

Abstract
This review describes the additions of allylmagnesium reagents to carbonyl compounds and to imines, focusing on the differences in reactivity between allylmagnesium halides and other Grignard reagents. In many cases, allylmagnesium reagents either react with low stereoselectivity when other Grignard reagents react with high selectivity, or allylmagnesium reagents react with the opposite stereoselectivity. This review collects hundreds of examples, discusses the origins of stereoselectivities or the lack of stereoselectivity, and evaluates why selectivity may not occur and when it will likely occur.
Graphical Abstract

1. Introduction
1.1. Allylmagnesium Reagents Exhibit Unique Reactivities
The additions of allylic nucleophiles to carbonyl compounds are powerful transformations because they form synthetically useful homoallylic alcohols. Although allylmetal reagents containing zinc, cerium, boron, titanium, and tin groups can be used in many instances,1 synthetic strategies often depend upon commercially available allylmagnesium reagents to introduce the synthetically useful allyl group (Scheme 1).2
Scheme 1.
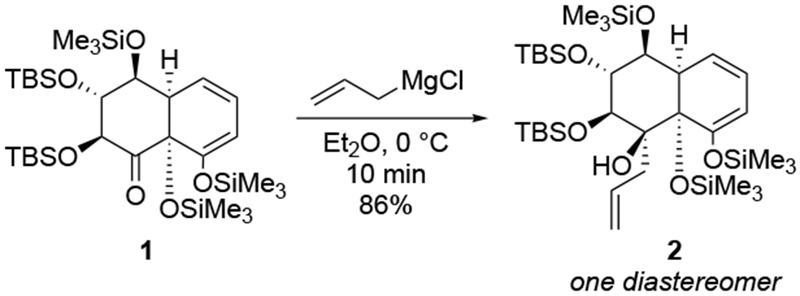
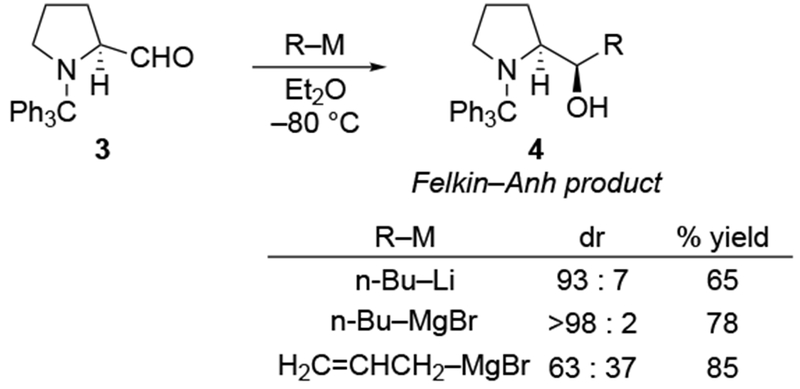
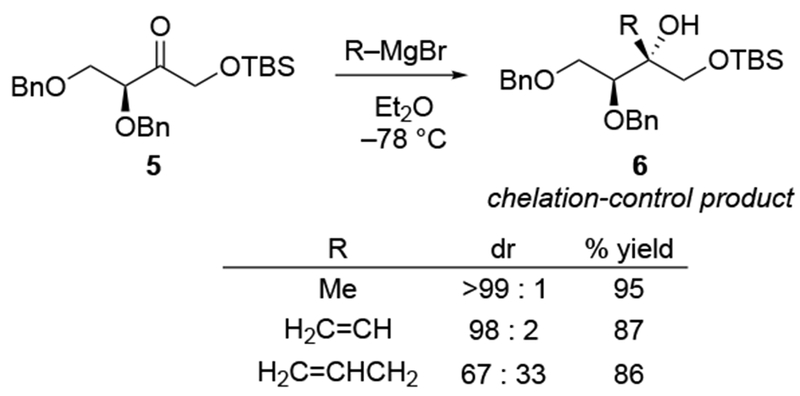
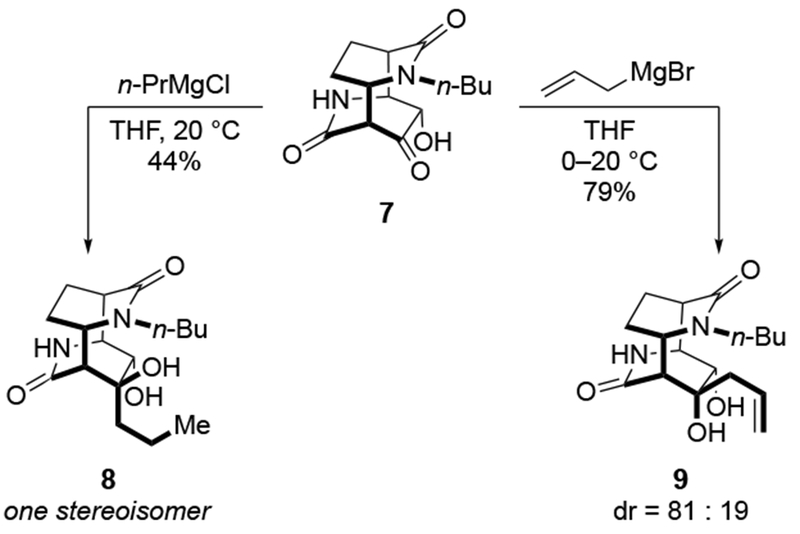
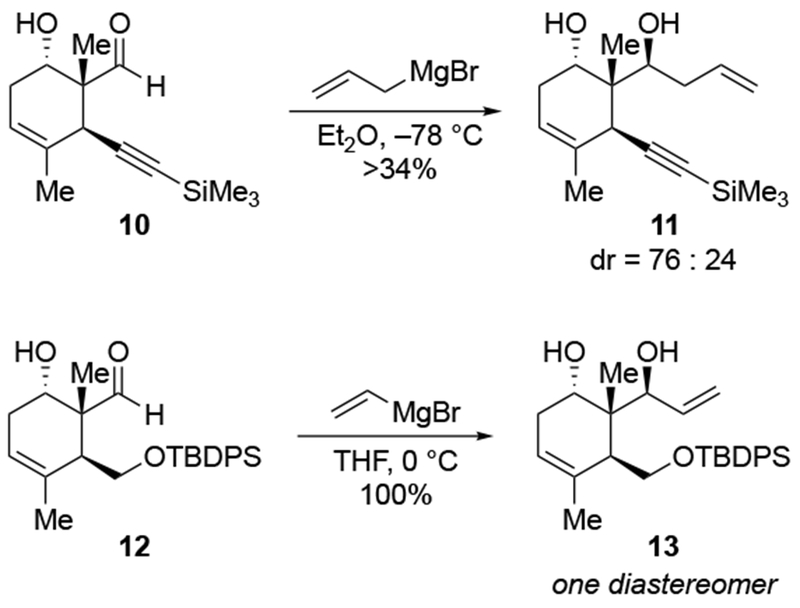
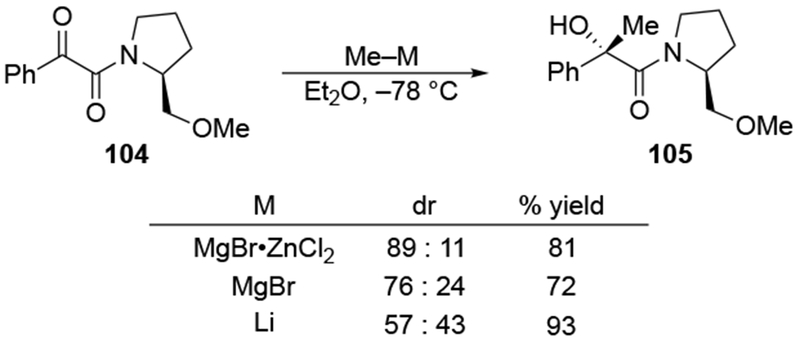
The reactions of allylmagnesium reagents, unfortunately, do not always follow models normally used to predict and explain the stereochemical outcomes of additions to carbonyl compounds. The Felkin–Anh or related models (for example, Scheme 23) and the chelation-control model (for example, Scheme 34) often fail to rationalize the product obtained in these transformations, although they can explain selectivities observed for additions of other organometallic nucleophiles. In some cases, allylmagnesium reagents react with opposite selectivity to other Grignard reagents5–7 (for example, Scheme 4).8 These problems can hinder efforts to develop stereoselective syntheses of natural products using allylmagnesium reagents, as illustrated for additions to structurally similar substrates 109 and 12,(Iwasaki et al. 2006, #301) which occur with contrasting selectivities (Scheme 5). Synthetic chemists often use allylation reactions because of the operational simplicity of the transformation, the commercial availability of the reagent, and the synthetic utility of the products,10–14 even when these reactions are not stereoselective or their outcome cannot be predicted.15,16
Scheme 2.
Scheme 3.
Scheme 4.
Scheme 5.
1.2. Purpose of the Review
This review documents the additions of allylmagnesium nucleophiles to chiral carbonyl compounds, imines, and related electrophiles to provide a guide to understanding when these reactions will likely occur with stereoselectivity and when they will likely not. The outcomes of the reactions described herein are analyzed using common stereochemical models and analysis of possible transition states. Because these models often fall short of explaining the outcomes of additions of allylmagnesium reagents, in some cases the analysis provided in the original papers will be supplemented with an analysis guided by our recent studies of the unusual reactivity of these reagents.17,18 The review focuses on examples reported since this topic was reviewed in 197119, although additions to chiral carbonyl compounds were not discussed in that review. The present review will emphasize more recent examples through 2018, particularly those applying to complex target synthesis, although some older work will be discussed for context. Comparisons to either different organomagnesium reagents or different allylmetal reagents have been provided in many cases to illustrate the unusual behavior of allylmagnesium reagents. Considering that other allylmetal reagents and their reactivities have been reviewed recently,20,21 that material will not be covered in depth. Generally, reactions that use allylmagnesium reagents directly, without transmetallation to other organometallic species, will be discussed, although some examples of such reactions will be included for comparison.
The purpose of this review is several-fold. It should inform chemists who see unexpected results with allylmagnesium reagents that their observations are not unique: many authors see divergent results for these reagents compared to other organomagnesium reagents. This review is also intended to explain why the selectivities might be different based upon the latest understanding of the mechanism of these reactions and the implications of that mechanism. With that information available, researchers should be able to report their results using mechanistically sound arguments and by comparing their observations to related work. Furthermore, the review intends to show that mechanistic arguments using transition state models are not infallible, and that the underlying assumptions governing their application must be considered thoughtfully. It is necessary, in light of mechanistic information about how additions of allylmagnesium reactions occur,18 that stereochemical analyses use the most recent and relevant information. Consequently, many stereochemical outcomes are reconsidered here based on those insights. This review is also intended to help synthetic chemists predict what might happen in planned reactions, so that synthetic approaches can be devised with the highest probability of success. Finally, this review pays respect to the contributions of the authors who are cited because they have contributed to our understanding of the important synthetic reactions that use allylmagnesium reagents.
1.3. Experimental Details
The experimental details of these reactions are important to consider. Information such as solvents and temperatures are provided to facilitate comparisons, considering the influence that some solvents22 and temperature23 can have on the outcomes of additions of allylic organomagnesium reagents. In most cases, such details were available either in the text or Supporting Information of any article, but, on occasion, such information was unavailable. Other cases clearly document that allylations and other types of additions were performed, but no details such as diastereoselectivity were provided, so no specific insight could be gleaned.24 When temperatures were not listed, it was assumed that the reactions were conducted at room temperature if that were implied; otherwise, temperatures were not listed. Yields are reported as greater than some value if a yield were reported over multiple steps that include the addition reaction (in most cases, only the addition reaction was shown for clarity). Diastereomeric ratios are provided, although in some cases authors only note a single product, so that fact is indicated in the Scheme. In several cases, the text, figures, and experimental results reported deviate slightly in issues such as solvent, temperature, product ratios, and yields. In such cases, the values from the experimental sections were used, although it should be stressed that such differences are minor and do not affect conclusions. Finally, stereochemical assignments in all cases are based upon the analysis provided by the authors of the individual papers and are assumed to be correct.
1.4. How to Use this Review
Although it would be ideal to read this review in its entirety, that idealistic view will only be possible for a reader with a considerable amount of time to devote to this subject. A reader could still appreciate the topic by selecting a specific topic from the Table of Contents and focusing on the examples therein. As the reader then generates questions regarding reaction mechanisms and reasons for stereochemical outcomes, then the introductory sections might be more useful.
2. Impact of the Mechanism of Addition Upon Stereoselectivity
2.1. Current Understanding of the Mechanism of Additions of Allylmagnesium Reagents to Carbonyl Compounds
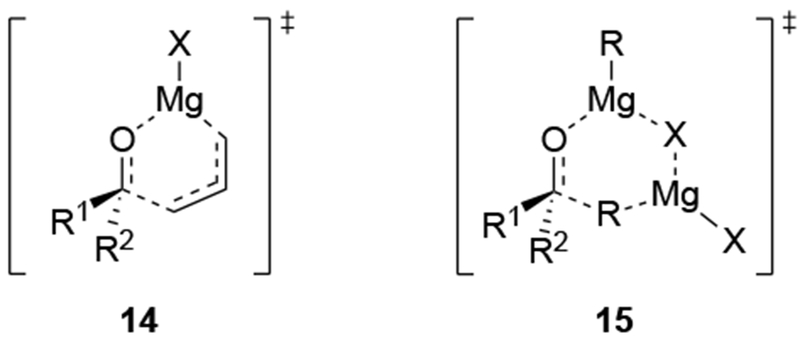
Recent studies provided insight into the mechanism of additions of allylmagnesium reagents to carbonyl compounds.18 Computational25 and experimental23 studies suggest that a concerted, six-membered-ring transition state is most likely responsible for the addition (i.e., 14, Scheme 6). This pathway, which was proposed many years ago based upon mechanistic studies with substituted allylic organomagnesium reagents,26 is unique among pathways for additions of organomagnesium reagents to carbonyl compounds. Non-allylic Grignard reagents react through different types of transition states, including four-center transition states and six-membered-ring transition states involving dimers of the reagents (i.e., 15).27 Considering the differences in addition mechanisms between allylic and non-allylic organomagnesium reagents, it may not be surprising that the stereochemical outcomes of addition reactions are different.
Scheme 6.
2.2. Diffusion-Controlled Additions
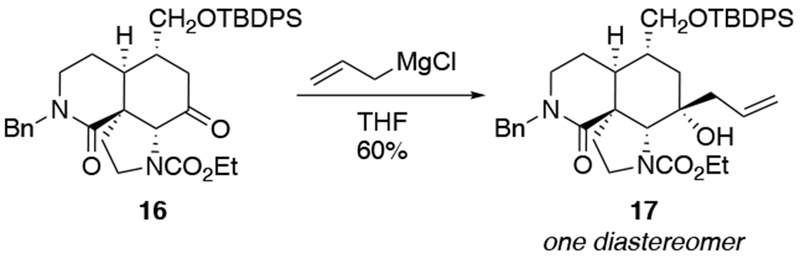
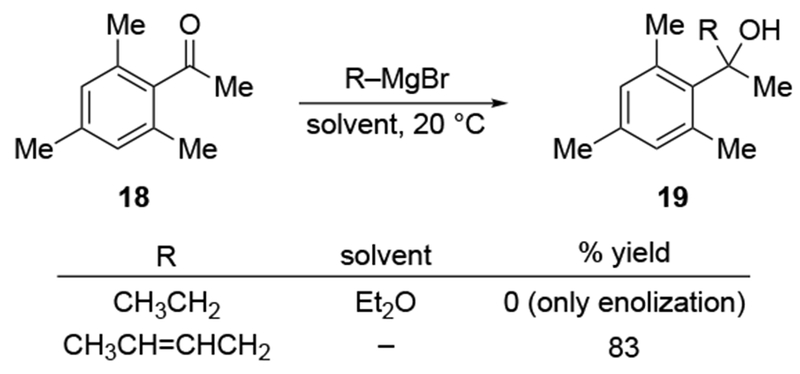
The most important mechanistic consequence of the six-membered-ring transition states through which allylmagnesium halides react is that these addition reactions proceed unusually quickly. Anecdotally, researchers noted the high reactivity of allylmagnesium reagents, a fact established carefully decades ago.28,29 This high reactivity allows allylmagnesium reagents to add to ketones for which no other organometallic nucleophile will add (Scheme 7)30 or to ketones that generally only undergo enolization (Scheme 8).31,32 In particular, additions of allylic Grignard reagents to unhindered carbonyl compounds likely occur at rates approaching the diffusion rate limit,28 although rates would be slower for additions to particularly hindered carbonyl compounds.17 This high reactivity can be synthetically useful and provides a strongly compelling reason to continue to use these reagents.
Scheme 7.
Scheme 8.
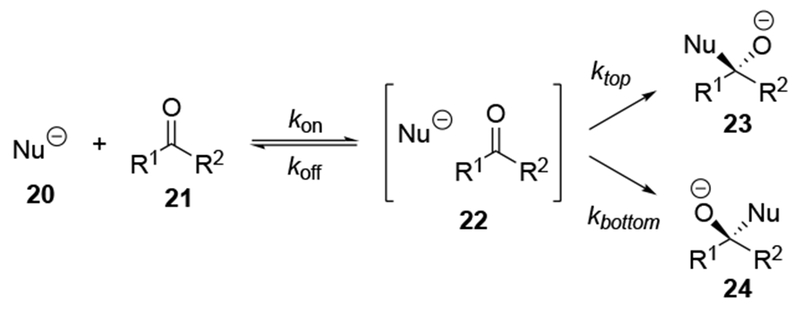
The high reactivity of allylmagnesium reagents comes at a cost of decreased stereoselectivity. This problem is best illustrated by considering the key steps involved in addition to a carbonyl compound (Scheme 9). Before the new carbon–carbon bond is formed, the nucleophile and electrophile must diffuse toward each other and form an encounter complex (i.e., 22) with a rate constant of kon. At that point, the nucleophile can attack the prochiral carbonyl compound (21) from either the top or bottom face, two processes that are competitive with the separation of encounter complex (the path labeled koff). As addition to the carbonyl group becomes faster with more reactive reagents, eventually the rates of ktop and kbottom become faster than the rate at which the two reaction partners can dissociate from each other (that is, ktop, kbottom >> koff). In this case of high reactivity, the transition state for bond formation no longer exclusively determines the selectivity of a reaction, but the manner in which the two components diffuse to one another (i.e., kon) will also influence diastereoselectivity. This change of the selectivity-controlling step from bond formation to diffusion has been documented for chemoselective additions to carbocations33 and stereoselective reactions of oxocarbenium ions.34–37 Therefore, as the rates of carbon–carbon bond formation approach the diffusion rate limit, the stereochemistry-determining step can be the approach of the reagents to each other (i.e., the step corresponding to kon), as illustrated in Scheme 9. Stereochemical models do not hold in this scenario because facial selectivity is determined before the carbon–carbon bond forming step.38 Therefore, any analysis of stereoselectivity must first consider whether these reactions might be diffusion-controlled or not. If the reaction were under diffusion control, then the diffusion step must be analyzed, not the bond-forming step.
Scheme 9.
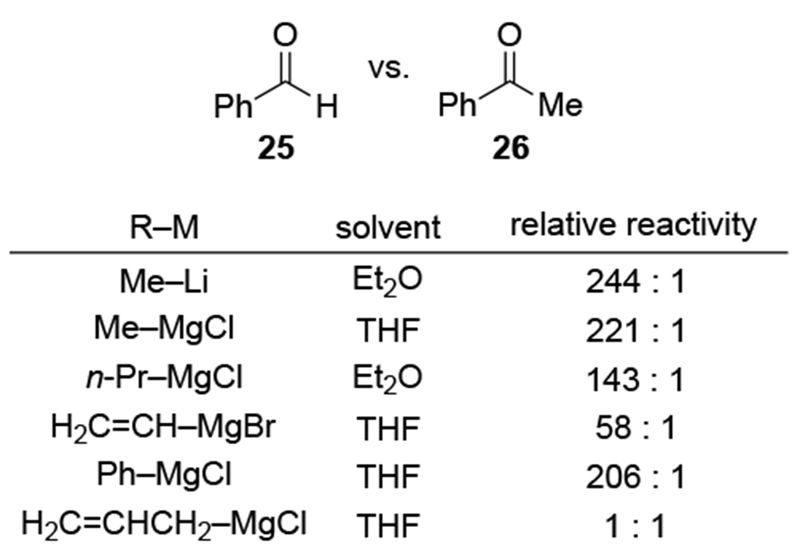
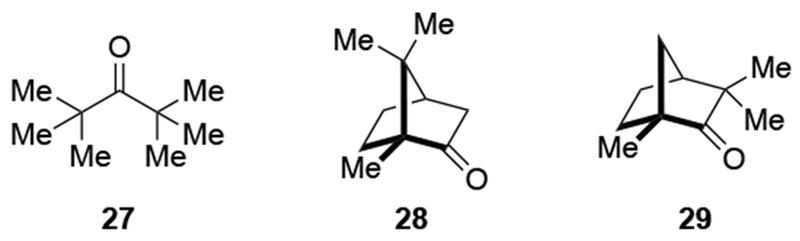
Recent studies suggest that additions of allylmagnesium reagents to aldehydes and most ketones occur at the diffusion rate limit.17,18 While most other organomagnesium reagents (and methyllithium) show dramatically different rates of addition to acetophenone and benzaldehyde, allylmagnesium reagents react with both at the same rate (Scheme 1017).28 These facts lead to the conclusion that many reactions of allylmagnesium reagents with ketones cannot be analyzed using transition state models, but, instead, must be analyzed by considering the diffusion step. Only in the case of highly hindered ketones, such as di-tert-butyl ketone (27), camphor (28), and fenchone (29, Scheme 11), do rates likely fall below the diffusion rate limit.17,23 The much slower addition of allylic Grignards to these hindered ketones is consistent with the fact that the reduction of di-tert-butyl ketone (27) by NaBH4 at 0 °C is over 5,000 times slower than the reduction of acetone.39 The rapid rate of addition of allylmagnesium reagents to most carbonyl compounds suggests that these reactions cannot be made faster by catalysis.40,41
Scheme 10.
Scheme 11.
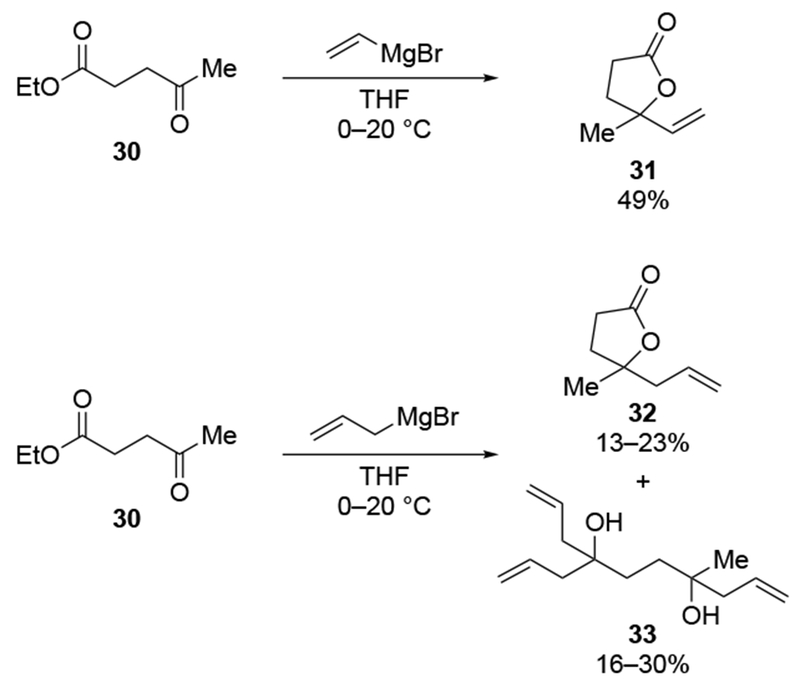
The lack of chemoselectivity resulting from the high rates at which allylmagnesium halides react presents a problem for multiply-substituted electrophiles commonly encountered in synthesis. Scheme 12 illustrates the problematic behavior of allylmagnesium bromide compared to vinylmagnesium bromide in such a system.42 Addition of vinylmagnesium bromide to compound 30, which contains both carbonyl and carboethoxy groups, resulted in addition to the carbonyl group followed by transesterification. Conversely, allylmagnesium bromide added to both the carbonyl and carboethoxy groups of compound 30 at comparable rates. Alternatively, the rate of addition to the carboethoxy group could be comparable to the rate of the intramolecular transesterification. Other evidence of the similar rates of additions of allylmagnesium reagents to aldehydes and esters have been reported.17 The ability of allylmagnesium reagents to add to esters rapidly can be useful for the analysis of triacylglyerols by facilitating deprotection of multiple ester groups,43 although it can complicate their application in natural product synthesis.44
Scheme 12.
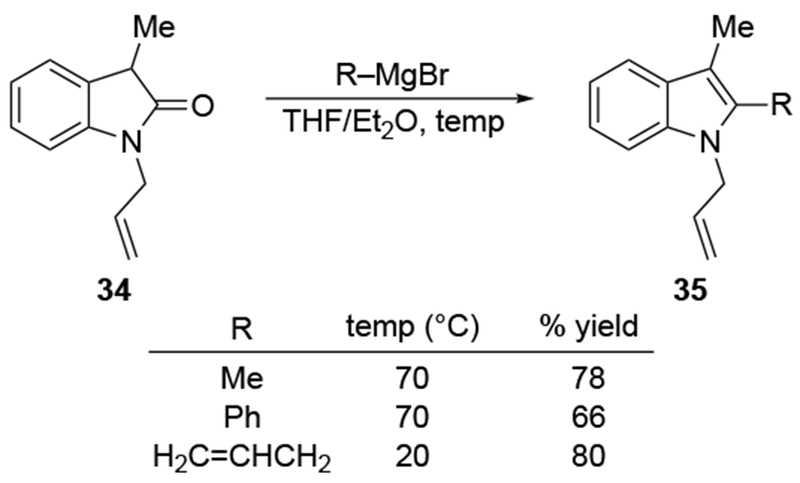
The high reactivity of allylmagnesium reagents even extends to the carboxamide functional group. In the addition to cyclic amide 34 (Scheme 13), allylmagnesium bromide reacted at room temperature, whereas higher temperatures were required to generate product 35 in good yield with the methyl and phenyl Grignard reagents.45 Multiple additions to amides are also not uncommon for allylmagnesium reagents, as illustrated in Scheme 14.46 It is notable that one of the two carboxamide groups of 36 does not react with the reagent, suggesting that chemoselectivity in allylic Grignard additions to complex molecules is possible. Other authors have noted similar behavior of allylmagnesium reagents with amides.47
Scheme 13.
Scheme 14.
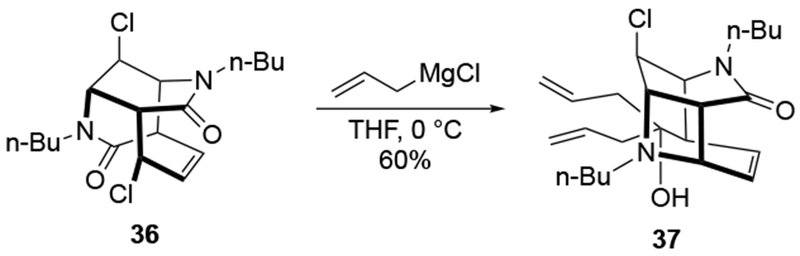
Even for reactions with seemingly simple electrophiles, the reactivity of allylmagnesium reagents can be quite useful (Scheme 15).48 Addition to aromatic aldehyde 38 using allylmagnesium bromide occurred in high yields, but efforts to achieve this allylation with allyltrimethylsilane and Lewis acids either gave no reaction or resulted in decomposition. This example illustrates the value of allylmagnesium reagents among the many reagents used to form allylated products.
Scheme 15.
2.3. Reversibility of Addition Reactions
For highly hindered carbonyl compounds, additions of some organomagnesium reagents can be reversible. The reversibility of additions of allylic magnesium reagents was first examined by Benkeser and co-workers, who monitored the isomerization of homoallylic magnesium alkoxides formed after addition of a crotyl Grignard reagent (Scheme 16).49,50 The observation of reversible reactions was established with sterically hindered ketones and long reaction times to achieve complete equilibration. In the case of addition of the smaller reagent, allylmagnesium bromide, to di-tert-butyl ketone (27), the reverse reaction required heating to reflux in THF.51 Other reversible additions are also slow: the reverse of the additions of benzylmagnesium reagents were performed at 140 °C for three to ten days.52 As a result, reversibility is not likely to occur, even in hindered systems, if reactions were quenched after the addition reaction.23 This protocol is normally followed by synthetic chemists.
Scheme 16.
2.4. Importance of Reaction Rate to Understanding Stereoselectivity
Competition experiments provide important insights into if and when stereoselectivity should be expected from reactions. Because the rate at which allylmagnesium nucleophiles react with carbonyl compounds approaches the diffusion limit, traditional rate measurements (or even stopped-flow measurements) cannot provide rate constants for these reactions.28,29 Such competition experiments can provide insight not only into chemoselectivity (Scheme 10 above) but also can reveal why, in some cases, additions of allylmagnesium reagents may not be diastereoselective. For example, competition experiments suggest why additions of allylmagnesium halides to α-alkoxy ketones in ethereal solvents are not generally diastereoselective (Scheme 17).22 Considering that addition through a chelated transition state should be faster than additions through a non-chelated transition state,53–55 the fact that addition of an allylmagnesium reagent to a chelating ketone is not faster than addition to a non-chelating ketone22 suggests that any explanations for stereochemical control involving chelation should be considered with some skepticism. Consequently, additions that can be analyzed using Felkin–Anh, Cieplak, Cornforth, and related models56,57 would also need to be evaluated as to whether or not the stereochemical outcome arises instead from diffusion-controlled behavior. Such studies would be relatively simple to perform in cases where sufficient quantities of the carbonyl compound were available, as is often the case in methodological studies. Information about relative reaction rates of reactions can be obtained using carefully controlled competition experiments.17 As a result of the recent relative rate studies of allylic Grignard reagents, the analyses reported in this review will necessarily make assumptions about relative rates based upon reasonable comparisons, as will be described below.
Scheme 17.
2.5. Additions to Imines
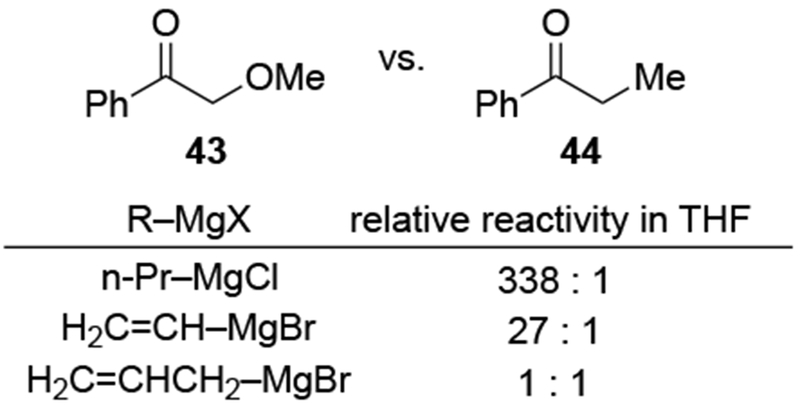
Although this review will focus on reactions of ketones and aldehydes, reactions of allylmagnesium reagents with imines and related electrophilic carbon–nitrogen double bonds will be discussed to provide comparisons. Much less is known about the mechanism of additions of allylmagnesium reagents to imines and other electrophiles with carbon–nitrogen double bonds. It is reasonable to assume that the six-membered ring transition state suggested above (14, Scheme 6) operates for additions of allylmagnesium reagents to imines. The rates of the reactions, however, must be considerably slower than additions to carbon–oxygen double bonds, although additions to activated imines such as N-sulfonylimines, could be even faster.58 As a result, stereochemical models such as the Felkin–Anh model may operate, so there may not be the same unpredictability or loss of stereoselectivity as observed with ketones and aldehydes. For example, addition of allylmagnesium bromide to chiral α-alkoxy aldimine 45 proceeded with high diastereoselectivity (Scheme 1859), as compared to the additions of allylmagnesium reagents to α-alkoxy aldehydes and ketones (as illustrated in Scheme 3).
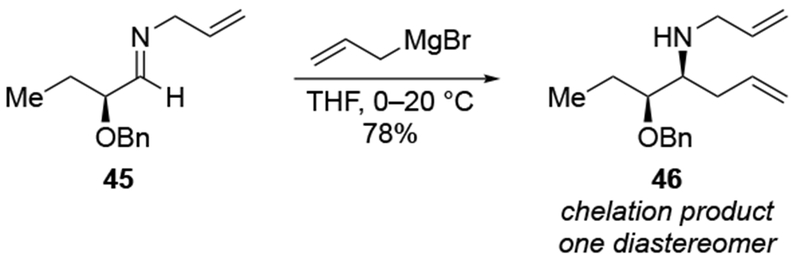
Scheme 18.
3. Caveats Regarding Stereochemical Models
Stereochemical models are powerful tools that help organic chemists rationalize and predict the stereochemical outcomes of reactions, but they need to be used wisely. Many stereochemical models are predicated on the assumption that the carbon–carbon bond-forming step is also the stereochemistry-determining step. As the rates of bond formation approach the diffusion limit, however, the bond-forming step may no longer determine the stereochemical outcome, and thus the model would no longer apply. Similarly, in some cases, reactions are diastereoselective, but it is not a difference in enthalpy that determines selectivity; differences in entropy in the corresponding transition states control diastereoselectivity.60–62
The failures of stereochemical models due to rapid reaction rates have been documented for the reactions of a family of electrophiles closely related to carbonyl compounds, namely oxocarbenium ions. The standard stereochemical models can fail to explain the outcomes of reactions of these electrophiles with some of the most reactive π-nucleophiles, Me3SiCN, and alcohols.34–37 When nucleophiles become more reactive, additions to these highly reactive oxocarbenium ions occur with decreased stereoselectivity (Scheme 19) likely because these reactions occur at rates approaching the diffusion limit and, therefore, the addition step is no longer the stereochemistry-determining step.36 While in this situation the electrophile is the highly reactive partner, the reactions of highly reactive allylmagnesium reagents with carbonyl compounds are likely to be analogous.22 As a result, stereochemical models need to be used wisely to prevent their being applied in settings in which their necessary conditions are not met. Such precise thinking requires discipline on the part of both authors and readers.
Scheme 19.
It is also imperative that any explanation of reactivity is logically sound. An example is illustrated for additions of nucleophiles to prochiral carbonyl compounds (Scheme 20). In this case, only two possible stereochemical products can be formed. Each of these products (resulting from addition to the Re or Si face) is generally ascribed to one of two different stereochemical models. The Cram chelate model can be used to explain how one product is formed, whereas the Felkin–Anh model would explain how the opposite stereoisomer would be formed.57 Haphazard use of these models can perpetuate a logical fallacy that confuses cause and effect. One could assume that if the product of chelation-control were favored, then the reaction must have proceeded through a transition state involving chelation-control. Conversely, if the diastereomer expected from the Felkin–Anh model were formed in excess, it could be proposed that the transition state for this reaction would involve the types of conformational preferences and interactions stipulated by that model. Although these explanations are consistent with the outcomes, they do not require that the particular mechanistic pathway described by the stereochemical model is operating.
Scheme 20.
This point is particularly clear for reactions generally analyzed using the Felkin–Anh model. This model is not the only explanation for formation of products with the same configuration. The original Felkin model,63 without the benefit of the insights applied by Anh and Eisenstein,64 also explained the formation of the same diastereomer (and other models have analyzed the same transition state differently65). The Felkin transition state, however, is just one of a number of possible transition states leading to the same product, and, in some cases, other models might be more consistent with the results, even though the same diastereomer of the product was formed.66 As a result, simply because a reaction yields the product expected from the proposed transition state model, that is insufficient to prove that this reaction proceeded through that transition state. Instead, the outcome is merely consistent with that transition state; other transition states could have led to the same outcome.
This issue is also relevant to product ratios. It is common in papers for authors to discuss the origin of the major product of a reaction, but a more sophisticated argument would need to explain the formation of minor products as well. If the major product were formed by, for example, a Felkin–Anh-like transition state, then application of the false logic described above would require that the minor product would necessarily arise from a chelation-controlled transition state. But such an argument is not universally applicable, particularly in cases where chelation would not be possible. Thus, it makes more sense that the minor product occurs from some higher-energy, Felkin–Anh-like transition state or possibly a completely different type of transition state altogether. It is conceivable that the final product ratio might be the result of a statistical average of several different pathways, based on their relative energies, as has been argued for additions to chiral carbonyl compounds67 and oxocarbenium ions68 and for the asymmetric hydrogenation of alkenes using chiral catalysts.69
These arguments would also apply to many other modes of stereochemical control. For the purposes of this review, we have analyzed the results of reactions of allylmagnesium halides with chiral carbonyl compounds and imines using four distinct models: the Cram chelation model (as formulated by Eliel and Frye,53–55 although with caveats obtained from more recent results22,70), the Felkin–Anh model, the torsional strain model71 associated with additions to cyclic ketones, and the concept of steric approach control.
This last model deserves additional introduction. Steric approach control addresses which face of a carbonyl group is attacked strictly based upon which face is more sterically accessible.72 What sets this model apart is that it is not analyzed in the same way as the other stereochemical models. Other models consider a number of possible transition states, analyze which transition state would have the lowest energy, and conclude that the reaction occurred through the lowest-energy transition state. Those models, by definition, assume a Curtin–Hammett kinetic scenario73 in which a number of different intermediates (such as encounter complexes and conformational isomers) are rapidly interconverting but the major fraction of molecules follows the lowest-energy pathway among all possible pathways.
Steric approach control, however, does not assume a Curtin–Hammett kinetic scenario. Instead, it focuses on what steric interactions develop upon bringing a reagent close to the starting material.74,75 Such steric interactions in the transition state can be sufficient to define selectivity. It is correct that steric approach is involved in the other modes of stereocontrol as well (as in determining which face is attacked once chelation had occurred or what interactions occur in the Felkin–Anh transition state once orbital interactions are optimized), but steric approach control requires only steric factors developing between the components of a reaction to explain selectivity. For example, additions to ketone 56 (Scheme 21) have been argued to be the result of steric approach control: the face of the carbonyl group that is preferentially attacked is determined by balancing the steric destabilization that would occur from attack of the opposing faces.76 This balance shifts as the size of the substituent on the nearby aromatic ring increases in size.
Scheme 21.
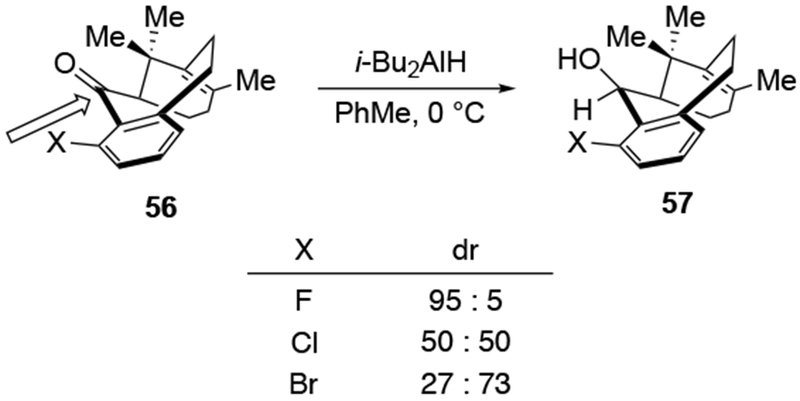
Although, on one level, it may be a semantic point to discuss steric approach as a separate mode of stereochemical control, it is a useful mode of thinking that enables different possibilities for analyzing stereochemistry. Analysis of the results in Scheme 21 illustrates how steric approach control can be used. Neither the Felkin–Anh model nor the chelation control model can explain these results adequately. It is also difficult to analyze these experiments by considering torsional effects in the transition states of addition, which should be similar for the three halogen-containing substrates. It is reasonable, however, to argue that the exo face of the carbonyl group becomes less accessible as the halogen atom increases in size, causing attack from that face to be slower.
Steric approach control can be a powerful mode of controlling stereochemistry, and it is worth acknowledging it as a possibility with the same importance as Felkin–Anh or chelation control models. A classic example of steric approach control involves the commonly used arguments to analyze approach to a π-system from the convex face of a bicyclic (or multicyclic) system. This “convex vs. concave” mode of analysis is an important stereochemical controlling element with a long history in organic chemistry.83,84
It should not be construed that steric approach control does not consider differences in energies of competing transition states. Rather, it evaluates which face of a molecule is more accessible to reagents. Those interactions are among the many factors that influence selectivity. Steric approach control, such as evaluating convex vs. concave faces, can be of secondary importance compared to other stereochemical control elements.85–87 Nevertheless, the analysis of which face of a π-system is more easily approached by a reagent can be useful when considering reactions in which the approach of the reagents (i.e., diffusion), and not the transition state for bond formation, is important for stereoselectivity. It is precisely this situation that may occur in the case of additions of allylmagnesium reagents to carbonyl compounds. The stereoselectivity of these reactions is no longer controlled by transition states that include bond formation, so, as rates approach the diffusion limit, transition-state models will no longer operate.38
4. Diastereoselectivities of Reactions with Carbonyl Compounds that Can Chelate
4.1. Stereochemical Control by the Chelation-Control Model
Chelation control is a commonly used method for controlling the stereochemistry of additions to carbonyl compounds. This approach is most useful in the case of α-substituted carbonyl compounds (particularly α-alkoxy ketones), but chelation can be observed with more remotely substituted substrates. In comparison, α-alkoxy aldehydes react with lower selectivity than ketones do, likely because of the generally higher reactivity of the aldehyde functional group.17,28,88
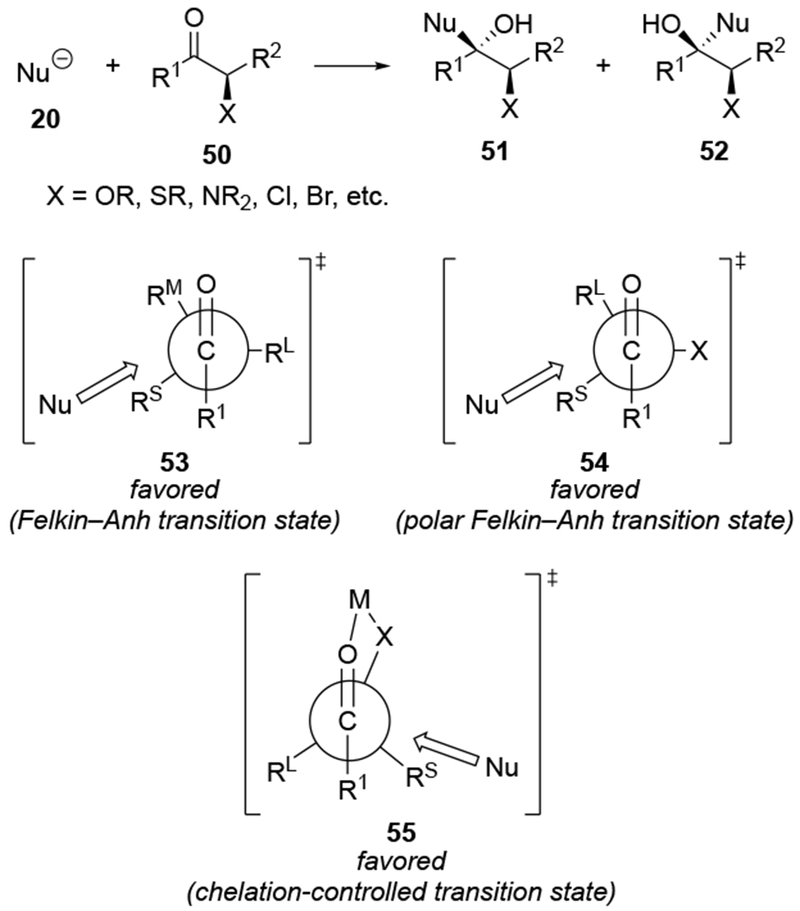
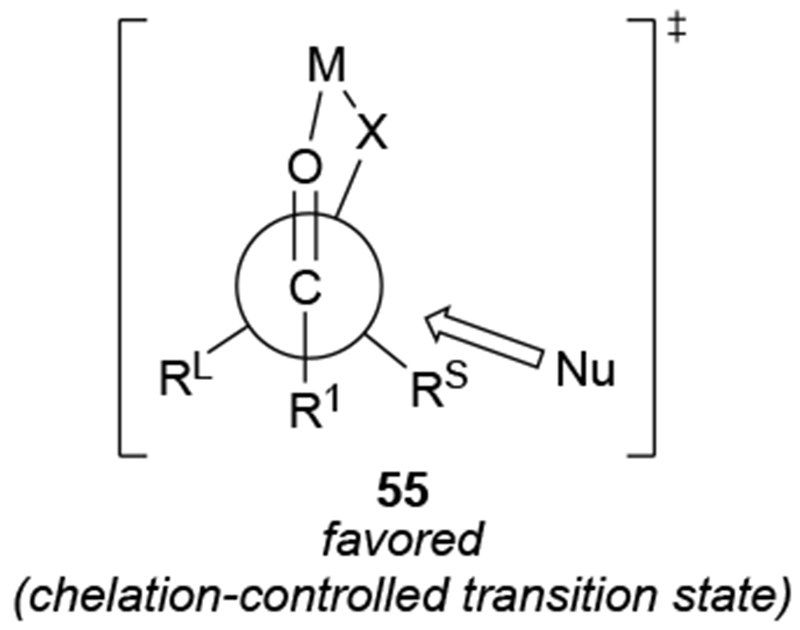
Chelation-controlled selectivity was not intended to denote an electronic difference between chelated and non-chelated transition states. As originally formulated, the model explained the high selectivity based upon the presence of a rigid intermediate with sterically differentiated faces in which the α-heteroatom and the carbonyl group are chelated to the organometallic reagent (Scheme 22).67 Approach to one face of the chelate 55 is sterically blocked by a larger substituent (in this case, Rl), so attack occurs from the less sterically hindered face. As a result, chelation control is a form of steric approach control: the difference in energy between the transition states of addition to one side of the chelated intermediate compared to the other is caused by the different steric destabilizations that develop between the incoming nucleophile and the substrate.
Scheme 22.
Chelation-controlled selectivity is not so simple to analyze, however. The contributions of Eliel, Frye, and co-workers are particularly important to the understanding of this model.53–55 Their studies revealed that chelated intermediate 58 is likely not the major intermediate present in solution in THF, one of the most common solvents used for reactions involving organomagnesium reagents. 1H NMR spectroscopy of ketones in the presence of MgBr2•(OEt2)2 (as a surrogate for the organomagnesium nucleophile) demonstrated that the chelated intermediate is a minor component in ethereal solution (Scheme 23).
Scheme 23.
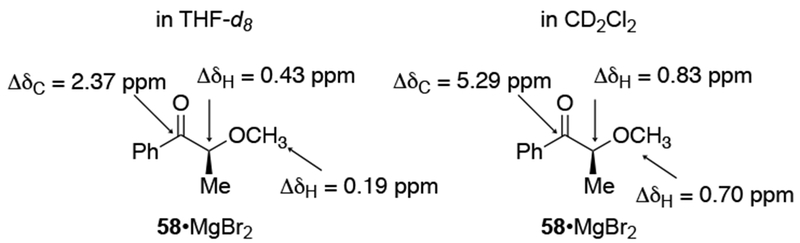
Solvent choice can make a significant difference in the quantity of the chelated intermediate. For example, complexation was much more pronounced in CD2Cl2 compared to THF-d8, as evidenced by the significant differences in chemical shifts observed upon addition of MgBr2 to an α-alkoxy ketone (Scheme 23).22 Other metal salts, such as EtZnCl, also formed significant quantities of chelated species with α-silyloxy ketones in CD2Cl2.89
The origin of stereoselectivity in systems where chelation is possible is more complex than just recognizing that chelation can occur. The difficulty is caused by the fact that many intermediates could be present in solution, such as those shown in Scheme 24, but the chelated intermediate, a minor component, must be more reactive than other species. Eliel, Frye, and co-workers measured reaction rates for additions of Me2Mg to ketones that could and could not form chelated intermediates.53,54 For α-methoxy ketones, reaction rates were up to 2,000 times higher than for ketones without a chelating group (Scheme 25).53 As a result, the chelation-control model is an example of a Curtin–Hammett kinetic scenario:73 a minor amount of a highly reactive species (i.e., the chelate 60) must undergo reaction with the organometallic reagent faster than any other species in solution reacts. Furthermore, the chelated carbonyl compound must react much faster considering the generally high stereoselectivities observed. It was suggested that the chelated carbonyl compound was more reactive because the carbon–carbon bond formation is unimolecular instead of bimolecular,54 a suggestion that has been supported computationally.90
Scheme 24.
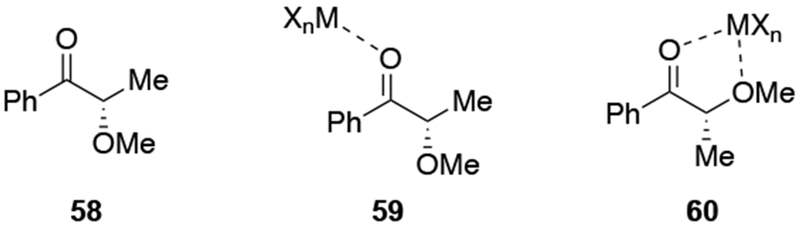
Scheme 25.
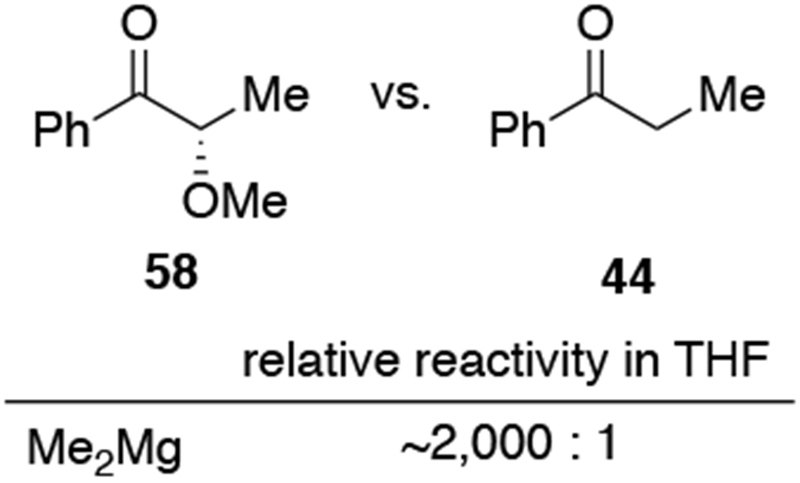
This conclusion, however, may only be the case for the monomeric dialkylmagnesium species examined in those studies. The structures of the more commonly used organomagnesium halides are likely to be more complicated (Scheme 26). The Schlenk equilibrium, which exchanges ligands on magnesium atoms, enables different types of organomagnesium reagents to be the nucleophilic species.91 Grignard reagents also readily form aggregates, depending on the solvent, concentration, halide, and nature of the organic group, all of which would complicate this picture significantly.91,92 Although the aggregation state of organomagnesium halides vary widely, multimeric species are plausible intermediates in which there is a synergy between the complexation of the carbonyl compound to one equivalent of a magnesium species and a second magnesium species.93–95 This type of mechanism parallels observations with organozinc reagents, which show chelation-controlled selectivity in the presence of dimeric zinc species.96
Scheme 26.
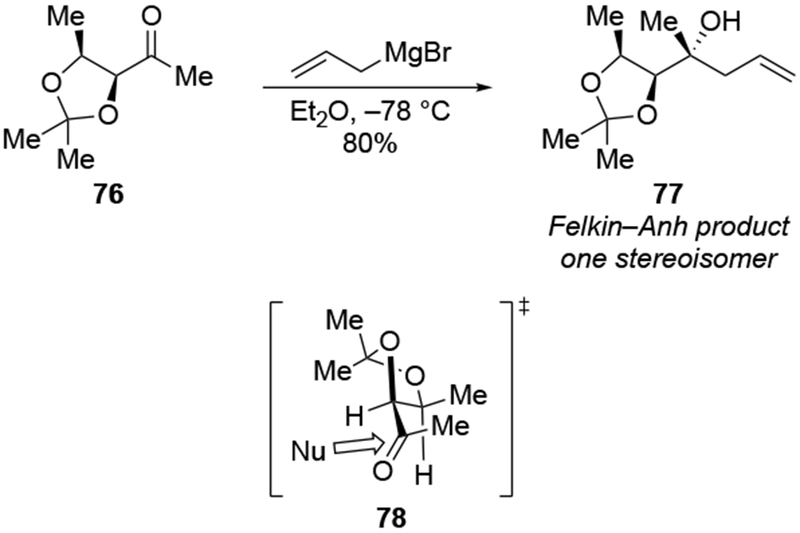
As suggested above, the use of non-ethereal solvents can change the stereochemical outcome dramatically. For example, whereas addition of methylmagnesium halides followed the chelation-control mode in ethereal solvents, addition of allylmagnesium reagents were generally poorly selective, as would be expected from a reaction whose stereochemistry-determining step was diffusion (Scheme 27).22 By contrast, additions in the non-complexing solvent CH2Cl2 were highly diastereoselective. In non-ethereal solvents such as CH2Cl2, the chelated form of the carbonyl compound was demonstrated to be more favored compared to ethereal solvents (Scheme 23),22 which might be expected to increase the amount of product formed from this intermediate.
Scheme 27.
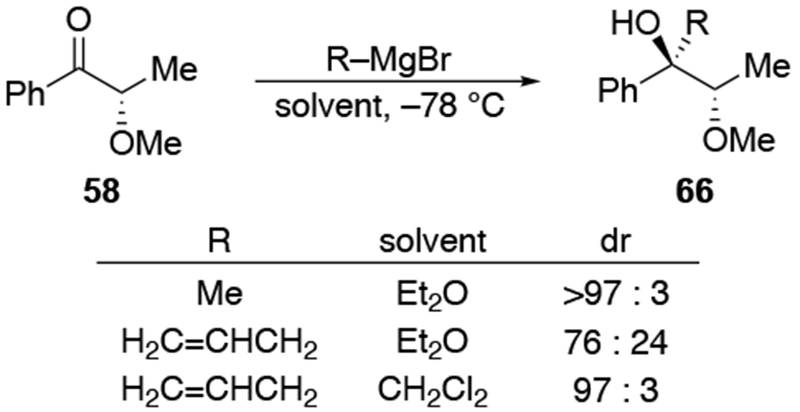
Other experiments, however, show that the observation of highly diastereoselective reactions with allylmagnesium halides in CH2Cl2 cannot be explained by the chelation control model as currently defined. That model includes the stipulation that chelation is accompanied by acceleration of the reaction rate,53,54 but no chelation-induced rate acceleration was observed for allylmagnesium halides (Scheme 28).22 The chelation-control model describing additions of Grignard reagents to α-alkoxy ketones can therefore be broadened to state that the product arising from addition to the favored face of the chelated intermediate should be the major product of a reaction in either of two scenarios: a small amount of the chelated intermediate is the most reactive species in solution, or the chelated intermediate must be the major species in solution. Therefore, the nature of the solvent needs to be considered in any analysis of additions of organometallic nucleophiles to ketones capable of chelation.
Scheme 28.
The nature of stereocontrol through chelation is likely even more complicated. One final example reinforces the general caveats noted about the stereochemical models used by organic chemists (Section 3). In studies of hydride reduction reactions, it was demonstrated that the product expected from chelation control was formed without any evidence that it was either a result of a highly reactive chelated intermediate or that the major species in solution was a chelated intermediate.70 Therefore, it is important to remember that all that is required for stereoselectivity is that the diastereotopic faces of the carbonyl compound are sufficiently differentiated.
4.2. Additions of Allylmagnesium Reagents to α-Substituted Carbonyl Compounds Capable of Chelation
Among the more consistent problems in using allylmagnesium reagents is their general reluctance to engage in chelation-controlled additions. This point is illustrated in Scheme 3 in the Introduction section. The striking difference in diastereoselectivity can be attributed to the high rates of additions of allylmagnesium reagents, which approach the diffusion rate limit. In this section, examples of chelation-controlled and non-chelation-controlled additions are documented. These examples will focus on reactions of allylmagnesium reagents; the studies of chelation-controlled additions by other organometallic reagents such as organotitanium or organozinc reagents have been reported elsewhere.97–100
4.2.1. Additions to Acyclic Ketones and Aldehydes
4.2.1.1. Additions to α-Alkoxy Ketones
As noted in the Introduction section (Section 1), additions of allylmagnesium reagents to chiral, α-alkoxy ketones are generally unselective. The examples shown in Scheme 29 below are illustrative.4,101 Addition of an alkyllithium reagent preferentially gave the Felkin–Anh product (albeit with low selectivity), likely because chelation to the lithium atom is generally weaker.102,103 On the other hand, most organomagnesium reagents reacted with high chelation-controlled stereoselectivity. Among the organomagnesium reagents investigated, however, only allylmagnesium bromide reacted with no stereoselectivity. Similar observations were made using other protecting group schemes.104
Scheme 29.
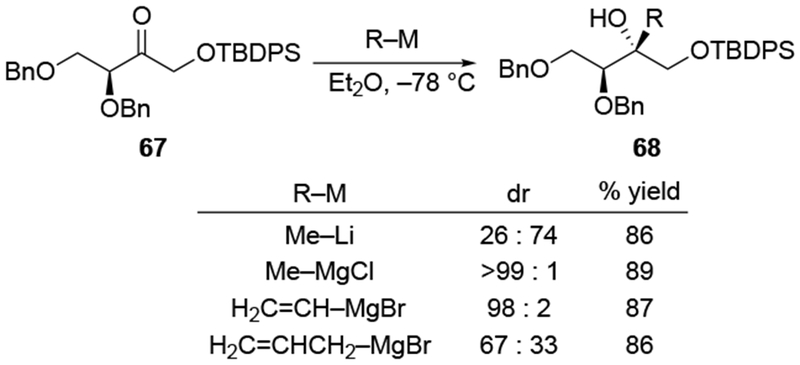
The contrasting reactivity of allylmagnesium reagents compared to other organomagnesium reagents is a general observation. Among several Grignard reagents examined for additions to ketone 69 and related substrates, allylmagnesium bromide was notable for its lack of chelation-controlled diastereoselectivity (Scheme 30).105
Scheme 30.
Even in conformationally controlled systems, these reactions are not generally stereoselective (Scheme 31).101 In this case, the low stereoselectivity was not a problem because the authors were interested in exploring the biological activity of different stereoisomers of the final product. That advantage does not apply to most situations, however.
Scheme 31.
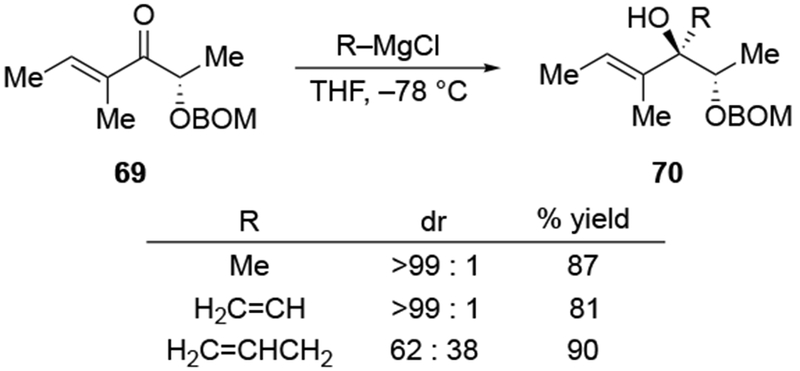
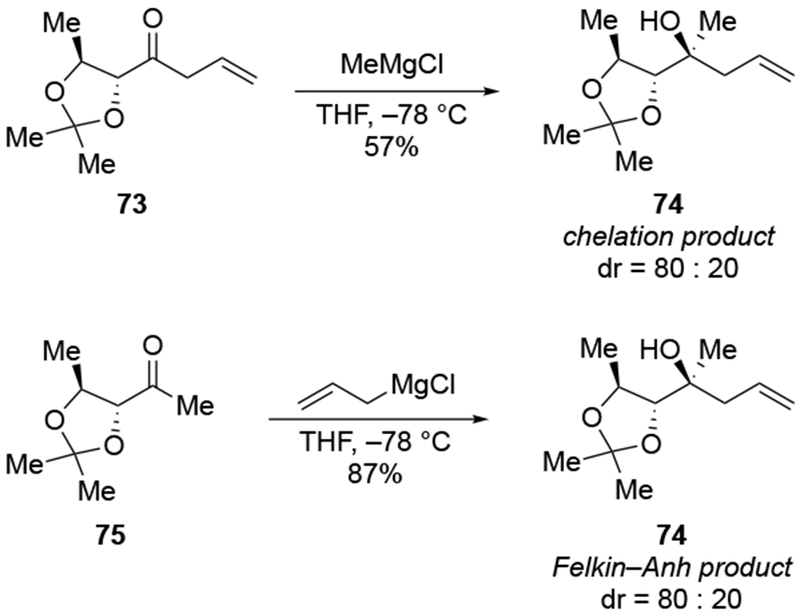
In some cases, addition of an allylmagnesium reagent occurred with the opposite stereoselectivity from what was expected (Scheme 32).106 Whereas addition of MeMgCl to the ketone 73 occurs with some selectivity for the product expected from chelation control, addition of allylmagnesium chloride to ketone 75 provided similar selectivity but for the opposite stereoisomer (i.e., the product of addition consistent with the Felkin–Anh model).
Scheme 32.
The importance of the overall structure of a compound and not just the potentially chelating α-stereocenter is illustrated by comparing the results shown in Scheme 32 to studies of a related ketone 76, Scheme 33 based upon the figure drawn in the paper and its conversion to a cyclic acetal).107 Addition to this ketone, the diastereomer of 75, gave much higher selectivity, but that reaction also did not form the chelation-control product: the transformation was highly Felkin–Anh selective. This reaction may be an example of a reaction selective for the Felkin–Anh product that does not proceed through a Felkin–Anh transition state, as discussed in Section 3. It is likely that the ketone exists predominantly in the conformation 78, which would minimize dipole interactions and avoid a potential syn-pentane-like interaction between the β-methyl group on the ring and the methyl group on the carbonyl carbon atom. Addition from the more accessible face would then give the observed product. This analysis also makes it easier to rationalize the fact that addition of a different organomagnesium reagent to ketone 76 occurred with the opposite sense of facial selectivity, giving the product expected from chelation control as a single stereoisomer (Scheme 34).108 The rate acceleration required of chelation control53,54 could be necessary for addition of an alkylmagnesium reagent to this hindered ketone. Addition of allylmagnesium halide would not need such rate acceleration for the reaction to proceed;22 addition could occur from the lowest-energy conformer.
Scheme 33.
Scheme 34.
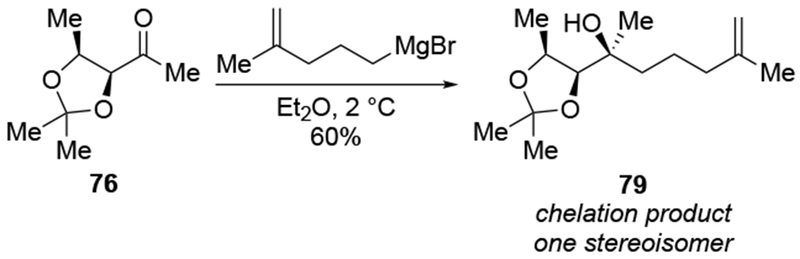
A similar comparison could be made with the addition to an imine, although these reactions will be discussed in more detail in a later section (Section 7). Addition of an allylzinc reagent to imine 80 gave the product expected from chelation control, whereas addition of the allylmagnesium reagent gave the Felkin–Anh product (Scheme 35).109
Scheme 35.
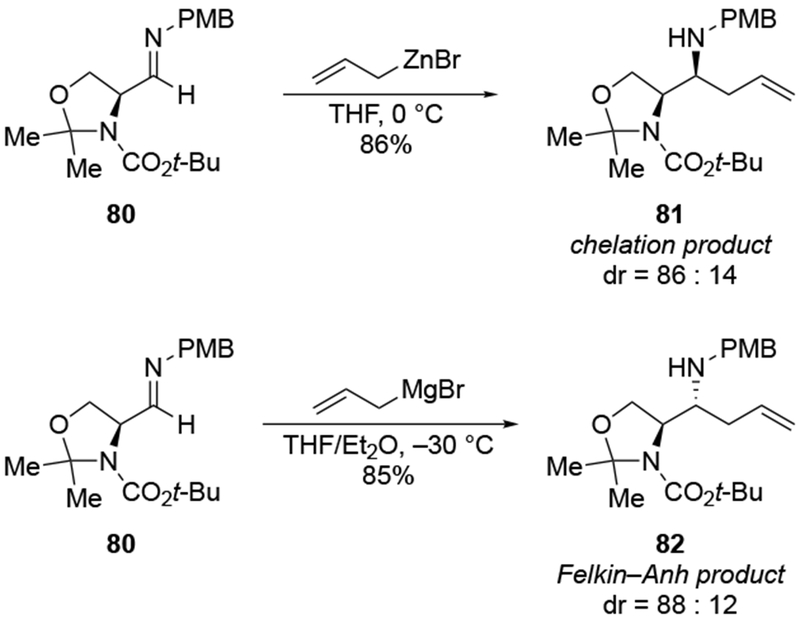
High selectivity for allylation can be observed in the case of α-hydroxy ketones, where the chelating group may be an OMgX group, not an OR group. The addition of an excess of allylmagnesium chloride to ketone 83 gave high selectivity for the product anticipated from chelation to the α-OMgCl group (Scheme 36).106 This type of addition has been seen previously (for example, with ketone 85, Scheme 37).110
Scheme 36.
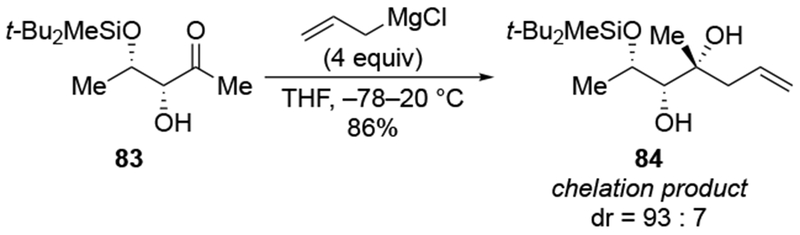
Scheme 37.
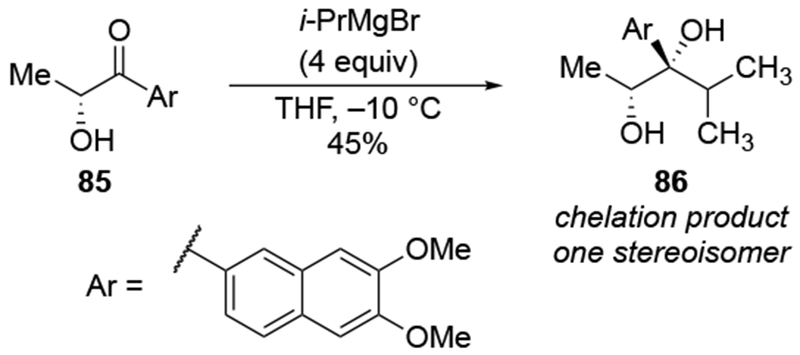
Analyzing these selectivities by assuming that deprotonation of the hydroxyl group occurred first followed by chelation-controlled addition may or may not be logical. The rate of deprotonation of an alcohol by an alkylmagnesium reagent is much faster than addition, but for allylmagnesium reagents, the rate of addition to a carbonyl group is competitive with proton transfer from a relatively acidic OH group.111 Consequently, it cannot be assumed that the nearby OH group has been converted to OMgX prior to addition to the carbonyl group for every molecule of the reaction mixture.
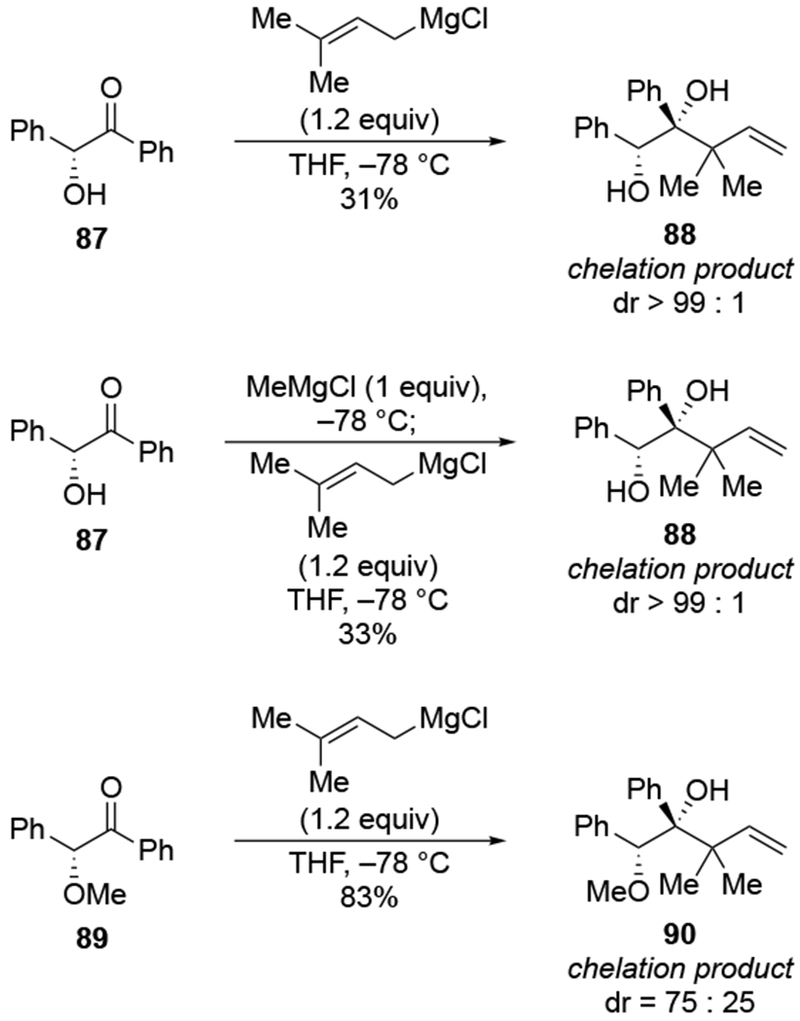
Evidence supporting the chelating ability of the OMgX group is illustrated by a comparison using prenylmagnesium chloride, a substituted allylmagnesium reagent (Scheme 38).23 Addition of one equivalent of prenylmagnesium chloride to the α-hydroxy ketone 87 gave high selectivity for the diol 88, which would be anticipated from chelation control (Scheme 38). The low yield of the product could result from competitive deprotonation of the hydroxyl group. A control experiment indicated that chelation to an OMgCI group is most likely responsible for selectivity. Initial deprotonation of the OH group was performed with MeMgCI. No addition of a methyl group occurred under these conditions, which is consistent with the fact that proton transfer should be much faster with an alkylmagnesium reagent.111 Subsequent addition of prenylmagnesium chloride to the resulting alkoxide gave the chelation-control product with similar selectivity and yield as in the original experiment. By comparison, prenylation of the corresponding α-methoxy ketone 89 occurred with low stereoselectivity.
Scheme 38.
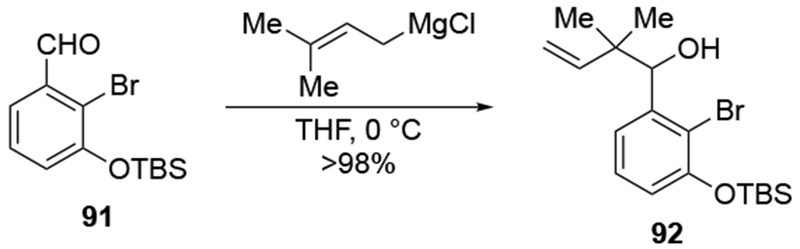
These experiments also suggest a reason why additions to α-hydroxy ketones are more stereoselective than to α-alkoxy ketones. It is likely that with a negative charge present on the molecule, addition to the carbonyl group could be relatively slow (that is, with a rate constant that is slower than the rate of separation of the encounter complex, as discussed in Section 2.2). Additionally, with the magnesium atom now tightly bound to the α-oxygen atom, complexation of the carbonyl group to the metal atom becomes a unimolecular process, not a bimolecular one. That chelation should lead to faster addition54 to the magnesium alkoxide and therefore stereoselectivity. It is also worth noting that the general regioselectivity exhibited by prenylmagnesium reagents and the presence of this functional group in natural products make this reagent an attractive nucleophile for use in synthesis (for example, Scheme 39112). Oxidation of alcohol 92, followed by asymmetric reduction of the ketone provided the stereoisomer needed for the synthetic route.
Scheme 39.
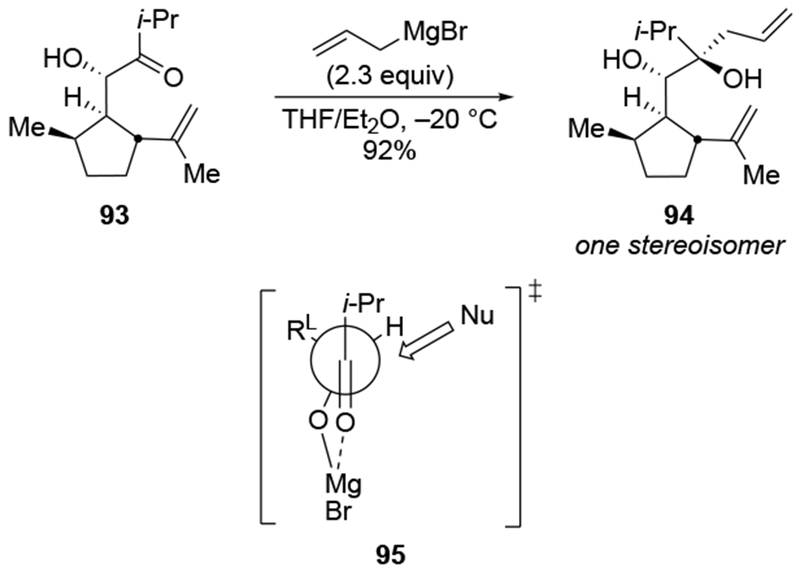
Addition of an allylmagnesium reagent to a more structurally complex α-hydroxy ketone also resulted in high stereoselectivity (Scheme 40).113 The stereoselectivity can again be rationalized by considering chelation to an α-OMgX group (as in 95).
Scheme 40.
4.2.1.2. Use of Non-Coordinating Solvents to Improve Selectivities of Allylations of α-Alkoxy Ketones
As noted above (Section 4.1), the use of non-ethereal solvents in reactions with allylmagnesium halides can give rise to more stereoselective reactions. Strictly speaking, however, these selectivities cannot be explained using the chelation-control model under Curtin-Hammett kinetics, as formulated by Eliel and Frye.53–55 Instead, stereoselectivity could arise because the major species in solution is the chelated intermediate.22 Allylations that are not selective in ethereal solvents show some improvement using halogenated solvents (Scheme 41).114 The selectivity for allylation of ketone 96 can be improved under carefully controlled conditions.22 The presence of additional magnesium salts under these conditions can improve the selectivity further, likely by increasing the proportion of a chelated species in solution compared to unchelated substrate. The selectivities under these conditions are comparable to those observed for the addition of allylstannanes with chelating Lewis acids.114 This combination of non-coordinating solvents and additional magnesium salts also can increase the stereoselectivity of additions of other organomagnesium reagents to α-alkoxy ketones.115
Scheme 41.
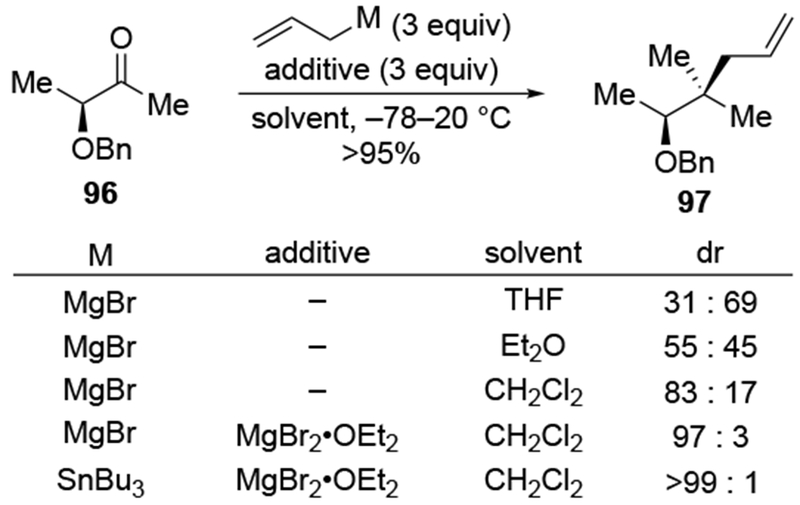
The use of halogenated solvents, although it should be considered as a possible solution, cannot solve all selectivity issues. For example, in a study using a chiral auxiliary to control selectivities for additions to a carbonyl group, additions of a number of organomagnesium reagents in CH2Cl2 with excess magnesium salts led to highly diastereoselective reactions (Scheme 42). Only in the case of allylmagnesium halides was this reaction unselective, even under these optimized conditions (the counterion was not specified in all cases).114 Another example of the failure of non-complexing solvents to solve the issues of low selectivity is illustrated for ketone 100, where allylmagnesium reagents once again reacted with uniquely low stereoselectivity (Scheme 43).116
Scheme 42.
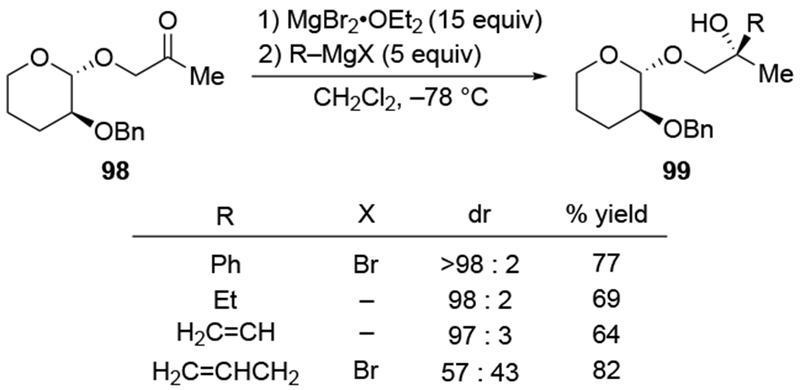
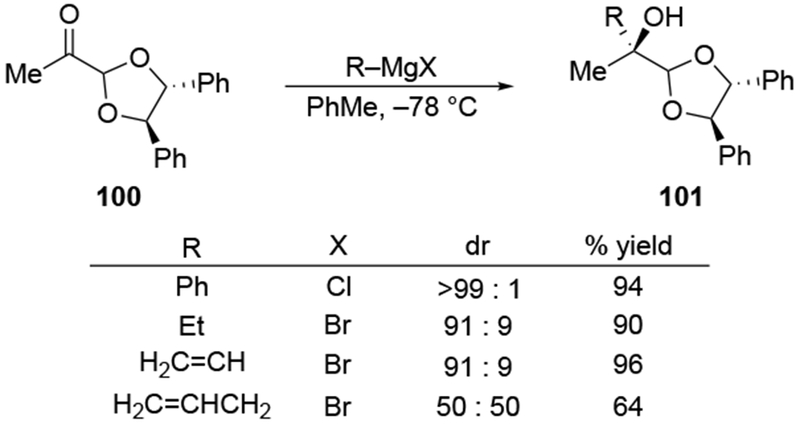
Scheme 43.
Related additions to an α-keto amide gave little diastereoselectivity for reactions of allylmagnesium halides (Scheme 44).117 Whereas selectivity could be achieved in some additions of allylmetal reagents in halogenated solvents, the addition of allylmagnesium bromide in Et2O was unselective (the other diastereomer was somewhat favored). Reactions of methylmagnesium and methyllithium reagents in Et2O with a similar substrate were similarly unselective, reacting with the same sense of selectivity as the allylmagnesium reagent (Scheme 45).118
Scheme 44.
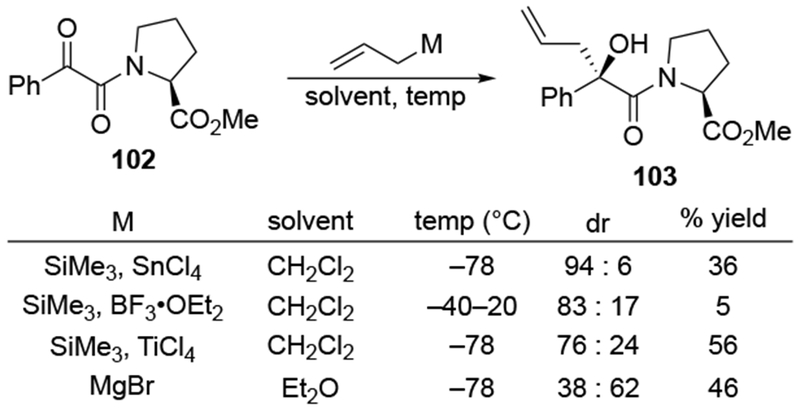
Scheme 45.
4.2.2. Additions to α-Alkoxy Acyl Silanes
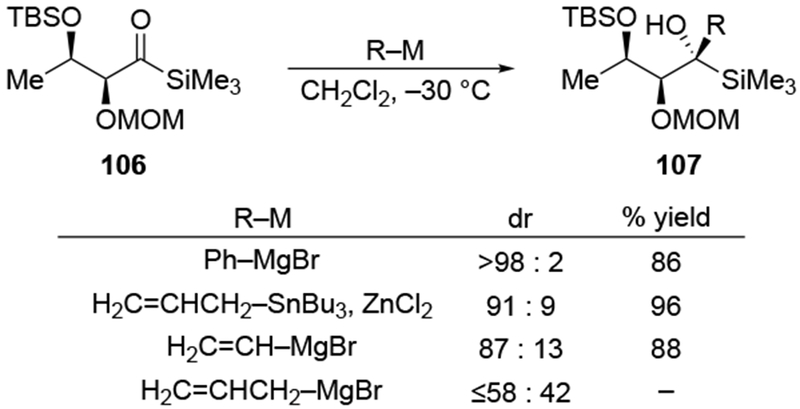
Just as with additions to α-alkoxy ketones, additions of most organomagnesium halides to α-alkoxy acyl silanes can also be achieved with chelation control (Scheme 46).119 The additions of allylmagnesium reagents, however, proceeded with little stereoselectivity. In this case, use of a different allylmetal reagent formed product 107 selectively. These results demonstrate that although acyl silanes are more electrophilic than the corresponding ketones,120 they appear to follow similar trends of diastereoselectivity.
Scheme 46.
4.2.3. Additions to β-Alkoxy Carbonyl Compounds
4.2.3.1. Additions to β-Alkoxy Ketones
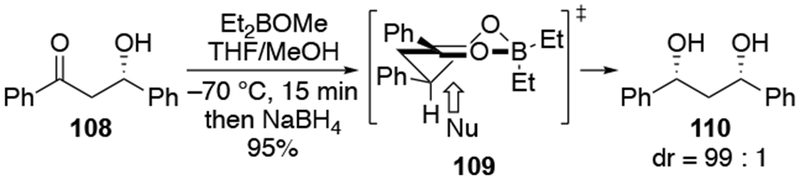
Additions of nucleophiles to β-alkoxy ketones using chelation control can be stereoselective. A particularly useful example is the stereoselective reductions of β-hydroxy ketones (Scheme 47).121 This reaction involves chelation of a boron reagent to the β-hydroxyl and carbonyl groups followed by reduction of the chelate 109 from the stereoelectronically favored face.122,123 Methods for performing chelation-controlled reductions of β-alkoxy ketones have also been reported.124
Scheme 47.
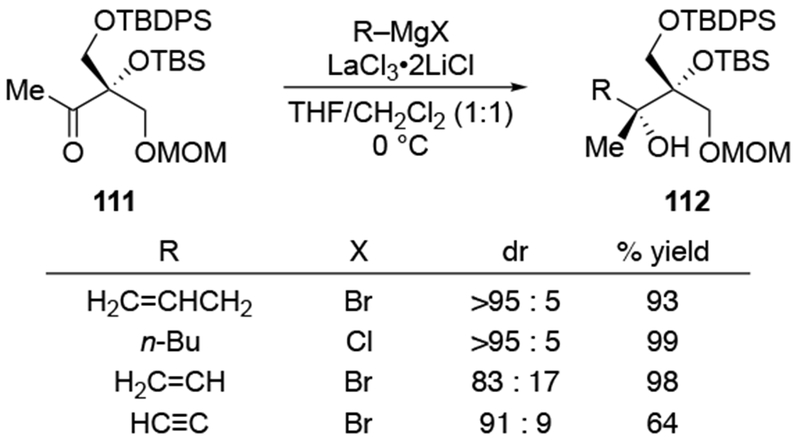
As a general rule, additions of Grignard reagents to β-alkoxy ketones are much less selective than the corresponding reactions of α-alkoxy ketones.100 Nevertheless, attempts to obtain the β-chelation product have been successful in some cases.125 In the presence of lanthanum salts, additions of organomagnesium reagents can be stereoselective in some cases (Scheme 48). Selectivity was also higher in mixtures of THF and CH2Cl2 instead of THF alone. No information was provided regarding the selectivities without the use of the metal salt and whether these conditions resulted in transmetallation to form an organolanthanum intermediate. The higher selectivity of the allylation reaction in this case may also reflect the fact that this ketone is highly hindered, so it should undergo additions more slowly, possibly below the diffusion rate limit.
Scheme 48.
Attempts to achieve chelation-controlled addition using an OMgX group at the β-position were not selective (Scheme 49).126,127 Additions of allylmagnesium halides to β-hydroxy ketones 113 and 115 resulted in low selectivity. This lack of selectivity appears to be general.128
Scheme 49.
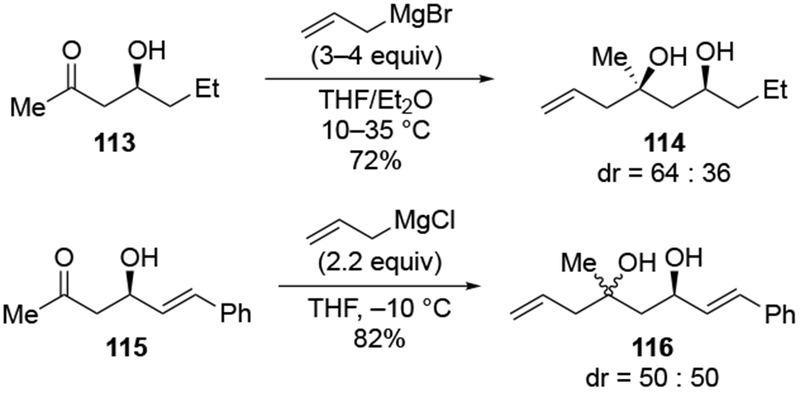
In more complex settings, however, there have been examples of remote complexation of an alkoxide anion (Scheme 50).129 In the case of hindered ketone 117, treatment with allylmagnesium bromide gave a complicated mixture of products, likely because the carbonyl and carboxyl groups reacted with allylmagnesium reagents at competitive rates.17 Deprotonation of the hydroxyl group also likely occurred at a similar rate.111 Transmetallation to zinc did allow for a stereoselective addition reaction, which the authors attributed to chelation of the remote alkoxy group of compound 117.129 It is also likely that the ketone adopts a conformation resembling 119 to minimize destabilizing steric interactions (i.e., allylic strain130) between the two carbonyl groups.
Scheme 50.
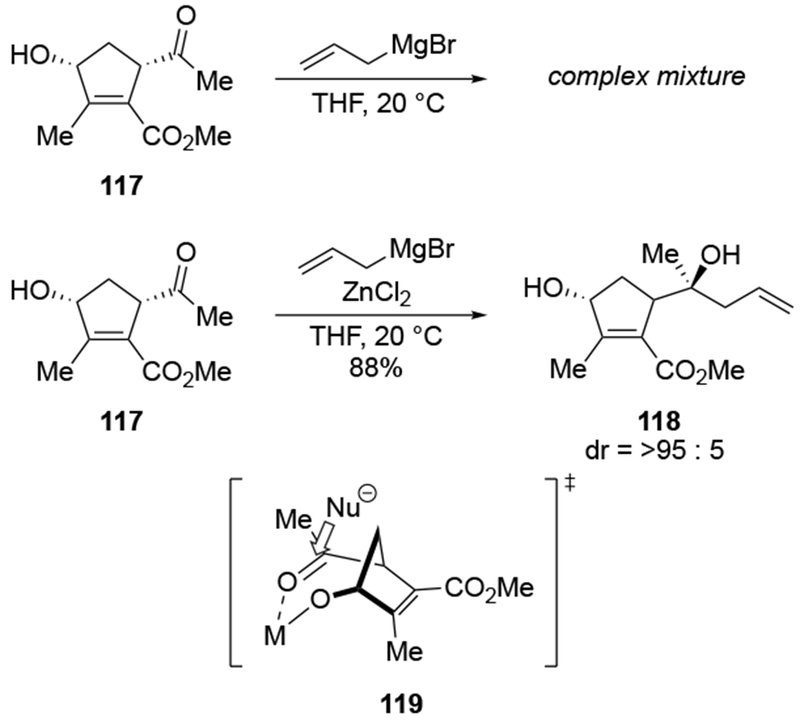
A case of β-chelation controlled addition was reported in the context of a chromium-mediated transformation (Scheme 51).131 This reaction incorporates the allyl fragment into the ring by an addition reaction onto β-alkoxy-substituted ketone 122. The product of that step, alkoxide 123, was attributed by the authors to be formed through the chelated transition state 126, where the nucleophile was delivered to the external face of carbonyl group, which faces away from the ring. Further transformations that close the ring to the five- and six-membered rings involve reactions of chromium carbenes.
Scheme 51.
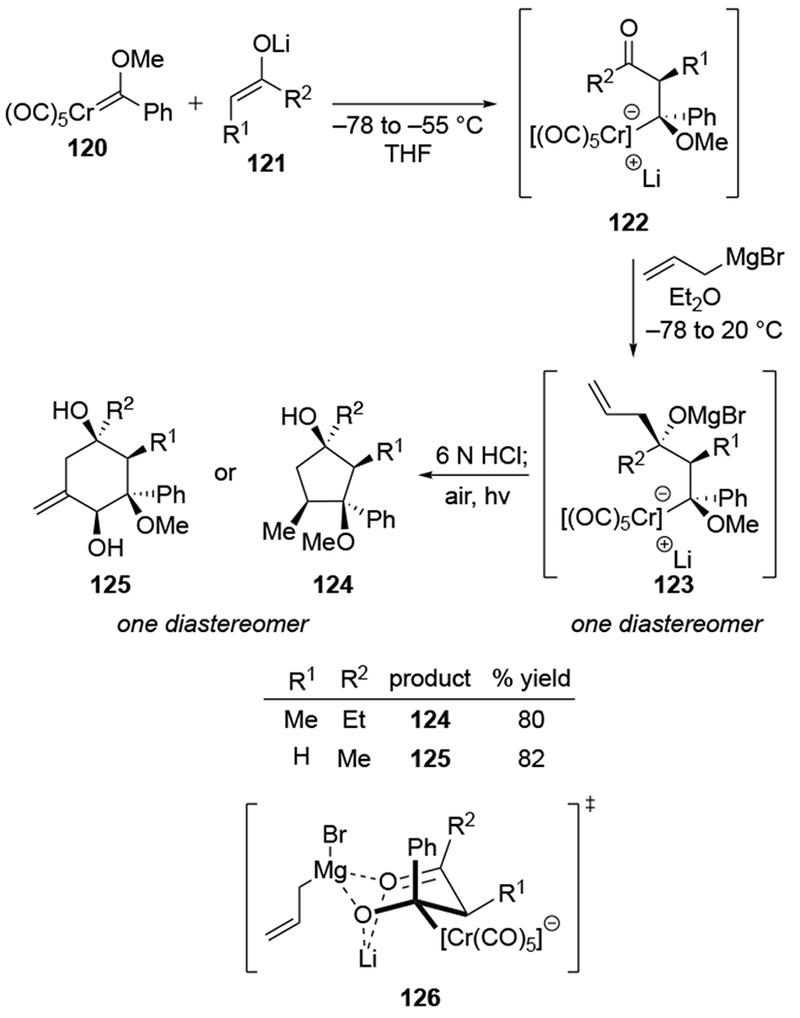
4.2.4. Additions to α-Substituted Aldehydes
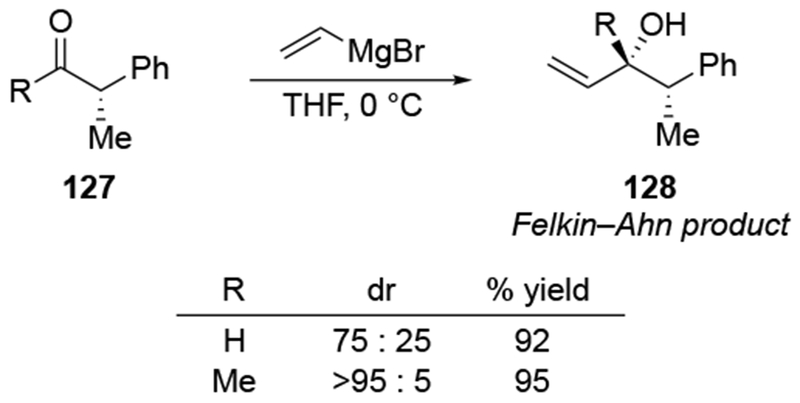
Generally, additions to chiral aldehydes are much less stereoselective than the corresponding additions to ketones, even for non-allylic Grignard reagents 22,132–134.This difference in selectivity likely relates to the significantly higher electrophilicity of aldehydes compared to ketones.17,135 The difference in selectivity can be illustrated by several examples using Grignard reagents. For example, Felkin–Anh selective additions to aldehydes are generally poorly selective, but additions to the corresponding ketones can be highly stereoselective (Scheme 52).136
Scheme 52.
A more complicated example is illustrated in Scheme 53.137 Addition of racemic alkyl Grignard reagent 129 to racemic aldehyde 130 (shown as only one enantiomer to simplify the discussion) gave a mixture of four diastereomeric products (each as its racemate). This lack of selectivity shows that the BnOCH2– protecting group, though capable of chelation, could not control the stereoselectivity of addition to the aldehyde. Upon oxidation to ketone 132, one of the stereocenters was removed, leaving only two diastereomers detectable by NMR spectroscopy. Addition of MeMgCl to ketone 132 formed only two detectable stereoisomers, which indicates that the new stereocenter was formed with high stereoselectively. The relative configuration of this product, which is consistent with chelation-controlled addition to the ketone directed by the α-BnOCH2O group, was established upon conversion of tertiary alcohol 133 into the natural product zoapatanol. The stereoselectivity of the third step (132 → 133) was not an artifact of the order of addition: reversing the order of the synthetic steps in which the Grignard reagents were added gave the tertiary alcohol 133 with the opposite configuration at the newly formed stereogenic center. That result arose from chelation-controlled alkylation of a methyl ketone intermediate (not shown). These results demonstrate that, even for alkylmagnesium reagents, chelation-controlled additions to ketones can be stereoselective while additions to aldehydes are not.
Scheme 53.

The above results reinforce observations involving allylmagnesium reagents (Scheme 54).138 Regardless of the substitution pattern on the aldehyde, stereoselectivity of addition to the α-alkoxy aldehyde was poor. The diastereoselectivity was comparable to that of a simple ketone that did not contain a chelating group (R1 = Ph, R2 = Me). Other examples confirm that additions of allylmagnesium reagents to α-alkoxy aldehydes proceed with low stereoselectivity (Scheme 55).139
Scheme 54.

Scheme 55.

Recent experiments suggest a possible explanation for why α-alkoxy aldehydes are not generally able to undergo chelation control with any Grignard reagent. Competition experiments using THF as solvent revealed that no rate acceleration occurred for additions to an aldehyde capable of forming a chelated intermediate (138) versus an aldehyde that cannot (139) for either allyl- or methylmagnesium chloride (Scheme 56). As discussed in Section 4.1, the rate acceleration observed for additions to β-alkoxy ketones was essential for diastereoselectivity because these systems operate under Curtin–Hammett control, where the majority of the product is formed through the lower-energy, chelated transition state. If rate acceleration were not present, the chelated transition state would no longer be favored, so addition could occur to any of the many forms of the aldehyde in solution (i.e., not complexed to RMgX (58), complexed to RMgX but not chelated (59), and chelated to RMgX (60), as illustrated in Scheme 24), resulting in low stereoselectivity.22 It is possible that, because reactions of allylmagnesium halides with α-alkoxy ketones are not chelation-controlled due to the high rate constants associated with addition reactions, reactions involving all organomagnesium halides with α-alkoxy aldehydes are generally not amenable to chelation control due to the higher reactivity of aldehydes compared to ketones.17,88
Scheme 56.

4.2.4.1. Use of Other Metals to Increase Selectivity of Allylation
A commonly used strategy to solve the problem of low stereoselectivity for allylation of aldehydes using allylmagnesium halides is to use other allylmetal reagents. Considering that this topic was reviewed in considerable detail,1,20 only some foundational studies will be described here. The high diastereoselectivity that can sometimes be obtained in reactions of α-alkoxy aldehydes with a number of different allylmetal reagents is illustrated in Scheme 57.97,138,140,141 Transmetallation can be effective for providing products with high chelation-controlled diastereoselectivity, and the use of allylsilanes or allylstannanes in the presence of Lewis acids provides another alternative. This solution can be general (Scheme 58).142 The preference for the Felkin–Anh product in the case of the allylboron nucleophile (pin = OCMe2CMe2O) reflects the fact that the boron atom cannot accommodate the extra coordination required for observing chelation-controlled addition.
Scheme 57.

Scheme 58.

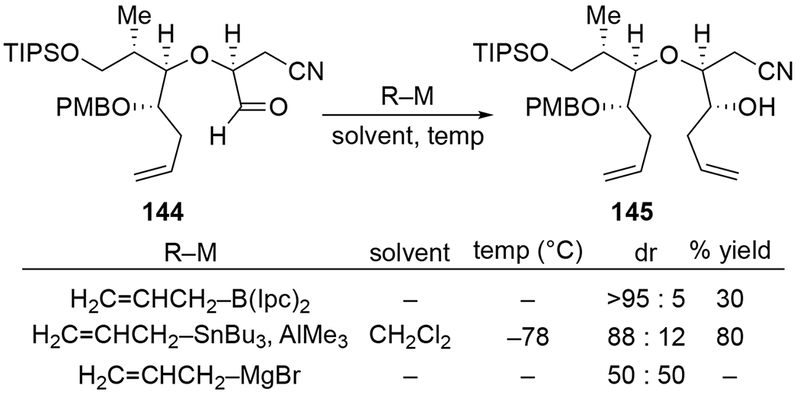
Related studies show that the use of other allylmetal reagents can be effective to control the diastereoselectivity of additions to α-alkoxy aldehydes (Scheme 59).143 Allylmagnesium bromide added to aldehyde 144, but the product was formed with little stereoselectivity (reaction conditions were not provided). Attempts at allylation using allyltrimethylsilane gave no reaction, but addition of allyltributylstannane with AlMe3 as a Lewis acid gave good diastereoselectivity. A common method to performing stereoselective allylations involves using a chiral boron reagent,144 but in this case that reagent did not add efficiently.
Scheme 59.
4.2.4.2. Use of Non-Coordinating Solvents and Salts to Improve Selectivities of Allylation
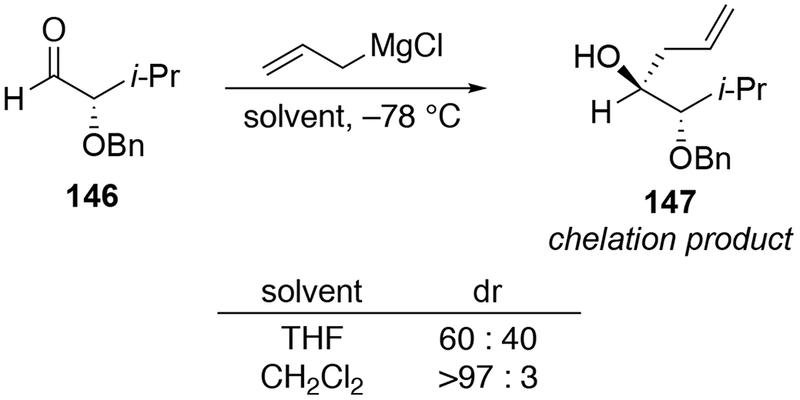
As with ketones, non-coordinating solvents such as CH2Cl2 can be used to obtain the chelation-controlled product in the absence of chelation-induced rate acceleration (Scheme 60).22 Addition of a suspension of allylmagnesium bromide in CH2Cl2 to aldehyde 146 was able to form alcohol 147, the product expected by chelation control, as a single diastereomer. By comparison, the addition in THF gave a mixture of diastereomers. As with α-alkoxy ketones, however, this protocol is not always successful (Scheme 61).145
Scheme 60.
Scheme 61.

Other examples of highly selective additions to α-alkoxy aldehydes using CH2Cl2 as the solvent have been reported. For both additions of vinyl- and allylmagnesium bromide to aldehyde 151, chelation-control products were formed stereoselectively (Scheme 62).146 Changing the protecting group on the hydroxyl group of 150 to a non-coordinating silyl ether led to high diastereoselectivity favoring the product with the opposite configuration (Scheme 121, Section 5.3).
Scheme 62.

Scheme 121.
As with ketones, when an α-alkoxy aldehyde is already in a noncoordinating solvent, the addition of metal salts can increase the diastereoselectivity of reactions with allylmagnesium halides.147 Whereas addition to 140 gave low selectivity without additives, the authors report high selectivity in the presence of a magnesium salt (Scheme 63).114,147 It should be noted that for the more hindered aldehyde 146 with an α-benzyloxy group (Scheme 60), the addition of magnesium salts were not necessary to observe high selectivity, but the use of CH2Cl2 as solvent was.
Scheme 63.

