

SUMMARY
Invariant natural killer T (iNKT) cells are potent immune cells for targeting cancer; however, their clinical application has been greatly hindered by their extremely low numbers in cancer patients. Here, we developed a proof-of-concept for hematopoietic stem cell-engineered iNKT (HSC-iNKT) cell therapy with the potential to provide therapeutic levels of iNKT cells for a patient’s lifetime. Using a human HSC engrafted mouse model and a human iNKT TCR gene engineering approach, we demonstrated the efficient and long-term generation of HSC-iNKT cells in vivo. These HSC-iNKT cells closely resembled endogenous human iNKT cells, could deploy multiple mechanisms to attack tumor cells, and effectively suppressed tumor growth in vivo in multiple human tumor xenograft mouse models. Preclinical safety studies showed no toxicity or tumorigenicity of the intended HSC-iNKT cell therapy. Collectively, these results demonstrated the feasibility, safety, and cancer therapy potential of the proposed HSC-iNKT cell therapy and laid a foundation for future clinical development.
In Brief
Yang and colleagues reported the preclinical development of a hematopoietic stem cell engineered-invariant natural killer T (HSC-iNKT) cell therapy. HSC-iNKT cell therapy has the potential to treat a broad range of cancers through providing cancer patients with therapeutic levels of iNKT cells for a lifetime.
Graphical Abstract
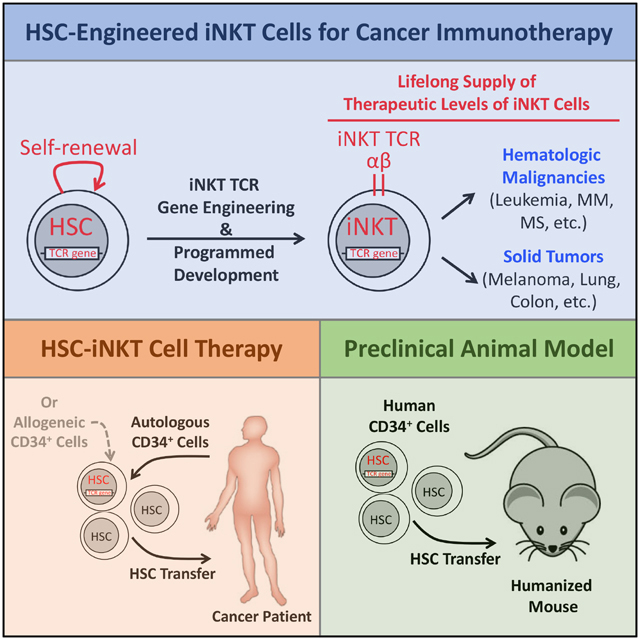
INTRODUCTION
Invariant natural killer T (iNKT) cells are a small population of αβ T lymphocytes highly conserved from mice to humans (Bendelac et al., 2007; Kronenberg and Gapin, 2002). These cells have several unique features, making them exceedingly attractive agents for developing cancer immunotherapy (King et al., 2018; Krijgsman et al., 2018; Lam et al., 2017). First, iNKT cells have a strong relevance to cancer. There is compelling evidence suggesting a significant role of iNKT cells in tumor surveillance in mice (Berzins et al., 2011; Vivier et al., 2012). In humans, iNKT cell frequencies are decreased in patients with solid tumors (including melanoma, colon, lung, breast, and head and neck cancers) and hematologic malignancies (including leukemia, multiple myeloma, and myelodysplastic syndromes), while increased iNKT cell numbers are associated with a better prognosis (Berzins et al., 2011; Krijgsman et al., 2018; Lam et al., 2017). Second, iNKT cells have the remarkable capacity to target multiple types of cancer independent of tumor antigen and major histocompatibility complex (MHC) restrictions (Fujii et al., 2013). iNKT cells recognize glycolipid antigens presented by non-polymorphic CD1d, which frees them from MHC restriction (Bendelac et al., 2007). Many tumor tissues express conserved glycolipid antigens that can be recognized by iNKT cells, although the nature of these glycolipids remain to be identified (Gapin, 2010; Mallevaey and Selvanantham, 2012; Wu et al., 2003). Third, iNKT cells can deploy multiple mechanisms to attack tumor cells, including direct killing of CD1d+ tumors and immune adjuvant effects such as activating NK cells, activating DCs and thereby stimulating cytotoxic T lymphocytes (CTLs), and inhibiting tumor-associated macrophages (TAMs) (Brennan et al., 2013; Cortesi et al., 2018; Fujii et al., 2013; Krijgsman et al., 2018; Song et al., 2009; Vivier et al., 2012).
Attracted by the potent and broad anticancer functions of iNKT cells, researchers have conducted a series of clinical trials utilizing iNKT cells to treat various forms of cancer, ranging from solid tumors to hematologic malignancies (Nair and Dhodapkar, 2017; Waldowska et al., 2017). These clinical trials have utilized α-galactosylceramide (αGC, a synthetic glycolipid ligand specifically stimulating iNKT cells) alone, or αGC-pulsed DCs alone or in combination with ex vivo-expanded patient iNKT cells, and have shown these treatments to be safe and well tolerated. Several recent trials reported encouraging antitumor immunity in patients with melanoma, non-small cell lung cancer, and head and neck squamous cell carcinomas, attesting to the therapeutic potential of iNKT cell-based immunotherapies (Exley et al., 2017; Kunii et al., 2009; Motohashi et al., 2006; Yamasaki et al., 2011). However, most other trials yielded unsatisfactory results. Overall, these trials have all worked through the direct stimulation or ex vivo-expansion of patients’ endogenous iNKT cells, thus yielding only short-term, limited clinical benefits to a small number of patients. The extremely low frequencies of iNKT cells in cancer patients (~0.001–0.1% in blood), as well as the rapid depletion of these cells post-stimulation, are considered the major factors limiting the success of these trials (Krijgsman et al., 2018). In order to unleash the full potential of iNKT cells for cancer immunotherapy, innovative therapies that can overcome these limitations are in high demand.
In this study, we aim to overcome the current limitations by genetically engineering human hematopoietic stem cells (HSCs) to produce iNKT cells targeting cancer (Figure S1A). Because of the longevity and self-renewal of HSCs, by adoptively transferring iNKT T cell receptor (TCR) gene-engineered HSCs into cancer patients, this new therapy has the potential to provide patients with therapeutic levels of iNKT cells for a lifetime (Figure S1A) (Morrison et al., 1995). This TCR-engineered HSC transfer approach takes advantage of two molecular mechanisms governing T cell development, TCR allelic exclusion and TCR instruction, to genetically program HSCs to produce T cells of designated antigen specificity and T cell subtype identity (Yang and Baltimore, 2005). Previously, we and others have successfully utilized this strategy to generate tumor antigen-specific CD4 helper and CD8 cytotoxic T cells of both mouse and human origins (Bettini et al., 2012; Giannoni et al., 2013; Vatakis et al., 2011; Yang and Baltimore, 2005). Human clinical trials are ongoing, testing NY-ESO-1 TCR-engineered HSC adoptive therapy for treating multiple cancers (Baltimore et al., 2010; PuigSaus et al., 2018). Recently, we successfully generated iNKT cells in mice through iNKT TCR-engineered bone marrow transfer and proved cancer therapy potential of the engineered iNKT cells in a mouse melanoma lung metastasis model (Smith et al., 2015). Based on these previous works, as well as the scientific rationale that human iNKT cells also follow a “TCR-instructed” developmental path similar to that of the mouse iNKT cells (Godfrey and Berzins, 2007), we hypothesized that it would be possible to engineer human HSCs with a human iNKT TCR gene to produce human iNKT cells targeting cancer. Here, we report the preclinical development of the proposed HSC-engineered iNKT cell therapy, demonstrating its feasibility, safety, and cancer therapy potential.
RESULTS
Cloning of Human iNKT TCR Genes
Single human iNKT cells were sorted from healthy donor peripheral blood mononuclear cells (PBMCs) using flow cytometry based on a stringent combination of surface markers, gated as hTCRαβ+6B11+hTCR Vβ11+hCD161hi (Figure 1A) (Bendelac et al., 2007). 6B11 is a monoclonal antibody that specifically recognizes the human iNKT TCR invariant alpha chain (hTCR Vα24-Ja18) (Chan et al., 2013; Montoya et al., 2007). We included hTCR Vβ11 staining to focus on the dominant Vβ11+ population of human iNKT cells (Bendelac et al., 2007). The sorted single human iNKT cells were then subjected to TCR cloning using an established single-cell TCR cloning technology (Figure 1B) (Smith et al., 2015). A validated pair of iNKT TCR α and β chain genes (thereafter collectively referred to as the human iNKT TCR gene) were selected for further development.
Figure 1. Cloning of Human Invariant Natural Killer T Cell Receptor (iNKT TCR) Genes and Construction of Lentiviral Gene Delivery Vectors.

(A-B) Cloning of human iNKT TCR genes using a single-cell RT-PCR approach. (A) FACS sorting of single human iNKT cells. (B) Representative DNA gel image showing the human TCR α and β chain PCR products from six sorted single iNKT cells.
(C) Schematics of the Lenti/iNKT, Lenti/iNKT-EGFP, and Lenti/iNKT-sr39TK vectors.
(D) FACS detection of intracellular expression of iNKT TCRs (identified as hTCR Vβb11+6B11+) in 293T cells transduced with the indicated iNKT TCR gene delivery lentivectors.
(E-G) Functional characterization of the Lenti/iNKT-sr39TK vector by transducing human PBMC T cells. (E) FACS detection of surface iNKT TCR expression on vector-transduced PBMC T cells. (F) ELISA analysis of IFN-γ production by vector-transduced PBMC T cells post-αGC stimulation (n = 3). (G) FACS quantification of the depletion of vector-transduced PBMC T cells post-GCV treatment (n = 4).
Representative of 2 experiments. See also Figure S1.
Construction of Human iNKT TCR Gene Delivery Vectors
A pMNDW lentiviral vector designated for HSC-based gene therapy was chosen to deliver the iNKT TCR gene (Figure 1C) (Cartier et al., 2009). Three vectors were constructed: 1) a Lenti/iNKT vector encoding the iNKT TCR gene, which is intended for eventual clinical usage; 2) a Lenti/iNKT-EGFP vector encoding the iNKT TCR gene together with an enhanced green fluorescent protein (EGFP) reporter gene, which allows for convenient tracking of vector-engineered HSCs and their progeny cells using flow cytometry and thus is valuable for preclinical studies; and 3) a Lenti/iNKT-sr39TK vector encoding the iNKT TCR gene together with an sr39TK suicide/PET imaging reporter gene, which allows for monitoring vector-engineered HSCs and their progeny cells using PET imaging and for depleting engineered cells through ganciclovir (GCV) administration in case of an adverse side effect, and thus is valuable for early clinical development (Figure 1C) (Black et al., 1996; Gschweng et al., 2014; Larson et al., 2017). The gene-delivery capacity of these lentivectors, as well as the functionality of the encoded human iNKT TCR gene and EGFP and sr39TK reporter genes, were studied by transducing 293T cells (Figure 1D) and primary human PBMC T cells (Figures 1E–1G) with individual lentivectors followed by functional tests. Notably, all three lentivectors mediated efficient expression of the human iNKT TCR gene (Figure 1D); the resulting transgenic human iNKT TCRs recognized αGC and responded to αGC stimulation, as evidenced by hCD1d/PBS-57 tetramer binding and induced IFN-γ production (Figures 1E and 1F); and the encoded sr39TK enabled efficient GCV-induced depletion of gene-engineered cells (Figure 1G). In the present study, these three lentivectors were utilized simultaneously or individually, depending on the purpose of a given experiment.
iNKT TCR Gene-Engineering of Human HSCs
iNKT TCR gene-delivery lentivectors were of high titer, and could transduce human CD34+ hematopoietic stem and progenitor cells (referred to as HSCs thereafter) of various donors robustly and efficiently (Figures S1B–S1D). Gene-engineering rate of HSCs could be conveniently adjusted through titrating lentivectors utilized for transduction, resulting in 0–50% iNKT TCR+ HSCs with an average vector copy number (VCN) of 0–3 per cell (Figure S1E).
Production of BLT-iNKT Humanized Mice
We utilized a BLT (human bone marrow-liver-thymus engrafted NOD/SCID/γc−/− mice) humanized mouse model, that supports the engraftment of human HSCs and supports the development of human T cells, to study the in vivo generation of HSC-engineered human iNKT cells (Lan et al., 2006; Melkus et al., 2006; Smith et al., 2016). Human HSCs, either mock-transduced or transduced with human iNKT TCR gene-delivery vectors, were adoptively transferred into NOD/SCID/γc−/− (NSG) mice engrafted with human thymus to produce standard BLT mice or iNKT TCR gene-engineered BLT mice (denoted as BLT or BLT-iNKT mice, respectively) (Figure 2A). These BLT and BLT-iNKT humanized mice were then utilized for further study.
Figure 2. Generation of Hematopoietic Stem Cell-Engineered Human iNKT (HSC-iNKT) Cells in BLT-iNKT Humanized Mice.

(A) Experimental design to generate HSC-iNKT cells in a BLT humanized mouse model.
