsdonk EP, van Tol A, Phillips MC, Rothblat GH. Cholesterol efflux from cells to immunopurified subfractions of human high density lipoprotein: Lp-ai and lp-ai/aii. J Lipid Res. 1991;32:1993–2000 [PubMed] [Google Scholar]
- 46.Luc G, Bard JM, Ferrières J, Evans A, Amouyel P, Arveiler D, Fruchart JC, Ducimetière P. Value of hdl cholesterol, apolipoprotein a-i, lipoprotein a-i, and lipoprotein a-i/a-ii in prediction of coronary heart disease: The prime study. Prospective epidemiological study of myocardial infarction. Arterioscler Thromb Vasc Biol. 2002;22:1155–1161 [DOI] [PubMed] [Google Scholar]
- 47.Bartel DP. Micrornas: Target recognition and regulatory functions. Cell. 2009;136:215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by micrornas: Are the answers in sight? Nat Rev Genet. 2008;9:102–114 [DOI] [PubMed] [Google Scholar]
- 49.Fabian MR, Sundermeier TR, Sonenberg N. Understanding how mirnas post-transcriptionally regulate gene expression. Prog Mol Subcell Biol. 2010;50:1–20 [DOI] [PubMed] [Google Scholar]
- 50.Filipowicz W, Sonenberg N. The long unfinished march towards understanding microrna-mediated repression. RNA. 2015;21:519–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hennessy EJ, van Solingen C, Scacalossi KR, Ouimet M, Afonso MS, Prins J, Koelwyn GJ, Sharma M, Ramkhelawon B, Carpenter S, Busch A, Chernogubova E, Matic LP, Hedin U, Maegdefessel L, Caffrey BE, Hussein MA, Ricci EP, Temel RE, Garabedian MJ, Berger JS, Vickers KC, Kanke M, Sethupathy P, Teupser D, Holdt LM, Moore KJ. The long noncoding rna chrome regulates cholesterol homeostasis in primate. Nat Metab. 2019;1:98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.