4.2.4.3. Allylations of α-Alkoxy Aldehydes
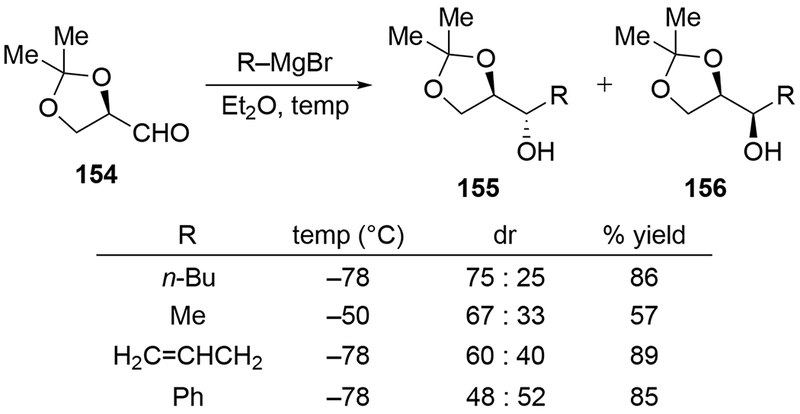
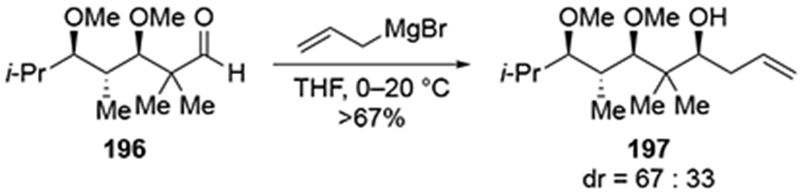
A common example of a substrate that is reluctant to undergo chelation-controlled, stereoselective additions by Grignard reagents is protected glyceraldehyde acetonide (154). Although a useful protecting group for 1,2- and 1,3-diols, the acetonide is not generally optimal for obtaining chelation-controlled addition products from α-alkoxy aldehydes (Scheme 64).99,148 Addition of a variety of Grignard reagents, including allylmagnesium bromide, to aldehyde 154 proceeded with low diastereoselectivity. This lack of selectivity was attributed to the presence of the second oxygen atom at the β-position, which could chelate through a six-membered ring transition state. As a result, better selectivities are usually observed with different protection schemes, particularly with a benzyl group at the α-oxygen atom.149,150 Additions to a more elaborated version of this aldehyde (157) were also unselective (Scheme 65).151
Scheme 64.
Scheme 65.
It is possible to observe highly stereoselective reactions of glyceraldehyde acetonide (159). Whereas no selectivity was observed above with allylmagnesium bromide (Scheme 64),148 additions of an allylzinc reagent to aldehyde 159 proceeded with high diastereoselectivity, favoring the product expected had Felkin–Anh control operated (Scheme 66).152
Scheme 66.

Generally, however, allylmagnesium reagents add to acetonide derivatives with low diastereoselectivity (Scheme 67).107 Regardless of the relative stereochemistry of the starting material, additions are unselective.
Scheme 67.

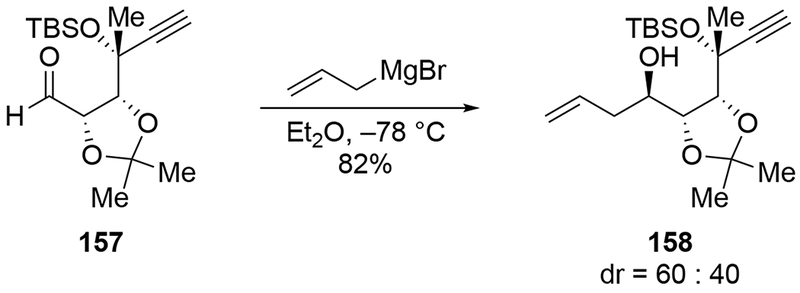
With other types of ketal-protected α,β-dihydroxy aldehydes, stereoselectivities were also low (Scheme 68).153. Despite this low stereoselectivity, the allylation reaction of aldehyde 165 has been used in natural product synthesis after separation of the desired stereoisomer from the undesired one.154,155
Scheme 68.
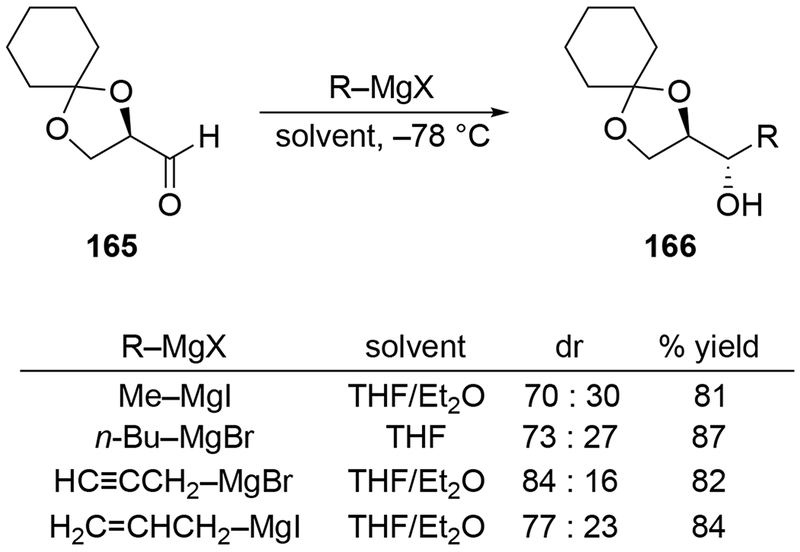
In some cases with potentially chelating alkoxy groups, allylation gave unexpected products (Scheme 69).156 Instead of forming the product expected from chelation control, addition to 167 resulted in low stereoselectivity in favor of the Felkin–Anh product. As noted earlier (Section 3), the formation of the Felkin–Anh product need not be the result of a Felkin–Anh transition state; other transition states could be responsible for formation of this product. Ultimately, achieving the desired stereochemical outcome required using organoindium reagents, which are generally useful for achieving chelation-controlled additions.157 A similar observation was made with aldehyde 170, which underwent allylation with allylmagnesium bromide unselectively (Scheme 70).158 The opposite diastereomer was obtained using an organozinc reagent.
Scheme 69.
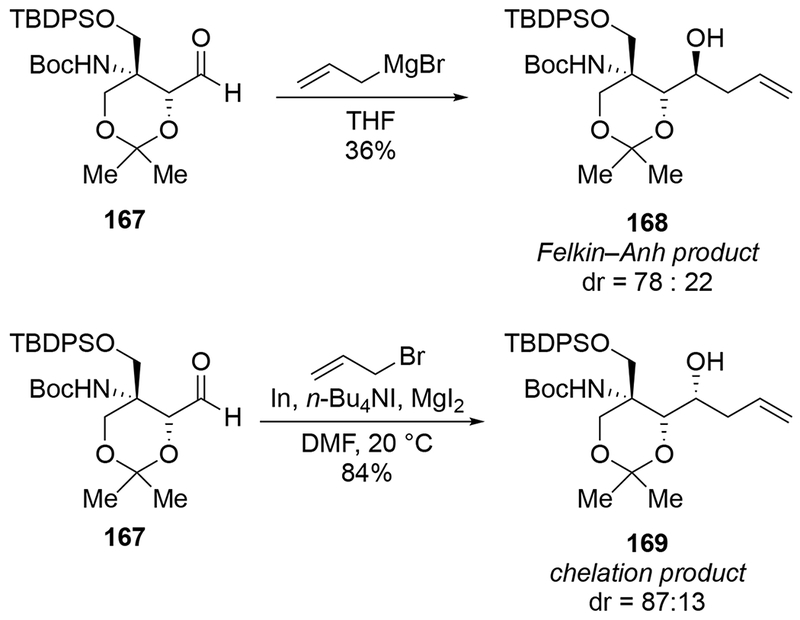
Scheme 70.
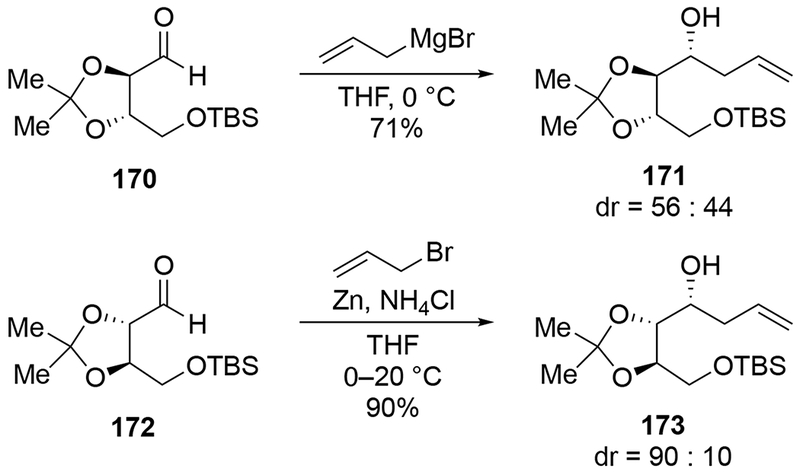
Other cyclic ethers bearing formyl groups also reacted unselectively. Additions to furanose-derived aldehyde 174 gave an inseparable mixture of diastereomers, which were carried on in a subsequent ring-closing metathesis reaction to prepare nucleoside analogues (Scheme 71).159 This type of furan generally gives low stereoselectivity for additions of nucleophiles.160,161 Low selectivity was also observed in the pyran series (Scheme 72).162 The pyran-substituted aldehyde 176 can react diastereoselectively: addition of methylmagnesium bromide was moderately diastereoselective (the stereochemistry of the major product was not assigned),163 and addition of an alkenyllithium reagent was highly stereoselective (Scheme 73).164
Scheme 71.

Scheme 72.

Scheme 73.