(B-D) Generation of HSC-iNKT cells in BLT-iNKT mice. (B) Time-course FACS monitoring of human immune cells (gated as hCD45+ cells), human αβ T cells (gated as hCD45+hTCRαβ+ cells), and human iNKT cells (gated as hCD45+hTCRαβ+6B11+ cells) in the peripheral blood of BLT-iNKT mice and control BLT mice post-HSC transfer (n = 9–10). (C) FACS detection of human immune cells in various tissues of BLT-iNKT and control BLT mice, at week 20 post-HSC transfer. (D) FACS detection of HSC-iNKT cells in various tissues of BLT-iNKT mice, at week 20 post-HSC transfer. hiNKT, human iNKT cells; hTc, conventional human αβ T cells (gated as hCD45+hTCRαβ+6B11− cells).
(E-G) Long-term production of HSC-iNKT cells in BLT-iNKT mice. (E) Experimental design. BM, total bone marrow cells harvested from the primary BLT-iNKT mice; Thy, human thymus implants collected from the primary BLT-iNKT mice. (F) FACS detection of HSC-iNKT cells in the peripheral blood of the secondary BLT-iNKT mice at week 16 after the secondary BM/Thy transfer. (G) Quantification of F (n = 4–5).
(H) Controlled production of HSC-iNKT cells in BLT-iNKT mice. BLT-iNKT mice were generated with PBSCs transduced with titrated amounts of Lenti/iNKT-sr39TK vector (4 × 108, 2 × 108, or 1 × 108 TU per 1 × 106 PBSCs). FACS quantification of human iNKT cells in the blood of indicated BLT-iNKT mice at week 16 post-HSC transfer were presented (n = 8–10).
(I) Table summarizing experiments that have successfully generated HSC-iNKT cells in the BLT human mouse model.
Representative of 2 (E-G, H) and over 10 (B-D) experiments.
Generation of HSC-iNKT Cells in BLT-iNKT Humanized Mice
We detected efficient and comparable reconstitution of human immune cells, in particular human ab T cells, in both BLT and BLT-iNKT mice (Figure 2B). BLT mice contained minimal numbers of endogenous human iNKT cells below detection level (Figure 2B). Numbers of human iNKT cells were greatly increased in BLT-iNKT mice; these iNKT cells started to appear in the peripheral blood of BLT-iNKT mice around 2 months post-HSC transfer, then peaked and stabilized around 3 months, indicating that they were derived from the iNKT TCR gene-engineered HSCs (Figure 2B). Analysis of BLT-iNKT mice revealed a tissue distribution of HSC-engineered human iNKT cells (referred to as HSC-iNKT cells thereafter) that was typical of human iNKT cells: besides circulating in blood, HSC-iNKT cells homed to lymphoid organs such as the bone marrow and spleen, as well as other peripheral organs such as the liver (Figures 2C and 2D) (Godfrey and Berzins, 2007). The in vivo production of HSC-iNKT cells was stable and long-lasting, evident in the persistence of high levels of iNKT cells in the primary BLT-iNKT mice for over 5 months after the initial HSC transfer (Figure 2B), and in the secondary BLT-iNKT mice after the secondary HSC transfer (Figures 2E–2G). These results indicate that the iNKT TCR gene-engineered HSCs retain their longevity and self-renewal capacity and are able to produce iNKT cells over time, highlighting the long-term therapeutic potential of the HSC-engineered iNKT cell approach. Moreover, by titrating the iNKT TCR gene-engineering rate of HSCs used for adoptive transfer, we could generate BLT-iNKT mice that contained gradient levels of human iNKT cells ranging from 5% to over 60% of total human ab T cells (Figures 2H and S1E). The capacity to control the outputs of engineered human iNKT cells could be very valuable for the clinical application of HSC-iNKT cell therapy, allowing dosing the engineered iNKT cells in cancer patients according to therapeutic and safety needs.
The method of generating human iNKT cells through TCR gene-engineering of HSCs is highly robust. In our experiments, we have succeeded in generating HSC-iNKT cells in BLT-iNKT mice engineered with all three iNKT TCR gene delivery lentivectors (Lenti/iNKT, Lenti/iNKT-EGFP, and Lenti/iNKT-sr39TK), using human CD34+ HSCs isolated from various cell sources of multiple donors (cord blood, fetal liver, and G-CSF-mobilized peripheral blood), and using human thymus implants collected from various tissue sources of multiple donors (fetal thymus and post-natal thymus) (Figure 2I). Notably, there was no need to match human CD34+ HSCs with human thymus implants for donors, or tissue sources, or human leukocytes antigens (HLAs), making the generation of BLT-iNKT mice highly adaptable (Figure 2I). The BLT-iNKT humanized mouse model therefore can be utilized as a powerful and versatile tool for the preclinical development of HSC-iNKT cell therapy, and for the study of human iNKT cell biology and other human iNKT cell-based immunotherapies. Importantly, the success of generating human iNKT cells through TCR gene engineering of G-CSF-mobilized peripheral blood CD34+ HSCs (referred to as PBSCs thereafter) from multiple human donors directly indicates the robustness and translational potential of the proposed HSC-iNKT cell therapy (Figures 2I and S1A). Because of their possible relevance to early clinical development, in this report, we focused on studying HSC-iNKT cells generated in BLT-iNKT mice produced with Lenti/iNKT-sr39TK vector-transduced PBSCs (and implanted with human fetal thymus), unless otherwise indicated.
Safety Study of HSC-iNKT Cell Therapy in BLT-iNKT Humanized Mice
Safety of the HSC-iNKT cell therapy was evaluated by long-term monitoring and terminal pathology analysis of the BLT-iNKT humanized mice in comparison with the control BLT mice (Figure S2). Over a period of 5 months, BLT-iNKT mice showed a steady body weight increase and a survival rate comparable to that of the control BLT mice (Figures S2A and S2B). Pathological analysis of the various tissues of the BLT-iNKT mice collected at the end of 5 months showed no increase of tissue inflammation, hematopoietic neoplasm, or non-hematopoietic neoplasm, comparable to that of the control BLT mice (Figure S2C). 5 months after HSC transfer, BLT mice gradually developed graft-versus-host disease (GvHD) and eventually died of this disease due to the xenoreactive nature of the engrafted human conventional αβ T cells (Lan et al., 2006; Melkus et al., 2006; Smith et al., 2016). Intriguingly, compared to the control BLT mice, BLT-iNKT mice were significantly protected from GvHD, evidenced by the reduced auto-activation of engrafted human conventional ab T cells in BLT-iNKT mice (Figures S2D–S2F) and the resulting elongated survival of these mice (Figure S2G). This phenomenon agreed with the clinical observation that in leukemia patients who received allogenic HSC transfer, levels of donor-derived iNKT cells positively correlated with the therapeutic effects of graft-versus-leukemia (GvL) and negatively correlated with the incidences and severity of GvHD, presumably attributed to the regulatory function of human iNKT cells (de Lalla et al., 2011; Haraguchi et al., 2004; Rubio et al., 2012). Reduced GvHD in BLT-iNKT mice suggest that, like native human iNKT cells, HSC-iNKT cells promote a favorable balance of GvL compared to GvHD. Taken together, these results demonstrate the safety of HSC-iNKT cell therapy in the BLT-iNKT pre-clinical animal model. The results also suggest a possible pro-GvL/anti-GvHD dual benefit of HSC-iNKT cell therapy in an allogeneic HSC transfer therapeutic setting (Figure S1A).
Biodistribution and Controlled Depletion of HSC-iNKT Cells in BLT-iNKT Humanized Mice Visualized by PET Imaging
Incorporation of an sr39TK suicide/PET imaging reporter gene in the human iNKT TCR gene delivery vector provides an additional safety control for HSC-iNKT cell therapy, allowing the depletion of gene-engineered human immune cells through GCV treatment in case the therapy needs to be terminated due to safety concerns (Figure 3A). It also provides an opportunity to visualize the biodistribution and in vivo dynamics of the gene-engineered human HSCs and their progeny cells in Lenti/iNKT-sr39TK vector-engineered BLT-iNKT mice (denoted as BLT-iNKTTK mice) using non-invasive PET imaging, a clinically applicable technology (Figure 3A). Using PET imaging combined with CT scan, we detected the distribution of gene-engineered human immune cells across the lymphoid tissues of BLT-iNKTTK mice, particularly in the bone marrow and spleen (Figure 3B). Treating BLT-iNKTTK mice with GCV effectively depleted gene-engineered human cells across the body (Figures 3C–3D and S3A–S3B). The depletion of gene-engineered human cells in bone marrow was confirmed by droplet digital PCR (Figure 3G). Importantly, the GCV-induced cell depletion was specific, evidenced by the selective depletion of HSC-iNKT cells but not the overall human immune cells in BLT-iNKTTK mice as measured by flow cytometry (Figures 3E–3F and S3C–S3D).
Figure 3. Biodistribution and Controlled Depletion of HSC-iNKT Cells in BLT-iNKT Humanized Mice Visualized by PET Imaging.

(A) Experimental design. BLT-iNKTTK, BLT-iNKT mice generated with Lenti/iNKT-sr39TK vector-transduced PBSCs.
(B) Biodistribution of vector-engineered human immune cells. Representative PET/CT images were presented. Note that there were background signals in the gastrointestinal tract (GI) and gallbladder of all mice. BM, bone marrow.
(C-D) PET/CT analysis of controlled depletion of vector-engineered human immune cells in BLT-iNKTTK mice via GCV treatment. (C) Representative PET/CT images. (D) Quantification of C (n = 4–5).
(E-F) FACS validation of controlled depletion of HSC-iNKT cells in BLT-iNKTTK mice via GCV treatment. (E) Representative FACS plots of bone marrow cells. (F) Quantification of E (n = 4–5).
(G) Droplet Digital PCR (ddPCR) validation of controlled depletion of vector-engineered human immune cells in BLT-iNKTTK mice via GCV treatment (n = 4–5). VCN, vector copy number.
Representative of 2 experiments. See also Figures S2 and S3.
Development of HSC-iNKT Cells in BLT-iNKT Humanized Mice
Utilizing the BLT-iNKT mouse model, we set out to study the in vivo development of HSC-iNKT cells. Analysis of the human thymus implants in BLT-iNKT mice detected the presence of developing HSC-iNKT cells; these cells followed a typical iNKT cell development path defined by CD4/CD8 co-receptor expression: from DN (double-negative) to DP (double-positive), then to SP (single-positive) or back to DN cells (Figure 4A and S4A) (Godfrey and Berzins, 2007). In the periphery, HSC-iNKT cells further upregulated memory T cell and NK cell markers (CD45RO and CD161, respectively), a signature of iNKT cell development resembling that of endogenous human iNKT cells (Figures 4B–4C and S4A–S4B) (Godfrey and Berzins, 2007).
Figure 4. Development, Phenotype, and Functionality of HSC-iNKT Cells.

(A-C) Development of HSC-iNKT cells. (A) FACS analysis of developing HSC-iNKT cells in the human thymus implants of BLT-iNKT mice. (B) FACS analysis of mature HSC-iNKT cells in the periphery (blood) of BLT-iNKT mice. (C) FACS analysis of control native human iNKT (PBMC-iNKT) cells in the blood of a representative healthy human donor.
(D-E) Allelic exclusion of endogenous TCRs in HSC-iNKT cells. (D) FACS plots showing the TCR Vβ usage by HSC-iNKT cells and HSC-TC cells harvested from the liver of BLT-iNKT mice. hTCR Vβs (FITC) and hTCR Vβs (PE) staining antibodies collectively stained human TCR Vβ 1, 2, 3, 4, 5.1, 5.2, 5.3, 7.1, 7.2, 8, 9, 12, 13.1, 13.2, 13.6, 16, 17, 18, 20, 21.3, and 23. (E) Quantification of D (n = 5).
(F) Phenotype of HSC-iNKT cells. FACS plots are presented, showing the surface markers of HSC-iNKT cells isolated from the spleen of BLT-iNKT mice, compared to those of endogenous human iNKT (PBMC-iNKT) and conventional αβ T (PBMC-Tc) cells isolated from healthy donor peripheral blood.
(G-H) Antigen responses of HSC-iNKT cells. Spleen cells of BLT-iNKT mice were cultured in vitro in the presence or absence of αGC for 7 days. (G) FACS quantification of HSC-iNKT cell expansion over time (n = 3). (H) ELISA analysis of cytokine production at day 7 (n = 3).
(I) Production of effector molecules by HSC-iNKT cells. BLT-iNKT mice spleen cells and healthy donor PBMCs were stimulated in vitro with αGC for 7 days. FACS plots are presented, showing the intracellular production of effector cytokines and cytotoxic molecules in HSC-iNKT cells compared to that in the PBMC-iNKT and PBMC-Tc cells.
Representative of 2 experiments. See also Figure S4.
We have previously shown that overexpression of a transgenic mouse iNKT TCR gene in mouse HSCs induced allelic exclusion and blocked the rearrangement of endogenous TCR genes in the resulting HSC-engineered mouse iNKT cells (Smith et al., 2015). Study of the human HSC-iNKT cells generated in BLT-iNKT mice revealed that these cells expressed the transgenic human iNKT TCRs (hTCR Vβ11+), but not the other human TCR Vβ chains analyzed in our experiments, indicating that allelic exclusion also occurred during the development of human HSC-iNKT cells (Figures 4D and 4E). Utilizing BLT-iNKT mice engineered with the Lenti/iNKT-EGFP vector (denoted as BLT-iNKTGFP mice), we studied the lineage differentiation of iNKT TCR-engineered human HSCs by tracking GFP+ human HSCs and their progeny human immune cells. Analysis of human αβ T cell population in the periphery of BLT-iNKTGFP mice revealed that GFP+ cells were exclusively human iNKT cells (Figures S4C and S4D). Analysis of bone marrow cells of BLT-iNKTGFP mice showed that GFP+ and GFP− cells comprised a similar composition of all lineages of human immune cells analyzed, including HSCs, T cells, B cells, myeloid cells, monocytes, dendritic cells, and NK cells (Figures S4E and S4F). Taken together, these results indicate that iNKT TCR-engineered human HSCs have the full potential to remain as long-term HSCs or to differentiate into various lineages of human immune cells; however, once going down the path of T cell development, the engineered T cell progenitor cells will commit to become iNKT cells but not other T cells following a TCR-instruction mechanism (Smith et al., 2015). Notably, because only T cells express the CD3 molecules that are required to support the surface display of TCRs and the downstream TCR signaling events, the expression of transgenic human iNKT TCRs is functionally restricted to the therapeutic HSC-iNKT cells without interfering with the activities of other lineages of human immune cells, which is a desirable feature for gene- and cell-based therapies.
Phenotype and Functionality of HSC-iNKT Cells
Next, we studied the phenotype and functionality of HSC-iNKT cells, in comparison with that of endogenous human iNKT cells and conventional αβ T cells isolated from the peripheral blood of healthy human donors (denoted as PBMC-iNKT and PBMC-Tc cells, respectively). HSC-iNKT cells displayed a surface phenotype closely resembling PBMC-iNKT cells but distinct from PBMC-Tc cells: they expressed a mixed pattern of CD4/CD8 co-receptors (DN, CD4 SP, and CD8 SP); they expressed high levels of NK cell markers (CD161 and NKG2D); they upregulated T cell memory and activation markers (CD45RO and CD69); and they down-regulated lymphoid organ homing marker (CD62L) and upregulated peripheral tissue and inflammatory site homing markers (CCR4, CCR5, and CXCR3) (Figure 4F). When stimulated with αGC, HSC-iNKT cells proliferated vigorously (Figure 4G). They secreted high levels of TH0/TH1 cytokines such as IFN-γ, limited amounts of TH2 cytokines such as IL-4, and minimal amounts of TH17 cytokines such as IL-17, indicating a TH0/TH1-prone effector function of these cells (Figure 4H). Intracellular staining showed that at the single-cell level, HSC-iNKT cells produced excess amounts of effector molecules, in particular pro-inflammatory cytokines (IFN-γ, TNF-α, and IL-2) and cytotoxic molecules (Perforin and Granzyme B), at levels comparable to that produced by the native PBMC-iNKT cells (Figure 4I). The capacity to produce excess amounts of pro-inflammatory and cytotoxic effector molecules is a signature of iNKT cells which is attractive to cancer therapy (King et al., 2018; Krijgsman et al., 2018; Lam et al., 2017). Analysis of transcription factor expression pattern revealed that HSC-iNKT cells expressed high levels of PLZF, the “master” transcription factor regulating iNKT cell development and functionality, resembling that of native iNKT cells while differing from that of native conventional αβ T cells and γδ T cells isolated from healthy donor peripheral blood (Figure S4G) (Kovalovsky et al., 2008; Savage et al., 2008).
Tumor-Attacking Mechanisms of HSC-iNKT Cells
One of the most attractive features of iNKT cells is that they can attack tumors through multiple mechanisms (Brennan et al., 2013; Fujii et al., 2013; Krijgsman et al., 2018; Vivier et al., 2012). We therefore studied HSC-iNKT cells for their capacity to deploy these various tumor-attacking mechanisms, including: 1) direct killing of CD1d+ tumor cells, 2) adjuvant effects on enhancing NK-mediated killing of tumor cells, 3) adjuvant effects on boosting dendritic cell and cytotoxic T lymphocyte (DC/CTL) antitumor activities, and 4) inhibition of tumor-associated macrophages (TAMs) (Figure 5A).
Figure 5. Tumor-Attacking Mechanisms of HSC-iNKT Cells.

(A) Diagram showing the possible mechanisms utilized by human iNKT cells to attack tumor cells. APC, antigen presenting cell; NK, natural killer cell; DC, dendritic cell; CTL, cytotoxic T lymphocyte; TAM, tumor-associated macrophage.
(B-E) Studying the direct killing of CD1d+ tumor cells by HSC-iNKT cells (tumor:iNKT ratio 1:10). (B) Experimental design to study CD1d-dependent killing of MM.1S-hCD1d-FG cells in the presence of αGC. (C) Tumor killing data from B (n = 3). (D) Experimental design to study CD1d-dependent killing of MM.1S-hCD1d-FG cells in the absence of αGC. (E) Tumor killing data from D (n = 3).
(F-I) Studying the adjuvant effects of HSC-iNKT cells on enhancing NK cell-mediated killing of tumor cells (tumor:NK:iNKT ratio 1:2:2). (F) Experimental design. (G) Tumor killing (n = 3). (H) FACS plots showing CD69 expression on NK cells. (I) Quantification of H (n = 3).
(J-N) Studying the adjuvant effects of HSC-iNKT cells on boosting DC/CTL antitumor reactions (DC:CTL:iNKT ratio 1:1:1). (J) Experimental design. (K) FACS plots showing CD86 expression on MoDCs. (L) Quantification of K (n = 3). (M) FACS plots showing the detection of ESO-T cells in the mixed cell culture. (N) Quantification of M (n = 3).
(O-S) Studying the inhibition of macrophages by HSC-iNKT cells (macrophage:iNKT ratio 1:1). Monocytes isolated from healthy donor PBMCs were studied. (O) Experimental design. (P) FACS plots showing CD69 expression on HSC-iNKT cells. (Q) Quantification of P (n = 3). (R) FACS plots showing the viability of monocytes. (S) Quantification of R (n = 3).
Representative of 2 experiments. See also Figures S5 and S6.
We utilized an in vitro tumor cell killing assay to study the direct killing of CD1d+ tumor cells. A human multiple myeloma (MM) cell line, MM.1S, was chosen as the model tumor target, because MM has a strong clinical relevance to iNKT cells, and because primary human MM tumor cells are CD1d+ and are proven targets of native human iNKT cell killing (Dhodapkar et al., 2003; Dhodapkar and Richter, 2011; Spanoudakis et al., 2009). Notably, most MM cell lines, including MM.1S, have lost CD1d expression (Dhodapkar et al., 2003). We therefore engineered MM.1S cells to overexpress CD1d. We also engineered these cells to overexpress the firefly luciferase (Fluc) and EGFP reporters to enable the sensitive measurement of tumor killing using luminescence reading or flow cytometry. The resulting MM.1S-FG and MM.1S-hCD1d-FG cell lines were then used for the in vitro tumor cell killing assay (Figures S5A and S5B). In the presence of aGC, HSC-iNKT cells killed tumor cells aggressively in an CD1d-dependant manner, attesting to the strong tumor-killing potency of HSC-iNKT cells triggered by CD1d/antigen recognition (Figures 5B–5C and S5C–S5D). Importantly, even without the addition of αGC, HSC-iNKT cells could still effectively kill MM.1S-hCD1d-FG tumor cells, although tumor killing was less aggressive and required the addition of IL-15 (Figures 5D and 5E). Besides its role as an essential homeostatic cytokine in supporting the peripheral maintenance of iNKT cells, IL-15 is critical in supporting the function of iNKT cells, especially when iNKT cells respond to sub-optimal antigen stimulations (Liu et al., 2012; Matsuda et al., 2002). The direct killing of MM.1S-hCD1d-FG tumor cells by HSC-iNKT cells was dependent on the presence of CD1d on tumor cells (Figures 5E), indicating that HSC-iNKT cells can directly kill CD1d+ tumor cells by recognizing tumor cell-derived lipid antigens presented by CD1d.
Next, we studied the capacity of HSC-iNKT cells for enhancing NK cell-mediated killing of tumor cells, utilizing an in vitro NK adjuvant effect assay (Figure 5F). A K562 human myelogenous leukemia cell line, which did not express HLA-I or CD1d, was engineered to express Fluc and EGFP and utilized as the model tumor target to assess NK cell-mediated killing (Figures S5E–S5G). αGC was used as a surrogate tumor-derived lipid antigen to stimulate HSC-iNKT cells. Antigen-stimulated HSC-iNKT cells significantly enhanced the activation (measured by CD69 upregulation) and tumor cell-killing efficacy of NK cells (Figures 5G–5I), likely through secreting NK cell-activation cytokines, including IFN-γ and IL-2 (Figure S5H) (Krijgsman et al., 2018; Vivier et al., 2012). Therefore, HSC-iNKT cells can function as an effective cellular adjuvant to enhance NK cell-mediated killing of tumor cells.
Subsequently, we evaluated the ability of HSC-iNKT cells for boosting DC- and CTL-mediated antitumor activities, using an in vitro DC/CTL adjuvant effect assay (Figure 5J). DC-stimulated CTL response to NY-ESO-1, a well-studied tumor antigen common in many cancers, was analyzed (Bethune et al., 2018). Monocyte-derived dendritic cells (MoDCs) were generated from HLA-A2+ healthy donor PBMCs (Figures S6A and S6B); these MoDCs expressed CD1d as well as HLA-A2 and thus could present aGC to stimulate HSC-iNKT cells, and also present NY-ESO-1 peptide (ESOp) to activate ESO-specific CD8+ CTLs (denoted as ESO-T cells) (Figures S6C–S6F). In the presence of aGC, HSC-iNKT cells efficiently promoted the maturation of MoDCs as evidenced by the upregulation of co-stimulatory marker CD86 on MoDCs (Figures 5K and 5L). MoDCs matured by HSC-iNKT cells showed significantly enhanced capacity to present ESOp tumor antigens, thereby inducing greatly increased activation and expansion of ESO-T cells as well as resulting in enhanced ESO-T cell mediated killing of target tumor cells (Figures 6M–6N and S6G–S6H). Therefore, HSC-iNKT cells can function as an effective cellular adjuvant to boost DC/CTL antitumor activities.
Figure 6. In Vivo Antitumor Efficacy of HSC-iNKT Cells Against Hematologic Malignancies in a Human Multiple Myeloma (MM) Xenograft Mouse Model.

(A-I) In vivo antitumor efficacy of HSC-iNKT cells was studied using an MM.1S-hCD1d-FG human MM xenograft NSG mouse model. HSC-iNKT cells derived from PBSCs of three different donors were studied, to verify the robustness of the HSC-iNKT cell therapy. Data from one representative donor were presented. (A) Experimental design. BLI, live animal bioluminescence imaging. (B) BLI images showing tumor loads in experimental mice over time. (C) Quantification of B (n = 6–8). TBL, total body luminescence. (D) FACS plots showing the detection of tumor cells (gated as GFP+ cells) in various tissues of experimental mice. (E) Quantification of D (n = 6–8). MNCs, mononuclear cells. (F) FACS plots showing the detection of HSC-iNKT cells in various tissues of experimental mice. (G) FACS plots showing the expression of CD62L and CD69 on HSC-iNKT cells isolated from the blood and bone marrow of tumor-bearing mice. (H-I) Quantification of G (n = 6).
(J-L) In vivo antitumor efficacy of HSC-iNKT cells was studied using a control MM.1S-FG human MM xenograft NSG mouse model. HSC-iNKT cells derived from PBSCs of a representative donor were studied. (J) Experimental design. (K) BLI images showing tumor loads in experimental mice over time. (L) Quantification of K (n = 4–5).
Representative of 3 (A-I) and 2 (J-L) experiments.
Lastly, we evaluated the potential of HSC-iNKT cells in inhibiting tumor-associated macrophages (TAMs), utilizing an in vitro macrophage inhibition assay (Figure 5O). Human CD14+ monocytes isolated from healthy donor PBMCs were used in this study (Figure S6I). These monocytes expressed CD1d (Figure S6J); when loaded with aGC as a surrogate tumor-derived lipid antigen, these monocytes stimulated HSC-iNKT cells (evidenced by upregulation of CD69 on HSC-iNKT cells; Figures 6P and 6Q) and were quickly killed by HSC-iNKT cells (Figures 6R and 6S). Therefore, HSC-iNKT cells have the potential to inhibit TAMs in a CD1d/antigen-dependent manner.
Taken together, these results indicate that HSC-iNKT cells are capable of deploying multiple mechanisms to attack tumor cells, attesting to their multifaceted cancer therapy potential.
In Vivo Antitumor Efficacy of HSC-iNKT Cells Against Hematologic Malignancies in a Human Multiple Myeloma Xenograft Mouse Model
In vivo antitumor efficacy of HSC-iNKT cells against hematologic malignancies was studied using a human multiple myeloma (MM) xenograft NSG mouse model (Figure 6A). The pre-established MM.1S-hCD1d-FG human MM cell line was utilized for this study (Figure S5A). MM.1S-hCD1d-FG cells were inoculated intravenously (i.v.) into NSG mice to establish disease, followed by i.v. injection of HSC-iNKT cells and monitoring of HSC-iNKT cell therapeutic effects (Figure 6A). This tumor model allowed the in vivo study of direct killing of CD1d+ blood cancer cells by HSC-iNKT cells (Figure S5A). HSC-iNKT cells showed a robust suppression of MM, as evidenced by a significant decrease of total body luminescence measured by BLI (Figures 6B and 6C). Terminal tissue collection and flow cytometry analysis confirmed a significant reduction of GFP+ tumor cells in multiple tissues, particularly in the bone marrow, which is the primary site of MM, and in the liver, which is another major MM site in this model (Figures 6D and 6E). Correspondingly, flow cytometry analysis detected the presence of HSC-iNKT cells in various tumor-residing tissues (Figure 6F). In particular, high numbers of HSC-iNKT cells homed to the areas of MM involvement, including bone marrow and liver (Figure 6F), where these HSC-iNKT cells displayed an activated phenotype (CD62LloCD69hi) correlating with their antitumor function (Figures 6G–6I). The antitumor efficacy of HSC-iNKT cells was robust and consistent for all three donors that we studied (Figure 6A). The suppression of MM in the MM.1S-CD1d-FG model was mediated by HSC-iNKT cells through a CD1d-dependent tumor recognition and suppression mechanism, because HSC-iNKT cells did not exhibit tumor suppression effects and were not enriched at tumor sites in the control MM.1S-FG model which lacked the expression of CD1d (Figures 6J–6L). These data demonstrated the in vivo antitumor efficacy of HSC-iNKT cells and support their therapeutic potential against hematologic malignancies.