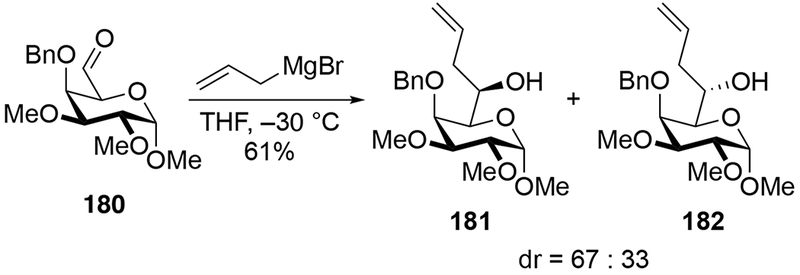
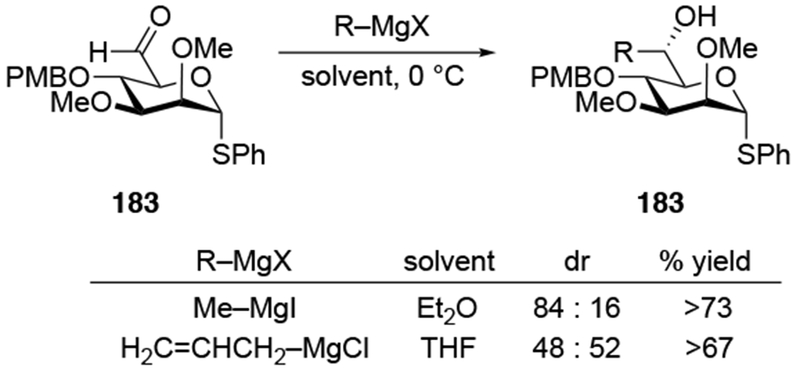
Addition of allylmagnesium halides to aldehydes derived from pyranosides show lower diastereoselectivities than additions of other organomagnesium reagents. Allylation of aldehyde 180 was not stereoselective (Scheme 74).165 Addition of methylmagnesium iodide to related aldehyde 183 gave product 184 in higher diastereoselectivity, however (Scheme 75).166 The difference in stereoselectivity was attributed to the difference in counterion for magnesium and for the difference in solvent. Comparing the results in Scheme 76 to the examples discussed in this review, however, suggest that the difference in selectivity for forming 184 is more likely the result of the nature of the nucleophile, with allylmagnesium reagents generally giving poor stereoselectivity in cases when other organomagnesium reagents are selective. Additionally, it has been demonstrated that the counterion and ethereal solvent does not significantly affect the diastereoselectivity of a reaction between Grignard reagents and α-alkoxy carbonyl compounds.22
Scheme 74.
Scheme 75.
Scheme 76.
4.2.4.4. Allylations of α-Nitrogen Atom-Substituted Aldehydes
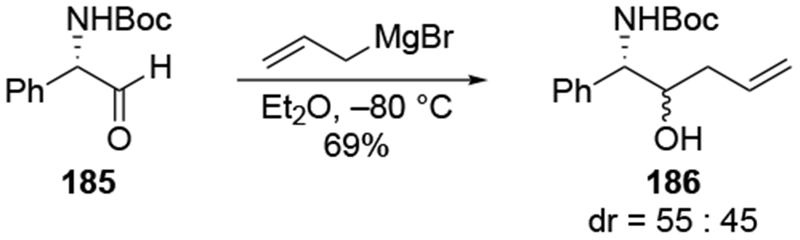
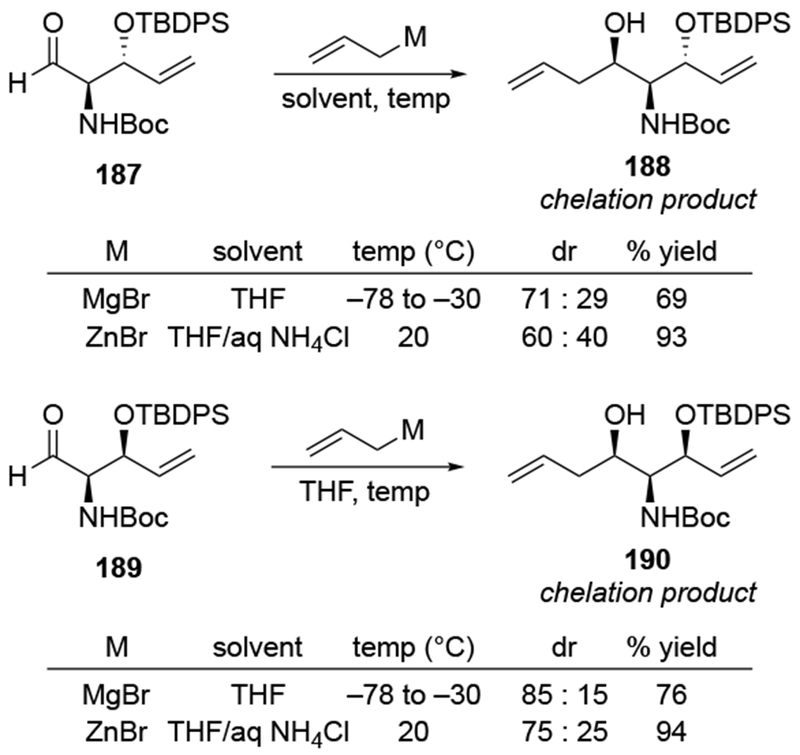
Similar efforts to achieve chelation-controlled addition of an allylmetal reagent using a nitrogen atom were met with limited success. Attempted allylation of a chiral aldehyde with a potentially chelating carbamoyl group (185) led to no selectivity (Scheme 76).167 Similarly, additions of either allylmagnesium or allylzinc reagents gave chelation-control product 188 in low diastereoselectivity (Scheme 77).168 In the case of diastereomeric ketone 189, the product expected by the chelation-control model (190) was formed with slightly higher stereoselectivity. In both of these examples, the allylmagnesium reagent formed the product more diastereoselectively than the allylzinc reagent, which is atypical (Scheme 70). These diastereoselectivities do not seem to be restricted to allylmetal reagents: additions of other organometallic reagents also proceeded with low diastereoselectivity (Scheme 78).169 Related reactions also occurred with low stereoselectivity.170, 171
Scheme 77.
Scheme 78.
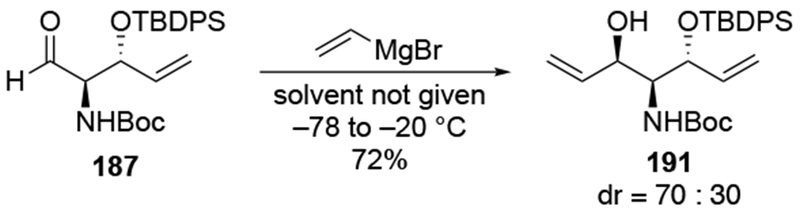
Reactions of amino sugars behaved much as additions with pyrans related to sugars (Scheme 79). Low selectivity was observed upon addition of allylmagnesium chloride to aldehyde 192.172 By contrast, allylation using allyltrimethylsilane with TiCl4 gave high diastereoselectivity. Reactivity of that nucleophile, however, depended upon the choice of Lewis acid.
Scheme 79.
4.2.5. Additions to β-Substituted Aldehydes
4.2.5.1. Additions to β-Alkoxy Aldehydes
Similar to the additions to β-alkoxy ketones discussed in Section 4.2.3.1, additions to β-alkoxy aldehydes generally proceed with little diastereoselectivity. For example, with or without a fully substituted α-carbon atom, which would cause considerable steric hindrance towards nucleophilic attack, stereoselectivity was low (Scheme 80).173 Low stereoselectivity was also observed for another sterically congested aldehyde (Scheme 81).174.
Scheme 80.
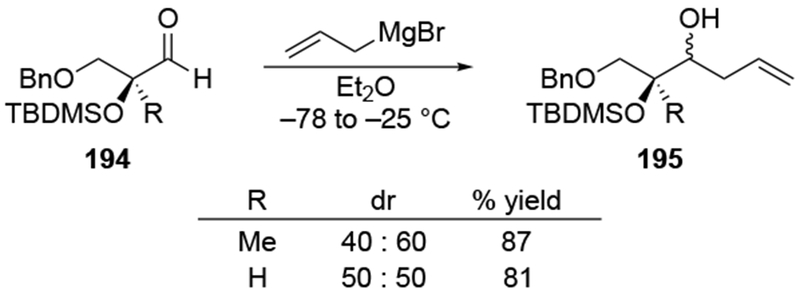
Scheme 81.
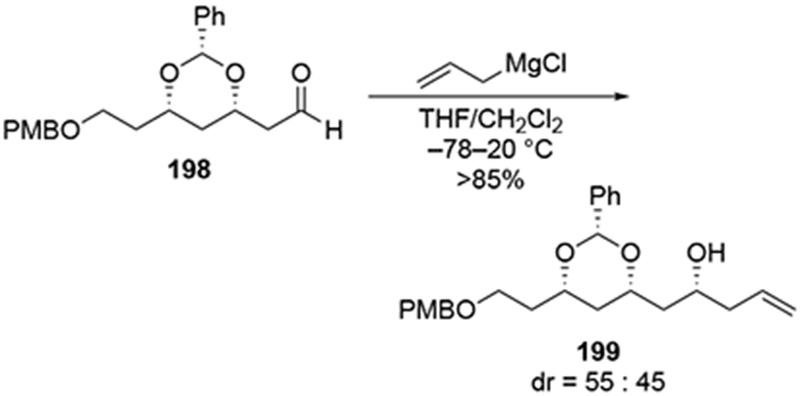
The challenges associated with allylation and the efforts to correct undesired configurations are illustrated for the β-alkoxy aldehyde 198 (Scheme 82).175,176 In the course of a natural product synthesis, it was necessary to achieve a diastereoselective synthesis of polyoxygenated products such as 199. The addition reactions of allylmagnesium halides were efficient, but they were not stereoselective. Efforts to repair this stereochemical course by oxidation of both diastereomers followed by hydride reduction were also ineffective, forming allylic alcohol 199 as an 80:20 mixture of diastereomers. The only reasonable solution was to employ chiral allyl silane reagents177 to control the new stereogenic center efficiently. Other authors have observed similarly low stereoselectivity in related systems, which could be repaired by an oxidation-reduction sequence.178 Stereochemical inversion through further elaboration to correct issues of stereochemistry is a common course of action made to remedy the low diastereoselectivity in reactions of allylmagnesium halides.
Scheme 82.
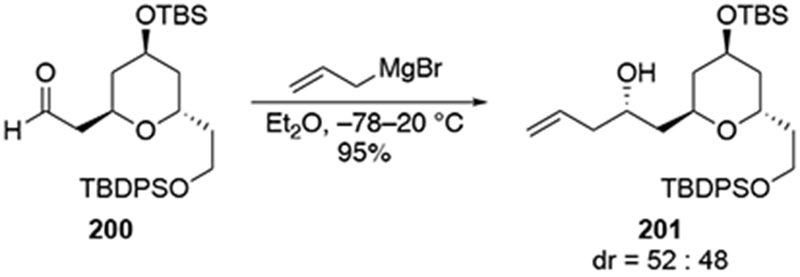
An aldehyde with a β-tetrahydropyranyl group also reacted with little selectivity in the presence of allylmagnesium bromide (Scheme 83).179 In the course of this synthesis, it was important to obtain this diastereomer selectively, but the authors note that efforts using chiral reagents to control the stereoselectivity were unsuccessful. In the end, a circuitous repair involved isolating the minor diastereomer and inverting the homoallylic configuration using a Mitsunobu reaction.180
Scheme 83.
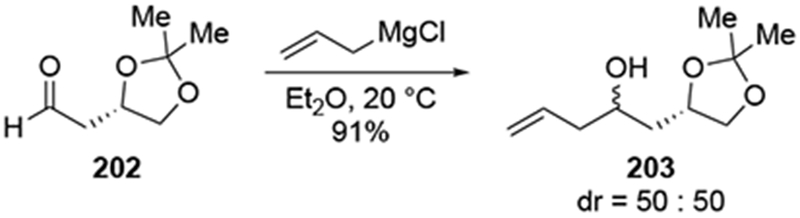
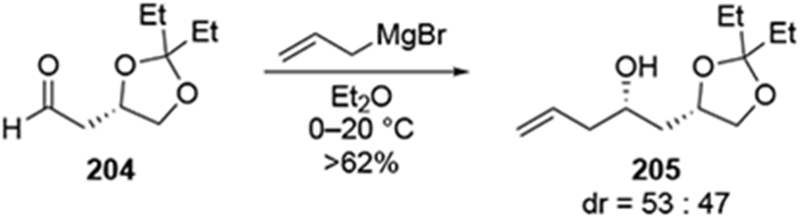
In the course of a natural product synthesis, it was shown that the allylation of a β-alkoxy aldehyde 202 by allylmagnesium chloride was unselective (Scheme 84).181 The undesired diastereomer was recycled to the desired diastereomer through a two-step sequence which inverted the configuration at the homoallylic position. Low diastereoselectivity was also observed in the addition of allylmagnesium bromide to a similar β-alkoxy aldehyde (204, Scheme 85).182 Although the use of chiral reagents (allylboranes, in this case) were effective at controlling the stereochemistry, the authors found it operationally easier, considering the scales of their reactions, to use the readily available allylmagnesium bromide despite the low diastereoselectivity that resulted. The two diastereomeric alcohols of 205 were first separated, and then the undesired stereoisomer was converted to the desired alcohol by a Mitsunobu inversion followed by ester hydrolysis.
Scheme 84.
Scheme 85.
Chelation-controlled additions to β-substituted aldehydes can be achieved using other metal salts as additives.183 The addition of ZnCl2 to the addition of aldehyde 206 led to a completely stereoselective reaction (Scheme 86). No experiment without the use of zinc salts was reported, so the outcome of addition of the magnesium reagent is not known. Although the authors did not comment on the origin diastereoselectivity, it is consistent with addition through a transition state resembling 208 where chelation occurs to the carbonyl oxygen atom in the β-position rather than the β-alkoxy group.
Scheme 86.
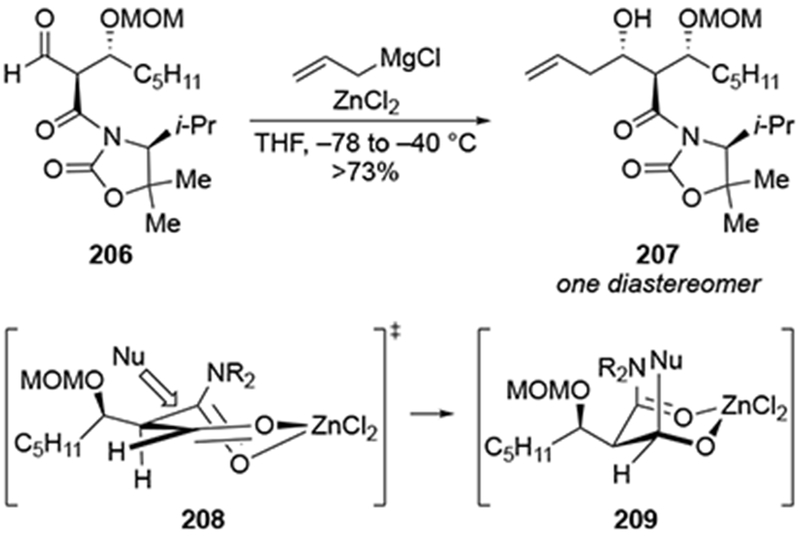
4.2.5.2. Additions to β-Silyloxy Aldehydes
Efforts toward the synthesis of a natural product and its analogues led to investigations of methods to obtain the product expected by chelation-control in the allylation of β-silyloxy aldehydes (Scheme 87).184 Chelation-controlled selectivity could not be achieved with the β-silyloxy aldehyde 210 using allylmagnesium or allylzinc reagents. By contrast, additions to aldehyde 212 were diastereoselective. The reaction with allylmagnesium chloride formed the diastereomer of product 213. This diastereomer is formally the product of addition through a Felkin–Anh transition state. Instead, this product was likely formed through a transition state resembling 214, which is consistent with the modified Cornforth model.185 This model is justified because aldehydes 210 and 212 resemble aldehydes investigated by Evans to understand how substituents at the α- and β-positions of carbonyl compounds influence the diastereoselectivities of addition reactions.186 Alternatively, in the presence of zinc salts, a chelate resembling 215 would be favored, leading to the illustrated product.
Scheme 87.
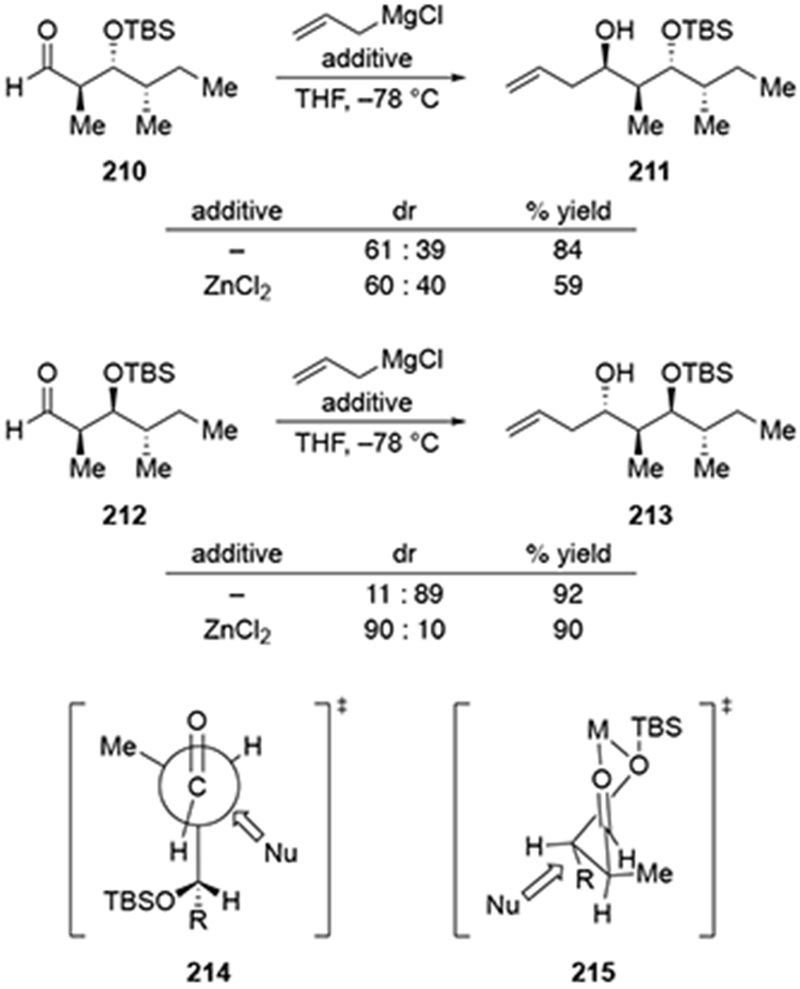
Addition to aldehyde 212 formed the desired diastereomer of 213 expected by the chelation-control model in the presence of ZnCl2. This reaction may or not involve transmetallation from magnesium to zinc, considering that the zinc salt was first combined with the aldehyde followed by addition of the organomagnesium reagent. As a result, transmetallation would need to compete with addition to the carbonyl group. Because complexation of alkylzinc halides to α-silyloxy ketones has been established,89,187 it is possible that product 213 could be obtained by chelation, as illustrated in transition state 214.
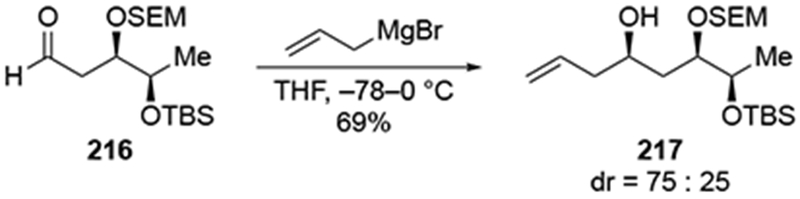
Additional experiments provide added justification for the above analysis. When a group is not present at the α-position to reinforce stereochemistry, as depicted with aldehyde 216 below (Scheme 88), allylation was less selective than the “matched” case (212) but more selective than the “mismatched” case (210) shown in Scheme 87.188 It is also worth noting that in this system the use of a chiral allylborane reagent or an allylstannane reagent in the presence of a Lewis acid also did not afford product 217 with high stereoselectivity.
Scheme 88.
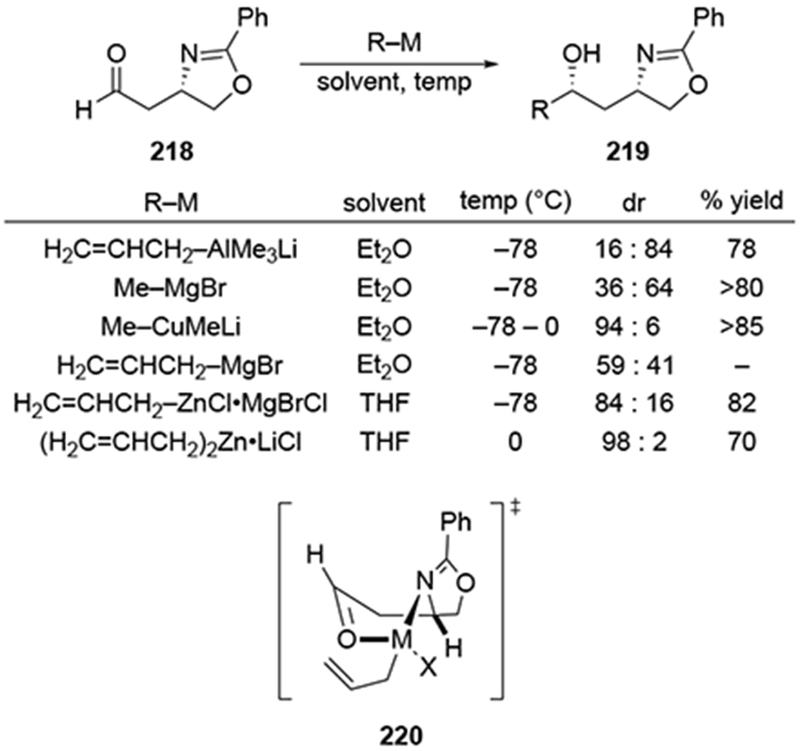
4.2.5.3. Additions to β-Nitrogen-substituted Aldehydes
An example of β-chelation was observed in the case of aldehyde 219, which contains a β-nitrogen atom capable of metal complexation (Scheme 89).189,190 Additions of both MeMgBr and allylmagnesium bromide led to low stereoselectivity. In contrast to reactions with simpler systems,191 the methyl cuprate reagent gave low diastereoselectivity. Selectivities for the allylation reaction could be improved by transmetallation to zinc. The stereochemical outcome for addition of the β-chelation product was different than for addition of an allylzinc reagent to a β-alkoxy carbonyl compound, however.192 It was argued that a boat-like chelate was favored in this system because the nitrogen atom is trigonal planar. Subsequent intramolecular delivery of the nucleophilic group would occur as illustrated in transition state 220. By comparison, the aluminate complex would react through a more standard cyclic chelating transition state, thus favoring the opposite product.
Scheme 89.
4.2.6. Additions to Aldehydes with Distant Chelating Groups
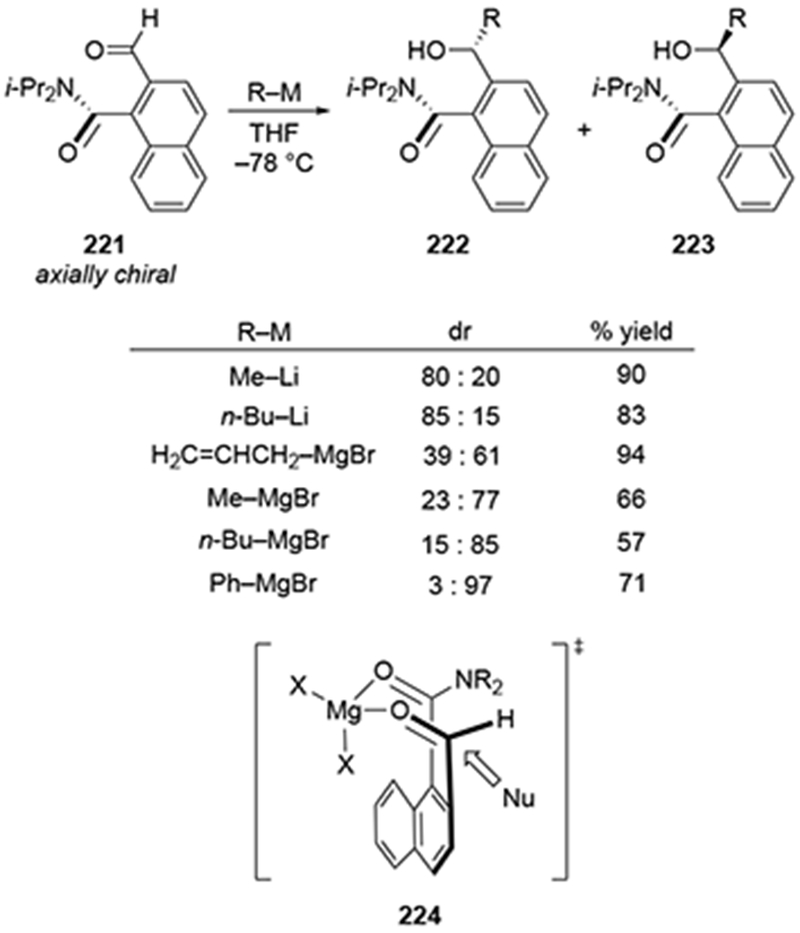
Groups other than alkoxy substituents can be used to control the stereoselectivity of addition to carbonyl compounds from remote positions using chelation. Axially chiral aldehyde 221 underwent diastereoselective reactions with a number of different nucleophiles (Scheme 90).193 In the case of organolithium reagents, it was believed that chelation was not possible, but in the presence of magnesium ions, chelation could occur (as illustrated in transition structure 224), leading to the diastereoselective formation of alcohol 222. Selectivity was moderate with methyl- and n-butylmagnesium reagents, but phenylmagnesium bromide formed 223 with a diastereomeric ratio of 97:3. As with other reactions, allylmagnesium reagents underwent this reaction with low diastereoselectivity.
Scheme 90.
4.3. Additions to Cyclic Ketones
4.3.1. Alkoxy-substituted Cyclic Ketones
The stereochemical outcome for nucleophilic addition to cyclic ketones is often influenced by distinct factors from their acyclic counterparts. Although they were among the early examples studied when stereochemical models such as the work of Felkin were originally formulated,194 these systems are more complicated than those of acyclic ketones. Bond rotation is restricted in acyclic ketones, and torsion angles and torsional strain exert strong influences upon the preferences for nucleophilic attack.71 Approach of the nucleophile to one face of the carbonyl could be favored judging by the steric environment, but torsional strain in the transition state could favor the opposing side. Also, distinct electronic effects can operate, leading to selectivities that need to be analyzed separately.65,195 It is not essential to master these subtleties to notice the disparate reactivity profile of allylmagnesium reagents compared to other Grignard reagents.
4.3.1.1. α-Oxygenated Six-Membered-Ring Ketones
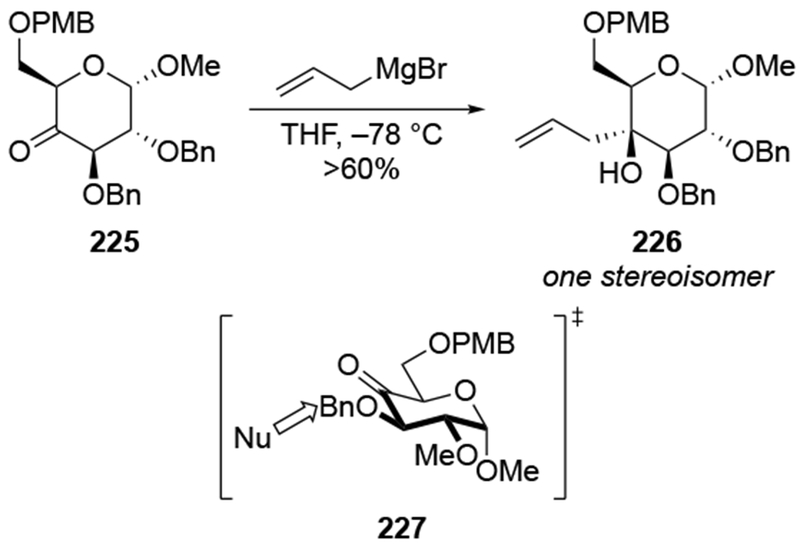
In some cases, the diastereoselectivities for allylations of alkoxy-substituted cyclohexanones can be quite high using Grignard reagents. Nucleophilic addition to highly substituted α-alkoxy ketone 225 proceeded with high selectivity for addition anti to the alkoxy group (Scheme 91).196 Whether this reaction is the result of chelation-controlled addition or simply addition to the lowest-energy conformer (227) from the sterically more accessible side cannot be established. Nevertheless, this example provides a good comparison for other highly functionalized, six-membered-ring ketones.
Scheme 91.
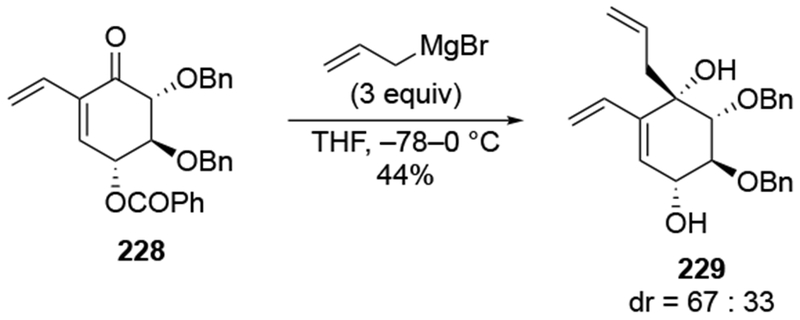
The presence of an α-alkoxy group is not sufficient to observe selectivity, however. Addition to substituted cyclohexenone 228 occurred with low selectivity for addition anti to the α-benzyloxy group, at the same time removing the benzoyl protecting group (Scheme 92).197 Nevertheless, the major isomer could be converted to a substrate that underwent an efficient intramolecular Diels–Alder reaction leading to the scaffold of a natural product.
Scheme 92.
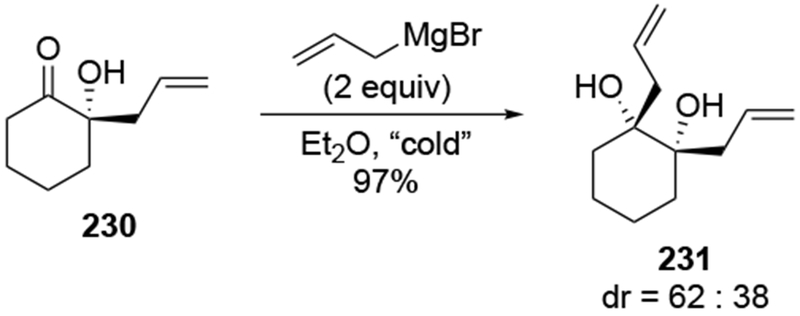
Another example suggests that allylation with chelation-controlled selectivity should not be expected in cyclic ketones. Ketone 230 (Scheme 93).198 contains an α-hydroxyl group that might be expected to undergo highly stereoselective reactions considering that the presence of hydroxyl groups can lead to high selectivity for the chelation-control product in acyclic systems (as illustrated by, for example, Scheme 36). Instead, low selectivity was observed. By comparison, additions of phenyl and methyl Grignard reagents to α-hydroxy ketone 232 were highly stereoselective (Scheme 94).199 likely by formation of chelate 234 and addition from the face not blocked by the axial phenyl group.
Scheme 93.
Scheme 94.
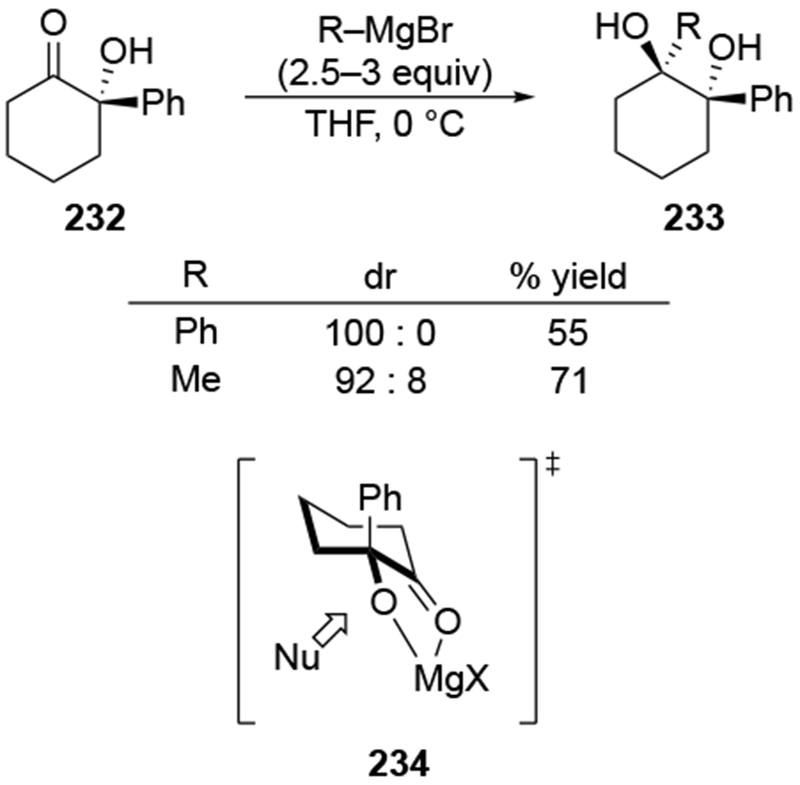
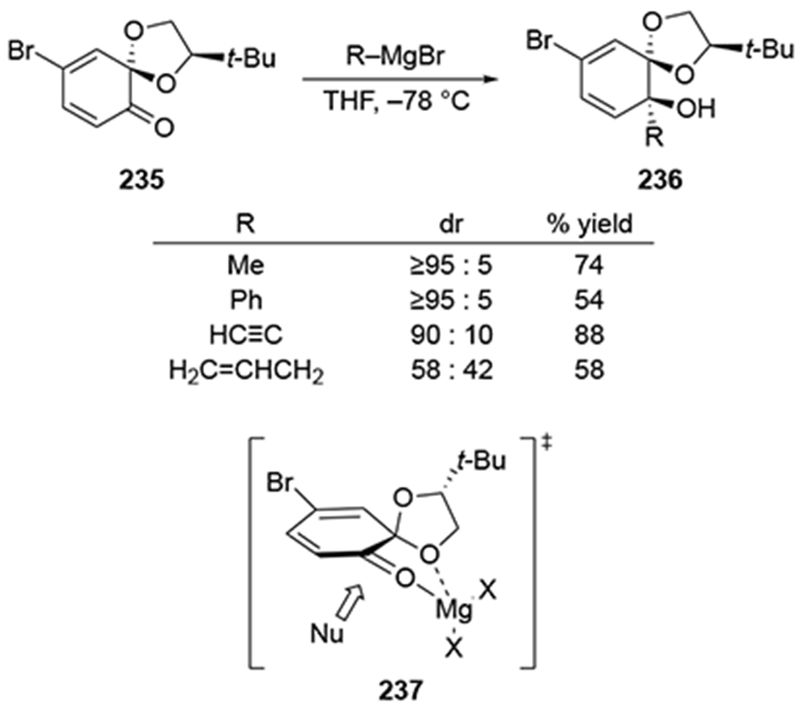
Although the examples shown in Schemes 91 and 92 did not include a direct comparison between an allylmagnesium reagent and other organomagnesium reagents, it appears that, just as for acyclic systems, allylmagnesium reagents add to cyclic ketones with low selectivity (Scheme 95).200 Additions of several organomagnesium bromides to ketone 235 occurred with high stereoselectivity, favoring a product that could arise through addition to the more accessible face of the multicyclic chelate 237. On the other hand, allylmagnesium bromide added with no stereoselectivity. This difference in selectivity mirrors other losses of diastereoselectivity with allylic organomagnesium reagents compared to nonallylic organomagnesium reagents.17,22,23 Considering that this cyclohexadienone structure is likely to be highly flattened, it might be anticipated that the two faces of the carbonyl group should show little stereochemical differentiation without chelation. Therefore, the allylmagnesium reagent, which does not benefit from rate acceleration conferred by chelation,22 added with little stereochemical discrimination.
Scheme 95.
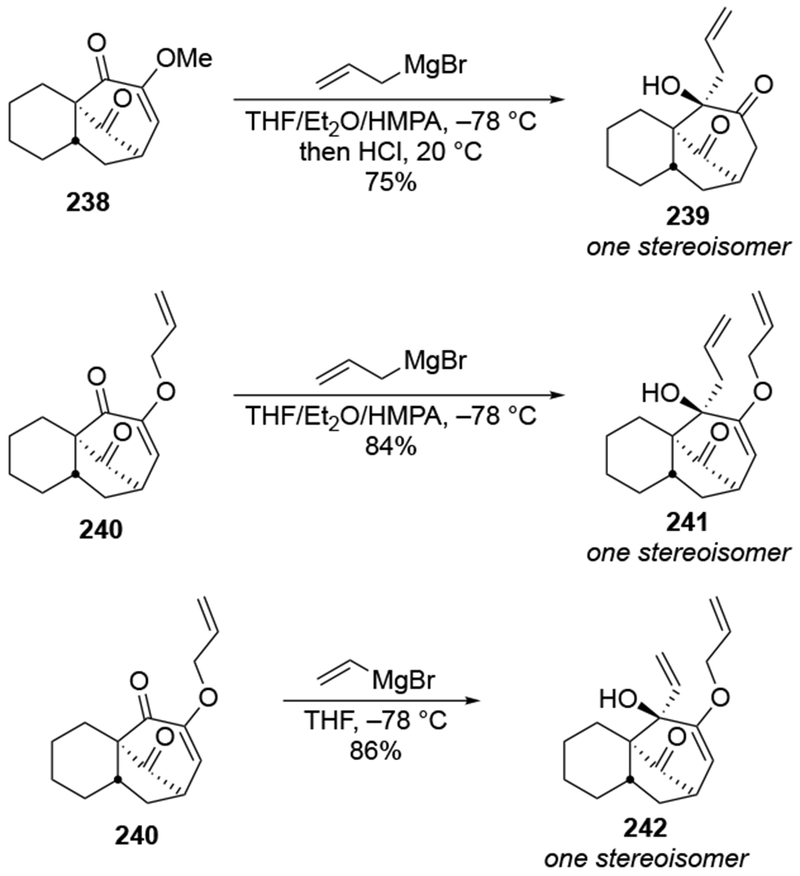
Chelation in bridged six-membered ring ketones has been invoked both to explain stereoselectivity and reactivity of ketones.201. For the bicyclic ketones 238 and 240, attack of allylmagnesium bromide occurred at the conjugated carbonyl group instead of the hindered carbonyl group on the one-atom bridge (Scheme 96). It was suggested that the addition to the conjugated carbonyl group was the result of chelation to the nearby alkoxy group, which would be consistent with the kinetic acceleration conferred by chelation.54 The authors did not discuss why HMPA was used as the solvent, which should complex tightly to magnesium, likely breaking up dimeric species202 and disrupting chelation.
Scheme 96.
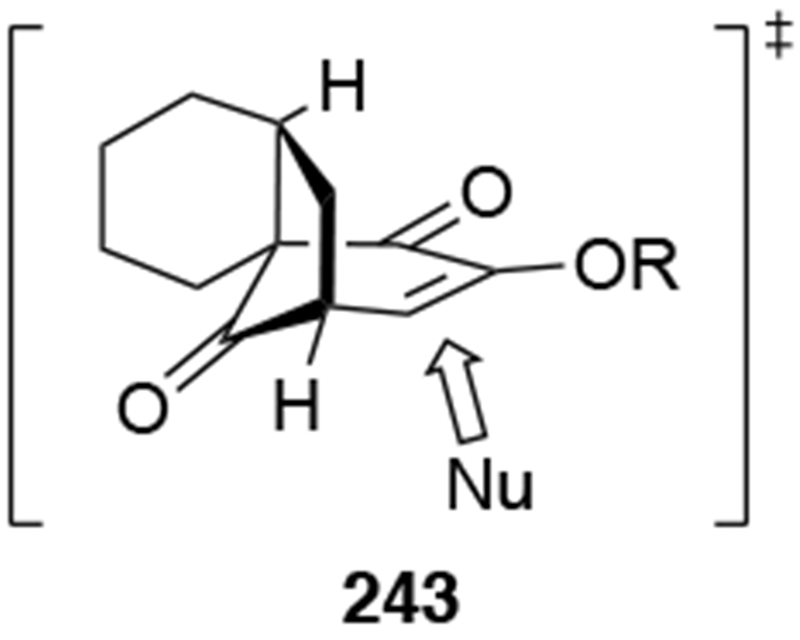
An alternative explanation for the chemoselectivity and stereoselectivity of this addition relies on the overall structure of this bicyclic ketone. The carbonyl group on the three-carbon bridge appears to be more sterically accessible than the carbonyl group on the one-carbon bridge (the reactions of allylmagnesium reagents with ketones with the carbonyl group in a bridge will be discussed in Section 6.8). Addition to the bottom face of this ketone, which should be less hindered considering that longer bridge with the additional ring occupies the top face, should lead to stereoselectivity (Scheme 97).
Scheme 97.
4.3.1.2. Additions to β-Hydroxycyclohexanones
The highly stereoselective additions of allylmagnesium chloride to cyclohexanones with β-hydroxyl groups illustrates that chelation by a hydroxyl group is possible (Scheme 98).203 The planned synthetic route required that the allyl groups were installed stereoselectivity so that subsequent ring-closing metathesis reactions of the two allyl groups could form additional rings. In general, these additions of allylmagnesium halides to the hindered cyclohexanones were stereoselective. It is possible that addition of an allylmagnesium reagent to the highly hindered cyclohexanone 244 is slower than diffusion and possibly slower than deprotonation of the hydroxyl group. While the reaction rates of allylmagnesium reagents with sterically crowded carbonyl compounds has been observed to decrease,17 competitive deprotonation versus addition has only been examined for relatively unhindered aldehydes and ketones.111 In the case of cyclohexanone 244, the strongly chelating OMgX group could control selectivity (as discussed in Section 4.2.1.1) through transition state 246. Addition of a substituted allylic organomagnesium reagent, crotylmagnesium chloride, to cyclohexanone 244 in the presence of CeCI3 (Scheme 99)203,204 also occurred with high stereoselectivity with respect to which face of the carbonyl group the reagent reacted, although control of the configuration at the exocyclic position was low (as observed in other systems and discussed in Section 6.2.1.4). With epimeric ketone 248, the reaction also showed high selectivity for addition anti to the hydroxyl group, which can be explained by chelation to this β-oxygen in the axial position (250, Scheme 100). It was also noted that addition to benzoate 251 favored the same diastereomer of product (Scheme 101),204 although with lower selectivity (and the acyl group had been removed in the reaction, which is consistent with the competitive rates of addition of allylmagnesium reagents to ketones and esters17).
Scheme 98.
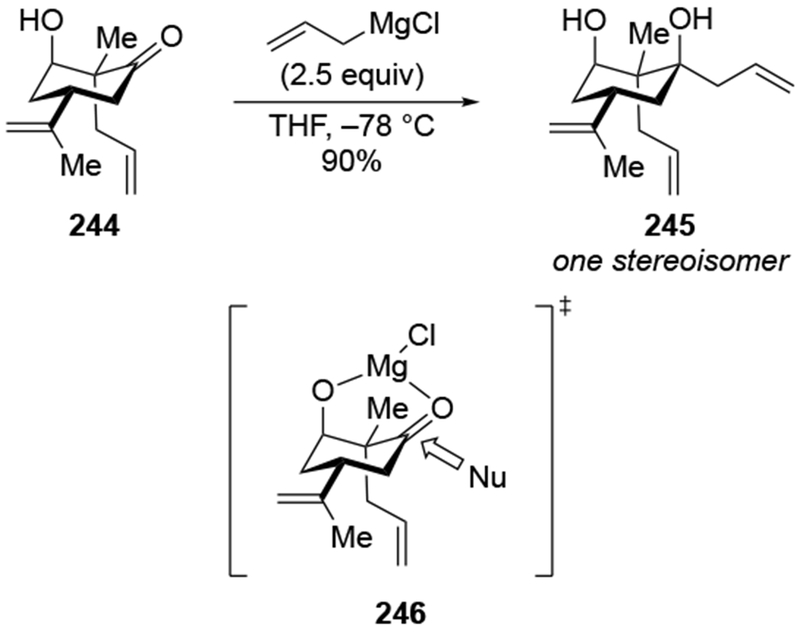
Scheme 99.
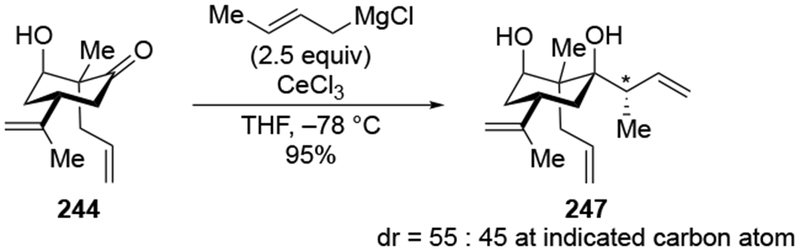
Scheme 100.
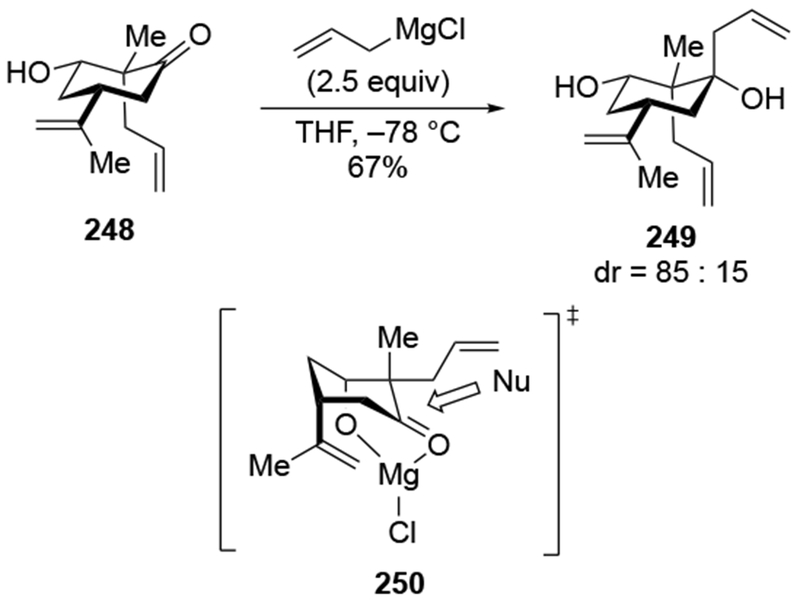
Scheme 101.
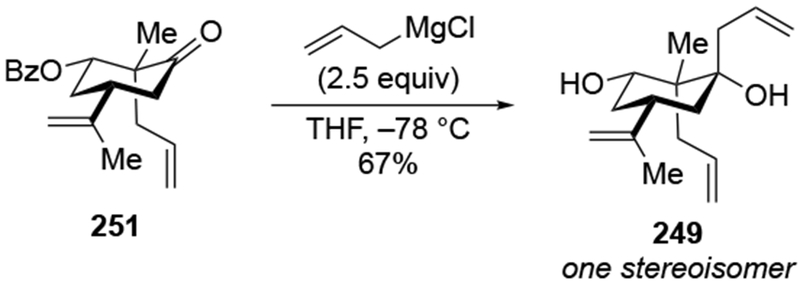
The high selectivity obtained in the examples shown above would be particularly surprising if controlled by chelation, considering that chelation would require a bridged bicyclic structure as an intermediate. It is not clear that a chelate can be formed considering that complexation would have to occur on the π-face of the carbonyl group, not in the σ-plane. It may be that the role of the OMgCl group is more electrostatic: in the transition state, partial negative charge on the carbonyl oxygen atom may be electrostatically stabilized by the nearby magnesium atom, lowering the energy of this transition state. Nevertheless, these examples illustrate that remote chelation may be possible.
4.3.1.3. Additions to γ-Substituted Cyclohexanones
The idea of remote chelation is not general. A remote amido group in an unhindered cyclohexanone could not control selectivity by chelation (Scheme 102).205 The low selectivity observed for addition to ketone 252 is similar to what was observed for 4-tert-butylcyclohexanone194 (which will be discussed in Section 6.2.1).
Scheme 102.
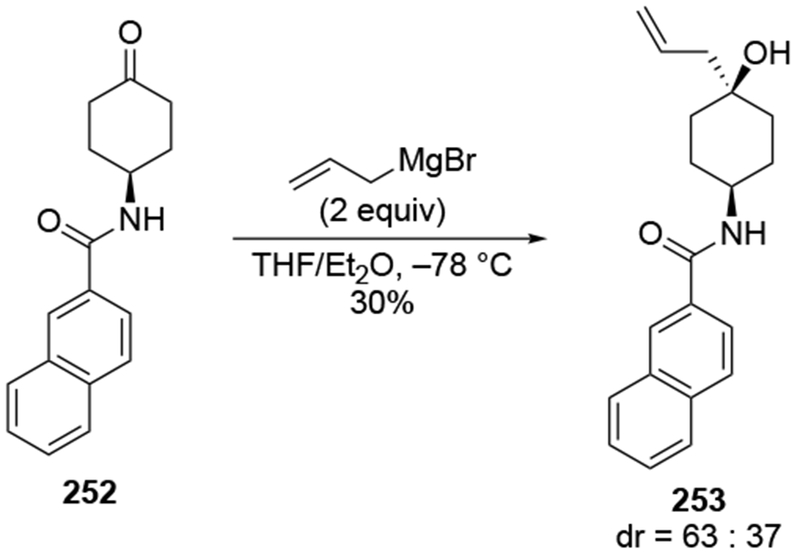
4.3.2. Additions to Alkoxy-substituted Five-membered Ring Ketones
As with highly substituted cyclohexanones bearing α-alkoxy groups (Scheme 91), additions of organomagnesium reagents to α-alkoxycyclopentanones occurred anti to the alkoxy group. In the case of ketone 254 (Scheme 103), only one diastereomer of product was observed upon additions of both vinyl- and allylmagnesium chloride.206 If the transformation shown below were controlled by chelation, it is not clear whether chelation using the α-benzyloxy or α-methoxy group would be more favorable. This stereoselective addition reaction was used in the development of a synthesis of nucleoside inhibitors (Scheme 104).207 This example provides additional evidence that stereochemical outcomes do not depend upon whether Et2O or THF is employed or what the counterion is for the organomagnesium reagent.22
Scheme 103.
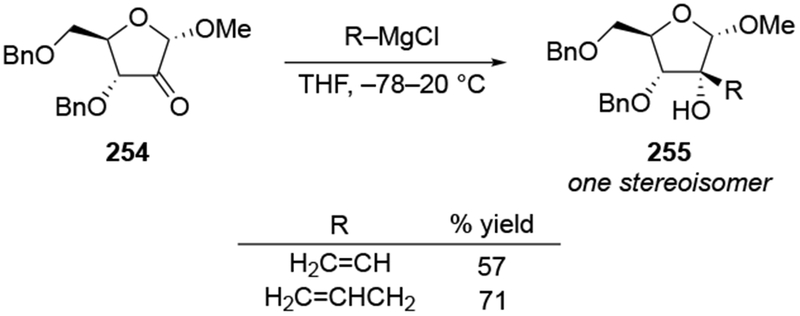
Scheme 104.
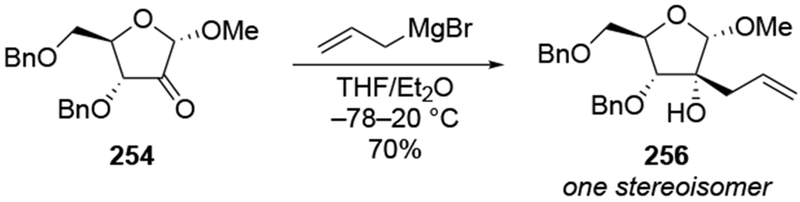
The stereochemical outcome is more difficult to explain in the case of a more highly substituted cyclopentanone, which was an intermediate in the synthesis of a carbohydrate (Scheme 105).208 Ketone 257 possesses an α-hydroxyl group, which should provide an effective chelating group (OMgX), but the diastereoselectivity for the addition to 257 using an allylmagnesium reagent was low. High selectivity was obtained using an allylic silane in the presence of a Lewis acid, which may chelate the carbonyl group and the hydroxyl group.
Scheme 105.
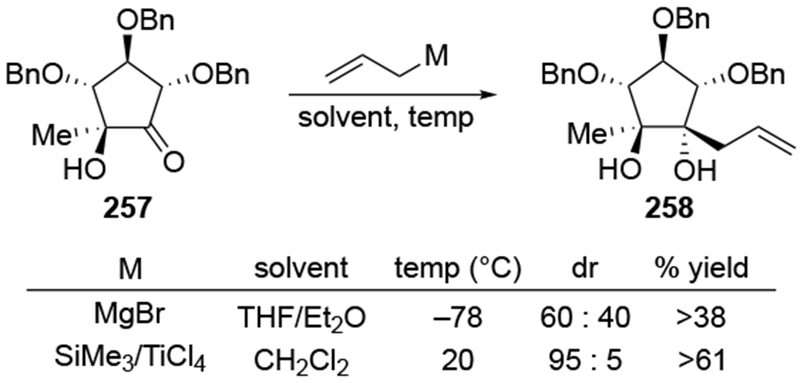
4.3.3. Additions to Alkoxy-substituted Four-membered Ring Ketones
Additions of allylmagnesium reagents to substituted cyclobutanones do not appear to follow a consistent trend. Addition to cyclobutanone 259, which contains a naphthylamido group, proceeded with low stereoselectivity (Scheme 106), which is similar to the selectivity observed for reactions of 4-substituted cyclohexanones (Scheme 102).205 In a later paper from the same team, the product of addition to carbamate analogue 261 was identified as a single stereoisomer (Scheme 107).209 This seeming contradiction may not be chemically significant. In the latter paper, unlike the former one, diastereoselectivity was not discussed explicitly, and the analytical data provided may describe only the desired stereoisomer, and not the stereoisomeric product.
Scheme 106.
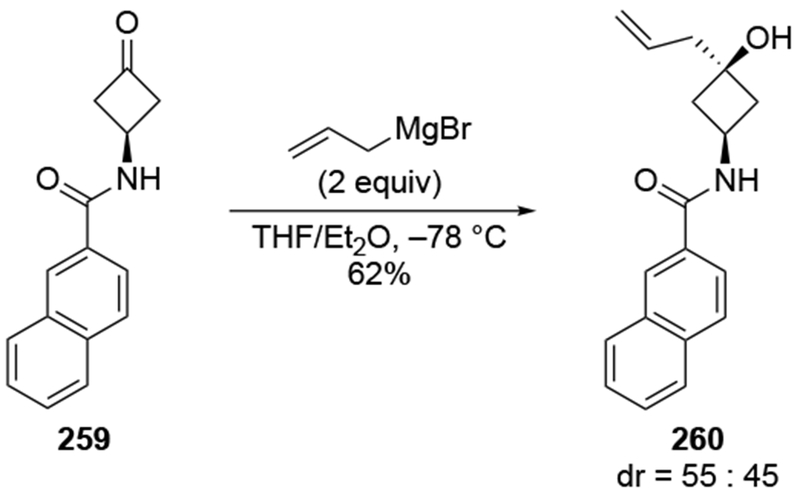
Scheme 107.
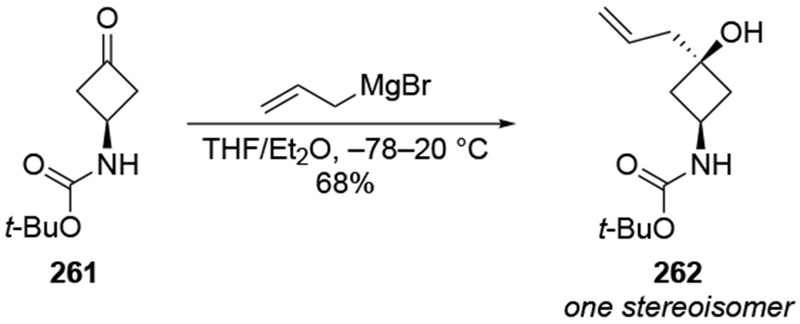
4.4. Additions of Allylmagnesium Halides to Cyclic Hemiacetals
Reactions of cyclic acetals add a mechanistic ambiguity to the general difficulties of analyzing addition reactions of allylic Grignard reagents. Hemiacetals exist in an equilibrium between the ring-closed form 263, which should be the favored structure,210 and ring-opened form 264, an aldehyde with a remote alkoxide group (Scheme 108). Although there might only be a small amount of the aldehyde form in solution, this intermediate would likely be the electrophile reacting with the Grignard reagent.
Scheme 108.
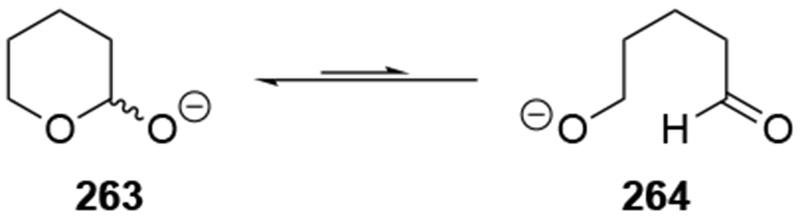
Experiments with a six-membered ring acetal are consistent with this analysis.211 For stereoselectivity to occur, addition to the ring-opened aldehyde form of 265 would need to be controlled by either a β-alkoxy group or a δ-OMgCl group. The β-alkoxy group would not be expected to control stereoselectivity (as discussed in Sections 4.2.5.1), and the more distant group could not, either.
Hemiacetals also undergo reactions faster with allylmagnesium halides than other reagents (Scheme 110).212 Attempts to open acetal 267 with either vinylmagnesium chloride or vinyllithium failed to give any product. By contrast, addition of allylmagnesium chloride gave a high yield of addition product 268, although with low diastereoselectivity. It is possible that only a small amount of the hindered aldehyde 269 is present, and thus the overall rate of addition would include both the rate constant for the carbon–carbon bond formation and the concentration of the electrophile, which would be quite low. The observation of any addition products would require a highly reactive nucleophile to trap the small amount of the aldehyde electrophile. The authors also note that the same reaction for a less hindered acetal (270) was successful with allylmagnesium chloride, but diastereoselectivity was again low for adduct 271 (Scheme 111).
Scheme 110.
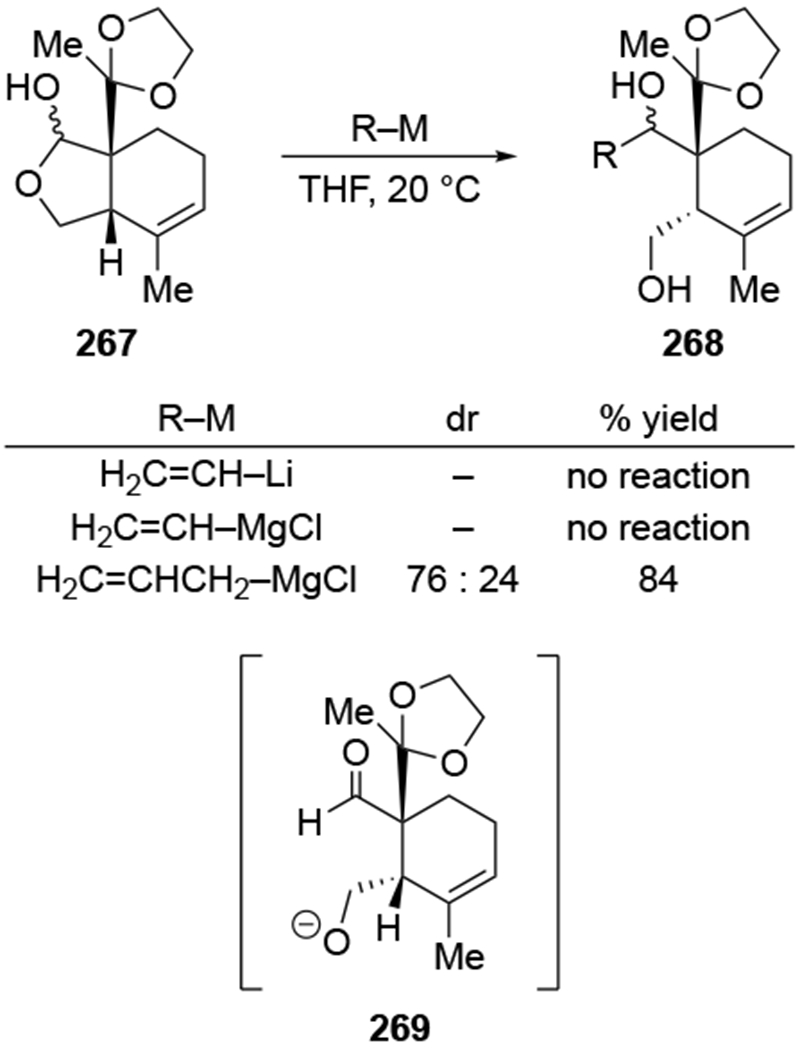
Scheme 111.
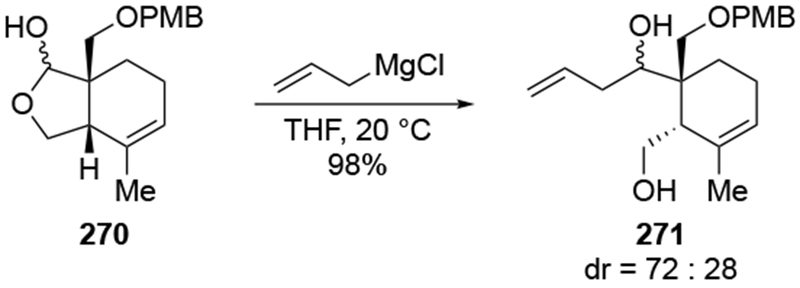
In more structurally rigid electrophiles, however, acetal substitution reactions with allylmagnesium reagents can be highly stereoselective (Scheme 112).213 Ring-opening of acetal 272 occurred with high stereoselectivity for several organomagnesium reagents, including allylmagnesium bromide. In this case, the bridged hemiacetal alkoxide may open more readily than for the acetal in Scheme 110 because of ring strain. The increased concentration of the electrophilic aldehyde form would then allow for additions of alkyl- and alkenylmagnesium reagents to proceed. Subsequent chelation through an intermediate resembling 274 may control the stereochemistry of the observed product. Such an intermediate bears some structural characteristics of the putative remote chelation by OMgX substituents discussed above (Section 4.2.1.1).
Scheme 112.
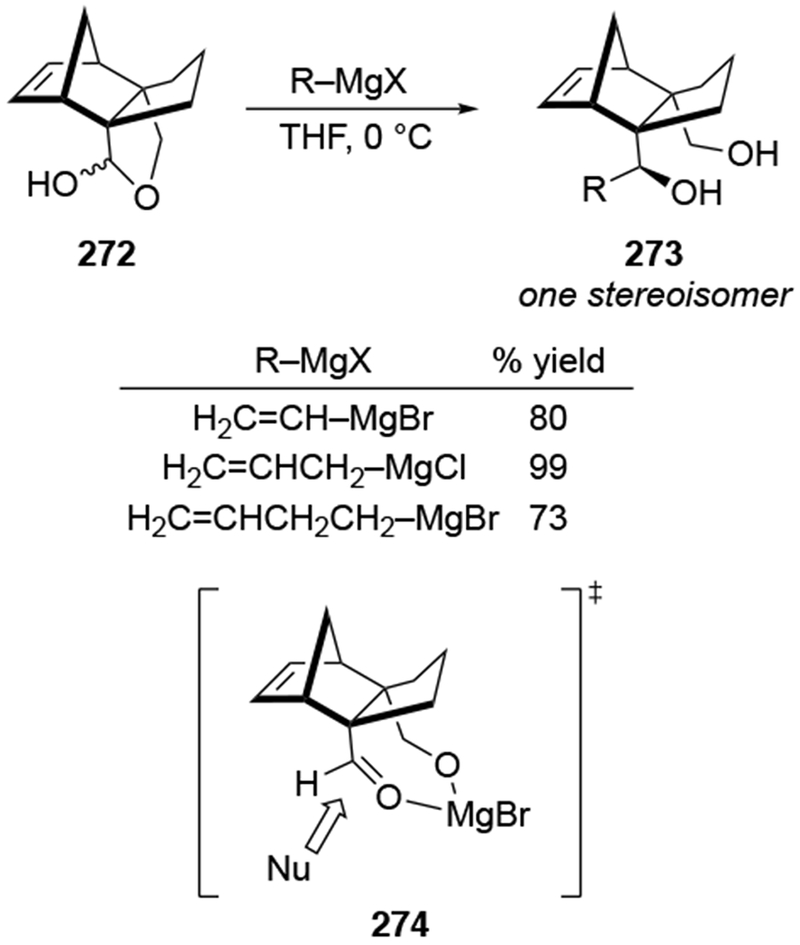
5. Diastereoselectivity of Reactions of Allylmagnesium Reagents with Carbonyl Compounds by Felkin–Anh Control
5.1. Felkin–Anh Stereoselectivity
The use of Felkin–Anh-type stereochemical models to explain selectivity must be accompanied by a similar level of analysis as was applied to the use of the chelation-control model (Section 4.1). The “anti-Felkin–Anh” product is, in a sense, the chelation-controlled product, so determining which model correctly describes the pathway through which a particular product is formed can be a subjective decision. Some assumptions upon which transition state models such as the Felkin–Anh model rely have been discussed in detail in Section 3 above, so they will not be elaborated here. In this section, reactions of α-chiral carbonyl compounds will be analyzed using the Felkin–Anh model, which will also include the polar Felkin–Anh variant (where RL is an electron-withdrawing group, not simply a sterically large group). These reactions could be analyzed differently using a myriad of the existing variants of the Felkin–Anh and related models,56,57 but the specific origin of selectivity will be less the focus than the similarities and differences exhibited by allylic and nonallylic organomagnesium reagents. As will emerge, the specific transition state determining the favored stereochemical outcome can be difficult to ascertain for many reactions. This section also focuses on reactions that likely do not involve chelation control, although, at times, the choice of stereochemical model to explain a reaction outcome can appear arbitrary.
Just as with chelation control, the Felkin–Anh model considers the influence of steric destabilization on which transition state is favored. In examples that do not include α-heteroatom-substituted carbonyl compounds, the group that occupies the RL position is determined by steric effects. In that case, the incoming nucleophile will approach the electrophile as far away from RL as possible to minimize steric destabilization in the transition state. Consequently, nucleophilic attack occurs from the face away from RL (Scheme 113).63 Two possible configurations place RL opposite the approach of the nucleophile (275 and 276). The lower energy transition state, 275, places the least sterically encumbered group in the RS position because the nucleophile will attack near that group, developing interactions that are somewhere between gauche and eclipsed.214 Consequently, steric approach considerations also play a significant role in determining which of the two diastereotopic faces of the carbonyl group is most reactive.
Scheme 113.
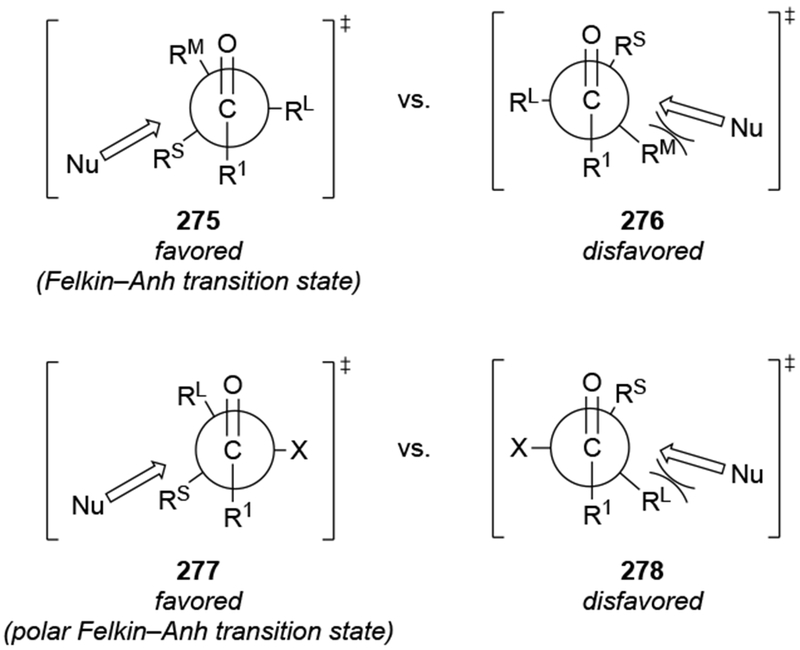
The transition state proposed by Felkin was confirmed by Anh and Eisenstein, who proposed that alignment of the π-system of the carbonyl group with the σ*C-RL orbital lowered the energy of the lowest unoccupied molecular orbital, thus favoring the Felkin transition state 277.64 These studies also provided insight into how the electronic nature of substituents can influence stereoselectivity. Electronic interactions involving the developing bond between the nucleophile and the carbon atom and orbitals associated with the C–X bond lead to transition states with different energies due to how favorable certain filled–unfilled orbitals interactions are in each potential transition state. For substrates containing a heteroatom at the α-chiral position, these orbital interactions define which group resides in the RL position.
5.2. Additions to α-Chiral Acyclic Ketones
One example of stereoselective addition of an allylmagnesium nucleophile to a chiral ketone can be attributed to a Felkin–Anh-type transition state. Allylation of ketone 279 using allylmagnesium chloride formed homoallylic alcohol 280 with high stereoselectivity (Scheme 114).215 The selectivity can be understood by examining transition state 281, with the phenyl group serving as the large group (RL). The selectivity of this reaction may not be exclusively due to 281 serving as the lowest energy transition state, however. The presence of two sterically large aromatic rings on both sides of the ketone likely causes the conformer 281 to be the lowest energy conformer in solution, resulting in only one available site of attack even at the diffusion rate limit. The idea that this conformation is favored is supported by X-ray crystal structures of α-phenyl-substituted phenyl ketones that adopt conformers resembling 281.216–220
Scheme 114.
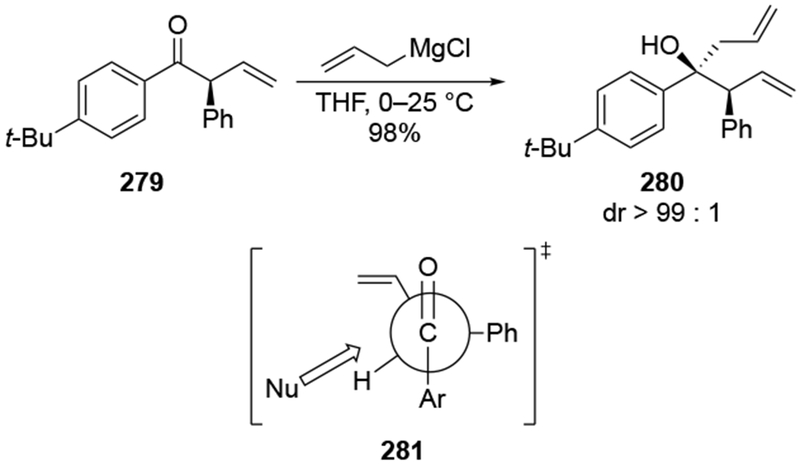
Analysis of another highly stereoselective reaction illustrates the subjectivity of interpreting stereochemical outcomes. Addition of allylmagnesium chloride to α-fluoroketone 282 proceeded with high stereoselectivity (Scheme 115).221 Similar selectivity was reported for addition of an aryllithium reagent to ketone 282. These examples could be analyzed only considering the steric size of the substituents, just as Scheme 114 was. The phenyl group could serve as RL, with attack over the small fluorine atom leading to the major product through transition state 285. Alternatively, the F substituent could adopt the position of RL (as in the polar Felkin–Anh model), leading to attack over the smaller benzyl group, as shown in transition state 286. The ability of a fluorine atoms to serve as the RL group in the polar Felkin–Anh model (by virtue of the relatively low energy of σ*C-F) may be operative, although other effects could operate as well.185,222–224 The results of the additions reported in Scheme 115 cannot distinguish between transition states 285 and 286 because both pathways would lead to the major product observed in these reactions.
Scheme 115.
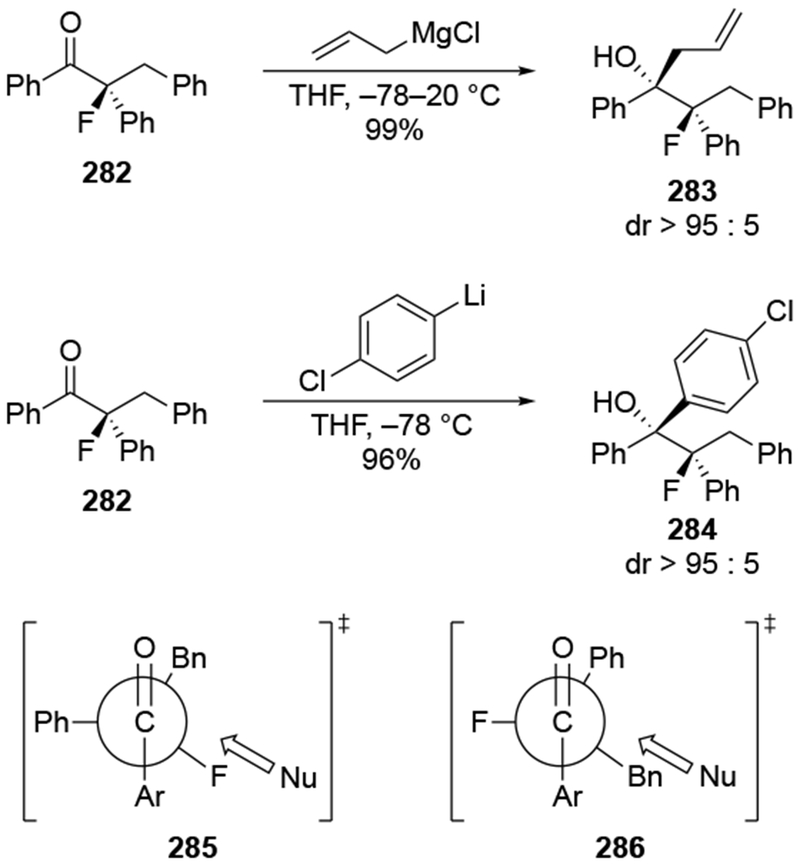
Less sterically congested substrates, however, underwent less stereoselective addition reactions of allylmagnesium reagents compared to additions of other organomagnesium reagents. For most organomagnesium reagents, addition to methyl ketone 287 was stereoselective, except for the addition of phenylmagnesium bromide, which was considered to be a small nucleophile (Scheme 116).136,225 The stereoselectivities of these reactions are consistent with the transition state 289. Additions of allylmagnesium reagents to methyl ketone 287 and trifluoromethyl ketone 291, however, were less stereoselective (Scheme 117; the use of ultrasound was likely employed to blend the reaction mixture).226 The presence of the reduction product 293 in the case of the trifluoromethyl ketone 291 was considered carefully. The authors provided evidence that these allylation reactions did not proceed by a single-electron transfer mechanism, a mechanism that is also unlikely based on mechanistic studies using radical clock substrates.227 Instead, they suggest that the reduction product emerges from a β-hydride transfer reaction from the allyl group, although this process was not observed in the reaction of the simple methyl ketone substrate.
Scheme 116.
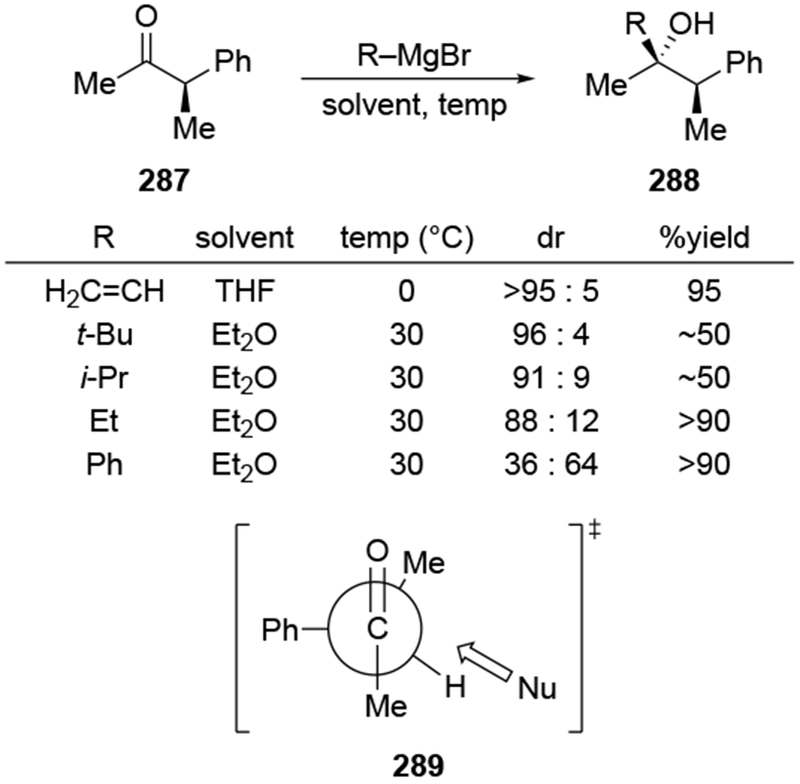
Scheme 117.
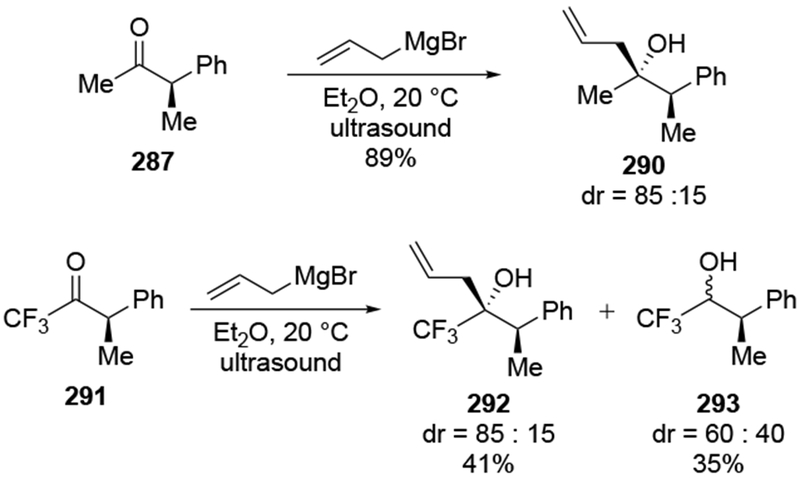
5.3. Additions to Chiral Exocyclic Ketones and Aldehydes
Additions of allylmagnesium halides to exocyclic chiral ketones can be diastereoselective (Scheme 118).228,229 The major product can be formed from a Felkin–Anh-like transition state resembling 296. It may be possible that the ketone adopts this conformation in the ground state to minimize steric interactions, just as observed in Scheme 114. In this conformation, only one face of the carbonyl group is more accessible. The stereoselectivity was responsive to solvents. Selectivity was generally higher in coordinating solvents (like DME and THF) and less selective in non-coordinating solvents (like toluene). The presence of excess magnesium salts resulted in only a modest increase in diastereoselectivity. The highest selectivities were achieved in the presence of Ti(Oi-Pr)4, which could involve transmetallation to form an allyltitanium reagent. Nevertheless, an advantage of the use of the Grignard reagent was that the presence of titanium salts complicated workup.228
Scheme 118.
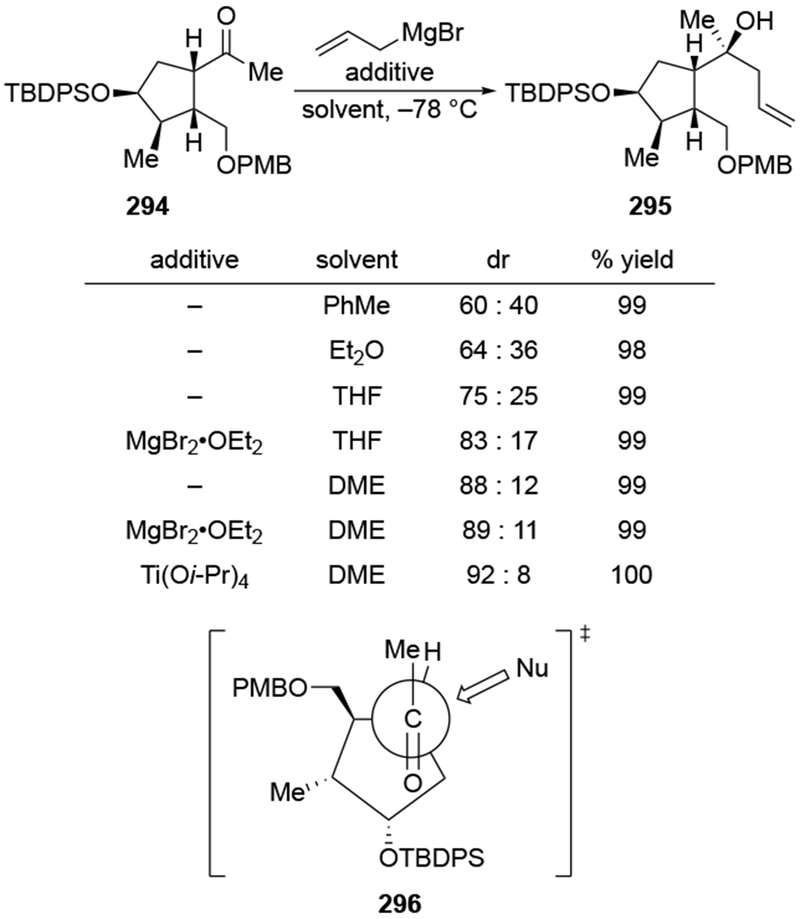
Addition of allylmagnesium halides to aldehyde 297 under conditions expected to favor chelation control instead gave the opposite stereoselectivity (Scheme 119).230 In general, additions of Grignard reagents to tetrahydrofuran-substituted aldehydes are not stereoselective unless chelation is suppressed, such as when non-coordinating solvents are used.231 In this case, Felkin–Anh selectivity can be high.232 In the example shown in Scheme 119, transmetallation to zinc did not result in a chelation-controlled addition, although it did mitigate the competing addition to the ester carbonyl group17 observed in the addition of allylmagnesium bromide to aldehyde 297. The use of an allyl silane as the nucleophile was ineffective, highlighting an important advantage of the more reactive allylmagnesium reagents. As with other examples, a circuitous repair was required to obtain the desired configuration. Alcohol 298 was oxidized to the corresponding ketone and then reduced with LiBH4 to give the desired diastereomer of alcohol 298.
Scheme 119.
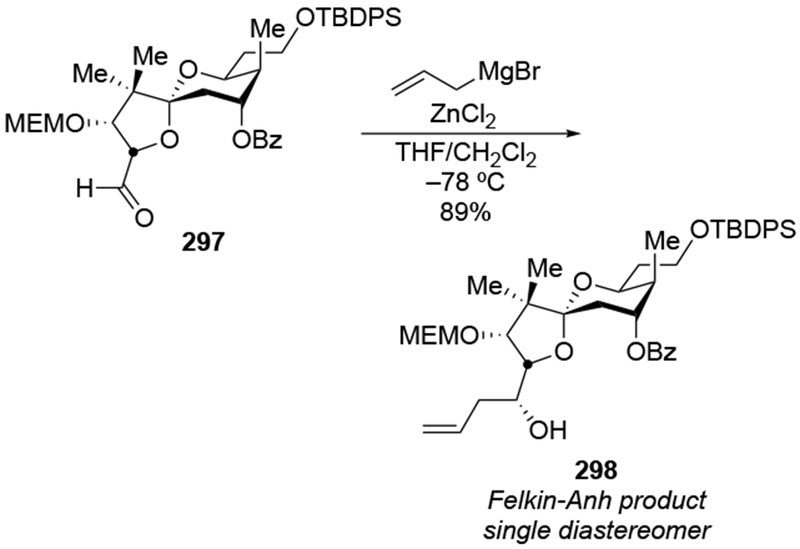
A selective addition to a bicyclic system substituted with a formyl group was reported, which demonstrates that selective additions can occur in these type of aldehydes (Scheme 120).11 This addition occurred to give magnesium alkoxide 300 with reasonable diastereoselectivity (86:14) after an intramolecular Diels–Alder reaction. The selectivity of this addition is consistent with what would be expected from the Felkin–Anh model (transition state 303, assuming that the tetrasubstituted carbon atom serves as RL). The configuration at this carbon atom proved to be a challenge that needed to be addressed. A subsequent reaction involved a Grob fragmentation, but the secondary hydroxyl group was in the wrong orientation for optimal orbital alignment. The configuration was inverted by a process of oxidation and reduction, which itself was not highly stereoselective, although further recycling was possible.
Scheme 120.
Felkin–Anh control can operate in formyl-substituted oxazolines. Although the oxazoline group can engage in chelation (Scheme 89), it can also serve as an electron-withdrawing group, as illustrated in Scheme 121.233,234 The major product in this reaction can be understood using the polar Felkin–Anh model where the nitrogen atom adopts the position of RL (306). Addition to hindered aldehyde 304 occurred with high diastereoselectivity only at low temperatures: at −50 °C, selectivity dropped precipitously to 70:30.
5.4. Additions of Allylmagnesium Halides to Chiral Cyclic Ketones Controlled by Felkin–Anh Selectivity
Although many examples of additions of allylmagnesium reagents to cyclic ketones have been reported, this section will focus on the few examples of Felkin–Anh selectivity (or lack thereof) where there is not a strong steric bias to determine diastereoselectivity. The examples noted here are striking because of the differences between the selectivities observed with allylmagnesium reagents and other organomagnesium nucleophiles. A more inclusive discussion of additions to cyclic ketones will be presented in Section 6.
Additions of organomagnesium reagents to α-trifluoromethylthio ketones illustrate the unique reactivity of allylmagnesium halides (Scheme 122; Ad = adamantyl).235 Addition of methylmagnesium bromide to ketone 307 proceeded with high stereoselectivity, favoring the product where the nucleophile added anti to the sulfur substituent, which would be expected based upon previous studies of Felkin–Anh selectivity.222 Allylation, on the other hand, proceeded with little stereoselectivity. This result is consistent with the argument that Felkin–Anh selectivity, which relies on a lower transition state energy for the Felkin–Anh pathway than for others, should be difficult to achieve if reaction rates approach the diffusion limit.
Scheme 122.
Reactions of the hindered ketone 309 provide more contrasts between allylic and nonallylic organomagnesium reagents (Scheme 120).5,6 Addition of allylmagnesium chloride provided a product that could be consistent with chelation control (i.e., addition of the allyl group anti to the potentially chelating α-methoxy group). Chelation in this substrate would be difficult, however, due to the axial orientation of the OMe group (as illustrated in structure 313). As a result, the preference for formation of diastereomer 310 could be interpreted as the result of Felkin–Anh-controlled addition anti to the axial α-methoxy group. On the other hand, addition of vinylmagnesium chloride occurred to the opposite face, which would require the endocyclic oxygen atom to control Felkin–Anh selectivity. By contrast, the less substituted ketone 311 reacted with both reagents with high diastereoselectivity anti to the axial α-methoxy group. These examples serve as a reminder that stereoselectivity can be controlled by a number of factors.
5.5. Additions of Allylmagnesium Halides to Chiral Acyclic Aldehydes
As noted above, sometimes it is arbitrary to decide what constitutes Felkin–Anh versus chelation-controlled selectivity, so this section focuses on reactions that are unlikely to involve chelation. As discussed in the Introduction (Section 1.1, Scheme 2), carbonyl compounds generally capable of reacting selectively through a Felkin–Anh transition state (as illustrated with alkylmetal nucleophiles) typically react with allylmagnesium reagents to give products with low diastereoselectivity.
The difference in selectivity when using an alkenylmagnesium reagent compared to an allylmagnesium reagent is illustrated by addition reactions to the simple aldehyde 314 (Scheme 124).146 Addition of vinylmagnesium bromide occurred with 89:11 diastereoselectivity, favoring the product expected from addition through Felkin–Anh transition state 316, where RL is the silyloxy group. By contrast, addition of allylmagnesium bromide gave low stereoselectivity (the configuration was determined after conversion to a cyclic product). Chelated transition states are unlikely, considering that, for most organometallic reagents, chelation to a bulky silyloxy group is normally disfavored236 (although chelation can be favored with zinc reagents98,187).
Scheme 124.
Two other examples confirm the generally low stereoselectivity of additions of allylmagnesium halides to substrates capable of reacting through a Felkin–Anh transition state. Addition of allylmagnesium bromide to aldehyde 317 provided a mixture of products, with a similar level of selectivity to that obtained in the addition to aldehyde 314 (Scheme 125).237 Similarly low stereoselectivity was observed for aldehyde 319 (Scheme 126).238 In both of these cases, the configuration of the major product can be considered to arise from a transition state resembling 316 above, with the branched alkyl group serving as RL. Although the diastereoselectivity for the Felkin–Anh isomer (320) was low, the reaction proceeded in high yield.238 The stereoisomers were separated, and the minor isomer was oxidized to the corresponding ketone, which was reduced by borane in the presence of a chiral oxazaborolidine catalyst to provide more of the desired diastereomer 320.239
Scheme 125.
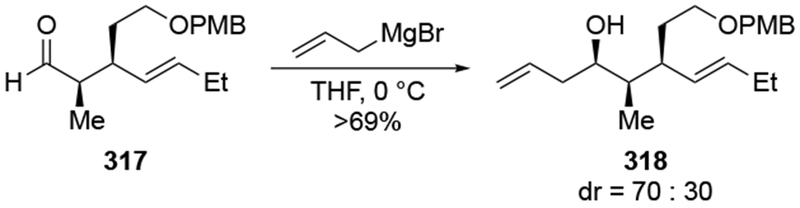
Scheme 126.
This protocol of oxidation of a mixture of stereoisomeric homoallylic alcohols followed by reduction is a general route to convert chiral aldehydes to homoallylic alcohols with high stereoselectivity. In the course of preparing the desired homoallylic alcohol 324 in a synthesis of a natural product, allylation of aldehyde 321 was performed (Scheme 127).240 This reaction provided the undesired stereoisomer 322 in high yield but with low diastereoselectivity. The diastereomers were separated, and the major, undesired stereoisomer (322) was oxidized to form ketone 323. Reduction of this ketone proceeded with stereoselectivity that would be predicted using the Felkin–Anh model to give the desired stereoisomer (324). It is unlikely that reduction of ketone 323 could occur by a β-chelation-controlled reduction with NaBH4 because this reagent does not generally conform to the chelation-control model.70
Scheme 127.
In the case of an α-amino aldehyde, addition of an allylmagnesium reagent occurred with high diastereoselectivity (Scheme 128).241 Addition to aldehyde 325 provided homoallylic alcohol 326, likely through a transition state resembling 327, where the NBn2 group adopts the position expected for RL.
Scheme 128.
The high selectivity of the above reaction contrasts with the generally low diastereoselectivity observed for similar reactions of α-amino aldehydes. In the course of developing methodology for obtaining Felkin–Anh-selective additions to α-amino aldehydes, Reetz and coworkers observed that allylmagnesium reagents were particularly unselective compared to other reagents (Scheme 129).242 Low selectivity was attributed to the high reactivity of allylmagnesium reagents without additional discussion.243 It should be noted, however, that an inverse correlation between reactivity and selectivity can only be expected when reaction rates approach the diffusion rate limit.244
Scheme 129.
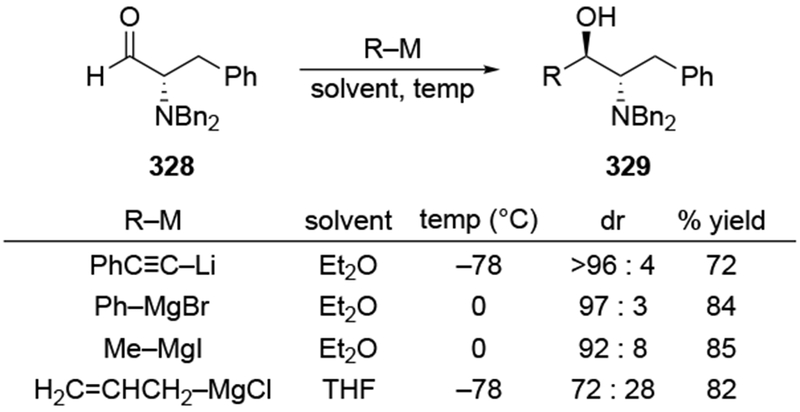
A similar trend of low Felkin–Anh selectivity for reactions of allylmagnesium reagents was observed with an Fmoc-protected α-amino aldehyde (Scheme 130).245 The selectivity was attributed to the large ortho ester group serving as RL in a Felkin–Anh type transition state. For several of the nucleophiles, selectivities for the Felkin–Anh product were improved by either lowering the temperature of the reaction or using a different solvent, but no improvements to the selectivity of the allylic nucleophile were demonstrated. It is also worth noting that control experiments showed that the carbamoyl group did not engage in chelation.
Scheme 130.
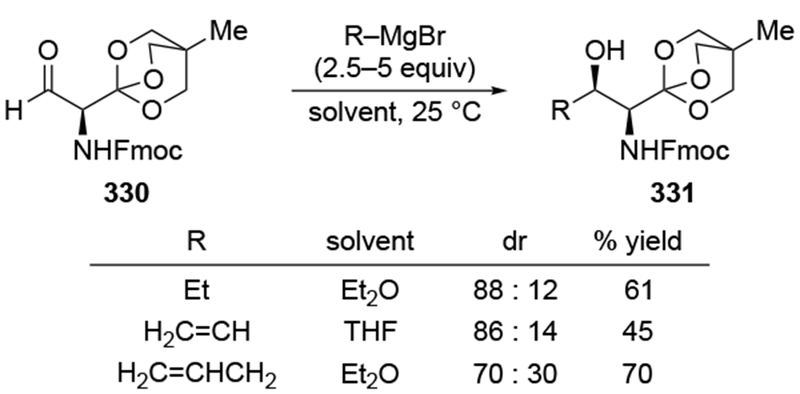
A related system highlighting the unusual reactivity profile of allylmagnesium halides involves the reactions of highly substituted aldehyde 332 (Scheme 131).246,247 In the course of synthesizing fluorinated peptide isosteres, the stereoselectivity of addition reactions of organomagnesium reagents were examined. Use of only one equivalent of a Grignard reagent such as methylmagnesium chloride generated none of product 333, which suggests that deprotonation of the carbamate group was faster than addition to the carbonyl group. Upon addition of an excess of Grignard reagent, selectivity was observed, which would be expected to result from chelation between the formyl group and the deprotonated carbamate, as illustrated in transition state 334. The authors optimized this reaction as an “inverse addition,” in which the aldehyde was added to the Grignard reagent.
Scheme 131.
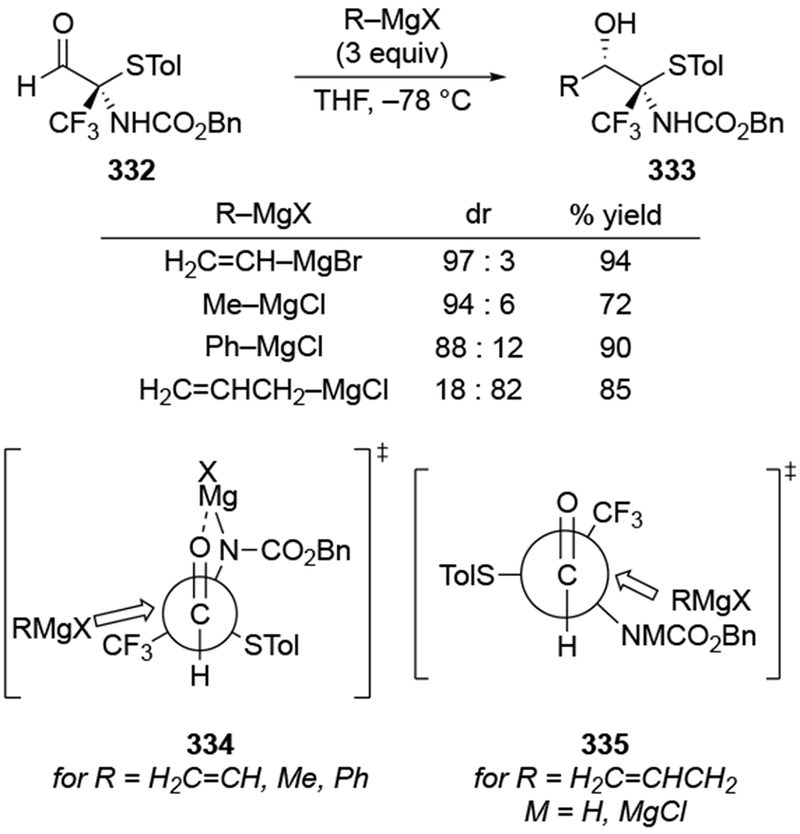
The results with allylmagnesium chloride differed from the results obtained with alkyl and alkenyl Grignard reagents. Use of only one equivalent of allylmagnesium chloride generated product with 50% conversion (as compared to no conversion). The major product, however, had the opposite configuration (i.e., the Felkin–Anh product). The yield was improved somewhat upon addition of excess allylmagnesium reagent (Scheme 131). The favored diastereomer formed in the allylation reaction is consistent with addition through a Felkin–Anh transition state resembling (335, in which the carbamoyl group may or may not be deprotonated. The fact that substantial product was formed with only one equivalent of the reagent suggests that addition may occur to either the protonated or deprotonated form of the electrophile, as also observed for a simple α-hydroxy ketone (Section 4.2.1.1). Instead of being controlled by chelation, the stereochemical outcome may be governed by the nature of the lowest energy conformer, with hyperconjugation stabilizing the conformer that maximizes overlap of the σC-S bond with the π-system of the carbonyl group.222,248. The anomalously low selectivity exhibited by allylmagnesium reagents was also illustrated in reactions of difluoromethyl aldehyde 336 with various organomagnesium reagents (Scheme 132).246,249
Scheme 132.
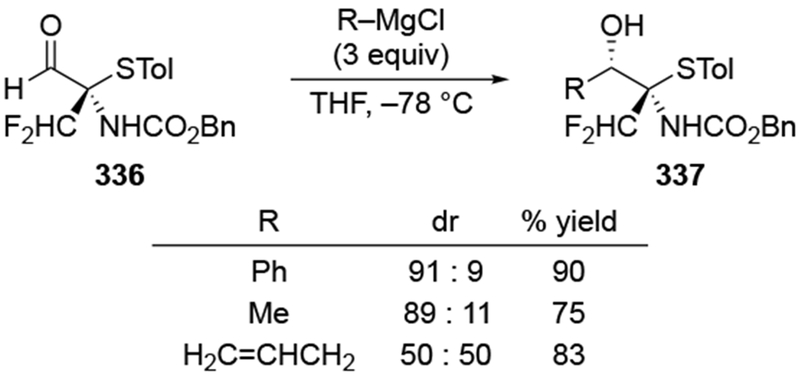
Achieving high Felkin–Anh selectivity can be challenging with many carbon nucleophiles. For example, addition to aldehyde 338 gave generally low Felkin–Anh selectivity (Scheme 133),250, presumably through a transition state where the dimethylamino-substituted carbon atom served as RL (340). With phenylmagnesium bromide, however, the chelation-control product was formed. Use of organozinc reagents instead of organomagnesium reagents with aldehyde 338 generally gave higher selectivity for the desired anti product through chelation.
Scheme 133.
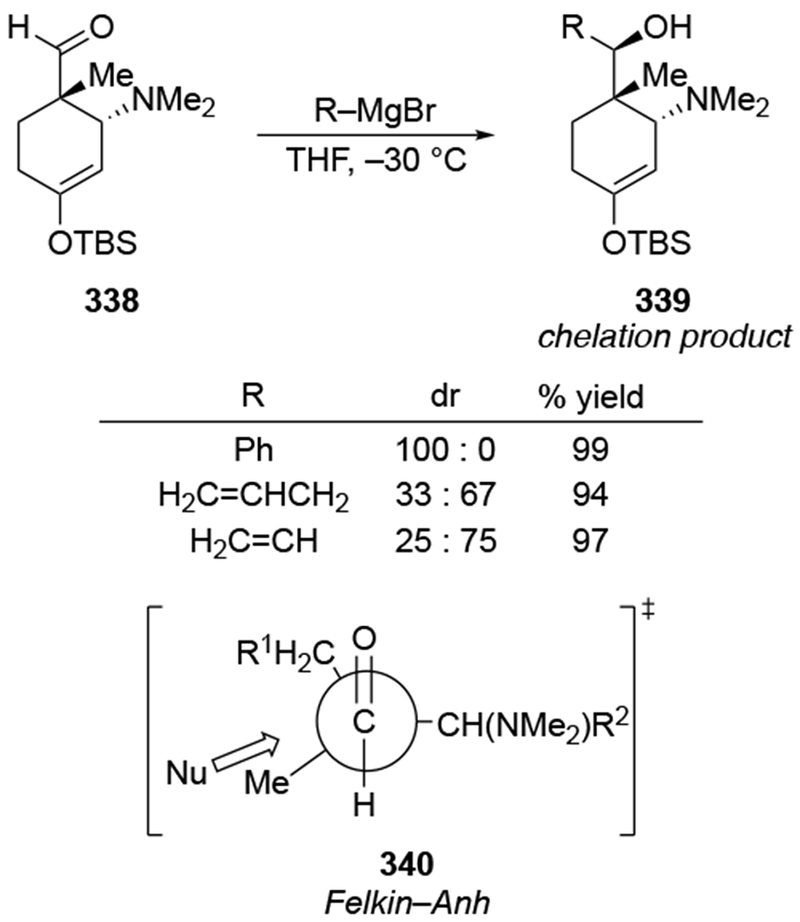
High selectivity for the product consistent with the Felkin–Anh model can be observed in a highly biased system, even with allylmagnesium reagents (Scheme 134).251 Addition of organomagnesium reagents to aldehyde 341 proceeded with high diastereoselectivity. It was argued that Felkin–Anh transition state 343 was responsible for selectivity.
Scheme 134.
An alternative explanation of the observed stereochemical outcome can be made based upon consideration of the X-ray crystallographic analysis of addition product 342 (R = Et). The conformation of that adduct closely resembles the transition state 344, with the alkyl group that was introduced pointing away from the ring system. The aldehyde may therefore adopt a ground-state conformation resembling the conformation of the electrophile in transition state 344 (Scheme 135), only allowing a nucleophilic attack from the opposite direction of the multicyclic ring system. This steric approach control argument, which may be required to explain the diastereoselectivity observed for allylmagnesium reagents despite their high reactivity, may be more general (as discussed in Section 6).
Scheme 135.
Less hindered aldehydes displayed lower Felkin–Anh selectivity in additions of allylmagnesium reagents. In the case of aldehyde 345, addition of allylmagnesium bromide formed product 346 as a 50:50 mixture of diastereomers (Scheme 136).252 The configuration that was required for a planned synthesis was achieved by oxidation of the product to form the cyclopropyl ketone, followed by reduction with a hindered borohydride reagent (which occurred with 90:10 stereoselectivity).
Scheme 136.
The previous discussion of chelation-controlled reactions (Section 4.2.4.3) established that additions to a formyl group attached to a cyclic ether ring did not benefit from chelation (Scheme 71).160 Instead, the major product for additions to substrates such as 347 is more consistent with the Felkin–Anh model, although the selectivity is low (Scheme 137). Related systems also show low selectivity, although a different explanation was provided.253 Similar low selectivity was observed for tetrahydrothiophene analogue (Scheme 138).254
Scheme 137.
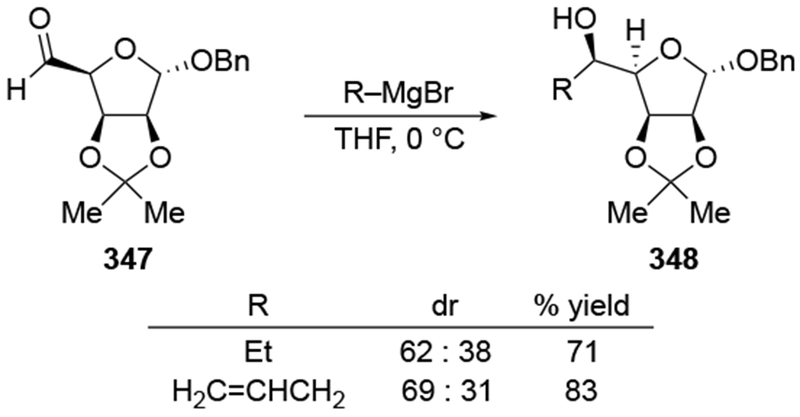
Scheme 138.
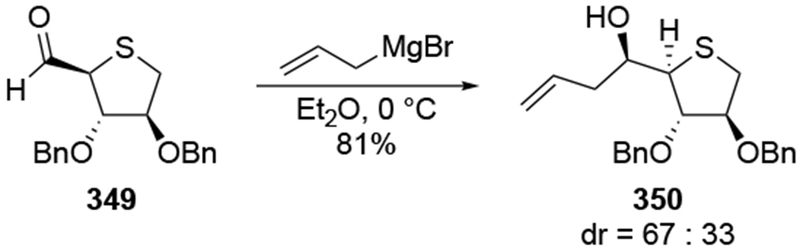
In the course of a planned synthesis of a natural product, stereoselective formation of homoallylic alcohol 352 was attempted by additions of a carbon nucleophile to the formyl pyrrolidine 351 (Scheme 139).255 Improved but not optimal selectivity could be achieved using an organozinc reagent. The observed major product did not appear to arise from a Felkin–Anh transition state: instead, it likely reflects the fact that the substrate adopted a low-energy conformation that would minimize syn-pentane-like interactions between the two oxygen atoms as illustrated in transition state 353. This conformation is adopted in the solid state by an aldehyde resembling 351.256
Scheme 139.
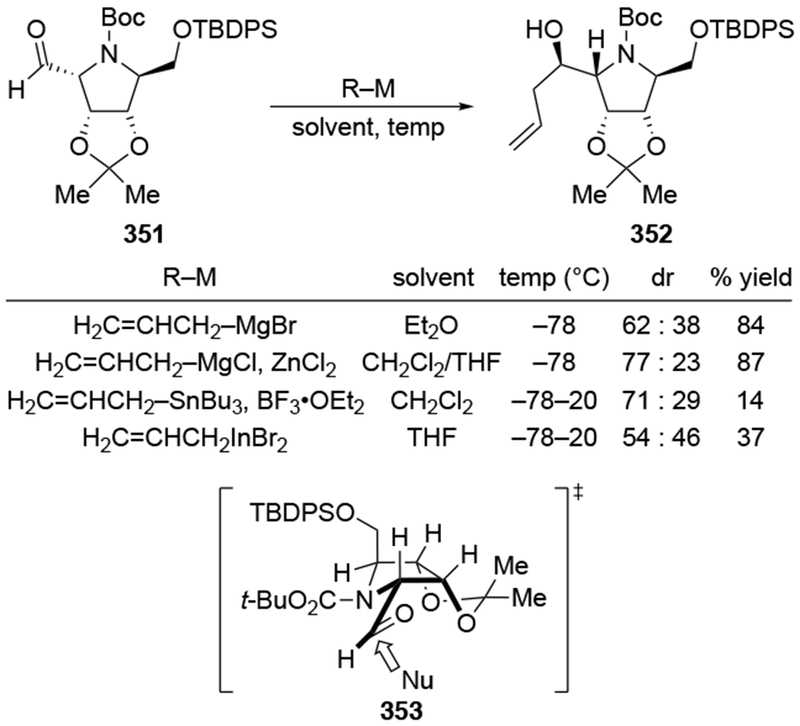
Additions to aldehydes bearing 2-pyrrolidine rings generally exhibit Felkin–Anh type selectivity, but not with allylmagnesium reagents. Additions to the triphenylmethyl-protected amine 3 gave high selectivity for additions of most organomagnesium reagents (Scheme 140).3 Stereoselectivity was markedly lower for addition of allylmagnesium bromide. For these reactions, the nitrogen atom should adopt the RL position, both electronically and sterically. Because of the constraints of the pyrrolidine ring, the carbonyl oxygen atom would need to either point over the ring (354) or away from it (355). Transition state 355 would normally not be favored because steric interactions develop between the nucleophile and the ring as it approaches along the Bürgi–Dunitz angle.257 Nevertheless, this conformation already possesses syn-pentane-like interactions between the group on the nitrogen atom and the carbonyl oxygen atom, which should further favor reaction through alternative transition state 355.
Scheme 140.
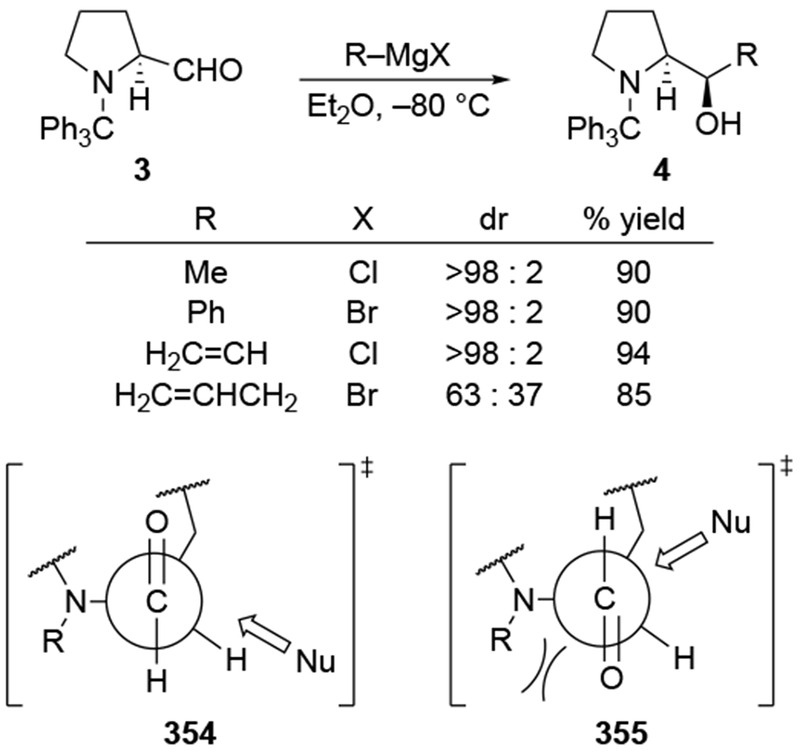
Additions of most organomagnesium reagents to the similar aldehyde 356 were also highly stereoselective (Scheme 141).258 The reaction of 356 with allylmagnesium bromide, however, occurred with lower selectivity. The authors invoked a chelated transition state between the oxygen atoms in the sulfonyl and carbonyl groups to explain the resulting stereoselectivity. Such chelation, however, is not required to explain this selectivity when one considers this example along with the work illustrated in Scheme 140. In that case, there is no sulfonyl group, and the diastereoselectivity is comparable.
Scheme 141.
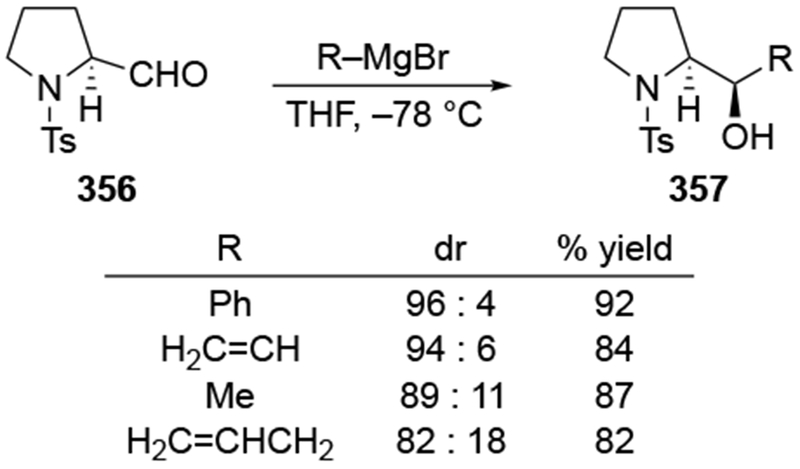
Other N-tosylpyrrolidine derivatives show similarly low selectivity for additions of allylmagnesium reagents. The synthesis of homoallylic alcohol 359 was pursued towards the synthesis of a pyrrolizidine alkaloid (Scheme 142),259 but the diastereoselectivity for addition of allylmagnesium bromide was low. The desired stereoisomer, formally the isomer expected from chelation control, was obtained with some selectivity using an allylstannane reagent.
Scheme 142.
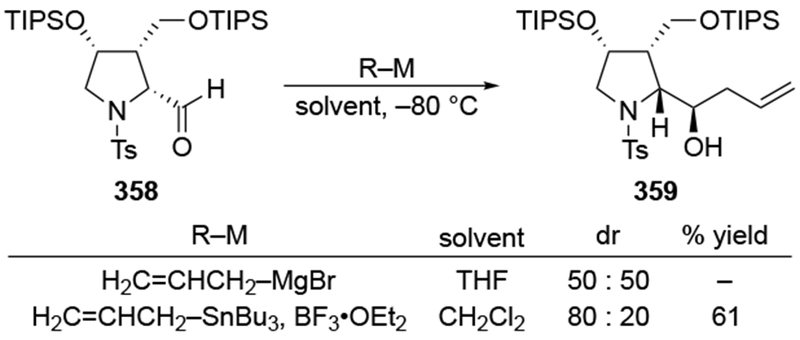
The preference for forming Felkin–Anh products in reactions of aldehydes bearing hindered pyrrolidines is also generalizable to substituted piperidines (Scheme 143).260 The same explanation as detailed above can be used to explain the highly selective stereochemical outcome in forming 361. These hindered carbonyl compounds may have strong conformational preferences in the ground state. It is likely that the formyl group adopts an axial orientation to minimize allylic strain with the substituent on nitrogen, and that a chair conformation such as 363 would be disfavored because it would create 1,3-diaxial interactions, leading to a more boat-like structure 362 being favored. The resulting axial formyl group is likely favored in a conformation that puts the hydrogen atom closer to the more sterically demanding ring. Attack from over the ring would be disfavored because that face would be sterically congested.
Scheme 143.
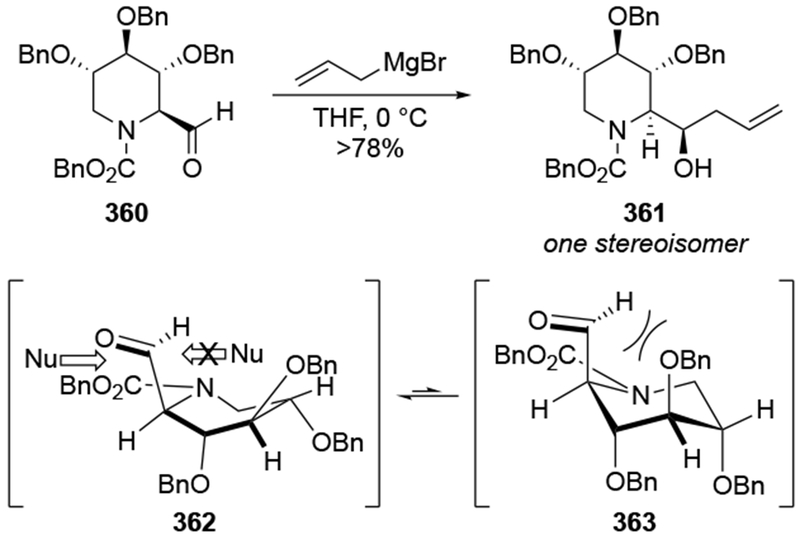
Steroids bearing acyloxy groups (20-keto steroids) generally undergo highly stereoselective additions of Grignard reagents (Scheme 144).261“263 Additions of allylmagnesium reagents were somewhat selective in this series, but selectivity could be improved by the use of allyltitanium reagents.263 The stereoselectivities of these reactions can be considered to arise from attack anti to the sterically large quaternary carbon atom using the Felkin–Anh model (366).
Scheme 144.
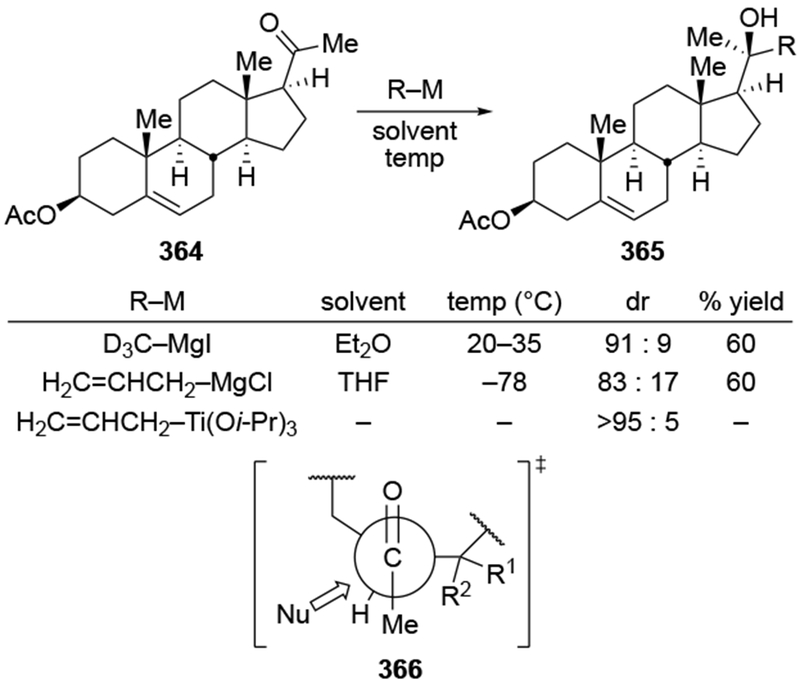
Other examples of additions of allylic magnesium reagents to related ketones have been reported. Addition of allyl- and methallylmagnesium halides to ketone 367 gave products with high stereoselectivity (Scheme 145).264 Additions of other reagents, such as heteroaryllithium reagents, were also highly stereoselective (for example, to form the furyl adduct). For many of these reagents, TMEDA was used because, without it, mixtures of products were formed, but it was not indicated whether these mixtures included other stereoisomers or only other products. Similarly, en route to vitamin D derivatives, addition of allylmagnesium bromide to ketone 369 occurred to form one major product (Scheme 146).265 Additions to related ketones with methylmagnesium reagents also proceed with high diastereoselectivity.262
Scheme 145.
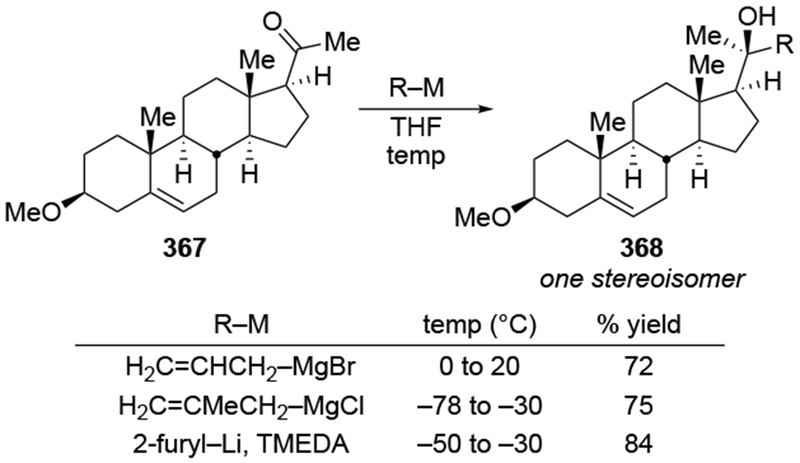
Scheme 146.
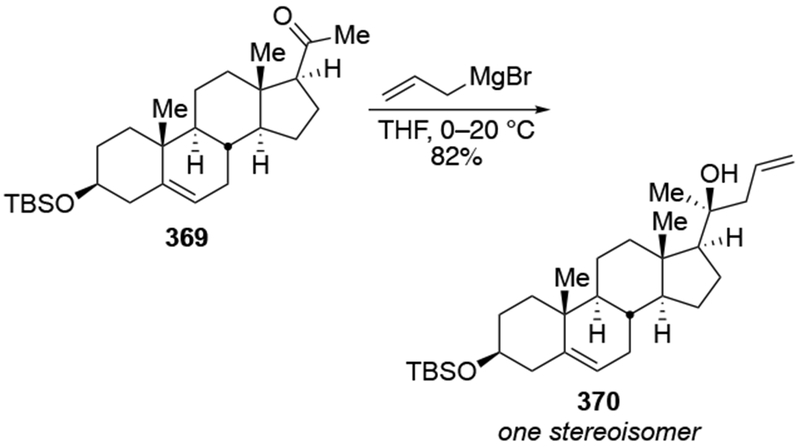
6. Diastereoselectivity of Reactions with Carbonyl Compounds by Steric Approach Control
6.1. Steric Approach Control and Stereoselectivity
As noted in the previous section, the developing steric interactions that occur upon approach of a nucleophile to an electrophile (i.e., steric approach control) play an important role in determining the stereoselectivity of nucleophilic attack onto carbonyl compounds. The case of chelation control provides a logical starting point for illustrating this concept. The fundamental role of chelation in heteroatom-substituted carbonyl compounds is to define a conformation. Once that conformation is defined, then steric approach control determines which face of the carbonyl group is attacked: the nucleophile will approach from the side that minimizes the number of steric interactions (Scheme 147). For chelation-controlled additions in non-coordinating solvents, chelation need play no other role than to differentiate the diastereotopic faces of the carbonyl compound.22 Conversely, chelates formed in coordinating solvents must be more reactive than non-chelated forms, because the chelate species is in such low abundance.53–55) One face of the chelate, however, must be more sterically accessible than the other to observe any stereoselectivity.
Scheme 147.
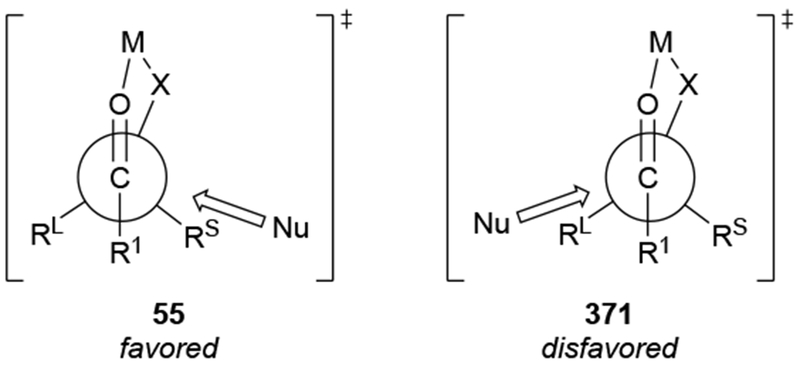