In Vivo Antitumor Efficacy of HSC-iNKT Cells Against Solid Tumors in a Human Melanoma Xenograft Mouse Model
In vivo antitumor efficacy of HSC-iNKT cells against solid tumors was studied using a human melanoma xenograft NSG mouse model (Figure 7A). An A375-hCD1d-hIL-15-FG human melanoma cell line and a control A375-hIL-15-FG melanoma cell line were generated for this study (Figures S7A and S7B). When co-cultured in vitro, HSC-iNKT cells effectively killed the A375-hCD1d-hIL-15-FG cells in a CD1d/antigen dependent manner (Figures S7C–S7F). For the in vivo study, A375-hIL-15-hCD1d-FG cells loaded with aGC were subcutaneously inoculated into NSG mice to form solid tumors, followed by i.v. injection of HSC-iNKT cells and monitoring of tumor growth (Figure 7A). We detected a significant suppression of tumor growth by HSC-iNKT cells, analyzed by time-course BLI (Figures 7B and 7C), measurement of tumor size over time (Figure 7D), and terminal tumor weight assessment (Figure 7E). Terminal tissue collection and flow cytometry analysis revealed a highly efficient infiltration of HSC-iNKT cells into solid tumors (Figure 7F). The targeted homing of HSC-iNKT cells to the tumor sites was impressive, as much smaller numbers of HSC-iNKT cells were detected in all other tissues examined (blood, spleen, and liver), likely due to the expression of inflammatory site-homing markers on HSC-iNKT cells (Figures 7F and 4F). HSC-iNKT cells isolated from tumors displayed a highly active phenotype (CD62LloCD69hi) compared to HSC-iNKT cells isolated from non-tumor tissues like the liver, corresponding with their antitumor activities (Figures 7G-7I). The suppression of solid tumor growth in the A375-hIL-15-hCD1d-FG model was mediated by HSC-iNKT cells through a CD1d-dependent direct killing mechanism, because HSC-iNKT cells did not induce tumor suppression in the control A375-hIL-15-FG model which lacked the expression of CD1d (Figures 7J–7L and S7G–S7H). Taken together, these in vivo results demonstrated the therapeutic potential of HSC-iNKT cells for treating solid tumors. In particular, the capacity of HSC-iNKT cells effectively trafficking to the tumor sites and infiltrating solid tumors is highly desirable for cancer immunotherapy.
Figure 7. In Vivo Antitumor Efficacy of HSC-iNKT Cells Against Solid Tumors in a Human Melanoma Xenograft Mouse Model.

(A-I) In vivo antitumor efficacy of HSC-iNKT cells was studied using an A375-hIL-15-hCD1d-FG human melanoma xenograft NSG mouse model. (A) Experimental design. (B) BLI images showing tumor loads in experimental mice over time. Note that beyond day 22, BLI signals were saturated and thus were not included for quantification. (C) Quantification of B (n = 5). (D) Measurements of tumor size over time (n = 5). (E) Measurements of tumor weight at the terminal harvest on day 30 (n = 5). (F) FACS plots showing the detection of HSC-iNKT cells in various tissues of experimental mice. (G) FACS plots showing the expression of CD62L and CD69 on HSC-iNKT cells isolated from the livers and tumors of tumor-bearing mice. (H-I) Quantification of G (n = 5).
(J-L) In vivo antitumor efficacy of HSC-iNKT cells was studied using a control A375-hIL-15-FG human melanoma xenograft NSG mouse model. (J) Experimental design. (K) BLI images showing tumor loads in experimental mice over time. Note that beyond day 21, BLI signals were saturated and thus were not included for quantification. (L) Quantification of K (n = 5).
Representative of 2 experiments. See also Figure S7.
DISCUSSION
HSC transplantation (HSCT) has a history of over half a century and has been utilized routinely to treat a variety of malignant and non-malignant hematopoietic disorders (Chabannon et al., 2018). Over the last decade, gene-engineered HSCT has undergone rapid development and has shown great promise for treating diseases like monogene-related immunodeficiencies (Chabannon et al., 2018). Safe and efficient gene delivery vectors have been developed, and robust HSC gene modification protocols have been established (Chabannon et al., 2018). In 2016, the first gene-engineered HSCT therapy product received marketing authorization from the European Medicines Agency, marking a major milestone and paving the way for this new class of cell therapy drugs (Chabannon et al., 2018). Therefore, both the technology and clinical platforms are ready to translate the intended HSC-iNKT cell therapy.
Generation of HSC-engineered iNKT cells requires a functional thymus. Aging-related degeneration of human thymus in many cancer patients therefore may be a concern (Lynch et al., 2009). Clinical studies of adult cancer patients receiving HSC transfer were reported previously, showing the reconstitution of iNKT cells in these patients (Beziat et al., 2010; Haraguchi et al., 2004). These clinical data support the likelihood that HSC-iNKT therapy can benefit aged cancer patients.
Increasing iNKT cells in cancer patients may have certain risks, including possible toxicity and interference of immune surveillance. The levels of iNKT cells achievable in cancer patients may also be limited by the availability of host supporting factors such as homeostatic cytokines. These concerns can be greatly alleviated through controlling the iNKT cell outputs via controlling the HSC gene-engineering rates (Figures 2H and S1E). For clinical trial design, an initial dose targeting ~5% iNKT cells in blood may be a rational starting point: 1) this level exists in healthy individuals and therefore should be physiological safe and possible to achieve (Chan et al., 2009); and 2) this level represents over 100–1,000 folds increase of iNKT cells in cancer patients and likely can exhibit certain therapeutic effects (Vivier et al., 2012). After the 5% initial dose proven feasible and tolerable, dose escalation (up or down) may follow. Of course, all need to be tested clinically. Notably, besides the dosage control, we have engineered an additional safety control for the intended HSC-iNKT cell therapy, by incorporating an sr39TK suicide gene in the iNKT TCR gene delivery vector (Figures 1C and 1G). This engineered safety control will allow the effective depletion of engineered iNKT cells (and their engineered progenitor HSCs) through GCV administration in case of a safety need (Figures 3 and S3).
HSC-iNKT cell therapy can certainly combine with other therapeutic modalities. For instance, adoptive transfer of aGC-loaded DCs into cancer patients has been demonstrated to be safe and effective in stimulating human iNKT cells in vivo; therefore, it is plausible to propose that a combination of HSC-iNKT cell therapy with DC/αGC vaccination would maximize the therapeutic potential of the engineered HSC-iNKT cells (Nair and Dhodapkar, 2017; Waldowska et al., 2017). Alternatively, iNKT cells in cancer patients have been shown to express high levels of T cell-exhaustion marker PD-1 (Kamata et al., 2016). It is therefore conceivable that a combination of HSC-iNKT cell therapy with the PD-1/PD-L1 blockade therapy may also produce synergistic therapeutic effects (Durgan et al., 2011). Moreover, recent studies showed promising cancer therapy potential of chimeric antigen receptor (CAR)-engineered iNKT cells; therefore further engineering HSC-iNKT cells to express CAR to enhance their tumor-targeting effectiveness may represent another attractive approach (Heczey et al., 2014; Rotolo et al., 2018; Tian et al., 2016).
In summary, here we report the pre-clinical development of an HSC-iNKT cell therapy that has the potential to treat a broad range of hematologic malignancies and solid tumors and that is ready to translate into clinical development. Our study also established a humanized BLT-iNKT mouse model that can be utilized as a valuable tool to study human iNKT cell biology and human iNKT cell-based immunotherapy.
STAR*METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for new reagents generated in this study may be directed to and will be fulfilled by the Lead Contact, Lili Yang (liliyang@ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
NOD.Cg-PrkdcSCIDIl2rgtm1Wjl/SzJ (NOD/SCID/IL-2Rγ−/−, NSG) mice were maintained in the animal facilities at the University of California, Los Angeles (UCLA). Six-to ten-week-old females were used for all experiments unless otherwise indicated. All animal experiments were approved by the Institutional Animal Care and Use Committee of UCLA.
Human Tumor Cell Lines
Human multiple myeloma (MM) cell line MM.1S, human chronic myelogenous leukemia cancer cell line K562, and human melanoma cell line A375 were all purchased from the American Type Culture Collection (ATCC). MM.1S and K562 cells were cultured in R10 medium. A375 cells were cultured in D10 medium.
To make stable tumor cell lines overexpressing human CD1d, human HLA-A2.1, human NY-ESO-1, human IL-15, and/or firefly luciferase and enhanced green fluorescence protein (Fluc-EGFP) dual-reporters, the parental tumor cell lines were transduced with lentiviral vectors encoding the intended gene(s) (Yang et al., 2008). 72h post lentivector transduction, cells were subjected to flow cytometry sorting to isolate gene-engineered cells for making stable cell lines. Six stable tumor cell lines were generated for this study, including MM.1S-FG, MM.1S-hCD1d-FG, A375-hIL-15-FG, A375-hIL-15-hCD1d-FG, A375-A2-ESO-FG, and K562-FG.
Human PBMCs, CD34+ HSCs, and Thymus Tissues
Human peripheral blood mononuclear cells (PBMCs), human fetal liver or cord blood CD34+ hematopoietic stem and progenitor cells (referred to as HSCs), and fetal thymus tissues were obtained from the CFAR Gene and Cellular Therapy Core Laboratory at UCLA, without identification information under federal and state regulations. Postnatal human thymus was obtained under IRB exemption as anonymized, discarded waste from patients undergoing cardiac surgery at Children’s Hospital Los Angeles (CHLA). G-CSF-mobilized healthy donor peripheral blood units were purchased from HemaCare or Cincinnati Children’s Hospital Medical Center (CCHMC), followed by isolation of CD34+ HSCs through magnetic-activated cell sorting using a CliniMACS (Miltenyi Biotec) according to the manufacturer’s instructions. For all isolates, the purity of CD34+ cells were more than 97% as evaluated by flow cytometry.
Materials and Reagents
α-Galactosylceramide (αGC, KRN7000) was purchased from Avanti Polar Lipids. Fluorochrome-conjugated hCD1d/PBS-57 tetramer reagents were provided by the NIH Tetramer Core Facility (Emory University, Atlanta, GA). Recombinant human IL-2, IL-3, IL-4, IL-7, IL-15, Flt3-Ligand, Stem Cell Factor (SCF), Thrombopoietin (TPO), and Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) were purchased from Peprotech.
X-VIVO™ 15 Serum-free Hematopoietic Cell Medium was purchased from Lonza. RPMI1640 and DMEM cell culture medium were purchased from Corning Cellgro. Fetal bovine serum (FBS) was purchased from Sigma. Medium supplements, including Penicillin-Streptomycine-Glutamine (P/S/G), MEM non-essential amino acids (NEAA), HEPES Buffer Solution, and Sodium Pyruvate, were purchased from Gibco. Beta-Mercaptoethanol (β-ME) was purchased from Sigma. Normocin was purchased from InvivoGen. Complete lymphocyte culture medium (denoted as C10 medium) was made of RPMI 1640 supplemented with FBS (10% vol/vol), P/S/G (1% vol/vol), MEM NEAA (1% vol/vol), HEPES (10 mM), Sodium Pyruvate (1 mM), β-ME (50 μM), and Normocin (100 μg/ml). Medium for culturing monocyte-derived dendritic cells (MoDC) and non-adherent tumor cells (denoted as R10 medium) was made of RPMI 1640 supplemented with FBS (10% vol/vol) and P/S/G (1% vol/vol). Adherent cell culture medium (denoted as D10 medium) was made of DMEM supplemented with FBS (10% vol/vol) and P/S/G (1% vol/vol).