This last statement raises the prospect for another form of stereocontrol. Conformational bias of reactive intermediates arising from sources other than complexation of metals might also lead to sterically differentiated faces. For example, hydrogen-bonding has been used to define the conformation of a reactant that in turn can lead to stereoselective reactions.266–268 Additionally, understanding stereochemical control can focus on considerations of what conformational isomer might be favored by allylic strain, and then consideration of the steric accessibility of the two faces of the molecule.130 The overall topography of a molecule, such as differentiation between convex versus concave faces of multicyclic molecules77,79–82 or the steric differences observed in double bonds contained within medium-ring systems,269 can control stereoselectivity. These effects have been used to control selectivity for a diverse range of reactions, including reactions of electrophiles,270 nucleophiles,271 and radicals272 (Scheme 148). Conformational analysis of a reactive species is a form of steric approach control: only one face of a prochiral functional group is sterically accessible to a reagent. The same analysis would hold to explain why large nucleophiles add to cyclohexanones from the equatorial position: although torsionally this conformational preference is disfavored, it would minimize steric interactions between the nucleophile and electrophile that would develop in the transition state of the reaction (for example, Scheme 149273).
Scheme 148.
Scheme 149.
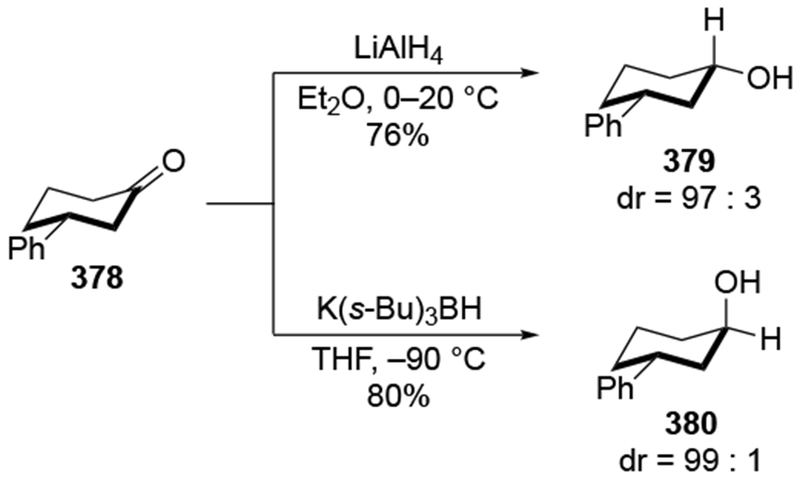
Steric approach control of stereoselectivity, combined with the emerging picture of how one can observe chelation control even without rate acceleration,22,70 suggests a way to understand why allylmagnesium reagents can sometimes react with carbonyl compounds with high stereoselectivity. As observed with chelation-controlled additions of allylmagnesium reagents to chiral α-alkoxy ketones in non-polar solvents, if the electrophile preferentially adopts a conformation where steric effects are different enough between the two faces of the electrophile, then a highly stereoselective reaction can ensue. It is precisely that issue that is explored in this section, where stereoselectivity cannot be analyzed neatly using either chelation control or Felkin–Anh control.
6.2. Additions of Allylmagnesium Halides to Monocyclic Ketones
6.2.1. Additions of Allylmagnesium Halides to Six-membered Ring Ketones
Additions of nucleophiles to six-membered ring ketones have been analyzed extensively, and differing explanations of the origin of stereoselectivity have been proposed. With small nucleophiles, both torsional and electronic factors seem to contribute to that selectivity.195 With larger nucleophiles, however, these reactions are dominated by steric approach control issues.
Considering that allylmagnesium halides are better described as large nucleophiles (in line with other organomagnesium and organolithium reagents) and not small nucleophiles (like LiAlH4), torsional analysis need not be the first concern. With the additions occurring at rates approaching the diffusion limit, steric approach control may be responsible for these additions. The following sections organize these reactions according to the type of ring and its substitution pattern.
6.2.1.1. Additions to 4-Substituted Six-membered Ring Ketones
A formative examination of additions to 4-substituted cyclohexanones included the additions of allylmagnesium reagents to 4-tert-butylcyclohexanone (381) compared to additions of other organomagnesium reagents (Scheme 150).194 Addition of propylmagnesium bromide proceeded with moderate stereoselectivity for attack from the equatorial face, as illustrated in transition state 383. This mode of attack was favored because of fewer developing steric interactions; axial attack would incur two gauche interactions. Smaller reagents (like LiAlH4), however, tend to add from the axial face, a preference that has been attributed, among other reasons, to torsional selectivity.195 The lower selectivity for allylmagnesium bromide was originally attributed to its smaller size compared to propylmagnesium reagents.
Scheme 150.
Fifty years of accumulated knowledge of these reactions suggest that this conclusion should be reevaluated. First, the difference in energy that corresponds to this selectivity is less than 1 kcal/mol, which could arise from many different factors. Second, based upon more recent results,17 the reaction of the ketone with allylmagnesium bromide likely occurs at rates approaching the diffusion rate limit. Consequently, almost no stereoselectivity would be expected because the two diastereotopic faces of the cyclohexanone would not be significantly sterically differentiated in the carbon–carbon bond-formation step.17 Furthermore, the mechanism of addition of allylmagnesium reagents to ketones is likely to be different than for other organomagnesium reagents,23,25 so direct comparisons between reactions are likely to be imperfect.
The advantages of using 4-tert-butylcyclohexanone (381) as a model ketone have led to the development of numerous variants of nucleophilic additions, including allylations. For example, other Grignard reagents have been examined, and those reactions also exhibit little diastereoselectivity (Scheme 151).274 Other authors have confirmed this low stereoselectivity with allylmagnesium halides and other Grignard reagents.275,276
Scheme 151.
Additions of substituted allylmetal reagents to 4-substituted cyclohexanones also occur with little facial stereoselectivity (Scheme 152).277 Although both methallylmagnesium and methallyllithium reagents reacted with little stereoselectivity, methallylzinc reagents exhibited somewhat higher selectivity for addition from the equatorial face. These results could be interpreted as indicating that both the methallylmagnesium and methallyllithium reagents reacted at the diffusion rate limit, but the methallylzinc reagent reacted more slowly.
Scheme 152.
The additions of other substituted allylmagnesium reagents provide additional support for the generally low stereoselectivity of these reactions.277 Addition of crotylmagnesium and crotyllithium reagents to ketone 381 proceeded with low facial selectivity, just as the corresponding methallyl reagents had (Scheme 153). The regioisomeric alcohols 386 were formed in small quantities (values were not given). Similar selectivities were observed for cinnamylmetal reagents (not shown). The regioselectivity of these reactions is consistent with what is known about the reactions of methallyl and cinnamyl nucleophiles. Typically, allylic magnesium reagents, such as crotylmagnesium reagents (Scheme 154),278 add to ketones and aldehydes to provide the product resembling 388, not 386.278,279‘ The origin of the selectivity for the product resembling 388 is preferential reaction through a six-membered transition state involving allylic transposition (i.e., transition state 14 in the Introduction, Section 2.1).18,23 This selectivity even holds for reactions of more highly substituted allylic magnesium reagents such as prenylmagnesium halides with relatively unhindered carbonyl compounds.23
Scheme 153.
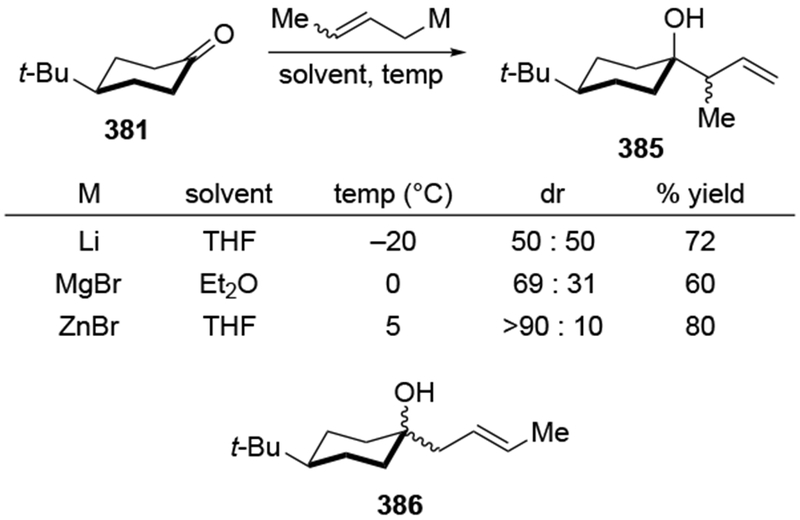
Scheme 154.
Addition of a tert-butyl-substituted allylic magnesium reagent also showed little facial selectivity (Scheme 155).280,281 These reactions showed some selectivity for addition at the less substituted end of the allylic magnesium reagent. The regiochemistry of the major product of addition (389) is consistent with the reagent reacting through the same type of transition state that would be expected for any organomagnesium reagent (i.e., transition states 391 and 392, without allylic transposition).23 In that respect, this allylic magnesium reagent behaves more like any other organomagnesium reagent: it should react at rates below the rate of diffusion. The minor regioisomer 390 would be formed through a cyclic transition state with allylic transposition resembling 393. The addition of this substituted allylic Grignard reagent through the cyclic transition state was highly stereoselective, potentially due to steric interactions between the two tert-butyl groups that arise in cyclic transition state 394.
Scheme 155.
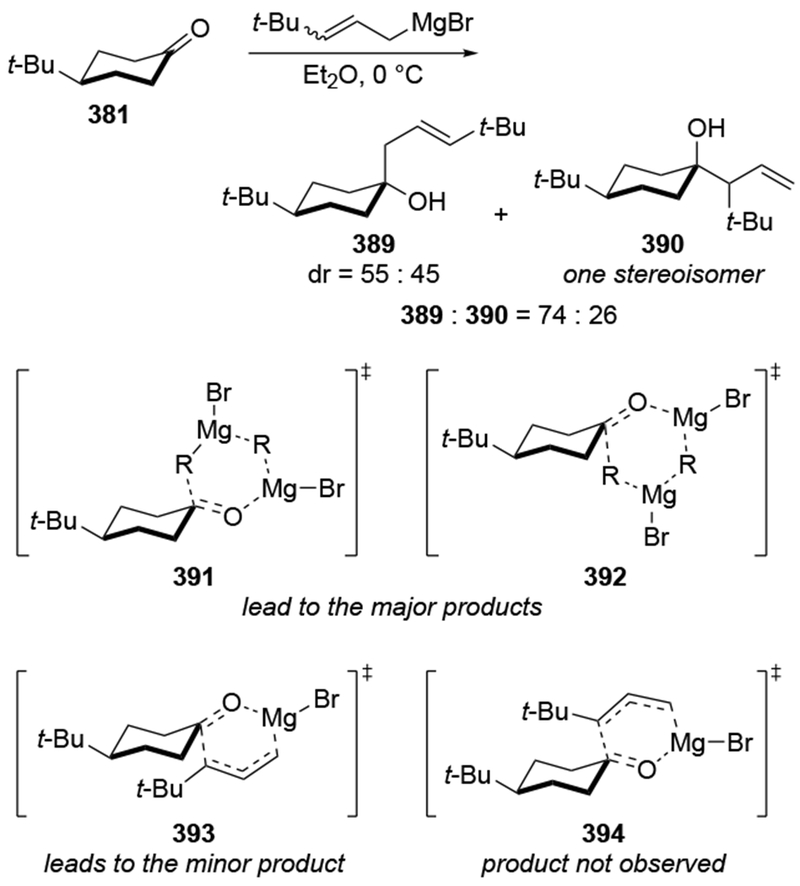
As a general rule, it is difficult to obtain high stereoselectivity for additions of organometallic reagents to 4-substituted cyclohexanones. For example, additions to the phenyl-substituted cyclohexanone 395 proceeded with low stereoselectivity regardless of the metal (Scheme 156).276,282 As noted earlier (Scheme 102, Section 4.3.1.3), the presence of a 4-NHCOR group also resulted in low stereoselectivity.205
Scheme 156.
One possible exception to this general rule appears in the addition to 4-carbomethoxycyclohexanone (397) to form product 398 (Scheme 157).283 Only one stereoisomer of the product was depicted in the paper, but no specific comment was made about the diastereoselectivity of its formation. No stereochemical proof for the structure of the product was included, nor was there any characterization of the product beyond a 1H NMR spectrum. The low yield for this reaction is consistent with the above analysis (Section 2.2) that the rates of addition to esters are comparable to those of aldehydes and ketones. A number of products may have been formed in this reaction, but only the desired product was isolated.
Scheme 157.
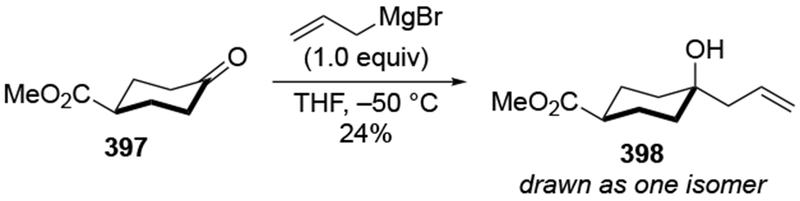
Additions to a 4,4-disubstituted cyclohexanone were also unselective (Scheme 158).284 This example illustrates not only the generally low selectivity of this family of cyclohexanones, but also the somewhat counterintuitive conformational preferences of geminally substituted six-membered rings,285 which might result in this ketone existing as a mixture of conformational isomers.
Scheme 158.
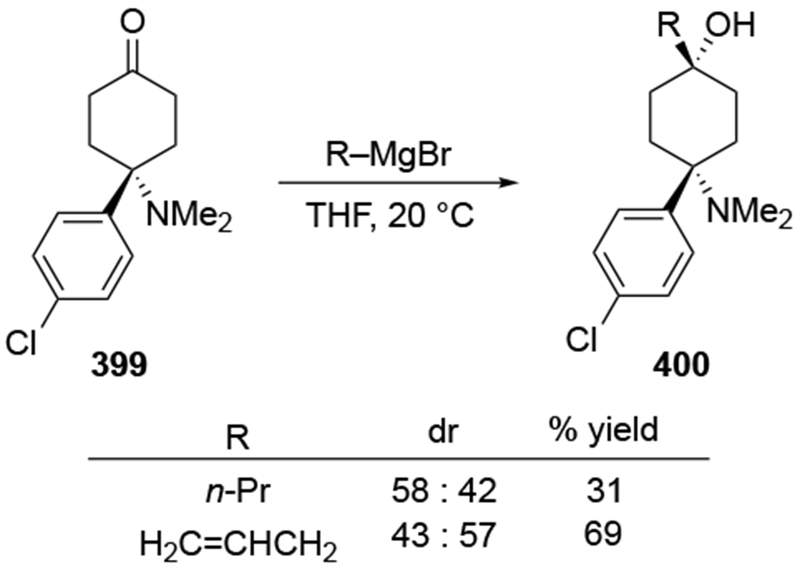
In contrast to the above examples, additions of allylmagnesium reagents to 4-substituted enones proceed with higher diastereoselectivity (Scheme 159).286 These reactions are likely to proceed by addition from the axial orientation because this approach permits appropriate orbital overlap with the carbonyl group, as shown in 403. Although this preference is not high, the selectivity of addition of allylmagnesium bromide is slightly higher than for addition of methyllithium.287
Scheme 159.
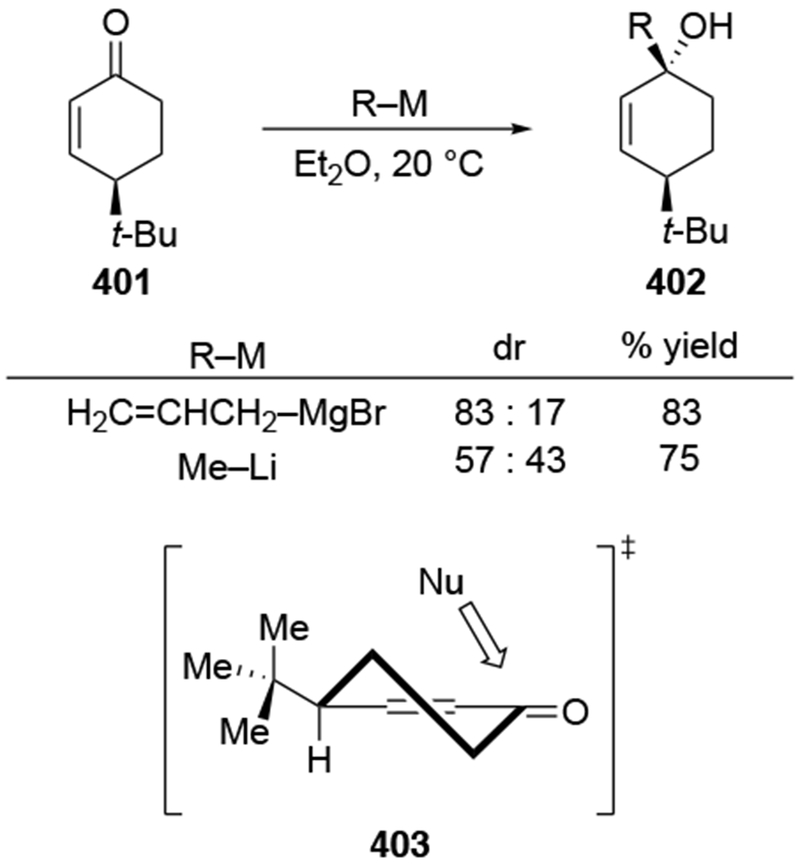
6.2.1.2. Additions to 3-Substituted Six-membered Ring Ketones
Like the 4-substituted systems, cyclohexanones substituted at C-3 generally do not react with Grignard reagents with high selectivity (Scheme 160).274 The low selectivity for the reactions of this substrate may reflect both the low bias of the cyclohexanone group to selectivity and the fact that both axial- and equatorial conformers should be present in solution.288 Consequently, multiple pathways could lead to each diastereomer of product. In the case of the allylmagnesium reagent, addition might also not be stereoselective because addition occurs at the diffusion rate limit: there might not be enough steric destabilization from the nucleophilic approach to decrease the rate constants for addition from either face.
Scheme 160.
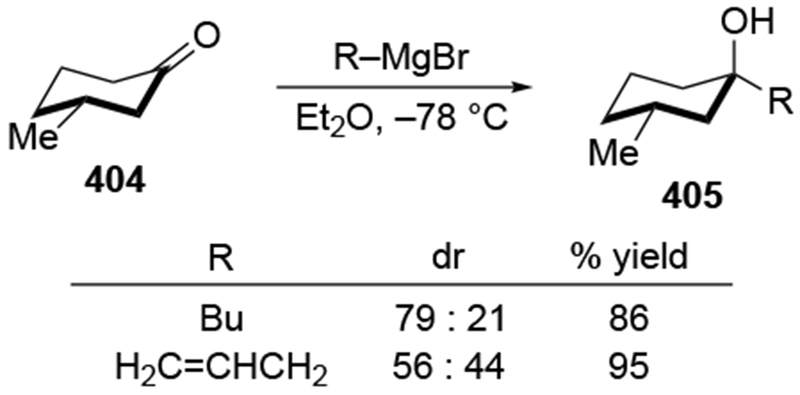
The importance of conformational control and reaction rate are supported by observations of the addition to hindered ketone 406 (Scheme 161).289 With the additional substituents compared to ketone 404, the conformational preference would be biased toward the conformer depicted in transition state 408 to minimize syn-pentane interactions. This conformer would then be likely to be attacked only from the equatorial face because of developing syn-pentane interactions in a transition state involving axial attack. Attack at this hindered axial face could occur at a rate below the diffusion rate limit, so stereoselectivity could result even if attack from one face were particularly fast.17,22
Scheme 161.
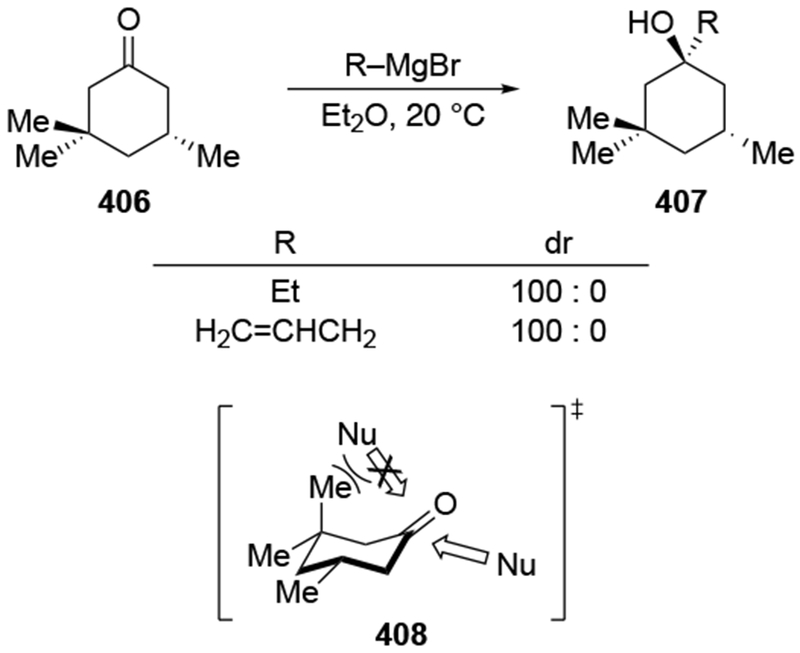
A similar explanation could hold for reactions of the geminally substituted tetrahydropyranone 409 (Scheme 162).290 The preferred conformation likely positions the alkyl group equatorial considering the high preference for the alkyl group to be in the equatorial position at C-2 of the pyranone291 as well as an anomeric effect with the axial methoxy group.292,293 There is also little steric penalty for placing an alkoxy group axial in the axial position on a 3-alkoxycyclohexanone.294 Attack from the axial face would develop a destabilizing syn-pentane-like 1,3-diaxial steric interaction,295 so attack would occur from the equatorial face. It should also be noted that this relative stereochemistry was observed for 3-OMgX-substituted cyclohexanones, which was attributed to chelation (Section 4.3.1.2, Schemes 98 and 100).
Scheme 162.
In the case of 3-substituted enone, diastereoselectivities can be quite high. In a study comparing the efficacy of different allylmetal reagents, it was shown that carvone (411) underwent highly selective allylation with allylmagnesium bromide (Scheme 163).286 No conditions for this reaction were given, but it is likely to similar to conditions employed with related substrates (Scheme 160).
Scheme 163.
A detailed study of the additions of allylic organomagnesium reagents to pulegone (413) also provides similar high selectivity (Scheme 164).296 With several of the reagents examined, the reactions were diastereoselective, favoring a 1,3-trans relationship between the nucleophile and the substituent on the β-stereocenter. Furthermore, most of the substituted allylic organomagnesium reagents reacted to give the products expected for reactions through a six-membered ring intermediate with allylic transposition (transition state 14, Section 2.1). Addition of the highly hindered reagent prenylmagnesium bromide, however, gave not only low diastereoselectivity for the γ-product 415 (Scheme 165), but also a significant number of products formed from conjugate addition. This result is not unique: another hindered allylic Grignard reagent gave similar results (Scheme 166). Computational studies of this reaction have been conducted.297
Scheme 164.
Scheme 165.
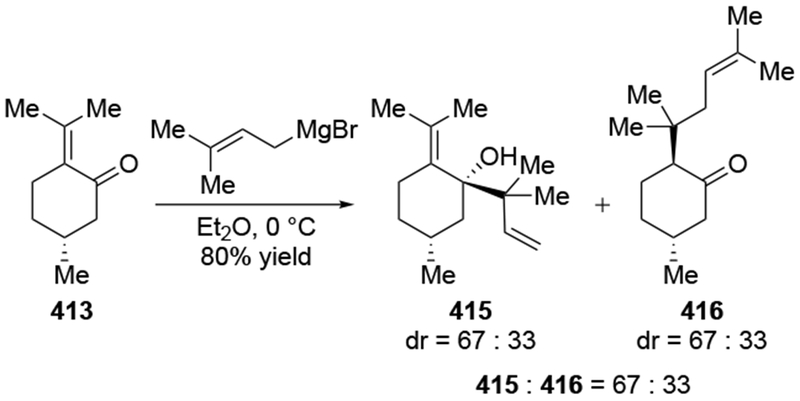
Scheme 166.
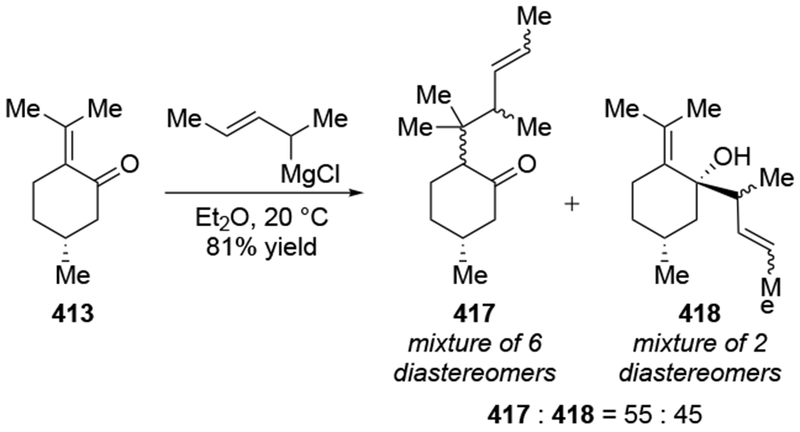
Additions to monosubstituted tetrahydropyranone derivatives are not generally diastereoselective for any organometallic agents. Addition of allylmagnesium bromide to ketone 419 proceeded with low 1,3-cis diastereoselectivity (Scheme 167).298 Addition of an aryllithium reagent to a similar ketone, 421, also proceeded with low stereoselectivity.299 By contrast, high 1,3-trans selectivity is expected for additions of alkylmetal reagents to dihydropyranones (that is, the unsaturated ketone) with this substitution pattern.300–302
Scheme 167.
6.2.1.3. Additions to 2-Substituted Six-membered Ring Ketones
As a general rule, additions of nucleophiles such as organomagnesium halides to 2-substituted cyclohexanones are more selective than for the other substituted cyclohexanones.71 Additions occur anti to the substituent to minimize steric interactions that develop in the transition state (Scheme 168).303 It should be noted, however, that additions of allylmagnesium halides occur with lower selectivity, which is consistent with their reactivity with other ketones and aldehydes.
Scheme 168.
The difference in selectivity between different reagents is also illustrated by additions to other 2-substituted cyclohexanones (Scheme 169).304 Whereas addition of a vinylmetal reagent occurs with high selectivity, the lower selectivity observed for allylation is similar to what was observed with the simple ethyl substituent in the previous case (Scheme 168).
Scheme 169.
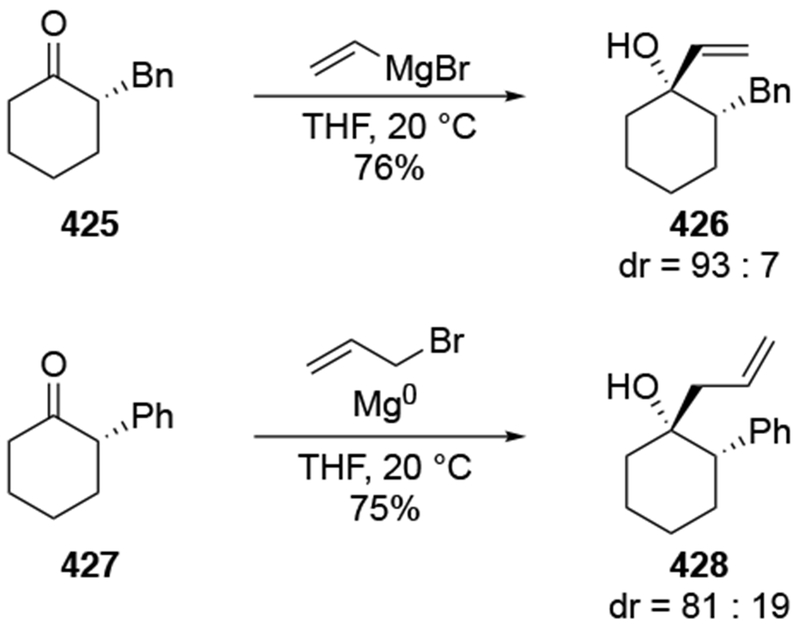
The modest diastereoselectivity for the allylation reactions of 2-substituted cyclohexanones has been confirmed for a number of substrates (Scheme 170).305 In particular, these studies indicate that the presence of a methoxy group does not alter the selectivity. This result is consistent with the generally low chelation-controlled selectivity observed by allylmagnesium reagents, as discussed in Section 2.2. For organomagnesium reagents, additions to 2-alkoxycyclohexanones are stereoselective, while additions of organolithium and organotitanium reagents are less selective (Scheme 171).306 Low selectivity was also observed in a case where there was potential for β-chelation (Scheme 172),307 although simple Grignard reagents did not react selectively either.308
Scheme 170.
Scheme 171.
Scheme 172.
The low selectivity exhibited for additions of allylmagnesium reagents may or not be problematic to synthetic chemists. In the case of addition to ketone 436 (Scheme 173), the stereochemical consequence of this low selectivity was irrelevant, considering that both stereoisomers could be converted to the final target by an elimination reaction.309 In other cases, the diastereomeric addition products needed to be separated (Scheme 174).310
Scheme 173.
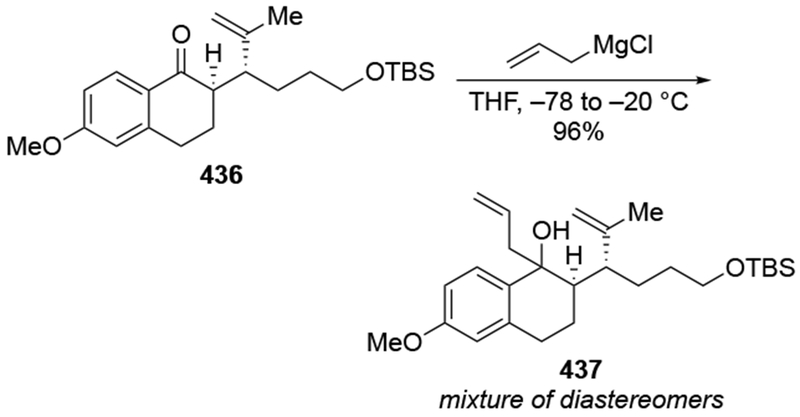
Scheme 174.
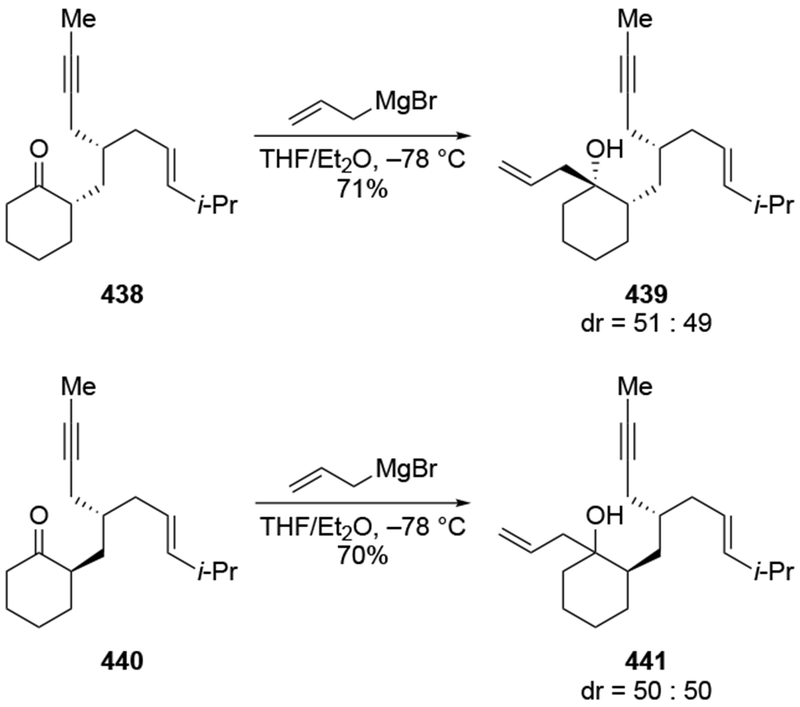
Additions to 2,2-disubstituted cyclohexanones also illustrate a dramatic difference in reactivity for allylmagnesium reagents compared to other Grignard reagents (Scheme 175).311 A variety of organomagnesium reagents added to ketone 442 with high stereoselectivity, but additions of allylmagnesium did not. It is worth noting that a crystal structure of a related ketone where the propargyl group was replaced by a benzylic group (shown as “R”) favored the conformation 444 where the carboxyethyl group adopted an axial orientation.312 The major addition products would arise from axial attack considering how hindered equatorial attack would be.
Scheme 175.
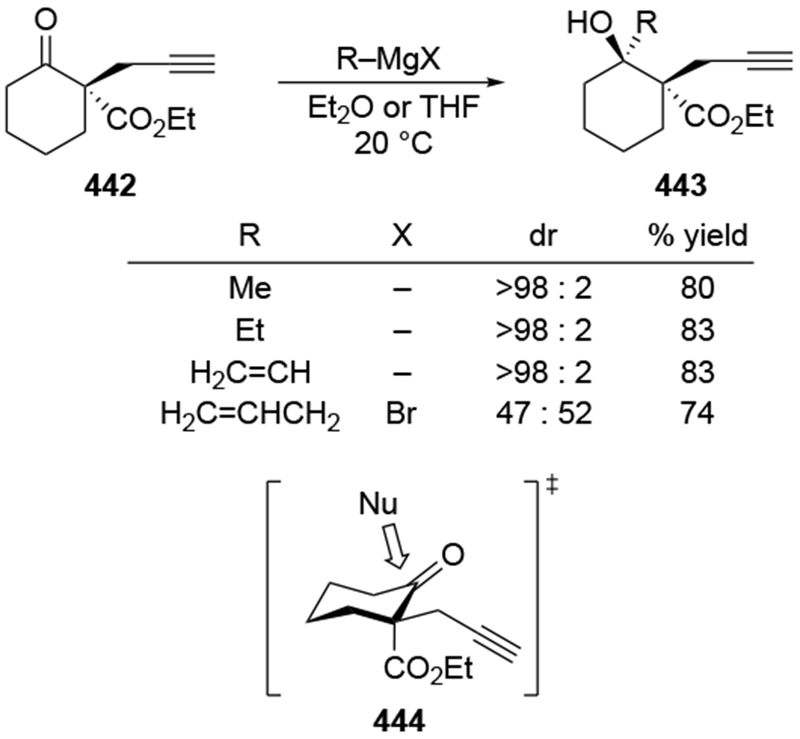
The above observation of low stereoselectivity for addition of allylmagnesium halides is not general, however. Additions of both vinyl- and allylmetal reagents to ketone 445 were highly stereoselective (Scheme 176).313 The addition of homoallylic magnesium reagents, however, were unsuccessful. This example might be more selective because the two substituents are larger than those in Scheme 175, with the larger group occupying the equatorial orientation. Approach of the nucleophile from the equatorial position would develop gauche interactions with both substituents, which could be avoided upon attack from the axial orientation, as shown in transition state 447.
Scheme 176.
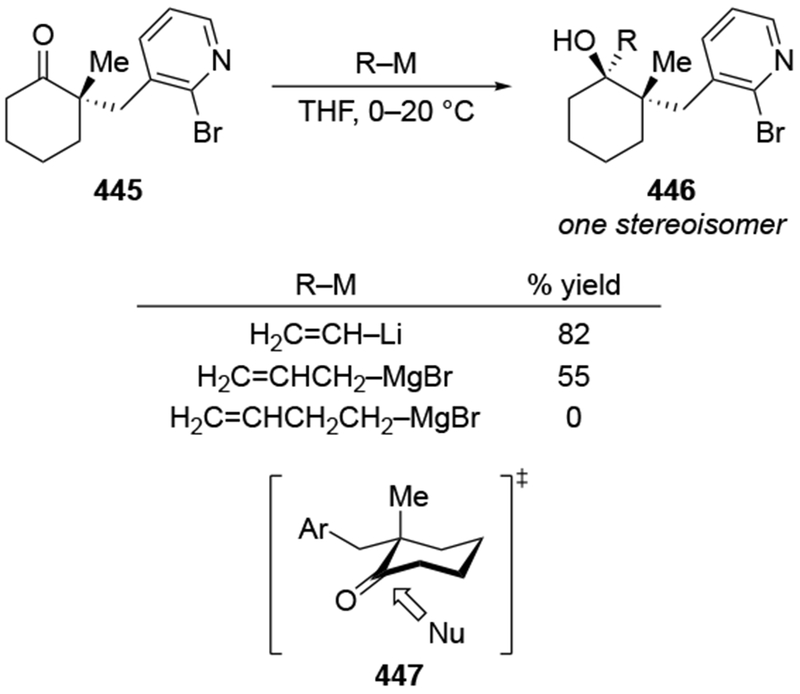
The additions to 2-substituted cyclohexanones with geminal substituents at the adjacent carbon atom were also highly stereoselective (Scheme 177).314 It was noted later that additions of homoallylic magnesium reagents were also highly stereoselective, but those reactions required the addition of CeCl3 to facilitate addition.315 Generally, CeCl3 is used to assist additions of alkylmagnesium reagents, particularly in the case of readily enolizable ketones.316 Just as with the 3,5,5-trisubstituted cyclohexanones (Scheme 161), the presence of an axial substituent at the geminally substituted carbon atom likely decreases the rate of addition from that face, so addition from the equatorial face would be favored (as illustrated in transition state 452). Similar highly selective reactions were observed in a related system (Scheme 178).313
Scheme 177.
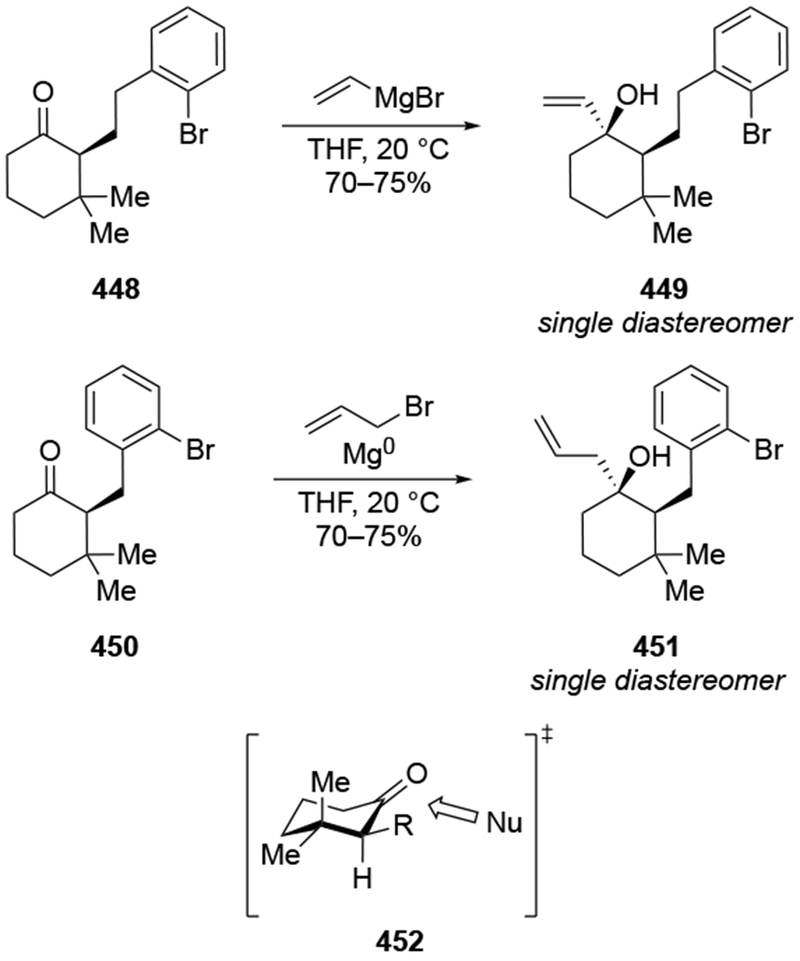
Scheme 178.
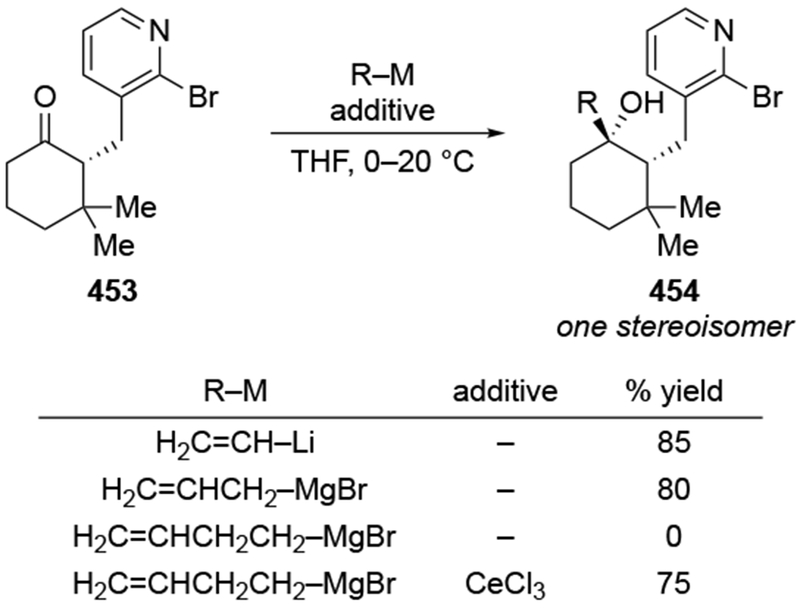
6.2.1.4. Additions to Six-membered Ring Ketones with Multiple Stereocenters
As with earlier examples, additions of allylmagnesium reagents to more sterically congested or conformationally constrained carbonyl compounds tend to give higher diastereoselectivity. This similarity between reactions of different types of organomagnesium reagents may occur because the rates of addition of allylmagnesium halides may be below the diffusion rate limit, at least for the diastereotopic face that leads to the minor product.
Menthone (455) is the canonical example of a multiply substituted six-membered ring where additions of various types of organomagnesium reagents, including allylmagnesium reagents, are highly stereoselective (Scheme 179).317 These results contrast with observations in the additions to simple 2-substituted ketones described above (Section 6.2.1.3). Other authors have also noted the highly stereoselective additions of allylic organomagnesium reagents to menthone.282,318,319 The high selectivity of these reactions may result from the influence of the isopropyl group. Two possible modes of approach to the carbonyl group (transition state 457) can be considered for attack on what is likely the lowest energy conformer.320 If the nucleophile attacked from the bottom face of the carbonyl group, the transition state would be destabilized by interactions between the ring and a methyl group, which would develop a syn-pentane interaction. Conversely, approach from the top face is less hindered. Crotylation is also highly stereoselective for addition to the carbonyl group. The moderate diastereoselectivity observed is a result of reaction through a six-membered transition state leading to a mixture of diastereomers at the exocyclic stereogenic center (Scheme 180).318
Scheme 179.
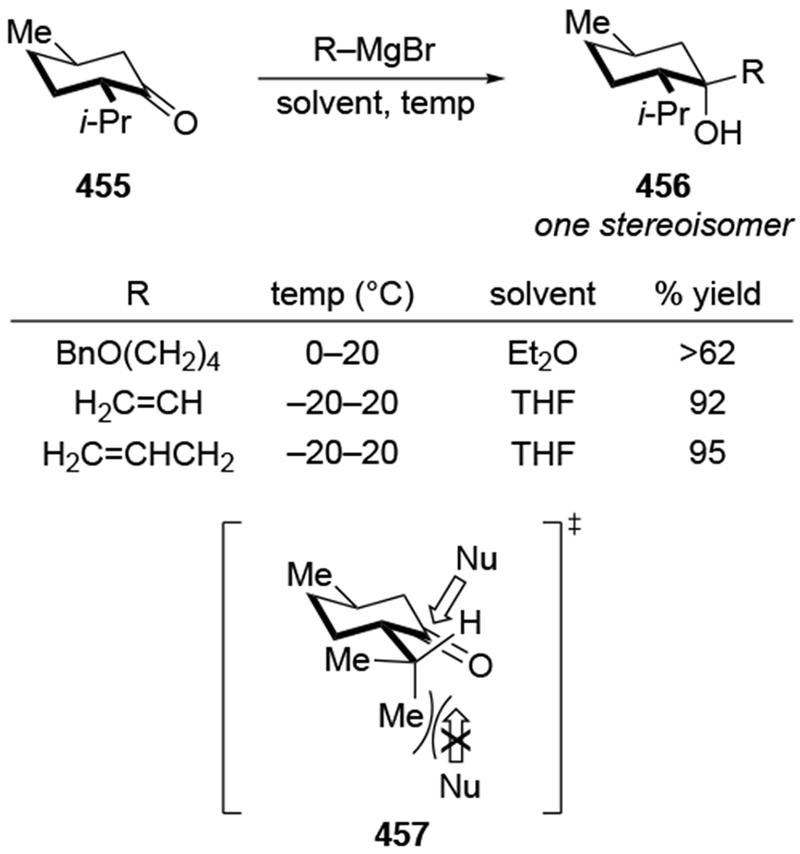
Scheme 180.
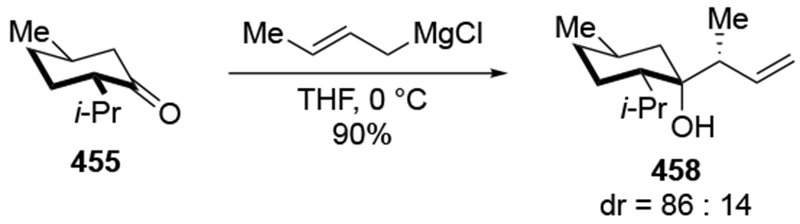
In the case of a more substituted cyclohexanone related to menthone, additions of allylmagnesium and aryllithium reagents were also highly diastereoselective, although reaction conditions were not provided (Scheme 181).321 The illustrated conformation of 459 could be favored, and developing 1,3-diaxial interactions with the axial methyl group and the isopropyl group would develop upon addition to the axial face of this conformer.
Scheme 181.
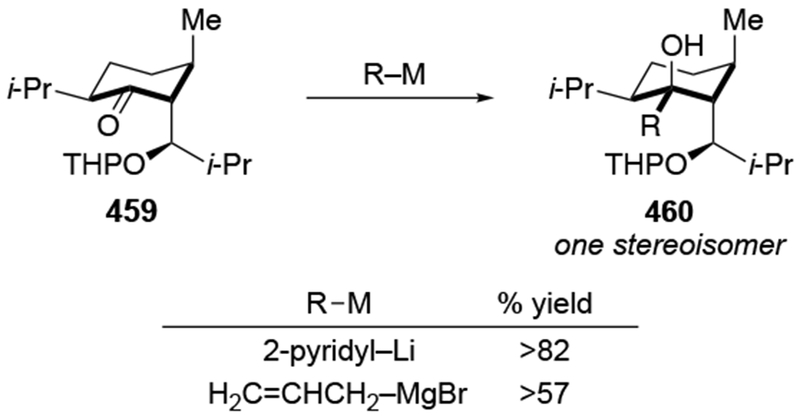
Conformational constraints imposed by other substituents on 2-alkoxycyclohexanones exert strong influences on chelation (Scheme 182).322 As noted previously (Scheme 171), chelation can play a large role in additions of some Grignard reagents to simple α-alkoxy cyclohexanones. When a chelating group is constrained to an axial position (as in 461), chelation is unlikely, so no selectivity was observed unless strongly chelating indium reagents were used (Scheme 182).157 By contrast, when the chelating group was fixed to be equatorial (as in 464), the additions of allylmagnesium reagents and alkylmagnesium reagents, among others, were highly stereoselective (Scheme 183). The additions of the allylindium reagents also appear to benefit from chelation-induced rate acceleration, which would be consistent with the standard view of chelation control (Section 4.1).53–55
Scheme 182.
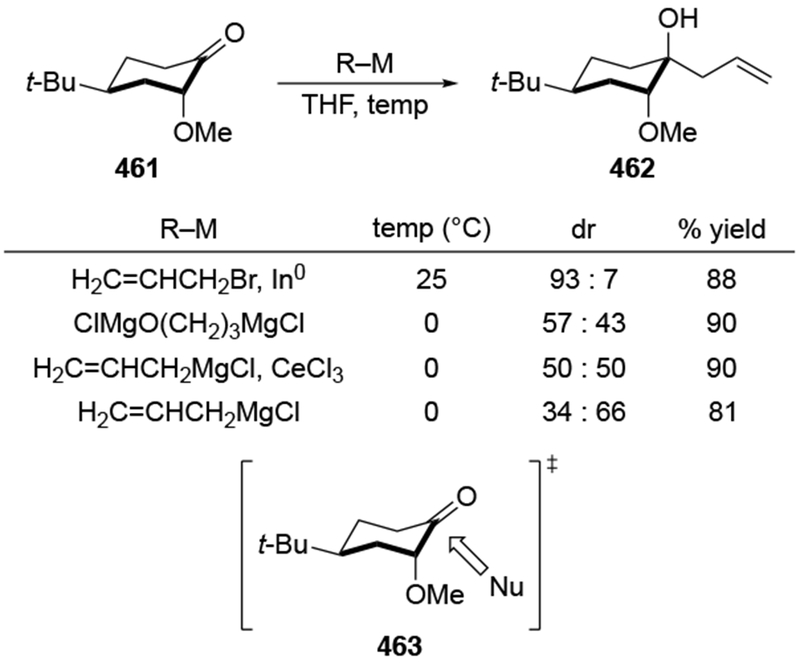
Scheme 183.
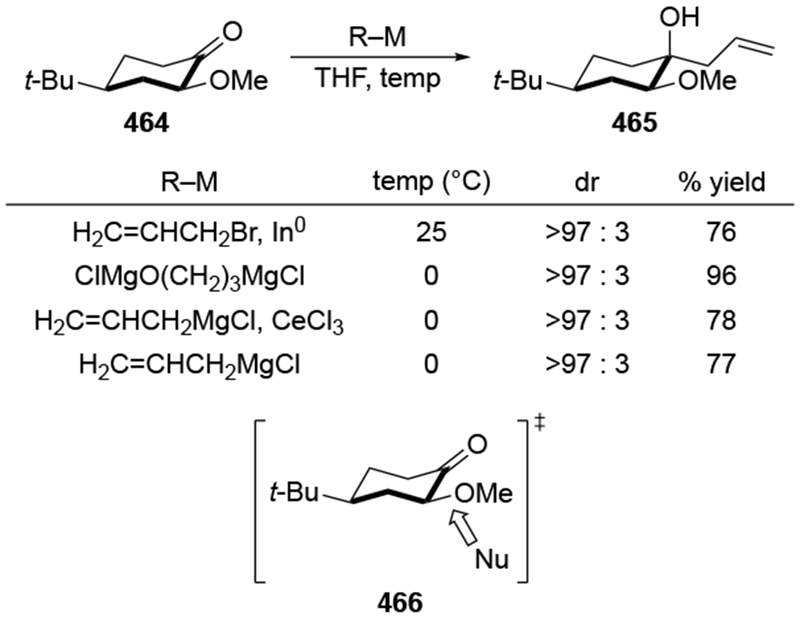
It should not be concluded that addition to a cyclohexanone with an axial group at C-2 leads to low stereoselectivity and addition to a cyclohexanone with an equatorial group leads to high selectivity, however. Addition of methyllithium to the cyclohexanone with an axial methyl group (467) gave predominantly one stereoisomer of adduct (Scheme 184).323 By contrast, addition of an alkynyllithium species to the compound with the equatorial methyl group (i.e., 469) gave a mixture of products.324
Scheme 184.
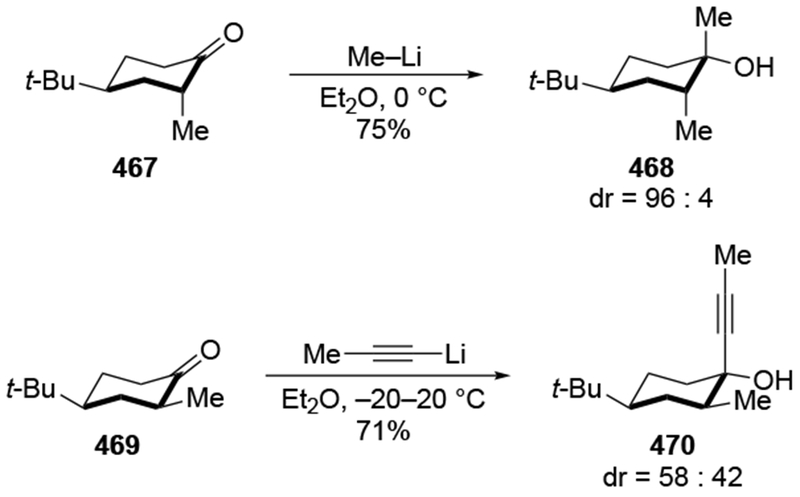
An inherent difference in selectivities between different types of Grignard reagents was noted for a number of cyclohexanones with a potential chelating group at C-2 (Scheme 185).322,325 The alkoxyalkyl Grignard reagent gave selectivity for what is argued to be the non-chelation product, whereas allylmagnesium reagents gave low selectivity. Ultimately, the use of the chelating allylindium reagent provided the solution for obtaining the chelation product as the complementary diastereomer to 472.
Scheme 185.
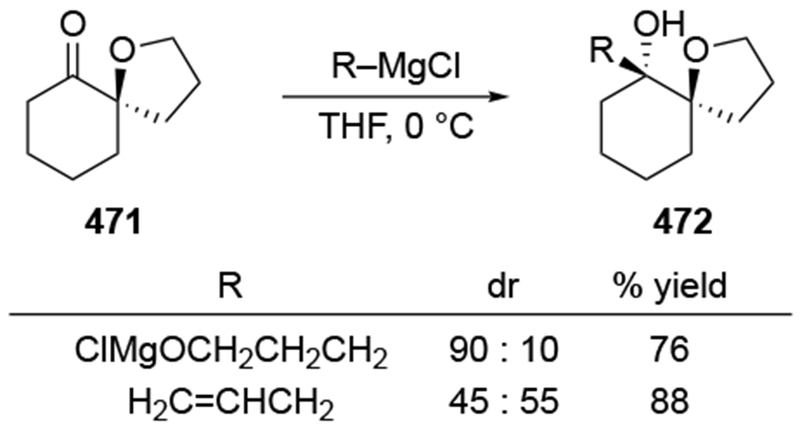
Chelation to a β-alkoxy group, however, is not a defining mode of selectivity, at least in a highly substituted cyclohexanone. Additions of organomagnesium reagents to a mixture of ketones 473 and 474 occur at the equatorial face of each conformationally constrained ketone, regardless of the presence of a chelating oxygen atom at the β-position (Scheme 186).326
Scheme 186.
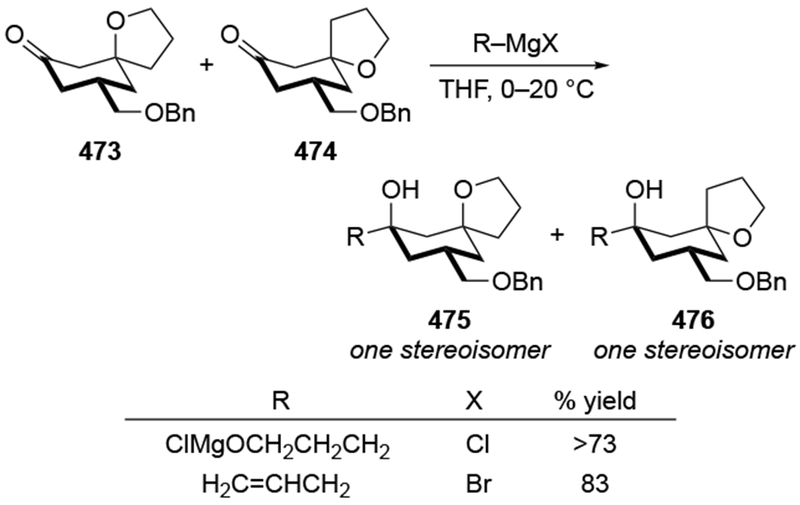
In the case of a β-hydroxy substituted variant, allylation also favored addition from the equatorial face (Scheme 187).326 As noted above (Scheme 186), the hydroxyl group is not necessary to encourage this mode of stereocontrol. It is worth noting, however, that chelation appeared to be important in the case of a different cyclohexanone (Scheme 99, Section 4.3.1.2).203
Scheme 187.
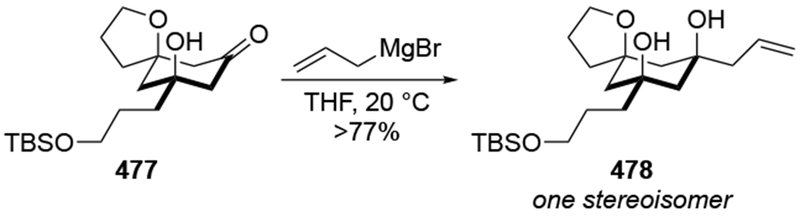
The relative orientation of substituents in a multiply substituted cyclohexanone can influence the stereoselectivities of additions to ketones (Scheme 188).327 For additions to a 2,3-trans-disubstituted cyclohexanone, addition of the homoallylic reagent was employed using a cerium salt (the counterion was not specified) to minimize enolization, but addition was faster than enolization with allylmagnesium reagents, as discussed in Section 2. The diastereoselectivities for both reactions, however, were modest. By contrast, addition of allylmagnesium bromide to the 2,3-cis-disubstituted ketone occurred with high diastereoselectivity, likely because the conformational preferences of the ketone magnify the differences between the diastereotopic faces. The conformer of 482 likely places the allyl group equatorially as shown.328 Addition to the axial position is now slower because of the presence of the axial ethyl group at C-3, leading to preferential equatorial attack.
Scheme 188.
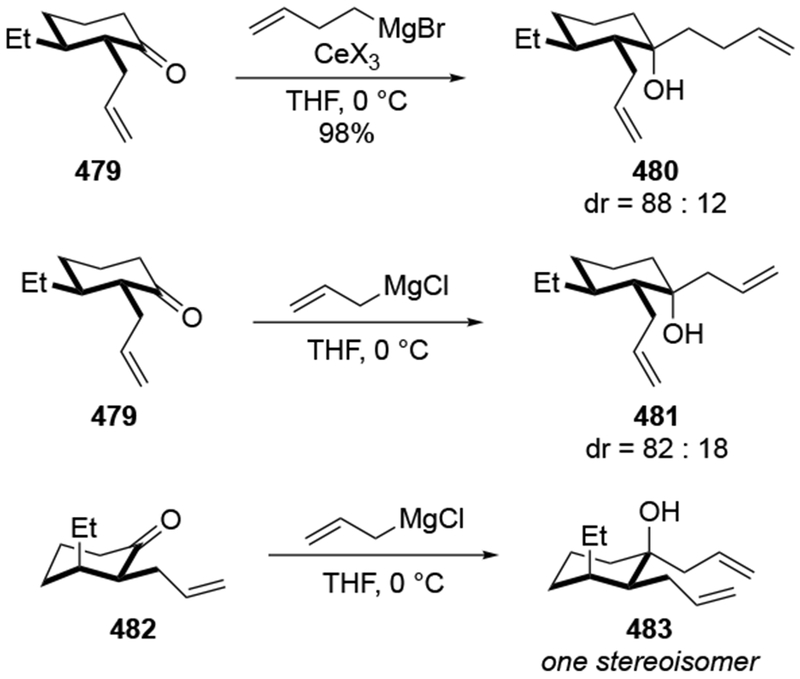
A highly substituted cyclohexenone also reacted with allylmagnesium with high selectivity (Scheme 189).329 Although the authors note that the product was formed with high stereoselectivity, the configuration was not assigned because it was not critical to the synthetic plan. It is likely that the major stereoisomer was formed upon attack as shown for 486, in analogy to observations with another cyclohexenone (Scheme 159).
Scheme 189.
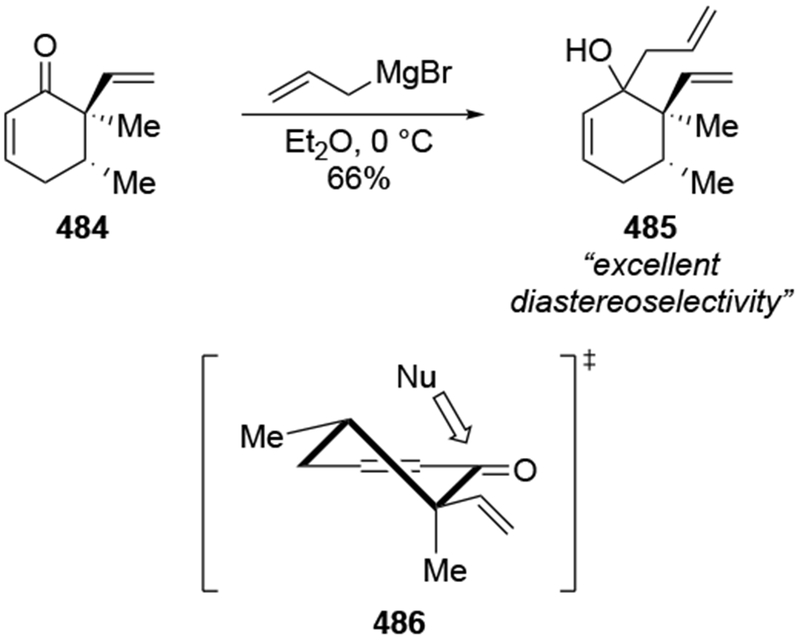
Additions to ketones with a number of alkoxy groups led to divergent diastereoselectivities depending upon the nucleophile that was employed (Scheme 190).330 Addition of methyllithium to ketone 487 occurred from the axial orientation to give exclusively alcohol 488. By contrast, addition of allylmagnesium bromide occurred with little selectivity. The same observation was made for a different ketone under similar conditions, forming alcohols 490 with the selectivity depending upon the nucleophile (Scheme 191).
Scheme 190.
Scheme 191.
Another example illustrates the importance of steric size of the substituent on stereoselectivity (Scheme 192). Unlike in other cases (Section 4.3.1.2), 203,204 addition of allylmagnesium bromide to the free β-hydroxy ketone 491 occurred with low diastereoselectivity.331 When the hydroxyl group was protected as the sterically large pivaloyloxy group, the addition occurred with high diastereoselectivity. It is likely that the conformation 493 is favored in the hindered carbonyl compound, and equatorial attack occurs to minimize developing 1,3-diaxial interactions that would occur upon attack from the axial orientation.
Scheme 192.
The difference in selectivity of additions of Grignard additions to a highly substituted cyclohexenone is striking (Scheme 193).7 This enone is likely to adopt the conformation 497 that minimizes 1,2-allylic strain by placing the oxygen atoms equatorial (a similar enone adopts this type of conformation in the solid state332). Whereas attack from the top face might be torsionally favored, it should be disfavored by a developing 1,3-diaxial interaction, whereas attack from the bottom face is more sterically accessible. It is possible that the more reactive allyl reagent responds only to the steric effects, whereas the other reagent is aided by chelation to the nearby oxygen atom.
Scheme 193.
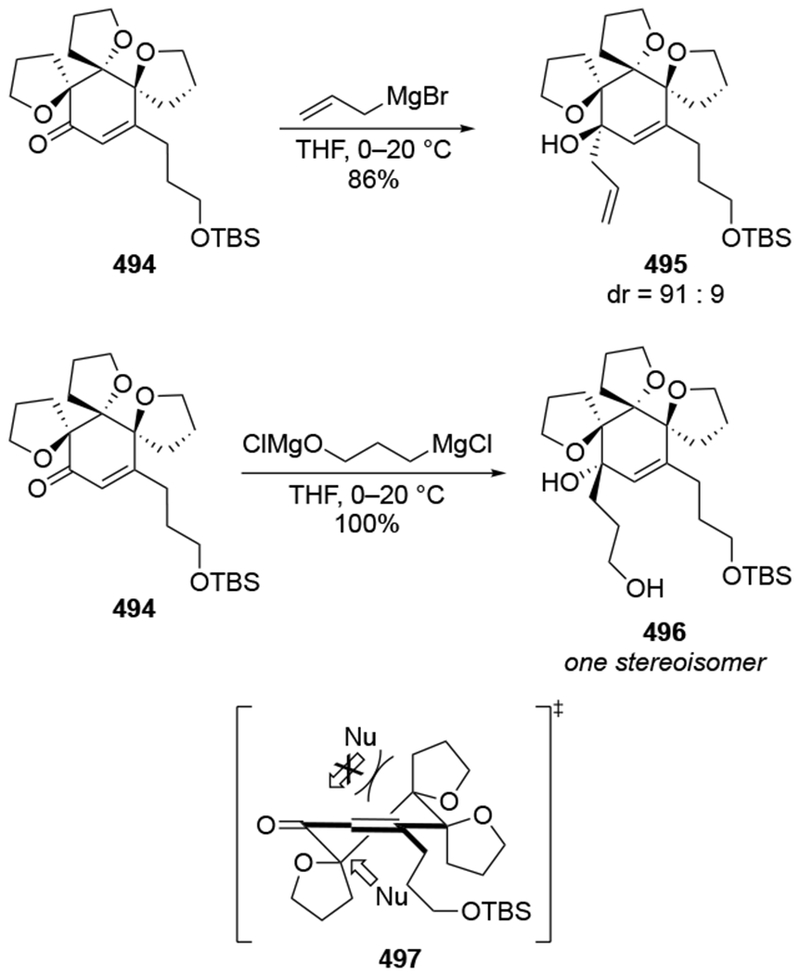
High selectivities were observed for additions to other cyclohexanones with an exocyclic alkylidene substituent. Additions of allylmagnesium reagents to ketone 498 occurred with the highest stereoselectivities of the nucleophiles examined (Scheme 194).333 The low yields observed for the other reagents reflect significant amounts of 1,4-addition, which was not observed for allylmagnesium reagents. Allylmagnesium reagents are generally more prone than other organomagnesium reagents to 1,2-addition than conjugate addition to unsaturated systems,19,334–336 which is consistent with the mechanistic analysis that they undergo addition through six-membered ring transition states with allylic transposition.18 The favored mode of addition in these cases was attributed to the goal of minimizing torsional strain upon nucleophilic attack (transition state 500), and it was argued that the six-membered ring transition state for allylation minimized the steric size of the allyl group. A related unsaturated cyclohexanone was also observed to react with high diastereoselectivity with allylmagnesium reagents with a similar sense of selectivity (Scheme 195).337 The ring likely adopts the conformer 503 to minimize allylic strain interactions that would be found in conformer 504, and addition to minimize torsional interactions would occur from the axial face.
Scheme 194.
Scheme 195.
6.2.2. Additions to Five-membered Ring Ketones
6.2.2.1. Additions to 2-Substituted Five-membered Ring Ketones
The literature on additions to five-membered ring ketones with substituents at C-2 is somewhat inconsistent. Cyclopentanones are considerably puckered,338 so controlling diastereoselectivity is possible by differentiating the two faces of the carbonyl group should be possible, just as it is for cyclohexanones. Additions of some nucleophiles to 2-methylcyclopentanone (505) can be selective, whereas others may not be (Scheme 196).71,339–341
Scheme 196.
Chelation-controlled additions also seem to proceed with little selectivity (Scheme 197).341,342 More recent examples show that methylation306 proceeded with lower selectivity than allylation (Scheme 198).343 Nevertheless, some additions of vinyllithium reagents to five-membered ketones can be diastereoselective, even in highly functionalized systems.344
Scheme 197.
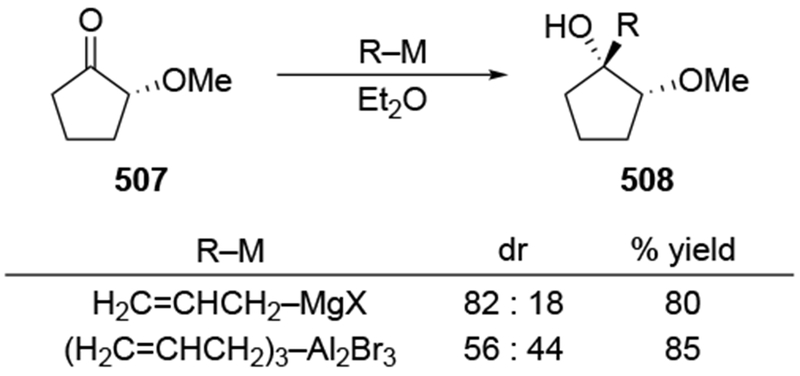
Scheme 198.
Additions to cyclopentanone derivative 513 illustrate some general trends (Scheme 199).313 Additions can be highly stereoselective, but they often need to be optimized for the appropriate choice of metal nucleophile. On the other hand, allylmagnesium reagents add readily with high stereoselectivity. Some Grignard reagents required transmetallation with CeCl3 to afford product. Similar cyclopentanones with pendant aryl-substituted alkyl groups show similar trends.315 A 2-aryl-substituted cyclopentanone has been shown to undergo highly stereoselective allylation (Scheme 200).345
Scheme 199.
Scheme 200.
The additions of nucleophiles to ketone 517 illustrate how phosphoryl groups can be used to obtain high diastereoselectivity in some cases (Scheme 201).346 Yields could be improved by first pre-mixing the Grignard reagent with BF3•Et2 prior to adding the ketone or using an allylzinc reagent, but, in the case of the cyclopentanone 517, addition of allylmagnesium itself was efficient. The steric influence of the phosphoryl group was also observed for six-membered ring ketones, forming products 519 and 520 as the major diastereomers. As an indication of the atypical reactivity of allylic organomagnesium reagents, the less reactive methyl, phenyl, and vinyl Grignard reagents only gave the products in small amounts under any conditions. Benzylmagnesium bromide also underwent addition, but not as efficiently as allylmagnesium chloride did.
Scheme 201.
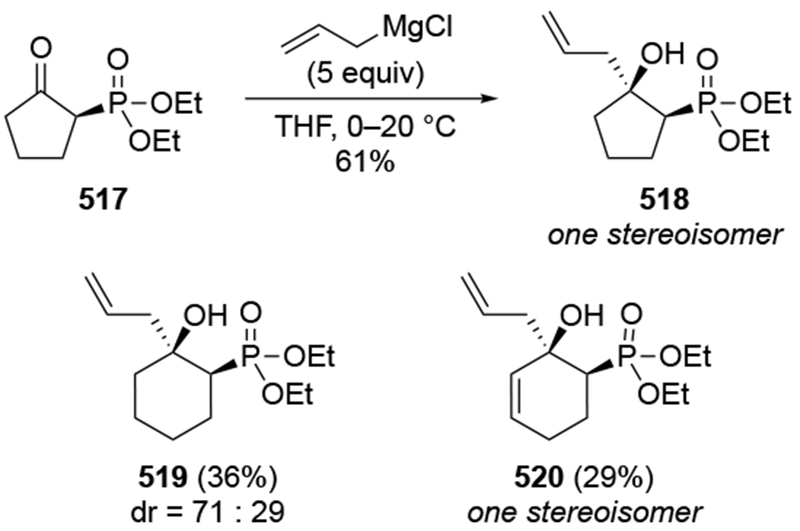
The selectivity for additions of allylmagnesium reagents to 2-substituted cyclopentanones is evident in more complex settings. Geminally substituted ketone 521 reacted stereoselectively with allylmagnesium bromide to give alcohol 522 (Scheme 202).347 This compound was used in a synthesis of a natural product. Addition likely occurred from the more accessible face of the equatorial conformer, as illustrated in transition state 523.
Scheme 202.
The presence of geminal stereocenters at C-2, however, can lead to low selectivity (Scheme 203).311 This trend is similar to what had been observed in the six-membered ring analogue of ketone 524 (i.e., ketone 442, Scheme 175), in which most organomagnesium reagents reacted with high selectivity, but allylmagnesium reagents showed lower selectivity.
Scheme 203.
A benzo-fused cyclopentanone with a geminally substituted stereocenter underwent highly selective additions with alkyl- and allylmagnesium halides (Scheme 204).348 It was suggested that the stereoselectivity arose from chelation between the ketone carbonyl group and the hindered carboalkoxyl group, so nucleophilic attack occurred from the face opposite the CF3 group. This selectivity, however, should be compared to the Felkin–Anh-selective additions to similar indanone derivatives with an SCF3 group, in which allylation proceeded with low diastereoselectivity (Scheme 122, Section 5.4).
Scheme 204.
6.2.2.2. Additions to 3-Substituted Five-membered Ring Ketones
The few reports available for additions of allylmagnesium nucleophiles to 3-substituted cyclopentanones show little selectivity for these reactions (Scheme 205).349,350 Similarly, additions of propylmagnesium bromide yielded a 55:45 mixture of stereoisomers.274 Additions of alkyllithium reagents to related substrates were also unselective.351 More highly substituted cyclopentanones with only one stereogenic center also did not undergo highly stereoselective additions (Scheme 206).352
Scheme 205.
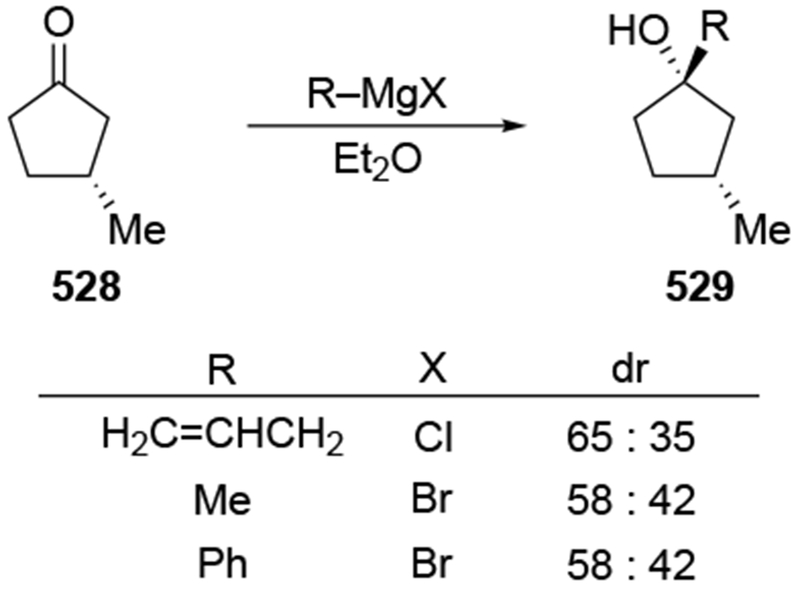
Scheme 206.
In the case of a cyclopentanone fused to an aromatic ring, stereoselectivity depended upon the nucleophile (Scheme 207).353 Addition of vinylmagnesium bromide occurred with high diastereoselectivity, presumably because the diastereotopic faces were significantly differentiated because of the ring fusion. Allylmagnesium bromide, however, added with little stereoselectivity.
Scheme 207.
6.2.2.3. Additions to Five-membered Ring Ketones with Multiple Stereocenters
Selectivity tends to be higher in cases where the ketone is more substituted, regardless of ring size. For example, addition of allylmagnesium chloride to a cyclopentanone with several potentially chelating groups occurred with high stereoselectivity (Scheme 103 and 104, Section 4.3.2). Another example of the reactivity of allylmagnesium reagents to a highly substituted cyclopentanone is illustrated by reactions of ketone 534, which were examined in the course of a synthesis of diterpenes (Scheme 208).354 Addition of an excess of allylmagnesium chloride formed the diol 535 as a mixture of diastereomers. The authors remarked that 10–20% of other compounds were separated from the major products, and these compounds were suspected to possess different configurations at the tertiary hydroxyl group (i.e., not complete selectivity of addition of the allylmagnesium reagent to the ketone functional group). It is worth noting, however, that other authors had difficulty even adding other types of carbon nucleophiles to ketones resembling 534, being successful only with sterically small alkynylcerium reagents.355
Scheme 208.
Selective allylations can be complicated when taking into account the similar reactivity of allylmagnesium reagents with electrophiles such as esters, aldehydes, and ketones.17 This selectivity has been achieved in carbohydrate systems with the use of a cerium reagent (Scheme 209).356 The similarity in outcome to the previously described system is notable (Scheme 103, Section 4.3.2), but no experiments without cerium salts were reported using ketone 536. The benzoyl group at the anomeric center migrated to the newly formed tertiary hydroxyl group, which resulted in having to re-protect the now epimerized anomeric center.
Scheme 209.
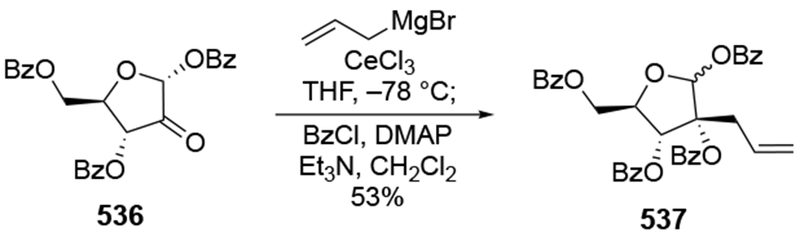
A reaction with a highly hindered five-membered ring ketone illustrates the utility of the high reactivity of allylmagnesium reagents (Scheme 210).357 Attempted additions of other carbon nucleophiles under a variety of conditions gave no addition products. Addition of allylmagnesium chloride, however, occurred at −78 °C to give the adduct as a single stereoisomer. The selectivity can be understood as involving nucleophilic attack from the top face of a low energy conformer of the ketone similar to that illustrated in Scheme 202 above.
Scheme 210.
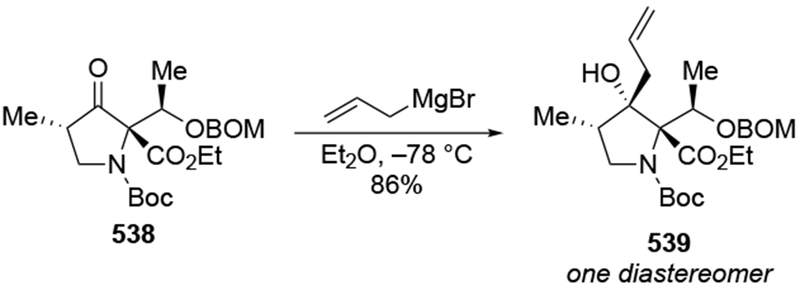
6.3. Additions to Four-membered Ring Ketones
As noted in the section describing chelation-controlled additions in cyclobutanones (Section 4.3.3), additions of allylmagnesium reagents to 3-substituted cyclobutanones proceed with low stereoselectivity. When a stereocenter is adjacent to the carbonyl group, however, these reactions can be stereoselective. For example, addition of allylmagnesium reagents to cyclobutanone 540 occurred with high anti selectivity (Scheme 211).358 This stereoselectivity is not general, however: related examples proceed with low diastereoselectivity (Scheme 212).359 Other authors report mixtures of stereoisomers upon additions of Grignard and organolithium reagents to such cyclobutanones.360 Some additions of Grignard reagents, however, proceeded with high diastereoselectivity provided that the two groups on the ring are sufficiently different in size (Scheme 213).361
Scheme 211.
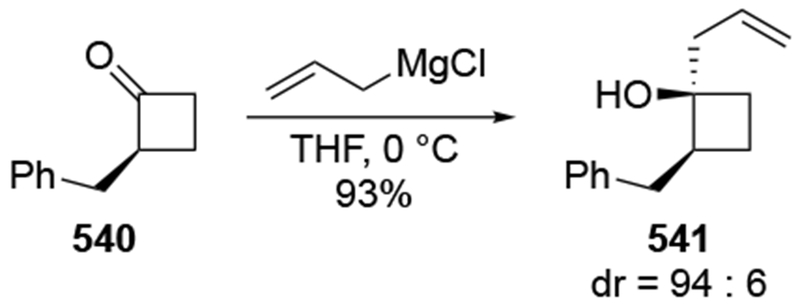
Scheme 212.
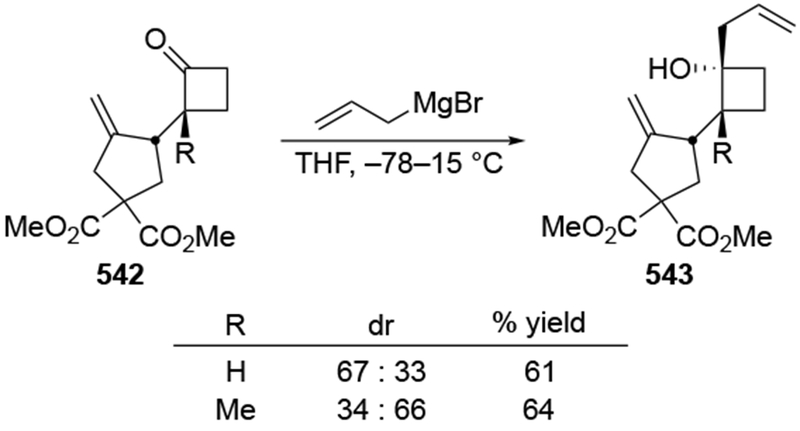
Scheme 213.
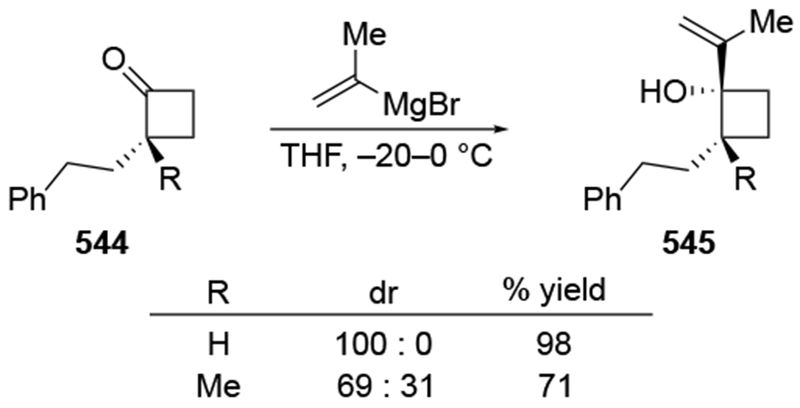
Reactions of cyclobutanediones proceed with some stereoselectivity. In the course of developing routes to terpene natural products, addition of allylmagnesium bromide to dione 547, prepared in situ from diol 546, gave mostly the cis-substituted products 548 and 549 (Scheme 214).362 The major adduct involved addition of the two allyl groups anti to the alkoxymethyl groups but syn to each other. This outcome could result from the conformational preferences of the starting ketone. The initial cyclobutanedione might be expected to adopt a conformation where a resident alkoxy group is positioned near the carbonyl group to maximize electrostatic effects.363 This hypothesis is supported by X-ray crystal structures of alkoxyalkyl-substituted cyclobutanones, which adopt conformations where the alkoxy groups approach the carbonyl carbon atoms.364–366 As a result, addition should occur from the face that is more exposed (i.e., away from the alkoxyalkyl group) as illustrated for transition structure 551 (Scheme 215). Once the first addition occurred, the resulting OMgBr group could form a chelate with the second carbonyl group (552). Addition to the second carbonyl group in the molecule would now be slower, considering that the molecule has negative charge. This addition could be slower than the diffusion rate limit, which would then allow the chelation to accelerate the addition (Section 4.1). That chelation would give the cis relationship between the two allyl groups. In the course of using these reactions in target-directed synthesis, it was noted that diastereoselectivity was higher using a substituted benzyl protecting group (Scheme 216).367
Scheme 214.
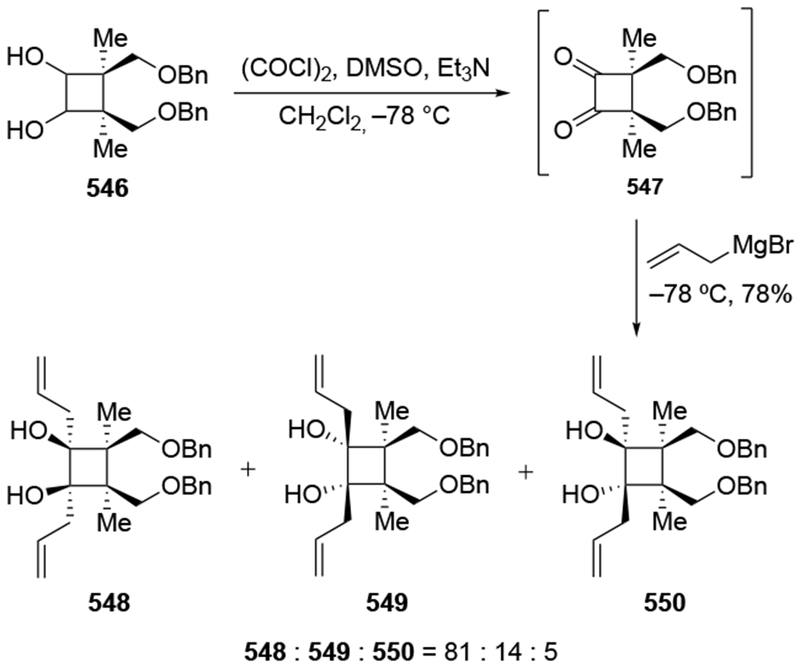
Scheme 215.
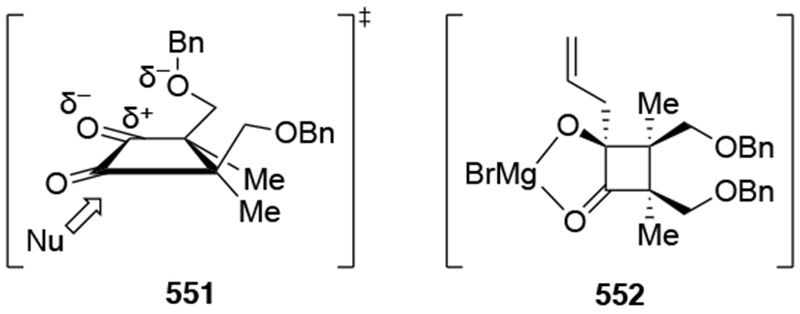
Scheme 216.
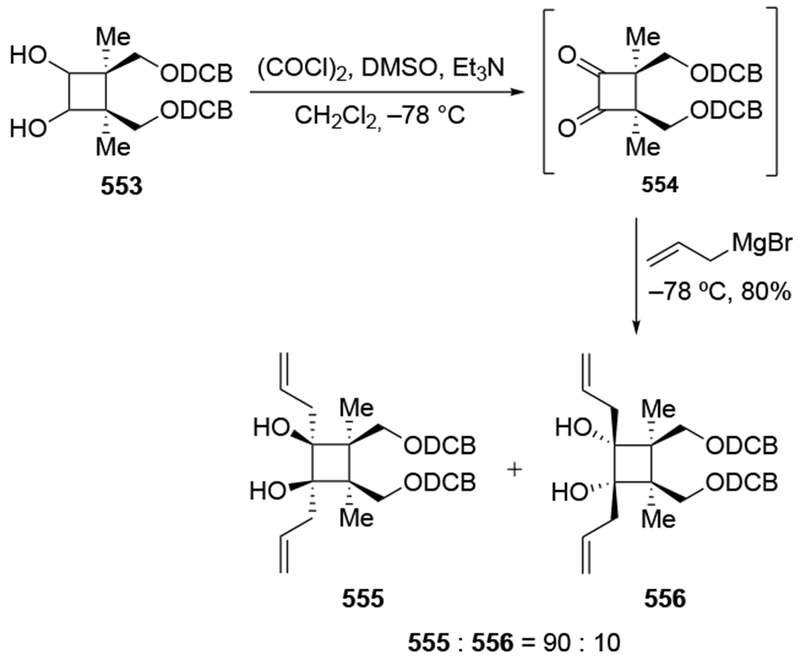
6.4. Additions to Seven-membered Ring Ketones
In general, additions of Grignard and alkyllithium reagents to 2-substituted cycloheptanones are highly stereoselective.368 This selectivity arises because additions avoid developing torsional strain in the transition state.368 Steric effects might also discourage attack from the face nearer to a substituent.
Allylmagnesium reagents also add to cycloheptanones with high diastereoselectivity. The results in Schemes 217315 and 218313 contrast with the low stereoselectivities observed for some additions of allylmagnesium reagents to 2-substituted five- and six-membered ring ketones (Sections 6.2.1.3 and 6.2.2.1, respectively). As with those other types of ketones, allylmagnesium reagents generally added more smoothly without the need to transmetallate to other metals such as cerium.
Scheme 217.
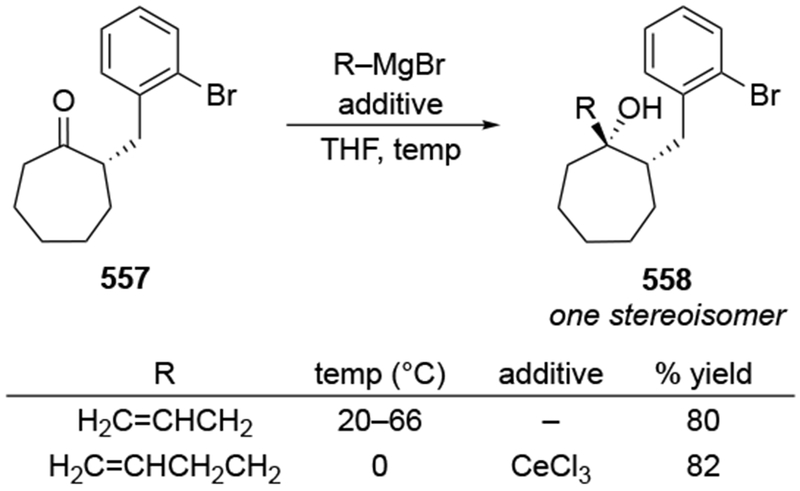
Scheme 218.
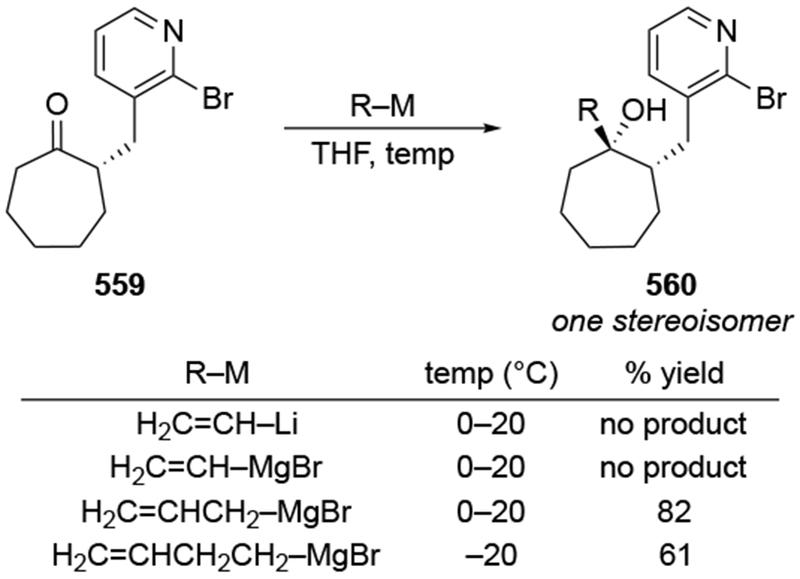
In contrast to additions to monosubstituted cycloheptanones, additions of Grignard reagents to 2,2-disubstituted cycloheptanones proceeded with generally low stereoselectivity, not just for allylmagnesium reagents (Scheme 219).311 Selectivities for additions to ketone 561 are lower than for the analogous six- and five-membered ring (Schemes 175, 176 and 203, respectively). The added flexibility of the seven-membered ring might contribute to this decreased selectivity.
Scheme 219.
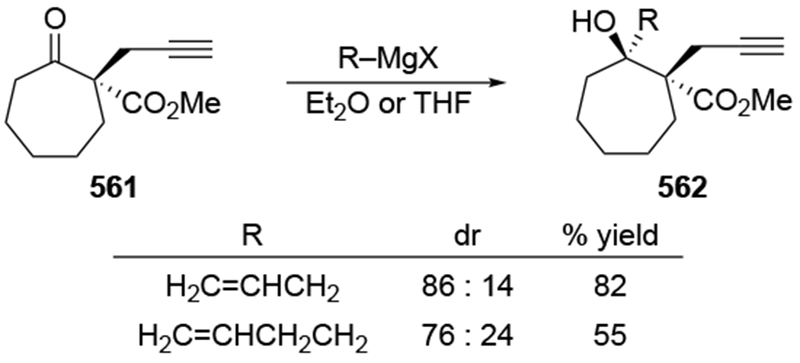
A geminally disubstituted cycloheptanone fused to an aromatic ring, however, underwent highly stereoselective allylation (Scheme 220).369 No explanation was given for why this reaction was stereoselective, nor was a comment made regarding why cerium salts were used, which are often used to suppress enolization or conjugate addition.370 The X-ray crystal structure of alcohol 564, however, provides some insight into the overall structure of this ring system. The lowest energy conformation of the ketone 563 likely resembles 565, where the branched group blocks the lower face. Nucleophilic attack from the more exposed top face would lead to the observed product.
Scheme 220.
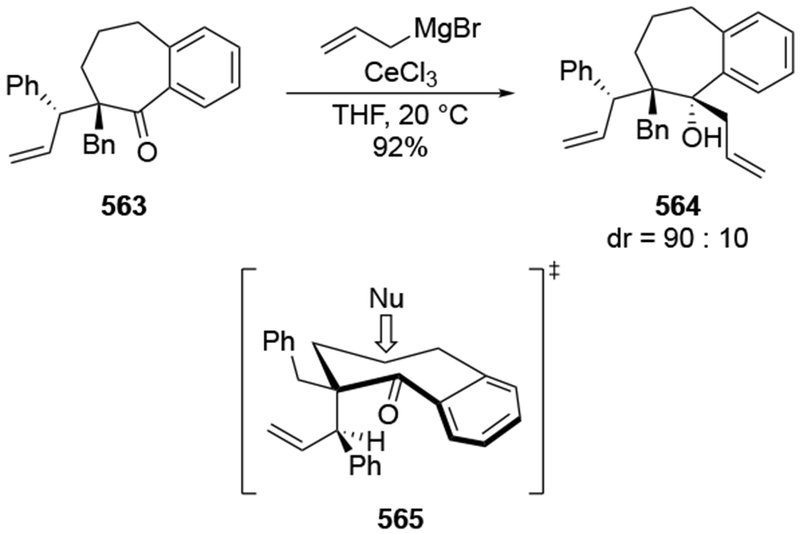
Additions of allylmagnesium reagents to disubstituted cycloheptanones can show some selectivity. The allylation of 2,3-disubstituted isomer 566 (Scheme 221)327 formed the product expected for additions to a 2-substituted cycloheptanone.368 The magnitude of the diastereoselectivity matches the selectivity observed for additions to a similar trans-substituted cyclohexanone (Scheme 188).
Scheme 221.
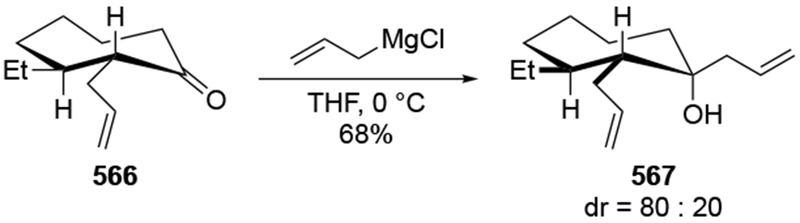
Addition of allylmagnesium bromide to a cis-2,7-disubstituted cycloheptanone was diastereoselective (Scheme 222).371 Ketone 568 was isolated as a mixture of diastereomers (80:20), with the major isomer characterized as the cis isomer. Addition of allylmagnesium bromide to that mixture gave one major isomer of product, identified by the authors as 569 because the allyl group should have added opposite to the two methyl groups. The minor isomer, 570, was assigned as the product arising from the minor amount of trans-substituted ketone present in the starting material.
Scheme 222.
As observed for a geminally substituted seven-membered ring ketone fused to an aromatic ring (Scheme 220), addition of allylmagnesium chloride to a vicinally substituted cycloheptanone fused to an aromatic ring occurred with high diastereoselectivity (Scheme 223).372 By comparison, addition to a related five-membered ring ketone exhibited low stereoselectivity (Scheme 207).
Scheme 223.
The reactivity of a highly substituted seven-membered ring ketone illustrates the high selectivity that can be observed with allylmagnesium reagents even when other reagents do not add (Scheme 224).373 Addition of allylmagnesium chloride to ketone 573 at −98 °C gave predominately one stereoisomer of tertiary alcohol 574. The selectivity can be explained by considering the X-ray crystal structure of ketone 573, which resembles conformer 575. The one face is blocked by one of the large tert-butyl groups, so attack occurred from the other face. It was noted that other Grignard reagents did not react with ketone 573; only reactions of allylmagnesium and LiAlH4 were observed, which is in accordance with the high reactivity of allylmagnesium reagents.
Scheme 224.
6.5. Additions to Eight-membered Ring Ketones
Nucleophilic additions to substituted cyclooctanones are not common reactions. Felkin–Anh type additions of organometallic reagents have been observed for cyclooctanones with α-SPh groups,374 but few examples of additions to alkyl-substituted cyclooctanones have been reported.375,376 Such reactions can be stereoselective: addition of an alkenyllithium reagent to a 2-substituted cyclooctanone proceeded with high selectively, although the yield was not reported (Scheme 225).377
Scheme 225.
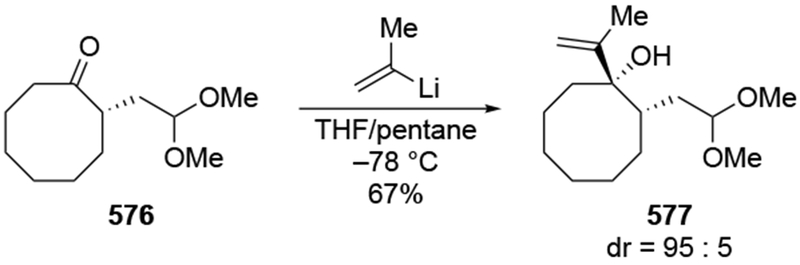
Few examples of allylations of substituted cyclooctanones have been reported. Addition of allylmagnesium chloride to a 2,3-disubstituted cyclooctanone gave low stereoselectivity, (Scheme 226), which was slightly lower selectivity than observed for the seven-membered ring system (Scheme 221).327 Across this series of six- to seven- to eight-membered rings, the selectivity for allylation gradually decreased.327 Similarly, reactions of 3-alkoxy-substituted cyclooctanones were not selective with any nucleophile, including allylmagnesium halides (Scheme 227).378 The generally low stereoselectivity in these substrates may reflect the number of potential conformers of a substituted cyclooctanone,379 which could allow nucleophiles to approach both faces of the carbonyl group.
Scheme 226.
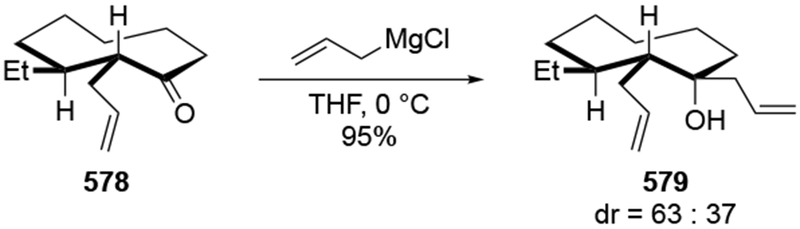
Scheme 227.
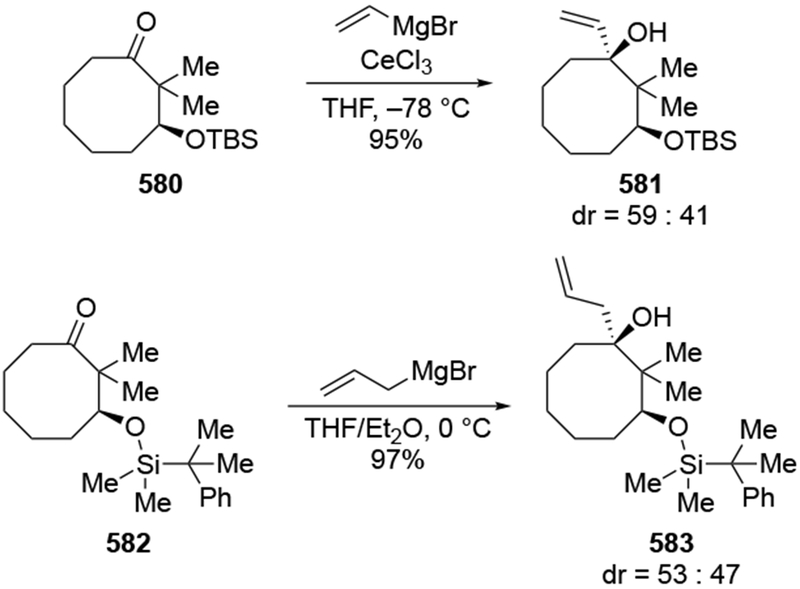
Reactions of conformationally constrained eight-membered ring ketones illustrate the relationship between the topography of an electrophile and the stereoselectivities of its reactions (Scheme 228).380 Two related ketones, 584 and 586, differing only in a substituent on an sp2-hybridized carbon atom, gave products with the opposite configuration upon exposure to nucleophiles. The difference in stereochemical outcomes can be traced to the conformational preferences of the starting ketone, as determined by X-ray crystallography. In the case of ketone 584, the conformer 588 with the Ph group equatorial was favored. Increasing the steric size of the group from SiMe3 to tert-butyl, however, changed the conformational preference to favor the pseudoaxial ketone 589 to minimize allylic strain. In both cases, nucleophilic attack from the more exposed convex face would lead to the observed products. The additions of the nucleophiles, therefore, involve identifying which face of the ketone is more exposed, as required by a steric approach control argument. The results with allylmagnesium halide can be explained by considering that even particularly fast reactions should also be governed by the relative ability of the nucleophile to approach the carbonyl group from the different faces.
Scheme 228.
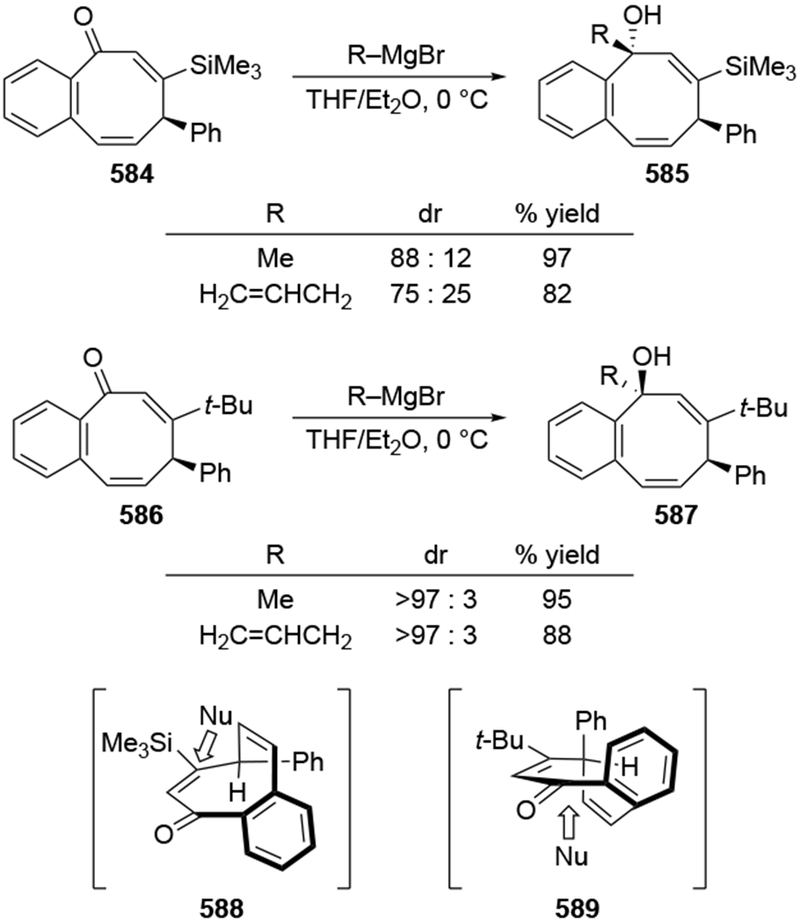
Additions to another eight-membered ring electrophile emphasize the importance of the conformation of a medium-sized ring and the stereochemical courses of its reactions.269 Although not an addition to a ketone, addition of allylmagnesium halides to the eight-membered ring bis(imine) 590 was controlled by the ability of the nucleophile to approach the electrophile (Scheme 229). The low energy conformation of the ring of 590 differentiates the faces of the ring, and nucleophiles can only add from the convex face.381 This substrate was prone to additions under single-electron additions, which could be operative in this case. It is worth noting, however, that addition of tert-butylmagnesium halides led to no reaction, but allylation was clean and highly diastereoselective.
Scheme 229.
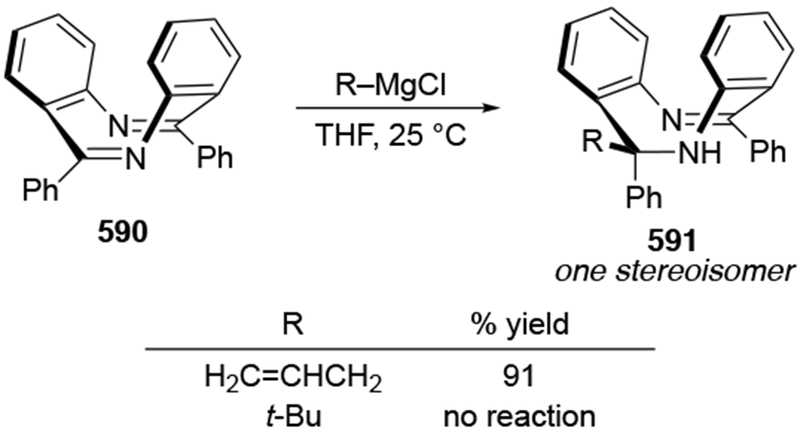
6.6. Additions to Nine-membered Ring Ketones
The same conformational control of stereoselective allylation can be observed in a conformationally constrained nine-membered ring trans-alkene with an endocyclic carbonyl group (Scheme 226).382 Addition of allylmagnesium chloride to either chiral, optically active alkene 592 or its derived epoxide 594 occurred with high diastereoselectivity. This stereochemical course can be explained by involving addition to the more exposed convex face of the chiral ketone. Reduction with NaBH4 also occurred from the same face.
6.7. Additions to Cyclic Ketones with Other Rings
As substrates become more complex by involving more rings, it becomes more difficult to categorize the type of substrate involved. The following sections focus on reactions of cyclic ketones where another ring is fused to it adjacent to the carbonyl group, where the ring is bridged, or where there are multiple rings involved. Readers wishing to know whether a planned addition of allylmagnesium to a cyclic ketone bearing other rings might be stereoselective should consult all sections to identify the best analogies to any proposed system.
6.7.1. Additions to Fused Cyclic Ketones
6.7.1.1. Additions to Fused Six-membered Ring Ketones
Reactions of a cyclohexanone with a fused epoxide ring (i.e., a fused [4.1.0]-bicyclic system) can lead to high diastereoselectivity for additions of various organometallic reagents (Scheme 231).383 With all nucleophiles examined, including allylmagnesium bromide, a single stereoisomer of product was formed. Other authors have observed similar selectivities with additions of nucleophiles to other epoxy ketones,384 although reactions of other epoxy ketones show low diastereoselectivity.385 This phenomenon was also observed for fused cyclopentanones (as will be shown in Section 6.7.1.2). The selectivity could result from attack from the more exposed convex face of the highly substituted fused bicyclic ring system. Electronic influences of the nearby epoxide ring cannot be discounted, although in general α,β-epoxy carbonyl compounds do not undergo highly diastereoselective addition reactions.386,387
Scheme 231.
Addition of an excess of allylmagnesium bromide to the highly substituted epoxy ketone 598 gave a single stereoisomer of the adduct 599 (Scheme 232).388. As with the above example (Scheme 231), the nucleophile added from the convex face. The authors proposed that a chelating interaction resembling 600 was responsible for selectivity. Such a chelated intermediate need not be invoked to explain the observed stereochemistry, however, considering the results shown in Scheme 231.
Scheme 232.
Additions to ketones residing in a cis-fused decalin-like ring system gave high stereoselectivity (Scheme 233).389 Addition occurred exclusively from the more accessible convex face of the fused system.
Scheme 233.
Additions of organometallic reagents to trans-fused decalin-derived ketones generally occur from the equatorial face. For example, addition of allylmagnesium bromide to the simple decalinone 603 gave the equatorial addition product as the major product (Scheme 234).282 The magnitude of this selectivity is not significantly different from selectivities observed for additions of methyllithium and methylmagnesium iodide (Scheme 235), showing a general preference for equatorial attack.390 The same preference for equatorial attack on a trans-decalin system was observed for addition of allylmagnesium bromide in a more highly substituted case (Scheme 236).391
Scheme 234.
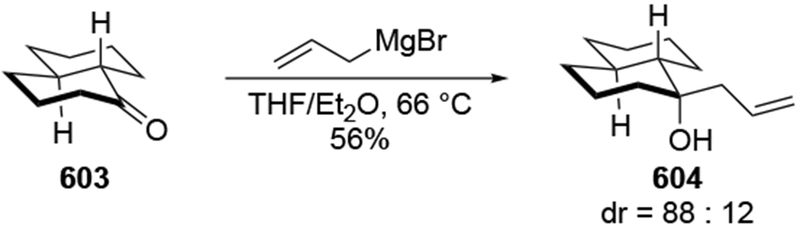
Scheme 235.
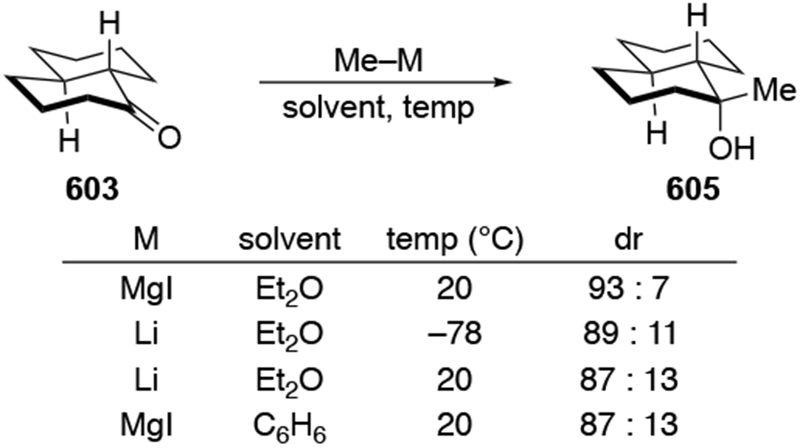
Scheme 236.
Attempts to obtain chelation-controlled allylation of a fused six-membered ring ketone were particularly difficult (Scheme 237).392 It was noted that the selectivity was sensitive to the amount of the ether that was present in the solutions of Grignard reagent employed. The reaction was optimized by removing all ether and performing the reaction in a mixture of toluene and CH2Cl2. Even so, only modest stereoselectivity was observed. The stereochemical outcome of this reaction was explained by considering a fused-ring chelate 610. As noted in Section 4.1, however, control of addition of allylmagnesium reagents by chelation is not as straightforward as chelation-controlled additions for other organomagnesium reagents.
Scheme 237.
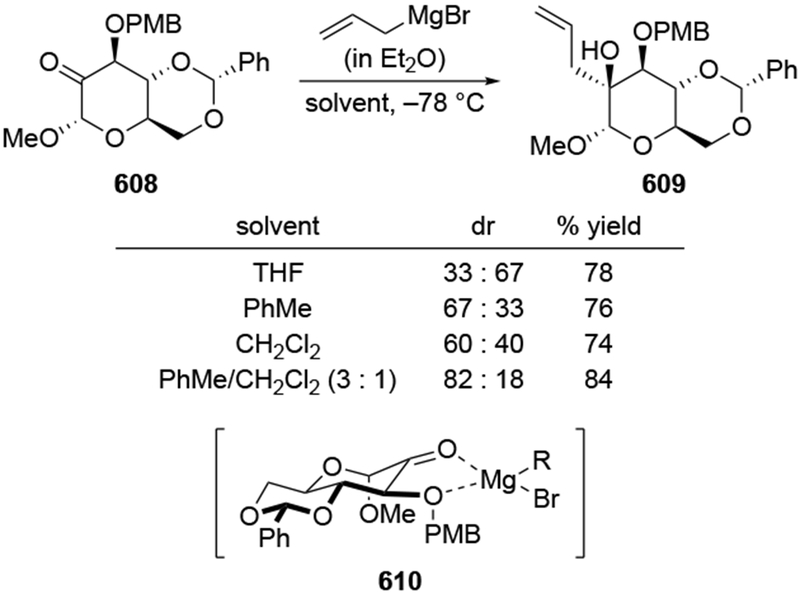
The stereoselectivities of allylations of related fused six-membered ring ketones are highly sensitive to the precise structure of the fused ring (Schemes 238–240).10,392 With the cis ring fusion in acetonide 611, addition gave high selectivity for addition from the convex face by transition state 613 (Scheme 238). In the case of another highly substituted fused cyclohexanone, additions were generally highly stereoselective for all organomagnesium reagents examined (Scheme 239). This selectivity was independent of protecting group at the adjacent α-oxygen atom (Scheme 240).
Scheme 238.
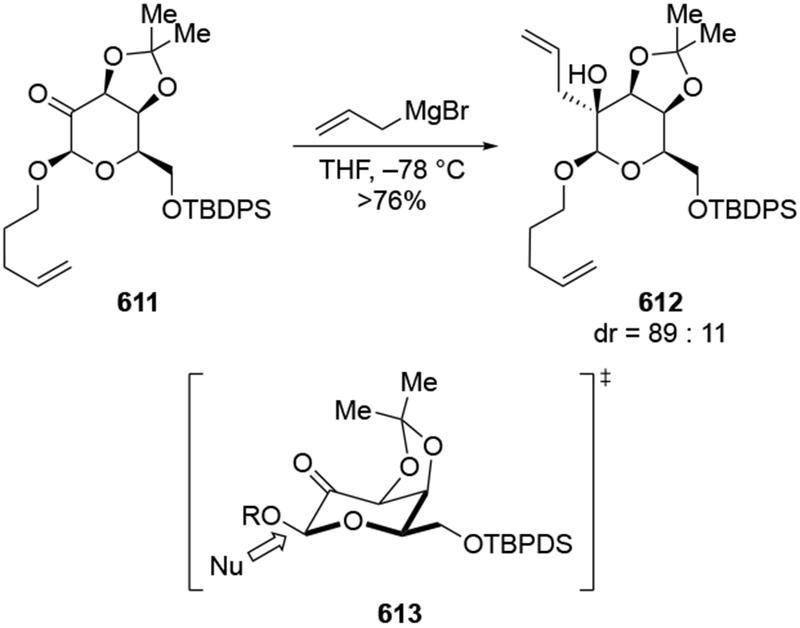
Scheme 240.
Scheme 239.
Allylation of a simple fused enone 618 shows the utility of allylmagnesium reagents in certain settings (Scheme 241).393 Initially, this reaction was attempted using allylsilanes or allylstannanes in the presence of a Lewis acid. Those efforts, however, led only to deprotection of the acetal. Addition of allylmagnesium chloride, however, proceeded in high yield with a level of diastereoselectivity that allowed for completion of the synthesis of a natural product.
Scheme 241.
Additions to other fused cyclohexenones gave similar stereoselectivities, even with other organometallic reagents. For example, allylation of cholest-3-one (620) gave the β-allylated product with 86:14 selectivity, although no reaction conditions were provided (Scheme 242).286 Addition of a vinyl anion derived from a glucal gave similar selectivities for alcohol 622 (the figure in this paper shows the opposite configuration C-14, but this figure is likely to be a typographical error considering that 4-cholestene-3-one 620 was used as the starting material for the synthesis of 622).394
Scheme 242.
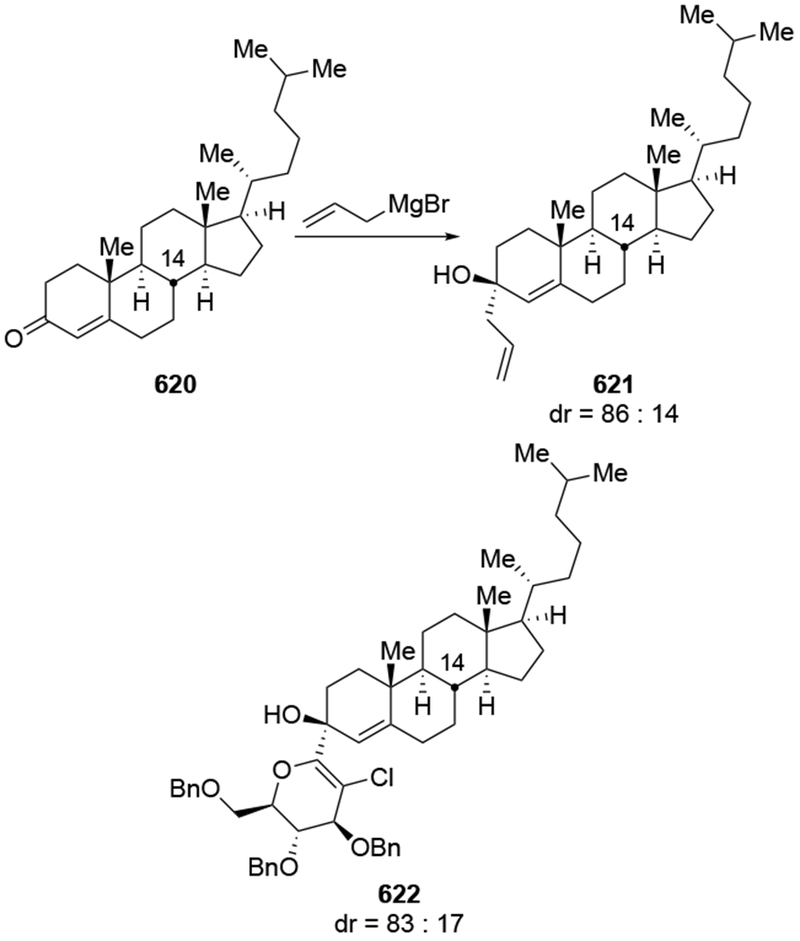
Reactions of a fused-ring vinylogous amide illustrate the high reactivity of allylmagnesium reagents (Scheme 243).395 Whereas additions of allyltrimethylsilane and allyltributylstannane in the presence of Lewis acids resulted in no reaction, addition of allylmagnesium reagents were successful, although details of this experiment were not provided. The use of allylic cuprates gave the 1,4-products as a mixture of diastereomers. Conjugate addition was ultimately successful using the more reactive396 methallyltributylstannane reagent with the iminium ion 625 generated in situ.
Scheme 243.
A related highly conjugated fused cyclohexanone underwent highly stereoselective additions with all organometallic reagents, including allylmagnesium halides (Scheme 244).397 This stereoselectivity can be attributed to the steric congestion at the carbonyl group so that additions of allylmagnesium reagents are unlikely to occur at rates approaching the diffusion rate limit. Attack from the more accessible face of the ketone, as illustrated in transition state 629, would lead to the observed product.
Scheme 244.
Reactions of a series of highly substituted steroid derivatives with the carbonyl group in the B ring occurred with high stereoselectivity. With a relatively simple ketone, addition from the more accessible α-face proceeded with modest selectivity (Scheme 245).398 Under these conditions, addition to the acetoxy group likely also occurred to remove the protecting group, considering that additions of allylmagnesium reagents to ketones and esters occur at competitive reaction rates (Scheme 12, Section 2.2). With the more substituted ketone 632, however, addition was more stereoselective (Scheme 246).399 The origin of this increased stereoselectivity is not obvious considering the structural similarity between the two compounds. Similar diastereoselectivities were reported for the formation of products 634 and 635.
Scheme 245.
Scheme 246.
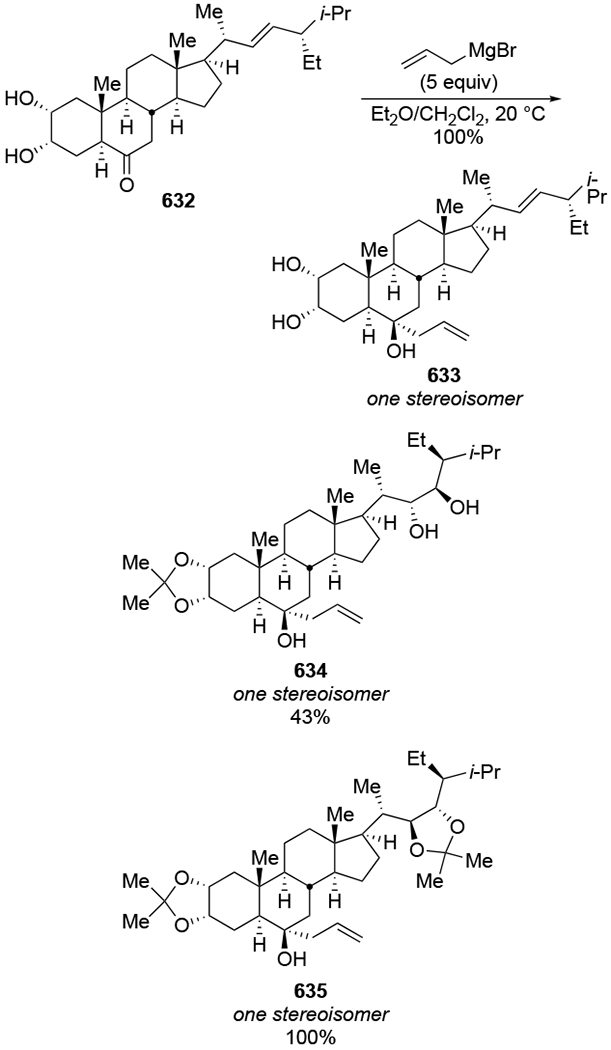