METHOD DETAILS
Lentiviral Vectors
Lentiviral vectors used in this study were all constructed from a parental lentivector pMNDW, that contains the MND retroviral LTR U2 region as an internal promoter and contains an additional truncated Woodchuck Responsive Element (WPRE) to stabilize viral mRNA (Giannoni et al., 2013; Smith et al., 2016). The pMNDW lentivector mediates high and stable expression of transgene in human HSCs and their progeny human immune cells (Cartier et al., 2009). The Lenti/iNKT vector was constructed by inserting into pMNDW a synthetic bicistronic gene encoding human iNKT TCRα-F2A-TCRβ; the Lenti/iNKT-EGFP vector was constructed by inserting into pMNDW a synthetic tricistronic gene encoding human iNKT TCRα-F2A-TCRβ-P2A-EGFP; the Lenti/iNKT-sr39TK vector was constructed by inserting into pMNDW a synthetic tricistronic gene encoding human iNKT TCRα-F2A-TCRβ-P2A-sr39TK; the Lenti/FG vector was constructed by inserting into pMNDW a synthetic bicistronic gene encoding Fluc-P2A-EGFP; the Lenti/CD1d vector was constructed by inserting into pMNDW a synthetic gene encoding human CD1d; the Lenti/IL-15-FG vector was constructed by inserting into pMNDW a synthetic tricistronic gene encoding human IL-15-F2A-Fluc-P2A-EGFP; the Lenti/ESO-sr39TK vector was constructed by inserting into pMNDW a synthetic tricistronic gene encoding human NY-ESO-1-specific TCRα-F2A-TCRβ-P2A-sr39TK; the Lenti/HLA-A2 vector was constructed by inserting into pMNDW a synthetic gene encoding human HLA-A2.1; and the Lenti/NY-ESO-1 vector was constructed by inserting into pMNDW a synthetic gene encoding human NY-ESO-1. The synthetic gene fragments were obtained from GenScript and IDT. Lentiviruses were produced using 293T cells, following a standard calcium precipitation protocol and an ultracentrifigation concentration protocol or a tandem tangential flow filtration concentration protocol as previously described (Cooper et al., 2011; Smith et al., 2016). Lentivector titers were measured by transducing HT29 cells with serial dilutions and performing digital qPCR, following established protocols (Cooper et al., 2011; Smith et al., 2016).
Antibodies and Flow Cytometry
Fluorochrome-conjugated antibodies specific for human CD45 (clone H130), TCRαβ (clone I26), CD4 (clone OKT4), CD8 (clone SK1), CD45RO (clone UCHL1), CD45RA (clone HI100), CD161 (clone HP-3G10), CD69 (clone FN50), CD56 (clone HCD56), CD62L (clone DREG-56), CD14 (clone HCD14), CD11b (clone ICRF44), CD11c (clone N418), CD20 (clone 2H7), HLA-A2 (clone BB7.2), CD1d (clone 51.1), PD-1 (clone EH12.2H7), CCR4 (clone L291H4), CCR5 (clone HEK/1/85a), CXCR3 (clone G025H7), NKG2D (clone 1D11), IFN-γ (clone B27), Granzyme B (clone QA16A02), Perforin (clone dG9), TNF-α (clone Mab11), IL-2 (clone MQ1–17H12), IL-4 (clone MP4–25D2) and IL-17 (clone BL168) were purchased from BioLegend; fluorochrome-conjugated antibodies specific for human CD34 (clone 581), TCR Vα24-Jα18 (clone 6B11) were purchased from BD Biosciences; fluorochrome-conjugated antibodies specific for human PLZF (alone 9E12), T-bet (clone 4B10), and CD1d (clone 1B1) were purchased from eBioscience. A fluorochrome-conjugated antibody specific for human Vβ11 was purchased from Beckman-Coulter. Human Fc Receptor Blocking Solution (TruStain FcX™) was purchased from BioLegend, while mouse Fc Block (anti-mouse CD16/32) was purchased from BD Biosciences. Fixable Viability Dye eFluor506 (e506) were purchased from affymetrix eBioscience. Cells were stained as previously described (Yang and Baltimore, 2005). Intracellular cytokines were measured using a Cell Fixation/Permeabilization Kit (BD Biosciences) according to the manufacturer’s instructions. Stained cells were analyzed using a MACSQuant Analyzer 10 flow cytometer (Miltenyi Biotec). FlowJo software was used to analyze the data.
Human iNKT Cell TCR Vβ Repertoire Analysis
Human iNKT cell TCR Vβ repertoire was analyzed through flow cytometry using an IOTest® Beta Mark TCR V beta Repertoire Kit (Beckman-Coulter), following the manufacturer’s instructions. Tube G was not used in staining because it included an antibody for Vβ11, which was stained separately. All other combined hTCR Vβs (FITC) and hTCR Vβs (PE) antibodies collectively stained for human TCR Vβ 1, 2, 3, 4, 5.1, 5.2, 5.3, 7.1, 7.2, 8, 9, 12, 13.1, 13.2, 13.6, 16, 17, 18, 20, 21.3, and 23.
ELISA
The ELISAs for detecting human cytokines were performed following a standard protocol from BD Biosciences. The capture and biotinylated antibody pairs for detecting human IFN-γ, IL-4, and IL-17 were purchased from BD Biosciences. The streptavidin-HRP conjugate was purchased from Invitrogen. Human IFN-γ, IL-4 and IL-17 standards were purchased from eBioscience. The tetramethylbenzidine (TMB) substrate was purchased from KPL. The samples were analyzed for absorbance at 450 nm using an Infinite M1000 microplate reader (Tecan).
Ganciclovir (GCV) In Vitro Killing Assay
Healthy donor PBMC T cells were cultured in vitro in C10 medium for 8 days, in the presence of 1 μg/ml anti-human CD3 (Clone HIT3a; LEAF™ purified; Biolegend), 1 μg/ml anti-human CD28 (Clone CD28.2; LEAF™ purified; Biolegend), and 10 ng/ml recombinant human IL-2. On days 2 and 3, concentrated Lenti/iNKT-sr39TK vectors were added into the cell culture. On day 4, titrated amounts of GCV (0–50 μM) were added into the cell culture. On day 8, selective killing of lentivector-transduced T cells (identified as hCD1d/PBS-57+ cells among total T cells) were analyzed using flow cytometry.
Single-Cell Human iNKT TCR Cloning
The single-cell iNKT TCR RT-PCR was performed based on an established protocol, with certain modifications (Smith et al., 2015). Single human iNKT cells were sorted from healthy donor PBMCs based on a stringent forum of surface markers (hTCRαβ+hTCR Vα24-Jα18+hTCR Vβ11+CD161+) using a FACSAria II flow cytometer (BD Biosciences). Single cells were sorted directly into PCR plates containing cell lysis buffer. The plates were then immediately flash frozen and stored at −80°C until use. Upon thawing, the cell lysate from each cell was split in half on the same PCR plate and processed directly into iNKT TCR cloning for both α and β chain genes using a OneStep RT-PCR kit (QIAGEN), following the manufacturer’s instructions and using the human iNKT TCR gene-specific primers. These primers were designed to amplify the ~200–300 bps spanning the CDR3 regions of the iNKT TCR α and β chain cDNAs and were customer-synthesized by IDT: for TCRα (FW primer: 5′-GCT CTC TGC ACA TCA CAG CCT CCC AG −3′; BW primer: 5′- CGG TGA ATA GGC AGA CAG ACT TGT CAC TG −3′) and for TCRβ (FW: 5′- CCA CAG AGA AGG GAG ATC TTT CCT CTG AGT C −3′; BW: 5′- CCT GTG GCC AGG CAC ACC AGT G −3′). Verified sequences (productive germline Vα24-Jα18-Cα assembly for TCRα and Vβ11-D/J/N-Cβ assembly for TCRβ) were used to construct the complete cDNA sequences encoding the TCR α and β chains from a single cell, based on information about human TCR genomic segments [the international ImMuno-GeneTics information system (IMGT), www.imgt.org]. The selected iNKT TCR α and β pair cDNAs were then synthesized as a single bicistonic gene, with codon optimization and an F2A sequence linking the TCRα and TCRβ cDNAs to enable their co-expression (GenScript).
Generation of BLT and BLT-iNKT Humanized Mice
BLT (human bone marrow-liver-thymus engrafted NSG mice) and BLT-iNKT (human iNKT TCR gene-engineered BLT mice) humanized mice were generated as previously described, with some modifications (Lan et al., 2006; Melkus et al., 2006; Smith et al., 2016). In brief, human CD34+ HSCs were cultured for no more than 48 hours in X-VIVO™ 15 Serum-free Hematopoietic Cell Medium (Lonza) containing recombinant human Flt3 ligand (50 ng/mL), SCF (50 ng/mL), TPO (50 ng/mL), and IL-3 (20 μg/mL) in non-tissue culture-treated plates coated with Retronectin (20 μg/ml) (Takara). Viral transduction, when applicable, was performed at 24 hours by adding concentrated lentivectors (Lenti/iNKT, Lenti/iNKT-EGFP, or Lenti/iNKT-sr39TK) directly to the culture medium. At around 48 hours, CD34+ cells were collected and i.v. injected into NSG mice (~0.5–1 × 106 cells per recipient) that had received 270 rads of total body irradiation. 1–2 fragments of human fetal or postnatal thymus (~1 mm3) were implanted under the kidney capsule of each recipient NSG mouse. The mice were maintained on trimethoprim/sulfmethoxazole (TMS) chow in a sterile environment for 8–12 weeks until analysis or use for further experiments. Unless otherwise indicated, data from BLT-iNKT mice produced with human fetal thymus and Lenti/iNKT-sr39TK vector-transduced PBSCs were presented.
Generation of Secondary BLT-iNKT Humanized Mice
Secondary BLT-iNKT mice were generated through harvesting total bone marrow cells and human thymus implants from primary BLT-iNKT mice followed by adoptive transfer into NSG recipient mice (Smith et al., 2016). Primary BLT-iNKT mice were produced with human fetal thymus and Lenti/iNKT-sr39TK vector-transduced PBSCs. Recipient NSG mice were pre-conditioned with 270 rads of total body irradiation. 10 × 106 primary BLT-iNKT mice bone marrow cells were i.v. injected into each NSG recipient mouse. Human thymus implants were dissected from the kidney capsule of primary BLT-iNKT mice, cut into fragments (~1 mm3), then surgically implanted under the kidney capsule of the NSG recipient mice (1–2 fragments per recipient). The mice were maintained on TMS chow in a sterile environment for 8–12 weeks until analysis or further experiments.
Safety Study of HSC-iNKT Cell Therapy in BLT-iNKT Mice
BLT-iNKT mice and control BLT mice were monitored for body weight and survival rate over a period of 5 months after HSC transfer. At the end of 5 months, mice were terminated and various tissues were collected, including bone marrow, spleen, Ileum, pancreas, kidney, lung, liver, heart, and brain. Tissues were fixed with 10% neutral-buffered formalin and embedded in paraffin for sectioning (5 mm thickness), followed by hematoxylin and eosin staining using standard procedures (UCLA Translational Pathology Core Laboratory). The sections were examined by a mouse pathologist (N.R.) who was blinded to the sample group assignments during initial scoring. Scores were assigned on the basis of a combination of criteria, including inflammation, hematopoietic neoplasm, and non-hematopoietic neoplasm. An ordinal scale scoring system was used: 0, no abnormal findings; 1, mild; 2, moderate; and 3, severe.
In another set of longer-term experiments, BLT-iNKT mice and control BLT mice were maintained for a period of over 8 months, to monitor graft-versus-host (GvH) reactions in these animals. Over time, mouse survival rates were recorded. At 6 months post-HSC transfer, a subset of animals were terminated and their tissues (e.g. liver) were collected and processed for flow cytometry analysis following established protocols (Smith et al., 2016). Activation status of engrafted human conventional αβ T cells (identified as hCD45+hTCRαβ+6B11− cells) was assessed through measuring T cell expression of CD45RA and CD45RO surface markers (CD45RAloCD45ROhi indicating T cell activation).
Biodistribution and Controlled Depletion of HSC-iNKT Cells in BLT-iNKT Humanized Mice Visualized by PET/CT Imaging
BLT-iNKTTK mice (BLT-iNKT mice generated using Lenti/iNKT-sr39TK vector-transduced PBSCs) were utilized for this study. Regular BLT mice were included as imaging controls. To study controlled depletion of vector-engineered human HSCs and their progeny immune cells, experimental animals received i.p. injection of nucleoside prodrug ganciclovir (GCV) for five consecutive days (50 mg/kg per injection per day). PET/CT imaging data were collected one day prior to GCV treatment, and 7 days post GCV treatment. PET/CT scans were performed using a microPET/CT system Genisys 8 (Sofie Bioscience). Mice were anesthetized using 1.5–2% isoflurane. 20 μCi of 18F-FHBG probes was administrated via tail vein. Acquisition of static PET images was started 60 min after probe injection. Maximum-likelihood expectation maximization with 60 iterations was used for PET image reconstruction. All images were corrected for photon attenuation. The CT acquisition parameters were 40 kVp, 190 mA, and 720 projections with an exposure time of 55 ms at each projection. For image analysis, PET/CT images were analyzed using OsiriX Imaging Software (Version 3.9.3; Pixmeo SARL).
HSC-iNKT Cell Phenotype and Functional Study
HSC-engineered human iNKT cells (denoted as HSC-iNKT cells) were isolated from the spleen of BLT-iNKT mice. Phenotype of HSC-iNKT cells was studied using flow cytometry, through analyzing cell surface markers including co-receptors (CD4 and CD8), NK cell markers (CD161 and NKG2D), memory/effector T cell markers (CD45RO and CD69), and homing markers (CD62L, CCR4, CCR5, and CXCR3). Native human iNKT cells and conventional T cells isolated from healthy donor PBMCs (denoted as PBMC-iNKT and PBMC-Tc cells, respectively) were included as FACS analysis controls.
Response of HSC-iNKT cells to antigen stimulation was studied by culturing BLT-iNKT spleen cells in vitro in C10 medium for 7 days, in the presence or absence of αGC (100 ng/ml). Proliferation of HSC-iNKT cells (identified as hCD45+hTCRαβ+6B11+ cells of total cell culture) was measured by cell counting and flow cytometry over time. Cytokine production was assessed by ELISA analysis of cell culture supernatants collected on day 7 (for human IFN-γ, IL-4, and IL-17).