Fusion of a six-membered ring ketone to a six-membered ring does not guarantee a stereoselective addition, however (Scheme 247).400 It is likely that ketone 636 is too flat to provide much differentiation between its diastereotopic faces. Conversely, addition to the ketone 638 was highly stereoselective at the newly formed stereogenic center. The carbonyl group that was attacked in this substrate is considerably more sterically congested, as evidenced by the low conversion to product upon addition of the nucleophile. This steric congestion likely prevents attack from the top face of the carbonyl group, leading to high diastereoselectivity.
Scheme 247.
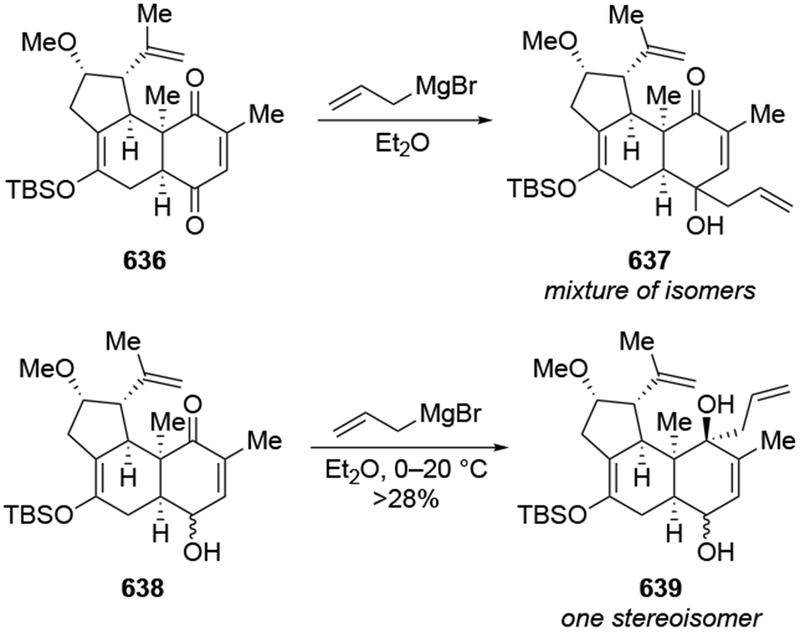
6.7.1.2. Additions to Fused Five-membered Ring Ketones
In contrast to reactions of fused six-membered ring ketones, additions of allylmagnesium reagents to fused five-membered ring systems have been studied less. One example, shown in Scheme 248,384 is analogous to observations for a six-membered ring fused system (Schemes 231–232, Section 6.7.1.1). Reactions of other Grignard reagents with ketone 640 gave similar diastereoselectivities.
Scheme 248.
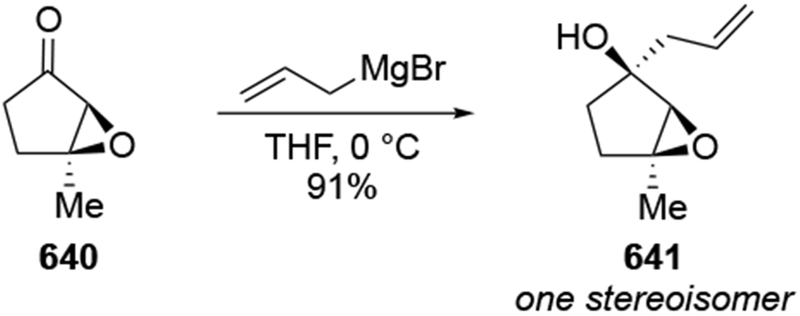
In the case of a multiply fused system, addition of allylmagnesium bromide was highly stereoselective (Scheme 249).401 The nucleophile approached ketone 642 from the side opposite to the fusion with the five-membered ring compared to the fusion with the four-membered ring, as illustrated in transition state 644. It is worth noting that the acyloxy group was also attacked by the reagent, as expected based upon the similar rates of additions of allylmagnesium reagents to ketones and esters (Scheme 12, Section 2.2).
Scheme 249.
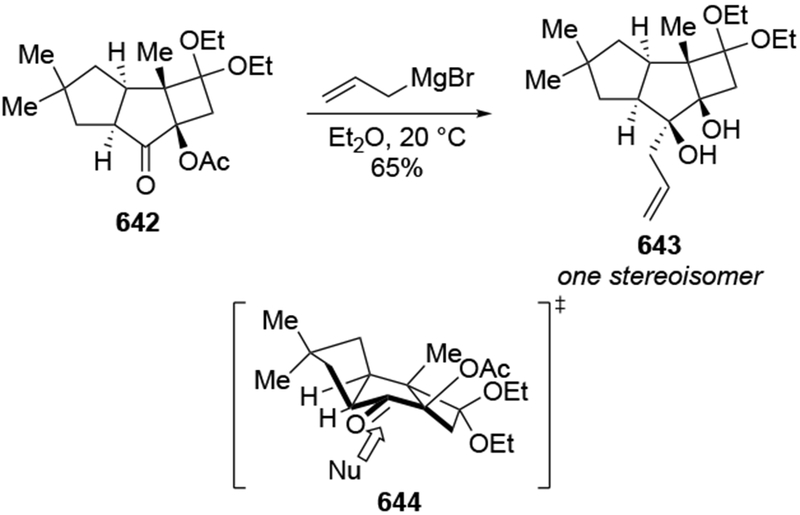
Cis-fused cyclopentanone 645 underwent additions with generally low diastereoselectivity for a number of organometallic reagents (Scheme 250).402 Computational studies suggested that lack of stereoselectivity in this case was the result of two modes of attack that could occur with similar rate constants. Attack from the exposed convex top face was expected for ketone 645 considering the highly stereoselective addition to fused cyclopentanone 647, which cannot chelate (Scheme 251). For ketone 645, however, it is possible that chelation to the methoxy group would promote addition from the more congested concave face. This chelation would involve a group at the γ-position, which is an unusual mode of chelation control that does not generally proceed with high stereoselectivity with magnesium reagents.403 Nevertheless, this mode of chelation might compete with the inherent preference for addition from the convex face, leading to an unselective reaction.
Scheme 250.
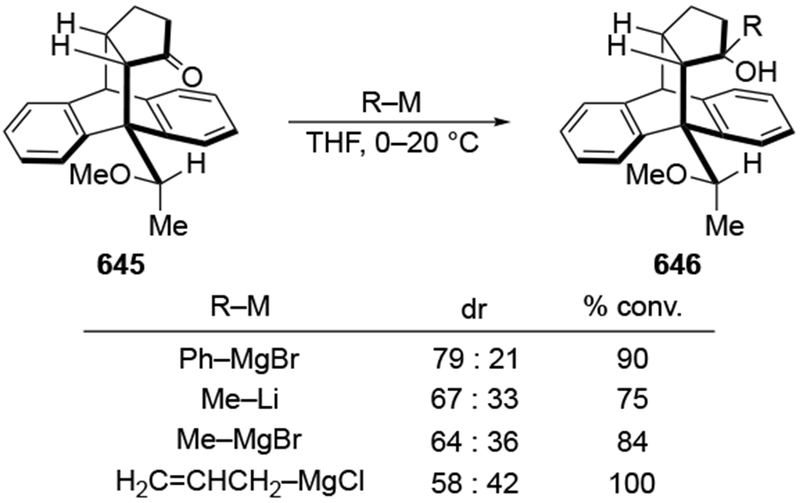
Scheme 251.
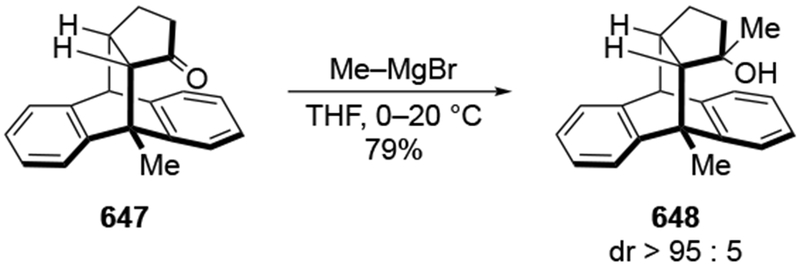
Steroid-derived fused cyclopentanones underwent highly diastereoselective reactions. Addition of allylmagnesium reagents occurred in a 1,2-fashion to enone 649 to give the α-allylated product 650 (Scheme 252).404 In later studies, it was found that selectivities for the 1,2-addition products were higher when these reagents were transmetallated to form the allylaluminum reagents (Scheme 253).405 High diastereoselectivity was observed for addition of these reagents from the α-face. These selectivities can be understood by examining the conformation about the enone ring. Attack from the β-face is blocked by the axial methyl group, so attack should be favored from the α-face (as illustrated by transition state 655, Scheme 254).
Scheme 252.
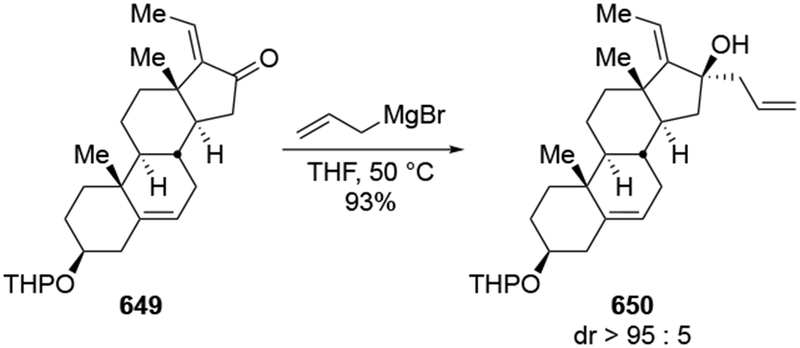
Scheme 253.
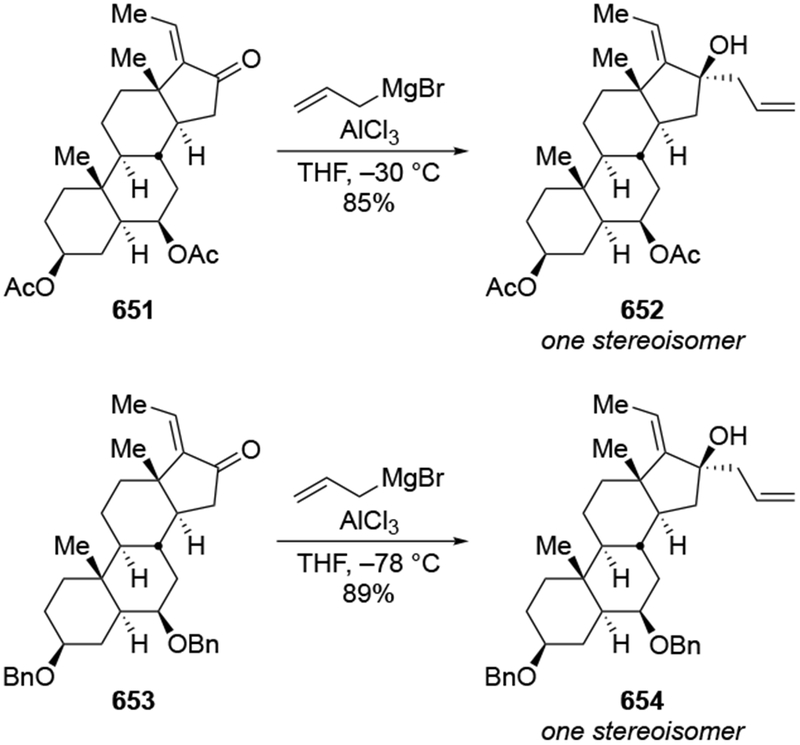
Scheme 254.
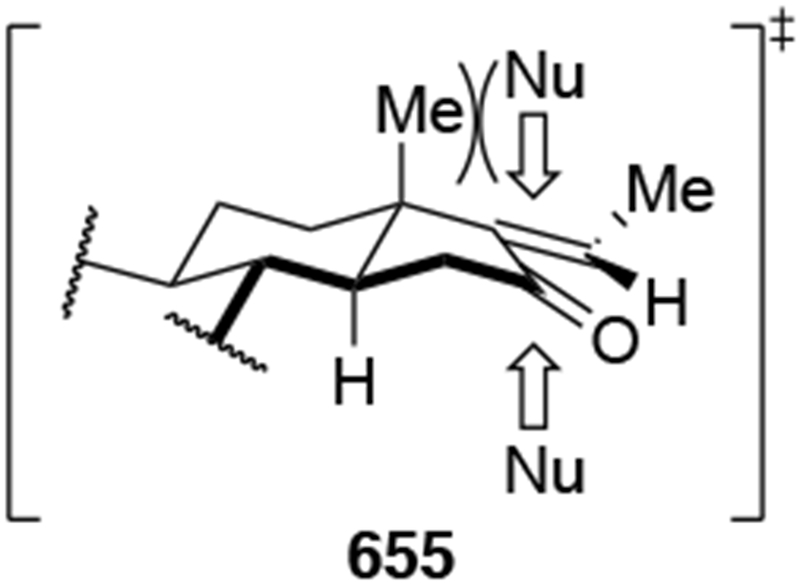
6.8. Additions to Bridged Bicyclic Ketones
Additions of allylmagnesium nucleophiles to bridged bicyclic ketones are generally diastereoselective. Because of the bridges of these compounds, they tend to be more sterically crowded than other ketones, with interactions resembling eclipsed or syn-pentane interactions developing upon addition from any face. These reactions are also not likely to occur at rates approaching the diffusion rate limit considering how sterically congested they are.17,23 The stereochemical preferences in these cases are relatively easy to predict and explain as occurring by preferential attack from what appears, at first glance, to be the more sterically accessible diastereotopic face.
6.8.1. Additions to Bridged Bicyclic Ketones with the Carbonyl Group in the One-Carbon Bridge
Additions to ketone 656 illustrate the generally high selectivity observed for additions of allylmagnesium halides to bridged bicyclic ketones with the carbonyl group in the one-carbon bridge (Scheme 255). In the course of a natural product synthesis, the allylation of ketone 656 was shown to proceed with preferential addition over the more flattened, less sterically encumbered face bearing the carbon–carbon double bond.406 It was essential in this case to install an allyl group, which was later elaborated to a tetrahydrofuran moiety by a hydroboration and oxidation sequence.
Scheme 255.
Similar additions to hindered bicyclic ketone 658 gave highly diastereoselective additions (Scheme 256).13 Under these conditions, the ester group was also removed, as would be expected from rapid addition also to this carbonyl group, as discussed in Section 2.2. Just as for ketone 656, attack occurred over the less hindered, more flattened bridge bearing one trigonal carbon atom. Similar observations were made with a related ketone 660 (Scheme 257).
Scheme 256.
Scheme 257.
Experiments conducted in the course of developing a library synthesis using microfluidics technology provide valuable insight into reactions of bicyclic ketones and illustrate the unique reactivity of allylmagnesium reagents (Scheme 258).12 Addition of alkyl-, alkenyl-, or arylmagnesium bromides to dione 662 gave only addition to the enone portion of the molecule. Addition occurred from the face with the smaller, less sterically demanding bridge (additions to ketones in longer bridges will be discussed in detail in the next section). No addition occurred at the bridging carbonyl group, however, indicating that additions to this carbonyl group are relatively slow.
Scheme 258.
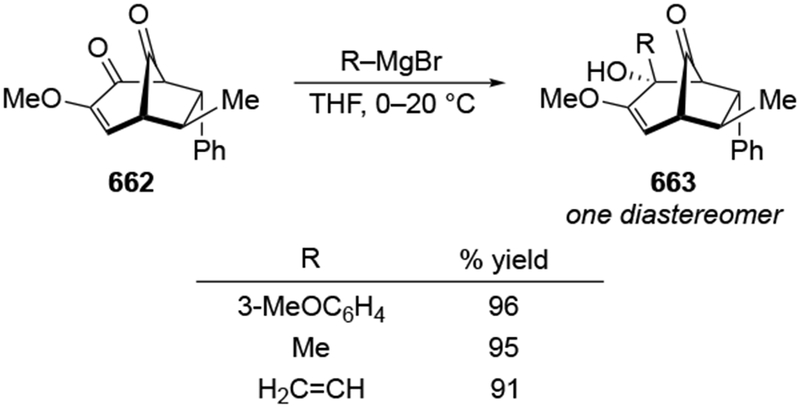
When a smaller nucleophile, an alkynylmagnesium reagent, was employed, addition to ketone 662 occurred to one or the other but not both of the two carbonyl groups (Scheme 259). This result suggests that steric approach had determined the diastereoselectivity. It is worth noting that the alkynylmagnesium reagent added only to one of the carbonyl groups of 662 and that addition occurred to the bridging carbonyl group from the less sterically encumbered face, again on the side with the trigonal planar carbon atoms. It is likely that once addition occurred to the bridging carbonyl group, it is not possible to add a second equivalent of reagent to the enone portion of the magnesium alkoxide 666 because both faces are sterically congested.
Scheme 259.
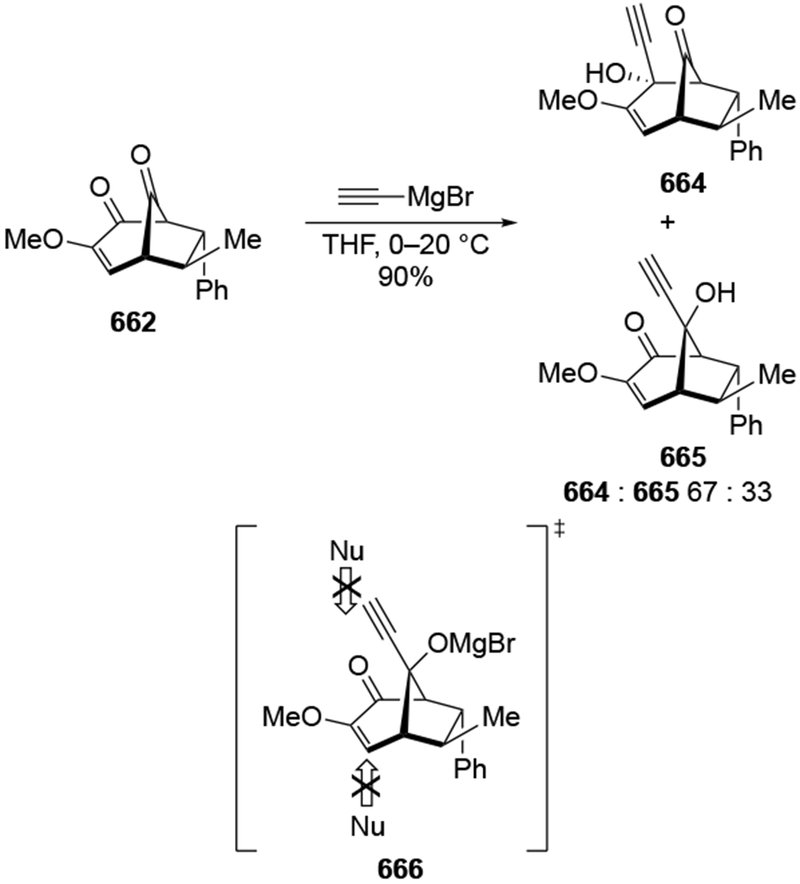
Addition of allylmagnesium bromide to ketone 662 gave completely different results (Scheme 260). This reaction gave diol 667, which resulted from addition to both carbonyl groups. Allylmagnesium bromide was the only nucleophile that added to both carbonyl groups. The configuration of 667, particularly on the one-carbon bridge, suggests that addition occurs in a specific order. Addition must first occur to the enone functional group from the less hindered face to give alkoxide 668. The subsequent step would involve allylation at the bridgehead carbonyl group. That the second addition does not occur near the trigonal planar atoms, as it had with the propargyl nucleophile (Scheme 259), may result because of the added hindrance of the endo allyl group. This argument is supported by the observation that addition of allylmagnesium bromide to ketone 669 (Scheme 261) occurred with the same sense of stereoselectivity as the second addition illustrated in Scheme 260.
Scheme 260.
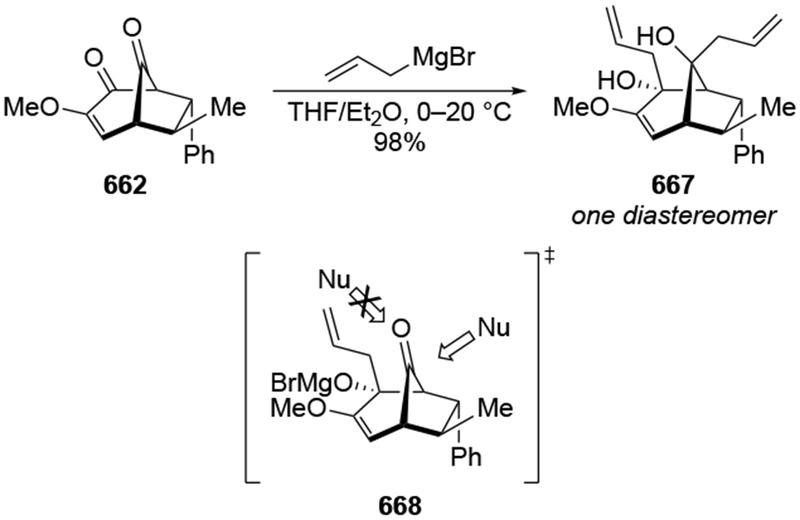
Scheme 261.
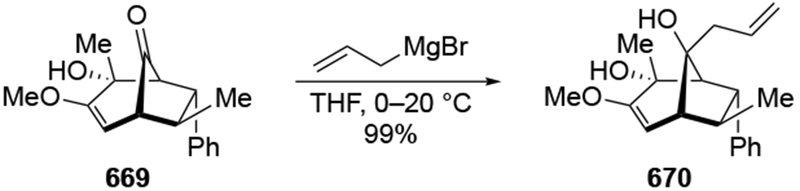
Not all additions to bridging carbonyl groups are stereoselective. In the course of a synthesis of an alkaloid, addition to ketone 671 was observed to occur with low stereoselectivity (Scheme 262).16 No comment was made regarding the choice of CH2Cl2 as the solvent, although the use of a non-coordinating solvent suggests that the authors were attempting to encourage chelation control.22 The low stereoselectivity observed is likely due to the fact that the two faces are not sterically strongly differentiated. Despite the low stereoselectivity, the allyl group was essential to the synthesis because it was converted to a carbonyl group that could be used for fragment coupling.
Scheme 262.
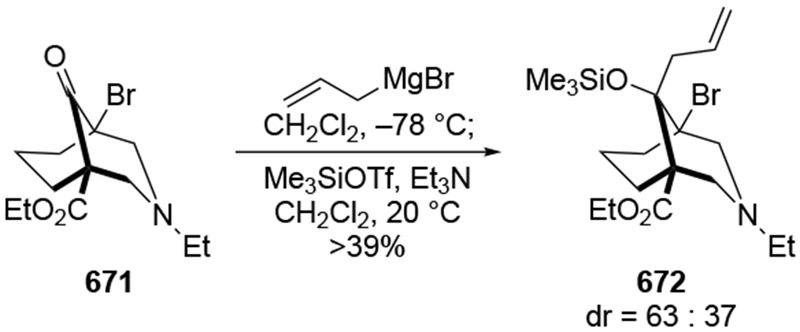
Similar issues of stereoselectivity were observed for bicyclic ketone 673, which also point to an issue of the heightened reactivity of allylmagnesium reagents (Schemes 263 and 264).407 Addition of ethylmagnesium bromide occurred slowly at low temperatures (−65 °C), so the reaction mixture needed to be warmed (Scheme 263). This higher temperature, however, caused some reduction to form alcohol 675, presumably by β-hydrogen atom transfer from the ethylmagnesium bromide, which can be a common pathway with hindered ketones and in certain solvents.408,409 The presence of the reduced products attests to the hindrance of the carbonyl group. The major product was formed by addition over the more flattened fused aromatic ring. By contrast, addition of allylmagnesium chloride occurred at −78°C with relatively low diastereoselectivity (Scheme 264), a point the authors suggest may be due to its enhanced reactivity (as noted in Section 2.2, reactivity-selectivity correlations can only be expected as reaction rates approach the diffusion limit244). The temperature needed to be controlled carefully with allylmagnesium reagents, however. If the reaction mixture were warmed to −25 °C, the product 677 was formed. This product results from both addition to the carbonyl group and addition to the pyridine ring, resulting in a nucleophilic aromatic substitution. As with the previous example, the allyl group was necessary to complete the synthesis, because it was used to form a ring to the nitrogen atom attached at the bridgehead carbon atom.
Scheme 263.
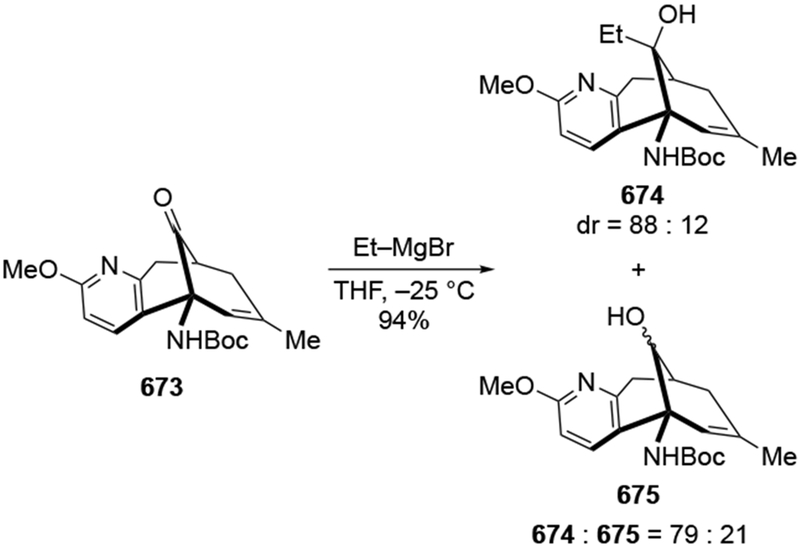
Scheme 264.
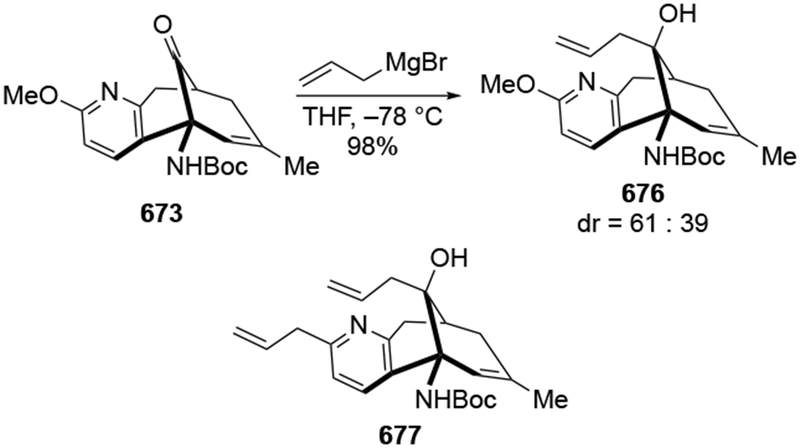
A similar substrate with a saturated ring also exhibited similar low stereoselectivity.(Scheme 265).410 The allyl group was used for a similar annulation protocol to form an additional ring.
Scheme 265.
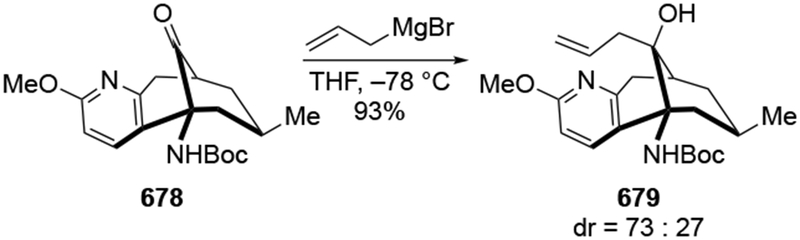
The reactions of the highly hindered bridged ketone 680 provide other examples of the dramatic differences of reactions with allylmagnesium halides compared to other reagents (Scheme 266).411 Addition of the Grignard reagent substituted with the OMgCl group gave preferential attack from the face bearing the two oxygen atoms poised to engage in chelation. Allylmagnesium bromide, on the other hand, added from the more accessible face that would develop fewer 1,3-diaxial interactions, as might be expected from such a highly reactive reagent. It should be noted that hydride reagents, including Zn(BH4)2, attacked from the same face as allylmagnesium bromide. The lack of chelation control with the zinc reducing reagent is different than its usual reactivity.412,413
Scheme 266.
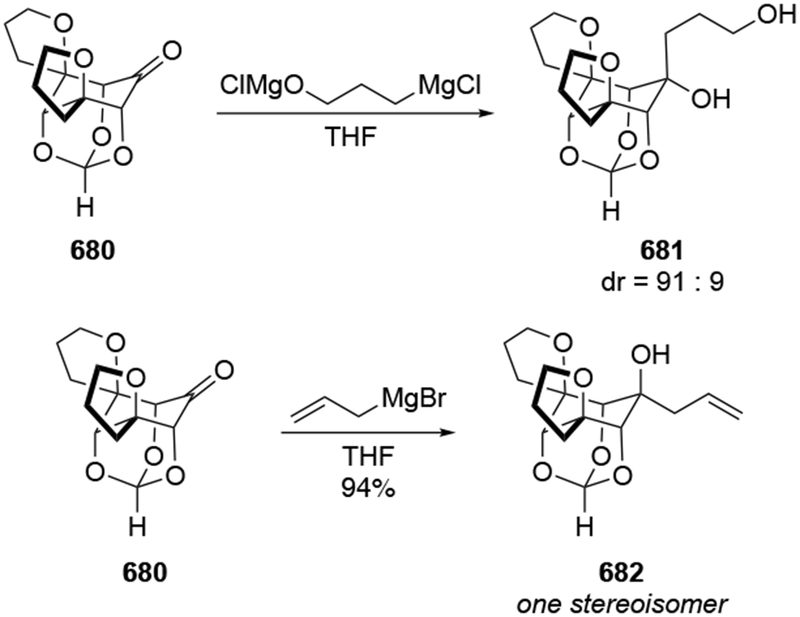
6.8.2. Additions to Bridged Bicyclic Ketones with the Carbonyl Group in the Two- or Three-Carbon Bridge
The archetypal bridged bicyclic system is the [2.2.1]-bicycloheptane system possessed by compounds such as camphor. The inherent selectivity of this system, however, is best illustrated by norcamphor (683), without the additional steric hindrance found in the terpene. In this case, addition occurred with high diastereoselectivity from the shorter, less sterically imposing bridge (Scheme 267).414 That this reaction is stereoselective argues that it is also not a reaction under diffusion control. This view is supported by the observation that this mode of stereoselectivity can be seen for additions of other organomagnesium reagents (Scheme 268).415
Scheme 267.
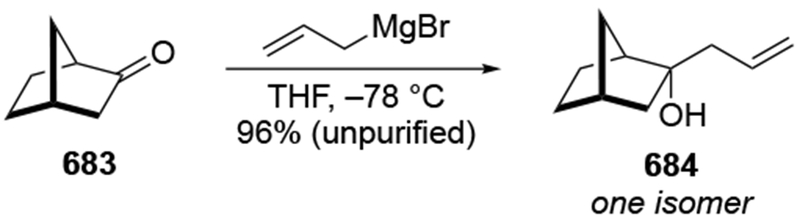
Scheme 268.
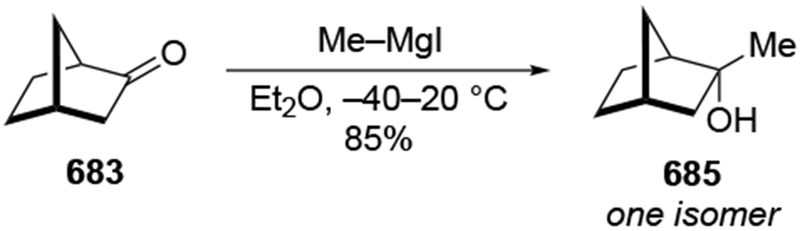
The same sense of diastereoselectivity was observed for a derivative of norcamphor (686, Scheme 269).416 The addition of these two methyl groups had no influence on the sense of stereoselectivity
Scheme 269.
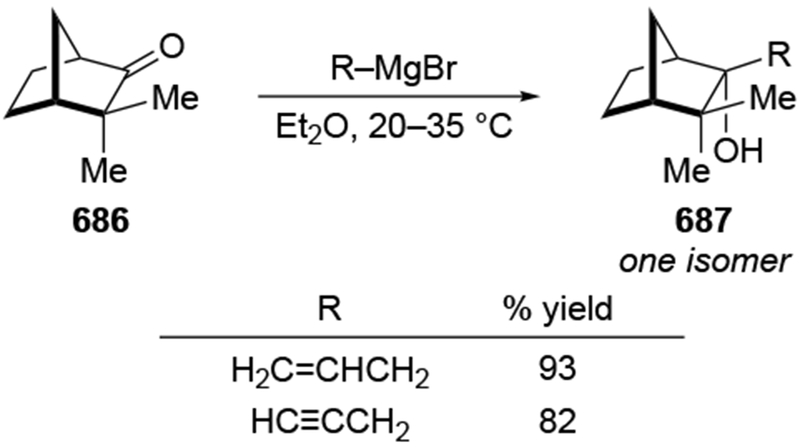
Additions to the even more hindered ketone fenchone (29) provide useful information. In this case, allylmagnesium reagents added again from the face opposite to the small bridge, as observed for norcamphor (Scheme 270).417,418 In this case, reactions of allylmagnesium operate at rates below the diffusion rate limit17,23 through the six-membered ring transition state with allylic transposition (i.e., 14, Section 2.1).23 On the other hand, addition of prenylmagnesium chloride occurred with high regioselectivity, favoring the product of addition at the α-carbon atom of the reagent, likely because addition at the γ-carbon atom is too sterically hindered (Scheme 271).23
Scheme 270.
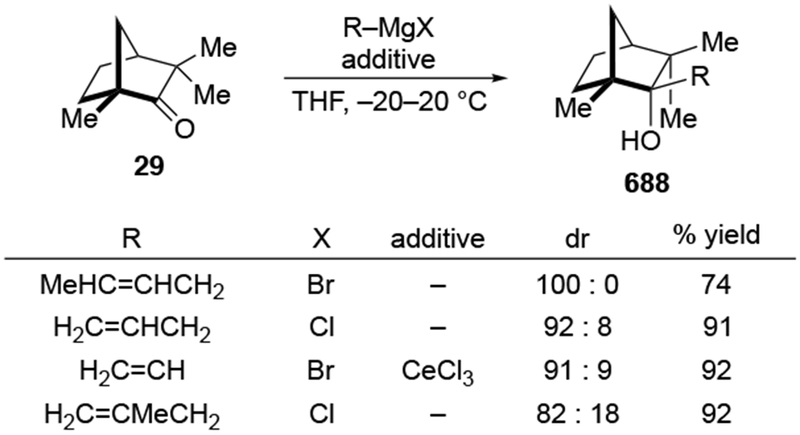
Scheme 271.
Nucleophilic additions to camphor occur with the opposite stereoselectivity, and this trend is general for a number of organometallic reagents (Scheme 272).419 In the original paper, the identity of the halide ion used was not given, but that has not generally been seen to affect selectivity even for phenomena such as chelation control, which might be more sensitive to the counterion.22 The high selectivity for the allylation of camphor has been confirmed by many researchers, even for substituted allylic organomagnesium reagents (for example, Scheme 273418).17,23,282,319,420,421 The selectivity for this addition is the opposite to that observed for norcamphor (Scheme 267–268). That difference is likely because the one-carbon atom bridge is now sterically demanding with the geminal substitution at this center. Control experiments demonstrate that these reactions are not occurring at rates that approach the diffusion rate limit,17 but still likely proceed through the six-membered ring transition state with allylic transposition (i.e., 14, Section 2.1).23 Addition of a particularly large organometallic nucleophile (Scheme 274) proceeded with lower diastereoselectivity,422 although with the same sense as observed for additions of phenyl and naphthyl Grignard reagents.423
Scheme 272.
Scheme 273.
Scheme 274.
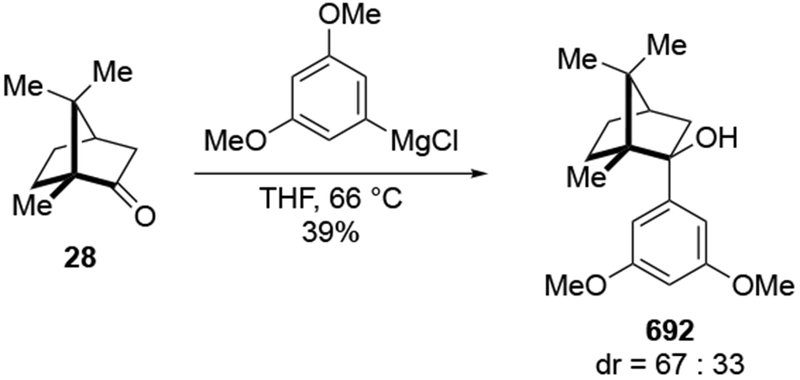
The reactions of camphorthione 693 illustrate a unique reactivity of allylmagnesium reagents compared to other organomagnesium reagents (Scheme 275).424 Addition of allylmagnesium bromide to thione 693 gave the exo-allylated isomer, which appears to involve addition from the more sterically hindered face. The mechanism of this reaction, however, is different than additions to ketones. Addition appears to first occur at the sulfur atom to form the anion 695, which then undergoes a [2,3]-sigmatropic rearrangement leading to the final product.425 Similarly, additions to thiofenchone (697) also occurred with allylmagnesium reagents, whereas no reaction was observed with isoprenylmagnesium bromide (Scheme 276). This observation is similar to the observation that additions to thionolactones were much faster and cleaner with allylmagnesium reagents than with other Grignard reagents.426
Scheme 275.
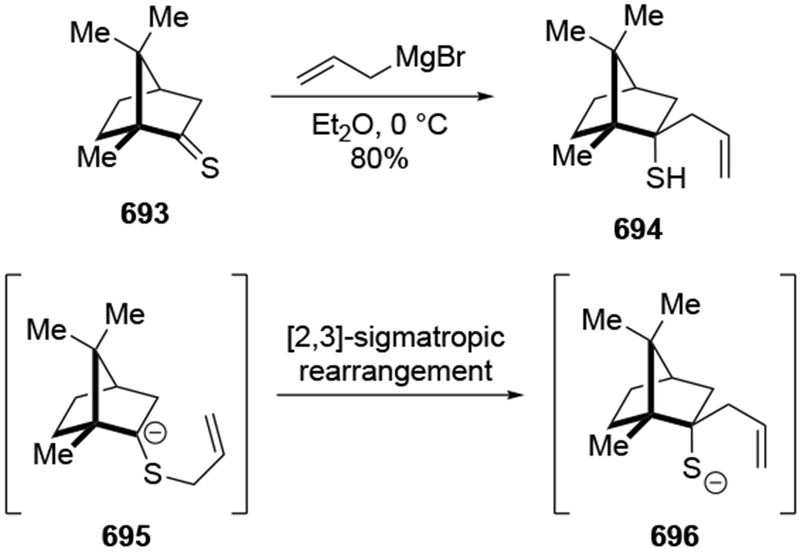
Scheme 276.
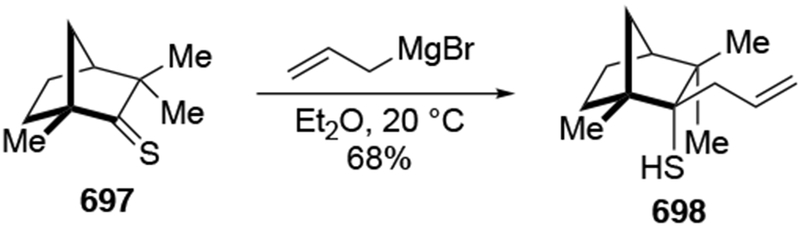
The selectivity for attack away from the geminally substituted bridge of camphor is also manifested in homologated versions of camphor. Addition of allylmagnesium chloride to homocamphor (699) occurred with high diastereoselectivity (Scheme 277).427 The same stereochemical course was observed for its hydroxylated variant (Scheme 278), suggesting that chelation in this case does not play a significant role in selectivity.428 That stereochemical course is also consistent for other organomagnesium reagents and alkyllithium reagents.
Scheme 277.
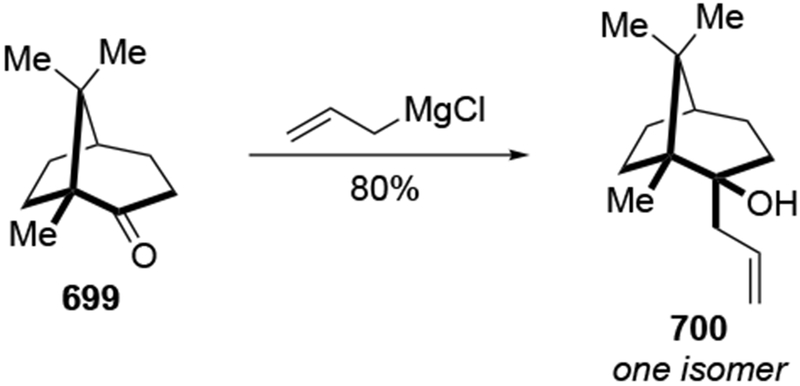
Scheme 278.
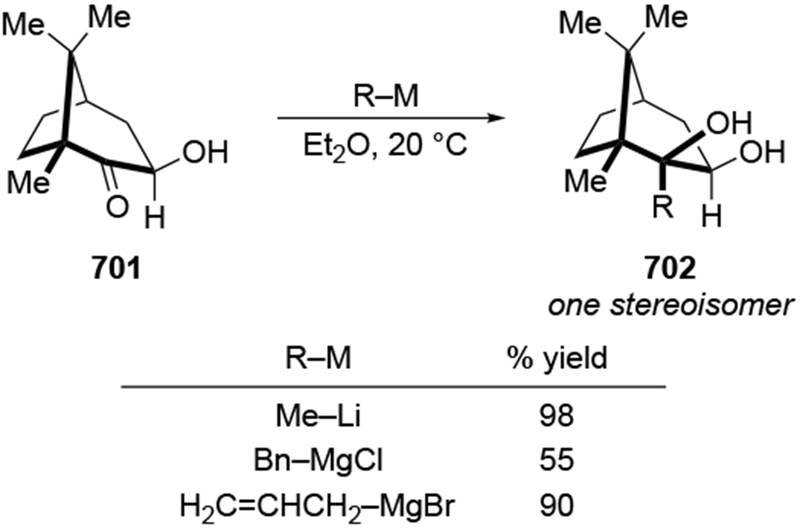
Even when the carbonyl group is not adjacent to the bridgehead atoms, additions can be highly stereoselective, even with allylmagnesium reagents. This point is illustrated with the bridged bicyclic ketones 703 (Scheme 279).429 In all cases, addition occurred from the exo face, on the side of the shorter bridge, with high stereoselectivity. The stereochemical courses of these reactions likely arise from preferential addition from the face that minimizes developing syn-pentane interactions with the bridges. The authors failed to observe additions of a number of organometallic reagents, including alkyllithium reagents and organomagnesium reagents, without the presence of CeCl3 as an additive, so these additives were used in all cases. It should be noted that other nucleophiles, such as allylstannanes, allylsilanes, allylindium reagents, and Reformatsky reagents, failed to add to these ketones.
Scheme 279.
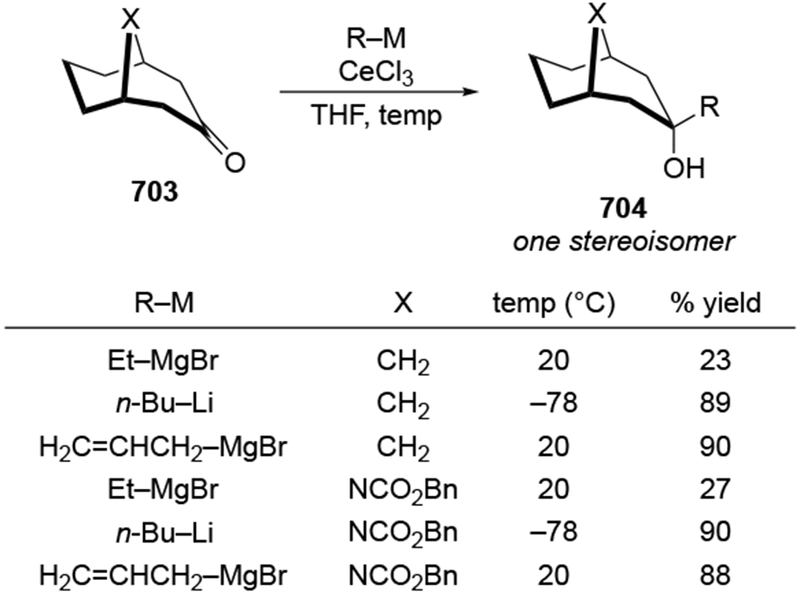
Other results indicate that addition of allylmagnesium reagents to these bicyclic ketones are unlikely to require CeCl3 to observe addition. For example, addition to the nitrogen-containing variant of these compounds can be achieved with just allylmagnesium reagents, in high stereoselectivity and in high yield (Scheme 280).430 The introduction of an allyl group was necessary because it was subsequently converted into a tetrahydrofuran ring. Other researchers have also used additions of allylmagnesium reagents to ketones such as 707 because the products can be elaborated to form compounds related to natural products (Scheme 281)431 or to pharmaceutical agents.432
Scheme 280.
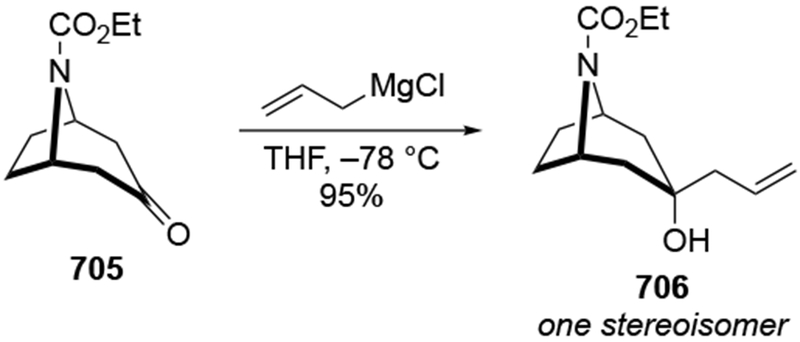
Scheme 281.
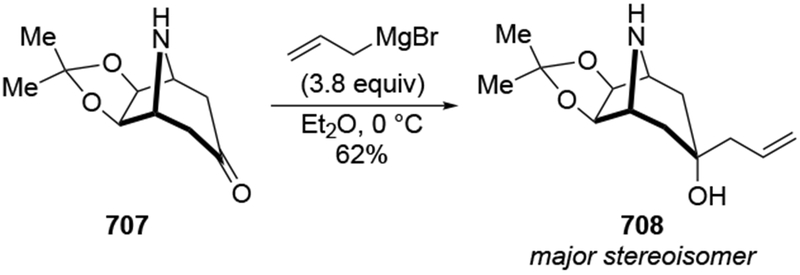
Several oxygen-containing bicyclic systems also underwent additions of allylmagnesium reagents with high stereoselectivity. Addition to bicyclic ketone 709 occurred from the same less-encumbered face (Scheme 282).433 Similar selectivity was observed for bicyclic ketone 711 (Scheme 283).434
Scheme 282.
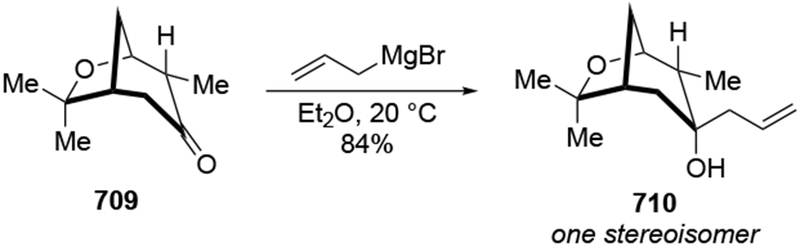
Scheme 283.
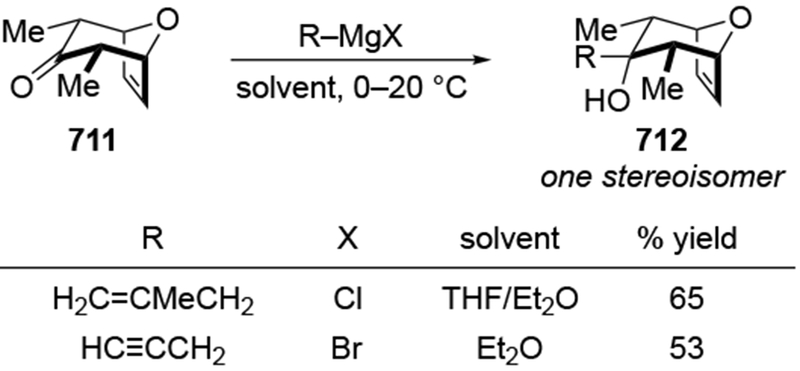
6.8.3. Additions to Bridged Bicyclic Ketones with Carbonyl Groups in the Larger Bridge
A family of oxygen-atom bridged bicyclic ketones was employed to prepare a system of stabilized carbocations (Scheme 284).435 Because of the subsequent steps involved, the configurations of the addition products were not described in detail. Nevertheless, additions of allylmagnesium bromide occurred efficiently in a number of cases. Addition of homoallylmagnesium bromide, unlike addition of allylmagnesium bromide, was accomplished in the presence of a lanthanide salt. Although no experiments were provided showing that these salts were necessary, they are often used to improve the efficiency of additions to hindered and enolizable ketones. 436
Scheme 284.
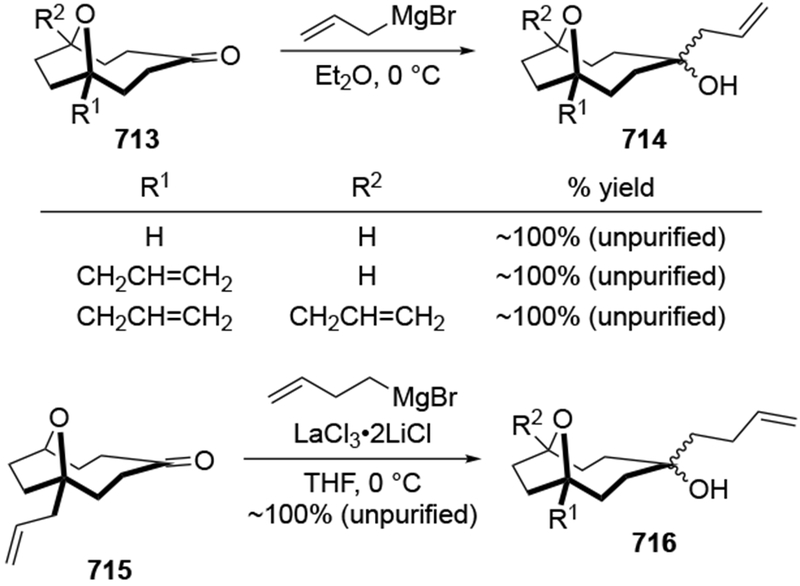
A multiply bridged system also gave reasonable selectivity for additions, even with allylmagnesium reagents (Scheme 285).437 In this system, the nucleophile added from the less sterically encumbered face away from the three-membered ring, as illustrated in transition state 719. It is worth comparing the selectivity in this series to the epoxide-fused ketones discussed earlier (Schemes 231, 232, and 248), which occur with similar senses of stereoselectivity.
Scheme 285.
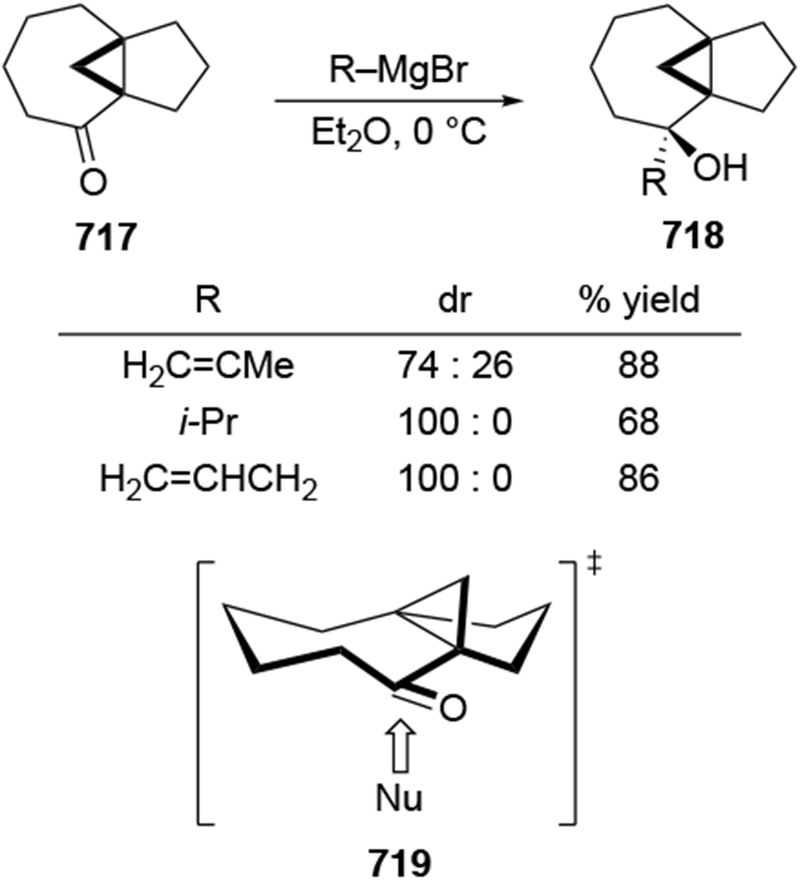
6.9. Additions to Multicyclic Ketones
This section collects examples that do not fit neatly into other cases. These are multicyclic ketones where the carbonyl group may be considered simply part of a fused ring or is part of a spiro ring, in a bridge of a series of bicyclic rings, or even in an acyclic chain attached to a multicyclic framework. As a result, these examples tend to be more associated with natural product synthesis, so there may be fewer comparisons between additions of allyl groups versus additions of alkyl groups if the allyl group were necessary for the synthesis. These systems also tend to be highly sterically hindered, so rates of reaction with allylmagnesium reagents are likely to be below the diffusion limit, leading to stereoselective reactions.
6.9.1. Multicyclic Ketones with Carbonyl Group in Fused Ring
Reactions of a hindered androstenone derivative with various allylic organomagnesium reagents provide interesting mechanistic information (Schemes 286–288).438 Addition of allylmagnesium reagents occurred only from the relatively unhindered bottom face (as illustrated by transition state 722, Scheme 286) compared to the top face, where addition would develop syn-pentane interactions.
Scheme 286.
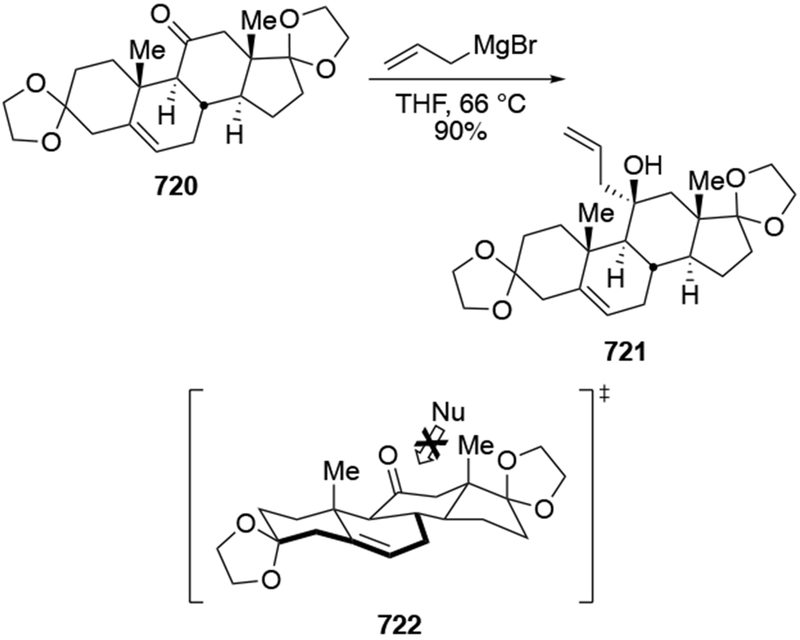
Scheme 288.
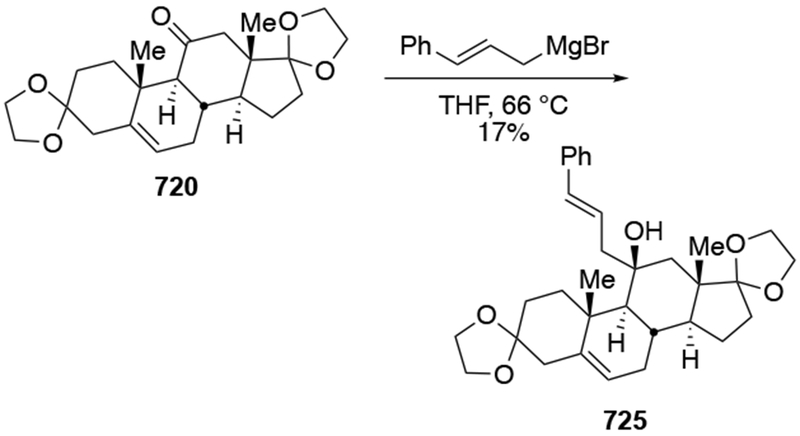
Additions of substituted allylic organomagnesium reagents give an indication of how hindered even the bottom face is (Scheme 287).438 Addition of crotylmagnesium reagents occurred exclusively from the bottom face, just as for allylmagnesium bromide. With this reagent, however, reasonable selectivity was observed for the anti stereoisomer 723. Normally, ketones need to be relatively hindered and the two groups well differentiated to see high anti:syn selectivity,278 suggesting that the bottom face is still sterically hindered. Upon increasing the steric size of the group on the nucleophile to a phenyl group, addition was slow and inefficient (Scheme 288). The major product, however, was the α-isomer, 725. Experiments with prenylmagnesium bromide (Me2C=CHCH2MgBr) showed similar trends of increasing amounts of the α-isomer as steric hindrance increases23 (as illustrated in Scheme 271). Those results were interpreted as reactions no longer following the six-membered ring transition state with allylic rearrangement (i.e., they would follow the same transition state as any other Grignard reagent, as discussed in Section 2.1).23
Scheme 287.
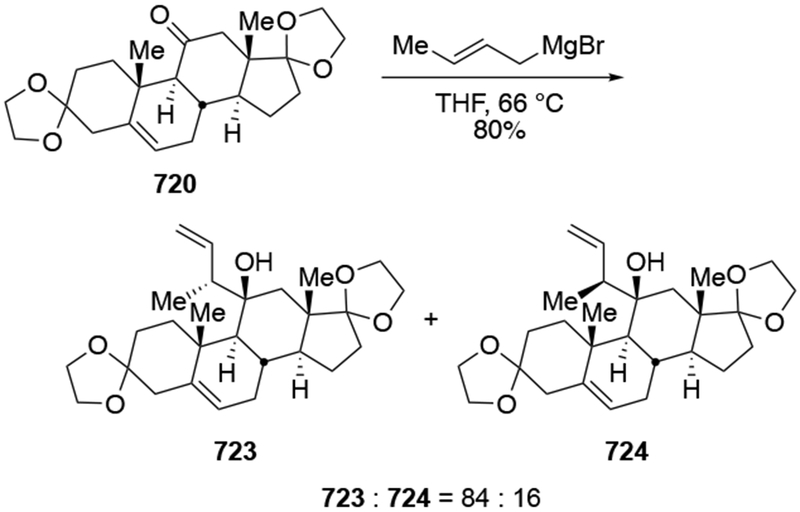
Because alcohol 727 was an important precursor in the pursuit of a natural product synthesis, efforts were focused on developing a stereoselective addition of an allyl nucleophile to ketone 726 (Scheme 289).439 Inspection of molecular models suggested that attack should occur from the more easily approached bottom face. Allylmagnesium bromide, however, added with little selectivity, although little experimental detail was provided. Ultimately, a chiral organozinc reagent was employed to obtain the desired stereochemistry to complete the synthesis (Scheme 290).440 As noted earlier (Section 4.2.5.1), this strategy of using a chiral reagent is often the best solution in difficult allylations. The use of an allylmetal reagent here was critical because the allyl group was necessary for a subsequent anionic oxy-Cope rearrangement,441 a powerful synthetic transformation in natural product synthesis.442
Scheme 289.
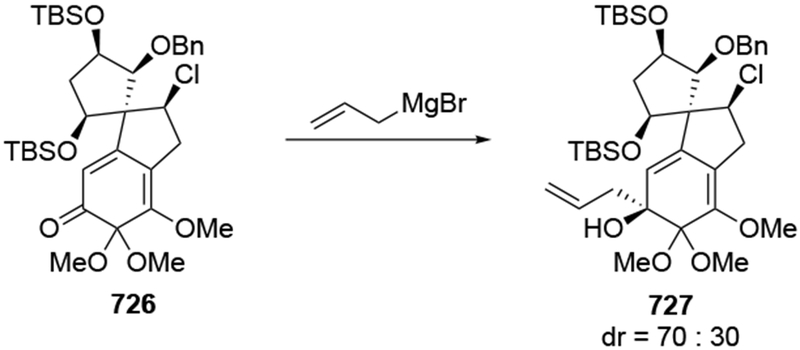
Scheme 290.
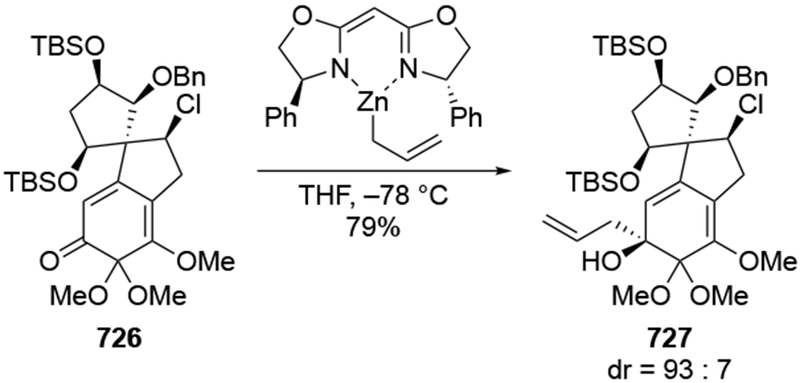
Cis-fused rings gave high selectivity for additions should the carbonyl group be near the ring fusion. This point is illustrated for a cyclohexanone fused to a five- and a seven-membered ring, which gave high stereoselectivity for addition of allylmagnesium bromide from the more accessible convex face (Scheme 291).443 This example also suggests that addition of allylmagnesium halides to carbon–nitrogen double bonds are much slower than additions to carbon–oxygen double bonds. A cycloheptanone fused cis to a five-membered ring also reacted with high stereoselectivity (Scheme 292).16 Alcohol 731 was prepared as part of a stereochemical proof of how all of the rings of a natural product had been joined. Because alcohol 731 was not a synthetic intermediate on the way to the final target, no details about this reaction were given.
Scheme 291.
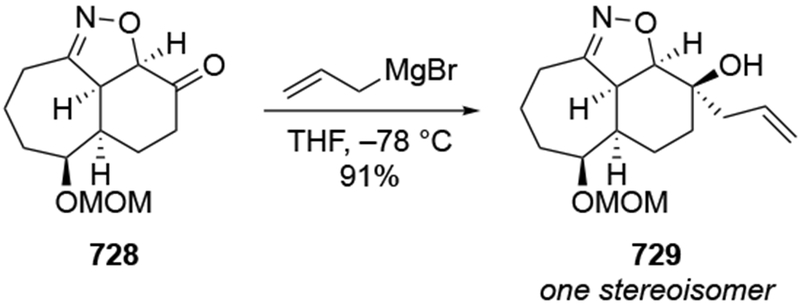
Scheme 292.
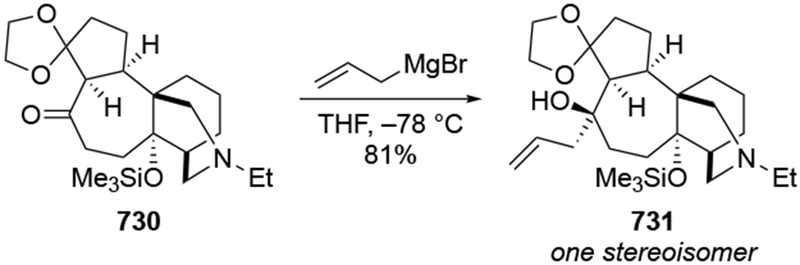
A cyclopentanone with sterically differentiated faces also exhibited high stereoselectivity in the additions of allylmagnesium reagents (Scheme 293).444 Stereoselective addition to ketone 732 and subsequent intramolecular acyl transfer formed carbamate 733 as a single stereoisomer. Addition was completely stereoselective to the convex face, even with the free amino group (734), as illustrated for the formation of alcohol 735. This allyl group was then used to anneal a six-membered ring onto the five-membered ring in the context of a synthesis of an alkaloid.
Scheme 293.
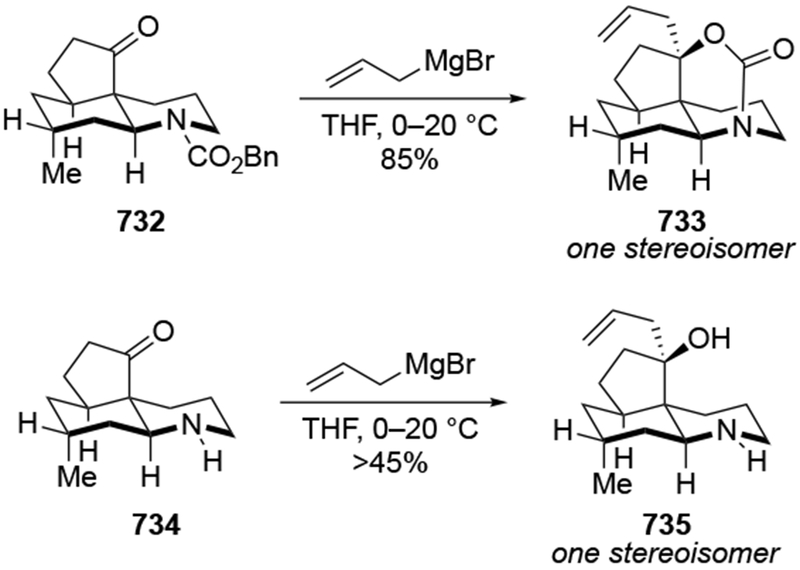
The reactions of a series of highly substituted ketones illustrate the powerful influence of a cis fused ring to control additions of allylmagnesium reagents from the convex face (Scheme 294).445 For enediones 736 and 738, addition of allylmagnesium bromide occurred opposite to the cis-fused bicyclic system. It should be noted that, even after the addition of the first allyl group, addition occurred from the same face (the new OMgBr group did not provide enough impetus for chelation to occur). On the other hand, for the more hindered dione 740, attack occurred from the face opposite the more sterically demanding bicyclic system. The authors used the combination of alkenes present in these diols to perform ring-closing metathesis reactions to form multicyclic products.
Scheme 294.
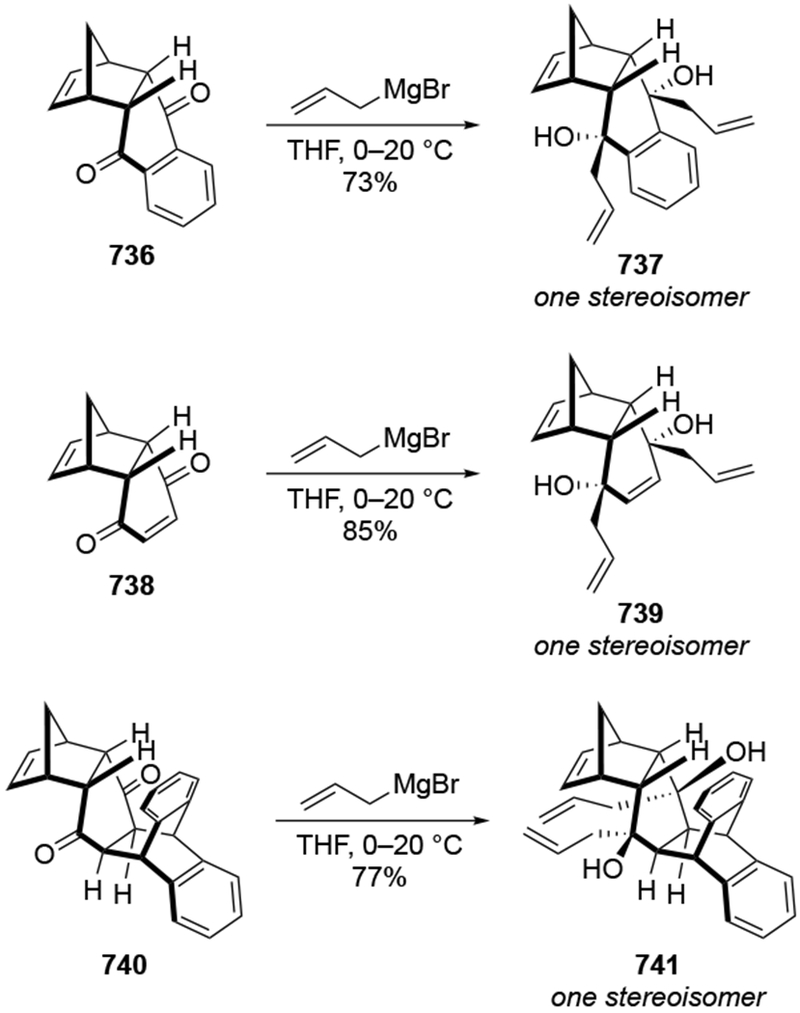
6.9.2. Multicyclic Ketones with the Carbonyl Group in a Bridging Ring
Multicyclic diones 742 and 744 are so sterically congested that their reactions exhibit distinct selectivities (Scheme 295).446 Addition to the symmetric diketone 742 occurred to only one of the two carbonyl groups even with an excess of reagent. The product was formed with high diastereoselectivity, indicating that attack occurred from the face away from the acetonide moiety at the top of the molecule. This stereochemical course, however, was undesired for the intended application in target-directed synthesis. Radical allylation was ultimately used to install this allyl group with the correct configuration. At a later point in the synthesis, the dione 744 was formed after introduction of the 2-propenyl group by addition of the corresponding organolithium reagent to a ketone, and then 744 was subjected to allylation. This reaction also involved attack away from the acetonide group, resulting in the formation of diol 745. In a later key step, the two vicinal allyl groups were subjected to ring-closing metathesis followed by hydrogenation to form a fused cyclohexane ring.
Scheme 295.
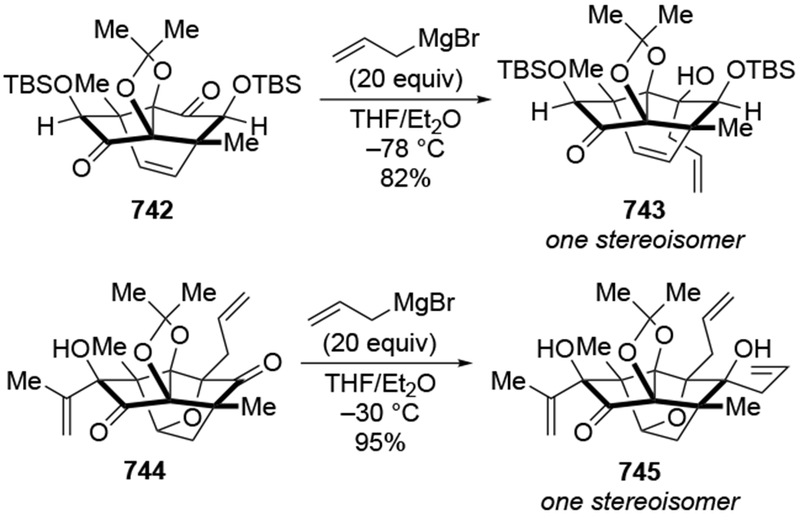
Other multicyclic ketones underwent highly stereoselective additions of nucleophiles, including allylmagnesium halides. Addition to ketone 746 occurred from the convex face of this bicyclic system (Scheme 296).434 A similar highly substituted bridged system also gave high stereoselectivity (Scheme 297).447 The use of CeCl3 in this reaction was not explained, although presumably it was employed to improve the selectivity for 1,2-addition.316
Scheme 296.
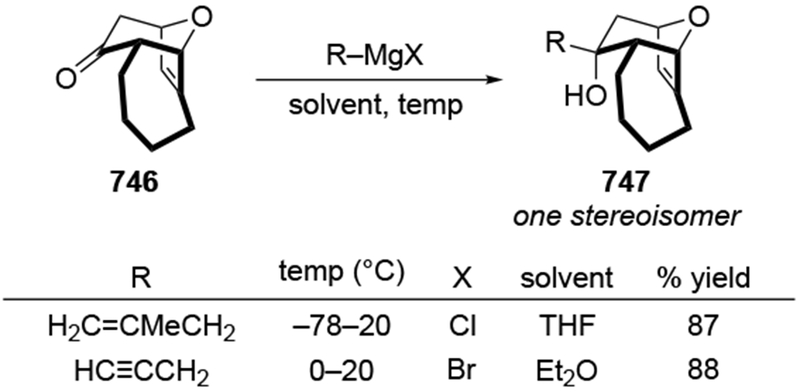
Scheme 297.
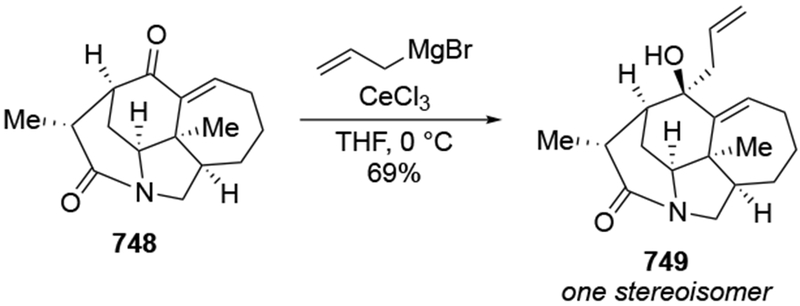
A series of highly substituted bridged bicyclic compounds was prepared that underwent highly stereoselective additions of allylmagnesium reagents (Scheme 298).448 In all cases, addition occurred from the more accessible face. The allyl groups were introduced because subsequent ring-closing metathesis reactions led to highly caged final products. It is worth noting the similarity in these structures compared to other highly caged carbonyl compounds (Scheme 4, Section 1.1).8
Scheme 298.
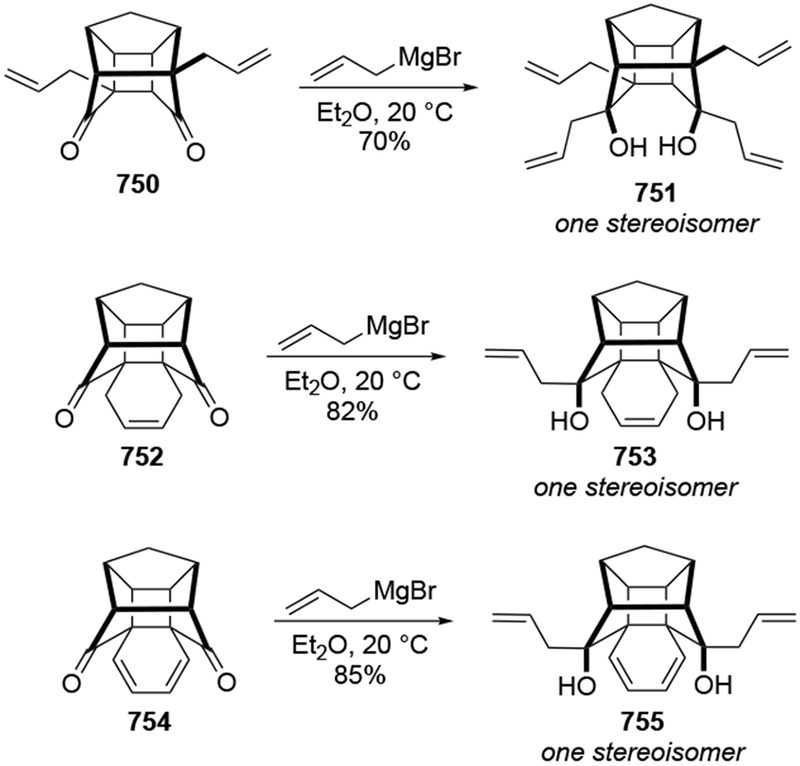
6.9.3. Multicyclic Ketones with the Carbonyl Group in a Spiro Ring
Hindered spirocyclic ketones generally undergo highly diastereoselective reactions because the carbonyl group is sterically hindered. This hindrance leads to additions that are likely to occur at rates that are below the diffusion limit. An example of this mode of attack is shown in Scheme 299.449 Addition of allylmagnesium bromide to the cyclopentanone 756 occurred with complete stereoselectivity from the face opposite the N–OBn group. Presumably the six-membered ring adopts a conformation that places the silyloxyethyl side chain in an equatorial position to minimize 1,3-diaxial interactions, which would position the carbonyl group such that one face is blocked by the substituted nitrogen atom (as in 758). The high selectivity in this case was important to the synthesis of an alkaloid because the allyl group was needed to close a five-membered ring with the nitrogen atom.
Scheme 299.
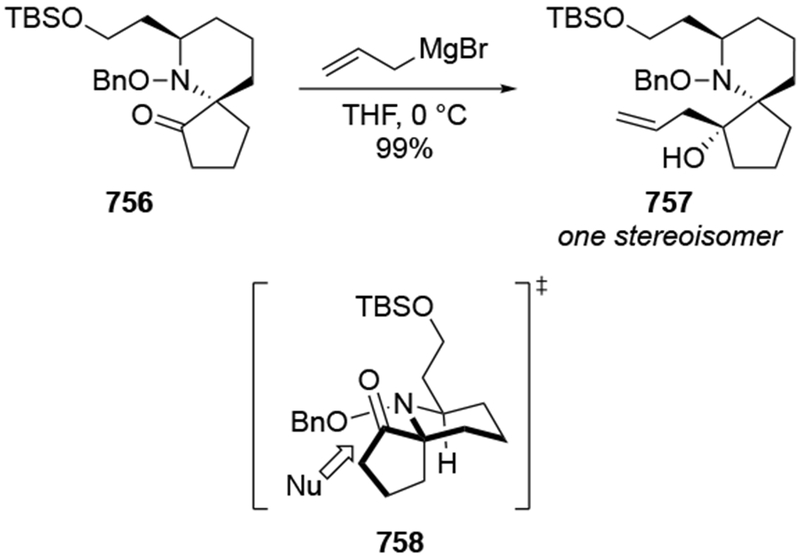
Reactions of the spirobicyclic ketone 759 illustrate the unique reactivity of organomagnesium reagents (Scheme 300).450. Addition of allylmagnesium reagents to ketone 759 proceed with low selectivity, presumably from the face away from any deprotonated nitrogen atom. Addition of the acetylide anion, however, gave high stereoselectivity for addition to the opposite face of the carbonyl group (i.e., from the same side as the nitrogen atom). This change in selectivity was explained as due to the size of the reagents, but the outcome may be the result of other factors. Addition of 2-propenylmagnesium bromide (or the corresponding organolithium reagent), also a large reagent, occurred with the same stereoselectivity as observed for the alkynyl anion, providing alcohol 761. These results may implicate chelation between the reagent and the nitrogen atom in the case of the alkynyl and propenyl reagents. As discussed in Section 4.1, allylmagnesium reagents are less susceptible to stereochemical control by chelation.
Scheme 300.
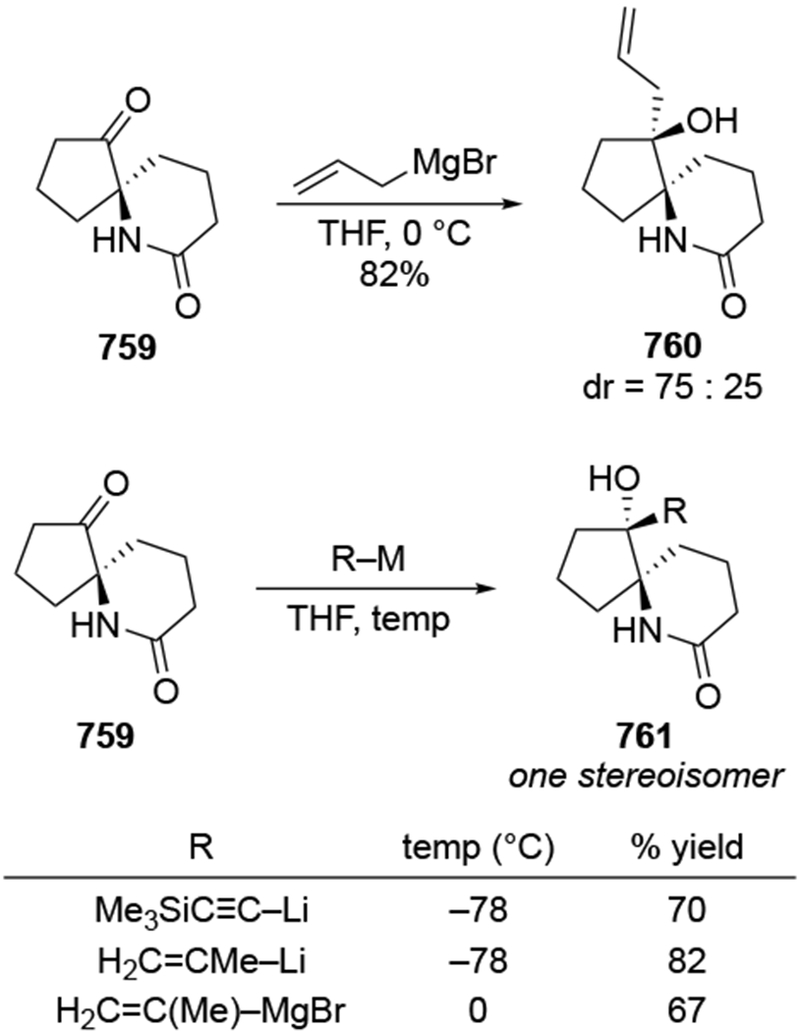
A hindered spirocyclic cyclopentanone underwent highly stereoselective allylation with allylmagnesium bromide (Scheme 301).451 The authors indicate that one stereoisomer of product was formed in this reaction, but they do not indicate the configuration. Conformational analysis for the ketone suggests that the product should be formed by attack from the back side of the conformation 764, where the front face is blocked by the substituted pyrrolidine. The carbonyl group of ketone 762 must be particularly sterically hindered: it underwent a total of 17 synthetic steps without need for protection. Among the conditions that this carbonyl group survived included exposure to phosphonate anions, hindered borohydride reducing agents, amines under reductive amination conditions, thiolate anions, and MCPBA. Even with the highly reactive allylmagnesium reagent, only slow conversion to the addition product was observed. Enolization, however, could be a competitive process.
Scheme 301.
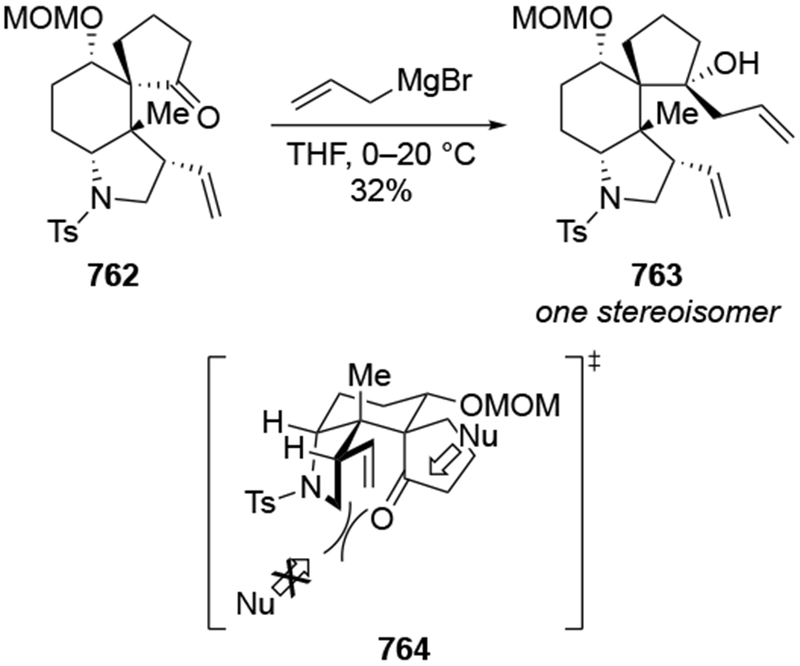
The selective additions of allylmagnesium reagents to spirocyclic ketones is useful in natural product synthesis (Scheme 302).14 Addition of allylmagnesium bromide to ketone 765 formed a single stereoisomer of adduct 766, with addition occurring from the face opposite to the nearby vinyl group. The allyl group was subsequently shortened to a two-carbon tether by ozonolysis, and this tether was then used to form the bridged bicyclic amine framework of the target molecules.
Scheme 302.
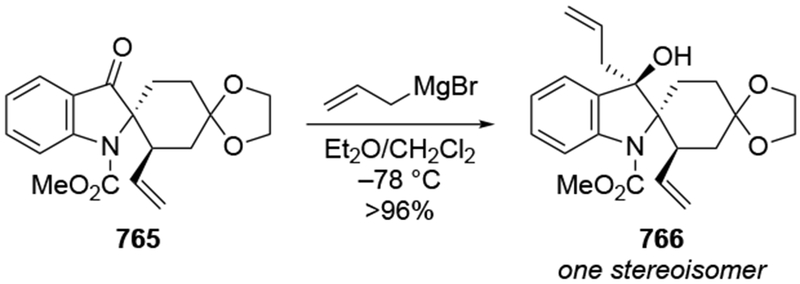
For the spirocyclic ketone 767, the carbonyl group is hindered enough that it might be anticipated that additions would be stereoselective (Scheme 303).452 Addition might be expected to occur from the face opposite to the substituted carbon atom of the spirocyclic six-membered ring. In this case, however, the ketone also contained a fused five-membered ring that will exert strong control on selectivity, just as it did with the other five-membered ring systems discussed above (Section 6.7.1.2). This effect appeared to be stronger: addition occurred with complete stereoselectivity to form the product where addition occurred from the face opposite to the adjacent fused ring, and thus toward the more substituted carbon atom of the adjacent spiro ring. As with other cases, the presence of an allyl group was required for the synthesis. It was shortened by one atom by ozonolysis then connected to the protected cyclohexanone ring by an intramolecular aldol addition.
Scheme 303.
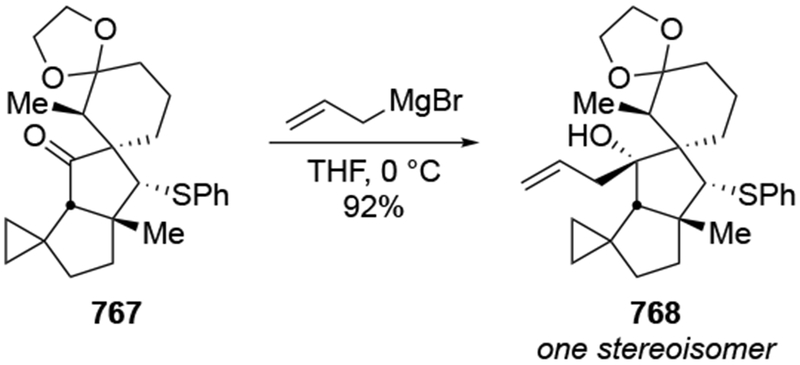
Additions of Grignard reagents to a family of spirocyclic diones raises questions about stereoselectivity that are difficult to address with the available information (Scheme 304).453 Allylation of dione 769 provided a mixture of 1,3-diols 770 and 771. Previous experiments reported by this group showed that the presence of the two trigonal carbon atoms in a spirocyclic ring could influence stereoselectivity of additions of methyllithium to ketones to moderate levels (approximately 85:15).454 This stereoselectivity, however, did not hold true for the more complex double-addition of the latter report. The reaction of the mono-addition product 773 appears to be contradictory (Scheme 305). This ketone, which was isolated from a mixture formed by adding fewer equivalents of the reagent to ketone 769 and isolating a small amount of it, gave, upon addition of the nucleophile, exclusively 771, and not 770, after column chromatography. This result indicates that diol 770, the 1,3-syn isomer, must come from the diastereoisomer of 773. That prediction is supported by a related system: addition to vinylated ketone 774 gave the 1,3-syn product 775. The same low selectivity observed with 769 was also observed with 776 and 779 (Scheme 306). These results reinforce the authors’ suggestion that electronic effects control addition to these rings,454 but they suggest that they operate only for the second addition and not for the first addition, which occurred unselectively. It is also worth mentioning that when vinylmagnesium bromide was added to the hindered mono-ketones, CeCl3 needed to be added. These additions were not diastereoselective (Scheme 307).
Scheme 304.
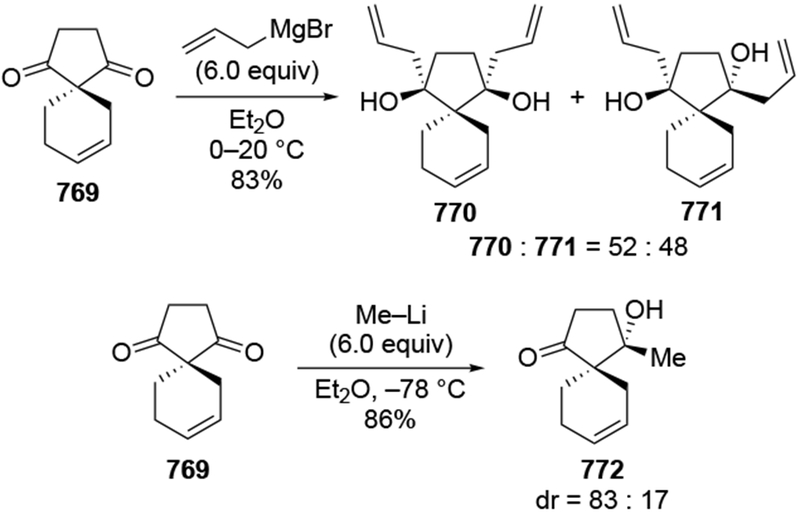
Scheme 305.
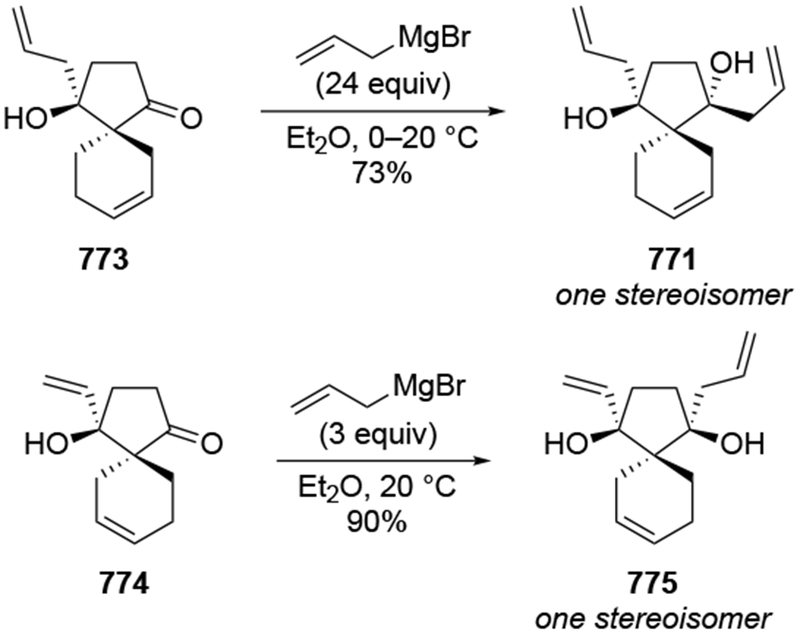
Scheme 306.
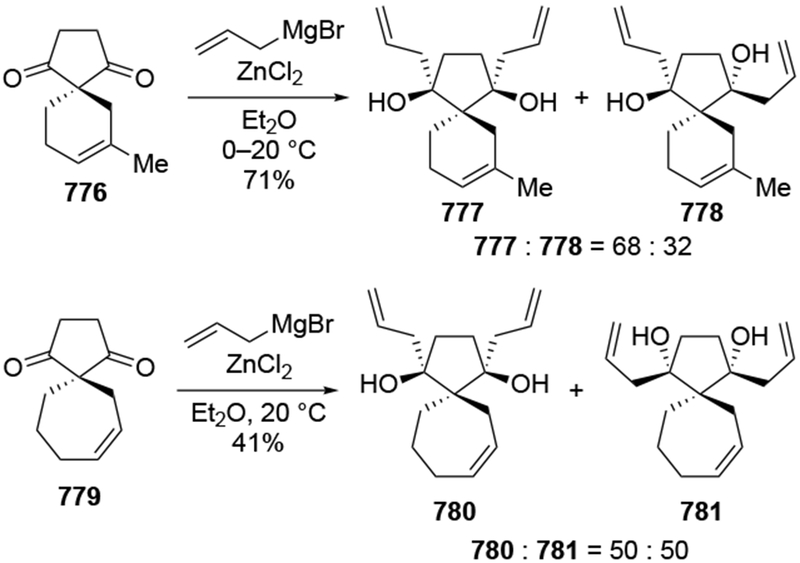
Scheme 307.
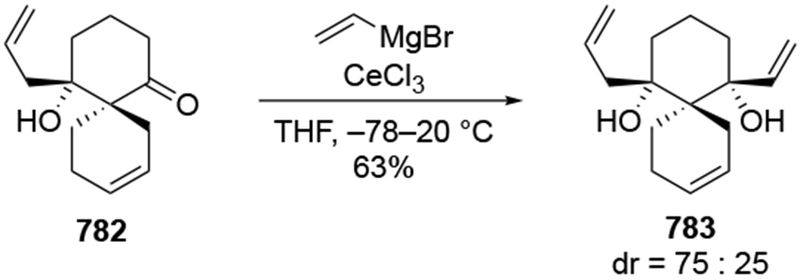
6.10. Additions to Carbonyl Groups Attached to Rings
Although some examples of additions to carbonyl groups attached to rings can be stereoselective, it is not clear that general trends can be established. The difficulty in obtaining stereoselective additions to exocyclic carbonyl groups is illustrated by additions explored in the course of a synthesis of a terpene natural product.455 Model aldehyde 784 underwent additions of allylmagnesium reagents with low stereoselectivity, but at least in the desired sense (Scheme 308). The low selectivity is consistent with the generally low stereoselectivity observed for additions to aldehydes, which is the result of reaction rates approaching the diffusion limit (as noted in Section 2.2). Addition of allylzinc bromide gave the undesired stereochemical outcome, which was the only product formed when an allyl silane was used as the nucleophile. Although the use of the allylmagnesium reagent would have been ideal, the sensitivity of the protecting scheme of the compound 787 used for the application in synthesis (Scheme 309) required the use of the zinc reagent (deprotection of the acetate would likely occur with allylmagnesium halides, as discussed in Section 2.2). The low diastereoselectivity for this allylation was repaired by subsequent oxidation of the resulting alcohol and reduction with LiAlH4, which occurred with 81:19 diastereoselectivity.
Scheme 308.
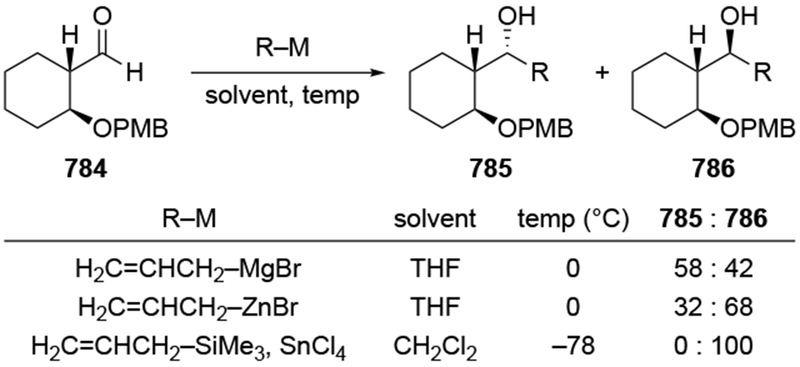
Scheme 309.
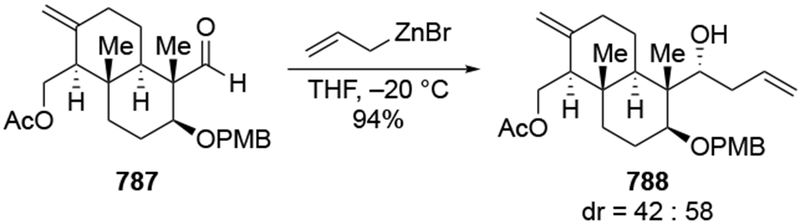
A related substrate also showed little diastereoselectivity upon addition of allylmagnesium bromide (Scheme 310).48 Whereas the mixture of alcohols 790 could be separated chromatographically, the authors found that it was easiest to separate diastereomeric alcohols 792 by silylation of the mixture of products. Only one of the two diastereomers was silylated, leading to simple separation by chromatography.
Scheme 310.
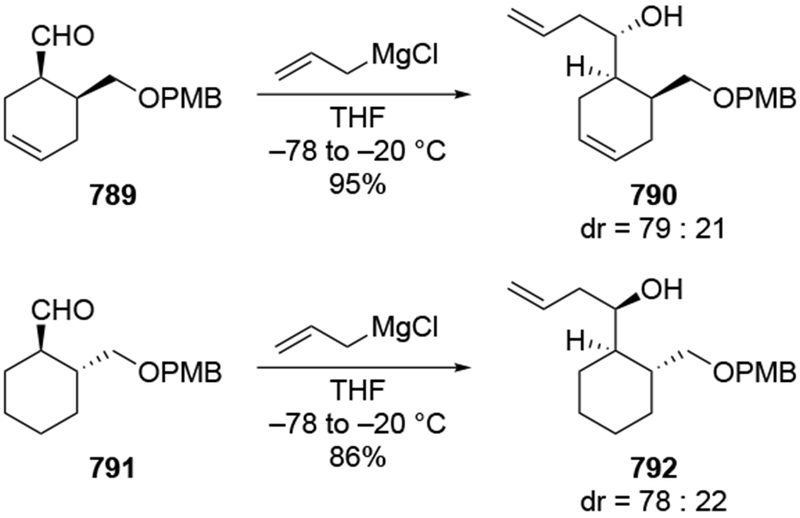
Generally, the additions of allylmagnesium reagents to such cyclohexyl-substituted aldehydes are not stereoselective. For example, in the process of developing potential drugs for the treatment of African sleeping sickness, routes to prepare cyclic peroxides and related structures as libraries of compounds were examined (Scheme 311).15 The synthetic route included the additions of allylic magnesium reagents to aldehydes such as 793 and 796. These reactions proceeded with little stereoselectivity.
Scheme 311.
A comparison between different nucleophilic additions to an exocyclic aldehyde is informative. Addition of vinylmagnesium chloride to an aldehyde formed the allylic alcohol 13 as a single diastereomer (Scheme 312).456 That selectivity was explained by invoking a long-range chelation with an OMgCl group, a species that, as discussed above, can be effective in chelation (as discussed in Section 4.2.1.1). Chelation may not even be necessary when considering that the low-energy conformation could be locked into a conformer resembling 799 (Scheme 313) to avoid eclipsing interactions and syn-pentane-like interactions. Regardless of the reason, however, it is notable that addition of allylmagnesium bromide to a similar aldehyde gave little selectivity for the desired isomer of alcohol 11 (Scheme 314).9 Selectivity was slightly improved by changing the solvent, but the ratios are nevertheless much lower than for formation of 13. The difference could result from the different sizes of the two groups (i.e., the silyloxymethyl group in 13 compared to the alkynyl group in (11). The relative populations of the two conformers could be different considering the small size of the alkyne group.9 Nevertheless, addition of allylmagnesium bromide did proceed with lower selectivity, thus complicating a synthetic scheme.
Scheme 312.
Scheme 313.
Scheme 314.
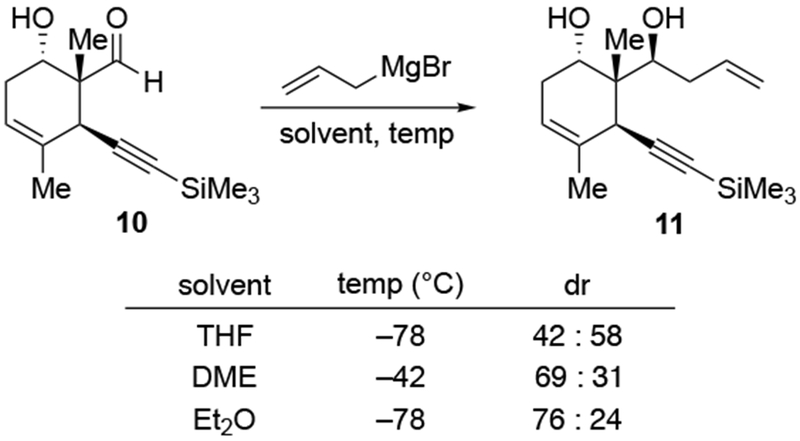
Understanding whether a selective nucleophilic addition results from chelation control or conformational control can be difficult. This issue is illustrated by additions to the chiral glyceraldehyde derivative 801 (Scheme 315).457 As noted in Section 4.2.4.3, additions to glyceraldehyde derivatives are not particularly selective. Additions to aldehyde 801, however, were highly stereoselective for a number of different organometallic reagents. The selectivity was attributed to chelation to the endocyclic oxygen atom through a chelate resembling 803. Such chelation should be enhanced considering that the non-polar solvent used would not compete with the chelation structure, as discussed previously (Section 4.2.4.2).
Scheme 315.
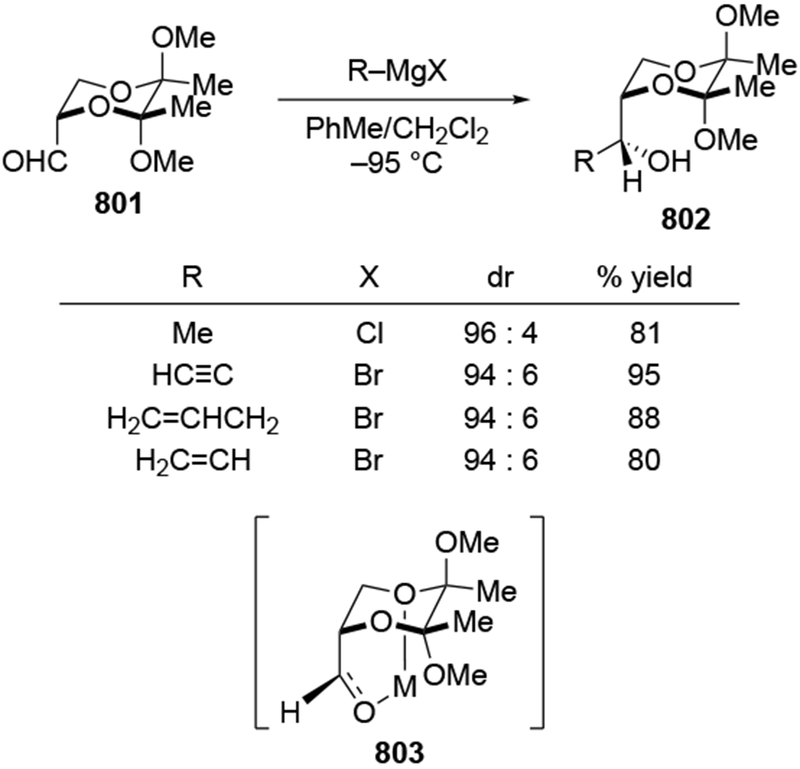
An alternative argument, based upon steric approach control, can be considered. The X-ray crystal structure of aldehyde 801 was reported, and the aldehyde’s conformation resembles the chelated form 803. This conformation is likely favored to minimize destabilizing interactions between the nearby axial methoxy group and the carbonyl oxygen atom (as shown in 804, Scheme 316). If this conformation were favored in solution, even a diffusion-controlled reaction like addition of allylmagnesium reagents should be highly stereoselective. The results with a related system support this analysis. Additions to the acetal 806 proceeded with generally lower stereoselectivity (Scheme 317).458 These acetals lacked the axial OMe groups present in aldehyde 801. The resulting stereoselectivities were somewhat lower, even under conditions optimized for chelation. It is likely that even though chelation should be favored using zinc atoms, the ring is less sterically constrained than for the ketal illustrated in Scheme 315.
Scheme 316.
Scheme 317.
A chiral aldehyde with the formyl group appended to a spirobicyclic system showed low stereoselectivity for allylation (Scheme 318).459 The stereoisomers could be separated, and the major isomer underwent an arene-olefin photocycloaddition to construct much of the core structure of a natural product. Recycling of the minor isomer was planned using an inversion sequence after this step.
Scheme 318.
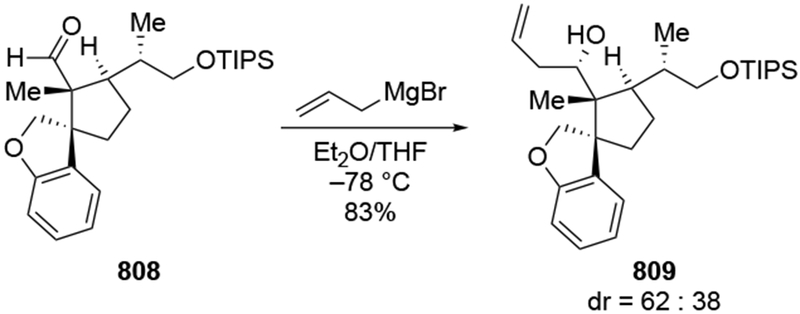
A ketone where the carbonyl group was exocyclic to a five-membered ring also underwent addition with low stereoselectivity, although reaction conditions were not provided (Scheme 319).460 The presence of the mixture was unimportant considering that this stereocenter was later removed by elimination.
Scheme 319.
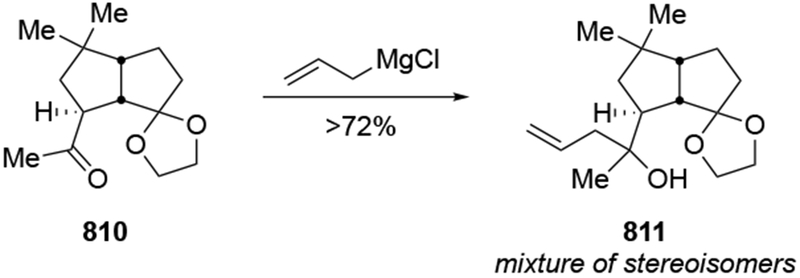
Two related aldehydes appended to six-membered rings also underwent reaction with allylmagnesium reagents with low stereoselectivity (Scheme 320).461 The lack of selectivity in the allylation of aldehyde 812 was irrelevant because both isomers were converted to the same final product with high stereoselectivity through an anionic oxy-Cope rearrangement 441,442
Scheme 320.
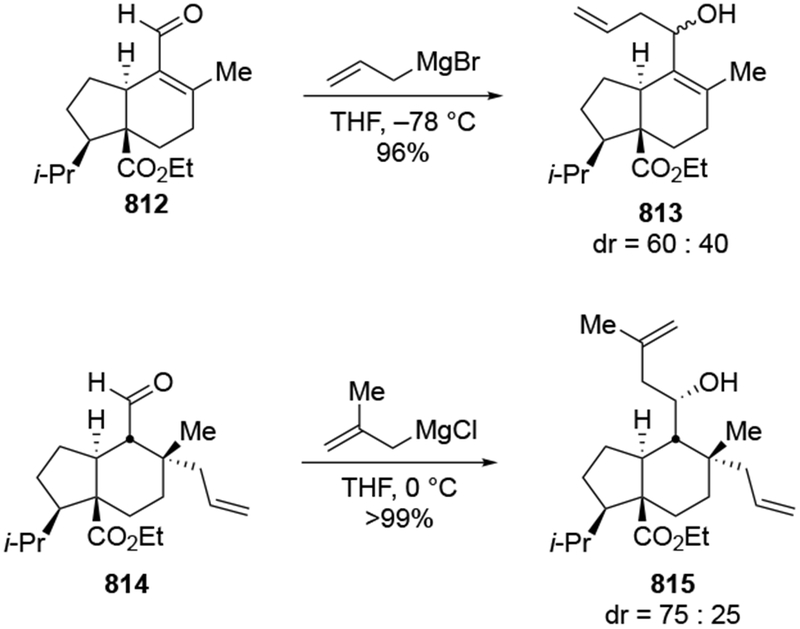
Additions to cyclopropyl-substituted aldehydes also indicate the difficulty of such additions (Scheme 321).462 The low selectivity in this case is general for a number of different Grignard reagents in both coordinating and non-coordinating solvents. The lack of stereoselectivity can be explained by the competing transition states illustrated in Scheme 322. Because the group cis to the carbonyl group is small, diastereoselectivity is low because both s-cis and s-trans-conformers are populated, and they undergo additions to the carbonyl group from each face. 56
Scheme 321.
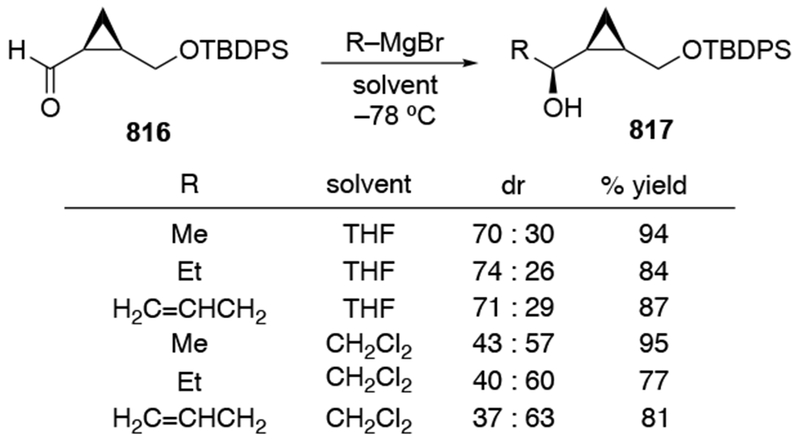
Scheme 322.
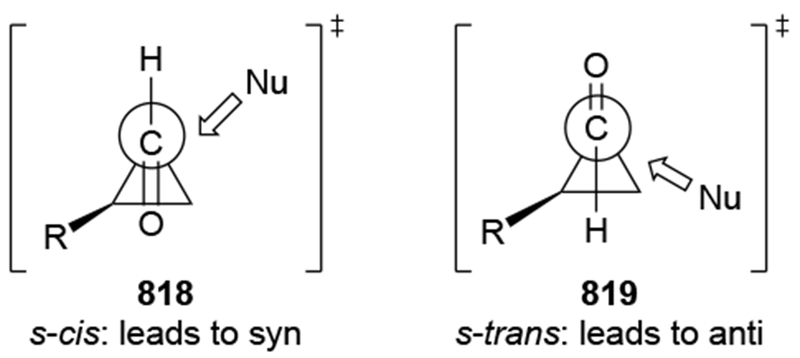
Additions to aldehydes appended to bridged systems generally do not show high degrees of selectivity. For example, additions to α-bromo aldehyde 820 were unselective (Scheme 323).463 Felkin–Anh selectivity was expected in this case due to the general behavior of α-bromocarbonyl compounds.464,465 Instead, no selectivity was observed for the additions of organomagnesium or organolithium reagents. The significantly different time courses of the two experiments may suggest that the addition of allylmagnesium bromide was faster, as would be expected (Section 2.2).
Scheme 323.
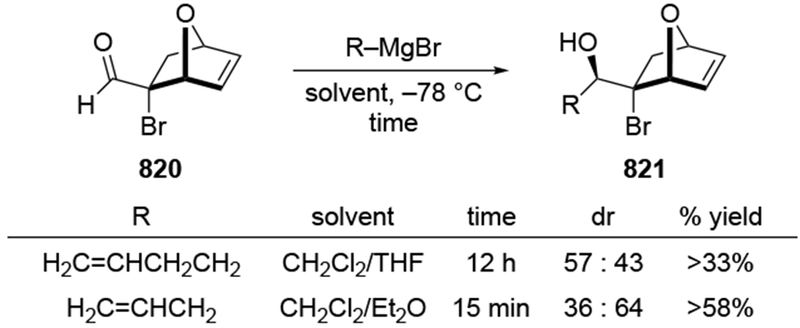
Even in a hindered exocyclic aldehyde, diastereoselectivity was modest (Scheme 324).466 Addition to the hindered β-alkoxy aldehyde 822 occurred with low diastereoselectivity, which was established after conversion to the resulting cyclic products by ring-closing metathesis. The aldehyde 822 resembles an aldehyde discussed earlier (Scheme 120). The resulting spirobicyclic compounds were designed to be analogues of paclitaxel, and some analogues showed promising biological activity.
Scheme 324.
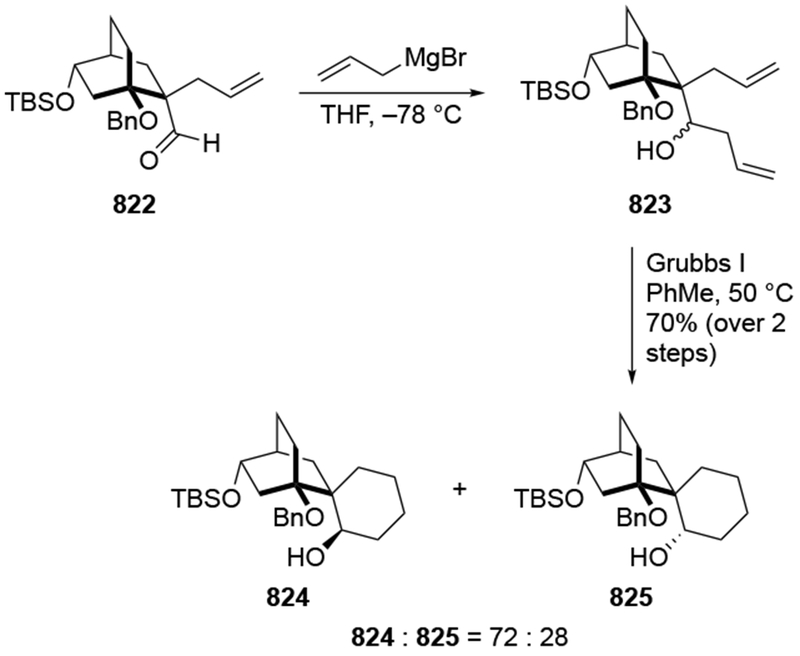
A related bicyclic ketone also underwent allylation with modest stereoselectivity, although the stereochemistry of the major isomer was not determined (Scheme 325).467 As with the system described in Scheme 324, the allyl group was used in a fragmentation reaction. This reaction broke the bridged bicyclic system to form a radical that closed on the allyl group, forming another ring.
Scheme 325.
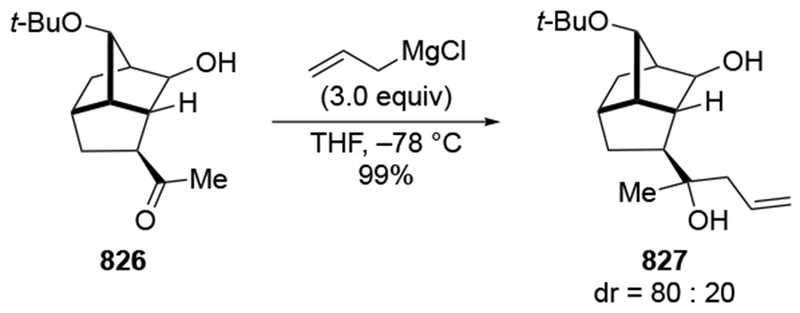
6.11. Additions to Other Acyclic Carbonyl Compounds
An important example in additions to acyclic carbonyl compounds underscores the importance of looking at the three-dimensional structure of a carbonyl compound to determine which face may be more prone to attack by nucleophiles (Scheme 326).468 This study showed that the diastereoselectivities of additions of organometallic reagents to a family of related ketones did not follow a simple pattern. Attempts to control the stereoselectivity on a protected, acyclic, polyoxygenated ketone (828) using chelation did not give good diastereoselectivity for any reagent except the hindered reagent 2-propenylmagnesium bromide. Upon changing the terminal protecting groups to acetonides, which were earlier shown to give poor chelation-controlled selectivity in this system, and incorporating an α-methoxy group adjacent to the carbonyl group (830), poor selectivity was observed for all reagents (Scheme 327). Simply changing the protecting group at the α-hydroxyl group to a benzyl group led to high stereoselectivity for all reagents examined except allylmagnesium bromide (Scheme 328). This pattern of high selectivity for other Grignard reagents but low selectivity for allylmagnesium halides is common, which, as discussed in Section 2.2, may result because the additions of this reagent occur near the diffusion rate limit and thus are unlikely to occur diastereoselectively.
Scheme 326.
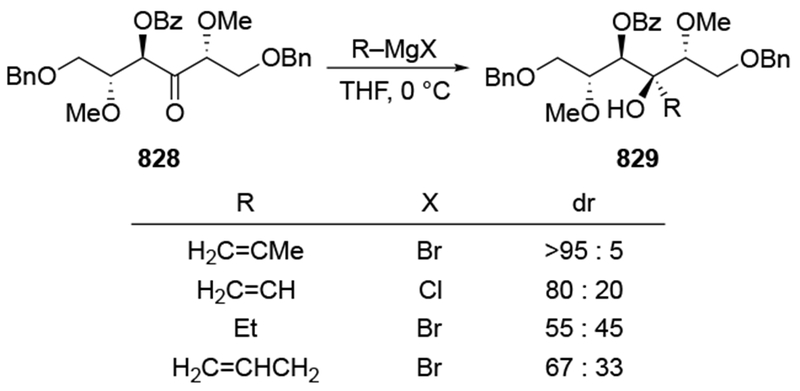
Scheme 327.
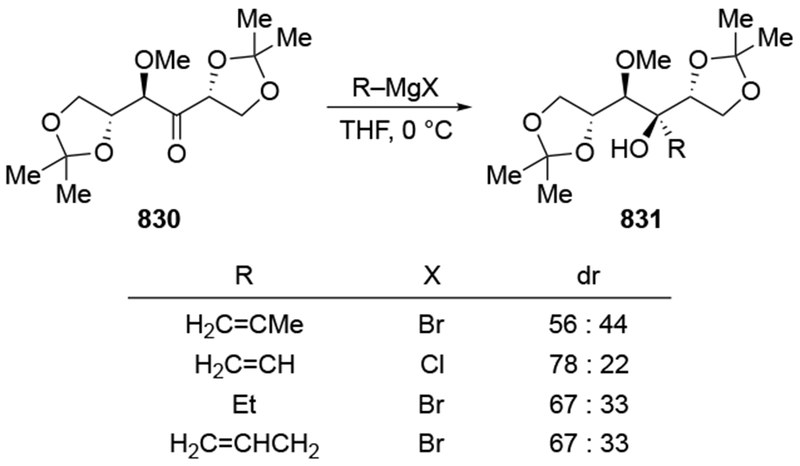
Scheme 328.
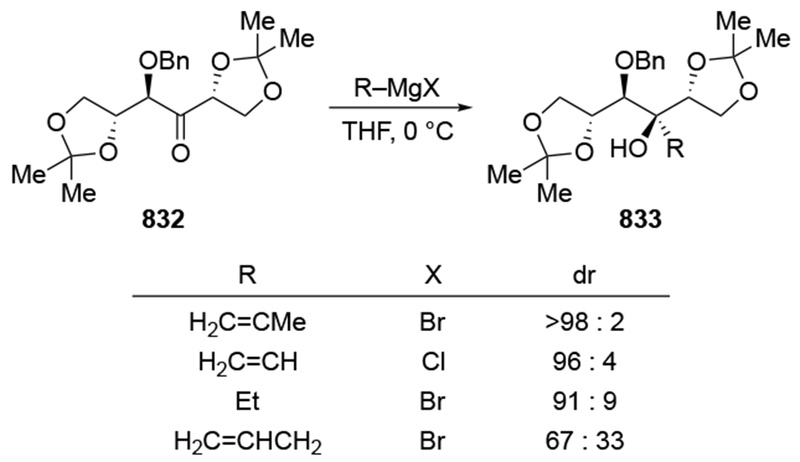
The results shown in Scheme 329, however, complicate the analysis. With the progressively larger groups on the α-oxygen atom of ketone 834, selectivity was high with every reagent examined. It was necessary to have two acetonide protecting groups to observe high selectivity with allylmagnesium bromide, but not for the other Grignard reagents, which likely under went chelation-controlled additions to α-alkoxy ketone 836 (Scheme 330).
Scheme 329.
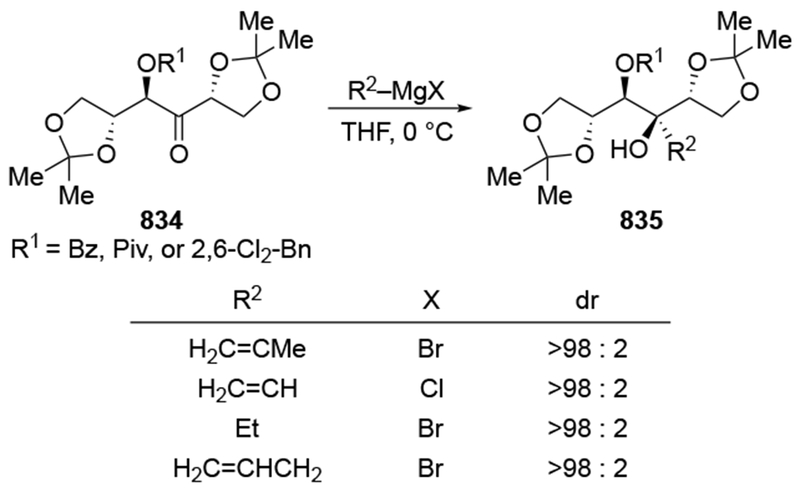
Scheme 330.
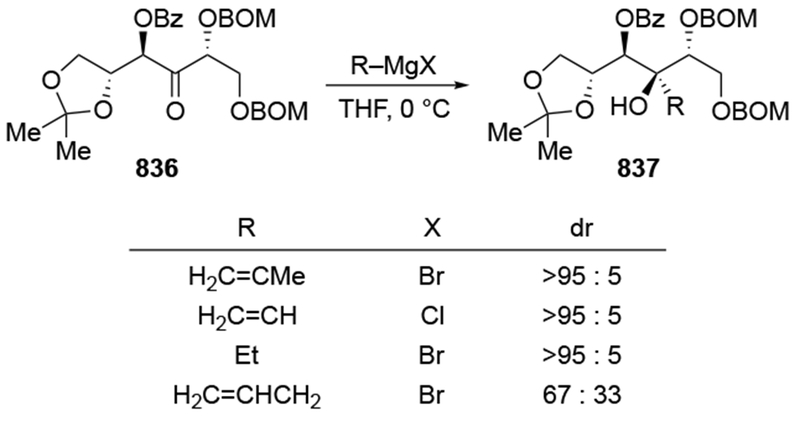
The authors noted that their studies on this class of substrates provided evidence that transition state models such as Felkin–Anh and chelation-control do not entirely govern the stereoselectivity of reactions. Instead, they interpreted the stereochemical course of these reactions to be governed by the ground state conformations of each substrate and steric approach control. This conclusion was supported by careful conformational analysis. Benzoyl-protected ketone 834 adopts a rigid conformation, the same in solution and in the solid phase, as illustrated in Scheme 331. That conformation exposes only one face of the carbonyl group to nucleophilic attack. Consequently, high stereoselectivity was observed for all nucleophiles, even for allylmagnesium bromide. This example illustrates that the high reactivity of allylmagnesium reagents is not incompatible with high selectivity.
Scheme 331.
Conformational effects and steric approach control could also operate in reactions of an acyclic aldehyde (Schemes 332–333).469 Addition of allylmagnesium bromide to the aldehyde 839 occurred with only modest diastereoselectivity (Scheme 332). Efforts to improve this stereoselectivity by using a chiral reagent were modestly successful. By contrast, addition of allylmagnesium bromide to the diastereomeric aldehyde 842 gave the product 843 as a single diastereomer (Scheme 333). This result cannot be readily accommodated by either the Felkin–Anh model or the chelation-control model, which would depend principally upon the neighboring stereocenters to control stereochemistry (and chelation-controlled additions of allylmagnesium reagents with aldehydes are unlikely to be successful22).
Scheme 332.
Scheme 333.
Just as with the examples illustrated in Schemes 326–330, the analysis of the conformational preferences of these aldehydes provides insight. Aldehyde 839, which reacts unselectively, likely adopts a conformation resembling 844, with the larger group placed in the position expected for a Felkin–Anh type transition state (Scheme 334). This conformation would also place the silyloxy group in the expected anti alignment to minimize destabilizing dipole interactions,186 but that orientation places the large alkyl group bearing R1 in the path of nucleophilic attack. The alternative Felkin–Anh type transition state would minimize that interaction but would not permit the minimization of unfavorable dipole interactions. On the other hand, the other diastereomer of aldehyde (842) would likely adopt a conformation resembling 845, which more closely resembles a Felkin–Anh transition state and minimizes both unfavorable steric interactions and dipole interactions. Support for this analysis can be found by analyzing an X-ray crystal structure of a chiral β-silyloxy aldehyde with similar substitution pattern (although different relative configuration at the β-carbon atom); that aldehyde adopts a conformation resembling 845.470
Scheme 334.
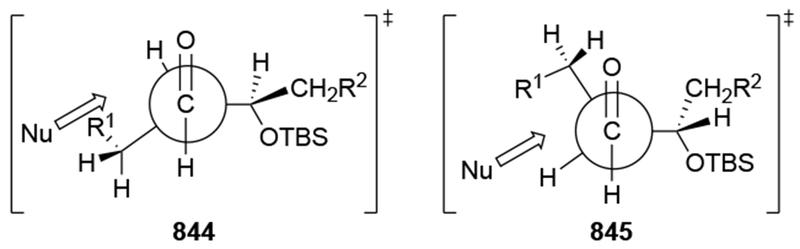
Another reaction that might be governed by conformational preferences and steric approach control is the addition of allylmagnesium bromide to ketone 846 (Scheme 335).471 The identity of the solvent appears to influence the propensity of the magnesium alkoxide intermediate to undergo an intramolecular nucleophilic substitution reaction, but it does not affect the diastereoselectivity.
Scheme 335.
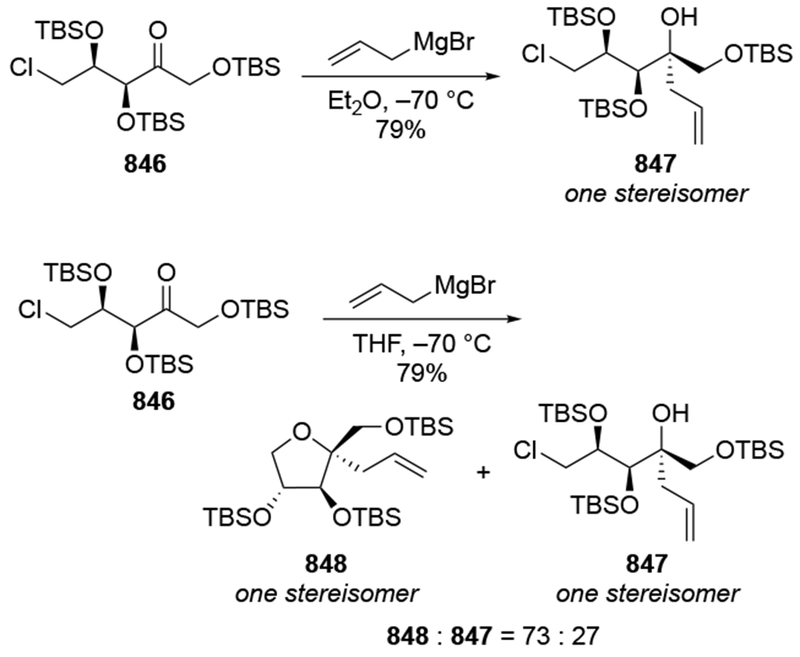
An analysis of the conformational preferences of this ketone suggest an explanation for the diastereoselectivity. Silyl ethers often prefer different conformations than alkyl ethers,472 and the presence of vicinal silyloxy groups could cause them to orient themselves anti to each other.473–475 This strong conformational preference would control the overall conformation of the molecule and thus the diastereoselectivity of its addition reactions. Based on the crystal structure of an α,β-bis(tert-butyldimethylsilyloxy)-substituted ketone,476 a conformation can be assigned to ketone 846 that minimizes steric interactions (Scheme 336). Attack on 849 would be blocked from the front face by the silyloxy groups, so attack from the more accessible back face would be favored, which would form the observed product. Similar selectivities have been observed with a related α,β-bis(tert-butyldimethylsilyloxy)-substituted ketone (850), including with methallylmagnesium reagents (Scheme 337), which were also attributed to strong conformational control exerted by the vicinal silyloxy groups.477
Scheme 336.
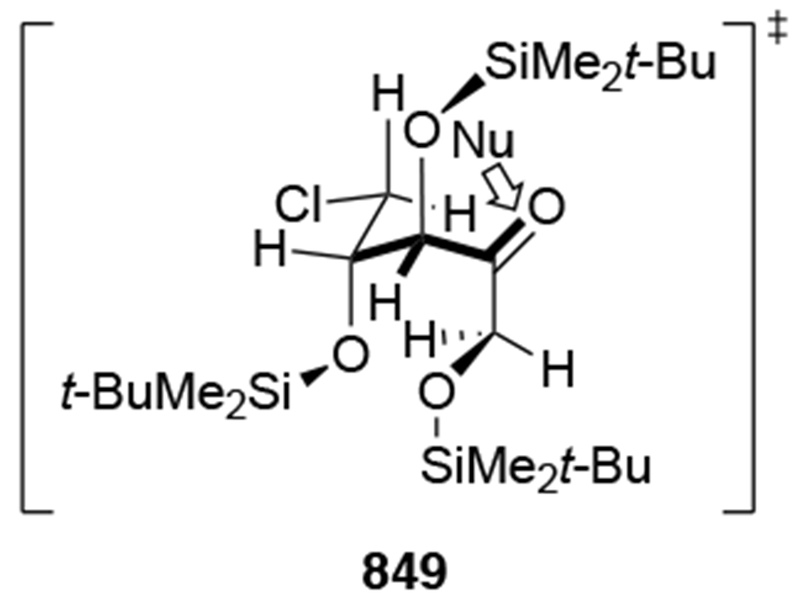
Scheme 337.
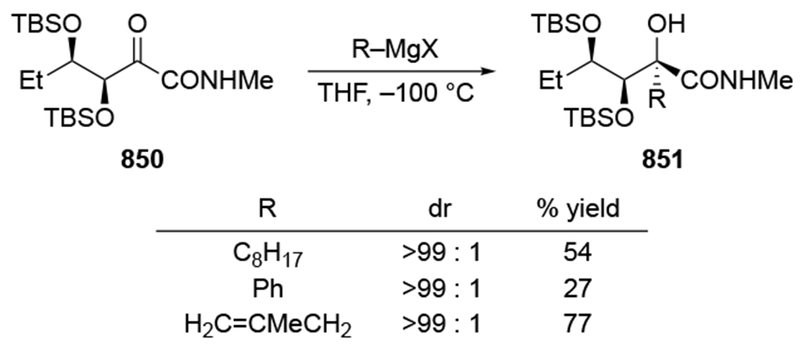
Simple β-substituted ketones generally do not react with stereoselectivity. For example, allylation of hindered ketone 852 was not diastereoselective (Scheme 338).478 It was not important to the synthetic endeavor, however: this stereocenter was destroyed in a subsequent SN1-like ring closure.
Scheme 338.
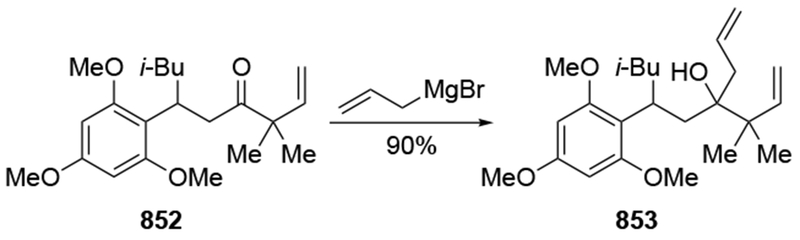
Addition of allylmagnesium halides to β-methyl-substituted ketones are generally not diastereoselective. Addition to 854 gave homoallylic alcohol 855 with low stereoselectivity (Scheme 339).479 Higher selectivity (75:25) could be achieved using chiral reagents, but, in either case, the isomers needed to be separated to complete the desired synthesis using electrochemical methods. Low stereoselectivity was also observed for addition of allylmagnesium reagents to β-methyl-substituted ketone 856 (Scheme 340).480
Scheme 339.
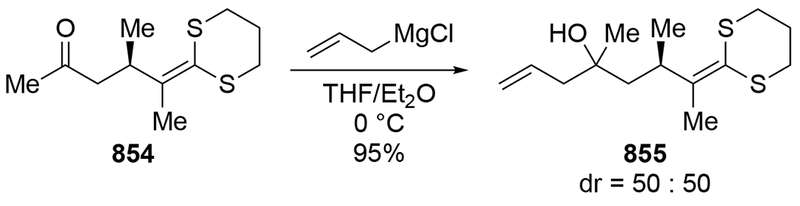
Scheme 340.
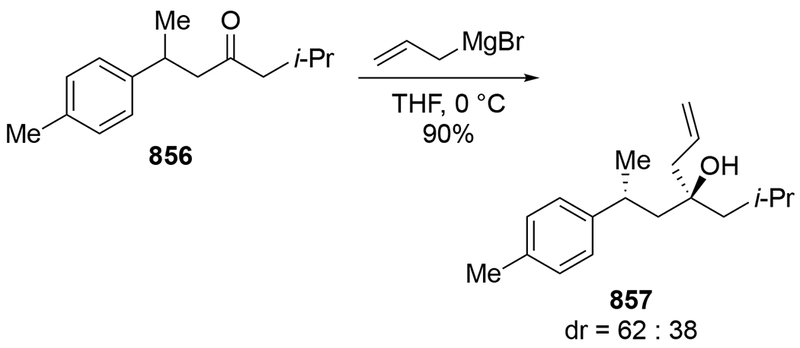
Another example of the significant difference between allylmagnesium reagents and other Grignard reagents was noted in the course of developing chiral auxiliaries for performing additions to aldehydes (Scheme 341).481 Whereas additions of reagents such as organomagnesium, organolithium, and Reformatsky reagents all gave the product with high diastereoselectivity, additions of allylmagnesium reagents gave low selectivity. The selectivity could be improved somewhat by changing the solvent or by first transmetallating to zinc.
Scheme 341.
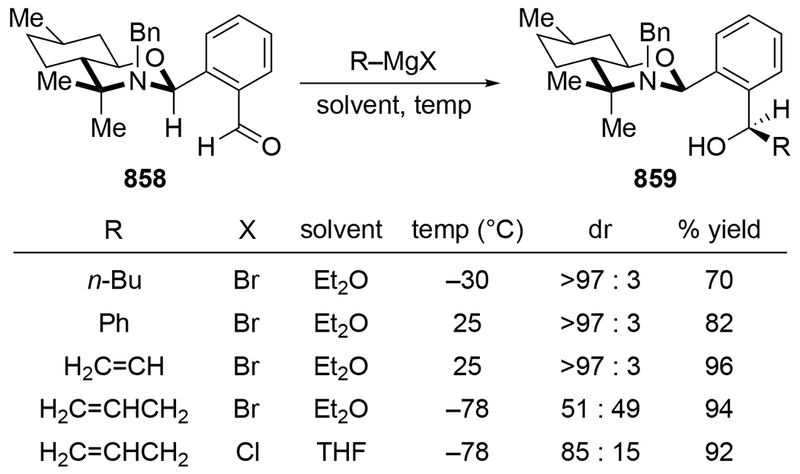
Even with allylic strain constraints, additions to enals with a γ-stereogenic center occurred with low diastereoselectivity. Additions of allylmagnesium bromide to aldehyde 860 gave a mixture of isomeric products (Scheme 342).482 Although control of the absolute stereochemistry could be achieved using reagent-based methods, purification was difficult. The most effective course of action was to use the commercially available allylmagnesium reagent, separate the diastereomeric alcohols 861, and convert the undesired isomer into the desired one using the Mitsunobu inversion protocol followed by hydrolysis.
Scheme 342.
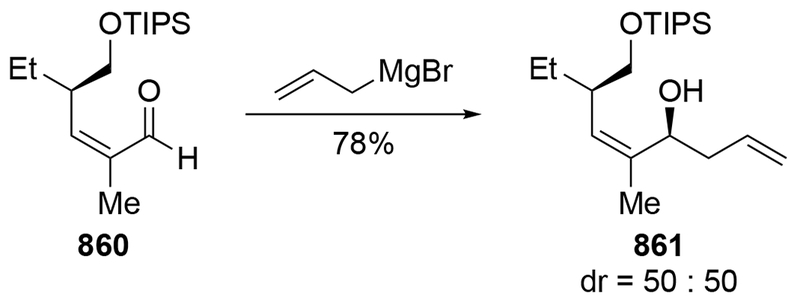
Similar substrates with sulfoximine chiral auxiliaries, however, show higher selectivity (Scheme 343).483,484 Additions to the carbonyl group of ketone 862 occurred with high diastereoselectivity. On the other hand, additions of allylmagnesium bromide to aldehyde 864 occurred with lower diastereoselectivity than additions of other organomagnesium reagents (Scheme 344).
Scheme 343.
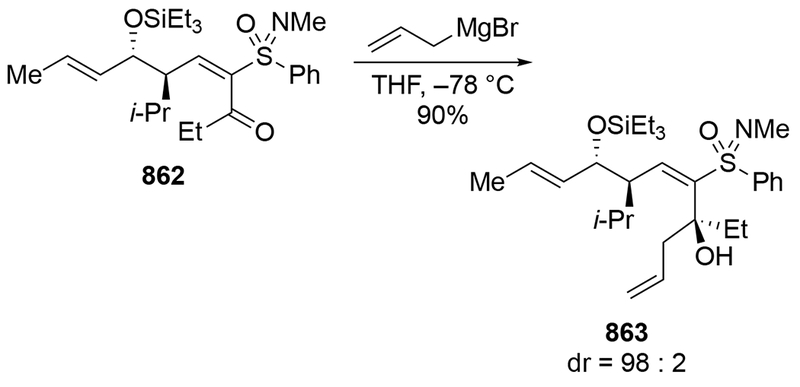
Scheme 344.
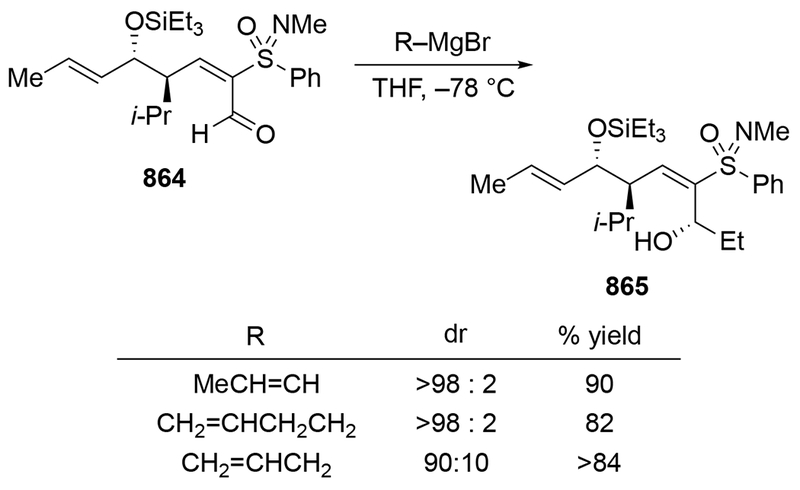
Substituted enals also did not react stereoselectively with allylmagnesium reagents (Scheme 345). Nevertheless, these unselective reactions were used in efforts to establish the configurations of natural products. The diastereomers of the addition products 867485 and 869486 were separated and individually converted to advanced intermediates that could be compared spectroscopically to natural products.
Scheme 345.
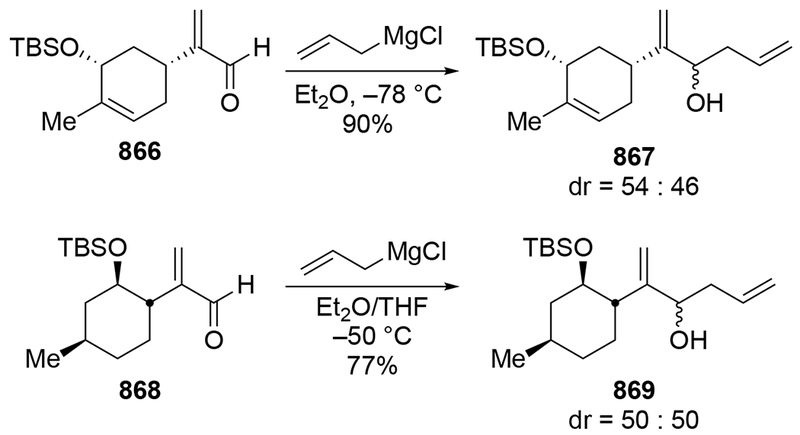
In the course of synthesizing new vitamin D derivatives, additions to hindered enone 870 were explored (Scheme 346).487 Although selectivity with allylmagnesium reagents was low, somewhat higher selectivity was observed with alkylmagnesium reagents. The authors attribute the facial selectivity to the conformational preferences of the ground state, as determined using molecular mechanics calculations. It is possible that conducting these additions at lower temperatures could have improved selectivity.
Scheme 346.
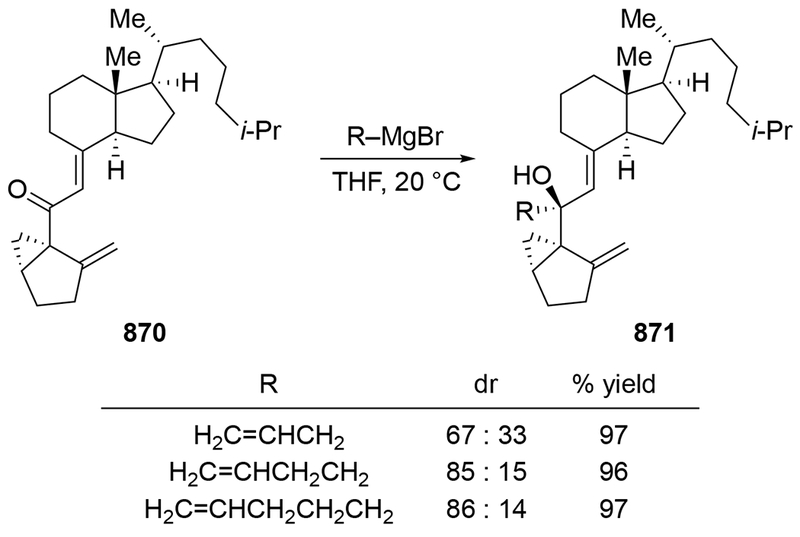
Additions of allylmagnesium halides to chiral aldehydes may not be stereoselective, but they can nevertheless be synthetically useful. Addition of allylmagnesium bromide to a mixture of diastereomeric aldehydes 872 led to a mixture of diastereomeric alcohols (Scheme 347).488 Neither of the stereocenters on this side-chain were important for the synthetic plan. After ring-closing metathesis involving the new allyl group, the secondary homoallylic hydroxyl group was oxidized to the give the corresponding ketone, and the stereocenter adjacent to that new carbonyl group was isomerized under basic conditions.
Scheme 347.
Two additional examples from the steroid literature also occur with similar low selectivity.489 Ketoaldehyde 874 reacted with prenylmagnesium chloride only at the formyl group to give a mixture of products. The regioselectivity of addition of the prenyl group contrasts with the general preference for prenylmagnesium reagents to give the γ-isomer, not the α-isomer.23 Similarly, additions to the α-hydroxyaldehyde 876, which is a mixture of diastereomers at the A-B ring fusion, also gave a mixture of stereoisomeric products. Evidently, the α-OMgX substituent did not control stereochemistry by chelation.
Additions of allylmagnesium reagents to aldehyde 878, with a complex side chain, occurred with low diastereoselectivity (Scheme 349).490 It was possible to obtain the product with the desired configuration by separating the stereoisomers of 879 and recycling the undesired one, through a sequence of oxidation and reduction. For aldehyde 880 (Scheme 350), however, addition was not stereoselective, and efforts to convert the mixture into the desired stereoisomer were unsuccessful.
Scheme 349.
Scheme 350.