Capacity of HSC-iNKT cells to produce effector molecules was studied in comparison with that of native PBMC-iNKT cells and PBMC-Tc cells. BLT-iNKT spleen cells and healthy donor PBMCs were cultured in vitro in C10 medium for 7 days with αGC stimulation (100 ng/ml). On day 7, cells were collected and analyzed for intracellular production of various pro-inflammatory cytokines (IFN-γ, TNF-α, and IL-2) and cytotoxic molecules (Perforin and Granzyme B) using a Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences) (Smith et al., 2015). The CD4/CD8 co-receptor expression patterns on HSC-iNKT cells and PBMC-iNKT cells pre- and post-αGC stimulation were studied using flow cytometry.
HSC-iNKT Cell In Vitro Expansion
Various tissues (spleen, lymph nodes, liver, bone marrow) were collected from BLT-iNKT mice, processed into single mononuclear cells, and pooled together for in vitro culture. Healthy donor PBMCs were loaded with αGC (by culturing 1 × 108 PBMCs in 5 ml C10 medium containing 5 mg/ml αGC for 1 hour), irradiated at 6,000 rads, then used to stimulate HSC-iNKT cells (denoted as αGC/PBMCs). To expand HSC-iNKT cells, pooled BLT-iNKT mouse tissue cells were mixed with αGC/PBMCs (ratio 1:1 or 1:1.5) and cultured in C10 medium for 7 days. Recombinant human IL-7 (10 ng/ml) and IL-15 (10 ng/ml) were added to cell cultures from day 2. On day 7, cell cultures were collected and HSC-iNKT cells were sorted out using flow cytometry (identified as hCD45+hTCRαβ+6B11+ cells). The sorted HSC-iNKT cells (>99% purify based on flow cytometry analysis) were expanded further with αGC/APCs and IL-7/IL-15 for another 7 to 14 days, then were aliquoted and frozen in LN2 storage tanks. For mechanistic and efficacy studies, HSC-iNKT cells were thawed from frozen stock and utilized for the intended assays.
HSC-iNKT Cell Tumor-Attacking Mechanism Study: In Vitro Direct Tumor Cell Killing Assay
MM.1S-FG or MM.1S-hCD1d-FG tumor cells (5–10 × 103 cells per well) were co-cultured with HSC-iNKT cells (ratio 1:10, unless otherwise indicated) in Corning 96-well clear bottom black plates for 24–48 hours, in X-VIVO™ 15 medium with or without the addition of αGC (100 ng/ml). At the end of culture, live tumor cells were quantified by adding D-Luciferin (150 μg/ml) (Caliper Life Science) to cell cultures and reading out luciferase activities using an Infinite M1000 microplate reader (Tecan) according to the manufacturer’s instructions. In tumor cell killing assays involving blocking CD1d, 10 μg/ml LEAF™ purified anti-human CD1d antibody (Clone 51.1, Biolegend) or LEAF™ purified mouse IgG2b κ isotype control antibody (Clone MG2b-57, Biolegend) was added to tumor cell cultures one hour prior to adding HSC-iNKT cells.
HSC-iNKT Cell Tumor-Attacking Mechanism Study: In Vitro NK Adjuvant Effect Assay
Primary human NK cells were isolated from healthy donor PBMCs through magnetic-activated cell sorting (MACS sorting), using an NK Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. K562-FG cells (5 × 104 cells per well) were co-cultured with NK cells and HSC-iNKT cells (ratio 1:2:2) in Corning 96-well clear bottom black plates for 24 hours, in C10 medium with or without the addition of αGC-loaded irradiated PBMCs as antigen presenting cells (APCs). Live tumor cells were quantified by adding D-Luciferin (150 μg/ml) (Caliper Life Science) to the cell cultures and reading out luciferase activities using an Infinite M1000 microplate reader (Tecan). NK cell activation status was monitored through measuring NK cell expression of CD69 using flow cytometry. Cytokine production from co-cultured NK and HSC-iNKT cells was analyzed through collecting cell culture supernatants and assaying cytokines (IFN-γ and IL-2) using ELISA.
HSC-iNKT Cell Tumor-Attacking Mechanism Study: In Vitro DC/CTL Adjuvant Effect Assay
CD1d+/HLA-A2+ human monocyte-derived dendritic cells (MoDCs) were generated by isolating CD14+ monocytes from HLA-A2+ healthy donor PBMCs using MACS sorting, followed by culturing monocytes in R10 medium supplemented with recombinant human GM-CSF (100 ng/ml) and IL-4 (20 ng/ml) for 4 days (Yang et al., 2008). NY-ESO-1 specific CD8+ human cytotoxic T lymphocytes (CTLs, or ESO-T cells) were generated through engineering human CD34+ HSCs with a TCR gene encoding a 1G4 TCR (HLA-A2-restricted, NY-ESO-1 tumor antigen-specific) and differentiating the TCR gene-engineered HSCs into CD8+ CTLs in an Artificial Thymic Organoid (ATO) culture, following an established protocol (Bethune et al., 2018; Seet et al., 2017). The resulting ESO-T CTLs were co-cultured with CD1d+/HLA-A2+ MoDCs in C10 medium for 3 days, with or without the addition of HSC-iNKT cells (cell ratio 1:1:1) and αGC (100 ng/ml). Activation of MoDCs was analyzed at 24 hours through measuring MoDC surface expression of CD86 using flow cytometry. Expansion of ESO-T cells was quantified at 72 hours through cell counting and flow cytometry analysis (ESO-T cells were gated as hTCRαβ+hTCR Vβ13.1+ cells of the total cell cultures). Tumor-killing potential of ESO-T cells was measured by adding A375-A2-ESO-FG tumor cells (1:1 ratio to input ESO-T cells) to the ESO-T/MoDC co-culture 24 hours post co-culture setup, and quantifying live tumor cells via luciferase activity reading in another 24 hours.
HSC-iNKT Cell Tumor-Attacking Mechanism Study: In Vitro Macrophage Inhibition Assay
CD14+ monocytes were isolated from healthy donor PBMCs through MACS sorting, followed by co-culturing with HSC-iNKT cells (ratio 1:1) for 24–48 hours in C10 medium with or without the addition of aGC (100 ng/ml). At the end of culture, cells were collected for flow cytometry analysis, to assess the activation of HSC-iNKT cells (gated as hTCRab+6B11+ cells of total cell culture) by measuring their expression of CD69, and to assess the viability of monocytes (gated as CD14+ cells of total cell culture) with the eBioscience Fixable Viability Dye eFluor 506 (e506).
Bioluminescence Live Animal Imaging (BLI)
BLI was performed using an IVIS 100 imaging system (Xenogen/PerkinElmer). Live animal imaging was acquired 5 minutes after intraperitoneal injection of D-Luciferin (1 mg per mouse). Imaging results were analyzed using a Living Imaging 2.50 software (Xenogen/PerkinElmer).
HSC-iNKT Cell In Vivo Antitumor Efficacy Study: MM.1S Human Multiple Myeloma Xenograft NSG Mouse Model
NSG mice were inoculated with 0.5–1 × 106 MM.1S-hCD1d-FG or MM.1S-FG cells intravenously (day 0) and were allowed to develop MM over the course of about 3 weeks. Prior to tumor inoculation (day 0), mice were pre-conditioned with 175 rads of total body irradiation. Three days post-tumor inoculation (day 3), mice received i.v. injection of vehicle (PBS) or 1 × 107 HSC-iNKT cells. Recombinant human IL-15 was supplemented to experimental animals through i.p injection to support the peripheral maintenance of HSC-iNKT cells twice per week starting from day 3 (500 ng per animal per injection). Over time, tumor loads in experimental animals were monitored using BLI, twice per week starting from day 2. At around week 3, mice were terminated. Various mouse tissues (blood, spleen, liver, and bone marrow) were harvested and processed for flow cytometry analysis to detect tissue-residing tumor cells (identified as GFP+ cells) and HSC-iNKT cells (identified as hCD45+6B11+ cells), following established protocols (Smith et al., 2016). Activation status of HSC-iNKT cells was monitored through measuring cell surface expression of CD62L and CD69 markers using flow cytometry.
HSC-iNKT Cell In Vivo Antitumor Efficacy Study: A375 Human Melanoma Xenograft NSG Mouse Model
NSG mice were inoculated with 1 × 106 aGC-loaded A375-hIL-15-hCD1d-FG cells or 1 × 106 αGC-loaded A375-hIL-15-FG cells subcutaneously (day 0) and were allowed to grow solid tumors over the course of approximately 4 weeks. Three days post-tumor inoculation (day 3), mice received 100 rads of total body irradiation followed by i.v. injection of vehicle (PBS) or 1 × 107 HSC-iNKT cells. Note that inclusion of a human IL-15 gene in the tumor cells aimed to provide human IL-15 within solid tumors in vivo, because the on-site presence of IL-15 has been indicated to be critical in maintaining iNKT cell survival in the hypoxic solid tumor microenvironment. Recombinant human IL-15 was also supplemented to experimental animals through i.p injection to support the peripheral maintenance of HSC-iNKT cells twice per week starting from day 3 (500 ng per animal per injection). Over time, tumor loads in experimental animals were monitored twice per week starting from day 2 by measuring total body luminescence using BLI (shown as TBL p/s), and by measuring tumor size using a Fisherbrand™ Traceable™ digital caliper (Thermo Fisher Scientific) (tumor volume calculated as (W(2) ×L)/2 mm3). At approximately week 4, mice were terminated. Solid tumors were retrived, weighted using a PA84 precision balance (Ohaus), then processed for flow cytometry analysis to detect tumor-infiltrating HSC-iNKT cells (identified as hCD45+6B11+ cells). Various mouse tissues (blood, spleen, and liver) were also collected and processed for flow cytometry analysis to detect tissue-residing HSC-iNKT cells (identified as hCD45+6B11+ cells), following established protocols (Smith et al., 2016). Activation status of HSC-iNKT cells was monitored through measuring cell surface expression of CD62L and CD69 markers using flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
A Prism 6 software (Graphpad) was used for all statistical analysis. Pairwise comparisons were made using a 2-tailed Student’s t test. Multiple comparisons were performed using an ordinary 1-way ANOVA, followed by Tukey’s multiple comparisons test. Kaplan-Meier survival curves were analyzed by log rank (Mantel-Cox) test adjusted for multiple comparisons. Data are presented as the mean ± SEM, unless otherwise indicated. In all figures and figure legends, “n” represents the number of samples or animals utilized in the indicated experiments. A P value of less than 0.05 was considered significant. ns, not significant; * P<0.05; ** P<0.01; *** P<0.001; **** P<0.0001.