Additions of allylmagnesium bromide to the related chiral aldehyde 882 also were not stereoselective (Scheme 351).491 The challenge of establishing the desired stereoisomer was eventually solved using chiral enolate chemistry.
Scheme 351.
7. Additions of Allylmagnesium Reagents to Imines and Related Substrates
7.1. Comparisons between Reactions of Imines and Reactions of Carbonyl Compounds
The analysis of reactions of imines and related substrates with allylmagnesium reagents provide useful comparisons to the related reactions of carbonyl compounds. Although both the carbon–oxygen and carbon–nitrogen double bonds are polarized bonds, the stereoselectivities of their reactions with carbon nucleophiles492 and the mechanisms of addition may or may not be the same. For example, in the case of iminium ions as electrophiles, any mechanism involving complexation of an organometallic nucleophile to the nitrogen atom is not possible considering that the nitrogen atom is positively charged. The additions of alkyl- and arylmagnesium reagents to simple imines are also likely to be slow.493–495 The reactivity of imines is also strongly influenced by the substituent on the nitrogen atom, which makes transition states more crowded. These groups can exert strong influences on the reactivity of the carbon–nitrogen double bond,58 and the stereochemical outcomes of the reactions, as illustrated for reactions with imines bearing chiral auxiliaries (Section 7.7). Considering that additions are generally slow, reactions with allylmagnesium reagents with imines may not be diffusion-controlled, although they are much faster than additions of alkylmagnesium reagents.336 Even with these differences, it is instructive to consider the reactions of compounds with carbon–nitrogen double bonds to provide comparisons to the reactions of carbon–oxygen double bonds discussed above.
This review will not cover all additions to imines. Instead, it will discuss the behavior of the carbon–nitrogen double bond with allylmagnesium reagents as a contrast to other reactions of this reagent. In general, reactions with imines do not show as dramatic differences in reaction outcomes as observed for reactions of carbonyl compounds with allylmagnesium reagents versus other Grignard reagents. As a result, only representative examples of additions will be presented. Other reviews have discussed in detail the diastereoselective reactions of imines with nucleophiles,492,496,497 including allyl nucleophiles.20,21
7.2. Mechanism of Additions of Allylmagnesium Reagents to the Carbon–Nitrogen Double Bond
In contrast to studies of the mechanism of additions of allylmagnesium reagents to carbon–oxygen double bonds,18 the mechanism of addition to carbon–nitrogen double bonds has not been examined in detail. Like the reactions of carbonyl compounds, however, these reactions may proceed with allylic transposition through a transition state resembling 884 (Scheme 352).498 This mechanism is supported by observations of additions of prenylmagnesium bromide to an activated imine, which gave preferentially the γ-product (Scheme 353),498 just as for additions to most aldehydes and ketones.23
Scheme 352.
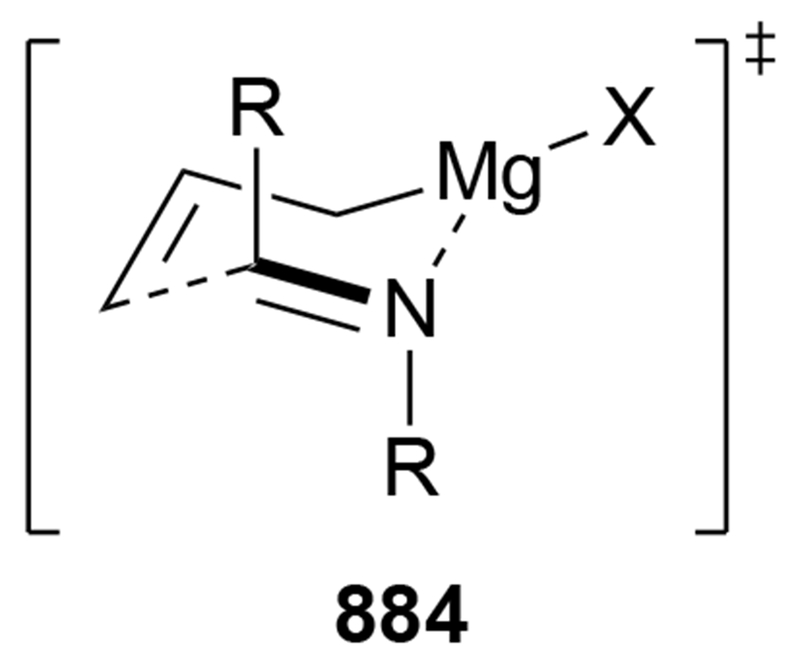
Scheme 353.
One study suggests that single-electron transfer pathways are unlikely, just as they are for most carbonyl compounds. A radical clock experiment, illustrated in Scheme 354,499 does not show evidence for radical intermediates (a similar experiment performed for additions of allylmagnesium reagents to alkyl aldehydes also showed no evidence for radical intermediates227). Addition of allylmagnesium bromide to imine 887 formed an amide intermediate, which eliminated the cyanide ion to form a new imine (888). This reaction occurred without ring-opening, which the authors suggest disproves the presence of radical intermediates. Addition of a second equivalent of the allylmagnesium reagent to imine 888 gave a diastereomeric mixture of products.
Scheme 354.
7.3. Additions of Allylmagnesium Reagents to Imines
7.3.1. Additions to Acyclic Imines
7.3.1.1. Additions to Acyclic Imines Capable of Chelation
Additions of Grignard reagents to imines with an α-alkoxy group can give products expected from chelation control. For example, additions of allylmagnesium reagents to simple imines proceed with chelation-controlled diastereoselectivity, as noted earlier (Scheme 18, Section 2.5).59 This observation, however, does not seem to be general. Additions to imine 890 occurred with high diastereoselectivity for most organomagnesium reagents (Scheme 355).500–504 Just as with α-alkoxy aldehydes, however (Section 4.2.4.3), additions of allylmagnesium reagents with imine 890 gave low stereoselectivity.503 No allylmetal reacted with high diastereoselectivity, however (Scheme 356).
Scheme 355.
Scheme 356.
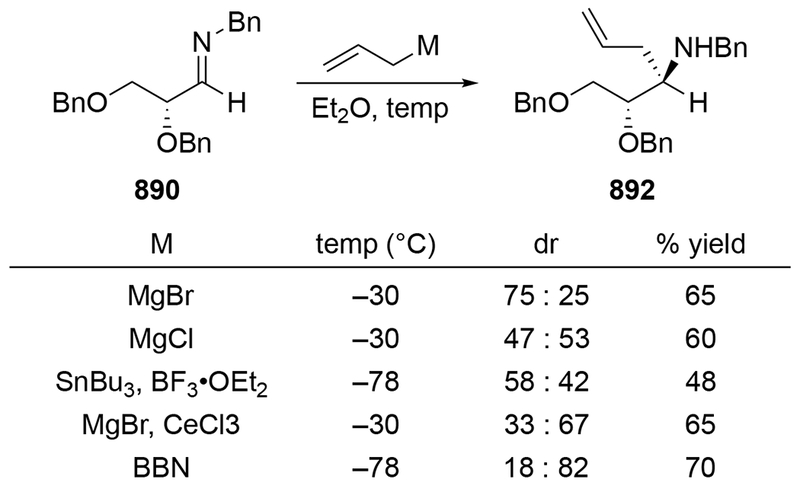
Installation of a group that should be more prone to chelation did not lead to high chelation-controlled diastereoselectivity (Scheme 357).505 The addition of allylmagnesium and allylzinc reagents gave moderate stereoselectivity for the chelation product. Additions of allyltitanium reagents were modestly selective for the Felkin–Anh product, which was strongly favored for the non-chelating boron reagent.
Scheme 357.
Addition of allylmagnesium bromide to more highly substituted substrates occurred with high diastereoselectivity (Scheme 358).506 This reaction was a key step in the synthesis of a conformationally constrained sialic acid derivative.
Scheme 358.
Additions of allylmetal nucleophiles to α-alkoxy and α-silyloxy imines behaved similarly (Scheme 359).507 Although the product from chelation control was favored, stereoselectivity was not particularly high, even with allylzinc reagents.
Scheme 359.
Additions of carbon nucleophiles to α-silyloxy silylimines, however, occurred with significantly different stereochemical outcomes depending upon the reagent used (Scheme 360).508 Imine 900 possesses a silyloxy group, which should not engage in chelation.236 Nevertheless, additions of alkylmetals like n-Bu–Li and PhCH2–MgCl both gave the product expected from chelation control. By contrast, additions of allylmagnesium reagents gave the Felkin–Anh product, even in the presence of zinc salts that would be expected to be favor chelation.
Scheme 360.
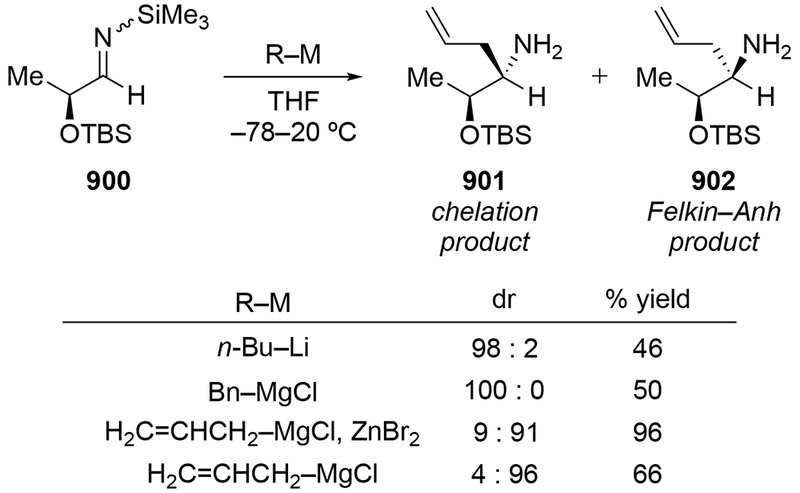
Allylations of imines with α-alkoxy groups as part of a ring gave only modest stereoselectivity (Scheme 361).509,510 Allylmagnesium reagents favored the expected chelation-controlled product, but the zinc reagents gave the opposite selectivity. These differences could involve chelation through different oxygen atoms, which might alter the outcome of addition.
Scheme 361.
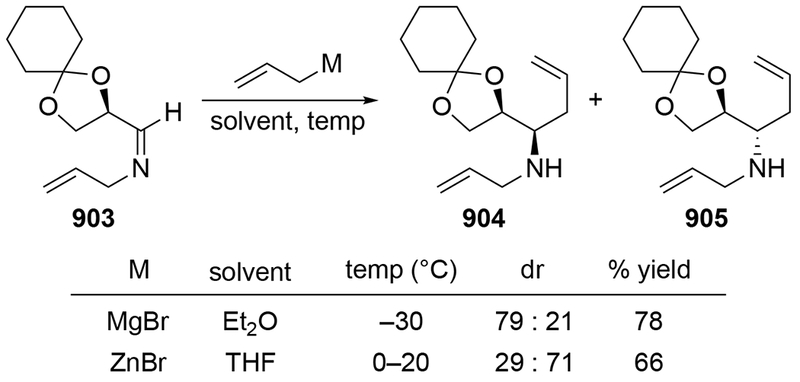
Additions to more highly substituted exocyclic imines, however, were highly diastereoselective. In contrast to additions to exocyclic aldehydes (for example, Scheme 71, Section 4.2.4.3), additions to exocyclic imines favored the product expected from chelation control with high stereoselectivity (Scheme 362).511 These reactions, which were used to prepare glycosidase inhibitors, parallel similar observations for additions of other organometallic nucleophiles to a related electrophile512
Scheme 362.
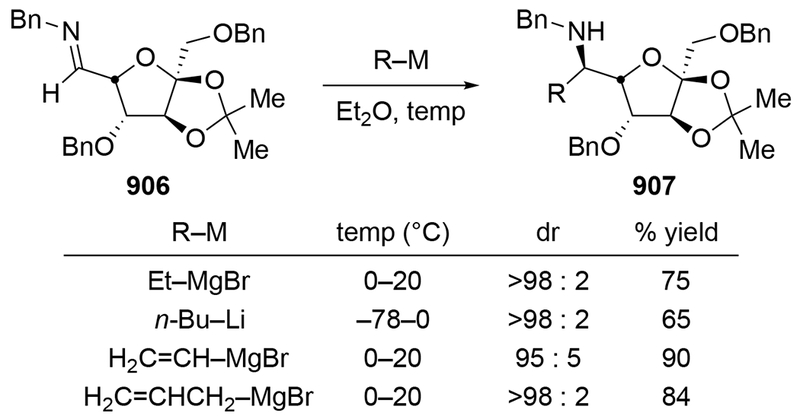
Reactions with another related substrate gave similar results (Scheme 363).513 Additions of both allyl- and homoallylmagnesium reagents gave the product as a single stereoisomer. Additions of vinylmagnesium reagents were reported to proceed with lower diastereoselectivity, although the selectivity was not given. Instead, addition of the vinyl nucleophile was conducted in the presence of a Lewis acid, which led to the opposite diastereomer, the product expected by considering the Felkin–Anh model (Scheme 364). This reversal of selectivity in the presence of BF3·Et2 was also observed with the aldimine 906 (Scheme 362).511
Scheme 363.
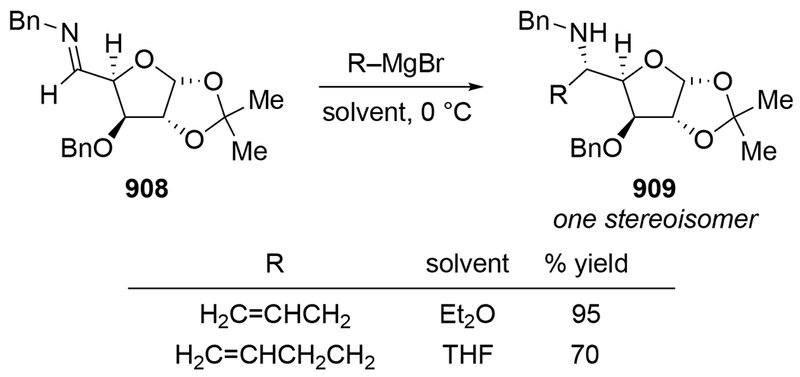
Scheme 364.
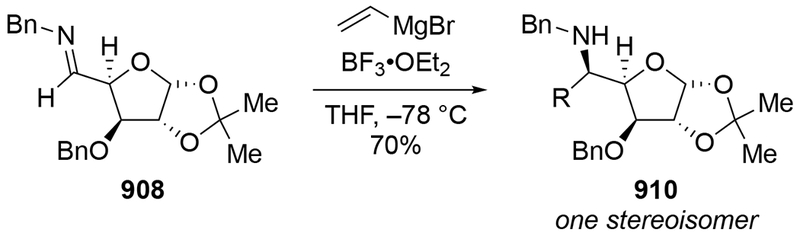
Chelation from a carbonyl group can also be used to control the stereoselectivity of additions to imines. Additions to the chiral α-imino ester 911, which was generated in situ from an α-bromo amine, proceeded with generally good diastereoselectivity (Scheme 365).514 Upon chelation between the imine nitrogen atom and the carbonyl group, attack would need to occur from the face away from the cumyl group. As observed for carbonyl compounds, however, addition of allylmagnesium reagents were much less stereoselective.
Scheme 365.
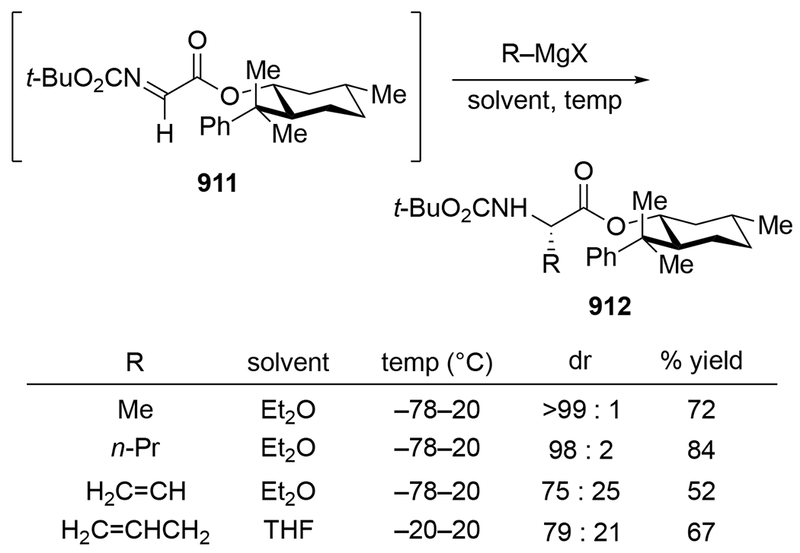
Just as with ketones and aldehydes (Sections 4.2.3.1 and 4.2.5.1, respectively), additions of carbon nucleophiles to imines with β-chelating groups did not lead to generally high stereoselectivity (Scheme 366).505 Among all the allylating reagents examined, allylmagnesium chloride gave the highest selectivity.
Scheme 366.
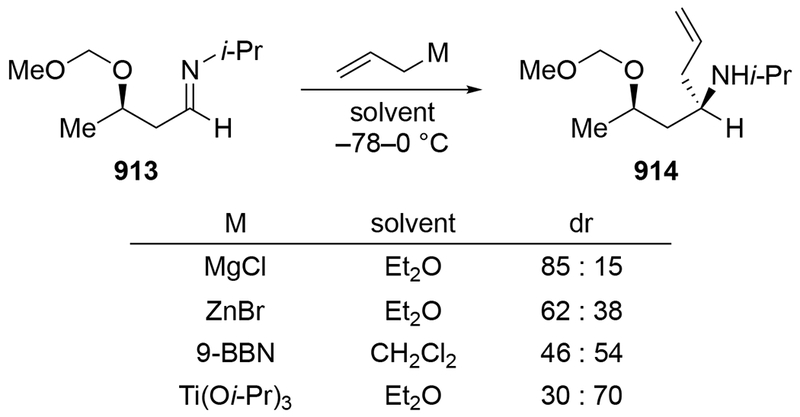
7.3.1.2. Additions to Acyclic Imines Not Capable of Chelation
The additions of allylmetal reagents to chiral imines with a stereocenter provide a clear comparison with reactions of related carbonyl compounds (Scheme 367).1,515 The stereoselectivity of allylations of imine 915 with allylmagnesium reagents was low. Addition of an allylboron reagent, however, proceeded with high diastereoselectivity.
Scheme 367.
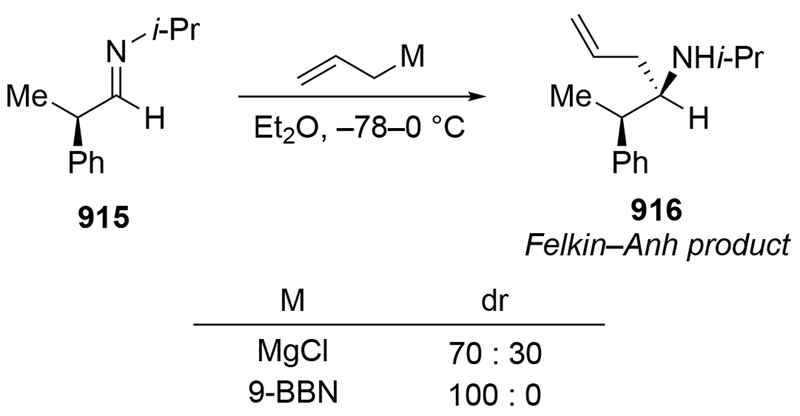
The analyses of the stereochemical courses of these reactions illustrate how the reactions of imines must be considered differently than reactions of aldehydes. At first analysis, the stereoselectivity observed for the addition of allylmagnesium halide is quite similar to the selectivity observed for addition to the corresponding aldehyde (Scheme 52). If these reactions proceed through a cyclic transition state with allylic transposition, as likely operates for additions to carbonyl compounds (i.e., 14, Section 2.1), the transition states develop other steric interactions with the substituent on the imine. The cyclic transition state with allylic transposition is illustrated by 884 (Scheme 352), assuming that the metal atom (boron or magnesium) possesses two groups, which could be substituents or solvent. In the case of the imine, the substituents must adopt axial orientations because the lone pair on the nitrogen atom would be complexed to the metal atom. This requirement brings the branched substituent close to the ligands on the metal center. The stereogenic center could adopt a conformation to minimize steric interactions with the groups on the metal, as illustrated for transition state 917 (Scheme 368). The larger ligands on the boron center likely contribute to the higher selectivity in that case.516 Transition state 917, is not, strictly speaking, a Felkin–Anh transition state, however, because of the arrangement of the atoms on the stereogenic center. To adopt a transition state more resembling the Felkin–Anh transition state (i.e., 918), steric interactions would develop with the axial group on the metal atom. This example provides an important caveat regarding stereochemical models: observation of a product expected from the Felkin–Anh model does not require that the transition state fits that model precisely. This point is discussed in detail in Section 3.
Scheme 368.
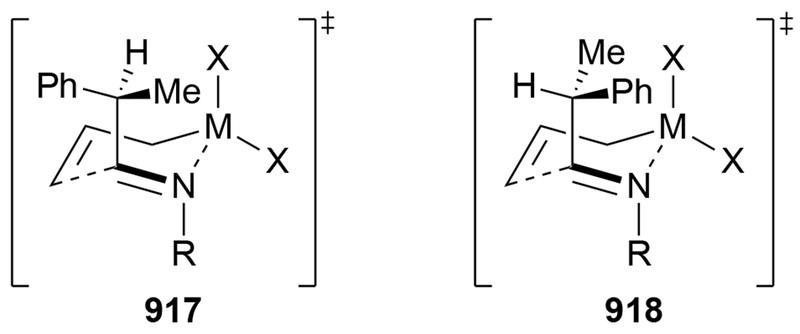
Additions to a family of N-tosylimines occurred with different stereoselectivity depending upon the nature of the organomagnesium reagent (Scheme 369).517 For both imine 919 and 921, additions of vinyl- and homoallylmagnesium reagents occurred with high diastereoselectivity that can be explained by invoking the Felkin–Anh-like transition state where the OR group adopts the RL position (923). Additions of allylmagnesium reagents, however, occurred with low diastereoselectivity.
Scheme 369.
7.3.2. Additions of Allylmagnesium Reagents to Exocyclic Imines
The low selectivity observed upon additions of Grignard reagents to 4-tert-butylcyclohexanone (Section 6.2.1.1) was also observed for their derived imines (Scheme 370).518 This example illustrates the challenges associated with reactions of imines and the utility of allylmagnesium reagents. Additions of other allylmetal reagents, including allylsilanes, allylstannanes, and allylzinc reagents, occurred only slowly or not at all. Furthermore, attempted additions of other organomagnesium reagents gave only products resulting from deprotonation of the imine.
Scheme 370.
The enhanced reactivity of allylmagnesium reagents compared to most other Grignard reagents is also illustrated for reactions of cyclohexanone sulfoximine 926 (Scheme 371).519 Benzyl- and allylmagnesium reagents were the only Grignard reagents that would add to this electrophile. It should be noted, however, that these reaction mixtures contained residual titanium salts because of how the sulfoximines were prepared. Consequently, the involvement of organotitanium reagents cannot be discounted.
Scheme 371.
Just as with five-membered ring ketones with highly differentiated faces (Section 6.2.2.3), the exocyclic amine 928 underwent highly diastereoselective addition reactions with allylmagnesium chloride (Scheme 372).520 The allyl group attacked from the face opposite the two alkyl groups, which could adopt pseudoaxial orientations to minimize allylic strain with the carbamoyl group.521 Attack on the carbon–nitrogen double bond from the more exposed face, as shown in 930, would form the observed product.
Scheme 372.
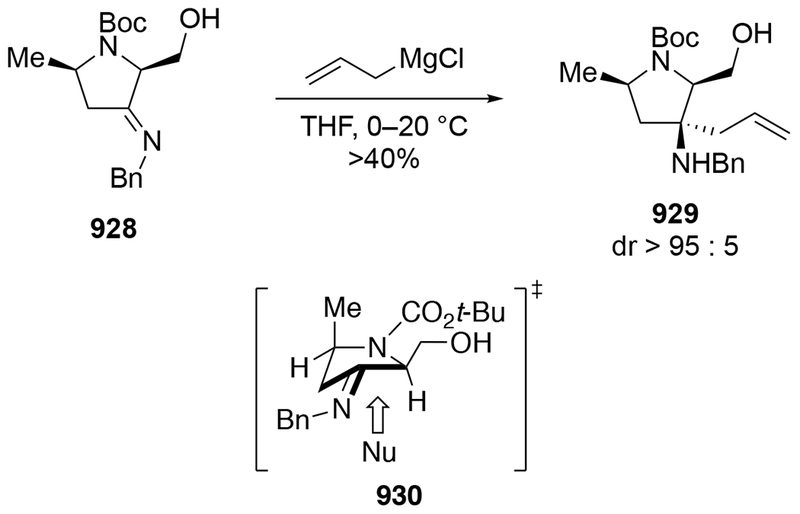
7.3.3. Additions of Allylmagnesium Reagents to Endocyclic Imines
Additions to cyclic imines are more easily analyzed than additions to acyclic ones. For example, addition to imine 931, formed in situ, resulted in high selectivity for the product 932 (Scheme 373).522 This outcome can be understood as arising from addition to the half-chair conformation of the imine along a trajectory that converts the half-chair into the chair form (i.e., 933→934, Scheme 374). This model follows the stereoelectronic and torsional model originally formulated to understand the reactions of iminium ions,122 which was later extended to reactions of oxocarbenium ions.123 The allyl group was cyclized onto the nitrogen atom to form the core structure of indolizidine natural products.
Scheme 373.
Scheme 374.
The utility of allylmagnesium reagents was demonstrated in the course of optimizing a method for the synthesis of quinolizidine alkaloids (Scheme 375).523 Addition of allylmagnesium chloride to imine 935, formed in situ, occurred with the same sense of stereoselectivity expected from Scheme 374. The use of allylmagnesium reagents in the presence of small quantities (as low as 23 mol%) of ZnCl2 gave slightly improved yields, but generating the allylzinc reagent stoichiometrically led to no reaction. Whereas the use of allylboranes gave similar yields, no allylation was observed using allylsilanes or allylstannanes in the presence of Lewis acids. Under the optimized conditions, crotylation with the zinc reagent, which likely proceeded through a cyclic transition state with allylic transposition similar to 884 (Scheme 352), also controlled stereoselectivity at the exocyclic stereocenter (Scheme 376). The configuration at the ring for 937 can be explained by assuming that the alkyl group adopts an equatorial orientation, leading to the cis-disubstituted ring; this selectivity is the same for the related oxocarbenium ions.524 Pyrrolidine 938 was converted to the alkaloid target by fusing a new ring onto the six-membered ring.
Scheme 375.
Scheme 376.
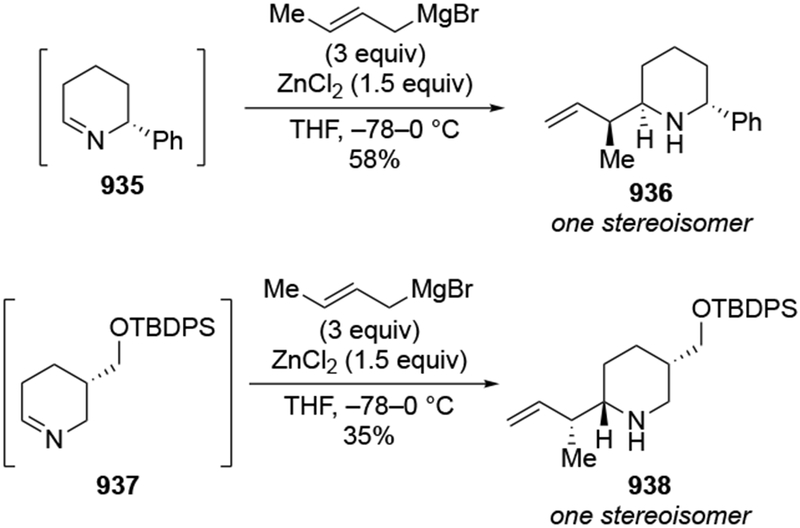
A more constrained six-membered ring imine reacted with lower stereoselectivity, however (Scheme 377).525 In the course of preparing potential anti-depressant drugs, additions of allylmetals to imine 939 were examined. Additions with allyllithium and allylmagnesium reagents were unselective, but addition of allylzinc bromide gave useful stereoselectivity. The allyl group was needed to form a fused pyrrolidine ring onto the tetrahydroisoquinoline framework.
Scheme 377.
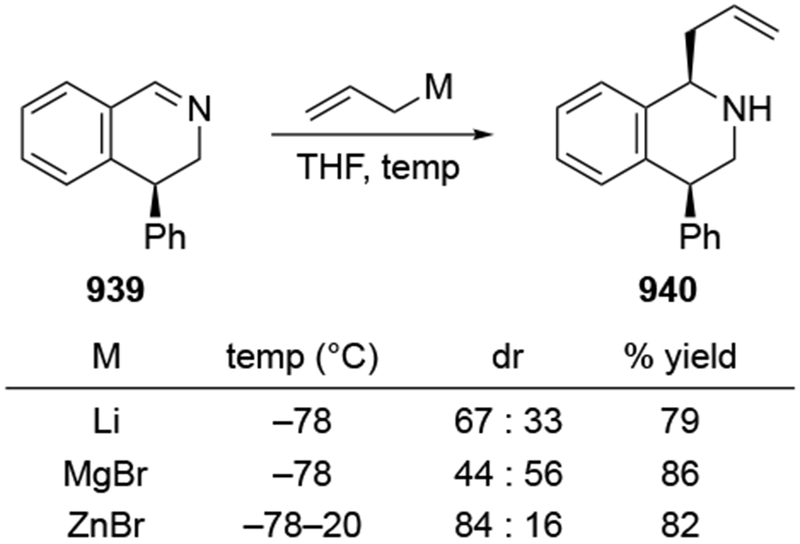
Addition of allylmagnesium chloride to a spirocyclic six-membered ring imine (941) occurred with high stereoselectivity (Scheme 378).526 This selectivity can be understood as the reaction occurring through a transition state resembling 933 (Scheme 374) with the more substituted atom of the cyclohexyl group adopting an equatorial position. Attack to the only accessible face along an axial trajectory would form the observed product.
Scheme 378.
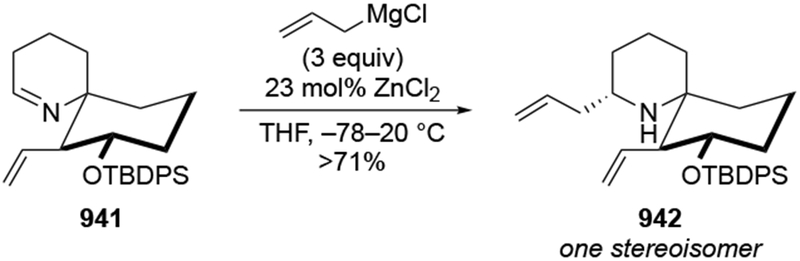
Additions to five-membered ring imines are also highly stereoselective, but the stereochemical outcomes of these reactions are somewhat difficult to reconcile. Addition of allylmagnesium chloride to imine 943 occurred with high diastereoselectivity for addition opposite the substituent (Scheme 379).527 Other additions of allylmagnesium halides to five-membered ring imines give similar stereoselectivities (Scheme 380).528 The stereochemical outcome is what might be expected by considering the substituent blocking one face so attack must occur from the other face. Additions to five-membered ring electrophiles with two trigonal atoms in the ring cannot be analyzed steric effects alone, however529,530.
Scheme 379.
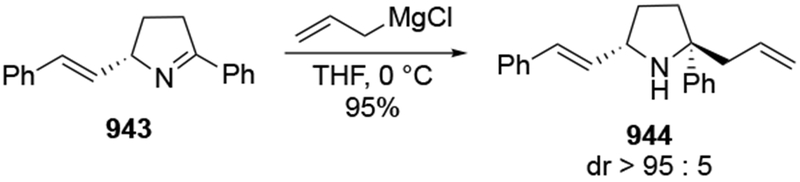
Scheme 380.
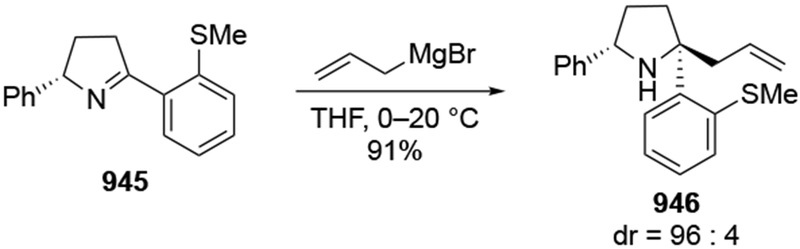
The difficulty of analysis of the outcomes in Schemes 379 and 380 emerges by comparison to the results in Scheme 81.531 Addition of allylmagnesium bromide to imine 947 gave good selectivity for addition syn to the substituent, not anti to it. The selectivity of this reaction can be considered to involve addition to the lowest-energy conformer of the imine from the stereoelectronically favored direction (i.e., from inside the envelope) through transition state 949 to give the major product (Scheme 382). This mode of attack is similar to the one that occurs for additions to five-membered ring oxocarbenium ions530,532 and iminium ions.533,534 It is not clear why this example proceeds with opposite stereoselectivity compared to Schemes 379 and 380.
Scheme 382.
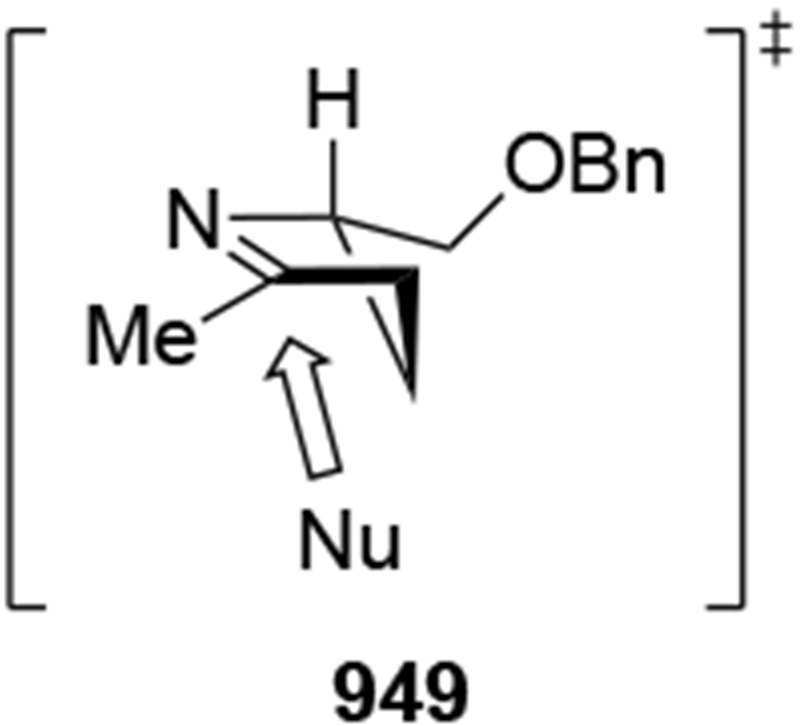
A similar syn-selective addition was observed in a related imine (Scheme 383).535 This example provides evidence that addition to imine 947 (Scheme 381) cannot involve chelation, because chelation of the reagent to the silyloxy group of imine 950 is unlikely.236 Nevertheless, the experiments shown in Schemes 381 and 383 are in conflict with those shown in Schemes 379 and 380.
Scheme 383.
Scheme 381.
Additions to a fused bicyclic five-membered ring imines were also highly diastereoselective (Scheme 384).536 These reactions required an excess of the organomagnesium reagent to generate the imine. Formation of the sulfonyl-stabilized carbanion caused a ring-opening reaction to generate the imine 955 (Scheme 385). This elimination resembles the decomposition of THF by alkyllithium reagents.537 The resulting imine was then trapped by the organometallic reagent from the convex face with high stereoselectivity.
Scheme 384.
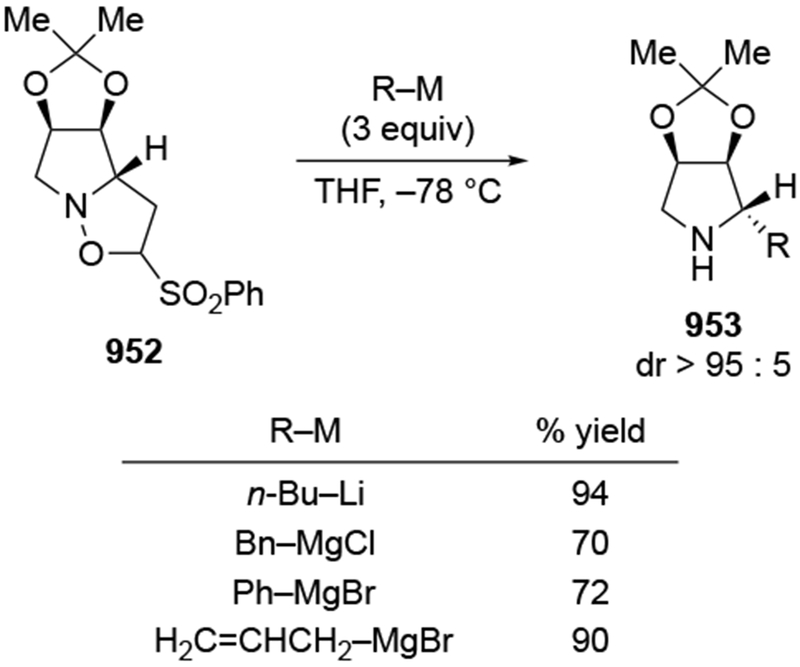
Scheme 385.
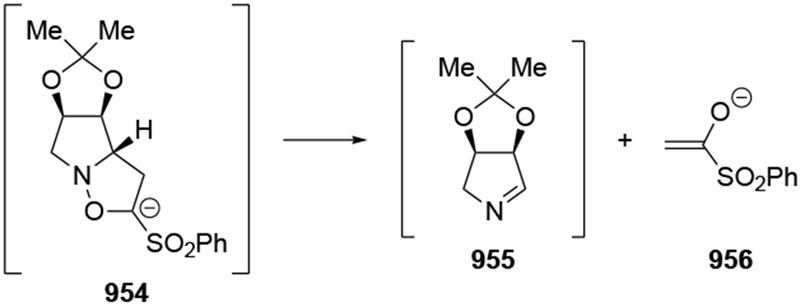
Additions to the indole derivative 957 provided an outcome that is difficult to accommodate by consideration of steric effects alone (Scheme 386).538 Addition to the indoline 957 occurred smoothly with allylmagnesium iodide to give the adduct 958. By contrast, additions of methylmagnesium iodide to indolines of this type were slow and occurred with low yields, and additions of benzylmagnesium reagents formed a number of products. Reactions with Grignard reagents with β-hydrogen atoms, such as ethyl, propyl, or butylmagnesium halides, did not give products. These results were explained as involving radical intermediates, a mechanism that has been discounted for additions of organomagnesium reagents, including allylmagnesium halides, to alkyl aldehydes.227 The configuration of the product involves coupling from the more hindered face. The stereochemical assignment, however, may be considered tentative: the two isomers were differentiated based upon their splitting patterns in their 1H NMR spectra, which may not be completely diagnostic.
Scheme 386.
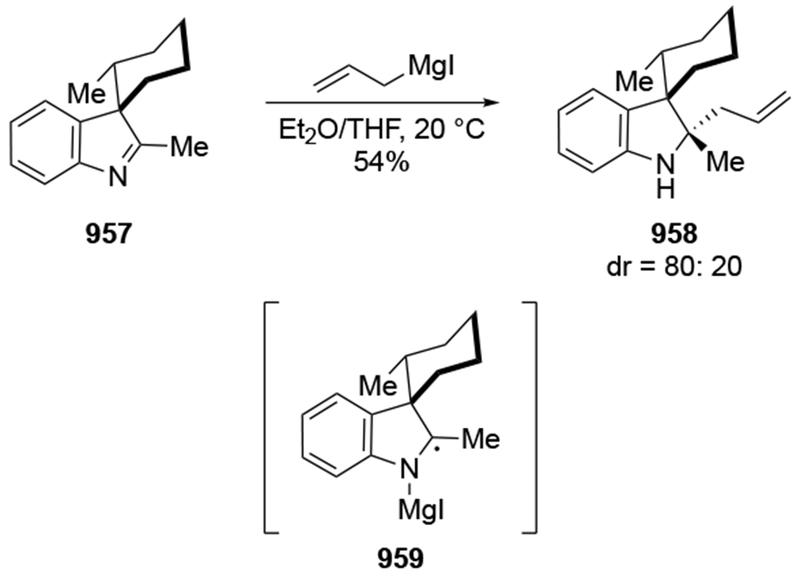
Additions to the seven-membered ring imine generated from N,O-acetal 960 led to different stereochemical preferences depending upon the reagent (Scheme 387).539 Reactions with allyl and phenylmagnesium reagents gave one stereoisomer of product, while the use of other Grignard reagents gave the opposite stereoisomer. The preferential formation of the isomer from a reagent such as methylmagnesium bromide could be argued to arise from chelation-controlled addition through magnesium alkoxide 961. The origin of the other stereoisomer was not explained, however. The sense of selectivity is similar to that observed for reactions of structurally similar seven-membered ring oxocarbenium ions,540 whose reactions are likely controlled by stereoelectronic and torsional effects.541
Scheme 387.
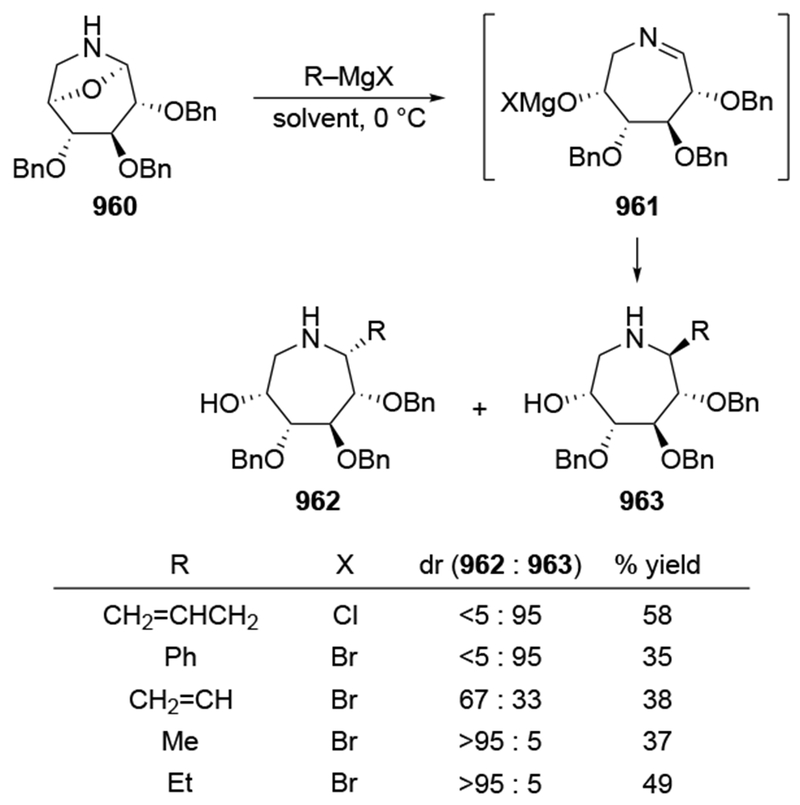
7.4. Additions of Allylmagnesium Reagents to Iminium Ions
A series of iminium ions, generated from the dihydropyridine 964, could be trapped with nucleophiles with good selectivity (Scheme 388).542,543 The iminium ion was generated by alkylating the enamine. Addition of an organometallic reagent then occurred with high diastereoselectivity, likely by minimizing torsional strain in the first-formed products.544 The allylcerium reagent derived from the allylmagnesium halide was employed to favor addition over proton transfer543
Scheme 388.
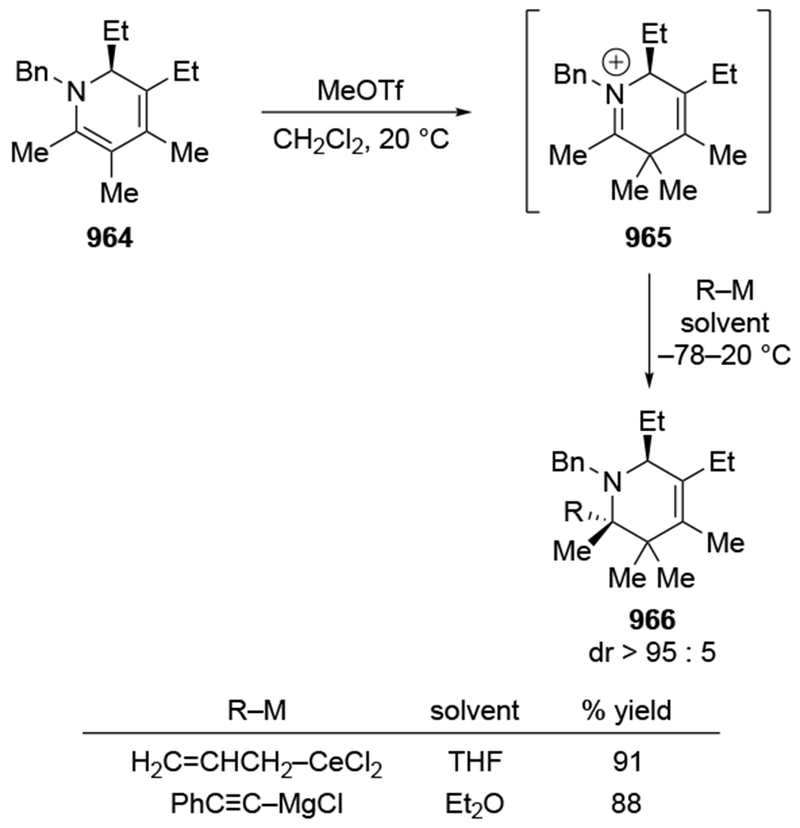
Fused bicyclic iminium ions also undergo stereoselective allylations (Scheme 389).545 The iminium ion 968, which was generated by abstraction of the cyano group with silver ion, could be observed spectroscopically. Trapping of the iminium ion with organometallic reagents, including allylmagnesium reagents, occurred from the convex face with high stereoselectivity. Additions of alkyllithium reagents were unsuccessful.
Scheme 389.
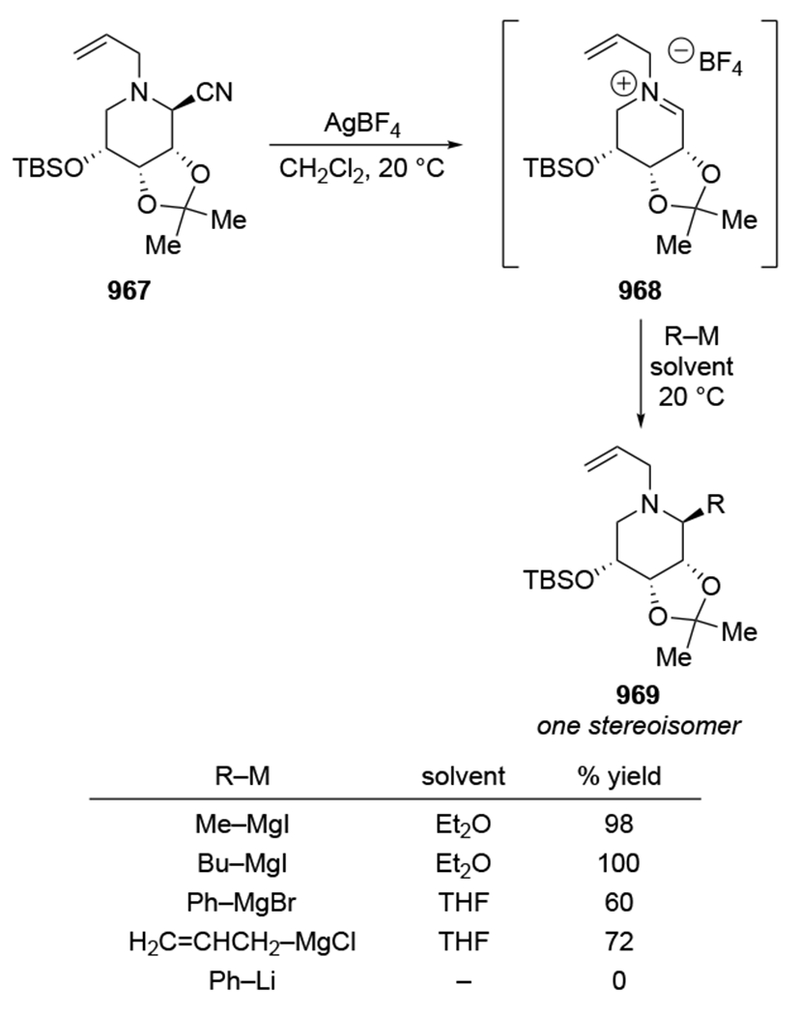
Additions to the fused bicyclic iminium 971 also occurred with high stereoselectivity (Scheme 390).546 The iminium ion was prepared in situ by first reduction of the bicyclic acetal to yield the hemiacetal followed by warming in the presence of the nucleophile. Only allylmagnesium bromide was examined because this group was essential to perform a subsequent ring closure to make the desired core structure of an alkaloid.
Scheme 390.
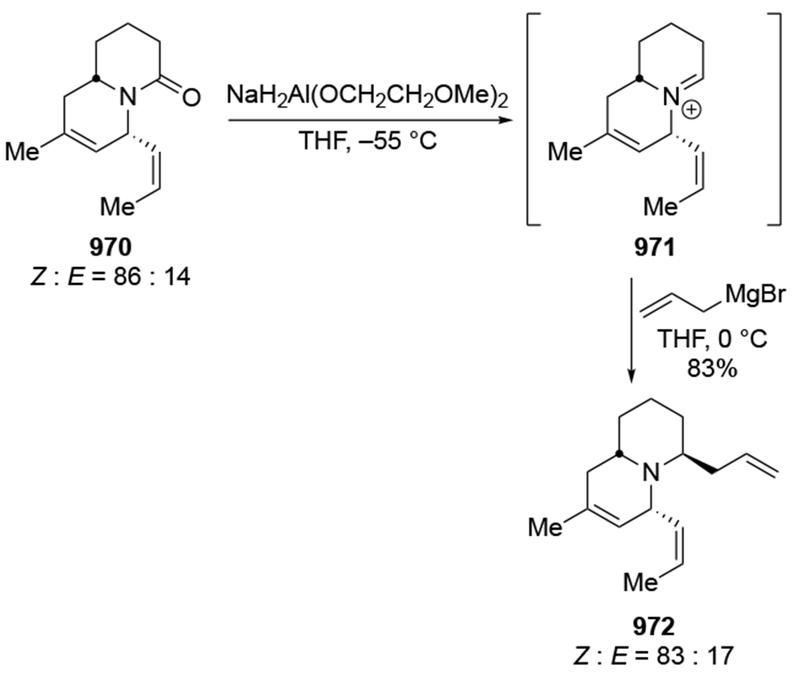
Additions of allylmagnesium reagents to multicyclic iminium ions can be highly stereoselective (Scheme 391).547 Addition of methallylmagnesium chloride occurred from the convex face to establish stereochemistry at a tetrasubstituted carbon stereocenter. This four-carbon chain was essential for the subsequent steps of a synthesis of an alkaloid natural product.
Scheme 391.
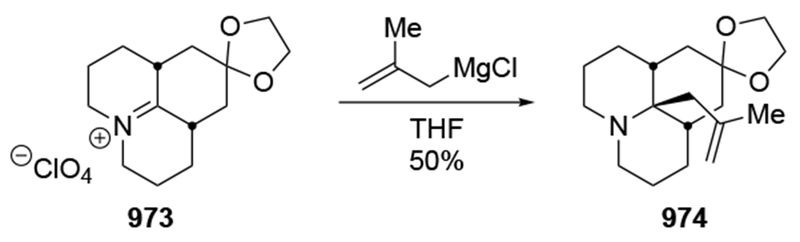
Additions to pyridinium ions can also be stereoselective with some organometallic nucleophiles, but not allylmagnesium halides (Scheme 392).548 Addition can form mixtures of regioisomers and stereoisomers. The use of methyllithium exhibited neither regio- nor stereoselectivity, but the use of alkylmagnesium bromides were both regio- and stereoselective. Additions of allylmagnesium bromide, however, were not highly regioselective. The major regioisomer (i.e., the regioisomer that formed faster), was the product of 1,4-addition, 976. This product was obtained with low stereoselectivity. By contrast, the minor regioisomer, the 1,2-addition product 977, was formed with high diastereoselectivity. This trend, in which the faster-formed regioisomer was made without diastereoselectively but the slower-formed regioisomer was made stereoselectively, was also observed for additions of allylic organomagnesium reagents to hindered ketones.23
Scheme 392.
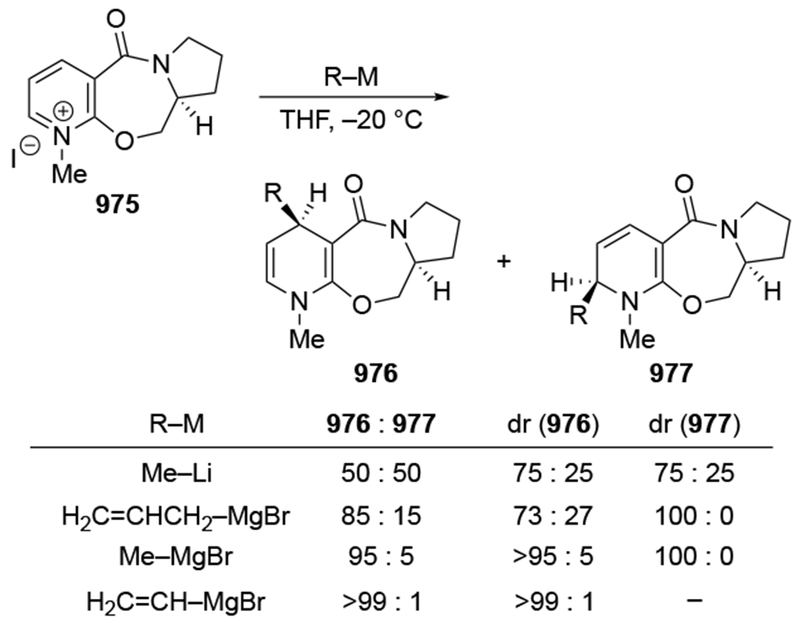
Five-membered ring iminium ions also underwent reactions with allylmagnesium reagents with some diastereoselectivity (Scheme 393).549 The only nucleophile that was used to trap these iminium ions was allylmagnesium bromide. The relative configuration of the product was not given.
Scheme 393.
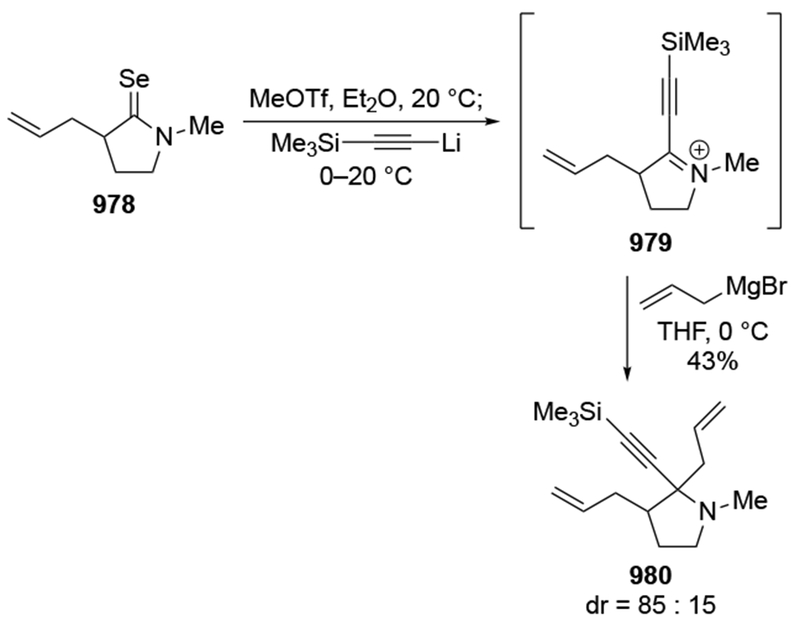
In the course of the synthesis of alkaloids that could be used for the treatment of cancer, the reactions of the five-membered ring iminium ion 982 were examined (Scheme 394).550 Whereas vinylmagnesium reagents reacted with high diastereoselectivity, allylmagnesium reagents reacted with a only a modest preference for the desired product. Because a two-carbon atom chain was needed for the proposed synthesis, use of the adduct 983 with the vinyl group would have been ideal. It was not possible to functionalize the double bond of 983 (R = CH=CH2) to obtain the desired product, however. As a result, the allylated compound 983 (R = CH2CH=CH2) was used to complete the synthesis. A carbon atom needed to be removed from the allyl chain by dihydroxylation and oxidative cleavage of the resulting diol. This chain was then used to close additional rings, which enabled the synthesis of the target molecule.
Scheme 394.
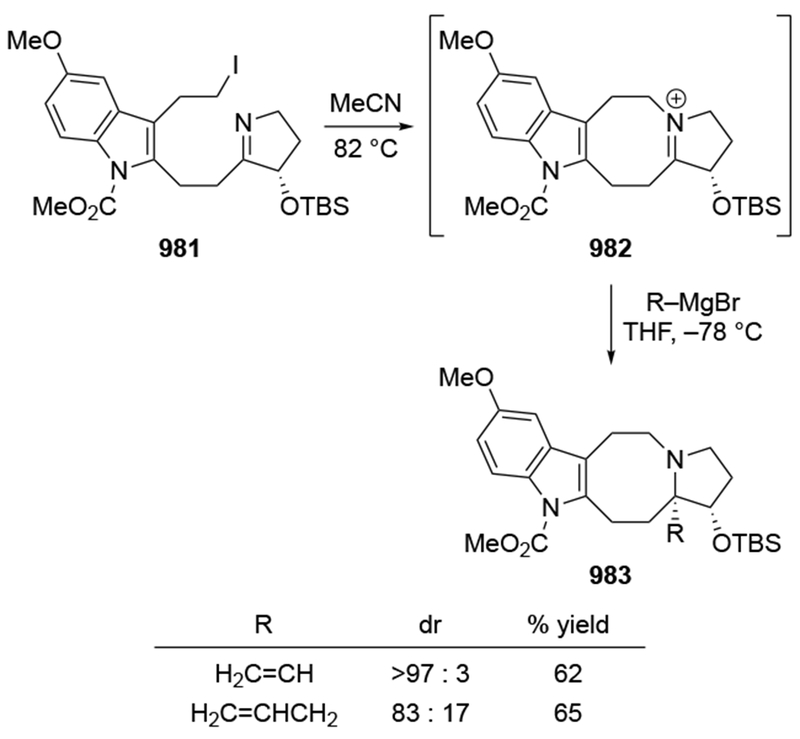
7.5. Additions of Allylmagnesium Reagents to Complexes of Imines with Lewis Acids and Metal Complexes
A transformation that is closely related to the reactions of nucleophiles with iminium ions are the reactions of nucleophiles with imines complexed to Lewis acids. Boron-containing Lewis acids have been employed commonly for reactions with organomagnesium reagents. For example, the reaction of 984, the boron complex of an imine, was highly diastereoselective for addition from the convex face (Scheme 395).551 The use of a chiral boron Lewis acid reagent (986) was capable of conferring high diastereoselectivity to additions of allylmagnesium bromide (Scheme 396), a transformation that enabled the enantioselective synthesis of pyrrolidines.552
Scheme 395.
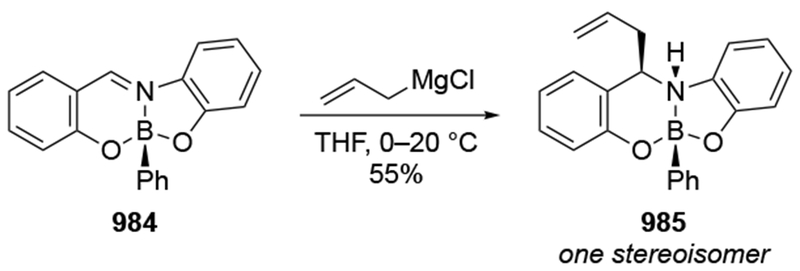
Scheme 396.
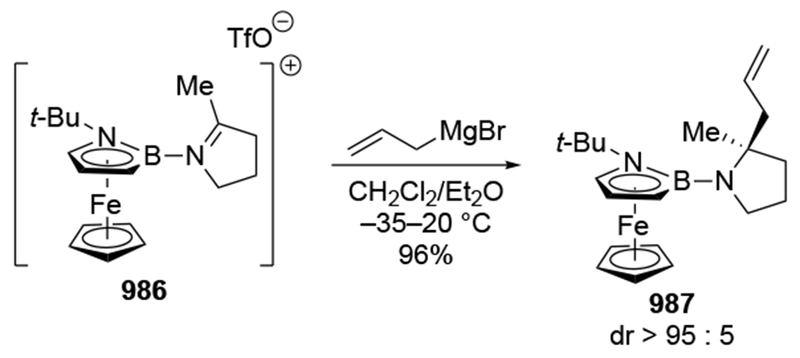
The use of complexation to a boron Lewis acid can change the stereoselectivity of an addition reaction. As discussed above (Scheme 381, Section 7.3.3), addition of allylmagnesium bromide to the imine 947 occurred with high stereoselectivity favoring addition from the face bearing the benzyloxymethyl group. After complexation to BF3•OEt2, however, the sense of diastereoselectivity was reversed (Scheme 397).531 It is likely that the Lewis acid-base complex adopts a conformation resembling 989 (Scheme 398) to minimize allylic strain, as has been observed for other nitrogen-containing five-membered ring species.521 Stereoelectronically favored attack from inside the envelope530,532,534 would lead to the observed product.
Scheme 397.
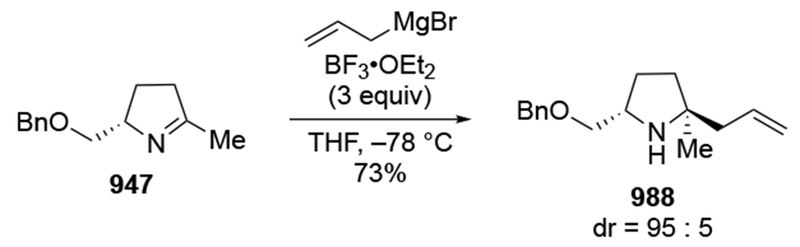
Scheme 398.
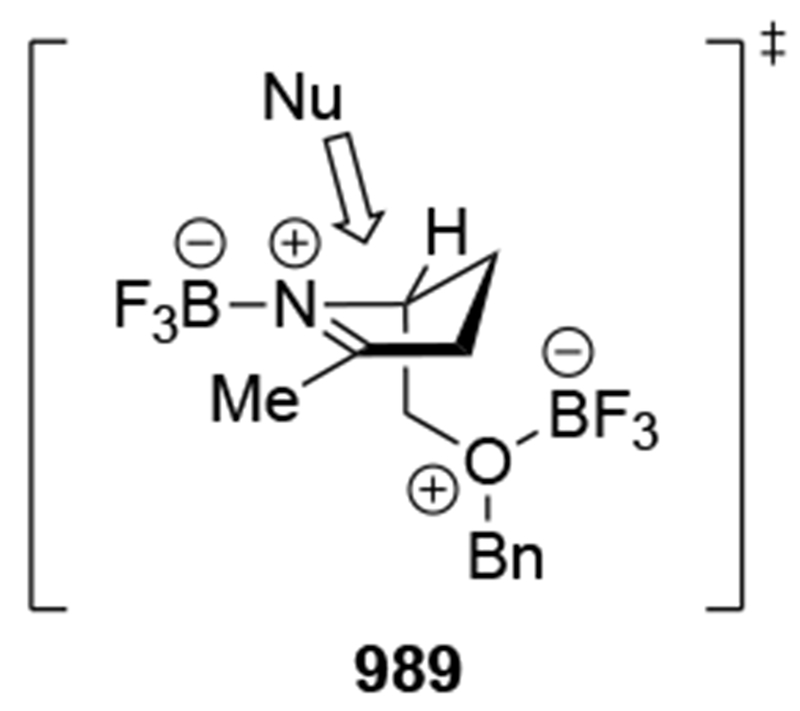
Additions of allylmagnesium reagents to complexes of amines with boron Lewis acids are also stereoselective with multicyclic imines (Scheme 399).553 The imine 990 was first complexed with the Lewis acid at −30 °C for 30 minutes, then allylmagnesium chloride was added to provide amine 991 a single stereoisomer. The product forms the cis-fused pyrrolidine ring by attack from the more exposed convex face.
Scheme 399.
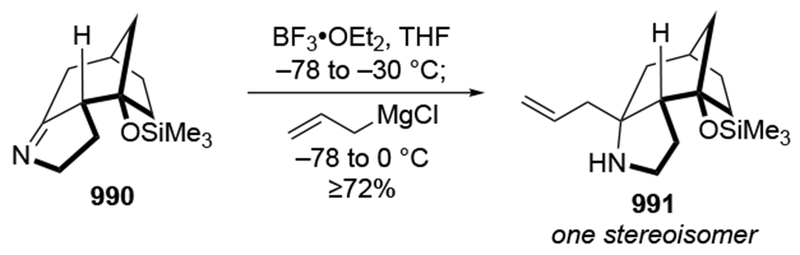
Transition metal complexes can also be used to activate imines for attack by carbon nucleophiles. The metal complex of an isoquinoline, 992, reacted with different diastereoselectivity depending upon the nucleophile employed (Scheme 400).554 Among the several organometallic reagents examined, including both Grignard reagents and organolithium reagents, allylmagnesium reagents gave the lowest stereoselectivity.
Scheme 400.
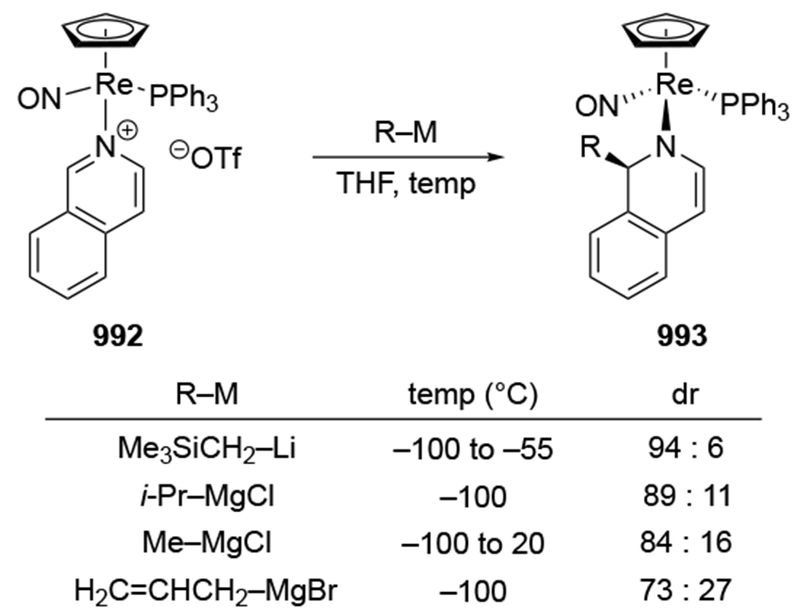
7.6. Additions of Allylmagnesium Reagents to Other C=N Double Bonds
7.6.1. Additions to Oximes
Although additions of allylmagnesium reagents to oxime derivatives are not common transformations, these reactions can be stereoselective. For example, addition to α-alkoxy oxime 994 proceeded with high stereoselectivity (Scheme 401).555 This stereochemical course can be understood by considering chelation between the OMe group and the oxime nitrogen (as illustrated in transition state 996). This degree of stereoselectivity is higher than generally seen with imines with α-alkoxy groups (Section 7.3.1.1).
Scheme 401.
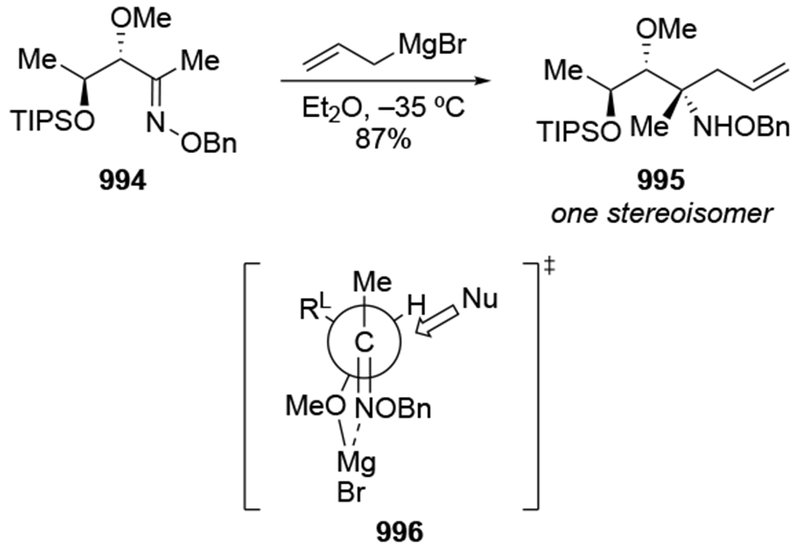
An oxime ether with the stereocenter more remote did not react as stereoselectively with allylmagnesium reagents, however (Scheme 402).556 Addition of allyllithium was stereoselective. The major product could be formed by addition to chelate 999 from the more exposed back face. Addition of allylmagnesium bromide, however, was unselective, and addition of a cerium reagent proceed with the opposite sense of selectivity.
Scheme 402.