DATA AND SOFTWARE AVAILABILITY
All data associated with this study are present in the paper or Supplemental Information.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human IFN-γ (ELISA, capture) | BD Biosciences | CAT#551221, RRID: AB_394099 |
| Anti-human IFN-γ (ELISA, detection) | BD Biosciences | CAT#554550, RRID: AB_395472 |
| Anti-human IL-4 (ELISA, capture) | BD Biosciences | CAT#554515; RRID: AB_398567 |
| Anti-human IL-4 (ELISA, detection) | BD Biosciences | CAT#554483; RRID: AB_395422 |
| Anti-human IL-2 (ELISA, capture) | BD Biosciences | CAT#554563; RRID: AB_398570 |
| Anti-human IL-2 (ELISA, detection) | BD Biosciences | CAT#555040; RRID: AB_395666 |
| Anti-human CD3 (Clone HIT3a; LEAF purified) | Biolegend | CAT#300314, RRID: AB_314050 |
| Anti-human CD28 (Clone CD28.2; LEAF purified) | Biolegend | CAT#302902, RRID: AB_314304 |
| Anti-human CD45 (Clone H130) | Biolegend | CAT#304026, RFID: AB_893337 |
| Anti-human TCR(alpha)(beta) (Clone I26) | Biolegend | CAT#306716, RRID: AB_1953257 |
| Anti-human CD4 (Clone OKT4) | Biolegend | CAT#317414, RRID: AB_571959 |
| Anti-human CD8 (Clone SK1) | Biolegend | CAT#344714, RRID: AB_2044006 |
| Anti-human CD45RO (Clone UCHL1) | Biolegend | CAT#304216, RRID: AB_493659 |
| Anti-human CD45RA (Clone HI100) | Biolegend | CAT#304126, RRID: AB_10708879 |
| Anti-human CD161 (Clone HP-3G10) | Biolegend | CAT#339928, RRID: AB_2563967 |
| Anti-human CD69 (Clone FN50) | Biolegend | CAT#310914, RRID: AB_314849 |
| Anti-human CD56 (Clone HCD56) | Biolegend | CAT#318304, RRID: AB_604100 |
| Anti-human CD62L (Clone DREG-56) | Biolegend | CAT#304822, RRID: AB_830801 |
| Anti-human CD14 (Clone HCD14) | Biolegend | CAT#325608, RRID: AB_830681 |
| Anti-human CD11b (Clone ICRF44) | Biolegend | CAT#301330, RRID: AB_2561703 |
| Anti-human CD11c (Clone N418) | Biolegend | CAT#337234, RRID: AB_2566656 |
| Anti-human CD20 (Clone 2H7) | Biolegend | CAT#555623, RRID: AB_395989 |
| Anti-human HLA-A2 (Clone BB7.2) | Biolegend | CAT#558570, RRID: AB_647220 |
| Anti-human CD1d (Clone 51.1) | Biolegend | CAT#350308, RRID: AB_10642829 |
| Anti-human PD-1 (Clone EH12.2H7) | Biolegend | CAT#329908, RRID: AB_940475 |
| Anti-human CCR4 (Clone L291H4) | Biolegend | CAT#359409, RRID: AB_2562430 |
| Anti-human CCR5 (Clone HEK/1/85a) | Biolegend | CAT#313705, RRID: AB_345305 |
| Anti-human CXCR3 (Clone G025H7) | Biolegend | CAT#306513, RRID: AB_2089652 |
| Anti-human NKG2D (Clone 1D11) | Biolegend | CAT#320812, RRID: AB_2234394 |
| Anti-human IFNγ (Clone B27) | Biolegend | CAT#506518, RRID: AB_2123321 |
| Anti-human Granzyme B (Clone QA16A02) | Biolegend | CAT#372204, RRID: AB_2687028 |
| Anti-human Perforin (Clone dG9) | Biolegend | CAT#308126, RRID: AB_2572049 |
| Anti-human TNFα (Clone Mab11) | Biolegend | CAT#502912, RRID: AB_315264 |
| Anti-human IL-2 (Clone MQ1–17H12) | Biolegend | CAT#500341, RRID: AB_2562854 |
| Anti-human IL-4 (Clone MP4–25D2) | Biolegend | CAT#500812, RRID: AB_315131 |
| Anti-human IL-17 (Clone BL168) | Biolegend | CAT#512334, RRID: AB_2563986 |
| Anti-human CD34 (Clone 581) | BD Biosciences | CAT#555822, RRID: AB_396151 |
| Anti-human TCR V(alpha)24-J(alpha)18 (Clone 6B11) | BD Biosciences | CAT#552825, RRID: AB_394478 |
| Anti-human TCR V(beta)11 | Beckman-Coulter | CAT#A66905 |
| Anti-human PLZF (Clone 9E12) | eBioscience | CAT#19-9322-82, RRID: AB_2637113 |
| Anti-human T-bet (Clone 4B10) | eBioscience | CAT#25-5825-80, RRID: AB_11041809 |
| Anti-mouse CD1d (Clone 1B1) | eBioscience | CAT#17-0011-82, RRID: AB_2573135 |
| Mouse lgG2b, κ isotype control antibody (Clone MPC-11) | Biolegend | CAT#400320 |
| Rat lgG2b, κ isotype control antibody (Clone eB149/10H5) | eBioscience | CAT#17-4031-82, RRID: AB_470176 |
| Human Fc Receptor Blocking Solution (TrueStain FcX) | Biolegend | CAT#422302 |
| Mouse Fc Block (anti-mouse CD16/32) | BD Biosciences | CAT#553142, RRID: AB_394657 |
| Anti-human CD1d antibody (Clone 51.1; LEAF purified) | Biolegend | CAT#350304, RRID: AB_10641291 |
| Mouse lgG2b, κ isotype control antibody (Clone MG2b-57; LEAF purified) | Biolegend | CAT#401212 |
| Tetramer/Dextramer | ||
| hCD1d/PBS-57 tetramer | NIH Tetramer Core Facility | N/A |
| HLA-A2/NY-ES0–1157–165 dextramer | This paper | N/A |
| Bacterial and Virus Strains | ||
| Lenti/iNKT | This paper | N/A |
| Lenti/iNKT-EGFP | This paper | N/A |
| Lenti/iNKT-sr39TK | This paper | N/A |
| Lenti/FG | This paper | N/A |
| Lenti/CD1d | This paper | N/A |
| Lenti/IL-15-FG | This paper | N/A |
| Lenti/ESO-sr39TK | This paper | N/A |
| Lenti/HLA-A2 | This paper | N/A |
| Lenti/NY-ESO-1 | This paper | N/A |
| Biological Samples | ||
| Human peripheral blood mononuclear cells (PBMCs) | UCLA | N/A |
| Human fetal liver | UCLA | N/A |
| Human cord blood CD34+ hematopoietic stem and progenitor cells (CB HSCs) | UCLA | N/A |
| Fetal thymus tissues | UCLA | N/A |
| Postnatal human thymus | CHLA | N/A |
| G-CSF-mobilized peripheral blood units | CCHMC | CAT#M001F-GCSF-3 |
| G-CSF-mobilized leukopak | HemaCare | CAT#M001CLPG-4-KIT |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Streptavidin-HRP conjugate | Invitrogen | CAT#SA10001 |
| Recombinant IL-2 (ELISA, standard) | eBioscience | CAT#570409 |
| Recombinant human IL-4 (ELISA, standard) | eBioscience | CAT#571809 |
| Recombinant human IL-17 (ELISA, standard) | eBioscience | CAT#570509 |
| Recombinant human IFN-γ (ELISA, standard) | eBioscience | CAT#570209 |
| Tetramethylbenzidine (TMB) | KPL | CAT#5120–0053 |
| Ganciclovir (GCV) | Sigma | CAT#ADV465749843 |
| α-Galactosylceramide (KRN7000) | Avanti Polar Lipids | SKU#867000P-1mg |
| Recombinant human IL-2 | Peprotech | CAT#200–02 |
| Recombinant human IL-3 | Peprotech | CAT#200–03 |
| Recombinant human IL-4 | Peprotech | CAT#200–04 |
| Recombinant human IL-7 | Peprotech | CAT#200–07 |
| Recombinant human IL-15 | Peprotech | CAT#200–15 |
| Recombinant human Flt3-Ligand | Peprotech | CAT#300–19 |
| Recombinant human SCF | Peprotech | CAT#300–07 |
| Human NY-ESO-1157–165 peptide | ThermoFisher | N/A |
| Recombinant human TPO | Peprotech | CAT#300–18 |
| Recombinant human GM-CSF | Peprotech | CAT#300–03 |
| X-VIVO 15 Serum-free Hematopoietic Cell Medium | Lonza | CAT#04–418Q |
| RPMI1640 cell culture medium | Corning Cellgro | CAT#10–040-CV |
| DMEM cell culture medium | Corning Cellgro | CAT#10–013-CV |
| Fetal Bovine Serum (FBS) | Sigma | CAT#F2442 |
| Penicillin-Streptomycine-Glutamine (P/S/G) | Gibco | CAT#10378016 |
| MEM non-essential amino acids (NEAA) | Gibco | CAT#11140050 |
| HEPES Buffer Solution | Gibco | CAT#15630056 |
| Sodium Pyruvate | Gibco | CAT#11360070 |
| Beta-Mercaptoethanol | Sigma | SKU#M6250 |
| Normocin | Invivogen | CAT#ant-nr-2 |
| Fixable Viability Dye eFluor506 | affymetrix eBioscience | CAT#65-0866-14 |
| Cell Fixation/Permeabilization Kit | BD Biosciences | CAT#554714 |
| RetroNectin recombination human fibronectin fragment, 2.5mg | Takara | CAT#T100B |
| 10% neutral-buffered formalin | Richard-Allan Scientific | CAT#5705 |
| D-Luciferin | Caliper Life Science | CAT#XR-1001 |
| Isoflurane | Zoetis | CAT#50019100 |
| Phosphate Buffered Saline (PBS) pH 7.4 (1X) | Gibco | CAT#10010–023 |
| Phorbol-12-myristate-13-acetate (PMA) | Calbiochem | CAT#524400 |
| lonomycin, Calcium salt, Streptomyces conglobatus | Calbiochem | CAT#407952 |
| Critical Commercial Assays | ||
| IOTest beta Mark TCR Vβ Repertoire Kit | Beckman-Coulter | CAT#IM3497 |
| OneStep RT-PCR kit | Qiagen | CAT#210212 |
| NK Cell Isolation Kit | Miltenyi Biotec | CAT#130-092-657 |
| Human CD14 Microbeads | Miltenyi Biotec | CAT# 130-050-201 |
| Fixation/Permeabilization Solution Kit | BD Sciences | CAT#55474 |
| Human IL-17A ELISA MAX Deluxe Kit | Biolegend | CAT#433915 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| Human multiple myeloma (MM) cell line MM.1S | ATCC | CRL-2974 |
| Human chronic myelogenous leukemia cancer cell line K562 | ATCC | CCL-243 |
| Human melanoma cell line A375 | ATCC | CRL-1619 |
| Human multiple myeloma (MM) cell line MM.1S-FG | This paper | N/A |
| Human multiple myeloma (MM) cell line MM.1S-hCD1d-FG | This paper | N/A |
| Human chronic myelogenous leukemia cancer cell line K562-FG | This paper | N/A |
| Human melanoma cell line A375-hlL-15-FG | This paper | N/A |
| Human melanoma cell line A375-hlL-15-hCD1d-FG | This paper | N/A |
| Human melanoma cell line A375-A2-ESO-FG | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcscid II2rgtm1Wjl/SzJ | The Jackson Laboratory | Stock #: 005557 |
| Human bone marrow-liver-thymus (BLT) engrafted NSG mice | This paper | N/A |
| Human iNKT TCR gene-engineered bone marrow-liver-thymus (BLT) mice | This paper | N/A |
| Secondary BLT-iNKT mice | This paper | N/A |
| BLT-iNKT mice generated using Lenti/iNKT-sr39TK vector-transduced PBMCs | This paper | N/A |
| Oligonucleotides | ||
| Primer: TCRα Forward: GCTCTCTGCACATCACAGCCTCCCAG | IDT | N/A |
| Primer: TCRβ Forward: CCACAGAGAAGGGAGATCTTTCCTCTGAGTC | IDT | N/A |
| Recombinant DNA | ||
| Vector: parental lentivector pMNDW | Giannoni et al., 2013 and Lan et al.,2006 | N/A |
| Software and Algorithms | ||
| FlowJo Software | FlowJo | https://www.flowio.com/solutions/flowio/downloads |
| OsiriX Imaging Software | OsiriX | https://www.osirix-viewer.com/ |
| Living Imaging 2.50 software | Xenogen/PerkinElmer | http://www.Derkinelmer.com/lab-croducts-and-services/resources/in-vivo-imaqinq-software-downloads.html |
| Prism 6 | Graphpad | https://www.ararjhrjad.com/scientific-software/orism/ |
| I-control 1.7 Microplate Reader Software | Tecan | https://www.selectscience.net/tecan/i-control-microplate-reader-software/81307 |
Highlights.
HSC-iNKT cell therapy can provide patients with a life-long supply of iNKT cells
HSC-iNKT cell therapy can potentially be utilized to treat a broad range of cancers
Preclinical study used a BLT humanized mouse model
Preclinical study demonstrated feasibility, safety, and cancer therapy potential
ACKNOWLEDGEMENTS
We gratefully thank D. Baltimore (California Institute of Technology) and J. Economou (UCLA) for advising this study. We thank the University of California, Los Angeles (UCLA) animal facility for providing animal support; the UCLA Translational Pathology Core Laboratory (TPCL) for providing histology support; the UCLA AIDS Institute/CFAR Virology Core/Gene and Cell Therapy Core/Humanized Mouse Core for providing human cells and tissues and humanized mice services; the UCLA BSCRC Flow Cytometry Core Facility for cell sorting support; the US National Institutes of Health Tetramer Core Facility for providing the hCD1d/PBS-57 tetramer reagents; N. Rozengurt (UCLA) for providing pathology analysis support; P. Wang (University of Southern California, USC) for critical reading of this manuscript; and E.L. Siegler for editing the manuscript. This work was supported by a Director’s New Innovator Award from the National Institutes of Health (NIH DP2 CA196335, to L.Y.), a Partnering Opportunity for Translational Research Projects Award from the California Institute for Regenerative Medicine (CIRM TRAN1–08533, to L.Y.), a Stem Cell Research Award from the Concern Foundation (to L.Y.), a Research Career Development Award from the STOP CANCER Foundation (to L.Y.), and a BSCRC-RHF Research Award from the Rose Hills Research Foundation (to L.Y.). D.J.S. is a predoctoral fellow supported by the UCLA Tumor Immunology Training Grant (USHHS Ruth L. Kirschstein Institutional National Research Service Award # T32 CA009056). J.Y. is a predoctoral fellow supported by the UCLA Broad Stem Cell Center (BSCRC) Predoctoral Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes 7 figures can be found with this article online at XXX.
DECLARATION OF INTERESTS
Y.Z., D.J.S, and L.Y. are inventors on patent(s) relating to this study filed by UCLA. D.J.S. and S.D.B. are currently employees of Kite, a Gilead Company. F.U. is currently an employee of PACT Pharma. S.H. is a consultant for Amgen and Merck. S.M.L. is on the speaker bureau for Takeda and is a consultant for Bristol-Meyers Squibb. J.R.H is a founder and board member of Isoplexis and PACT Pharma. C.G.R. is a founder and stockholder of Sofie Biosciences and Trethera Corporation. A.R. is a consultant for Amgen, Bristol-Meyers Squibb, Chugai, Genentech-Roche, Merck-MSD, Novartis, and Sanofi; a scientific advisory board member and stockholder for Advaxis, Apricity, Arcus, Bioncotech, Compugen, CytomX, Five Prime, FLX-Bio, ImaginAb, Isoplexis, Kite-Gilead, Merus, and Rgenix; and a co-founder and scientific advisory board member of Lutris, PACT Pharma, and Tango therapeutics. D.B.K. is an inventor on intellectual property licensed by UCLA to Orchard Therapeutics and is a member of their Scientific Advisory Board; D.B.K. is also a scientific advisory board member of Allogene Therapeutics. O.N.W. is a consultant, stockholder, and/or board member with Trethera Corporation, Kronos Biosciences, Sofie Biosciences, and Allogene Therapeutics. All other authors declare no competing interests. None of the declared companies contributed to or directed any of the research reported in this article.
REFERENCES
- Baltimore D, Witte ON, Yang L, Economou J, and Ribas A (2010). Overcoming barriers to programming a therapeutic cellular immune response to fight melanoma. Pigment Cell Melanoma Res. 23, 288–289. [DOI] [PubMed] [Google Scholar]
- Bendelac A, Savage PB, and Teyton L (2007). The biology of NKT cells. Annu. Rev. Immunol 25, 297–336. [DOI] [PubMed] [Google Scholar]
- Berzins SP, Smyth MJ, and Baxter AG (2011). Presumed guilty: natural killer T cell defects and human disease. Nat. Rev. Immunol 11, 131–142. [DOI] [PubMed] [Google Scholar]
- Bethune MT, Li XH, Yu J, McLaughlin J, Cheng D, Mathis C, Moreno BH, Woods K, Knights AJ, Garcia-Diaz A, et al. (2018). Isolation and characterization of NY-ESO-1-specific T cell receptors restricted on various MHC molecules. Proc. Natl. Acad. Sci. USA 115, E10702–E10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettini ML, Bettini M, and Vignali DA (2012). T-cell receptor retrogenic mice: a rapid, flexible alternative to T-cell receptor transgenic mice. Immunology 136, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beziat V, Nguyen S, Exley M, Achour A, Simon T, Chevallier P, Sirvent A, Vigouroux S, Debre P, Rio B, et al. (2010). Shaping of iNKT cell repertoire after unrelated cord blood transplantation. Clin. Immunol 135, 364–373. [DOI] [PubMed] [Google Scholar]
- Black ME, Newcomb TG, Wilson HM, and Loeb LA (1996). Creation of drug-specific herpes simplex virus type 1 thymidine kinase mutants for gene therapy. Proc. Natl. Acad. Sci. USA 93, 3525–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PJ, Brigl M, and Brenner MB (2013). Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol 13, 101–117. [DOI] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, et al. (2009). Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326, 818–823. [DOI] [PubMed] [Google Scholar]
- Chabannon C, Kuball J, Bondanza A, Dazzi F, Pedrazzoli P, Toubert A, Ruggeri A, Fleischhauer K, and Bonini C (2018). Hematopoietic stem cell transplantation in its 60s: A platform for cellular therapies. Sci. Transl. Med 10, eaap9630. [DOI] [PubMed] [Google Scholar]
- Chan AC, Serwecinska L, Cochrane A, Harrison LC, Godfrey DI, and Berzins SP (2009). Immune characterization of an individual with an exceptionally high natural killer T cell frequency and her immediate family. Clin. Exp. Immunol 156, 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WK, Rujkijyanont P, Neale G, Yang J, Bari R, Das Gupta N, Holladay M, Rooney B, and Leung W (2013). Multiplex and genome-wide analyses reveal distinctive properties of KIR+ and CD56+ T cells in human blood. J. Immunol 191, 1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AR, Patel S, Senadheera S, Plath K, Kohn DB, and Hollis RP (2011). Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. J. Virol. Methods 177, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesi F, Delfanti G, Grilli A, Calcinotto A, Gorini F, Pucci F, Luciano R, Grioni M, Recchia A, Benigni F, et al. (2018). Bimodal CD40/Fas-Dependent Crosstalk between iNKT Cells and Tumor-Associated Macrophages Impairs Prostate Cancer Progression. Cell Rep. 22, 3006–3020. [DOI] [PubMed] [Google Scholar]
- de Lalla C, Rinaldi A, Montagna D, Azzimonti L, Bernardo ME, Sangalli LM, Paganoni AM, Maccario R, Di Cesare-Merlone A, Zecca M, et al. (2011). Invariant NKT cell reconstitution in pediatric leukemia patients given HLA-haploidentical stem cell transplantation defines distinct CD4+ and CD4-subset dynamics and correlates with remission state. J. Immunol 186, 4490–4499. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, and Krasovsky J (2003). A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J. Exp. Med 197, 1667–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhodapkar MV, and Richter J (2011). Harnessing natural killer T (NKT) cells in human myeloma: progress and challenges. Clin. Immunol 140, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durgan K, Ali M, Warner P, and Latchman YE (2011). Targeting NKT cells and PD-L1 pathway results in augmented anti-tumor responses in a melanoma model. Cancer Immunol. Immunother 60, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, Zeng W, Mizukami Y, Clark J, Nemer D, et al. (2017). Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial. Clin. Cancer Res 23, 3510–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Shimizu K, Okamoto Y, Kunii N, Nakayama T, Motohashi S, and Taniguchi M (2013). NKT cells as an ideal anti-tumor immunotherapeutic. Front. Immunol 4, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gapin L (2010). iNKT cell autoreactivity: what is ‘self’ and how is it recognized? Nat. Rev. Immunol 10, 272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoni F, Hardee CL, Wherley J, Gschweng E, Senadheera S, Kaufman ML, Chan R, Bahner I, Gersuk V, Wang X, et al. (2013). Allelic exclusion and peripheral reconstitution by TCR transgenic T cells arising from transduced human hematopoietic stem/progenitor cells. Mol. Ther 21, 1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey DI, and Berzins SP (2007). Control points in NKT-cell development. Nat. Rev. Immunol 7, 505–518. [DOI] [PubMed] [Google Scholar]
- Gschweng EH, McCracken MN, Kaufman ML, Ho M, Hollis RP, Wang X, Saini N, Koya RC, Chodon T, Ribas A, et al. (2014). HSV-sr39TK positron emission tomography and suicide gene elimination of human hematopoietic stem cells and their progeny in humanized mice. Cancer Res. 74, 5173–5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi K, Takahashi T, Hiruma K, Kanda Y, Tanaka Y, Ogawa S, Chiba S, Miura O, Sakamaki H, and Hirai H (2004). Recovery of Valpha24+ NKT cells after hematopoietic stem cell transplantation. Bone Marrow Transplant. 34, 595–602. [DOI] [PubMed] [Google Scholar]
- Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, Gao X, Guo L, Yvon E, Hicks J, et al. (2014). Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 124, 2824–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata T, Suzuki A, Mise N, Ihara F, Takami M, Makita Y, Horinaka A, Harada K, Kunii N, Yoshida S, et al. (2016). Blockade of programmed death-1/programmed death ligand pathway enhances the antitumor immunity of human invariant natural killer T cells. Cancer Immunol. Immunother 65, 1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King LA, Lameris R, de Gruijl TD, and van der Vliet HJ (2018). CD1d-Invariant Natural Killer T Cell-Based Cancer Immunotherapy: alpha-Galactosylceramide and Beyond. Front. Immunol 9, 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, Chua K, Eidson M, Kim HJ, Im JS, et al. (2008). The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Front. Immunol 9, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijgsman D, Hokland M, and Kuppen PJK (2018). The Role of Natural Killer T Cells in Cancer-A Phenotypical and Functional Approach. Front. Immunol 9, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg M, and Gapin L (2002). The unconventional lifestyle of NKT cells. Nat. Rev. Immunol 2, 557–568. [DOI] [PubMed] [Google Scholar]
- Kunii N, Horiguchi S, Motohashi S, Yamamoto H, Ueno N, Yamamoto S, Sakurai D, Taniguchi M, Nakayama T, and Okamoto Y (2009). Combination therapy of in vitro-expanded natural killer T cells and alpha-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma. Cancer Sci. 100, 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam PY, Nissen MD, and Mattarollo SR (2017). Invariant Natural Killer T Cells in Immune Regulation of Blood Cancers: Harnessing Their Potential in Immunotherapies. Front. Immunol 8, 1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan P, Tonomura N, Shimizu A, Wang S, and Yang YG (2006). Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood 108, 487–492. [DOI] [PubMed] [Google Scholar]
- Larson SM, Truscott LC, Chiou TT, Patel A, Kao R, Tu A, Tyagi T, Lu X, Elashoff D, and De Oliveira SN (2017). Pre-clinical development of gene modification of haematopoietic stem cells with chimeric antigen receptors for cancer immunotherapy. Hum. Vaccin. Immunother 13, 1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, Guo L, Heczey A, Asgharzadeh S, Kim E, et al. (2012). IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J. Clin. Invest 122, 2221–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HE, Goldberg GL, Chidgey A, Van den Brink MR, Boyd R, and Sempowski GD (2009). Thymic involution and immune reconstitution. Trends Immunol. 30, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallevaey T, and Selvanantham T (2012). Strategy of lipid recognition by invariant natural killer T cells: ‘one for all and all for one’. Immunology 136, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda JL, Gapin L, Sidobre S, Kieper WC, Tan JT, Ceredig R, Surh CD, and Kronenberg M (2002). Homeostasis of V alpha 14i NKT cells. Nat. Immunol 3, 966–974. [DOI] [PubMed] [Google Scholar]
- Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, Wege AK, Haase AT, and Garcia JV (2006). Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med 12, 1316–1322. [DOI] [PubMed] [Google Scholar]
- Montoya CJ, Pollard D, Martinson J, Kumari K, Wasserfall C, Mulder CB, Rugeles MT, Atkinson MA, Landay AL, and Wilson SB (2007). Characterization of human invariant natural killer T subsets in health and disease using a novel invariant natural killer T cell-clonotypic monoclonal antibody, 6B11. Immunology 122, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Uchida N, and Weissman IL (1995). The biology of hematopoietic stem cells. Annu. Rev. Cell Dev. Biol 11, 35–71. [DOI] [PubMed] [Google Scholar]
- Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, Shimizu N, Horiguchi S, Okamoto Y, Fujii S, et al. (2006). A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer Res 12, 6079–6086. [DOI] [PubMed] [Google Scholar]
- Nair S, and Dhodapkar MV (2017). Natural Killer T Cells in Cancer Immunotherapy. Front. Immuno 8, 1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Saus C, Parisi G, Garcia-Diaz A, Krystofinski PE, Sandoval S, Zhang R, Champhekar AS, McCabe J, Cheung-Lau GC, Truong NA, et al. (2018). IND-Enabling Studies for a Clinical Trial to Genetically Program a Persistent Cancer-Targeted Immune System. Clin. Cancer Res 25, 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, Xiao X, Goudevenou K, Pitcher DS, Petevi K, et al. (2018). Enhanced Anti-lymphoma Activity of CAR19-iNKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell 34, 596–610 e511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio MT, Moreira-Teixeira L, Bachy E, Bouillie M, Milpied P, Coman T, Suarez F, Marcais A, Sibon D, Buzyn A, et al. (2012). Early posttransplantation donor-derived invariant natural killer T-cell recovery predicts the occurrence of acute graft-versus-host disease and overall survival. Blood 120, 2144–2154. [DOI] [PubMed] [Google Scholar]
- Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, Lantz O, and Bendelac A (2008). The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity 29, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seet CS, He C, Bethune MT, Li S, Chick B, Gschweng EH, Zhu Y, Kim K, Kohn DB, Baltimore D, et al. (2017). Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 14, 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Lin LJ, Moon H, Pham AT, Wang X, Liu S, Ji S, Rezek V, Shimizu S, Ruiz M, et al. (2016). Propagating Humanized BLT Mice for the Study of Human Immunology and Immunotherapy. Stem Cells Dev. 25, 1863–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Liu S, Ji S, Li B, McLaughlin J, Cheng D, Witte ON, and Yang L (2015). Genetic engineering of hematopoietic stem cells to generate invariant natural killer T cells. Proc. Natl. Acad. Sci. USA 112, 1523–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, Ara T, Silverman AM, DeClerck YA, Seeger RC, et al. (2009). Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J. Clin. Invest 119, 1524–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanoudakis E, Hu M, Naresh K, Terpos E, Melo V, Reid A, Kotsianidis I, Abdalla S, Rahemtulla A, and Karadimitris A (2009). Regulation of multiple myeloma survival and progression by CD1d. Blood 113, 2498–2507. [DOI] [PubMed] [Google Scholar]
- Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, Guo L, Xu X, Torikai H, Mo Q, et al. (2016). CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J. Clin. Invest 126, 2341–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatakis DN, Koya RC, Nixon CC, Wei L, Kim SG, Avancena P, Bristol G, Baltimore D, Kohn DB, Ribas A, et al. (2011). Antitumor activity from antigen-specific CD8 T cells generated in vivo from genetically engineered human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 108, E1408–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Ugolini S, Blaise D, Chabannon C, and Brossay L (2012). Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol 12, 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldowska M, Bojarska-Junak A, and Rolinski J (2017). A brief review of clinical trials involving manipulation of invariant NKT cells as a promising approach in future cancer therapies. Eur. J. Immunol 42, 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DY, Segal NH, Sidobre S, Kronenberg M, and Chapman PB (2003). Cross-presentation of disialoganglioside GD3 to natural killer T cells. J. Exp. Med 198, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki K, Horiguchi S, Kurosaki M, Kunii N, Nagato K, Hanaoka H, Shimizu N, Ueno N, Yamamoto S, Taniguchi M, et al. (2011). Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin. Immunol 138, 255–265. [DOI] [PubMed] [Google Scholar]
- Yang L, and Baltimore D (2005). Long-term in vivo provision of antigen-specific T cell immunity by programming hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 102, 4518–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Yang H, Rideout K, Cho T, Joo KI, Ziegler L, Elliot A, Walls A, Yu D, Baltimore D, et al. (2008). Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat. Biotechnol 26, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or Supplemental Information.