Addition of allylmagnesium to the oxime ether 1000 with a nearby 8-phenmenthol ester group proceeded with low diastereoselectivity (Scheme 403).557 This result is similar to the observation of low selectivity for addition of allylmagnesium to the corresponding N-acylimine (Scheme 365, Section 7.3.1.1).514 In the presence of zinc salts, however, high diastereoselectivity, which is consistent with a chelate, was observed. In general, additions of organomagnesium reagents did not occur or occurred in low yields unless the zinc salts were employed.557
Scheme 403.
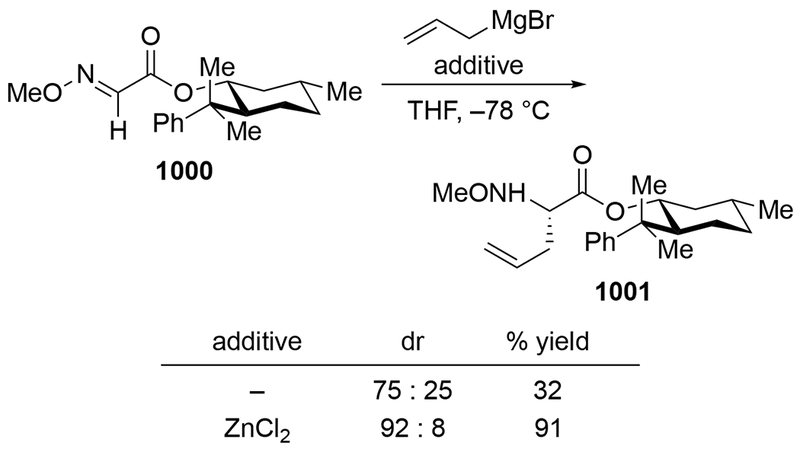
Additions of organometallic nucleophiles to five-membered cyclic oximes (i.e., isoxazolines), provided a stereoselective method for preparing β-amino acid analogues (Scheme 404).558,559 The oxime ethers were complexed to BF3 before addition of the nucleophile, as discussed in Section 7.5. These reactions were successful with allyl and benzylic Grignard reagents, but other Grignard reagents (i.e., alkyl and aromatic Grignard reagents) would not add. If the free hydroxyl group were protected, however, alkyllithium reagents could be employed to achieve the addition reaction (Scheme 405).
Scheme 404.
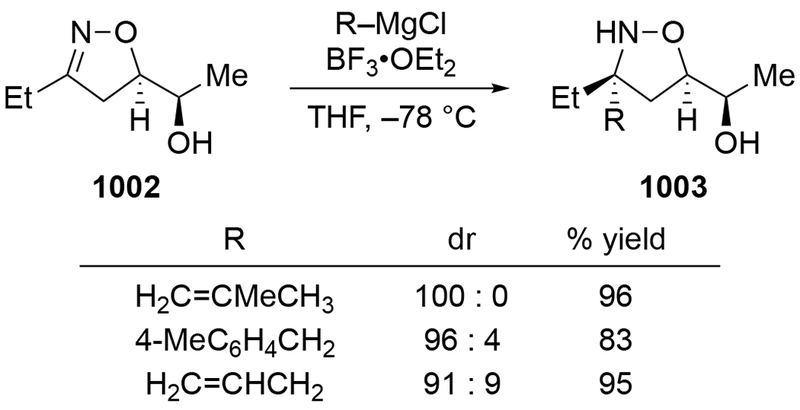
Scheme 405.
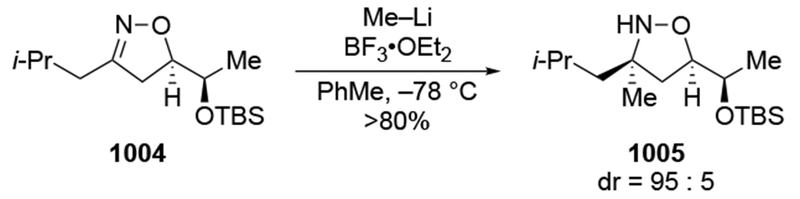
Addition of allylmagnesium bromide to a related isoxazoline proceeded with low stereoselectivity (Scheme 406).560 The low selectivity in this case may arise from the similar sizes between the geminal phenyl and trifluorethyl groups that do not differentiate between the faces. The configuration depicted for 1006 was provided in the Supporting Information of this paper, although it was not clear how this configuration was assigned.
Scheme 406.
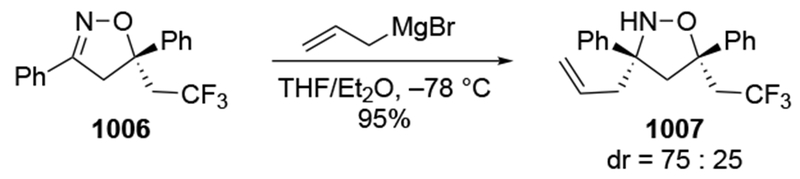
An unusual rearrangement was observed in the case of addition to cyclic oxime 1008 (Scheme 407).561 Addition occurred at the carbon–nitrogen double bond, but then a ring-opening reaction occurred. It was speculated that a proton-transfer reaction must occur from anion 1010 to form the allylic carbanion 1011 (Scheme 408). The addition to the oxime must be diastereoselective. Subsequent nucleophilic substitution at the nitrogen atom would form the resulting aziridine. Conversions of oximes into aziridines have been observed under similar conditions, although the mechanism is likely to be different.562
Scheme 407.
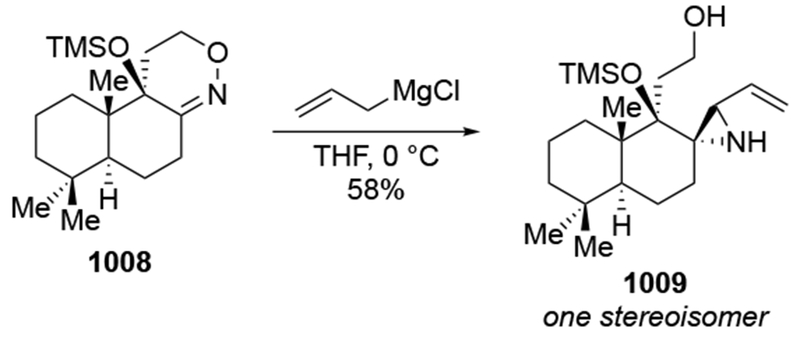
Scheme 408.
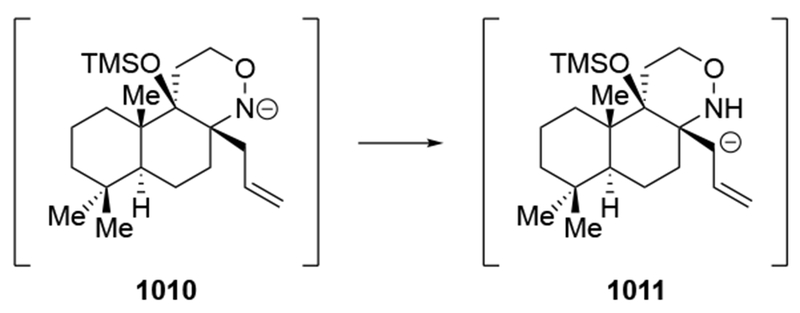
7.6.2. Additions to Amidines
Additions to amidines have not been examined extensively. Upon addition of allylmagnesium reagents to amidine 1012, an intramolecular nucleophilic displacement occurred to form aziridine 1013 as a single diastereomer (Scheme 409).233 Methylmagnesium iodide, however, did not add. This result underscores the difference in reactivity between allylmagnesium reagents and other Grignard reagents.
Scheme 409.
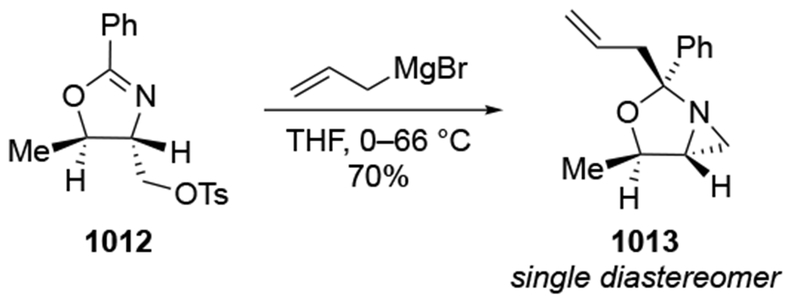
7.6.3. Additions to Hydrazones
Additions of organomagnesium reagents to hydrazone 1014 with allylmagnesium reagents were developed as a method to prepare chiral α-amino aldehydes (Scheme 410).563 The diastereoselectivity of the reaction was highly dependent upon the solvent. Nevertheless, the selectivities with allylmagnesium reagents were low; only upon transmetallation to the allyltitanium reagent could stereoselectivity be achieved. The authors ascribed the selectivity to a chelation-controlled addition through a transition state resembling 1016 (Scheme 411).
Scheme 410.
Scheme 411.
7.6.4. Additions to Nitrones
Nitrones possess strongly electrophilic C=N double bonds by virtue of the substituents on the nitrogen atom,495 so it is not surprising that organomagnesium reagents add to nitrones at low temperature. Depending upon the substitution pattern, these reactions can be highly stereoselective. Additions of allylmagnesium or allyllithium reagents to the glyceraldehyde acetonide-derived nitrones were not stereoselective (Scheme 412).564 Transmetallation or addition of Lewis acids changed the sense of diastereoselectivity. By comparison, additions to the α-amino analogue 1020 (Scheme 413) occurred with high diastereoselectivity. These selectivities have been explained as the result of additions to the top face of a chelate resembling 1023 (Scheme 414).565
Scheme 412.
Scheme 413.
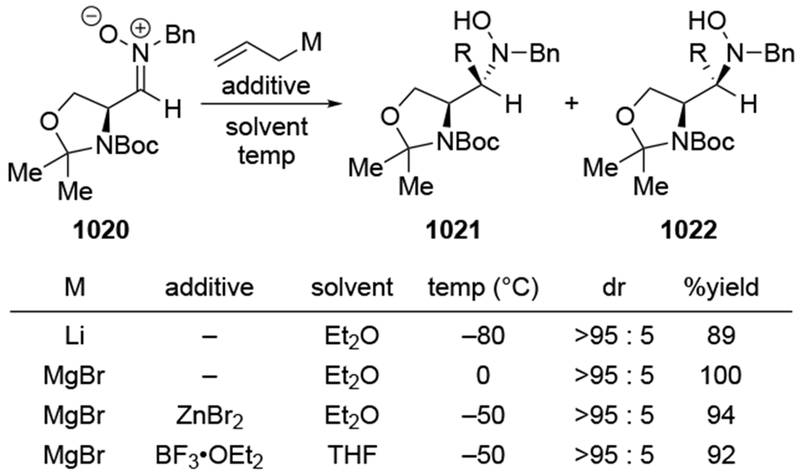
Scheme 414.
Additions of alkylmagnesium reagents to cyclic nitrone 1024 were generally highly diastereoselective (Scheme 415).566 Additions occurred anti to the large silyloxy group with complete diastereoselectivity. By contrast, addition of allylmagnesium bromide was less stereoselective. This low stereoselectivity could reflect the high electrophilicity of the nitrones, which possess relatively low-energy unoccupied orbitals.567 Consequently, reaction rates could approach the diffusion rate limit, thus diminishing diastereoselectivity.22,23
Scheme 415.
Additions to five-membered ring nitrones, on the other hand, were highly diastereoselective. Additions to highly substituted nitrone 1026 occurred from the α-face (Scheme 416).568 This sense of selectivity is similar to what was observed for additions of Grignard reagents to the related imine, although that imine did not react with consistently high stereoselectivity (Scheme 417).569
Scheme 416.
Scheme 417.
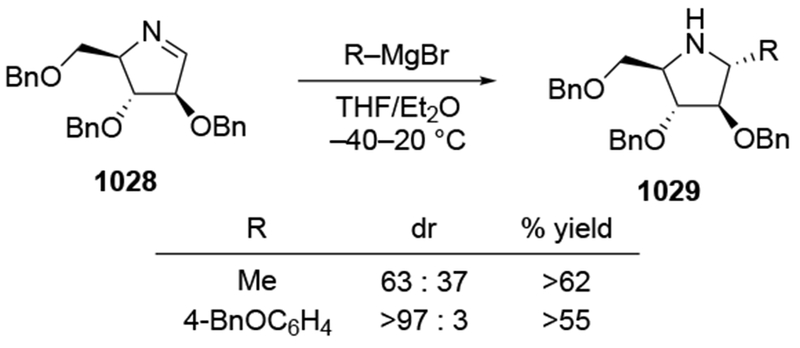
Additions to bicyclic nitrones, however, were generally highly diastereoselective. In the case of a fused bicyclic nitrone, addition occurred from the more accessible convex face (Scheme 418).570 The addition product was subsequently converted to an aza-sugar analogue. Similar outcomes were observed with a structurally related bicyclic nitrone (Scheme 419).571
Scheme 418.
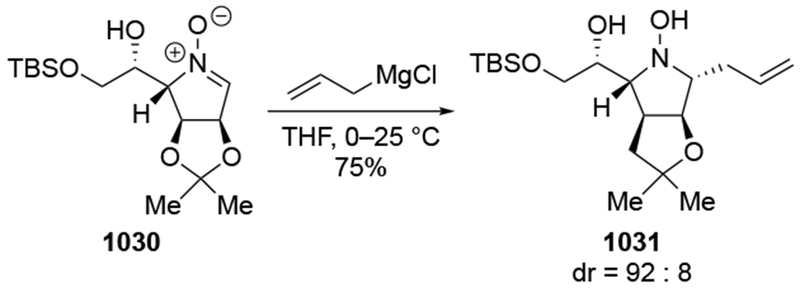
Scheme 419.
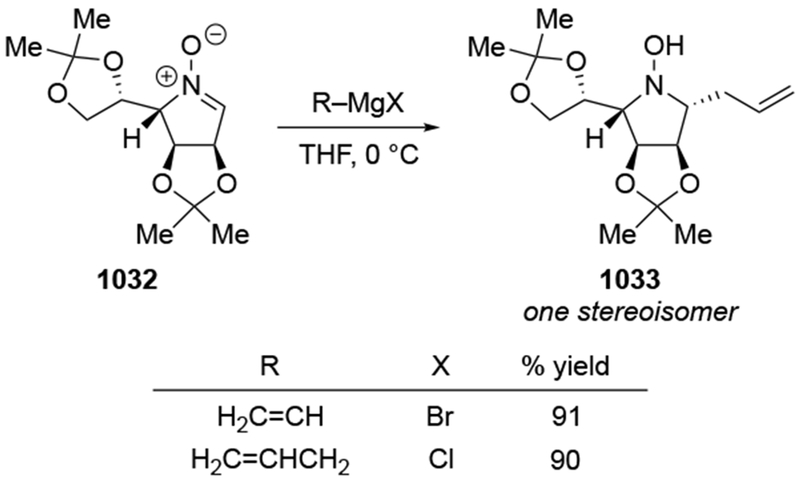
In the process of developing inhibitors of glycosyl transferases, the allylations of nitrones 1034 were examined (Scheme 420).572 In all cases, additions were highly stereoselective, even for monocyclic nitrones.
Scheme 420.
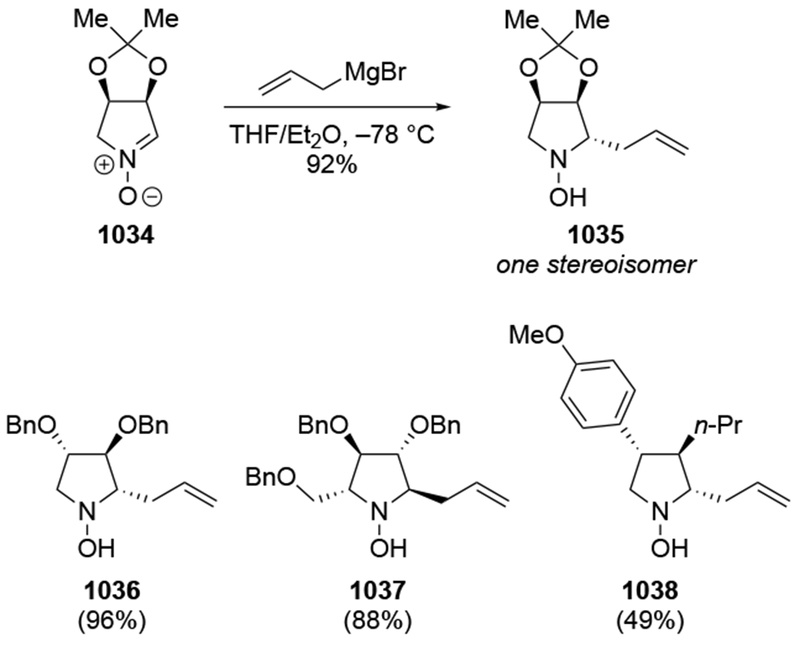
Additions to all five-membered ring nitrones, however, are not stereoselective with allylmagnesium reagents. Addition to nitrone 1039, which resembles nitrone 1036 above, proceeded with high selectivity with vinylmagnesium bromide, but not allylmagnesium bromide (Scheme 421).573
Scheme 421.
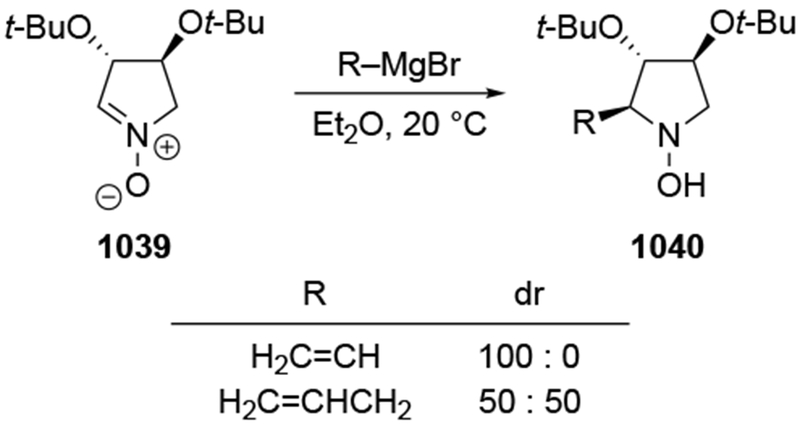
Additions to exocyclic nitrones occurred with different stereochemical outcomes depending upon which reagent was used.574 These addition reactions occurred twice to form two new stereogenic centers adjacent to the nitrogen atom (Scheme 422). Addition of a nucleophile to nitrone 1041 generates a second nitrone, 1043. That nitrone can be trapped by second equivalent of the nucleophile to form the final product 1044. With most organomagnesium reagents, the reaction gave three stereoisomers of product, with the major products having the same relative configurations (Scheme 423). Additions of allylmagnesium reagents, however, resulted in only two isomers of products with the opposite relative stereochemical outcome. The selectivity for only two isomers is caused by the highly stereoselective addition of the second equivalent of the allylmagnesium reagent, whereas the other Grignard reagents reacted less selectively.
Scheme 422.
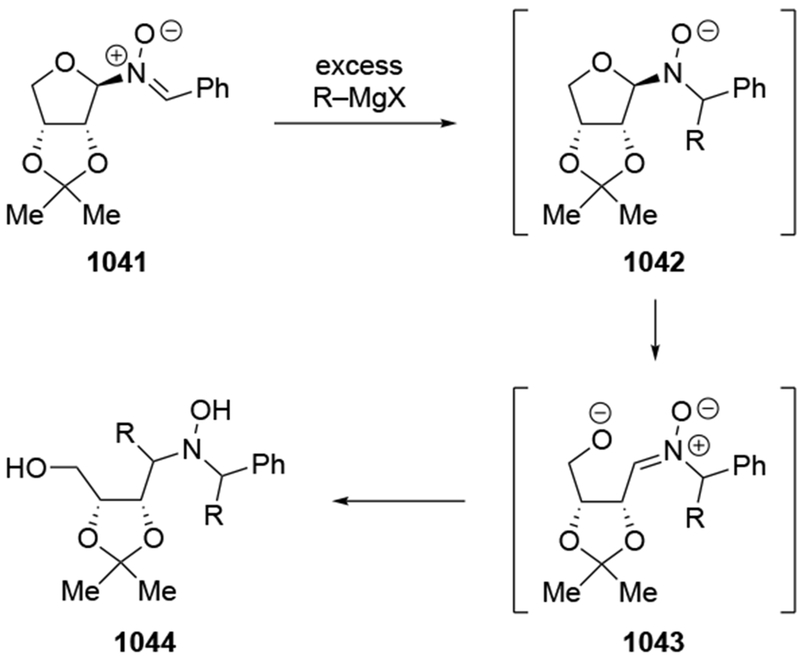
Scheme 423.
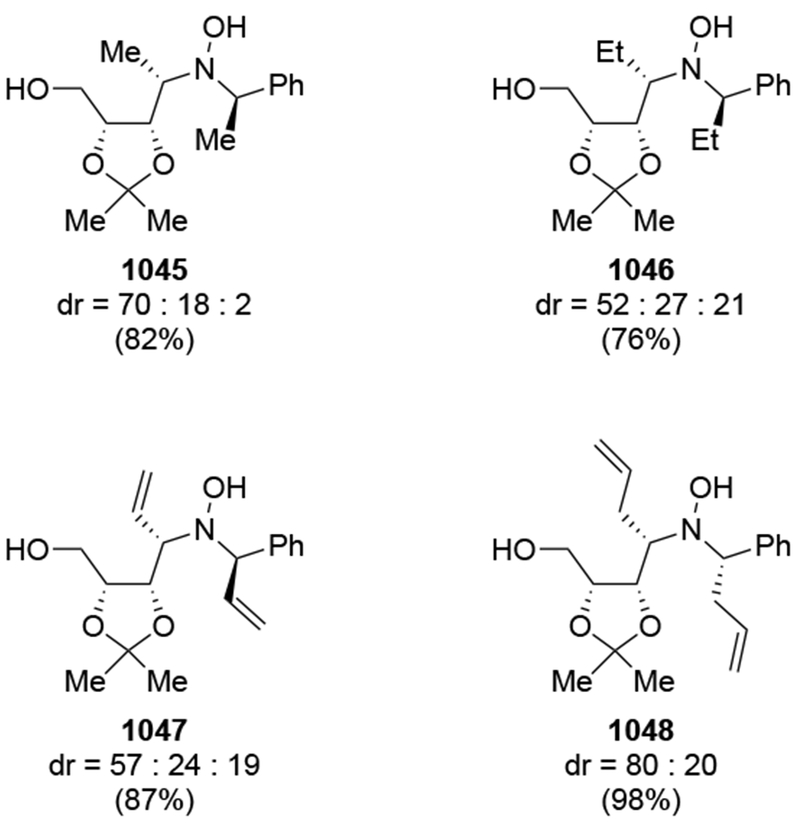
7.7. Additions of Allylmagnesium Reagents with Imines with Chiral Auxiliaries on Nitrogen
7.7.1. Additions to Sulfinyl Imines
Among the methods developed to install chiral auxiliaries on nitrogen atoms to control the stereoselectivities of addition reactions,492 sulfinyl imines have proven to be the most broadly applicable (Scheme 424).498 This method represents a useful application of allylmagnesium reagents. Additions of allylmagnesium bromide to aryl-substituted sulfinyl imines gave high diastereoselectivity. Additions of reagents such as n-butyllithium or vinylmagnesium bromide were unsuccessful, however, because those reagents deprotonated the carbon atom adjacent to the imine. These results led the authors to suggest that the additions of allylmagnesium reagents must follow a different mechanism than for other organometallic reagents. They proposed that addition of allylmagnesium bromide occurred through a six-membered ring transition state with allylic transposition like 1051, but with complexation to the oxygen atom of the sulfinyl imine (Scheme 425). Others researchers have also found additions of allylmagnesium reagents to these arylsulfinyl imines to be useful for the synthesis of chiral amines.575
Scheme 424.
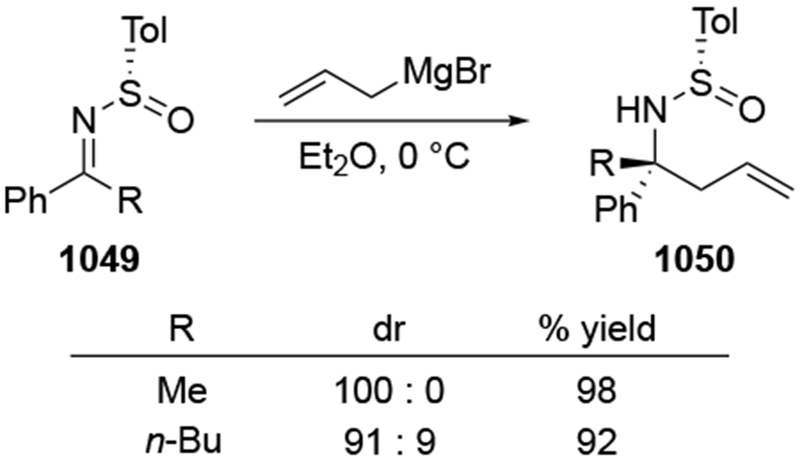
Scheme 425.
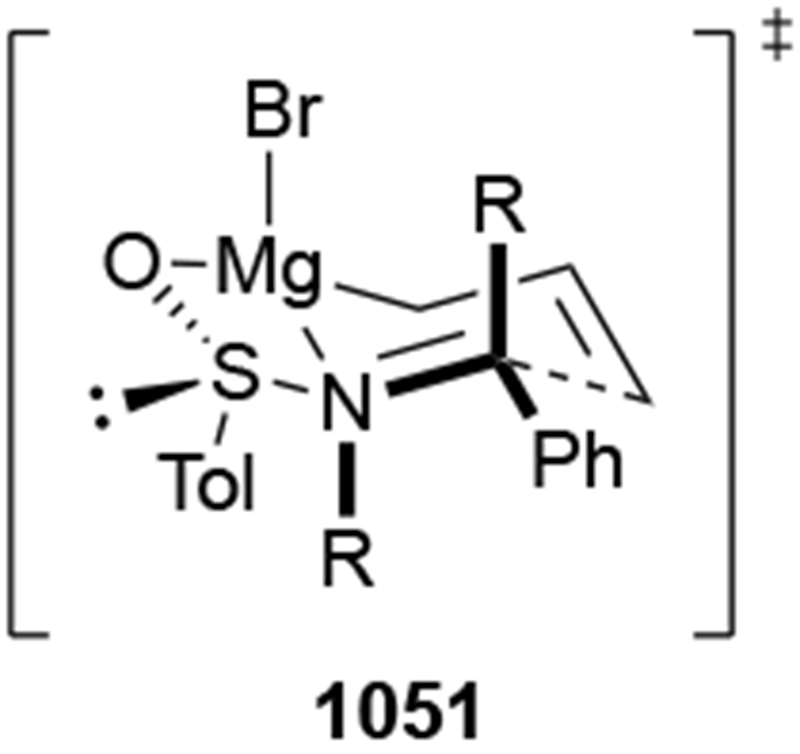
Nucleophilic addition to camphor-based sulfinyl imines was also found to be a useful method for the synthesis of chiral amines (Scheme 426).576 Of all the nucleophiles examined, among the most stereoselective was allylmagnesium bromide. It was suggested that methylmagnesium iodide was behaving as a larger nucleophile by aggregation, and that addition of allylmagnesium halides added by a mechanism involving allylic transposition, as observed for the other systems.
Scheme 426.
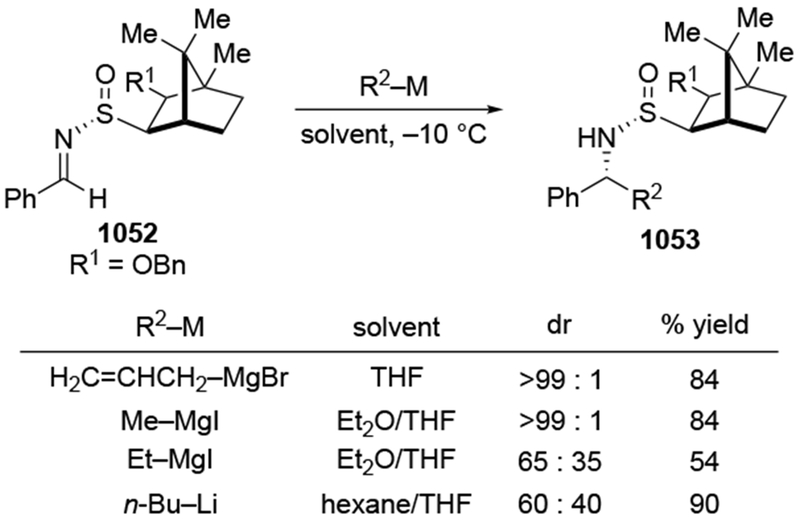
The tert-butyl variant of the sulfinyl imine has been used more frequently.577 This system also gives high selectivity for allylation (Scheme 427).578 The allylation reaction is a generally useful reaction for preparing homoallylic amines, as evidenced by its application to the synthesis of potential drug candidates (Scheme 428).579
Scheme 427.
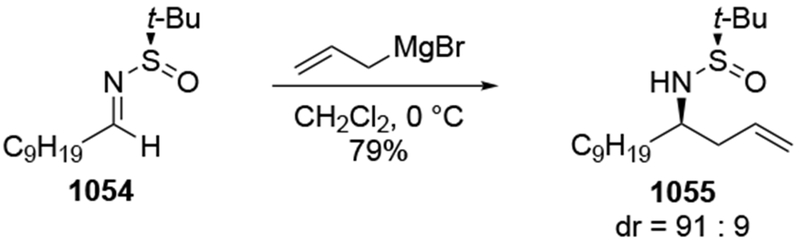
Scheme 428.
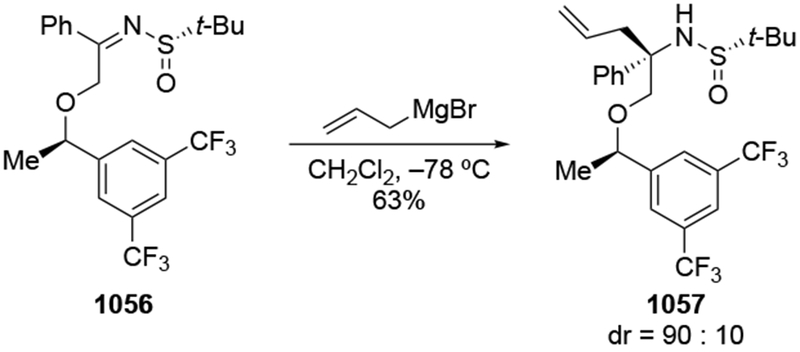
A practical and useful modification of this allylation has also been developed (Scheme 429).580 The cumyl sulfinyl imine enables purification simply by extraction. For this substrate, stereoselectivity for allylation is considerably higher at room temperature than it is at −78 °C, which suggests a difference in entropy between the transition states leading to the different products.581
Scheme 429.
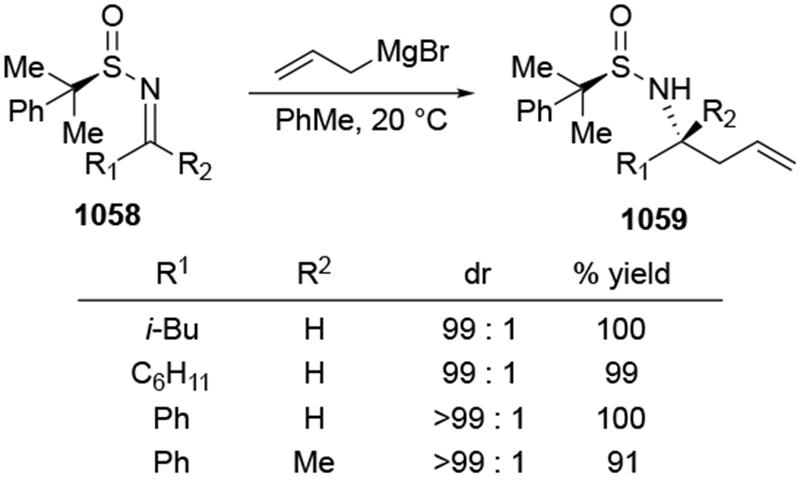
Some discrepancies in the literature about outcomes of allylation reactions of sulfinyl imines suggest that one should study the reported examples carefully before using this transformation. In the course of a synthesis of lactam derivatives, the allylation of sulfinimides was used to prepare starting materials with high optical purity (Scheme 430).582 Other authors had performed the same reaction with the enantiomeric starting material and reported the opposite stereochemical outcome (Scheme 431).583 It is likely that the structure of 1063 had been misassigned.582 The discrepancy in the level of stereoselectivity, however, is harder to explain, although it should be noted that the reaction conditions are quite different.
Scheme 430.
Scheme 431.
Analysis of the stereochemical outcome of addition to the ketone-derived sulfinyl imine is also not necessarily straightforward. Addition to imine 1064 was reported to proceed with modest stereoselectivity (Scheme 432).584 The two different enantiomers of 1064 were also reported to give different isomeric mixtures of 1065, although that discrepancy was attributed to changes to the isolation step for the products. Different results had been reported by other researchers (Scheme 433).583
Scheme 432.
Scheme 433.
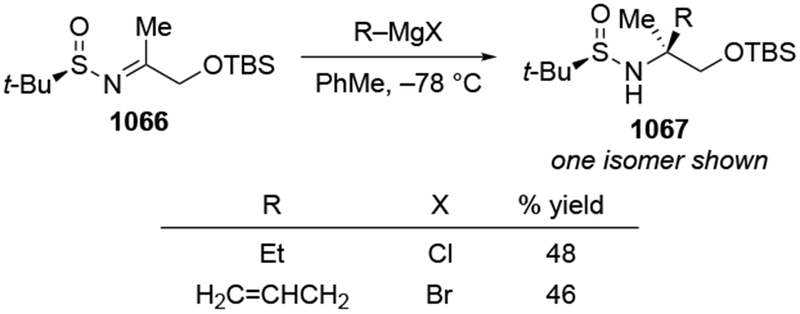
The additions to chiral sulfinyl imines have been applied to the synthesis of fluorinated amines. In the course of the synthesis of fluorinated amino acids, it was discovered that N,O-acetal precursors were superior for these reactions compared to the unstable fluorinated imine (Scheme 434).585 Additions of allylic organomagnesium reagents exhibited higher diastereoselectivity than additions of other organometallic agents, which was attributed to a chelated transition state similar to 1051 (Scheme 425).498,577,586 In a system with enolizable protons, addition was accompanied by small amounts of a condensation product, 1073 (Scheme 435). Addition to the chiral sulfinyl imine 1074 were not particularly diastereoselective, however (Scheme 436).587
Scheme 434.
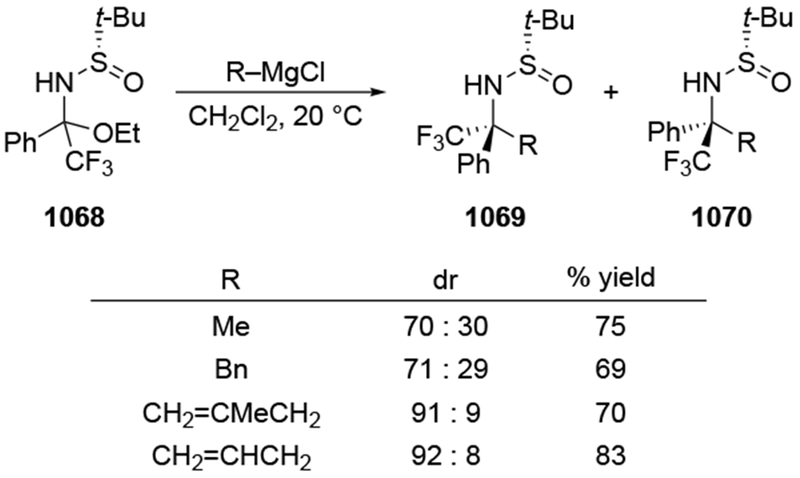
Scheme 435.
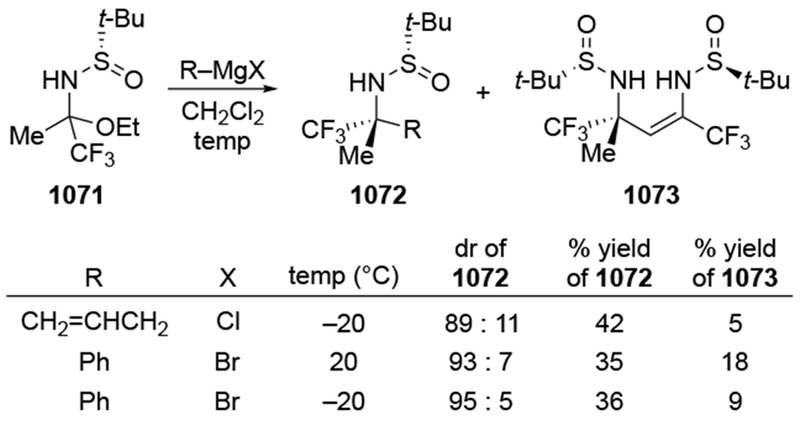
Scheme 436.
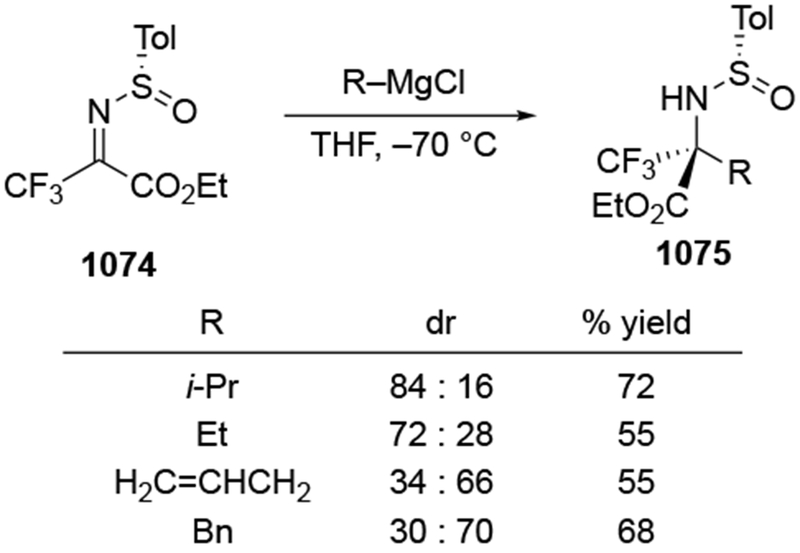
Chiral sulfinyl imines can also be used to prepare monofluorinated amines (Scheme 437).588 The method involved diastereoselective fluorination of anions of sulfinyl imidates followed by reduction to form sulfinyl imines such as 1076. Addition of the allylzincate reagent was highly stereoselective. In the absence of the zinc reagent, the diastereoselectivity was lower (dr = 83:17).
Scheme 437.
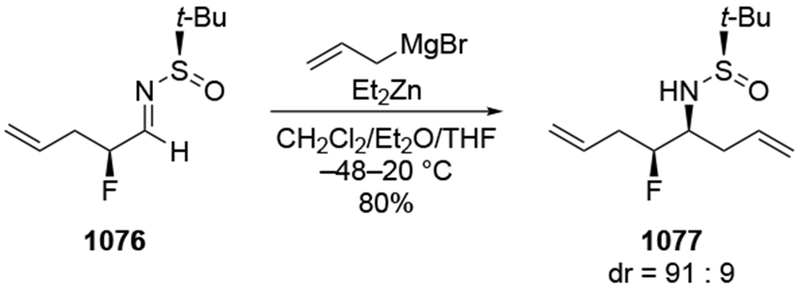
The allylation of sulfinyl imines can be useful in target-directed synthesis. For example, allylation of 1078 formed the amine 1079 after deprotection (Scheme 438).589 Synthesis of precursors to alkaloids can also be achieved using this reaction (Scheme 439).590,591 In both of these cases, the allyl group was used to form a six-membered ring with the homoallylic nitrogen atom by ring-closing metathesis. The allylation reaction has also been used to prepare amines that have been examined for their use for the synthesis of macrolide antibiotic analogues (Scheme 440).592 Allylations of an aromatic aldehyde progressed with good stereoselectivity (Scheme 441).593,594 The resulting amino group can then be cyclized by displacement of the chloride to form enantiomerically enriched tetrahydroisoquinolines, where the allyl group can be elaborated to close additional rings. This variant of the reaction has also proven to be useful for the preparation of precursors to alkaloids (Scheme 442).595 Addition of allylmagnesium bromide to 1088 in the presence of magnesium bromide provided reasonable selectivity for the desired product 1089. A simple purification by trituration enabled the isolation of the product with high selectivity (dr > 98:2).
Scheme 438.
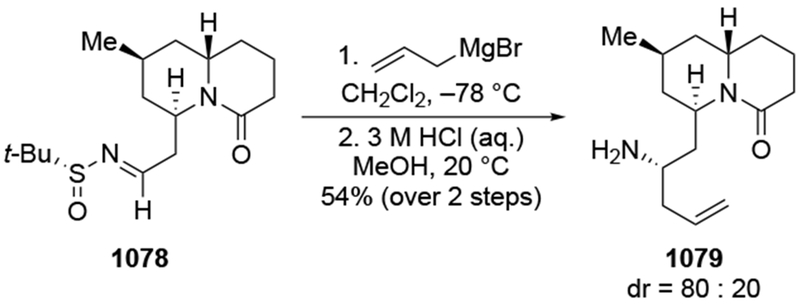
Scheme 439.
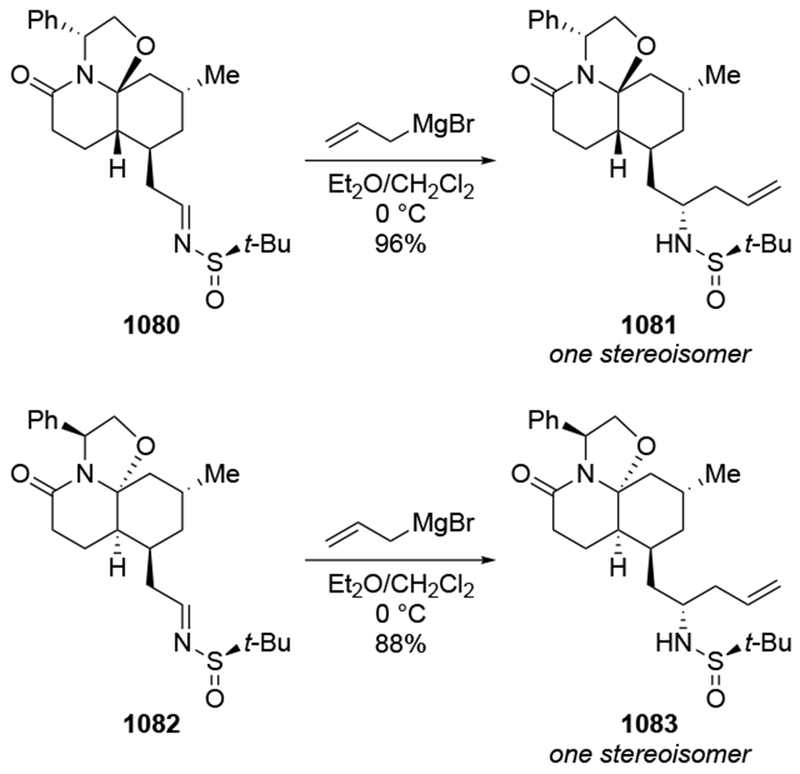
Scheme 440.
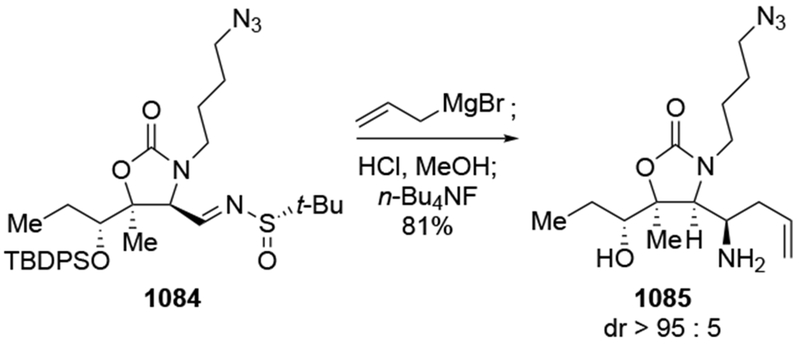
Scheme 441.
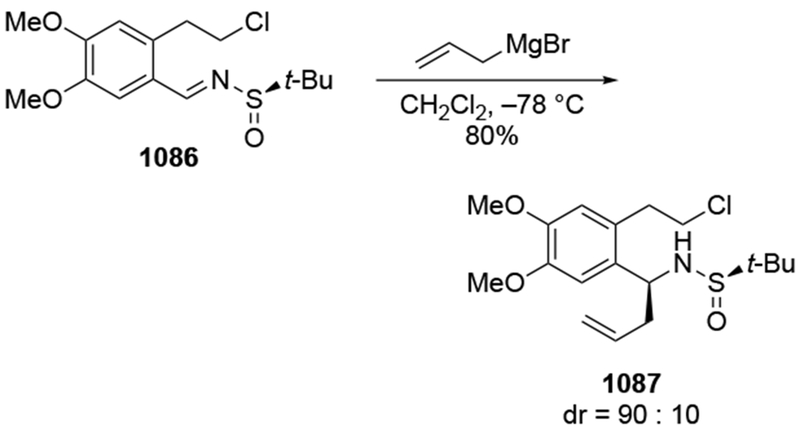
Scheme 442.
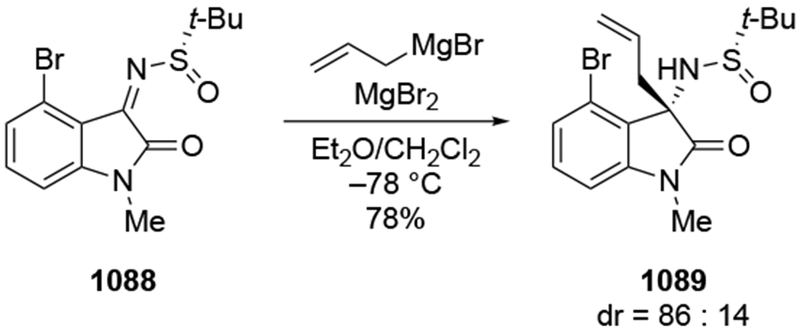
7.7.2. Additions to Imines with Other Chiral Auxiliaries on Nitrogen
Although the sulfinyl imines have emerged as the most common chiral auxiliaries for the additions of allylmagnesium reagents to imines (Section 7.7.1), other auxiliaries have been used in different cases. Imines derived from chiral amines have been used extensively for the stereoselective additions of allylmagnesium reagent. One example demonstrates a contrast with an experiment discussed in the context of the mechanism of addition (Scheme 354, Section 7.2). Whereas addition to the cyclopropyl-substituted imine gave poor diastereoselectivity for the double-addition reaction (Scheme 354), addition to the corresponding isopropyl derivative 1090 gave high stereoselectivity (Scheme 443).499 Additional experiments with the putative imine intermediate 1092 provided evidence for the presence of this intermediate. No explanation for the differences in selectivity for an isopropyl group and a cyclopropyl group was provided.
Scheme 443.
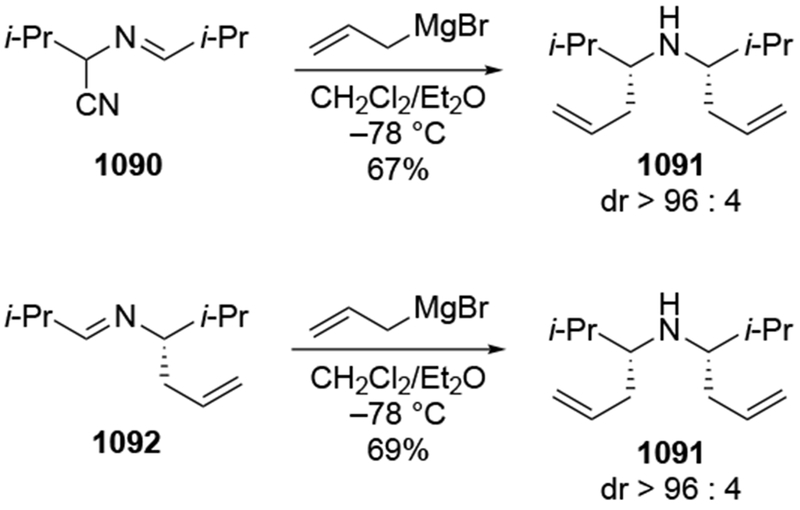
Other approaches have been developed to refine the stereoselective additions to the carbon–nitrogen double bond. One set of chiral auxiliaries that was employed involves the readily available α-methylbenzyl imine 1093 (Scheme 444).516 As with the additions to simple imines of α-chiral aldehydes (Scheme 367, Section 7.3.1.2), additions of allylboron reagents proceeded in generally higher selectivity than addition of allylmagnesium reagents. A transition state resembling 1095 was suggested, presumably with addition to the imine through a six-membered ring transition state 884 (Scheme 352). Other types of organometallic reagents can also be useful (Scheme 445).596
Scheme 444.
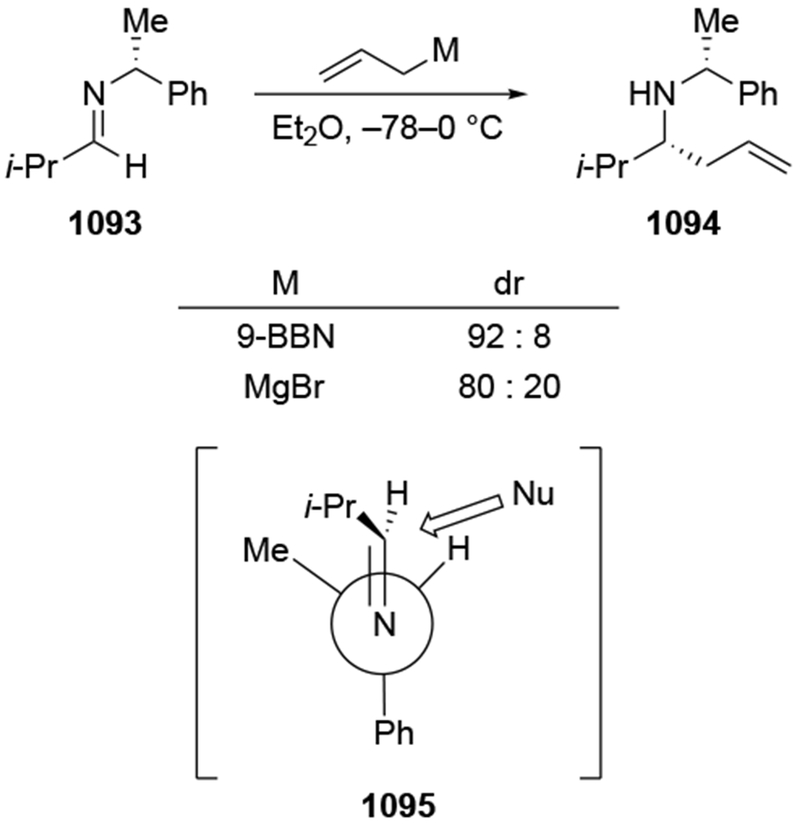
Scheme 445.
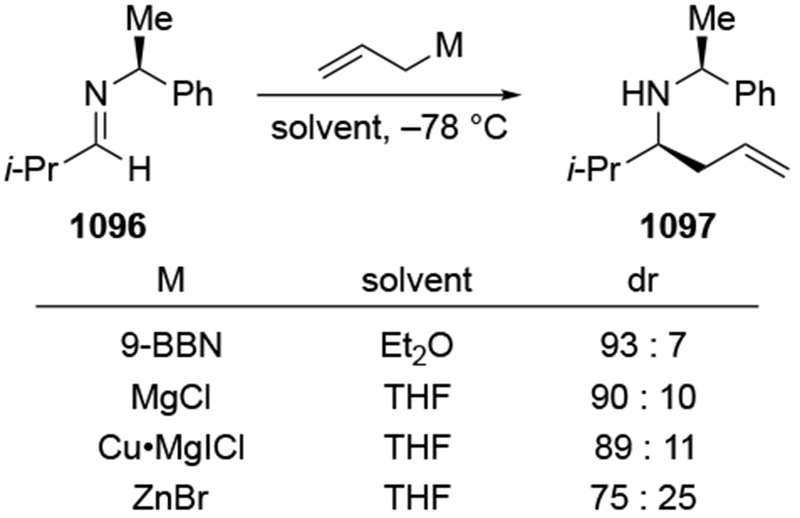
A modification of this method was introduced with a methoxy group on the aromatic ring (Scheme 446).597 The methoxy group was intended to provide another point of contact for the metal to reinforce the preference for a conformation resembling 1095 (Scheme 444). This modification allowed for the reactions of allylic organomagnesium reagents to be highly diastereoselective. It should be noted that the addition of the prenyl reagent gave the product with high γ-selectivity, suggesting that these reactions proceed by a transition state involving allylic transposition, as observed for additions to sulfinyl imines (Scheme 425, Section 7.7.1). This reaction has also been applied to the synthesis of 1,2-diamines (Scheme 447).598,599
Scheme 446.
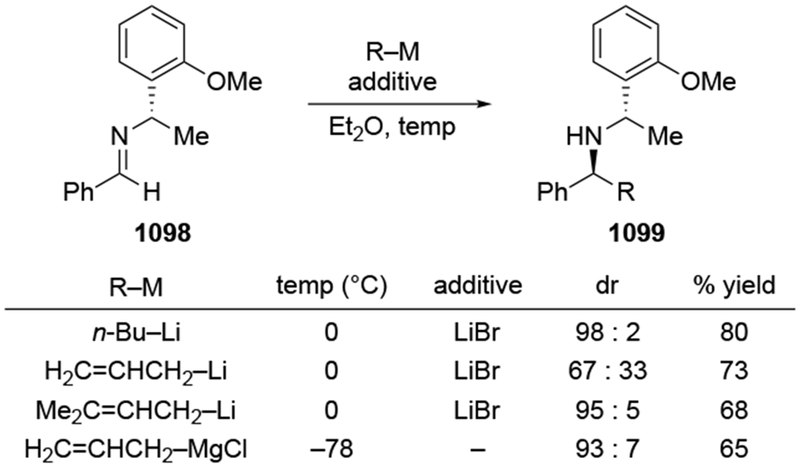
Scheme 447.
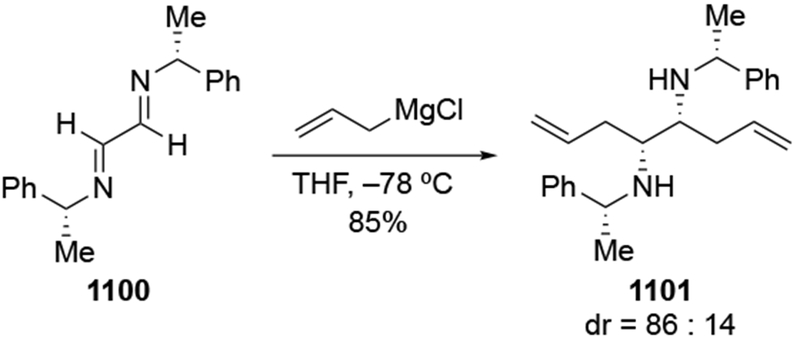
The presence of the chiral auxiliary on nitrogen, however, may not override substrate control (Scheme 448).600 Regardless of the configuration of the auxiliary, the stereoselectivity of addition to an α-epoxy imine was dominated by the chelating effect of the epoxide functional group. This stereochemical course can be reversed in the presence of BF3•Et2, which cannot engage in chelation (Scheme 449).
Scheme 448.
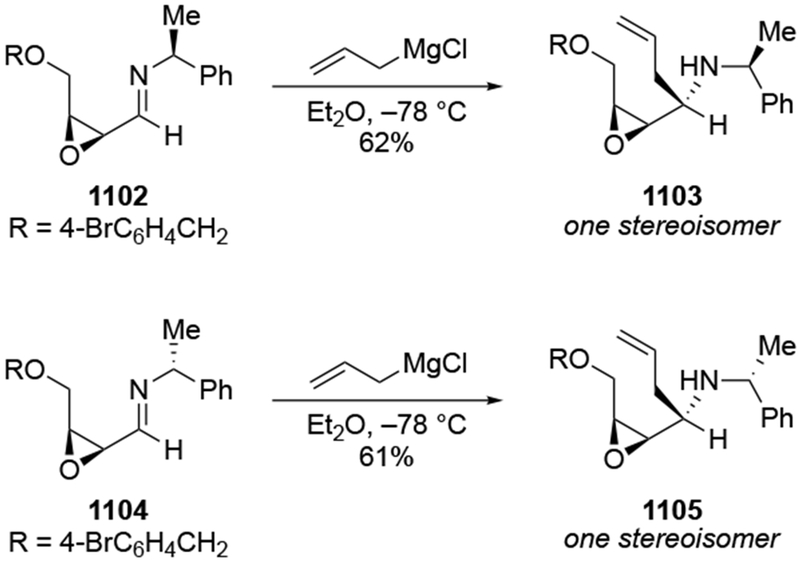
Scheme 449.
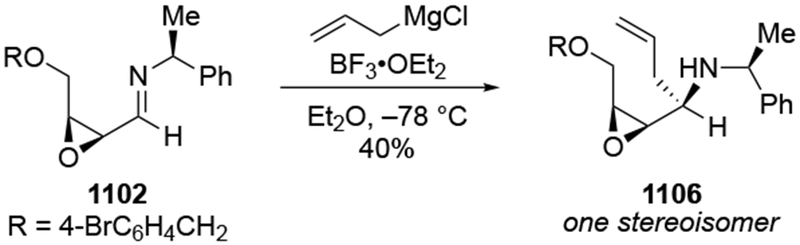
In other cases, the sense of the substrate control can be either matched or mismatched601 with the control exerted by the reagents. There are other cases where the stereochemical control elements, along with the preferences of reagents, are more in balance. With the imine 1107 with a potentially chelating α-alkoxy group, addition of allylmagnesium reagents were unselective, whereas additions of allylboranes were selective (Scheme 450).503 With the other diastereomer, however, the allylmagnesium reagents added stereoselectively, but the borane did not (Scheme 451). These selectivities can be explained by considering the two modes of stereocontrol (Scheme 452). Chelation would be most stereoselective when the larger benzyloxymethyl group and the phenyl group would occupy the same face (as illustrated by 1111); addition would occur away from these groups. Conversely, using the borane reagent, the reaction would proceed by a Felkin–Anh type transition state, and selectivity would be maximized when the larger phenyl group would be oriented away from the approaching nucleophile (as illustrated by 1112).503
Scheme 450.
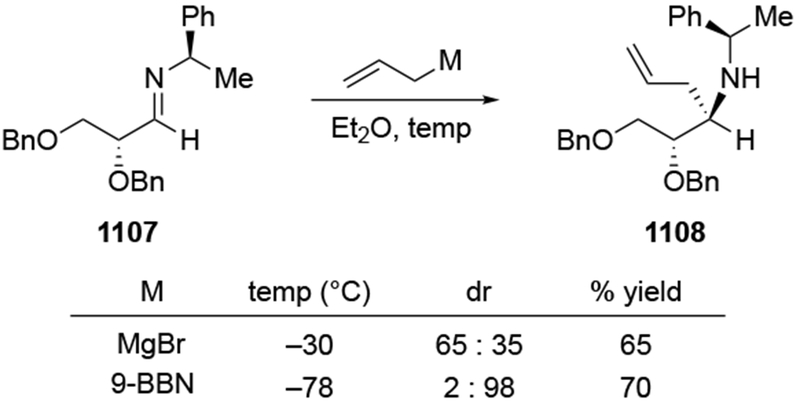
Scheme 451.
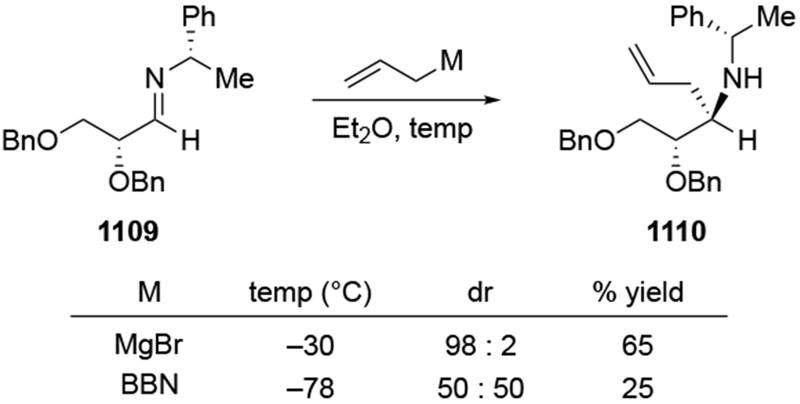
Scheme 452.
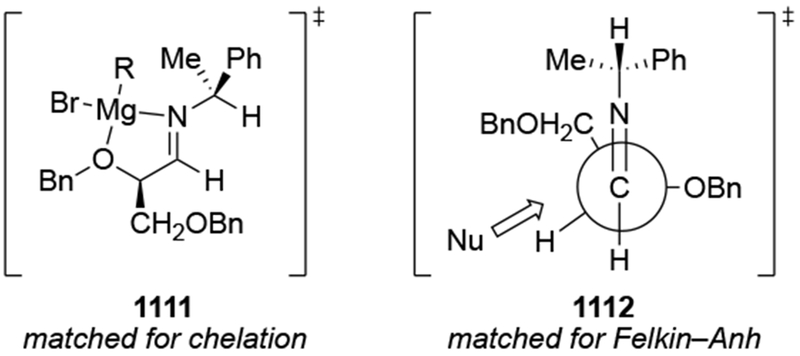
Additional results with chiral imines also indicate the high reactivity of allylmagnesium reagents. Upon addition of allylmagnesium chloride to the imine 1113 containing also ester and pyridine functional groups, modest diastereoselectivity for addition to the imine was observed (Scheme 453).602 Addition to the heteroaromatic ring and the ester functional group were also observed. The diastereoselectivity observed with the magnesium and zinc reagents was argued to involve a transition state (1115) where the metal atom was complexed to the imine nitrogen atom, the pyridine nitrogen atom, and the ester group (Scheme 454). Opposite diastereoselectivity was observed with an allyltin reagent, which should not engage the carboxyl group in chelation, as illustrated in transition state 1116.
Scheme 453.
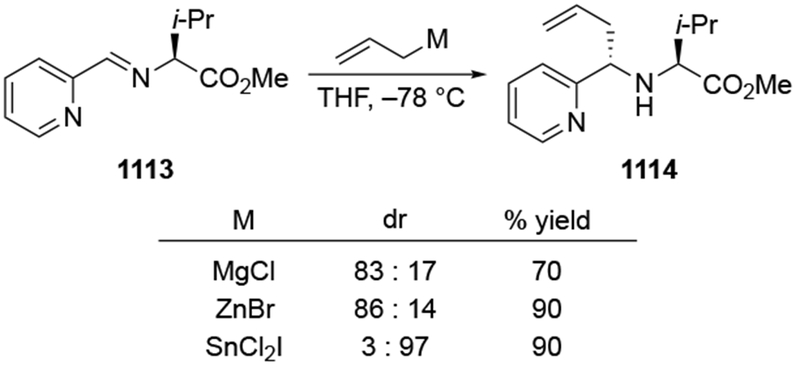
Scheme 454.
Other chiral imines also underwent stereoselective additions with a number of organomagnesium reagents. Addition to chiral imine 1117 gave high selectivity for amine 1118 (Scheme 455).603 It is likely that the difficulty of addition of some Grignard reagents could result from competitive deprotonation of the starting material. The presence of magnesium salts was not necessary to observe high stereoselectivity, although they did increase it somewhat. The selectivities of these reactions can be considered to occur through chelate 1119, which would minimize allylic strain. Addition from the face opposite the larger phenyl group would lead to the observed product.
Scheme 455.
Chiral hydrazones have also been examined for their applications for preparing chiral amines (Scheme 456).604 Organocerium reagents were used in this case because of a general lack of success with reagents such as Grignard reagents.605
Scheme 456.
Efforts to use a chiral diol-derived auxiliary illustrate the differences between other Grignard reagents and allylmagnesium halides (Scheme 457).606 Alkyllithium reagents added to imine 1122 in high yields, but other reagents, such as organomagnesium, organocerium, organozinc, and organocopper reagents, would not add. Only one Grignard reagent added: allylmagnesium bromide. The diastereoselectivity of this reaction was low, however.
Scheme 457.
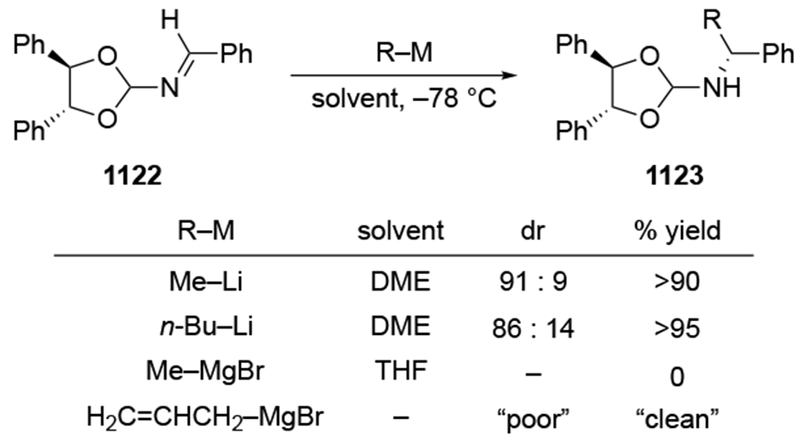
Additions of allylmagnesium halides to oxime ethers were less stereoselective than additions of alkyllithiums (Scheme 458).607 Although a direct comparison to other organomagnesium reagents was not made with this substrate, additions to a related substrate, unsaturated oxime 1126, also showed lower diastereoselectivity for the Grignard reagents, but not as low as for allylmagnesium bromide with oxime 1124.
Scheme 458.
Sulfenimines can also be useful chiral auxiliaries for preparing chiral amines (Scheme 459).576 Diastereoselectivity was sensitive to the nature of the group on the camphor-based auxiliary, with the free alcohol giving the product with the highest diastereoselectivity. This stereoselectivity was attributed to complexation of the magnesium atom to both the sulfur atom and the anionic oxygen atom, as illustrated in 1130. The higher selectivity may be attributable to the strong complexation with the OMgBr group, as was observed for chelation in α-hydroxy ketones (Section 4.2.1.1). No other organometallic nucleophiles were examined with this auxiliary; more studies were made on the sulfinyl imines, as discussed in Section 7.7.1.
Scheme 459.
An alternative chiral auxiliary for imines involves a phosphorus-based auxiliary (Scheme 460).608 Earlier studies with this auxiliary employed isolated imines,609 but this study used an N,O-acetal prepared in situ. Additions occurred with several organomagnesium reagents, but only with allylmagnesium bromide were these reactions highly diastereoselective, likely due to the cyclic transition state of allylation. This transition state allows for complexation to the oxygen atom on the phosphorous atom, as illustrated in 1133. The reaction mixture likely contains Ti(Oi-Pr)4 or other titanium salts because of the two-step, one-flask method for the synthesis of the imine. It is unlikely that the salts are important in the case of allylmagnesium reagents because the results are similar to those obtained using an earlier protocol.609
Scheme 460.
Additions to nitrones with chiral substituents on the nitrogen atom were also stereoselective (Schemes 461 and 462).610 In the case of additions of most Grignard reactions, additions to chiral nitrones 1134 were highly stereoselective. Addition of allylmagnesium reagents, however, proceeded with low diastereoselectivity. The stereoselectivity patterns, however, were odd: Addition of methylmagnesium reagents to nitrone 1136, however, gave products with the opposite stereoselectivity compared to the reactions of other reagents (Scheme 462). No explanation for this phenomenon was reported.
Scheme 461.
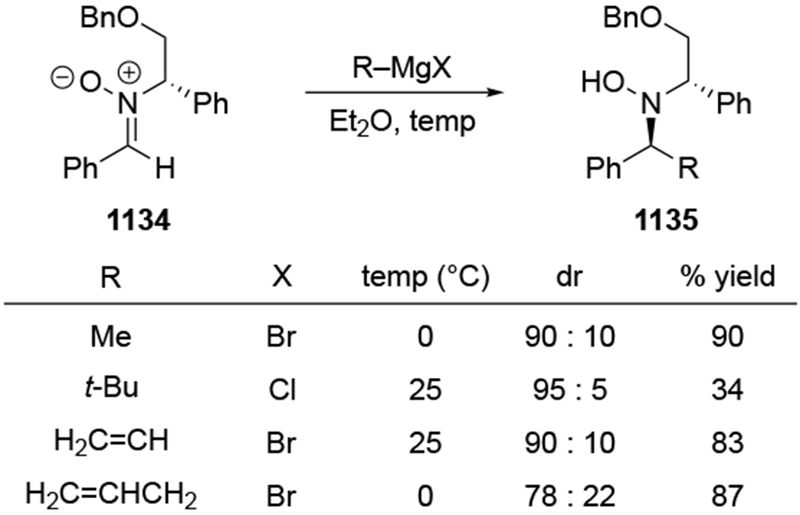
Scheme 462.
8. Conclusions
Allylmagnesium nucleophiles are useful reagents for introducing the allyl functional group into a variety of organic compounds. They are inexpensive, convenient to use, and highly reactive. It is this reactivity, however, that renders them unpredictable in synthetic endeavors. This review collected and analyzed hundreds of examples of the additions of these reagents to C=O and C=N double bonds, highlighting under what circumstances additions may be stereoselective, and when they may not.
Although in some cases allylmagnesium reagents react much as other organomagnesium reagents do, most often, they react differently. For example, chelation control cannot be expected for allylmagnesium reagents in ethereal solvents because they do not conform to the formal chelation-control model. Therefore, even when the product expected from chelation control is the major product, that does not mean that the reaction proceeded by the chelation control model as rigorously formulated by Eliel.53,54 Instead, other factors may be responsible for selectivity.
To be able to use allylmagnesium reagents more effectively, it is important to know why they react the way they do. A picture to explain that reactivity is emerging.18 While the details of transition state structure and possible intermediates provide some insight into the different patterns of reactivity exhibited by allylmagnesium reagents compared to other organomagnesium compounds, those details are not the most important factor underlying the difference in reactivity. The most important factor is that, for most aldehydes and ketones (unless they are unusually sterically hindered), the rate constants for addition of allylmagnesium reagents appear to be so fast that the carbon–carbon bond forming step is not the stereochemistry-determining step. Instead, it is the step that brings the nucleophile and electrophile together (i.e., diffusion, as discussed in Section 2.2 and Scheme 9) that determines the stereoselectivity. If the reagent approaches the carbonyl compound from the Re face, for example, the new bond will be formed on the Re face. For many carbonyl compounds, this diffusion step is random: both faces will be approached with equal probability, so diastereoselectivity will be low. On the other hand, for some carbonyl compounds where the faces are highly differentiated (such as for multicyclic ketones where the carbonyl group is embedded in a fused, bridged, or spirocyclic ring), the rate constants for approach to the different faces could be quite different, leading to diastereoselectivity. Even some acyclic compounds can exist in conformations that bury a carbonyl group, so that only one face of the carbon–oxygen double bond might be accessible, again resulting in diastereoselective reactions of allylmagnesium reagents. These cases underscore the point that careful consideration of the conformational preferences of the substrate, and how approach of the nucleophile may be faster on one face than the other, can give insight into how reactions might occur. Finally, in systems that are so hindered (such as carbonyl compounds flanked by tetrasubstituted carbon atoms17,23), rates of carbon–carbon bond formation can be below the diffusion rate limit. In these cases, again, diastereoselectivity will be observed because reaction outcomes can be determined by the relative energies of competing transition states, which is central to models such as the chelation-control model and the Felkin–Anh and related models.
Arriving at some of the conclusions regarding the reactivity of allylmagnesium reagents described in this article has taken assembling a considerable amount of information from disparate sources with different levels of detail. For example, sometimes additives such CeCl3 or ZnCl2 were used in reactions without any comment, so it is not possible to know what happens in the absence of these reagents. In other cases, non-ethereal solvents were used in reactions without any indication of what happened in ethereal solvents. Sometimes, stereochemical information was not provided. Stereochemical assignments can also be misleading, even in cases of well-studied reactions such as reductions of 2-substituted cyclohexanones, where different papers might suggest different stereochemical outcomes (as illustrated by comparing the following two papers discussing a different type of reaction than discussed here611,612). It would have been helpful for researchers to provide that information, or in other cases, when an allylmagnesium compound was used, to provide insight into what happened (if anything) with other reagents. Nevertheless, even without ideal information of that kind, a picture emerges of the unique reactivity of allylmagnesium reagents.
A question that might be raised is whether these reagents, given their unpredictability, are useful in modern organic synthesis. We believe that the answer is yes, as evidenced by the hundreds of creative applications of it in this Review. In many cases, they are the only reagents that are capable of reacting with highly hindered carbonyl compounds, and it is in just those cases, where the selectivity can be predicted to be the highest. Researchers should be aware of the details about the unusually high rates of reactions of allylmagnesium reagents, however, and consider carefully the implications of that rate.
Scheme 109.
Scheme 123.
Scheme 230.
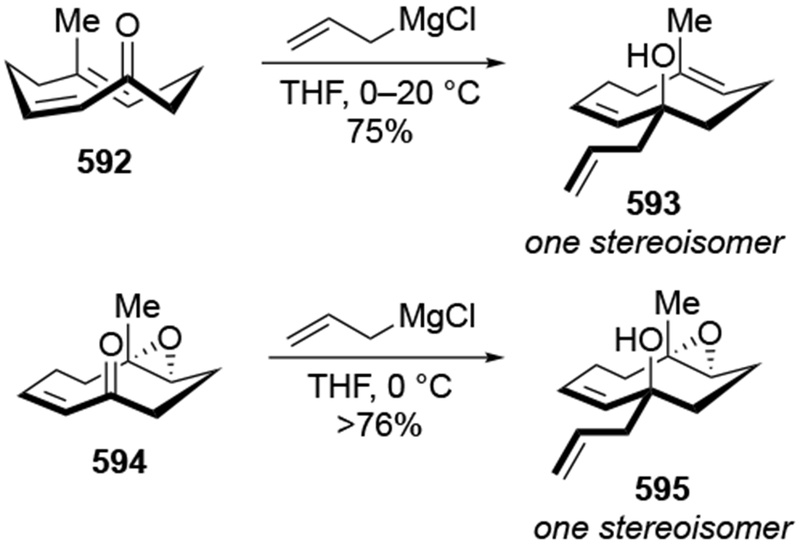
Scheme 348.
12. Acknowledgment
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for partial support during the preparation of this manuscript (57206-ND1). Additional support was provided by the National Institutes of Health, National Institute of General Medical Sciences (GM-129286). K.A.W. thanks the Global Research Initiatives, NYU and NYU Florence, who graciously provided a fellowship and research facilities that allowed for time for analysis and writing of portions of the manuscript.
Biographies
Biographical Sketches
Nicole D. Bartolo obtained her B.S. in Biomedical Sciences and Biochemistry at Marist College in 2015. She is currently a Ph.D. student studying with Professor Woerpel, where her research focuses on understanding the reactions of allylmagnesium reagents with carbonyl compounds and mines. She has also investigated the principals governing the stereoselectivities of reductions of ketones bearing chelating groups.
Dr. Jacquelyne A. Read obtained her B.S. in Chemistry at Wheaton College (IL) in 2012. She then moved to New York University and studied the diffusion-controlled reactivity of allylmagnesium halides under the direction of Professor Woerpel, completing her Ph.D. in 2018. She is now a National Institutes of Health (NIH) postdoctoral fellow in the laboratory of Professor Matthew Sigman at the University of Utah, where her research focuses on applying computational tools to understand non-covalent interactions driving selectivity in asymmetric catalysis.
Professor Elizabeth M. Valentin received her Ph.D. from the University of Puerto Rico, Rio Piedras in 2013, studying with Professor José A. Prieto. Her dissertation focused on applying epoxide-based strategies for the synthesis of polypropionate chains. She then pursued postdoctoral research with Professor Woerpel at New York University. She is currently an Assistant Professor of Chemistry at Susquehanna University.
Professor Keith Woerpel received his B.S. with Highest Distinction from the University of Virginia. He earned A.M. and Ph.D. degrees from Harvard University with Professor David Evans. He then was an NIH postdoctoral fellow at the University of California, Berkeley, with Professor Robert Bergman. He was appointed to the faculty at the University of California, Irvine, where he ascended to the rank of Professor. He is currently the Margaret and Herman Sokol Professor of Medicinal Chemistry at New York University. His research interests involve developing stereochemical models to understand complex reactions, exploring the unusual reactivities of silylenes and strained cyclic alkenes, and devising new methods for the synthesis of biologically active organic peroxides.
Footnotes
This review is dedicated to Professor David Evans (Harvard University), for his creativity in discovering synthetically useful stereoselective reactions and for his commitment to understanding the origins of stereochemical control.
The authors declare no competing financial interest.
13. References
- (1).Yamamoto Y; Asao N Selective Reactions Using Allylic Metals. Chem. Rev 1993, 93, 2207–2293. [Google Scholar]
- (2).Tatsuta K; Fukuda T; Ishimori T; Yachi R; Yoshida S; Hashimoto H; Hosokawa S The First Total Synthesis of Hibarimicinone, a Potent v-Src Tyrosine Kinase Inhibitor. Tetrahedron Lett. 2012, 53, 422–425. [Google Scholar]
- (3).Bejjani J; Chemla F; Audouin M N-Tritylprolinal: An Efficient Building Block for the Stereoselective Synthesis of Proline-Derived Amino Alcohols. J. Org. Chem 2003, 68, 9747–9752. [DOI] [PubMed] [Google Scholar]
- (4).Carda M; González F; Rodríguez S; Marco JA Highly Diastereoselective Additions of Organometallic Reagents to 1-O-Silylated 3,4-Di-O-Benzyl-l-Erythrulose Derivatives. Tetrahedron: Asymmetry 1993, 4, 1799–1802. [Google Scholar]
- (5).Holt DJ; Barker WD; Jenkins PR; Davies DL; Garratt S; Fawcett J; Russell DR; Ghosh S Ring-Closing Metathesis in Carbohydrate Annulation. Angew. Chem. Int. Ed 1998, 37, 3298–3300. [DOI] [PubMed] [Google Scholar]
- (6).Holt DJ; Barker WD; Jenkins PR; Panda J; Ghosh S Stereoselective Preparation of Enantiomerically Pure Annulated Carbohydrates Using Ring-Closing Metathesis. J. Org. Chem 2000, 65, 482–493. [DOI] [PubMed] [Google Scholar]
- (7).Paquette LA; Bolin DG; Stepanian M; Branan BM; Mallavadhani UV; Tae J; Eisenberg SWE; Rogers RD Intramolecular Oxymercuration of Stereoisomeric Cyclohexyl-Belted Poly(spirotetrahydrofuranyl) Platforms. J. Am. Chem. Soc 1998, 120, 11603–11615. [Google Scholar]
- (8).Lee Y.-g.; McGee KF Jr.; Chen J; Rucando D; Sieburth SM A [4 + 4] 2-Pyridone Approach to Taxol. 3. Stereocontrol During Elaboration of the Cyclooctane. J. Org. Chem 2000, 65, 6676–6681. [DOI] [PubMed] [Google Scholar]
- (9).Moon NG; Harned AM A Concise Synthetic Route to the Stereotetrad Core of the Briarane Diterpenoids. Org. Lett 2015, 17, 2218–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ley SV; Abad-Somovilla A; Anderson JC; Ayats C; Bänteli R; Beckmann E; Boyer A; Brasca MG; Brice A; Broughton HB et al. The Synthesis of Azadirachtin: A Potent Insect Antifeedant. Chem. Eur. J 2008, 14, 10683–10704. [DOI] [PubMed] [Google Scholar]
- (11).Maimone TJ; Voica A-F; Baran PS A Concise Approach to Vinigrol. Angew. Chem. Int. Ed 2008, 47, 3054–3056. [DOI] [PubMed] [Google Scholar]
- (12).Goodell JR; McMullen JP; Zaborenko N; Maloney JR; Ho C-X; Jensen KF; Porco JA Jr.; Beeler AB Development of an Automated Microfluidic Reaction Platform for Multidimensional Screening: Reaction Discovery Employing Bicyclo[3.2.1]octanoid Scaffolds. J. Org. Chem 2009, 74, 6169–6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Plummer CW; Wei CS; Yozwiak CE; Soheili A; Smithback SO; Leighton JL Design, Development, Mechanistic Elucidation, and Rational Optimization of Tandem Ireland Claisen/Cope Rearrangement Reaction for Rapid Access to the (Iso)Cyclocitrinol Core. J. Am. Chem. Soc 2014, 136, 9878–9881. [DOI] [PubMed] [Google Scholar]
- (14).Nakajima M; Arai S; Nishida A Total Syntheses of (+)-Grandilodine C and (+)-Lapidilectine B and Determination of Their Absolute Stereochemistry. Angew. Chem. Int. Ed 2016, 55, 3473–3476. [DOI] [PubMed] [Google Scholar]
- (15).Oguri H; Hiruma T; Yamagishi Y; Oikawa H; Ishiyama A; Otoguro K; Yamada H; Ōmura S Generation of Anti-trypanosomal Agents through Concise Synthesis and Structural Diversification of Sesquiterpene Analogues. J. Am. Chem. Soc 2011, 133, 7096–7105. [DOI] [PubMed] [Google Scholar]
- (16).Tabuchi T; Urabe D; Inoue M Construction of the Fused Pentacycle of Talatisamine via a Combination of Radical and Cationic Cyclizations. J. Org. Chem 2016, 81, 10204–10213. [DOI] [PubMed] [Google Scholar]
- (17).Read JA; Woerpel KA Allylmagnesium Halides Do Not React Chemoselectively Because Reaction Rates Approach the Diffusion Limit. J. Org. Chem 2017, 82, 2300–2305. [DOI] [PubMed] [Google Scholar]
- (18).Bartolo ND; Read JA; Valentín EM; Woerpel KA Reactions of Allylmagnesium Halides with Carbonyl Compounds: Reactivity, Structure, and Mechanism. Synthesis 2017, 49, 3237–3246. [Google Scholar]
- (19).Benkeser RA The Chemistry of Allyl and Crotyl Grignard Reagents. Synthesis 1971, 347–358. [Google Scholar]
- (20).Yus M; González-Gómez JC; Foubelo F Diastereoselective Allylation of Carbonyl Compounds and Imines: Application to the Synthesis of Natural Products. Chem. Rev 2013,113, 5595–5698. [DOI] [PubMed] [Google Scholar]
- (21).Yus M; González-Gómez JC; Foubelo F Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev 2011, 111, 7774–7854. [DOI] [PubMed] [Google Scholar]
- (22).Read JA; Yang Y; Woerpel KA Additions of Organomagnesium Halides to α-Alkoxy Ketones: Revision of the Chelation-Control Model. Org. Lett 2017, 19, 3346–3349. [DOI] [PubMed] [Google Scholar]
- (23).Bartolo ND; Woerpel KA Mechanistic Insight into Additions of Allylic Grignard Reagents to Carbonyl Compounds. J. Org. Chem 2018, 83, 10197–10206. [DOI] [PubMed] [Google Scholar]
- (24).Bronk BS; Letavic MA; Bertsche CD; George DM; Hayashi SF; Kamicker BJ; Kolosko NL; Norcia LJ; Rushing MA; Santoro SL et al. Synthesis, Stereochemical Assignment and Biological Activity of a Novel Series of C-4” Modified Aza-Macrolides. Bioorg. Med. Chem. Lett 2003, 13, 1955–1958. [DOI] [PubMed] [Google Scholar]
- (25).Henriques AM; Monteiro JGS; Barbosa AGH Multi-Configuration Spin-Coupled Description of Organometallic Reactions: A Comparative Study of the Addition of RMBr (M = Mg and Zn) to Acetone. Theor. Chem. Acc 2017, 136, 4. [Google Scholar]
- (26).Wilson KW; Roberts JD; Young WG Allylic Rearrangements. XXVIII. The Reaction of Butenylmagnesium Bromide with Hindered Ketones. J. Am. Chem. Soc 1950, 72, 218–219. [Google Scholar]
- (27).Evans DA Stereoselective Organic Reactions: Catalysts for Carbonyl Addition Processes. Science 1988, 240, 420–426. [DOI] [PubMed] [Google Scholar]
- (28).Holm T Use of Competition Kinetics with Fast Reactions of Grignard Reagents. J. Org. Chem 2000, 65, 1188–1192. [DOI] [PubMed] [Google Scholar]
- (29).Holm T Electron Transfer from Alkylmagnesium Compounds to Organic Substrates. Acta Chem. Scand. B 1983, 37, 567–584. [Google Scholar]
- (30).Borer BC; Deerenberg S; Bieräugel H; Pandit UK The First Synthesis of the ABCD Ring System of Manzamine A. Construction of the Macrocyclic Ring D. Tetrahedron Lett. 1994, 35, 3191–3194. [Google Scholar]
- (31).Young WG; Roberts JD The Reaction of Butenylmagnesium Bromide with Acetomesitylene. J. Am. Chem. Soc 1944, 66, 2131. [Google Scholar]
- (32).Kohler EP; Baltzly R Steric Hindrance in Certain Mesitylenic Ketones. J. Am. Chem. Soc 1932, 54, 4015–4026. [Google Scholar]
- (33).Ammer J; Nolte C; Mayr H Free Energy Relationships for Reactions of Substituted Benzhydrylium Ions: From Enthalpy over Entropy to Diffusion Control. J. Am. Chem. Soc 2012, 134, 13902–13911. [DOI] [PubMed] [Google Scholar]
- (34).Shenoy SR; Smith DM; Woerpel KA Nucleophilic Additions of Trimethylsilyl Cyanide to Cyclic Oxocarbenium Ions: Evidence for the Loss of Stereoselectivity at the Limits of Diffusion Control. J. Am. Chem. Soc 2006, 128, 8671–8677. [DOI] [PubMed] [Google Scholar]
- (35).Krumper JR; Salamant WA; Woerpel KA Continuum of Mechanisms for Nucleophilic Substitutions of Cyclic Acetals. Org. Lett 2008, 10, 4907–4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Krumper JR; Salamant WA; Woerpel KA Correlations Between Nucleophilicities and Selectivities in the Substitutions of Tetrahydropyran Acetals. J. Org. Chem 2009, 74, 8039–8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Beaver MG; Woerpel KA Erosion of Stereochemical Control with Increasing Nucleophilicity: O-Glycosylation at the Diffusion Limit. J. Org. Chem 2010, 75, 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mayr H; Breugst M; Ofial AR Farewell to the HSAB Treatment of Ambident Reactivity. Angew. Chem. Int. Ed 2011, 50, 6470–6505. [DOI] [PubMed] [Google Scholar]
- (39).Brown HC; Ichikawa K The Rates of Reaction of Sodium Borohydride with Some Representative Ketones. J. Am. Chem. Soc 1962, 84, 373–376. [Google Scholar]
- (40).Datilus V; AbdulKarim M; Patrizio A; Kaur P Ter-pyridine Catalyzed Allylation of Aldehydes and Ketones under Metal-Free Condition. Tetrahedron Lett. 2016, 57, 2778–2781. [Google Scholar]
- (41).Zong H; Huang H; Liu J; Bian G; Song L Added-Metal-Free Catalytic Nucleophilic Addition of Grignard Reagents to Ketones. J. Org. Chem 2012, 77, 4645–4652. [DOI] [PubMed] [Google Scholar]
- (42).Niidu A; Paju A; Müürisepp AM; Järving I; Kailas T; Pehk T; Lopp M Stereoselective Synthesis of 1-Methyl-1,2- and 1,3-Cyclopentanediols via γ-Lactones. Chem. Heterocycl. Compd 2013, 48, 1751–1760. [Google Scholar]
- (43).Becker CC; Rosenquist A; Hølmer G Regiospecific Analysis of Triacylglycerols Using Allyl Magnesium Bromide. Lipids 1993, 28, 147–149. [Google Scholar]
- (44).Liao X; Wu Y; De Brabander JK Total Synthesis and Absolute Configuration of the Novel Microtubule-Stabilizing Agent Peloruside A. Angew. Chem. Int. Ed 2003, 42, 1648–1652. [DOI] [PubMed] [Google Scholar]
- (45).Mandal T; Chakraborti G; Karmakar S; Dash J Divergent and Orthogonal Approach to Carbazoles and Pyridoindoles from Oxindoles via Indole Intermediates. Org. Lett 2018, 20, 4759–4763. [DOI] [PubMed] [Google Scholar]
- (46).Lim Y-H; Li T; Chen P; Schreiber P; Kuznetsova L; Carroll PJ; Lauher JW; Sieburth SM Asymmetry by Electrophilic Rearrangement of Symmetric 2-Pyridone Photodimers. Org. Lett 2005, 7, 5413–5415. [DOI] [PubMed] [Google Scholar]
- (47).Esteve J; Jiménez C; Nebot J; Velasco J; Romea P; Urpí F Highly Stereoselective Titanium-Mediated Aldol Reactions from Chiral α-Silyloxy Ketones. A Reliable Tool for the Synthesis of Natural Products. Tetrahedron 2011, 67, 6045–6056. [Google Scholar]
- (48).Gündemir-Durmaz T; Schmid F; El Baz Y; Häusser A; Schneider C; Bilitewski U; Rauhut G; Garnier D; Baro A; Laschat S Truncated Borrelidin Analogues: Synthesis by Sequential Cross Metathesis/Olefination for the Southern Fragment and Biological Evaluation. Org. Biomol. Chem 2016, 14, 8261–8269. [DOI] [PubMed] [Google Scholar]
- (49).Benkeser RA; Broxterman WE The Reaction of Crotylmagnesium Bromide with Hindered Ketones. The First Examples of a Reversible Grignard Reaction. J. Am. Chem. Soc 1969, 91, 5162–5163. [Google Scholar]
- (50).Benkeser RA; Siklosi MP; Mozdzen EC Reversible Grignard and Organolithium Reactions. J. Am. Chem. Soc 1978, 100, 2134–2139. [Google Scholar]
- (51).Benkeser RA; Siklosi MP The First Documented Reversible Addition of Allylmagnesium Bromide to a Ketone. J. Org. Chem 1976, 41, 3212–3213. [Google Scholar]
- (52).Christensen SH; Holm T; Madsen R The Retro Grignard Addition Reaction Revisited: The Reversible Addition of Benzyl Reagents to Ketones. Tetrahedron 2014, 70, 1478–1483. [Google Scholar]
- (53).Chen X; Hortelano ER; Eliel EL; Frye SV Are Chelates Truly Intermediates in Cram’s Chelate Rule? J. Am. Chem. Soc 1990, 112, 6130–6131. [Google Scholar]
- (54).Chen X; Hortelano ER; Eliel EL; Frye SV Chelates as Intermediates in Nucleophilic Additions to Alkoxy Ketones According to Cram’s Rule (Cyclic Model). J. Am. Chem. Soc 1992, 114, 1778–1784. [Google Scholar]
- (55).Eliel EL; Frye SV; Hortelano ER; Chen X; Bai X Asymmetric Synthesis and Cram’s (Chelate) Rule. Pure Appl. Chem 1991, 63, 1591–1598. [Google Scholar]
- (56).Mengel A; Reiser O Around and beyond Cram’s Rule. Chem. Rev 1999, 99, 1191–1223. [DOI] [PubMed] [Google Scholar]
- (57).Huryn DM 1.01 Carbanions of Alkali and Alkaline-Earth Cations: (II) Selectivity of Carbonyl Addition Reactions In Comprehensive Organic Synthesis II; Knochel P; Molander GA, Eds.; Elsevier: Amsterdam, 2014, p. 1–26. [Google Scholar]
- (58).Appel R; Mayr H Quantification of the Electrophilic Reactivities of Aldehydes, Imines, and Enones. J. Am. Chem. Soc 2011, 133, 8240–8251. [DOI] [PubMed] [Google Scholar]
- (59).Venkataiah M; Fadnavis NW A Novel Stereoselective Synthesis of (−)-β-Conhydrine from (R)-2,3-O-Cyclohexylidine Glyceraldehyde. Tetrahedron 2009, 65, 6950–6952. [Google Scholar]
- (60).Cainelli G; Galletti P; Giacomini D; Orioli P Solvent and Temperature Effect in Aldol Condensation between the Lithium Enolate of tert-Butyl Acetate and 2-Phenyl Propanal: Enthalpy and Entropy Contribution. Tetrahedron Lett. 2001, 42, 7383–7385. [Google Scholar]
- (61).Lombardo M; Fabbroni S; Trombini C Entropy-Controlled Selectivity in the Vinylation of a Cyclic Chiral Nitrone. An Efficient Route to Enantiopure Polyhydroxylated Pyrrolidines. J. Org. Chem 2001, 66, 1264–1268. [DOI] [PubMed] [Google Scholar]
- (62).Cainelli G; Giacomini D; Galletti P; Orioli P Dynamic Solvation Effects in Ethylmagnesium Bromide Addition to (2S)-O-(tert-Butyldimethylsilyl)lactal. Eur. J. Org. Chem 2001, 4509–4515. [Google Scholar]
- (63).Chérest M; Felkin H; Prudent N Torsional Strain Involving Partial Bonds. The Stereochemistry of the Lithium Aluminum Hydride Reduction of Some Simple Open-Chain Ketones. Tetrahedron Lett. 1968, 9, 2199–2204. [Google Scholar]
- (64).Anh NT; Eisenstein O Theoretical Interpretation of 1–2 Asymmetric Induction. The Importance of Antiperiplanarity. Nouv. J. Chim 1977, 1, 61–70. [Google Scholar]
- (65).Cieplak AS Stereochemistry of Nucleophilic Addition to Cyclohexanone. The Importance of Two-Electron Stabilizing Interactions. J. Am. Chem. Soc 1981, 103, 4540–4552. [Google Scholar]
- (66).Evans DA; Siska SJ; Cee VJ Resurrecting the Cornforth Model for Carbonyl Addition: Studies on the Origin of 1,2-Asymmetric Induction in Enolate Additions to Heteroatom-Substituted Aldehydes. Angew. Chem. Int. Ed 2003, 42, 1761–1765. [DOI] [PubMed] [Google Scholar]
- (67).Cram DJ; Kopecky KR Studies in Stereochemistry. XXX. Models for Steric Control of Asymmetric Induction. J. Am. Chem. Soc 1959, 81, 2748–2755. [Google Scholar]
- (68).Tietze LF; Kinzel T; Schmatz S Origin of Syn/Anti Diastereoselectivity in Aldehyde and Ketone Crotylation Reactions: A Combined Theoretical and Experimental Study. J. Am. Chem. Soc 2006, 128, 11483–11495. [DOI] [PubMed] [Google Scholar]
- (69).Guan Y; Ingman VM; Rooks BJ; Wheeler SE AARON: An Automated Reaction Optimizer for New Catalysts. J. Chem. Theory Comput 2018, 14, 5249–5261. [DOI] [PubMed] [Google Scholar]
- (70).Bartolo ND; Hornstein AL; Zhao AY; Woerpel KA Diastereoselectivities in Reductions of α-Alkoxy Ketones Are Not Always Correlated to Chelation-Induced Rate Acceleration. Synthesis 2019, 51, 296–302. [Google Scholar]
- (71).Ashby EC; Laemmle JT Stereochemistry of Organometallic Compound Addition to Ketones. Chem. Rev 1975, 75, 521–546. [Google Scholar]
- (72).Dauben WG; Fonken GJ; Noyce DS The Stereochemistry of Hydride Reductions. J. Am. Chem. Soc 1956, 78, 2579–2582. [Google Scholar]
- (73).Seeman JI Effect of Conformational Change on Reactivity in Organic Chemistry. Evaluations, Applications, and Extensions of Curtin-Hammett/Winstein-Holness Kinetics. Chem. Rev 1983, 83, 83–134. [Google Scholar]
- (74).Wigfield DC; Phelps DJ Deuterium Isotope Effects in the Reduction of Cyclohexanones. The Concepts of Steric Approach Control and Product Development Control. Can. J. Chem 1972, 50, 388–394. [Google Scholar]
- (75).Wipke WT; Gund P Simulation and Evaluation of Chemical Synthesis. Congestion: A Conformation-Dependent Function of Steric Environment at a Reaction Center. Application with Torsional Terms to Stereoselectivity of Nucleophilic Additions to Ketones. J. Am. Chem. Soc 1976, 98, 8107–8118. [Google Scholar]
- (76).Shea KJ; Higby RG; Gilman JW Stereochemistry of Hydride Reductions. The Tricyclo[9.3.1.03,8]Pentadecane (Taxane) Ring System. Tetrahedron Lett. 1990, 31, 1221–1224. [Google Scholar]
- (77).Ito M; Clark CW; Mortimore M; Goh JB; Martin SF Biogenetically Inspired Approach to the Strychnos Alkaloids. Concise Syntheses of (±)-Akuammicine and (±)-Strychnine. J. Am. Chem. Soc 2001, 123, 8003–8010. [DOI] [PubMed] [Google Scholar]
- (78).Frankowski KJ; Golden JE; Zeng Y; Lei Y; Aubé J Syntheses of the Stemona Alkaloids (±)-Stenine, (±)-Neostenine, and (±)-13-Epineostenine Using a Stereodivergent Diels-Alder/Azido-Schmidt Reaction. J. Am. Chem. Soc 2008, 130, 6018–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Li Y; Chen Z-X; Xiao Q; Ye Q-D; Sun T-W; Meng F-K; Ren W-W; You L; Xu L-M; Wang Y-F et al. Diastereoselective Total Synthesis of (±)-Schindilactone A, Part 2: Construction of the Fully Functionalized CDEFGH Ring System. Chem. Asian J 2012, 7, 2334–2340. [DOI] [PubMed] [Google Scholar]
- (80).Ma X; Tang Q; Ke J; Zhang J; Wang C; Wang H; Li Y; Shao H Straightforward and Highly Diastereoselective Synthesis of 2,2-Di-Substituted Perhydrofuro[2,3-b]-pyran (and Furan) Derivatives Promoted by BiCl3. Chem. Commun 2013, 49, 7085–7087. [DOI] [PubMed] [Google Scholar]
- (81).Knight BJ; Stache EE; Ferreira EM Complementary Stereochemical Outcomes in Proline-Based Self-Regenerations of Stereocenters. Org. Lett 2014, 16, 432–435. [DOI] [PubMed] [Google Scholar]
- (82).Bedell TA; Hone GAB; Valette D; Yu J-Q; Davies HML; Sorensen EJ Rapid Construction of a Benzo-Fused Indoxamycin Core Enabled by Site-Selective C-H Functionalizations. Angew. Chem. Int. Ed 2016, 55, 8270–8274. [DOI] [PubMed] [Google Scholar]
- (83).Velluz L; Valls J; Mathieu J Spatial Arrangement and Preparative Organic Synthesis. Angew. Chem. Int. Ed. Engl 1967, 6, 778–789. [Google Scholar]
- (84).Woodward RB; Bader FE; Bickel H; Frey AJ; Kierstead RW The Total Synthesis of Reserpine. Tetrahedron 1958, 2, 1–57. [Google Scholar]
- (85).Lopez SA; Pourati M; Gais H-J; Houk KN How Torsional Effects Cause Attack at Sterically Crowded Concave Faces of Bicyclic Alkenes. J. Org. Chem 2014, 79, 8304–8312. [DOI] [PubMed] [Google Scholar]
- (86).Soteras I; Lozano O; Escolano C; Orozco M; Amat M; Bosch J; Luque FJ Structure-Directed Reversion in the π-Facial Stereoselective Alkylation of Chiral Bicyclic Lactams. J. Org. Chem 2008, 73, 7756–7763. [DOI] [PubMed] [Google Scholar]
- (87).Meyers AI; Seefeld MA; Lefker BA; Blake JF; Williard PG Stereoselective Alkylations in Rigid Systems. Effect of Remote Substituents on π-Facial Additions to Lactam Enolates. Stereoelectronic and Steric Effects. J. Am. Chem. Soc 1998, 120, 7429–7438. [Google Scholar]
- (88).Brown HC; Wheeler OH; Ichikawa K Chemical Effects of Steric Strains—XIII: Kinetics of the Reaction of Sodium Borohydride with Carbonyl Groups—a Convenient Tool for Investigating the Reactivities of Aldehydes and Ketones. Tetrahedron 1957, 1, 214–220. [Google Scholar]
- (89).Stanton GR; Johnson CN; Walsh PJ Overriding Felkin Control: A General Method for Highly Diastereoselective Chelation-Controlled Additions to α-Silyloxy Aldehydes. J. Am. Chem. Soc 2010, 132, 4399–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Mori S; Nakamura M; Nakamura E; Koga N; Morokuma K Theoretical Studies on Chelation-Controlled Carbonyl Addition. Me2Mg Addition to α- and β-Alkoxy Ketones and Aldehydes. J. Am. Chem. Soc 1995, 117, 5055–5065. [Google Scholar]
- (91).Walker FW; Ashby EC Composition of Grignard Compounds. VI. Nature of Association in Tetrahydrofuran and Diethyl Ether Solutions. J. Am. Chem. Soc 1969, 91, 3845–3850. [Google Scholar]
- (92).Ashby EC; Laemmle J; Neumann HM Mechanisms of Grignard Reagent Addition to Ketones. Acc. Chem. Res 1974, 7, 272–280. [Google Scholar]
- (93).Safont VS; Moliner V; Oliva M; Castillo R; Andrés J; González F; Carda M A Theoretical Study of Addition of Organomagnesium Reagents to Chiral α-Alkoxy Carbonyl Compounds. J. Org. Chem 1996, 61, 3467–3475. [Google Scholar]
- (94).Mori T; Kato S Grignard Reagents in Solution: Theoretical Study of the Equilibria and the Reaction with a Carbonyl Compound in Diethyl Ether Solvent. J. Phys. Chem. A 2009, 113, 6158–6165. [DOI] [PubMed] [Google Scholar]
- (95).Yamazaki S; Yamabe S A Computational Study on Addition of Grignard Reagents to Carbonyl Compounds. J. Org. Chem 2002, 67, 9346–9353. [DOI] [PubMed] [Google Scholar]
- (96).Stanton GR; Koz G; Walsh PJ Highly Diastereoselective Chelation-Controlled Additions to α-Silyloxy Ketones. J. Am. Chem. Soc 2011, 133, 7969–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Reetz MT; Keβeler K; Schmidtberger S; Wenderoth B; Steinbach R Chelation-or Non-Chelation-Control in Stereoselective Reactions of Titanium Reagents with Chiral Alkoxy Carbonyl Compounds. Angew. Chem. Suppl 1983, 1511–1526. [Google Scholar]
- (98).Fan X; Walsh PJ Chelation-Controlled Additions to Chiral α- and β-Silyloxy, α-Halo, and β-Vinyl Carbonyl Compounds. Acc. Chem. Res 2017, 50, 2389–2400. [DOI] [PubMed] [Google Scholar]
- (99).Reetz MT Chelation or Non-Chelation Control in Addition Reactions of Chiral α- and β-Alkoxy Carbonyl Compounds. Angew. Chem. Int. Ed. Engl 1984, 23, 556–569. [Google Scholar]
- (100).Reetz MT Structural, Mechanistic, and Theoretical Aspects of Chelation-Controlled Carbonyl Addition Reactions. Acc. Chem. Res 1993, 26, 462–468. [Google Scholar]
- (101).El Bkassiny S; N’Go I; Sevrain CM; Tikad A; Vincent SP Synthesis of a Novel UDP-Carbasugar as UDP-Galactopyranose Mutase Inhibitor. Org. Lett 2014, 16, 2462–2465. [DOI] [PubMed] [Google Scholar]
- (102).Still WC; McDonald JH III. Chelation-Controlled Nucleophilic Additions. 1. A Highly Effective System for Asymmetric Induction in the Reaction of Organometallics with α-Alkoxyketones. Tetrahedron Lett. 1980, 21, 1031–1034. [Google Scholar]
- (103).Singh SM; Oehlschlager AC Chelation vs. Non-Chelation Control in Addition Reactions of Ethylmetallic Reagents to Acrolein Dimer. Can. J. Chem 1988, 66, 209–213. [Google Scholar]
- (104).Marco JA; Carda M; González F; Rodríguez S; Castillo E; Murga J Diastereoselectivity in Organometallic Additions to the Carbonyl Group of Protected Erythrulose Derivatives. J. Org. Chem 1998, 63, 698–707. [DOI] [PubMed] [Google Scholar]
- (105).Mulzer J; List B Highly Stereoselective Synthesis of Tetrasubstituted Alkenes via [2,3]-Wittig Rearrangement. Tetrahedron Lett. 1994, 35, 9021–9024. [Google Scholar]
- (106).Smaltz DJ; Švenda J; Myers AG Diastereoselective Additions of Allylmetal Reagents to Free and Protected syn-α,β-Dihydroxyketones Enable Efficient Synthetic Routes to Methyl Trioxacarcinoside A. Org. Lett 2012, 14, 1812–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Fronza G; Fuganti C; Grasselli P; Pedrocchi-Fantoni G; Zirotti C On the Steric Course of Addition of Diallylzinc onto α,β-Dialkoxy Chiral Carbonyl Compounds. Stereospecific Synthesis of 2,6-Dideoxysugars of the l-Series. Tetrahedron Lett. 1982, 23, 4143–4146. [Google Scholar]
- (108).Fuganti C; Grasselli P; Servi S Synthesis of (−)-Frontalin from the (2S,3R)-Diol prepared from α-Methylcinnamaldehyde and Fermenting Baker’s Yeast. J. Chem. Soc., Perkin Trans. 1 1983, 241–244. [Google Scholar]
- (109).Saha N; Chatterjee B; Chattopadhyay SK δ,E-Unsaturated α,β-Diamino Acids as Building Blocks for the Asymmetric Synthesis of Diverse α,β-Diamino Acids. J. Org. Chem 2015, 80, 1896–1904. [DOI] [PubMed] [Google Scholar]
- (110).Matsunaga N; Kaku T; Ojida A; Tasaka A Synthetic Studies on (1S)-1-(6,7-Dimethoxy-2-naphthyl)-1-(1H-imidazol-4-yl)-2-methylpropan-1-ol as a Selective C17,20-Lyase Inhibitor. Tetrahedron: Asymmetry 2004, 15, 2021–2028. [Google Scholar]
- (111).Osztrovszky G; Holm T; Madsen R Ultrafast Grignard Addition Reactions in the Presence of Water. Org. Biomol. Chem 2010, 8, 3402–3404. [DOI] [PubMed] [Google Scholar]
- (112).Ghosh AK; Kassekert LA Enantioselective Synthesis of Both Epimers at C-21 in the Proposed Structure of Cytotoxic Macrolide Callyspongiolide. Org. Lett 2016, 18, 3274–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Zhang J; Zheng S; Peng W; Shen Z Total Synthesis of (−)-Englerin A. Tetrahedron Lett. 2014, 55, 1339–1341. [Google Scholar]
- (114).Charette AB; Benslimane AF; Mellon C The Tetrahydropyranyl Group as a Chiral Auxiliary for the Nucleophilic Addition to α-Alkoxy Ketones. Tetrahedron Lett. 1995, 36, 8557–8560. [Google Scholar]
- (115).Yang Q; Draghici C; Njardarson JT; Li F; Smith BR; Das P Evolution of an Oxidative Dearomatization Enabled Total Synthesis of Vinigrol. Org. Biomol. Chem 2014, 12, 330–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Akhoon KM; Myles DC Highly Diastereoselective Additions to Chiral α-Keto Acetals. J. Org. Chem 1997, 62, 6041–6045. [Google Scholar]
- (117).Soai K; Ishizaki M Asymmetric Synthesis of Functionalized Tertiary Homoallyl Alcohols by Diastereoselective Allylation of Chiral α-Keto Amides Derived from (S)-Proline Esters: Control of Stereochemistry Based on Saturated Coordination of Lewis Acid. J. Org. Chem 1986, 51, 3290–3295. [Google Scholar]
- (118).Fujisawa T; Ukaji Y; Funabora M; Yamashita M; Sato T Stereocontrolled Addition Reaction of Organometallics to Chiral α-Keto Amides. Bull. Chem. Soc. Jpn 1990, 63, 1894–1897. [Google Scholar]
- (119).Cirillo PF; Panek JS Diastereoselectivity in Nucleophilic Addition Reactions to (α,β-Dialkoxyacyl)Silanes: An Operationally Useful Route to Optically Active 1,2,3-syn-Triols. J. Org. Chem 1990, 55, 6071–6073. [Google Scholar]
- (120).Patrocínio AF; Moran PJS. Acylsilanes and Their Applications in Organic Chemistry. J. Braz. Chem. Soc 2001, 12, 7–31. [Google Scholar]
- (121).Chen K-M; Hardtmann GE; Prasad K; Repič O; Shapiro MJ 1,3-Syn Diastereoselective Reduction of β-Hydroketones Utilizing Alkoxydialkylboranes. Tetrahedron Lett. 1987, 28, 155–158. [Google Scholar]
- (122).Stevens RV Nucleophilic Additions to Tetrahydropyridinium Salts. Applications to Alkaloid Syntheses. Acc. Chem. Res 1984, 17, 289–296. [Google Scholar]
- (123).Deslongchamps P Stereoelectronic Effects in Organic Chemistry. Pergamon: New York, 1983, p. 209–221. [Google Scholar]
- (124).Sarko CR; Guch IC; DiMare M Chelation-Controlled Protocol for the Diastereoselective Reduction of Ketones. J. Org. Chem 1994, 59, 705–706. [Google Scholar]
- (125).Loertscher BM; Young PR; Evans PR; Castle SL Diastereoselective Synthesis of Vicinal Tertiary Diols. Org. Lett 2013, 15, 1930–1933. [DOI] [PubMed] [Google Scholar]
- (126).Abate A; Brenna E; Fronza G; Fuganti C; Gatti FG; Serra S; Zardoni E Preparation of the Enantiomerically Enriched Isomers of the Odorous Cyclic Ethers Clarycet®, Florol®, and Rhubafuran® by Enzymatic Catalysis. Helv. Chim. Acta 2004, 87, 765–780. [Google Scholar]
- (127).Abate A; Brenna E; Costantini A; Fuganti C; Gatti FG; Malpezzi L; Serra S Enzymatic Approach to Enantiomerically Pure 5-Alken-2,4-Diols and 4-Hydroxy-5-Alken-2-Ones: Application to the Synthesis of Chiral Synthons. J. Org. Chem 2006, 71, 5228–5240. [DOI] [PubMed] [Google Scholar]
- (128).Yoshida K; Toyoshima T; Imamoto T Efficient Synthetic Routes to Aromatic Compounds Using Ring-Closing Olefin Metathesis Followed by Dehydration, Oxidation, and Tautomerization. Chem. Commun 2007, 3774–3776. [DOI] [PubMed] [Google Scholar]
- (129).Jouanneau M; Bonepally KR; Jeuken A; Tap A; Guillot R; Ardisson J; Ferezou JP; Prunet J Diastereoselective Synthesis of an Advanced Intermediate of Thapsigargin and Other 6,12-Guaianolides Using a RCEYM Strategy. Org. Lett 2018, 20, 2176–2180. [DOI] [PubMed] [Google Scholar]
- (130).Hoffmann RW Allylic 1,3-Strain as a Controlling Factor in Stereoselective Transformations. Chem. Rev 1989, 89, 1841–1860. [Google Scholar]
- (131).Barluenga J; Pérez-Sánchez I; Rubio E; Flórez J. Three/Four/Five-Component Diastereoselective Cyclization of Fischer Carbene Complexes, Lithium Enolates, and Allylmagnesium Bromide. Angew. Chem. Int. Ed 2003, 42, 5860–5863. [DOI] [PubMed] [Google Scholar]
- (132).Yang W-Q; Kitahara T Total Synthesis of a Nonclassical Bioactive Acetogenin, (+)-Muconin. Tetrahedron 2000, 56, 1451–1461. [Google Scholar]
- (133).Suzuki A; Sasaki M; Nakagishi T; Ueda T; Hoshiya N; Uenishi J.-i Construction of Iterative Tetrahydrofuran Ring Units and Total Synthesis of (+)-Goniocin. Org. Lett 2016, 18, 2248–2251. [DOI] [PubMed] [Google Scholar]
- (134).Denmark SE; Yang S-M Total Synthesis of (+)-Brasilenyne. Application of an Intramolecular Silicon-Assisted Cross-Coupling Reaction. J. Am. Chem. Soc 2004, 126, 12432–12440. [DOI] [PubMed] [Google Scholar]
- (135).Entemann CE Jr.; Johnson JR The Relative Reactivity of Various Functional Groups toward a Grignard Reagent. J. Am. Chem. Soc 1933, 55, 2900–2903. [Google Scholar]
- (136).Fleming I; Lewis JJ The Diastereoselectivity of Electrophilic Attack on Trigonal Carbon Adjacent to a Stereogenic Centre: Diastereoselective Protonation, Epoxidation and Acylation of Allylsilanes. J. Chem. Soc., Perkin Trans. 1 1992, 3267–3275. [Google Scholar]
- (137).Nicolaou KC; Claremon DA; Barnette WE Total Synthesis of (±)-Zoapatanol. J. Am. Chem. Soc 1980, 102, 6611–6612. [Google Scholar]
- (138).Chan TH; Li CJ A Concise Synthesis of (+)-Muscarine. Can. J. Chem 1992, 70, 2726–2729. [Google Scholar]
- (139).Mohan P; Fuertes MJ Synthesis of Vinyliodides: Progress toward the Total Synthesis of a Jatrophane Diterpene. Tetrahedron Lett. 2013, 54, 3919–3925. [Google Scholar]
- (140).Reetz MT; Kesseler K; Jung A Concerning the Role of Lewis Acids in Chelation Controlled Addition to Chiral Alkoxy Aldehydes. Tetrahedron Lett. 1984, 25, 729–732. [Google Scholar]
- (141).Kiyooka S.-i.; Heathcock CH. Acyclic Stereoselection. 20. High Diastereofacial Selectivity in the Stannic Chloride Mediated Reactions of Allylsilanes with Chiral α- and β-Alkoxy Aldehydes. Tetrahedron Lett. 1983, 24, 4765–4768. [Google Scholar]
- (142).Williams DR; Klingler FD Stereoselective Allylation for Preparation of l-Hexose Derivatives. Tetrahedron Lett. 1987, 28, 869–872. [Google Scholar]
- (143).Crimmins MT; Ellis JM; Emmitte KA; Haile PA; McDougall PJ; Parrish JD; Zuccarello JL Enantioselective Total Synthesis of Brevetoxin A: Unified Strategy for the B, E, G, and J Subunits. Chem. Eur. J 2009, 15, 9223–9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Brown HC; Randad RS; Bhat KS; Zaidlewicz M; Racherla US Chiral Synthesis via Organoboranes. 24. S-allylbis(2-isocaranyl)borane as a Superior Reagent for the Asymmetric Allylboration of Aldehydes. J. Am. Chem. Soc 1990, 112, 2389–2392. [Google Scholar]
- (145).Green ME; Rech JE; Floreancig PE Stereochemical Assignment of the C1-C6 Fragment of Psymberin by Synthesis and Natural Product Degradation. Org. Lett 2005, 7, 4117–4120. [DOI] [PubMed] [Google Scholar]
- (146).Schmidt B; Biernat A The Tandem Ring-Closing Metathesis-Isomerization Approach to 6-Deoxyglycals. Chem. Eur. J 2008, 14, 6135–6141. [DOI] [PubMed] [Google Scholar]
- (147).Yadav JS; Swamy T; Reddy BVS; Ravinder V Stereoselective Synthesis of C19-C27 Fragment of Bryostatin 11. Tetrahedron Lett. 2014, 55, 4054–4056. [Google Scholar]
- (148).Mulzer J; Angermann A On the Steric Course of the Addition of Some Organometallic Reagents to (R)-2,3-Isopropylidene Glyceraldehyde. Synthesis of Optically Active α-Benzyloxy Aldehydes, Alcohols, Carboxylic Acids and 1,2-Diols. Tetrahedron Lett. 1983, 24, 2843–2846. [Google Scholar]
- (149).Mead K; Macdonald TL Metal Ion Controlled Addition to α,β-Dialkoxy Carbonyl Compounds. J. Org. Chem 1985, 50, 422–424. [Google Scholar]
- (150).Reetz MT; Kesseler K Chelation- and Non-Chelation-Controlled Additions to 2-O-Benzyl-3-O-(tert-butyldimethylsilyl)glyceraldehyde. J. Org. Chem 1985, 50, 5434–5436. [Google Scholar]
- (151).Fürst R; Lentsch C; Rinner U Synthetic Studies Towards an Advanced Precursor of the Jatrophane Diterpene Pl-4. Synthesis 2013, 46, 357–367. [Google Scholar]
- (152).Nagaiah K; Sreenu D; Srinivasa Rao R; Yadav JS Stereoselective Synthesis of the Phytotoxic Nonenolide Herbarumin-I from l-Ascorbic Acid. Tetrahedron Lett. 2007, 48, 7173–7176. [Google Scholar]
- (153).Chattopadhyay A; Mamdapur VR (R)-2,3-O-Cyclohexylideneglyceraldehyde, a Versatile Intermediate for Asymmetric Synthesis of Chiral Alcohol. J. Org. Chem 1995, 60, 585–587. [DOI] [PubMed] [Google Scholar]
- (154).Bommagani S; Thodupunuri P; Sharma GVM. Attempts toward the Synthesis of Mupirocin-H. Arkivoc 2017, (iv), 20–33. [Google Scholar]
- (155).Schmidt B; Biernat A Synthesis of Enantiomerically Pure Dihydrofurans and Dihydropyrans from Common Precursors Using RCM and Tandem RCM–Isomerization. Synlett 2007, 2375–2378. [Google Scholar]
- (156).Nambu H; Noda N; Niu W; Fujiwara T; Yakura T Stereoselective Total Synthesis of Myriocin Using Rhodium(II)-Catalyzed C-H Amination Followed by Alkylation. Asian J. Org. Chem 2015, 4, 1246–1249. [Google Scholar]
- (157).Paquette LA; Mitzel TM Addition of Allylindium Reagents to Aldehydes Substituted at Cα or Cβ with Heteroatomic Functional Groups. Analysis of the Modulation in Diastereoselectivity Attainable in Aqueous, Organic, and Mixed Solvent Systems. J. Am. Chem. Soc 1996, 118, 1931–1937. [Google Scholar]
- (158).Prasad KR; Gutala P Total Synthesis and Determination of the Absolute Configuration of 5,6-Dihydro-α-pyrone Natural Product Synargentolide B. J. Org. Chem 2013, 78, 3313–3322. [DOI] [PubMed] [Google Scholar]
- (159).Hřebabecký H; Dračinský M; Procházková E; Šála M; Mackman R; Nencka R Control of α/β Anomer Formation by a 2′,5′ Bridge: Toward Nucleoside Derivatives Locked in the South Conformation. J. Org. Chem 2017, 82, 11337–11347. [DOI] [PubMed] [Google Scholar]
- (160).Mirabella S; Fibbi G; Matassini C; Faggi C; Goti A; Cardona F Accessing 2-Substituted Piperidine Iminosugars by Organometallic Addition/Intramolecular Reductive Amination: Aldehyde vs. Nitrone Route. Org. Biomol. Chem 2017, 15, 9121–9126. [DOI] [PubMed] [Google Scholar]
- (161).Ghosh AK; Cappiello J; Shin D Ring-Closing Metathesis Strategy to Unsaturated γ- and δ-Lactones: Synthesis of Hydroxyethylene Isostere for Protease Inhibitors. Tetrahedron Lett. 1998, 39, 4651–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Fuwa H; Ichinokawa N; Noto K; Sasaki M Stereoselective Synthesis of 2,6-Cis-Substituted Tetrahydropyrans: Brønsted Acid-Catalyzed Intramolecular Oxa-Conjugate Cyclization of α,β-Unsaturated Ester Surrogates. J. Org. Chem 2012, 77, 2588–2607. [DOI] [PubMed] [Google Scholar]
- (163).Fuwa H; Noji S; Sasaki M Studies toward the Total Synthesis of Gambieric Acids: Stereocontrolled Synthesis of a DEFG-Ring Model Compound. J. Org. Chem 2010, 75, 5072–5082. [DOI] [PubMed] [Google Scholar]
- (164).Alvarez E; Pérez R; Rico M; Rodríguez RM; Martín JD Simple Designs for the Construction of Complex Trans-Fused Polyether Toxin Frameworks. A Convergent Strategy Based on Hydroxy Ketone Cyclization of C-Linked Oxacycles. J. Org. Chem 1996, 61, 3003–3016. [DOI] [PubMed] [Google Scholar]
- (165).Moumé-Pymbock M; Furukawa T; Mondal S; Crich D Probing the Influence of a 4,6-O-Acetal on the Reactivity of Galactopyranosyl Donors: Verification of the Disarming Influence of the trans-gauche Conformation of C5–C6 Bonds. J. Am. Chem. Soc 2013, 135, 14249–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Dharuman S; Crich D Determination of the Influence of Side-Chain Conformation on Glycosylation Selectivity Using Conformationally Restricted Donors. Chem. Eur. J 2016, 22, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Yamazaki N; Atobe M; Kibayashi C Enantioselective Synthesis of NK-1 Receptor Antagonists (+)-CP-99,994 and (+)-CP-122,721. Tetrahedron Lett. 2002, 43, 7979–7982. [Google Scholar]
- (168).Bandyopadhyay A; Mitra P; Chattopadhyay S Diversity-Oriented Synthesis of Aminocyclohexitols from Garner’s Aldehyde. Synthesis 2013, 45, 536–544. [Google Scholar]
- (169).Chattopadhyay SK; Bandyopadhyay A A Stereo-Divergent Route to Aminocyclopentitol Derivatives. Tetrahedron Lett. 2011, 52, 3942–3944. [Google Scholar]
- (170).Abe J; Nagai Y; Higashikuni R; lida K; Hirokawa T; Nagai H; Kominato K; Tsuchida T; Hirata M; Inada M et al. Synthesis of Vitamin D3 Derivatives with Nitrogen-Linked Substituents at A-ring C-2 and Evaluation of Their Vitamin D Receptor-Mediated Transcriptional Activity. Org. Biomol. Chem 2012, 10, 7826–7839. [DOI] [PubMed] [Google Scholar]
- (171).Myeong I-S; Lee Y-T; Lee S-H; Jung C; Kim J-S; Park S-H; Kang J; Lee SJ; Ye I-H; Ham W-H Stereoselective Allylation Reactions of Acyclic and Chiral α-Amino-β-Hydroxy Aldehydes. Tetrahedron: Asymmetry 2017, 28, 1053–1060. [Google Scholar]
- (172).Hamana H; Ikota N; Ganem B Chelate Selectivity in Chelation-Controlled Allylations. A New Synthesis of Castanospermine and Other Bioactive Indolizidine Alkaloids. J. Org. Chem 1987, 52, 5492–5494. [Google Scholar]
- (173).Bartlett N; Gross L; Peron F; Asby DJ; Selby MD; Tavassoli A; Linclau B Stereocontrol by Quaternary Centres: A Stereoselective Synthesis of (−)-Luminacin D. Chem. Eur. J 2014, 20, 3306–3310. [DOI] [PubMed] [Google Scholar]
- (174).Roldán S; Cardona A; Conesa L; Murga J; Falomir E; Carda M; Marco JA Synthesis and Biological Evaluation of Simplified Pironetin Analogues with Modifications in the Side Chain and the Lactone Ring. Org. Biomol. Chem 2016, 15, 220–232. [DOI] [PubMed] [Google Scholar]
- (175).Hunter TJ; Wang Y; Zheng J; O’Doherty GA Asymmetric Iterative Hydration of Polyene Strategy to Cryptocaryols a and B. Synthesis 2016, 48, 1700–1710. [Google Scholar]
- (176).Hunter TJ; Zheng J; O’Doherty GA Approach to the Synthesis of the C1-C11 and C14–C18 Portion of Leucascandrolide A. Org. Chem. Front 2016, 3, 1120–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Kinnaird JWA; Ng PY; Kubota K; Wang X; Leighton JL Strained Silacycles in Organic Synthesis: A New Reagent for the Enantioselective Allylation of Aldehydes. J. Am. Chem. Soc 2002, 124, 7920–7921. [DOI] [PubMed] [Google Scholar]
- (178).Smith CM; O’Doherty GA Enantioselective Syntheses of Cryptocarya Triacetate, Cryptocaryolone, and Cryptocaryolone Diacetate. Org. Lett 2003, 5, 1959–1962. [DOI] [PubMed] [Google Scholar]
- (179).Kartika R; Taylor RE Electrophile-Induced Ether Transfer: Stereoselective Synthesis of 2,4,6-Trisubstituted Tetrahydropyrans. Angew. Chem. Int. Ed 2007, 46, 6874–6877. [DOI] [PubMed] [Google Scholar]
- (180).Kumara Swamy KC; Bhuvan Kumar NN; Balaraman E; Pavan Kumar KVP Mistunobu and Related Reactions: Advances and Applications. Chem. Rev 2009, 109, 2551–2651. [DOI] [PubMed] [Google Scholar]
- (181).Kocieński PJ; Yeates C; Street SDA; Campbell SF The 3,4-Dihydro-2H-pyran Approach to (+)-Milbemycin β3. Part 1. An Alternative Synthesis of (2S,4S,6R,8R,9S)-2-Formylmethyl-4-(dimethyl-t-butylsilyloxy)-8,9-dimethyl-1,7-dioxaspiro[5.5]undecane. J. Chem. Soc., Perkin Trans. 1 1987, 2183–2187. [Google Scholar]
- (182).Honjo E; Kutsumura N; Ishikawa Y; Nishiyama S Synthesis of a Spiroacetal Moiety of Antitumor Antibiotic Ossamycin by Anodic Oxidation. Tetrahedron 2008, 64, 9495–9506. [Google Scholar]
- (183).Brun E; Bellosta V; Cossy J Synthesis of the Acyclic Carbon Skeleton of Filipin III. J. Org. Chem 2016, 81, 8206–8221. [DOI] [PubMed] [Google Scholar]
- (184).Huang W; Ren R-G; Dong H-Q; Wei B-G; Lin G-Q Diverse Synthesis of Marine Cyclic Depsipeptide Lagunamide A and Its Analogues. J. Org. Chem 2013, 78, 10747–10762. [DOI] [PubMed] [Google Scholar]
- (185).Cee VJ; Cramer CJ; Evans DA Theoretical Investigation of Enolborane Addition to α-Heteroatom-Substituted Aldehydes. Relevance of the Cornforth and Polar Felkin–Anh Models for Asymmetric Induction. J. Am. Chem. Soc 2006, 128, 2920–2930. [DOI] [PubMed] [Google Scholar]
- (186).Evans DA; Dart MJ; Duffy JL; Yang MG A Stereochemical Model for Merged 1, 2- and 1,3-Asymmetric Induction in Diastereoselective Mukaiyama Aldol Addition Reactions and Related Processes. J. Am. Chem. Soc 1996, 118, 4322–4343. [Google Scholar]
- (187).Stanton GR; Kauffman MC; Walsh PJ Diastereoselective Chelation-Controlled Additions to β-Silyloxy Aldehydes. Org. Lett 2012, 14, 3368–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Ball M; Gregson T; Omori H; Thomas EJ Syntheses of C17–C27 Fragments of 20-Deoxybryostatins for Assembly Using Julia and Metathesis Reactions. Org. Biomol. Chem 2017, 15, 2740–2767. [DOI] [PubMed] [Google Scholar]
- (189).McGarvey GJ; Williams JM; Hiner RN; Matsubara Y; Oh T l-Aspartic Acid in Acyclic Stereoselective Synthesis. Synthetic Studies on Amphotericin B. J. Am. Chem. Soc 1986, 108, 4943–4952. [Google Scholar]
- (190).Overly KR; Williams JM; McGarvey GJ Studies toward the Application of Oxazoline-Epoxide Equivalency in 1,3-Asymmetric Induction. Tetrahedron Lett. 1990, 31, 4573–4576. [Google Scholar]
- (191).Still WC; Schneider JA Chelation-Controlled Nucleophilic Additions. 2. A Highly Effective System for Asymmetric Induction in the Reaction of Organometallics with β-Alkoxyaldehydes. Tetrahedron Lett. 1980, 21, 1035–1038. [Google Scholar]
- (192).Nagaoka H; Kishi Y Further Synthetic Studies on Rifamycin S. Tetrahedron 1981, 37, 3873–3888. [Google Scholar]
- (193).Clayden J; Westlund N; Wilson FX Asymmetric Induction Using Atropisomers: Diastereoselective Additions to 2-Acyl-1-Naphthamides. Tetrahedron Lett. 1996, 37, 5577–5580. [Google Scholar]
- (194).Chérest M; Felkin H Torsional Strain Involving Partial Bonds. The Steric Course of the Reaction between Allyl Magnesium Bromide and 4-T-Butyl-Cyclohexanone. Tetrahedron Lett. 1968, 9, 2205–2208. [Google Scholar]
- (195).Gung BW Structure Distortions in Heteroatom-Substituted Cyclohexanones, Adamantanones, and Adamantanes: Origin of Diastereofacial Selectivity. Chem. Rev 1999, 99, 1377–1386. [DOI] [PubMed] [Google Scholar]
- (196).Cardona F; D’Orazio G; Silva AMS; Nicotra F; La Ferla B Synthesis of glyco-Fused Bicyclic Compounds; Conformationally Constrained Scaffolds and Useful Polyfunctional Building Blocks. Eur. J. Org. Chem 2014, 2549–2556. [Google Scholar]
- (197).Hempel JE; Engers DW; Sulikowski GA Synthetic Studies Directed toward the AB Decalin Common to HMP-Y1 and Hibarimicinone. Tetrahedron Lett. 2014, 55, 2157–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Marvell EN; Cheng JC-P Intramolecular Ene Reactions of 1,2-Diallylcyclohexanes.J.Org. Chem 1980, 45, 4511–4514. [Google Scholar]
- (199).Fujiwara T; Tsuruta Y; Arizono K.-i.; Takeda T Oxidative Cleavage of Vic-Diols Using Copper(II) Bromide-Lithium t-Butoxide: A New Route to Unsymmetrical 1,5- and 1,6-Diketones. Synlett 1997, 962–964. [Google Scholar]
- (200).El Assal M; Peixoto PA; Coffinier R; Garnier T; Deffieux D; Miqueu K; Sotiropoulos J-M; Pouységu L; Quideau S Synthesis of Scyphostatin Analogues through Hypervalent Iodine-Mediated Phenol Dearomatization. J. Org. Chem 2017, 82, 11816–11828. [DOI] [PubMed] [Google Scholar]
- (201).Liu Y; Zhang Y; Wang X; Fu S; Liu B Synthesis of Polycyclic Frameworks through Iron-Catalyzed Intramolecular [5+2] Cycloaddition. Synlett 2018, 29, 1978–1982. [Google Scholar]
- (202).Clegg W; Craig FJ; Henderson KW; Kennedy AR; Mulvey RE; O’Neil PA; Reed D Solid State Structures and Dynamic Solution Equilibria of Bis(dibenzylamido)magnesium Complexes: Aggregation Dependence on Stoichiometry and Denticity of Donor Solvent. Inorg. Chem 1997, 36, 6238–6246. [Google Scholar]
- (203).Mehta G; Kumaran RS A Sequential RCM/Fragmentation Protocol Towards Chiral, Stereodefined Medium Ring Sesquiterpenoids. A Carvone Route to E- and Z-Germacrenes. Tetrahedron Lett. 2005, 46, 8831–8835. [Google Scholar]
- (204).Kumaran RS; Mehta G A Versatile, RCM Based Approach to Eudesmane and Dihydroagarofuran Sesquiterpenoids from (−)-Carvone: A Formal Synthesis of (−)-Isocelorbicol. Tetrahedron 2015, 71, 1718–1731. [Google Scholar]
- (205).Chen J; Collins GT; Levant B; Woods J; Deschamps JR; Wang S CJ-1639: A Potent and Highly Selective Dopamine D3 Receptor Full Agonist. ACS Med. Chem. Lett 2011, 2, 620–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Li N-S; Lu J; Piccirilli JA Efficient Synthesis of Methyl 3,5-Di-O-benzyl-α-d-ribofuranoside and Application to the Synthesis of 2’C-β-Alkoxymethyluridines. Org. Lett 2007, 9, 3009–3012. [DOI] [PubMed] [Google Scholar]
- (207).Jonckers THM; Vandyck K; Vandekerckhove L; Hu L; Tahri A; Van Hoof S; Lin T-I; Vijgen L; Berke JM; Lachau-Durand S et al. Nucleotide Prodrugs of 2’-Deoxy-2’-Spirooxetane Ribonucleosides as Novel Inhibitors of the HCV NS5B Polymerase. J. Med. Chem 2014, 57, 1836–1844. [DOI] [PubMed] [Google Scholar]
- (208).Adinolfi M; Barone G; Iadonisi A; Mangoni L; Manna R Synthesis of Caryose, the Carbocyclic Monosaccharide Component of the Lipopolysaccharide from Pseudomonas caryophylli. Tetrahedron 1997, 53, 11767–11780. [Google Scholar]
- (209).Chen J; Jiang C; Levant B; Li X; Zhao T; Wen B; Luo R; Sun D; Wang S Pramipexole Derivatives as Potent and Selective Dopamine D3 Receptor Agonists with Improved Human Microsomal Stability. ChemMedChem 2014, 9, 2653–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Baer S; Brinkman EA; Brauman JI Hemiacetal Anions: A Model for Tetrahedral Reaction Intermediates. J. Am. Chem. Soc 1991, 113, 805–812. [Google Scholar]
- (211).Vaithegi K; Prasad KR Enantiospecific Total Synthesis of the Putative Structure of Cryptopyranmoscatone B2. Tetrahedron 2018, 74, 2627–2633. [Google Scholar]
- (212).Guthertz A; Lusseau J; Desvergnes V; Massip S; Landais Y An Approach toward the Synthesis of the Spiroimine Fragment of 13-Desmethylspirolide C and Gymnodimine A. Chem. Eur. J 2019, 25, 1553–1560. [DOI] [PubMed] [Google Scholar]
- (213).Datta R; Bose S; Viththlbhai PB; Ghosh S An Expeditious Approach to Highly Functionalized Angularly Fused 5–5–n Ring Systems through Ring Opening–Ring Closing Metathesis of Norbornene Derivatives. Tetrahedron Lett. 2014, 55, 3538–3540. [Google Scholar]
- (214).Bürgi HB; Dunitz JD From Crystal Statics to Chemical Dynamics. Acc. Chem. Res 1983, 16, 153–161. [Google Scholar]
- (215).Sämann C; Knochel P A Convenient Synthesis of α-Substituted β,γ-Unsaturated Ketones and Esters via the Direct Addition of Substituted Allylic Zinc Reagents Prepared by Direct Insertion. Synthesis 2013, 45, 1870–1876. [Google Scholar]
- (216).Lou S; Fu GC Nickel/Bis(oxazoline)-Catalyzed Asymmetric Kumada Reactions of Alkyl Electrophiles: Cross-Couplings of Racemic α-Bromoketones. J. Am. Chem. Soc 2010, 132, 1264–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Ariel S; Trotter J 2-Cyclohexyl-1-(4-methoxyphenyl)-2-phenylethanone. Acta Cryst. C 1986, C42, 485–487. [Google Scholar]
- (218).Sun Q; Yao CJ; König B A Triphenylphosphine Mediated Photo-Rearrangement and Methanol Addition of Aryl Chalcones to 1-Propanones. Photochem. Photobiol. Sci 2015, 14, 948–952. [DOI] [PubMed] [Google Scholar]
- (219).Kanomata K; Toda Y; Shibata Y; Yamanaka M; Tsuzuki S; Gridnev ID; Terada M Secondary Stereocontrolling Interactions in Chiral Brønsted Acid Catalysis: Study of a Petasis–Ferrier-Type Rearrangement Catalyzed by Chiral Phosphoric Acids. Chem. Sci 2014, 5, 3515–3523. [Google Scholar]
- (220).Takahashi T; Nishihara Y; Hara R; Huo S; Kotora M Highly Regio- and Diastereo-Selective Carbon–Carbon Bond Formation Reaction of Unsymmetrical Zirconacyclopentanes Using Copper Salt. Chem. Commun 1997, 1599–1600. [Google Scholar]
- (221).Liang Y; Fu GC Catalytic Asymmetric Synthesis of Tertiary Alkyl Fluorides: Negishi Cross-Couplings of Racemic α,α-Dihaloketones. J. Am. Chem. Soc 2014, 136, 5520–5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Billings SB; Woerpel KA Nucleophilic Substitution Reactions of Sulfur-Substituted Cyclohexanone Acetals: An Analysis of the Factors Controlling Stereoselectivity. J. Org. Chem 2006, 71, 5171–5178. [DOI] [PubMed] [Google Scholar]
- (223).Pattison G Conformational Preferences of α-Fluoroketones May Influence Their Reactivity. Beilstein J. Org. Chem 2017, 13, 2915–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Wong SS; Paddon-Row MN Theoretical Evidence in Support of the Anh-Eisenstein Electronic Model in Controlling π-Facial Stereoselectivity in Nucleophilic Additions to Carbonyl Compounds. J. Chem. Soc., Chem. Commun 1990, 456–458. [Google Scholar]
- (225).Alvarez-Ibarra C; Arjona O; Pérez-Ossorio R; Pérez-Rubalcaba A; Quiroga ML; Santesmases MJ Stereoselectivity in the Condensation Reactions of 1-Phenylethyl Alkyl and Phenyl Ketones with Organometallic Reagents. J. Chem. Soc., Perkin Trans. 2 1983, 1645–1648. [Google Scholar]
- (226).Felix C; Laurent A; Mison P Mise en Évidence d’une Réactivité Spécifique des Trifluorométhylcétones vis-à-vis du Bromure d’Allylmagnésium. J. Fluorine Chem 1995, 70, 71–82. [Google Scholar]
- (227).Otte DAL; Woerpel KA Evidence that Additions of Grignard Reagents to Aliphatic Aldehydes Do Not Involve Single-Electron-Transfer Processes. Org. Lett 2015, 17, 3906–3909. [DOI] [PubMed] [Google Scholar]
- (228).Andrews SP; Ball M; Wierschem F; Cleator E; Oliver S; Hogenauer K; Simic O; Antonello A; Hünger U; Smith MD et al. Total Synthesis of Five Thapsigargins: Guaianolide Natural Products Exhibiting Sub-Nanomolar SERCA Inhibition. Chem. Eur. J 2007, 13, 5688–5712. [DOI] [PubMed] [Google Scholar]
- (229).Ley SV; Antonello A; Balskus EP; Booth DT; Christensen SB; Cleator E; Gold H; Högenauer K; Hünger U; Myers RM et al. Synthesis of the Thapsigargins. Proc. Natl. Acad. Sci. USA 2004, 101, 12073–12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Scarlato GR; DeMattei JA; Chong LS; Ogawa AK; Lin MR; Armstrong RW Asymmetric Synthesis of Calyculin C. 1. Synthesis of the C1–C25 Fragment. J. Org. Chem 1996, 61, 6139–6152. [DOI] [PubMed] [Google Scholar]
- (231).Mowat J; Kang B; Fonovic B; Dudding T; Britton R Inverse Temperature Dependence in the Diastereoselective Addition of Grignard Reagents to a Tetrahydrofurfural. Org. Lett 2009, 11, 2057–2060. [DOI] [PubMed] [Google Scholar]
- (232).Amouroux R; Ejjiyar S; Chastrette M Diastereoselectivite dans la Reaction des Organomagnesiens sur le Tetrahydrofurfural et son gem-Diacetate en Presence de HMPT. Acces aux Diols-1,2 Erythro. Tetrahedron Lett. 1986, 27, 1035–1038. [Google Scholar]
- (233).Fronza G; Mele A; Pedrocchi-Fantoni G; Pizzi D; Servi S Addition of Activated Grignard Reagents to 2-Phenyloxazolines. J. Org. Chem 1990, 55, 6216–6219. [Google Scholar]
- (234).Fronza G; Fuganti C; Pedrocchi-Fantoni G Synthesis of the Four Configurational Isomers of a Protected Form of N-Trifluoroacetyl-4-C-methyl-2,4,6-trideoxy-4-amino-l-hexose from l-Threonin. J. Carbohydr. Chem 1989, 8, 85–101. [Google Scholar]
- (235).Wang X; Yang T; Cheng X; Shen Q Enantioselective Electrophilic Trifluoromethylthiolation of β-Ketoesters: A Case of Reactivity and Selectivity Bias for Organocatalysis. Angew. Chem. Int. Ed 2013, 52, 12860–12864. [DOI] [PubMed] [Google Scholar]
- (236).Reetz MT; Hüllmann M Non-chelation-controlled Nucleophilic Addition to Chiral α-Siloxyketones. J. Chem. Soc., Chem. Commun 1986, 1600–1602. [Google Scholar]
- (237).Sulake RS; Chen C Total Synthesis of (+)-Antroquinonol and (+)-Antroquinonol D. Org. Lett 2015, 17, 1138–1141. [DOI] [PubMed] [Google Scholar]
- (238).Kita M; Oka H; Usui A; Ishitsuka T; Mogi Y; Watanabe H; Tsunoda M; Kigoshi H Total Synthesis of Mycalolides A and B through Olefin Metathesis. Angew. Chem. Int. Ed 2015, 54, 14174–14178. [DOI] [PubMed] [Google Scholar]
- (239).Corey EJ; Helal CJ Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew. Chem. Int. Ed 1998, 37, 1986–2012. [DOI] [PubMed] [Google Scholar]
- (240).Liu H-M; Chang C-Y; Lai Y-C; Yang M-D; Chang C-Y An Efficient Synthesis of the C27–C45 Fragment of Lagunamide A, a Cyclodepsipeptide with Potent Cytotoxic and Antimalarial Properties. Tetrahedron: Asymmetry 2014, 25, 187–192. [Google Scholar]
- (241).Kikuchi H; Horoiwa S; Kasahara R; Hariguchi N; Matsumoto M; Oshima Y Synthesis of Febrifugine Derivatives and Development of an Effective and Safe Tetrahydroquinazoline-Type Antimalarial. Eur. J. Med. Chem 2014, 76, 10–19. [DOI] [PubMed] [Google Scholar]
- (242).Reetz MT; Drewes MW; Schmitz A Stereoselective Synthesis of β-Amino Alcohols from Optically Active α-Amino Acids. Angew. Chem. Int. Ed. Engl 1987, 26, 1141–1143. [Google Scholar]
- (243).Reetz MT New Approaches to the Use of Amino Acids as Chiral Building Blocks in Organic Synthesis. Angew. Chem. Int. Ed. Engl 1991, 30, 1531–1546. [Google Scholar]
- (244).Mayr H; Ofial AR The Reactivity–Selectivity Principle: An Imperishable Myth in Organic Chemistry. Angew. Chem. Int. Ed 2006, 45, 1844–1854. [DOI] [PubMed] [Google Scholar]
- (245).Blaskovich MA; Evindar G; Rose NGW; Wilkinson S; Luo Y; Lajoie GA Stereoselective Synthesis of Threo and Erythro β-Hydroxy and β-Disubstituted-β-Hydroxy α-Amino Acids. J. Org. Chem 1998, 63, 3631–3646. [Google Scholar]
- (246).Volonterio A; Bravo P; Corradi E; Fronza G; Meille SV; Vergani B; Zanda M Unusual Nonchelation Controlled Allylation of a N-Monoprotected α-Amino Aldehyde: Stereoselective Entry to Nonracemic Trifluoromethyl Dipeptide Isosteres. J. Fluorine Chem 2001, 108, 245–252. [Google Scholar]
- (247).Volonterio A; Bravo P; Capelli S; Meille SV; Zanda M N-Cbz-Trifluoropyruvaldehyde N,S-Ketal: Absolute Stereochemistry and Addition of Grignard Reagents. Highly Stereoselective Entry to Trifluoro Analogues of Ephedra Alkaloids. Tetrahedron Lett. 1997, 38, 1847–1850. [Google Scholar]
- (248).Basso EA; Kaiser C; Rittner R; Lambert JB Axial/Equatorial Proportions for 2-Substituted Cyclohexanones. J. Org. Chem 1993, 58, 7865–7869. [Google Scholar]
- (249).Volonterio A; Vergani B; Crucianelli M; Zanda M; Bravo P (R)-Trifluoro- and Difluoropyruvaldehyde N,S-Ketals: Chiral Synthetic Equivalents of β-Trifluoro and β-Difluoro α-Amino Aldehydes. J. Org. Chem 1998, 63, 7236–7243. [DOI] [PubMed] [Google Scholar]
- (250).Soucy RL; Kozhinov D; Behar V Remarkable Diastereoselectivity in the Addition of Allylic and Unsaturated Diorganozinc Reagents to β-(N,N-Dialkylamino)-aldehydes. J. Org. Chem 2002, 67, 1947–1952. [DOI] [PubMed] [Google Scholar]
- (251).Christoffers J; Scharl H; Frey W; Baro A Transformation of an Optically Active Decahydro-6-isoquinolone Scaffold: Perfect Felkin–Anh Diastereoselectivity. Org. Lett 2004, 6, 1171–1173. [DOI] [PubMed] [Google Scholar]
- (252).Mohapatra DK; Durugkar KA Studies Towards the Total Synthesis of Halicholactone and Neohalicholactone: A Stereoselctive (sic) Synthesis of C1-C13 Fragment. Arkivoc 2004, (i), 146–155. [Google Scholar]
- (253).Ren H; Xie W; Xiong B; Shen J; Chen G; Chen Y-L Stereoselective Addition of Grignard Reagents to (2-Methyl-5-tert-butyl)phenyl 1-Thio-β-d-Ribopentodialdo-1,4-Furanoside Derivative. Tetrahedron 2017, 73, 2290–2304. [Google Scholar]
- (254).Kumar NS; Pinto BM Synthesis and Conformational Analysis of Bicyclic Sulfonium Salts. Structures Related to the Glycosidase Inhibitor Australine. J. Org. Chem 2006, 71, 2935–2943. [DOI] [PubMed] [Google Scholar]
- (255).Kitamura Y; Koshino H; Nakamura T; Tsuchida A; Nitoda T; Kanzaki H; Matsuoka K; Takahashi S Total Synthesis of the Proposed Structure for Pochonicine and Determination of Its Absolute Configuration. Tetrahedron Lett. 2013, 54, 1456–1459. [Google Scholar]
- (256).Arndt H-D; Polborn K; Koert U Stereoselective Synthesis of a Terpyrrolidine Unit, a Potential Building Block for Anion Recognition. Tetrahedron Lett. 1997, 38, 3879–3882. [Google Scholar]
- (257).Bürgi HB; Dunitz JD; Shefter E Geometrical Reaction Coordinates. II. Nucleophilic Addition to a Carbonyl Group. J. Am. Chem. Soc 1973, 95, 5065–5067. [Google Scholar]
- (258).Chaudhuri S; Parida A; Ghosh S; Bisai A Highly Stereoselective Syntheses of Proline-Derived Vicinal Amino Alcohols through Grignard Addition onto N-Tosylprolinal. Synlett 2015, 27, 215–220. [Google Scholar]
- (259).Basu S; Kandiyal PS; Ampapathi RS; Chakraborty TK Ti(III)-Mediated Radical Cyclization of Epoxy-β-Aminoacrylate in the Synthesis of the Substituted Pyrrolidine Core of Necine Bases: Synthesis of 2-epi-Rosmarinecine. RSC Adv. 2013, 3, 13630–13634. [Google Scholar]
- (260).Cheng B; Li Y-X; Jia Y-M; Yu C-Y Concise Synthesis of 1-epi-Castanospermine. Chin. Chem. Lett 2017, 28, 1688–1692. [Google Scholar]
- (261).Makino T; Shibata K; Rohrer DC; Osawa Y Steroid Conformations in Solid and Solution: Stereoselectivity of Grignard Addition to 20-Keto Steroids. J. Org. Chem 1978, 43, 276–280. [Google Scholar]
- (262).Lu Y; Chen J; Janjetovic Z; Michaels P; Tang EKY; Wang J; Tuckey RC; Slominski AT; Li W; Miller DD Design, Synthesis, and Biological Action of 20R-Hydroxyvitamin D3. J. Med. Chem 2012, 55, 3573–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Reetz MT; Steinbach R; Westermann J; Peter R; Wenderoth B Stereoselective Addition of Organotitanium Reagents to Carbonyl Compounds. Chem. Ber 1985, 118, 1441–1454. [Google Scholar]
- (264).Vitellozzi L; McAllister GD; Genski T; Taylor RJK. Organometallic Routes to Novel Steroids Containing Heterocyclic C-17 Side-Chains. Synthesis 2015, 48, 48–56. [Google Scholar]
- (265).Lin Z; Marepally SR; Ma D; Myers LK; Postlethwaite AE; Tuckey RC; Cheng CY; Kim T-K; Yue J; Slominski AT et al. Chemical Synthesis and Biological Activities of 20S,24S/R-Dihydroxyvitamin D3 Epimers and Their 1a-Hydroxyl Derivatives. J. Med. Chem 2015, 58, 7881–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Hanessian S; Yang H; Schaum R Hydrogen-Bonding as a Stereocontrolling Element in Free-Radical C-Allylation Reactions: Vicinal, Proximal, and Remote Asymmetric Induction in the Amino Acid Series. J. Am. Chem. Soc 1996, 118, 2507–2508. [Google Scholar]
- (267).Sieburth SM; McGee KF Jr.; Al-Tel TH Fusicoccin Ring System by [4 + 4] Cycloaddition. Control of Diastereoselectivity through Hydrogen Bonding. J. Am. Chem. Soc 1998, 120, 587–588. [Google Scholar]
- (268).Tripathy R; Carroll PJ; Thornton ER Diene–Dienophile Hydrogen-Bonding Control of Diels–Alder Reactions with Dienes Bearing a Remote Stereogenic Center. Amplification of the Receptor Nature of Dienes by Conformational Tuning Leading to High Diastereofacial Discrimination. J. Am. Chem. Soc 1991, 113, 7630–7640. [Google Scholar]
- (269).Still WC; Galynker I Chemical Consequences of Conformation in Macrocyclic Compounds. An Effective Approach to Remote Asymmetric Induction. Tetrahedron 1981, 37, 3981–3996. [Google Scholar]
- (270).Liu Q; Deng Y; Smith AB III. Total Synthesis of (−)-Nahuoic Acid Ci (B,,). J. Am. Chem. Soc 2017, 139, 13668–13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Dwulet NC; Zolfaghari TA; Brown ML; Cannon JS Diastereoselective Synthesis of Unnatural Amino Acids by Alkylation of α-tert-Butanesulfinamide Auxiliary-Bound Enolates. J. Org. Chem 2018, 83, 11510–11518. [DOI] [PubMed] [Google Scholar]
- (272).Stork G; Sher PM; Chen H-L Radical Cyclization-Trapping in the Synthesis of Natural Products. A Simple, Stereocontrolled Route to Prostaglandin F2α. J. Am. Chem. Soc 1986, 108, 6384–6385. [Google Scholar]
- (273).Llona-Minguez S; Mackay SP Stereoselective Synthesis of Carbocyclic Analogues of the Nucleoside Q Precursor (PreQ0). Beilstein J. Org. Chem 2014, 10, 1333–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Maruoka K; Itoh T; Sakurai M; Nonoshita K; Yamamoto H Amphiphilic Reactions by Means of Exceptionally Bulky Organoaluminum Reagents. Rational Approach for Obtaining Unusual Equatorial, Anti-Cram, and 1,4 Selectivity in Carbonyl Alkylation. J. Am. Chem. Soc 1988, 110, 3588–3597. [Google Scholar]
- (275).Reetz MT; Haning H Ligand Effects in Selective Addition Reactions of Organoindium Reagents with Carbonyl Compounds. J. Organomet. Chem 1997, 541, 117–120. [Google Scholar]
- (276).Gaschler MM; Andia AA; Liu H; Csuka JM; Hurlocker B; Vaiana CA;Heindel DW; Zuckerman DS; Bos PH; Reznik E et al. FINO2 Initiates Ferroptosis through GPX4 Inactivation and Iron Oxidation. Nat. Chem. Biol 2018, 14, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Mladenova M; Blagoev B; Gaudemar M; Gaudemar-Bardone F; Lallemand JY Structure Du Reactif D’ivanov Et De Quelques Organometalliques Issus Des Derives Phenylacetiques—II: Stereochimie De La Condensation Avec La t-Butyl-4 Cyclohexanone. Tetrahedron 1981, 37, 2157–2163. [Google Scholar]
- (278).Sjöholm RE Diastereoselectivity in the Addition of Crotylmagnesium Bromide to Unsymmetrical Ketones. Acta Chem. Scand 1990, 44, 82–89. [Google Scholar]
- (279).Zair T; Santelli-Rouvier C; Santelli M Stereochemistry of Addition of Allylic Grignard-Reagents to α,β-Ethylenic Ketones. J. Org. Chem 1993, 58, 2686–2693. [Google Scholar]
- (280).Felkin H; Frajerman C Competitive Rate Studies for Some Reactions Involving the Allyl, Butenyl, αγ-Dimethylallyl, and Propyl Grignard Reagents. The Mechanism of the Reactions of Allylic Grignard Reagents with Electrophilic Substrates. Tetrahedron Lett. 1970, 11, 1045–1048. [Google Scholar]
- (281).Chárest M; Felkin H The Influence of Steric and Torsional Strain on the Steric Course of Additions to Cyclohexanones. The Reaction Between 4-t-Butylcyclohexanone and t-Butylallyl Magnesium Bromide. Tetrahedron Lett. 1971, 12, 383–386. [Google Scholar]
- (282).Vasconcelos RS; Silva LF Jr.; Giannis A Synthesis of Tetrahydrofurans by Cyclization of Homoallylic Alcohols with Iodine/Iodine(III). J. Org. Chem 2011, 76, 1499–1502. [DOI] [PubMed] [Google Scholar]
- (283).LaMarche MJ; Leeds JA; Amaral A; Brewer JT; Bushell SM; Deng G; Dewhurst JM; Ding J; Dzink-Fox J; Gamber G et al. Discovery of LFF571: An Investigational Agent for Clostridium difficile Infection. J. Med. Chem 2012, 55, 2376–2387. [DOI] [PubMed] [Google Scholar]
- (284).Lednicer D; VonVoigtlander PF; Emmert DE 4-Amino-4-arylcyclohexanones and Their Derivatives: A Novel Class of Analgesics. 2. Modification of the Carbonyl Function. J. Med. Chem 1981, 24, 404–408. [DOI] [PubMed] [Google Scholar]
- (285).Eliel EL; Manoharan M Conformational Analysis. 40. Conformation of 1-Methyl-1-phenylcyclohexane and the Conformational Energies of the Phenyl and Vinyl Groups. J. Org. Chem 1981,46, 1959–1962. [Google Scholar]
- (286).Zhao L; Burnell DJ The Stereochemistry of 1,2-Additions of Allylmagnesium, Allylindium, and Allylbismuth to Cyclohexenones. Tetrahedron Lett. 2006, 47, 3291–3294. [Google Scholar]
- (287).Masamune T; Matsue H; Fujii M The Reduction of Stereoisomeric 6-tert-Butyl-3-methoxy-3-methylcyclohexenes with Lithium in Ethylamine. Bull. Chem. Soc. Jpn 1972, 45, 1812–1816. [Google Scholar]
- (288).Devlin FJ; Stephens PJ Conformational Analysis Using ab Initio Vibrational Spectroscopy: 3-Methylcyclohexanone. J. Am. Chem. Soc 1999, 121, 7413–7414. [Google Scholar]
- (289).Landor SR; O’Connor PW; Tatchell AR; Blair I Asymmetric Synthesis. Part VI. Reactions of 3,3,5-Trimethylcyclohexanone with Grignard Reagents and Configurational Interrelationships of 1-Alkyl(alkenyl and alkynyl)-3,3,5-trimethylcyclohexanols and Compounds Derived from 1-Hydroxy-3,3,5-trimethylcyclohexanecarbonitriles. J. Chem. Soc., Perkin Trans. 1 1973, 473–478. [Google Scholar]
- (290).Adam J; Klein R; Grabley S; Hammann P Chiral Building Blocks from Streptomyces-2. Stereoselective Transformation of Streptenol A into 3-Methyl-δ-Lactones. Tetrahedron 1995, 51, 8247–8258. [Google Scholar]
- (291).Eliel EL; Hargrave KD; Pietrusiewicz KM; Manoharan M Conformational Analysis. 42. Monosubstituted Tetrahydropyrans. J. Am. Chem. Soc 1982, 104, 3635–3643. [Google Scholar]
- (292).Juaristi E; Cuevas G Recent Studies of the Anomeric Effect. Tetrahedron 1992, 48, 5019–5087. [Google Scholar]
- (293).Gung BW; Wolf MA; Mareska DA; Karipides A Origin of the Conformational Mobility for 4-Tetrahydropyranone: An ab Initio MO Study. J. Org. Chem 1994, 59, 4899–4903. [Google Scholar]
- (294).Gorthey LA; Vairamani M; Djerassi C Optical Rotatory Dispersion Studies. 138. Synthesis and Conformational Analysis of β-Heteroatom-Substituted Cyclohexanones. J. Org. Chem 1985, 50, 4173–4182. [Google Scholar]
- (295).Ohno K; Yoshida H; Watanabe H; Fujita T; Matsuura H Conformational Study of 1-Butanol by the Combined Use of Vibrational Spectroscopy and ab Initio Molecular Orbital Calculations. J. Phys. Chem 1994, 98, 6924–6930. [Google Scholar]
- (296).El Idrissi M; Santelli M Stereochemistry of Addition of Allyl Grignard Reagents to (R)-(+)-Pulegone and Other α,β-Ethylenic Ketones. J. Org. Chem 1988, 53, 1010–1016. [Google Scholar]
- (297).Benhallam R; Zair T; Jarid A; Ibrahim-Ouali M Theoretical Study of the Stereochemistry of Crotylmagnesium Chloride’s Addition on a Series of Cyclic and Acylic Enones. J. Mol. Struct. (Theochem) 2003, 626, 1–17. [Google Scholar]
- (298).Ghosh AK; Krishnan K; Walters DE; Cho W; Cho H; Koo Y; Trevino J; Holland L; Buthod J Structure Based Design: Novel Spirocyclic Ethers as Nonpeptidal P2-Ligands for HIV Protease Inhibitors. Bioorg. Med. Chem. Lett 1998, 8, 979–982. [DOI] [PubMed] [Google Scholar]
- (299).Alcaraz M-L; Atkinson S; Cornwall P; Foster AC; Gill DM; Humphries LA; Keegan PS; Kemp R; Merifield E; Nixon RA et al. Efficient Syntheses of AZD4407 via Thioether Formation by Nucleophilic Attack of Organometallic Species on Sulphur. Org. Proc. Res. Dev 2005, 9, 555–569. [Google Scholar]
- (300).Ley SV; Humphries AC; Eick H; Downham R; Ross AR; Boyce RJ; Pavey JBJ; Pietruszka J Total Synthesis of the Protein Phosphatase Inhibitor Okadaic Acid. J. Chem. Soc., Perkin Trans. 1 1998, 3907–3912. [Google Scholar]
- (301).Peh G; Floreancig PE Convergent One-Pot Oxidative [n + 1] Approaches to Spiroacetal Synthesis. Org. Lett 2015, 17, 3750–3753. [DOI] [PubMed] [Google Scholar]
- (302).Trost BM; Gunzner JL; Dirat O; Rhee YH Callipeltoside A: Total Synthesis, Assignment of the Absolute and Relative Configuration, and Evaluation of Synthetic Analogues. J. Am. Chem. Soc 2002, 124, 10396–10415. [DOI] [PubMed] [Google Scholar]
- (303).Battioni J-P; Chodkiewicz W Stéréochimie de l’Addition d’Oganométalliques sur la Éthyl-2 Cyclohexanones. C. R. Chim 1970, 271. [Google Scholar]
- (304).Barentsen HM; Talman EG; Piet DP; Cornelisse J Intramolecular Meta Photocycloaddition of Conformationally Restrained 5-Phenylpent-1-enes. Part I: Bichromophoric Cyclohexane Derivatives. Tetrahedron 1995, 51, 7469–7495. [Google Scholar]
- (305).Negri JT; Paquette LA Electrostatic Modulation of Hydroxyl Group Ionization in Acidic Media. Evidence for the Competitive Operation of Intramolecular Sn2 Reactions. J. Am. Chem. Soc 1992, 114, 8835–8841. [Google Scholar]
- (306).Maruoka K; Oishi M; Shiohara K; Yamamoto H Methylaluminum Bis(4-substituted-2,6-di-tert-butylphenoxide) as an Efficient Nonchelating Lewis Acid: Application to Asymmetric Diels-Alder Reaction and Diastereoselective Alkylation to Alkoxy Carbonyl Substrates. Tetrahedron 1994, 50, 8983–8996. [Google Scholar]
- (307).Freeman-Cook KD; Halcomb RL Synthesis of Bicyclic Ketals Related to Zaragozic Acid by a Novel Photoannulation. Tetrahedron Lett. 1996, 37, 4883–4886. [Google Scholar]
- (308).Di Bussolo V; Catelani G; Mastrorilli E; Di Bugno C; Giorgi R Convenient Diastereospecific Synthesis of a Rociverine Precursor and Its Resolution by Lipase-Catalyzed Transesterification. Tetrahedron: Asymmetry 1996, 7, 3585–3592. [Google Scholar]
- (309).Kaluza NM; Schollmeyer D; Nubbemeyer U Total Synthesis of (−)-C/D-cis-Dehydro-3-O-methyl-estradiols. Eur. J. Org. Chem 2016, 357–366. [Google Scholar]
- (310).Aldegunde MJ; Castedo L; Granja JR Toward an Efficient Synthesis of Taxane Analogs by Dienyne Ring-Closing Metathesis. Org. Lett 2008, 10, 3789–3792. [DOI] [PubMed] [Google Scholar]
- (311).Taleb A; Lahrech M; Hacini S; Thibonnet J; Parrain J-L Grubbs Catalyst Promoted Oxacycloisomerization: An Easy Route to Bicyclic Dihydrofurans and Tricyclic Acetals. Synlett 2009, 1597–1600. [Google Scholar]
- (312).Reddy C; Babu SA; Aslam NA; Rajkumar V Construction of Functionalized Carbocycles Having Contiguous Tertiary Carbinol and All-Carbon Stereogenic Centers. Eur. J. Org. Chem 2013, 2362–2380. [Google Scholar]
- (313).Maiti S; Drew MGB; Mukhopadhyay R; Achari B; Banerjee AK Convenient Formation of Six- to Nine-Membered Carbocyclic Rings by 2-Pyridyl Radical Cyclization: A Generalized Synthesis of Pyridine-Fused Linear Tricyclic Systems. Synthesis 2005, 3067–3078. [Google Scholar]
- (314).Ghosh K; Ghosh AK; Ghatak UR Highly Regioselective 8-endo-Aryl Radical Cyclisation: A New Synthetic Route to Decahydrodibenzo-[a,d]- and -[a,e]-Cyclooctenols. J. Chem. Soc., Chem. Commun 1994, 629. [Google Scholar]
- (315).Ghosh K; Ghatak UR A Novel Route to Functionalized Linearly Benzannulated Medium Ring Carbocyclics through Regioselective Aryl Radical Cyclization. J. Indian Inst. Sci 2001, 81, 239–264. [Google Scholar]
- (316).Imamoto T; Takiyama N; Nakamura K; Hatajima T; Kamiya Y Reactions of Carbonyl Compounds with Grignard Reagents in the Presence of Cerium Chloride. J. Am. Chem. Soc 1989, 111, 4392–4398. [Google Scholar]
- (317).Fuganti C; Joulain D; Maggioni F; Malpezzi L; Serra S; Vecchione A 3-Alkyl-p-menthan-3-ol Derivatives: Synthesis and Evaluation of Their Physiological Cooling Activity. Tetrahedron: Asymmetry 2008, 19, 2425–2437. [Google Scholar]
- (318).Nokami J; Ohga M; Nakamoto H; Matsubara T; Hussain I; Kataoka K The First and Highly Enantioselective Crotylation of Aldehydes via an Allyl-Transfer Reaction from a Chiral Crotyl-Donor. J. Am. Chem. Soc 2001, 123, 9168–9169. [DOI] [PubMed] [Google Scholar]
- (319).Schmidt B; Staude L Unexpected Transfer Hydrogenation of C–C-Double Bonds During Tandem-RCM-Isomerization Reactions. J. Organomet. Chem 2006, 691, 5218–5221. [Google Scholar]
- (320).Schmitz D; Shubert VA; Betz T; Schnell M Exploring the Conformational Landscape of Menthol, Menthone, and Isomenthone: A Microwave Study. Front. Chem 2015, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Gardiner JM; Crewe PD; Smith GE; Veal KT; Pritchard RG; Warren JE Synthesis, Stereostructure, and Conformations of Novel Bi- and Trifunctional (+)-Isomenthone Derivatives. Org. Lett 2003, 5, 467–470. [DOI] [PubMed] [Google Scholar]
- (322).Paquette LA; Lobben PC π-Facial Diastereoselection in the 1,2-Addition of Allylmetal Reagents to 2-Methoxycyclohexanone and Tetrahydrofuranspiro-(2-cyclohexanone). J. Am. Chem. Soc 1996, 118, 1917–1930. [Google Scholar]
- (323).Fang J-M; Sun S-F; Rei M-H Stereochemistry of Nucleophilic Reductions of 2-Methyl-4-t-butylcyclohexanones. Further Support for the Linear Combination of SSC and PSC Stereochemical Models. J. Chem. Soc., Perkin Trans. 2 1989, 747–750. [Google Scholar]
- (324).Blunt JW; Hartshorn MP; Soong LT; Munro MHG Reactions of Propargyl Alcohols. V. Lithium Aluminium Hydride Reduction of Some C1-Epimeric 4-t-Butyl-1-prop-1′-ynylcyclohexanols. Aust. J. Chem 1982, 35, 2519–2528. [Google Scholar]
- (325).Paquette LA; Stepanian M; Mallavadhani UV; Cutarelli TD; Lowinger TB; Klemeyer HJ Definition of Several Control Elements Relevant to the Stereodefined Serial Elaboration of Belted Poly(spirotetrahydrofurans) Fitted with a Cyclohexane Core. J. Org. Chem 1996, 61, 7492–7507. [DOI] [PubMed] [Google Scholar]
- (326).Paquette LA; Tae J; Hickey ER; Trego WE; Rogers RD Preorganized Ligand Arrays Based on Spirotetrahydrofuranyl Motifs. Synthesis of the Stereoisomeric 1,8,14-Trioxatrispiro[4.1.4.1.4.1]Octadecanes and the Contrasting Conformational Features and Ionic Binding Capacities of These Belted lonophores. J. Org. Chem 2000, 65, 9160–9171. [DOI] [PubMed] [Google Scholar]
- (327).Naasz R; Arnold LA; Minnaard AJ; Feringa BL Enantioselective Synthesis of Bicyclic Compounds Catalytic 1,4-Addition-Ring Closing Metathesis. Chem. Commun 2001,735–736. [Google Scholar]
- (328).Pfeffer PE; Osman SF Nuclear Magnetic Resonance Studies of cis- and trans-2,3-Dimethylcycloalkanones. J. Org. Chem 1972, 37, 2425–2428. [Google Scholar]
- (329).Ople RS; Handore KL; Kamat NS; Reddy DS A Total Synthesis of (−)-Nardoaristolone B. Eur. J. Org. Chem 2016, 3804–3808. [Google Scholar]
- (330).Qian X; Sujino K; Otter A; Palcic MM; Hindsgaul O Chemoenzymatic Synthesis of α-(1→3)-Gal(NAc)-Terminating Glycosides of Complex Tertiary Sugar Alcohols. J. Am. Chem. Soc 1999, 121, 12063–12072. [Google Scholar]
- (331).Kobayashi T; Yamanoue K; Abe H; Ito H Diastereoselective Total Synthesis of (±)-Toxicodenane A. Eur. J. Org. Chem 2017, 6693–6699. [Google Scholar]
- (332).Paquette LA; Lowinger TB; Bolin DG; Branan BM trans,Anti,trans-Tetra(Spirotetrahydrofuranyl)Cyclohexane-1,2-Dione. Stereocontrolled Synthesis and Definition of Its Susceptibility to Photoisomerization. J. Org. Chem 1996, 61, 7486–7491. [DOI] [PubMed] [Google Scholar]
- (333).Koreeda M; You Z High Stereoselectivity in the 1,2-Nucleophilic Additions to a Hindered 2-Alkylidenecyclohexanone: An Example of Predominant Axial Attack by Sterically Demanding Nucleophiles. J. Org. Chem 1989, 54, 5195–5198. [Google Scholar]
- (334).Amoah E; Dieter RK Regioselective 1,4-Conjugate Addition of Grignard Reagents to α,β–γ,δ-Dienones and α,β–γ,δ-Dienyl Thiol Esters. J. Org. Chem 2017, 82, 2870–2888. [DOI] [PubMed] [Google Scholar]
- (335).Chen B; Lu Z; Chai G; Fu C; Ma S An Efficient Double 1,2-Addition Reaction of 2,3-Allenoates with Allyl Magnesium Chloride. J. Org. Chem 2008, 73, 9486–9489. [DOI] [PubMed] [Google Scholar]
- (336).Gilman H; Eisch J 1,2-Addition of Allylmagnesium Bromide to N-Diphenylmethyleneaniline and Structurally Related Systems. J. Am. Chem. Soc 1957, 79, 2150–2153. [Google Scholar]
- (337).Kito M; Sakai T; Shirahama H; Miyashita M; Matsuda F Construction of 8-Membered Ring Skeleton of Vinigrol via SmI2-Promoted Barbier Coupling. Synlett 1997, 219–220. [Google Scholar]
- (338).Esteban AL; Galache MP; Diez E; Altona C; Smits GF The Cyclopentanone Ring Puckering. J. Mol. Struct 1986, 142, 379–382. [Google Scholar]
- (339).Battioni J-P; Chodkiewicz W; Cadiot P Stéréochimie Des Méthyl-2 Cyclopentanols Tertiares. Étude En Résonance Magnétique Nucléaire D’oxydes De Phosphine Alleniques. C. R. Chim 1967, 264, 991–994. [Google Scholar]
- (340).Battioni J-P; Capmau M-L; Chodkiewicz W Stéréochimie de l’Addition d’Organométalliques sur la Methyl-2 Cyclopentanone. Bull. Soc. Chim. Fr 1969, 976–981. [Google Scholar]
- (341).Courtois G; Miginiac L The Reactivity of Allylic Organometallic Compounds of Lithium, Sodium, Magnesium, Zinc, Cadmium and Aluminium: Recent Advances. J. Organomet. Chem 1974, 69, 1–44. [Google Scholar]
- (342).Guillerm-Dron D; Capmau M-L; Chodkiewicz W Stéréochimie de l’Addition d’Organométalliques sur la Méthoxy-2 Cyclopentanone. C. R. Chim 1971, 273, 759–762. [Google Scholar]
- (343).Nishida A; Miyoshi I; Ogasawara Y; Nagumo S; Kawahara N; Nishida M; Takayanagi H The Novel Skeletal Rearrangement of Cyclopentanones into Hydroazulenones via a Radical Process and Its Application to the Formal Synthesis of Damsinic Acid. Tetrahedron 2000, 56, 9241–9257. [Google Scholar]
- (344).Hirst GC; Johnson TO Jr.; Overman LE First Total Synthesis of Lycopodium Alkaloids of the Magellanane Group. Enantioselective Total Syntheses of (−)-Magellanine and (+)-Magellaninone. J. Am. Chem. Soc 1993, 115, 2992–2993. [Google Scholar]
- (345).Tong G; Liu Z; Li P Stereocontrolled Construction of the Tricyclic Framework of Tiglianes and Daphnanes by an Oxidative Dearomatization Approach. Org. Lett 2014, 16, 2288–2291. [DOI] [PubMed] [Google Scholar]
- (346).Lentsch LM; Wiemer DF Addition of Organometallic Nucleophiles to β-Keto Phosphonates. J. Org. Chem 1999, 64, 5205–5212. [DOI] [PubMed] [Google Scholar]
- (347).Donohoe TJ; O’Riordan TJ; Peifer M; Jones CR; Miles TJ Asymmetric Synthesis of the Fully Elaborated Pyrrolidinone Core of Oxazolomycin A. Org. Lett 2012, 14, 5460–5463. [DOI] [PubMed] [Google Scholar]
- (348).Deng Q-H; Wadepohl H; Gade LH Highly Enantioselective Copper-Catalyzed Electrophilic Trifluoromethylation of β-Ketoesters. J. Am. Chem. Soc 2012, 134, 10769–10772. [DOI] [PubMed] [Google Scholar]
- (349).Battioni J-P; Chodkiewicz W Stéréochimie de l’Addition d’Oganométalliques sur les Éthyl-2, Méthyl-3 et Tertiobutyl-3 Cyclopentanones. Bull. Soc. Chim. Fr 1971, 1824–1833. [Google Scholar]
- (350).Laemmle J; Ashby EC; Roling PV Stereoselective Organometallic Alkylation Reactions. II. Organomagnesium and Organoaluminum Addition to Ketones Having Varied Steric Requirements. New Concept of Stereochemical Control. J. Org. Chem 1973, 38, 2526–2534. [Google Scholar]
- (351).Gomtsyan A; Schmidt RG; Bayburt EK; Gfesser GA; Voight EA; Daanen JF; Schmidt DL; Cowart MD; Liu H; Altenbach RJ et al. Synthesis and Pharmacology of (Pyridin-2-yl)methanol Derivatives as Novel and Selective Transient Receptor Potential Vanilloid 3 Antagonists. J. Med. Chem 2016, 59, 4926–4947. [DOI] [PubMed] [Google Scholar]
- (352).Gu Y-Q; Zhang P-P; Fu J-K; Liu S; Lan Y; Gong J-X; Yang Z Regio- and Stereoselective Syntheses of 7-Oxabicyclo[2.2.1]Heptanes via a Gold(I)-Catalyzed Cycloisomerization of Alkynediols: Asymmetric Total Synthesis of Farnesiferol C. Adv. Synth. Catal 2016, 358, 1392–1397. [Google Scholar]
- (353).Vickerman KL; Stanley LM Catalytic, Enantioselective Synthesis of Polycyclic Nitrogen, Oxygen, and Sulfur Heterocycles via Rh-Catalyzed Alkene Hydroacylation. Org. Lett 2017, 19, 5054–5057. [DOI] [PubMed] [Google Scholar]
- (354).Srikrishna A; Nagaraju G; Ravi G An Enantiospecific Approach to the 5–8-5 Tricyclic System of Basmanes. Synlett 2010, 3015–3018. [Google Scholar]
- (355).Gund M; Déry M; Amzallag V; Spino C Dialkoxycarbenes in (4 + 1) Cycloadditions: Application to the Synthesis of Carotol. Org. Lett 2016, 18, 4280–4283. [DOI] [PubMed] [Google Scholar]
- (356).Babu BR; Keinicke L; Petersen M; Nielsen C; Wengel J 2′-Spiro Ribo- and Arabinonucleosides: Synthesis, Molecular Modelling and Incorporation into Oligodeoxynucleotides. Org. Biomol. Chem 2003, 1, 3514–3526. [DOI] [PubMed] [Google Scholar]
- (357).Donohoe TJ; Chiu JYK; Thomas RE Synthesis of the Pyrrolidinone Core of KSM-2690 B. Org. Lett 2007, 9, 421–424. [DOI] [PubMed] [Google Scholar]
- (358).Xia J; Nie Y; Yang G; Liu Y; Gridnev ID; Zhang W Ir-Catalyzed Asymmetric Hydrogenation of α-Alkylidene β-Lactams and Cyclobutanones. Chin. J. Chem 2018, 36, 612–618. [Google Scholar]
- (359).Wu Z; Leboeuf D; Retailleau P; Gandon V; Marinetti A; Voituriez A Enantioselective Gold(I)-Catalyzed Rearrangement of Cyclopropyl-Substituted 1,6-Enynes into 2-Oxocyclobutyl-Cyclopentanes. Chem. Commun 2017, 53, 7026–7029. [DOI] [PubMed] [Google Scholar]
- (360).Trost BM; Jungheim LN 1-(Arylthio)cyclopropanecarboxaldehydes. Conjunctive Reagents for Secoalkylation. J. Am. Chem. Soc 1980, 102, 7910–7925. [Google Scholar]
- (361).Bernard AM; Frongia A; Secci F; Piras PP 2,2-Dimethyl Cyclopentanones by Acid Catalyzed Ring Expansion of Isopropenylcyclobutanols. A Short Synthesis of (±)-α-Cuparenone and (±)-Herbertene. Chem. Commun 2005, 3853–3855. [DOI] [PubMed] [Google Scholar]
- (362).Inoue M; Sato T; Hirama M Total Synthesis of Merrilactone A. J. Am. Chem. Soc 2003, 125, 10772–10773. [DOI] [PubMed] [Google Scholar]
- (363).Smith DM; Woerpel KA Electrostatic Interactions in Cations and Their Importance in Biology and Chemistry. Org. Biomol. Chem 2006, 4, 1195–1201. [DOI] [PubMed] [Google Scholar]
- (364).Pottie IR; Crane SN; Gosse AL; Miller DO; Burnell DJ Geminal Acylation of α-Heterosubstituted Cyclohexanones and Their Ketals. Can. J. Chem 2010, 88, 1118–1124. [Google Scholar]
- (365).Crane SN; Burnell DJ The BF3·Et2O-Catalyzed Reaction of 1,2-Bis[(trimethylsilyl)oxy]cyclobutene and Analogues with Aromatic Ketones. J. Org. Chem 1998, 63, 1352–1355. [Google Scholar]
- (366).Miesch L; Welsch T; Miesch M Stereoselective Cross Aldol Condensation of Bicyclo[3.2.0]alkanones. Org. Biomol. Chem 2013, 11, 4025–4029. [DOI] [PubMed] [Google Scholar]
- (367).Inoue M; Lee N; Kasuya S; Sato T; Hirama M; Moriyama M; Fukuyama Y Total Synthesis and Bioactivity of an Unnatural Enantiomer of Merrilactone A: Development of an Enantioselective Desymmetrization Strategy. J. Org. Chem 2007, 72, 3065–3075. [DOI] [PubMed] [Google Scholar]
- (368).Ando K; Condroski KR; Houk KN; Wu Y-D; Ly SK; Overman LE Stereoselectivities of Nucleophilic Additions to Cycloheptanones. Experimental and Theoretical Studies and General Purpose Force Field for the Prediction of Nucleophilic Addition Stereoselectivities. J. Org. Chem 1998, 63, 3196–3203. [Google Scholar]
- (369).Chen W; Chen M; Hartwig JF Diastereo- and Enantioselective Iridium-Catalyzed Allylation of Cyclic Ketone Enolates: Synergetic Effect of Ligands and Barium Enolates. J. Am. Chem. Soc 2014, 136, 15825–15828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Liu H-J; Shia K-S; Shang X; Zhu B-Y Organocerium Compounds in Synthesis. Tetrahedron 1999, 55, 3803–3830. [Google Scholar]
- (371).Bhanot OS; Das TK; Gupta I; Suri HS; Dutta PC Synthetic Studies on Terpenoids. 5. Synthesis of γ- and δ-Lactones from β-(2,7-Dimethyl-1,2-dihydroxycycloheptyl)propionic Acid. J. Org. Chem 1977, 42, 1623–1627. [Google Scholar]
- (372).Gelis C; Levitre G; Merad J; Retailleau P; Neuville L; Masson G Highly Diastereo- and Enantioselective Synthesis of Cyclohepta[b]indoles by Chiral-Phosphoric-Acid-Catalyzed (4+3) Cycloaddition. Angew. Chem. Int. Ed 2018, 57, 12121–12125. [DOI] [PubMed] [Google Scholar]
- (373).Clark TB; Woerpel KA Formation and Reactivity of Silacyclopropenes Derived from Siloxyalkynes: Stereoselective Formation of 1,2,4-Triols. Org. Lett 2006, 8, 4109–4112. [DOI] [PubMed] [Google Scholar]
- (374).Hannaby M; Warren S Dehydration of Alcohols with an Adjacent Phenylthio (PhS) Group. Tetrahedron Lett 1985, 26, 3133–3136. [Google Scholar]
- (375).Avasthi K; Salomon RG A Copper(I)-Catalyzed Photobicyclization Route to exo-1,2-Polymethylene- and 7-Hydroxynorbornanes. Nonclassical 2-Bicyclo[3.2.0]heptyl and 7-Norbornyl Carbenium Ion Intermediates. J. Org. Chem 1986, 51, 2556–2562. [Google Scholar]
- (376).Moosavi SM; Beddoes RS; Watt CIF Base-Catalysed Ring Openings of 1,2-Diphenylcycloalkanols Having Five-, Six-, Seven- and Eight-Membered Rings. J. Chem. Soc., Perkin Trans. 2 1997, 1585–1596. [Google Scholar]
- (377).Gahman TC; Overman LE Stereoselective Synthesis of Carbocyclic Ring Systems by Pinacol-Terminated Prins Cyclizations. Tetrahedron 2002, 58, 6473–6483. [Google Scholar]
- (378).Mukaiyama T; Shiina I; Kimura K; Akiyama Y; Iwadere H Synthesis of AB Ring Model System of Taxol via Allylation of 8-Membered Ring Compound and Intramolecular Aldol Condensation. Chem. Lett 1995, 24, 229–230. [Google Scholar]
- (379).Rozada TC; Gauze GF; Rosa FA; Favaro DC; Rittner R; Pontes RM; Basso EA The Conformational Analysis of 2-Halocyclooctanones. Spectrochim. Acta A: Mol. Biomol. Spectrosc 2015, 137, 176–184. [DOI] [PubMed] [Google Scholar]
- (380).Eccleshare L; Lozada-Rodriguez L; Cooper P; Burroughs L; Ritchie J; Lewis W; Woodward S Diversification of ortho-Fused Cycloocta-2,5-dien-1-one Cores and Eight- to Six-Ring Conversion by σ Bond C–C Cleavage. Chem. Eur. J 2016, 22, 12542–12547. [DOI] [PubMed] [Google Scholar]
- (381).Eisch JJ; Yu K; Rheingold AL Steric Factors in the Single-Electron Transfer Carbolithiation and Transannular Reduction of 6,12-Diphenyldibenzo[b,f]-[1,5]-diazocine by Organolithium Reagents. Eur. J. Org. Chem 2014, 818–832. [Google Scholar]
- (382).Zhang Y; Lotesta SD; Emge TJ; Williams LJ Structure and Reactivity of a Chiral Cyclononadienone. Tetrahedron Lett. 2009, 50, 1882–1885. [Google Scholar]
- (383).Runcie KA; Taylor RJK A Short and Efficient Route to Novel Scyphostatin Analogues. Org. Lett 2001, 3, 3237–3239. [DOI] [PubMed] [Google Scholar]
- (384).Dechoux L; Agami C; Doris E; Mioskowski C Metalated Epoxides as Carbenoids. Competing C–H and C=C Insertion in α-Alkoxy Epoxide Systems. Tetrahedron 2003, 59, 9701–9706. [Google Scholar]
- (385).Snape TJ Application of the Semi-Pinacol Rearrangement Towards the Generation of Alkenyl-Substituted Quaternary Carbon Centres. Org. Biomol. Chem 2006, 4, 4144–4148. [DOI] [PubMed] [Google Scholar]
- (386).Takeda Y; Matsumoto T; Sato F Synthesis of Optically Active β,γ-Epoxy Alcohols and Secondary Allylic Alcohols via Diastereoselective Addition of α-(Trimethylsilyl)-α,β-Epoxy Aldehydes with Organometallic Compounds. J. Org. Chem 1986, 51, 4728–4731. [Google Scholar]
- (387).Howe GP; Wang S; Procter G Stereoselective Additions to α,β-Epoxy-Aldehydes; the Formation of “Non-Chelation Controlled” Products. Tetrahedron Lett. 1987, 28, 2629–2632. [Google Scholar]
- (388).Kuwata K; Suzuki M; Inami Y; Hanaya K; Sugai T; Shoji M Stereoselective Synthesis of Scyphostatin Hydrophilic Moiety. Tetrahedron Lett. 2014, 55, 2856–2858. [Google Scholar]
- (389).Wahl MH; Jandl C; Bach TA [2 + 2] Photocycloaddition-Fragmentation Approach toward the Carbon Skeleton of cis-Fused Lycorine-type Alkaloids. Org. Lett 2018, 20, 7674–7678. [DOI] [PubMed] [Google Scholar]
- (390).Catanoso G; Vecchi E To What Extent Does the Substituent Conformation Influence the Kinetics of Addition Reactions on 5X-Bicyclo[4.4.0]decan-2-ones? Tetrahedron 2003, 59, 5555–5561. [Google Scholar]
- (391).Corey EJ; Roberts BE Total Synthesis of Dysidiolide. J. Am. Chem. Soc 1997, 119, 12425–12431. [Google Scholar]
- (392).Cleator E; McCusker CF; Steltzer F; Ley SV Grignard Additions to 2-Uloses: Synthesis of Stereochemically Pure Tertiary Alcohols. Tetrahedron Lett. 2004, 45, 3077–3080. [Google Scholar]
- (393).Yadav JS; Goreti R; Pabbaraja S; Sridhar B Short Route to Platencin. Org. Lett 2013, 15, 3782–3785. [DOI] [PubMed] [Google Scholar]
- (394).Boyd E; Hallett MR; Jones RVH; Painter JE; Patel P; Quayle P; Waring AJ The Lithiation of 2-Chloroglucal Derivatives. Tetrahedron Lett. 2006, 47, 8337–8341. [Google Scholar]
- (395).Tsukanov SV; Comins DL Total Synthesis of Alkaloid 205B. J. Org. Chem 2014, 79, 9074–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Hagen G; Mayr H Kinetics of the Reactions of Allylsilanes, Allylgermanes, and Allylstannanes with Carbenium Ions. J. Am. Chem. Soc 1991, 113, 4954–4961. [Google Scholar]
- (397).Johnston CA; Wilkie RP; Krauss H; Neal AR; Slawin AMZ; Lebl T; Westwood NJ Polycyclic Ethers and an Unexpected Dearomatisation Reaction During Studies Towards the Bioactive Alkaloid, Perophoramidine. Tetrahedron 2018, 74, 3339–3347. [Google Scholar]
- (398).Edelsztein VC; Di Chenna PH; Burton G Synthesis of C-C Bonded Dimeric Steroids by Olefin Metathesis. Tetrahedron 2009, 65, 3615–3623. [Google Scholar]
- (399).Phutdhawong WS; Ruensamran W; Phutdhawong W Synthesis and Preliminary Evaluation of Dimeric-28-Homobrassinosteroids for Plant Growth Regulators. Steroids 2016, 116, 38–44. [DOI] [PubMed] [Google Scholar]
- (400).Zhao L; Burnell DJ Synthesis of the Tetracyclic Core of the Kempanes by a Ring Closing Metathesis Strategy. Org. Lett 2006, 8, 155–157. [DOI] [PubMed] [Google Scholar]
- (401).Matsumoto T; Miyano K; Kagawa S; Yű S; Ogawa J.-i.; Ichihara A Total Synthesis of dl-Illudol. Tetrahedron Lett. 1971, 12, 3521–3524. [Google Scholar]
- (402).Adams H; Jones S; Meijer AJHM; Najah Z; Ojea-Jiménez I; Reeder AT Diels–Alder Reactions and Transformations of 2-Cyclopenten-1-one with a Chiral Anthracene Template. Tetrahedron: Asymmetry 2011, 22, 1620–1625. [Google Scholar]
- (403).Tomooka K; Okinaga T; Suzuki K; Tsuchihashi G.-i. Lactols in Stereoselection 1. Highly Selective 1,4- and 1,5-Asymmetric Induction. Tetrahedron Lett. 1987, 28, 6335–6338. [Google Scholar]
- (404).Koreeda M; Tanaka Y; Schwartz A Stereochemically Controlled Synthesis of Steroid Side Chains: Synthesis of Desmosterol. J. Org. Chem 1980, 45, 1172–1174. [Google Scholar]
- (405).Matovic NJ; Stuthe JM; Challinor VL; Bernhardt PV; Lehmann RP; Kitching W; De Voss JJ The Truth About False Unicorn (Chamaelirium luteum): Total Synthesis of 23R,24S-Chiograsterol B Defines the Structure and Stereochemistry of the Major Saponins from this Medicinal Herb. Chem. Eur. J 2011, 17, 7578–7591. [DOI] [PubMed] [Google Scholar]
- (406).Zhao X-H; Zhang Q; Du J-Y; Lu X-Y; Cao Y-X; Deng Y-H; Fan C-A Total Synthesis of (±)-Lycojaponicumin D and Lycodoline-Type Lycopodium Alkaloids. J. Am. Chem. Soc 2017, 139, 7095–7103. [DOI] [PubMed] [Google Scholar]
- (407).Ding R; Fu J-G; Xu G-Q; Sun B-F; Lin G-Q Divergent Total Synthesis of the Lycopodium Alkaloids Huperzine A, Huperzine B, and Huperzine U. J. Org. Chem 2014, 79, 240–250. [DOI] [PubMed] [Google Scholar]
- (408).Tuulmets A; Sassian M Reactions of Partially Solvated Grignard Reagents with a Ketone. J. Organomet. Chem 1999, 586, 145–149. [Google Scholar]
- (409).Singer MS; Salinger RM; Mosher HS Kinetic Study of the Reduction of Di-t-butyl Ketone with t-Butylmagnesium Compounds. J. Org. Chem 1967, 32, 3821–3826. [Google Scholar]
- (410).Fu J-G; Xu G-Q; Ding R; Lin G-Q; Sun B-F Asymmetric Total Synthesis of Lycopodium Alkaloids α-Obscurine, N-Desmethyl-α-obscurine, β-Obscurine and N-Desmethyl-β-obscurine. Org. Chem. Front 2016, 3, 62–65. [Google Scholar]
- (411).Paquette LA; Tae J A Head-to-Head Comparison of α- and β-Alkoxy Effects on Stereoselectivity. Nucleophilic Additions to a Cyclohexanone Substituted with Five Axial C–O Bonds. Tetrahedron Lett. 1999, 40, 5971–5974. [Google Scholar]
- (412).Takahashi T; Miyazawa M; Tsuji J Stereoselective Reduction of α-Alkoxy Acetylenic Ketones with Zinc Borohydride and K-Selectride. Tetrahedron Lett. 1985, 26, 5139–5142. [Google Scholar]
- (413).Nakata T; Tanaka T; Oishi T Stereoselective Reduction of α-Hydroxy Ketones. Tetrahedron Lett. 1983, 24, 2653–2656. [Google Scholar]
- (414).Curran DP Reduction of Δ2-Isoxazolines. 3. Raney-Nickel Catalyzed Formation of β-Hydroxy Ketones. J. Am. Chem. Soc 1983, 105, 5826–5833. [Google Scholar]
- (415).Wendeborn S; Nussbaumer H; Schaetzer J; Winkler T Efficient Synthesis of Novel Bridgehead-Substituted Bicyclo[3.2.1]octane-2,4-diones. Synlett 2010, 1966–1968. [Google Scholar]
- (416).Kreiser W; Janitschke L Erste Totalsynthese Von (−)-„Alben”. Chem. Ber 1979, 112, 408–422. [Google Scholar]
- (417).Dimitrov V; Bratovanov S; Simova S; Kostova K Cerium(III) Chloride as Catalytic and Stoichiometric Promoter of the Quantitative Addition of Organometallic Reagents to (+)-Camphor and (−)-Fenchone. Tetrahedron Lett. 1994, 35, 6713–6716. [Google Scholar]
- (418).Dimitrov V; Simova S; Kostova K Highly Effective and Practical Stereoselective Synthesis of New Homoallylic Alcohols with (+)-Camphor and (−)-Fenchone Skeleton. Tetrahedron 1996, 52, 1699–1706. [Google Scholar]
- (419).Capmau ML; Chodkiewicz W; Cadiot P Stereochimie de l’Addition d’Organometalliques sur le (+) Camphre. Tetrahedron Lett. 1965, 6, 1619–1624. [Google Scholar]
- (420).Bunnelle WH; Rafferty MA; Hodges SL Aldol-Equivalent Elaboration of Sterically Hindered Ketiones: Methallylmagnesium Chloride as a Synthon for Acetone Enolate. J. Org. Chem 1987, 52, 1603–1605. [Google Scholar]
- (421).Sezer S; Gümrükçü Y; Şahin E; Tanyeli C Stereoselective Synthesis of Spirocyclic Cyclopentapyrans by the Pauson–Khand Reaction on Camphor Tethered Enynes. Tetrahedron: Asymmetry 2008, 19, 2705–2710. [Google Scholar]
- (422).Duclos RI Jr.; Lu D; Guo J; Makriyannis A Synthesis and Characterization of 2-Substituted Bornane Pharmacophores for Novel Cannabinergic Ligands. Tetrahedron Lett. 2008, 49, 5587–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Bergdahl M; Nilsson M; Olsson T; Stern K Highly Diastereoselective Additions of Organocopper Reagents to 2-exo-Bornyl Crotonates. Tetrahedron 1991, 47, 9691–9702. [Google Scholar]
- (424).Lin C-E; Richardson SK; Wang W; Wang T; Garvey DS Preparation of Functionalized Tertiary Thiols and Nitrosothiols. Tetrahedron 2006, 62, 8410–8418. [Google Scholar]
- (425).Metzner P; Vialle J; Vibet A Exclusive Thiophilic Addition of Organo-Magnesium Compounds to a Thioketone. Tetrahedron Lett. 1976, 17, 4295–4296. [Google Scholar]
- (426).Nicolaou KC; McGarry DG; Somers PK; Kim BH; Ogilvie WW; Yiannikouros G ; Prasad CVC; Veale CA; Hark RR Synthesis of Medium-Sized Ring Ethers from Thionolactones. Application to Polyether Synthesis. J. Am. Chem. Soc 1990, 112, 6263–6276. [Google Scholar]
- (427).Büchi G; Macleod WD Jr. Synthesis of Patchouli Alcohol. J. Am. Chem. Soc 1962, 84, 3205–3206. [Google Scholar]
- (428).Yang T-F; Chang C-Y; Lin W-F Rearrangement of Acyclic-Alkyl [3.2.1 ]Bicyclic Alcohols to Free Methylene Dimethyl Alkyl [2.2.2]Bicyclic Octanes: Application to Preparation of Stereospecific Novel Sesquiterpene and Terpenoids. Tetrahedron Lett. 2015, 56, 801–804. [Google Scholar]
- (429).Momose T; Takizawa S; Kirihara M Bicyclo[3.3.1]nonanes as Synthetic Intermediates. XXIII. A Breakthrough by Lanthanoid Mediation in Nucleophilic Addition of Carbanions to the Inert “Fork Head” Carbonyl in Bicyclo[3.3.1]nonan-3-ones. Synth. Commun 1997, 27, 3313–3320. [Google Scholar]
- (430).Dow RL; Schneider SR; Paight ES; Hank RF; Chiang P; Cornelius P; Lee E; Newsome WP; Swick AG; Spitzer J et al. Discovery of a Novel Series of 6-Azauracil-Based Thyroid Hormone Receptor Ligands: Potent, TRβ Subtype-Selective Thyromimetics. Bioorg. Med. Chem. Lett 2003, 13, 379–382. [DOI] [PubMed] [Google Scholar]
- (431).Büchi G; Fliri H; Shapiro R Synthesis of Betalains. J. Org. Chem 1978, 43, 4765–4769. [Google Scholar]
- (432).Wilkinson SC; Lozano O; Schuler M; Pacheco MC; Salmon R; Gouverneur V Electrophilic Fluorocyclization of Allyl Silanes. Angew. Chem. Int. Ed 2009, 48, 7083–7086. [DOI] [PubMed] [Google Scholar]
- (433).Lochyński S; Frĉkowiak B; Siemieniuk A; Pîtkowski K; Ciunik Z; Wawrzeήczyk C Synthesis and Stereochemistry of Some New Derivatives of cis-Dihydropinol. Tetrahedron: Asymmetry 1999, 10, 1033–1039. [Google Scholar]
- (434).Grainger RS; Owoare RB Selective 1,5-Alkylidenecarbene Insertion Reactions on [3.2.1] Oxabicyclic Ethers: A New Approach toward the AB Ring System of Ingenol. Org. Lett 2004, 6, 2961–2964. [DOI] [PubMed] [Google Scholar]
- (435).Stoyanov ES; Gunbas G; Hafezi N; Mascal M; Stoyanova IV; Tham FS; Reed CA The R3O+•••H+ Hydrogen Bond: Toward a Tetracoordinate Oxadionium(2+) Ion. J. Am. Chem. Soc 2012, 134, 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (436).Krasovskiy A; Kopp F; Knochel P Soluble Lanthanide Salts (LnCl3·2 LiCl) for the Improved Addition of Organomagnesium Reagents to Carbonyl Compounds. Angew. Chem. Int. Ed 2006, 45, 497–500. [DOI] [PubMed] [Google Scholar]
- (437).Cossy J; BouzBouz S; Laghgar M; Tabyaoui B Selective Cyclopropylcarbinyl Rearrangement of Tricyclo[5.3.1.0]undecanols Induced by Pyridinium Chlorochromate. Tetrahedron Lett. 2002, 43, 823–827. [Google Scholar]
- (438).Lecomte V; Stéphan E; Vaissermann J; Jaouen G Are the 11-Oxo-Steroids Really So Hindered Towards Organometallic Compounds? Steroids 2004, 69, 17–21. [DOI] [PubMed] [Google Scholar]
- (439).Li F; Tartakoff SS; Castle SL Total Synthesis of (−)-Acutumine. J. Am. Chem. Soc 2009, 131, 6674–6675. [DOI] [PubMed] [Google Scholar]
- (440).Nakamura M; Hirai A; Sogi M; Nakamura E Enantioselective Addition of Allylzinc Reagent to Alkynyl Ketones. J. Am. Chem. Soc 1998, 120, 5846–5847. [Google Scholar]
- (441).Evans DA; Golob AM [3,3]Sigmatropic Rearrangements of 1,5-Diene Alkoxides. The Powerful Accelerating Effects of the Alkoxide Substituent. J. Am. Chem. Soc 1979, 97, 4765–4766. [Google Scholar]
- (442).Paquette LA Recent Applications of Anionic Oxy-Cope Rearrangements. Tetrahedron 1997, 53, 13971–14020. [Google Scholar]
- (443).Xuan J; Pan S; Zhang Y; Ye B; Ding H Construction of the Tricyclic Core of Steenkrotin-type Diterpenoids via Intramolecular [3 + 2] Cycloaddition. Org. Biomol. Chem 2015, 13, 1643–1646. [DOI] [PubMed] [Google Scholar]
- (444).Ishida H; Kimura S; Kogure N; Kitajima M; Takayama H Total Synthesis of (±)-Lycoposerramine-R, a Novel Skeletal Type of Lycopodium Alkaloid. Tetrahedron 2015, 71, 51–56. [Google Scholar]
- (445).Kotha S; Ravikumar O Ring-Rearrangement-Metathesis Approach to Polycycles: Substrate-Controlled Stereochemical Outcome During Grignard Addition. Eur. J. Org. Chem 2016, 3900–3906. [Google Scholar]
- (446).Urabe D; Nagatomo M; Hagiwara K; Masuda K; Inoue M Symmetry-Driven Synthesis of 9-Demethyl-10,15-dideoxyryanodol. Chem. Sci 2013, 4, 1615. [Google Scholar]
- (447).Sasano Y; Koyama J; Yoshikawa K; Kanoh N; Kwon E; Iwabuchi Y Stereocontrolled Construction of ABCD Tetracyclic Ring System with Vicinal All-Carbon Quaternary Stereogenic Centers of Calyciphylline a Type Alkaloids. Org. Lett 2018, 20, 3053–3056. [DOI] [PubMed] [Google Scholar]
- (448).Kotha S; Dipak MK Design and Synthesis of Novel Bis-Annulated Caged Polycycles via Ring-Closing Metathesis: Pushpakenediol. Beilstein J. Org. Chem 2014, 10, 2664–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Matsumura Y; Aoyagi S; Kibayashi C A Formal Total Synthesis of (±)-Halichlorine and (±)-Pinnaic Acid. Org. Lett 2004, 6, 965–968. [DOI] [PubMed] [Google Scholar]
- (450).Hilmey DG; Gallucci JC; Paquette LA Neighboring Effect of the Lactam Functionality in Select Reactions of 6-Azaspiro[4.5]decane-1,7-dione. Tetrahedron 2005, 61, 11000–11009. [Google Scholar]
- (451).Shao H; Bao W; Jing Z-R; Wang Y-P; Zhang F-M; Wang S-H; Tu Y-Q Construction of the [6,5,7,5] Tetracyclic Core of Calyciphylline a Type Alkaloids via a Tandem Semipinacol Rearrangement/Nicholas Reaction. Org. Lett 2017, 19, 4648–4651. [DOI] [PubMed] [Google Scholar]
- (452).Hou S-H; Tu Y-Q; Wang S-H; Xi C-C; Zhang F-M; Wang S-H; Li Y-T; Liu L Total Syntheses of the Tetracyclic Cyclopiane Diterpenes Conidiogenone, Conidiogenol, and Conidiogenone B. Angew. Chem. Int. Ed 2016, 55, 4456–4460. [DOI] [PubMed] [Google Scholar]
- (453).Gao F; Stamp CT; Thornton PD; Cameron TS; Doyle LE; Miller DO; Burnell DJ Selective Formation of Angular Tricyclic Compounds by Ruthenium-Mediated Ring-Rearrangement Metathesis. Chem. Commun 2012, 48, 233–235. [DOI] [PubMed] [Google Scholar]
- (454).Liu P-Y; Burnell DJ Facial Selectivity in Nucleophilic Reactions of Spirocyclic Ketones Can Be Controlled by a Distant, Orthogonal Double Bond. J. Chem. Soc., Chem. Commun 1994, 1183–1184. [Google Scholar]
- (455).Oikawa M; Hashimoto R; Sasaki M Asymmetric Synthesis and in vivo Biological Inactivity of the Right-Hand Terpenoid Fragment of Terpendole E. Eur. J. Org. Chem 2011,538–546. [Google Scholar]
- (456).Iwasaki J; Ito H; Nakamura M; Iguchi K A Synthetic Study of Briarane-Type Marine Diterpenoid, Pachyclavulide B. Tetrahedron Lett. 2006, 47, 1483–1486. [Google Scholar]
- (457).Michel P; Ley SV Butane-2,3-diacetals of Glyceraldehyde: A Stable Alternative to Glyceraldehyde Acetonide. Angew. Chem. Int. Ed 2002, 41, 3898–3901. [DOI] [PubMed] [Google Scholar]
- (458).Aube J; Mossman CJ; Dickey S (2s,3s,5s)- and (2s,3s,5r)-5-Carboxaldehyde-2,3-Diphenyl-1,4-Dioxane as Surrogates for Optically Pure 2,3-O-Isopropylideneglyceraldehyde in Asymmetric Synthesis. Tetrahedron 1992, 48, 9819–9826. [Google Scholar]
- (459).Shi H; De S; Wang Q; Gao S; Wang X; Chen C Construction of the 5,6,7-Tricyclic Skeleton of Lancifodilactone F. Tetrahedron Lett 2015, 56, 3225–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Tsunoda T; Kabasawa Y; Itô S; Kodama M Total Synthesis of “(±)-Senoxydene”. Tetrahedron Lett. 1984, 25, 773–776. [Google Scholar]
- (461).Reber KP; Xu J; Guerrero CA Synthesis of Mulinane Diterpenoids. J. Org. Chem 2015, 80, 2397–2406. [DOI] [PubMed] [Google Scholar]
- (462).Kazuta Y; Abe H; Yamamoto T; Matsuda A; Shuto S The Unusual 1,4-Chelation-Controlled Nucleophilic Addition to Aldehydes with High Stereoselectivity. A Systematic Study of Stereoselectivity in the Addition Reaction of Carbon Nucleophiles to cis-Substituted Cyclopropanecarbaldehydes. Tetrahedron 2004, 60, 6689–6703. [Google Scholar]
- (463).Hughes JME; Gleason JL A Bio-Inspired Cascade and a Late-Stage Directed sp3 C–H Lithiation Enables a Concise Total Synthesis of (−)-Virosaine A. Tetrahedron 2018, 74, 759–768. [Google Scholar]
- (464).Barluenga J; Llavona L; Bernad PL; Concellón JM. Preparation of Disubstituted Epichlorohydrins with Total Diastereoselectivity. Transformation of α-Bromocarbonyl Compounds into Allyl Alcohols. Tetrahedron Lett. 1993, 34, 3173–3176. [Google Scholar]
- (465).Rosenberg RE; Kelly WJ Felkin–Anh Is Not Enough. J. Phys. Org. Chem 2015, 28, 47–56. [Google Scholar]
- (466).Manner S; Oltner VT; Oredsson S; Ellervik U; Frejd T Spiro-bicyclo[2.2.2]octane Derivatives as Paclitaxel Mimetics. Synthesis and Toxicity Evaluation in Breast Cancer Cell Lines. Org. Biomol. Chem 2013, 11, 7134–7144. [DOI] [PubMed] [Google Scholar]
- (467).Lautens M; Blackwell J The Utility of the Homo Diels–Alder Reaction in the Synthesis of Natural Product Skeletons: A Cycloaddition–Fragmentation–Cyclization Sequence to Diquinanes and Triquinanes. Synthesis 1998, 537–546. [Google Scholar]
- (468).Mulzer J; Pietschmann C; Buschmann J; Luger P Diastereoselective Grignard Additions to O-Protected Polyhydroxylated Ketones: A Reaction Controlled by Groundstate Conformation? J. Org. Chem 1997, 62, 3938–3943. [Google Scholar]
- (469).Sutherlin DP; Armstrong RW Synthesis of 12 Stereochemically and Structurally Diverse C-Trisaccharides. J. Org. Chem 1997, 62, 5267–5283. [Google Scholar]
- (470).Poigny S; Nouri S; Chiaroni A; Guyot M; Samadi M Total Synthesis and Determination of the Absolute Configuration of Coscinosulfate. A New Selective Inhibitor of Cdc25 Protein Phosphatase. J. Org. Chem 2001, 66, 7263–7269. [DOI] [PubMed] [Google Scholar]
- (471).Schmid W; Whitesides GM A New Approach to Cyclitols Based on Rabbit Muscle Aldolase (RAMA). J. Am. Chem. Soc 1990, 112, 9670–9671. [Google Scholar]
- (472).Marzabadi CH; Anderson JE; Gonzalez-Outeirino; Gaffney PRJ; White CGH; Tocher DA; Todaro LJ. Why Are Silyl Ethers Conformationally Different from Alkyl Ethers? Chair-Chair Conformational Equilibria in Silyloxycyclohexanes and Their Dependence on the Substituents on Silicon. The Wider Roles of Eclipsing, of 1,3-Repulsive Steric Interactions, and of Attractive Steric Interactions. J. Am. Chem. Soc 2003, 125, 15163–15173. [DOI] [PubMed] [Google Scholar]
- (473).Yamada H; Nakatani M; Ikeda T; Marumoto Y Stable Axial-Rich Conformation of Pyranoses Derived from l-Rhamnose and d-Mannose. Tetrahedron Lett. 1999, 40, 5573–5576. [Google Scholar]
- (474).Tius MA; Busch-Petersen J Stereochemical Control in the Oxymercuration of 5-Alken-1-ols. Tetrahedron Lett. 1994, 35, 5181–5184. [Google Scholar]
- (475).Pedersen CM; Marinescu LG; Bols M Glycosyl Donors in ‘Unusual’ Conformations – Influence on Reactivity and Selectivity. C. R. Chim 2011, 14, 17–43. [Google Scholar]
- (476).Hara A; Morimoto R; Ishikawa Y; Nishiyama S Synthesis of the C7–26 Fragment of Amphidinolides G and H. Org. Lett 2011, 13, 4036–4039. [DOI] [PubMed] [Google Scholar]
- (477).Yoda H; Kitayama H; Takabe K; Kakehi A Diastereomer Differentiation During 1,2-Addition of Grignard Reagents under Rotamer Distribution Control. Tetrahedron: Asymmetry 1993, 4, 1759–1762. [Google Scholar]
- (478).Lal K; Zarate EA; Youngs WJ; Salomon RG Total Synthesis Necessitates Revision of the Structure of Robustadials. J. Am. Chem. Soc 1986, 108, 1311–1312. [Google Scholar]
- (479).Liu B; Duan S; Sutterer AC; Moeller KD Oxidative Cyclization Based on Reversing the Polarity of Enol Ethers and Ketene Dithioacetals. Construction of a Tetrahydrofuran Ring and Application to the Synthesis of (+)-Nemorensic Acid. J. Am. Chem. Soc 2002, 124, 10101–10111. [DOI] [PubMed] [Google Scholar]
- (480).El Karroumi J; El Haib A; Manoury E; Benharref A; Daran JC; Gouygou M; Urrutigoïty M Selectivity Controlled by Ligand Tuning in the Palladium-Catalysed Cyclocarbonylation: Synthesis of New γ and δ Lactones from a Natural Sesquiterpene. J. Mol. Cat. A: Chem 2015, 401, 18–26. [Google Scholar]
- (481).Pedrosa R; Sayalero S; Vicente M A Direct Efficient Diastereoselective Synthesis of Enantiopure 3-Substituted-Isobenzofuranones. Tetrahedron 2006, 62, 10400–10407. [Google Scholar]
- (482).Taylor RE; Jin M Toward a Total Synthesis of Peloruside A: Enantioselective Preparation of the C8-C19 Region. Org. Lett 2003, 5, 4959–4961. [DOI] [PubMed] [Google Scholar]
- (483).Lejkowski M; Gais H-J; Banerjee P; Vermeeren C Asymmetric Modular Synthesis of Highly Functionalized Medium-Sized Carbocycles and Lactones via Ring-Closing Metathesis of Sulfoximine-Substituted Trienes. J. Am. Chem. Soc 2006, 128, 15378–15379. [DOI] [PubMed] [Google Scholar]
- (484).Lejkowski M; Banerjee P; Schüller S; Münch A; Runsink J; Vermeeren C; Gais H-J Asymmetric Synthesis of Densely Functionalized Medium-Ring Carbocycles and Lactones through Modular Assembly and Ring-Closing Metathesis of Sulfoximine-Substituted Trienes and Dienynes. Chem. Eur. J 2012, 18, 3529–3548. [DOI] [PubMed] [Google Scholar]
- (485).Kobayashi K; Kunimura R; Takagi H; Hirai M; Kogen H; Hirota H; Kuroda C Total Synthesis of Highly Oxygenated Bisabolane Sesquiterpene Isolated from Ligularia lankongensis: Relative and Absolute Configurations of the Natural Product. J. Org. Chem 2018, 83, 703–715. [DOI] [PubMed] [Google Scholar]
- (486).Hirai M; Miyazaki R; Mitsui K; Kiuchi K; Onuki H; Hirota H; Kuroda C Synthesis and NMR Spectroscopic Elucidation of Four Diastereoisomers of Oxygenated Bisabolane Side Chain. Helv. Chim. Acta 2015, 98, 1035–1060. [Google Scholar]
- (487).Sokolowska K; Sicinski RR Synthesis of Novel Vitamin D3 Analog with an Additional Ring Annulated to A and seco-B Rings. Steroids 2014, 87, 67–75. [DOI] [PubMed] [Google Scholar]
- (488).Yamashita S; Ishihara Y; Morita H; Uchiyama J; Takeuchi K; Inoue M; Hirama M Stereoselective 6-exo Radical Cyclization Using cis-Vinyl Sulfoxide: Practical Total Synthesis of CTX3C. J. Nat. Prod 2011, 74, 357–364. [DOI] [PubMed] [Google Scholar]
- (489).Segal’ GM; Sydykov ZS.; Torgov IV. Partial Synthesis of 2-Desoxyecdysone and 2-Desoxyecdysterone, Insect Molting Hormones. Russ. Chem. Bull 1982, 31, 285–292. [Google Scholar]
- (490).Crimmins MT; Zhang Y; Williams PS Approach to the Synthesis of Briarane Diterpenes through a Dianionic Claisen Rearrangement and Ring-Closing Metathesis. Org. Lett 2017, 19, 3907–3910. [DOI] [PubMed] [Google Scholar]
- (491).Williams DR; Klein JC Intramolecular Diels-Alder (IMDA) Studies toward the Synthesis of Australifungin. Stereocontrol in the Acetate Aldol Reaction of β,β’-Branched Aldehydes. Org. Lett 2016, 18, 420–423. [DOI] [PubMed] [Google Scholar]
- (492).Alvaro G; Savoia D Addition of Organometallic Reagents to Imines Bearing Stereogenic N-Substituents. Stereochemical Models Explaining the 1,3-Asymmetric Induction. Synlett 2002, 0651–0673. [Google Scholar]
- (493).Stork G; Dowd SR A New Method for the Alkylation of Ketones and Aldehydes: The C-Alkylation of the Magnesium Salts of N-Substituted Imines. J. Am. Chem. Soc 1963, 85, 2178–2180. [Google Scholar]
- (494).Gandon V; Bertus P; Szymoniak J Zirconium-Catalyzed Ethylmagnesiation of Imines – Scope and Mechanism. Eur. J. Org. Chem 2001,3677–3681. [Google Scholar]
- (495).Bloch R Additions of Organometallic Reagents to C=N Bonds: Reactivity and Selectivity. Chem. Rev 1998, 98, 1407–1438. [DOI] [PubMed] [Google Scholar]
- (496).Enders D; Reinhold U Asymmetric Synthesis of Amines by Nucleophilic 1,2-Addition of Organometallic Reagents to the CN-Double Bond. Tetrahedron: Asymmetry 1997, 8, 1895–1946. [Google Scholar]
- (497).Mailyan AK; Eickhoff JA; Minakova AS; Gu Z; Lu P; Zakarian A Cutting-Edge and Time-Honored Strategies for Stereoselective Construction of C-N Bonds in Total Synthesis. Chem. Rev 2016, 116, 4441–4557. [DOI] [PubMed] [Google Scholar]
- (498).Hua DH; Miao SW; Chen JS; Iguchi S Stereoselective Addition Reactions of Chiral N-Benzylidene-p-toluenesulfinamides. Asymmetric Syntheses of β- and γ-Amino Acids. J. Org. Chem 1991, 56, 4–6. [Google Scholar]
- (499).Pearson WH; Aponick A; Dietz AL Synthesis of N,N-Bis(3-butenyl)amines from 2-Azaallyl Dication Synthetic Equivalents and Conversion to 2,3,6,7-Tetrahydroazepines by Ring-Closing Metathesis. J. Org. Chem 2006, 71, 3533–3539. [DOI] [PubMed] [Google Scholar]
- (500).Badorrey R; Cativiela C; Díaz-de-Villegas MD; Díez R; Gálvez JA Study of the Reactions between Vinylmagnesium Bromide and Imines Derived from (R)-Glyceraldehyde – The Key Step in the Stereodivergent Synthesis of Conveniently Protected, Enantiopure syn- and anti-2-Amino-1,3,4-butanetriol Derivatives. Eur. J. Org. Chem 2003, 2268–2275. [Google Scholar]
- (501).Cativiela C; Diaz-de-Villegas MD; Gálvez J Stereoselective Synthesis of α-Hydroxy-β-Amino Acids Using d-Glyceraldehyde as the Homochiral Source. Tetrahedron: Asymmetry 1996, 7, 529–536. [Google Scholar]
- (502).Badorrey R; Cativiela C; Díaz-de-Villegas MD; Gálvez J Reversal of the Stereochemical Course of the Addition of Phenylmagnesium Bromide to N-Benzylimines Derived from R-Glyceraldehyde Depending on the O-Protecting Group and Its Application to the Synthesis of Both Enantiomers of Phenylglycine. Tetrahedron 1997, 53, 1411–1416. [Google Scholar]
- (503).Badorrey R; Cativiela C; Díaz-de-Villegas MD; Díez R; Gálvez JA Stereodivergent Addition of Allylmetal Reagents to Imines Derived from (R)-2,3-Di-O-benzylglyceraldehyde by Appropriate Selection of Metal and Double Stereodifferentiation. Eur. J. Org. Chem 2002, 3763–3767. [Google Scholar]
- (504).Badorrey R; Cativiela C; Díaz-de-Villegas M. a. D.; Gálvez JA Highly Convergent Stereoselective Synthesis of Chiral Key Intermediates in the Synthesis of Palinavir from Imines Derived from l-Glyceraldehyde. Tetrahedron 2002, 58, 341–354. [Google Scholar]
- (505).Yamamoto Y; Komatsu T; Maruyama K Enantiodivergent 1,2- and 1,3-Asymmetric Induction in α- and β-Alkoxyimines via Metal Tuning and Stereodifferentiation. J. Chem. Soc., Chem. Commun 1985, 814–816. [Google Scholar]
- (506).Liu K-G; Zhou H-B; Wu Y-L; Yao Z-J Synthesis of a New Stable Conformationally Constrained 2,7-Anhydrosialic Acid Derivative. J. Org. Chem 2003, 68, 9528–9531. [DOI] [PubMed] [Google Scholar]
- (507).Lee Y-T; Jung C; Myeong I-S; Lee S-H; Kim J-S; Ham W-H Stereoselective Allylation of α-Hydroxy Aldimines and Its Application to the Formal Synthesis of (−)-β-Conhydrine. Tetrahedron 2018, 74, 506–511. [Google Scholar]
- (508).Cainelli G; Giacomini D; Mezzina E; Panunzio M; Zarantonello P N-Metallo Imines: A New Approach to α-Amino Alcohols from Aldehydes. Tetrahedron Lett. 1991, 32, 2967–2970. [Google Scholar]
- (509).Chattopadhyay SK; Biswas T; Biswas T Complementary Routes to Both Enantiomers of Pipecolic Acid and 4,5-Dihydroxypipecolic Acid Derivatives. Tetrahedron Lett. 2008, 49, 1365–1369. [Google Scholar]
- (510).Saha N; Chattopadhyay SK Enantiodivergency and Enantioconvergency in the Synthesis of the Dendrobate Alkaloid 241D. J. Org. Chem 2012, 77, 11056–11063. [DOI] [PubMed] [Google Scholar]
- (511).Godin G; Compain P; Masson G; Martin OR A General Strategy for the Practical Synthesis of Nojirimycin C-Glycosides and Analogues. Extension to the First Reported Example of an Iminosugar 1-Phosphonate. J. Org. Chem 2002, 67, 6960–6970. [DOI] [PubMed] [Google Scholar]
- (512).Bordier A; Compain P; Martin 0R.; Ikeda K; Asano N First Stereocontrolled Synthesis and Biological Evaluation of 1,6-Dideoxy-l-nojirimycin. Tetrahedron: Asymmetry 2003, 14, 47–51. [Google Scholar]
- (513).Chandrasekhar B; Rao BV; Rao KVM; Jagadeesh B A Short and Common Stereoselective Approach to 5/6, 6/6, 6/7 Bicyclic Aza Sugars. Tetrahedron: Asymmetry 2009, 20, 1217–1223. [Google Scholar]
- (514).Hamon DPG; Massy-Westropp RA; Razzino P The Asymmetric Synthesis of α-Amino Acids via the Addition of Grignard Reagents to Imine Derivatives. Tetrahedron 1992, 48, 5163–5178. [Google Scholar]
- (515).Yamamoto Y; Komatsu T; Maruyama K Very High 1,2-Asymmetric Induction in the Reaction of Allyl-9-BBN with Certain Imines. Evidence for a Stereoelectronic Effect to Enhance the Cram Selectivity. J. Am. Chem. Soc 1984, 106, 5031–5033. [Google Scholar]
- (516).Yamamoto Y; Nishi S; Maruyama K; Komatsu T; Ito W Very High 1,2- and 1,3-Asymmetric Induction in the Reactions of Allylic Boron Compounds with Chiral Imines. J. Am. Chem. Soc 1986, 108, 7778–7786. [DOI] [PubMed] [Google Scholar]
- (517).Fu J; Shen H; Chang Y; Li C; Gong J; Yang Z Concise Stereoselective Synthesis of Oxaspirocycles with 1-Tosyl-1,2,3-triazoles: Application to the Total Syntheses of (±)-Tuberostemospiroline and (±)-Stemona-lactam R. Chem. Eur. J 2014, 20, 12881–12888. [DOI] [PubMed] [Google Scholar]
- (518).Wright DL; Schulte JP II; Page MA An Imine Addition/Ring-Closing Metathesis Approach to the Spirocyclic Core of Halichlorine and Pinnaic Acid. Org. Lett 2000, 2, 1847–1850. [DOI] [PubMed] [Google Scholar]
- (519).Caldwell JJ; Collins I Rapid Synthesis of 4-Benzyl-4-aminopiperidines by Addition of Grignard Reagents to N-(1-Boc-Piperidin-4-ylidene)-tert-butanesulfinyl Imine. Synlett 2006, 2565–2568. [Google Scholar]
- (520).Brioche J; Meyer C; Cossy J Synthesis of Functionalized Allenamides from Ynamides by Enolate Claisen Rearrangement. Org. Lett 2013, 15, 1626–1629. [DOI] [PubMed] [Google Scholar]
- (521).Seebach D; Lamatsch B; Amstutz R; Beck AK; Dobler M; Egli M; Fitzi R; Gautschi M; Herradón B; Hidber PC et al. Structure and Reactivity of Five- and Six-Ring N,N-, N,O-, and O,O-Acetals: A Lesson in Allylic 1,3-Strain (A1,3 Strain). Helv. Chim. Acta 1992, 75, 913–934. [Google Scholar]
- (522).Coia N; Mokhtari N; Vasse J-L; Szymoniak J Diastereoselective Access to Nonracemic 2-cis-Substituted and 2,6-cis-Disubstituted Piperidines. Org. Lett 2011, 13, 6292–6295. [DOI] [PubMed] [Google Scholar]
- (523).Itabashi S; Shimomura M; Sato M; Azuma H; Okano K; Sakata J; Tokuyama H One-Pot Reductive Allylation of Amides by Using a Combination of Titanium Hydride and an Allylzinc Reagent: Application to a Total Synthesis of (−)-Castoramine. Synlett 2018, 29, 1786–1790. [Google Scholar]
- (524).Ayala L; Lucero CG; Romero JAC; Tabacco SA; Woerpel KA Stereochemistry of Nucleophilic Substitution Reactions Depending Upon Substituent: Evidence for Electrostatic Stabilization of Pseudoaxial Conformers of Oxocarbenium Ions by Heteroatom Substituents. J. Am. Chem. Soc 2003, 125, 15521–15528. [DOI] [PubMed] [Google Scholar]
- (525).Taketoshi A; Hosoda N; Yamaguchi Y; Asami M Diastereoselective Allylation of 3,4-Dihydro-4-phenylisoquinoline. Convenient Methods for the Preparation of cis- and trans-1-Allyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline. Bull. Chem. Soc. Jpn 2007, 80, 2418–2424. [Google Scholar]
- (526).Sato M; Azuma H; Daigaku A; Sato S; Takasu K; Okano K; Tokuyama H Total Synthesis of (−)-Histrionicotoxin through a Stereoselective Radical Translocation-Cyclization Reaction. Angew. Chem. Int. Ed 2017, 56, 1087–1091. [DOI] [PubMed] [Google Scholar]
- (527).Race NJ; Bower JF Palladium Catalyzed Cyclizations of Oxime Esters with 1,2-Disubstituted Alkenes: Synthesis of Dihydropyrroles. Org. Lett 2013, 15, 4616–4619. [DOI] [PubMed] [Google Scholar]
- (528).Majhail MK; Ylioja PM; Willis MC Direct Synthesis of Highly Substituted Pyrroles and Dihydropyrroles Using Linear Selective Hydroacylation Reactions. Chem. Eur. J 2016, 22, 7879–7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (529).Schmitt A; Reissig H-U A Highly Diastereoselective Route to Disubstituted Tetrahydrofuran Derivatives by Substitution of γ-Lactols with Silylated Nucleophiles. Synlett 1990, 40–42. [Google Scholar]
- (530).Larsen CH; Ridgway BH; Shaw JT; Woerpel KA A Stereoelectronic Model to Explain the Highly Stereoselective Reactions of Nucleophiles with Five-Membered-Ring Oxocarbenium Ions. J. Am. Chem. Soc 1999, 121, 12208–12209. [Google Scholar]
- (531).Arai T; Abe H; Aoyagi S; Kibayashi C Total Synthesis of (+)-Cylindricine C. Tetrahedron Lett. 2004, 45, 5921–5924. [Google Scholar]
- (532).Larsen CH; Ridgway BH; Shaw JT; Smith DM; Woerpel KA Stereoselective C-Glycosylation Reactions of Ribose Derivatives: Electronic Effects of Five-Membered Ring Oxocarbenium Ions. J. Am. Chem. Soc 2005, 127, 10879–10884. [DOI] [PubMed] [Google Scholar]
- (533).Bur SK; Martin SF Vinylogous Mannich Reactions: Some Theoretical Studies on the Origins of Diastereoselectivity. Org. Lett 2000, 2, 3445–3447. [DOI] [PubMed] [Google Scholar]
- (534).van Rijssel ER; Goumans TPM; Lodder G; Overkleeft HS; van der Marel GA; Codée JDC Chiral Pyrroline-Based Ugi-Three-Component Reactions Are under Kinetic Control. Org. Lett 2013, 15, 3026–3029. [DOI] [PubMed] [Google Scholar]
- (535).Zhai H; Wickenden JG; Sammis GM Cyclizations of Aminyl Radicals Generated from Substoichiometric Stannane. Synlett 2010, 3035–3038. [Google Scholar]
- (536).Flores M; García-García P; Garrido NM; Marcos IS; Sanz-González F; Díez D Domino Elimination/Nucleophilic Addition in the Synthesis of Chiral Pyrrolidines. J. Org. Chem 2013, 78, 7068–7075. [DOI] [PubMed] [Google Scholar]
- (537).Jung ME; Blum RB Generation of the Enolate of Acetaldehyde from Non-Carbonyl Substances and Its C-Alkylation, O-Acylation and O-Silylation. Tetrahedron Lett. 1977, 18, 3791–3794. [Google Scholar]
- (538).Rodriguez JG; Urrutia A; de Diego JE; Martinez-Alcazar MP; Fonseca I Synthesis of 2’-Alkylspiro[2-X-cyclohexan-1,3’-3’H-indole] (X = H; X = CH3) by an Unexpected Reaction between an Organomagnesium Halide and 2’-Methylspiro[2-X-cyclohexan-1,3’-3’H-indole]. X-ray Structure of a Fluorescent Dimeric Compound. J. Org. Chem 1998, 63, 4332–4337. [Google Scholar]
- (539).Mondon M; Fontelle N; Désiré J; Lecornué F; Guillard J; Marrot J; Blériot Y Access to l- and d-Iminosugar C-Glycosides from a d-gluco-Derived 6-Azidolactol Exploiting a Ring Isomerization/Alkylation Strategy. Org. Lett 2012, 14, 870–873. [DOI] [PubMed] [Google Scholar]
- (540).Castro S; Fyvie WS; Hatcher SA; Peczuh MW Synthesis of α-d-Idoseptanosyl Glycosides Using an S-Phenyl Septanoside Donor. Org. Lett 2005, 7, 4709–4712. [DOI] [PubMed] [Google Scholar]
- (541).Beaver MG; Buscagan TM; Lavinda O; Woerpel KA Stereoelectronic Model to Explain Highly Stereoselective Reactions of Seven-Membered-Ring Oxocarbenium-Ion Intermediates. Angew. Chem. Int. Ed 2016, 55, 1816–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (542).Duttwyler S; Chen S; Lu C; Mercado BQ; Bergman RG; Ellman JA Regio- and Stereoselective 1,2-Dihydropyridine Alkylation/Addition Sequence for the Synthesis of Piperidines with Quaternary Centers. Angew. Chem. Int. Ed 2014, 53, 3877–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (543).Duttwyler S; Chen S; Takase MK; Wiberg KB; Bergman RG; Ellman JA Proton Donor Acidity Controls Selectivity in Nonaromatic Nitrogen Heterocycle Synthesis. Science 2013, 339, 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Chen S; Chan AY; Walker MM; Ellman JA; Houk KN π-Facial Selectivities in Hydride Reductions of Hindered Endocyclic Iminium Ions. J. Org. Chem 2019, 84, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (545).Hu X-G; Daryl Ariawan A; Hunter L A d-Ribose-Derived α-Amino Nitrile as a Versatile Intermediate for the Collective Synthesis of Piperidine-Type Iminosugar C-Glycosides. Tetrahedron Lett. 2014, 55, 7222–7225. [Google Scholar]
- (546).Alujas-Burgos S; Oliveras-González C; Alvarez-Larena A; Bayón P; Figueredo M. Iterative Synthetic Strategy for Azaphenalene Alkaloids. Total Synthesis of (−)-9aepi-Hippocasine. J. Org. Chem 2018, 83, 5052–5057. [DOI] [PubMed] [Google Scholar]
- (547).Ayer WA; Bowman WR; Cooke GA; Soper AC Synthesis of Lycopodium Alkaloids. Part 2. A Method for the Construction of Substituted cis-trans-Hexahydrojulolidines. Tetrahedron Lett. 1966, 7, 2021–2026. [Google Scholar]
- (548).Schultz AG; Flood L; Springer JP Regio- and Stereoselective Control in the Addition of Grignard Reagents to the Pyridine Ring System. J. Org. Chem 1986, 51, 838–841. [Google Scholar]
- (549).Shibahara F; Suzuki M; Kubota S; Fukunaga T; Udagawa T; Murai T Selenolactams as Synthetic Intermediates for the Synthesis of Polycyclic Amines via Seleno-Claisen Rearrangements. J. Org. Chem 2018, 83, 3078–3089. [DOI] [PubMed] [Google Scholar]
- (550).Jin S; Gong J; Qin Y Total Synthesis of (−)-Lundurine A and Determination of Its Absolute Configuration. Angew. Chem. Int. Ed 2015, 54, 2228–2231. [DOI] [PubMed] [Google Scholar]
- (551).López-Ruiz H; Mera-Moreno I.; Rojas-Lima S; Santillán R; Farfán N Stereoselective Addition of Allylmagnesium Chloride to the C=N Bond of [4.3.0] Boron Heterobicycles. Tetrahedron Lett. 2008, 49, 1674–1677. [Google Scholar]
- (552).Liu S-Y; Lo MM-C; Fu GC The Synthesis of an Enantiopure Planar-Chiral Lewis Acid Complex via Kinetic Resolution and Its Application in Stereoselective Additions to Imines. Tetrahedron 2006, 62, 11343–11349. [Google Scholar]
- (553).Movassaghi M; Chen B Stereoselective Intermolecular Formal [3+3] Cycloaddition Reaction of Cyclic Enamines and Enones. Angew. Chem. Int. Ed 2007, 46, 565–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (554).Richter-Addo GB; Knight DA; Dewey MA; Arif AM; Gladysz JA New Strategies for Enantioselective Syntheses of 1-Alkyl- and 1,4-Dialkyl-1,2,3,4-tetrahydroisoquinolines: Diastereoselective Additions of Nucleophiles and Electrophiles to Isoquinoline Mediated by an Easily Resolved and Recycled Chiral Transition Metal Auxiliary. J. Am. Chem. Soc 1993, 115, 11863–11873. [Google Scholar]
- (555).Nicolaou KC; Rodriguez RM; Mitchell HJ; Suzuki H; Fylaktakidou KC; Baudoin O; van Delft FL Total Synthesis of Everninomicin 13,384–1—Part 1: Retrosynthetic Analysis and Synthesis of the A1B(A)C Fragment. Chem. Eur. J 2000, 6, 3095–3115. [DOI] [PubMed] [Google Scholar]
- (556).Ukaji Y; Kume K; Watai T; Fujisawa T Diastereofacial Discrimination in the Reaction of Chiral Alkoxymethyl Oxime Ethers with Allyl Metallic Reagents. Chem. Lett 1991, 20, 173–176. [Google Scholar]
- (557).Hatano M; Yamashita K; Ishihara K C- and N-Selective Grignard Addition Reactions of α-Aldimino Esters in the Presence or Absence of Zinc(II) Chloride: Synthetic Applications to Optically Active Azacycles. Org. Lett 2015, 17, 2412–2415. [DOI] [PubMed] [Google Scholar]
- (558).Buhrlage SJ; Chen B; Mapp AK A Flexible Synthetic Route to Isoxazolidine β-Proline Analogs. Tetrahedron 2009, 65, 3305–3313. [Google Scholar]
- (559).Fuller AA; Chen B; Minter AR; Mapp AK Succinct Synthesis of β-Amino Acids via Chiral Isoxazolines. J. Am. Chem. Soc 2005, 127, 5376–5383. [DOI] [PubMed] [Google Scholar]
- (560).Li X-T; Gu Q-S; Dong X-Y; Meng X; Liu X-Y A Copper Catalyst with a Cinchona-Alkaloid-Based Sulfonamide Ligand for Asymmetric Radical Oxytrifluoromethylation of Alkenyl Oximes. Angew. Chem. Int. Ed 2018, 57, 7668–7672. [DOI] [PubMed] [Google Scholar]
- (561).Suzuki H; Aoyagi S Total Synthesis of (−)-Chamobtusin A. Org. Lett 2012, 14, 6374–6376. [DOI] [PubMed] [Google Scholar]
- (562).Freeman JP Less Familiar Reactions of Oximes. Chem. Rev 1973, 73, 283–292. [Google Scholar]
- (563).Alexakis A; Lensen N; Tranchier J-P; Mangeney P Reactivity and Diastereoselectivity of Grignard Reagents toward the Hydrazone Functionality in Toluene Solvent. J. Org. Chem 1992, 57, 4563–4565. [Google Scholar]
- (564).Merino P; Delso I; Mannucci V; Tejero T High Stereocontrol in the Allylation of Chiral Non-Racemic α-Alkoxy and α-Amino Nitrones. Tetrahedron Lett 2006, 47, 3311–3314. [Google Scholar]
- (565).Merino P; Tejero T Understanding the High Diastereofacial Discrimination in Nucleophilic Additions to Nitrones: The First ab Initio Study on the Nucleophilic Addition Reactions of Chiral Nitrones with Grignard Reagents. Tetrahedron 2001, 57, 8125–8128. [Google Scholar]
- (566).Archibald G; Lin C-P; Boyd P; Barker D; Caprio V A Divergent Approach to 3-Piperidinols: A Concise Syntheses of (+)-Swainsonine and Access to the 1-Substituted Quinolizidine Skeleton. J. Org. Chem 2012, 77, 7968–7980. [DOI] [PubMed] [Google Scholar]
- (567).Párez P; Domingo LR; Aurell MJ; Contreras R Quantitative Characterization of the Global Electrophilicity Pattern of Some Reagents Involved in 1,3-Dipolar Cycloaddition Reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar]
- (568).Delso I; Tejero T; Goti A; Merino P Sequential Nucleophilic Addition/Intramolecular Cycloaddition to Chiral Nonracemic Cyclic Nitrones: A Highly Stereoselective Approach to Polyhydroxynortropane Alkaloids. J. Org. Chem 2011, 76, 4139–4143. [DOI] [PubMed] [Google Scholar]
- (569).Szcześniak P; Stecko S; Staszewska-Krajewska O; Furman B Sugar-Derived Cyclic Imines: One-Pot Synthesis and Direct Functionalization. Tetrahedron 2014, 70, 1880–1888. [Google Scholar]
- (570).Bande OP; Jadhav VH; Puranik VG; Dhavale DD; Lombardo M Stereo-controlled Approach to Pyrrolidine Iminosugar C-Glycosides and 1,4-Dideoxy-1,4-imino-l-allitol Using a d-Mannose-derived Cyclic Nitrone. Tetrahedron Lett. 2009, 50, 6906–6908. [Google Scholar]
- (571).Kaliappan KP; Das P; Chavan ST; Sabharwal SG A Versatile Access to Calystegine Analogues as Potential Glycosidases Inhibitors. J. Org. Chem 2009, 74, 6266–6274. [DOI] [PubMed] [Google Scholar]
- (572).Ghirardello M; Perrone D; Chinaglia N; Sádaba D; Delso I; Tejero T; Marchesi E; Fogagnolo M; Rafie K; van Aalten DMF et al. Analogues as Inhibitors of O- GlcNAc Transferase (OGT): Spectroscopic, Computational, and Biological Studies. Chem. Eur. J 2018, 24, 7264–7272. [DOI] [PubMed] [Google Scholar]
- (573).Cardona F; Moreno G; Guarna F; Vogel P; Schuetz C; Merino P; Goti A New Concise Total Synthesis of (+)-Lentiginosine and Some Structural Analogues. J. Org. Chem 2005, 70, 6552–6555. [DOI] [PubMed] [Google Scholar]
- (574).Bonanni M; Marradi M; Cicchi S; Faggi C; Goti A Double Addition of Grignard Reagents to N-Glycosyl Nitrones: A New Tool for the Construction of Enantiopure Azaheterocycles. Org. Lett 2005, 7, 319–322. [DOI] [PubMed] [Google Scholar]
- (575).Koriyama Y; Nozawa A; Hayakawa R; Shimizu M Reversal of Diastereofacial Selectivity in the Nucleophilic Addition Reaction to Chiral N-Sulfinimine and Application to the Synthesis of Indrizidine 223AB. Tetrahedron 2002, 58, 9621–9628. [Google Scholar]
- (576).Yang T-K; Chen R-Y; Lee D-S; Peng W-S; Jiang Y-Z; Mi A-Q; Jong T-T Application of New Camphor-Derived Mercapto Chiral Auxiliaries to the Synthesis of Optically Active Primary Amines. J. Org. Chem 1994, 59, 914–921. [Google Scholar]
- (577).Cogan DA; Liu G; Ellman J Asymmetric Synthesis of Chiral Amines by Highly Diastereoselective 1,2-Additions of Organometallic Reagents to N-tert-Butanesulfinyl Imines. Tetrahedron 1999, 55, 8883–8904. [Google Scholar]
- (578).Bertrand MB; Wolfe JP A Concise Stereoselective Synthesis of Preussin, 3-epi-Preussin, and Analogues. Org. Lett 2006, 8, 2353–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (579).Xiao D; Wang C; Palani A; Reichard G; Aslanian R; Shih N-Y; Buevich A Two Complementary, Diversity-Driven Asymmetric Syntheses of a 2,2-Disubstituted Piperidine NK1 Antagonist. Tetrahedron: Asymmetry 2006, 17, 2596–2598. [Google Scholar]
- (580).Pindi S; Wu J; Li G Design, Synthesis, and Applications of Chiral N-2-Phenyl-2-propyl Sulfinyl Imines for Group-Assisted Purification (GAP) Symmetric Synthesis. J. Org. Chem 2013, 78, 4006–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (581).Singleton DA; Wang Y; Yang HW; Romo D Mechanism and Origin of Stereoselectivity in Lewis Acid Catalyzed [2+2] Cycloadditions of Ketenes with Aldehydes. Angew. Chem. Int. Ed 2002, 41, 1572–1575. [DOI] [PubMed] [Google Scholar]
- (582).Han SJ; Stoltz BM A Mild and Efficient Approach to Enantioenriched α-Hydroxyethyl α,β-Unsaturated γ-Lactams. Tetrahedron Lett. 2016, 57, 2233–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (583).Yendapally R; Lee RE Design, Synthesis, and Evaluation of Novel Ethambutol Analogues. Bioorg. Med. Chem. Lett 2008, 18, 1607–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (584).Bretzke S; Scheeff S; Vollmeyer F; Eberhagen F; Rominger F; Menche D Modular Synthesis of the Pyrimidine Core of the Manzacidins by Divergent Tsuji–Trost Coupling. Beilstein J. Org. Chem 2016, 12, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (585).Grellepois F; Ben Jamaa A; Gassama A Diastereoselective Addition of Organomagnesium and Organolithium Reagents to Chiral Trifluoromethyl N-tert-Butanesulfinyl Hemiaminals. Eur. J. Org. Chem 2013, 6694–6701. [Google Scholar]
- (586).Hua DH; Lagneau N; Wang H; Chen J Asymmetric Synthesis of α-Amino Acids via Chiral N-Alkylidenesulfinamides. Tetrahedron: Asymmetry 1995, 6, 349–352. [Google Scholar]
- (587).Asensio A; Bravo P; Crucianelli M; Farina A; Fustero S; Soler JG; Meille SV; Panzeri W; Viani F; Volonterio A et al. Synthesis of Nonracemic α-Trifluoromethyl α-Amino Acids from Sulfinimines of Trifluoropyruvate. Eur. J. Org. Chem 2001, 1449–1458. [Google Scholar]
- (588).Huang W; Yao Y; Xu Y-J; Lu C-D Diastereoselective α-Fluorination of N-tert-Butanesulfinyl Imidates. J. Org. Chem 2018, 83, 14777–14785. [DOI] [PubMed] [Google Scholar]
- (589).Nishikawa Y; Kitajima M; Kogure N; Takayama H A Divergent Approach for the Total Syntheses of Cernuane-type and Quinolizidine-type Lycopodium Alkaloids. Tetrahedron 2009, 65, 1608–1617. [Google Scholar]
- (590).Pinto A; Piccichè M; Griera R; Molins E; Bosch J; Amat M Studies on the Synthesis of Phlegmarine-Type Lycopodium Alkaloids: Enantioselective Synthesis of (−)-Cermizine B, (+)-Serratezomine E, and (+)-Luciduline. J. Org. Chem 2018, 83, 8364–8375. [DOI] [PubMed] [Google Scholar]
- (591).Pinto A; Griera R; Molins E; Fernandez I; Bosch J; Amat M Access to Enantiopure 5-, 7-, and 5,7-Substituted cis-Decahydroquinolines: Enantioselective Synthesis of (−)-Cermizine B. Org. Lett 2017, 19, 1714–1717. [DOI] [PubMed] [Google Scholar]
- (592).Seiple IB; Zhang Z; Jakubec P; Langlois-Mercier A; Wright PM; Hog DT; Yabu K; Allu SR; Fukuzaki T; Carlsen PN et al. A Platform for the Discovery of New Macrolide Antibiotics. Nature 2016, 533, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (593).Reddy NSS; Reddy BJM; Reddy BVS A Convergent and Stereoselective Total Synthesis of (−)-Crispine A, (−)-Benzo[a]quinolizidine and (−)-Salsolidine. Tetrahedron Lett. 2013, 54, 4228–4231. [Google Scholar]
- (594).Reddy NSS; Reddy AS; Yadav JS; Reddy BVS The Stereoselective Total Synthesis of (−)-Dihydrotetrabenazine. Tetrahedron Lett. 2012, 53, 6916–6918. [Google Scholar]
- (595).Lathrop SP; Pompeo M; Chang WT; Movassaghi M Convergent and Biomimetic Enantioselective Total Synthesis of (−)-Communesin F. J. Am. Chem. Soc 2016, 138, 7763–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (596).Alvaro G; Boga C; Savoia D; Umani-Ronchi A Diastereoselective Addition of Allylmetal Compounds to Imines Derived from (S)-1-Phenylethanamine. J. Chem. Soc., Perkin Trans. 1 1996, 875–882. [Google Scholar]
- (597).Hashimoto Y; Kobayashi N; Kai A; Saigo K Highly Diastereoselective Addition of Organometallic Reagents to Chiral Imines Derived from 1-(2-Methoxyphenyl)ethylamine. Synlett 1995, 961–962. [Google Scholar]
- (598).Neumann WL; Rogic MM; Dunn TJ The Stereoselective Synthesis of Functionalized Vicinal Diamine Systems by the Double Allylation Reactions of “Protected” 1,2-Bis-Imine Precursors. Tetrahedron Lett. 1991, 32, 5865–5868. [Google Scholar]
- (599).Alvaro G; Grepioni F; Savoia D Synthesis and X-Ray Crystal Structure of N,N-Bis[(S)-1-phenylethyl]-(R,R)-4,5-diamino-1,7-octadiene. J. Org. Chem 1997, 62, 4180–4182. [DOI] [PubMed] [Google Scholar]
- (600).Beresford KJM; Howe GP; Procter G Asymmetric Synthesis via Nucleophilic Addition to α,β-Epoxyimines. Tetrahedron Lett. 1992, 33, 3355–3358. [Google Scholar]
- (601).Masamune S; Choy W; Petersen JS; Sita LR Double Asymmetric Synthesis and a New Strategy for Stereochemical Control in Organic Synthesis. Angew. Chem. Int. Ed. Engl 1985, 24, 1–30. [Google Scholar]
- (602).Alvaro G; Savoia D Addition of Allylmetal Reagents to the Imine Derived from 2-Pyridine Carboxaldehyde and Methyl (S)-Valinate. Synthesis of (S)- and (R)-1-(2-pyridyl)-3-butenamine. Tetrahedron: Asymmetry 1996, 7, 2083–2092. [Google Scholar]
- (603).Steinig AG; Spero DM Highly Diastereoselective Addition of Grignard Reagents to Aliphatic, Enolizable N-Alkylketimines and 2,2-Disubstituted 1,3-Oxazolidines. Asymmetric Synthesis of the Antidepressant Cericlamine. J. Org. Chem 1999, 64, 2406–2410. [Google Scholar]
- (604).Denmark SE; Nicaise O Organocerium Additions to Chiral α,α-Dialkoxy Hydrazones: Asymmetric Synthesis of N-Protected α-Amino Acetals and α-Amino Aldehydes. Synlett 1993, 359–361. [Google Scholar]
- (605).Denmark SE; Weber T; Piotrowski DW Organocerium Additions to SAMP-Hydrazones: General Synthesis of Chiral Amines. J. Am. Chem. Soc 1987, 109, 2224–2225. [Google Scholar]
- (606).Boezio AA; Solberghe G; Lauzon C; Charette AB Orthoacylimines: A New Class of Chiral Auxiliaries for Nucleophilic Addition of Organolithium Reagents to Imines. J. Org. Chem 2003, 68, 3241–3245. [DOI] [PubMed] [Google Scholar]
- (607).Moody CJ; Gallagher PT; Lightfoot AP; Slawin AMZ Chiral Oxime Ethers in Asymmetric Synthesis. 3. Asymmetric Synthesis of (R)-N-Protected α-Amino Acids by the Addition of Organometallic Reagents to the ROPHy Oxime of Cinnamaldehyde. J. Org. Chem 1999, 64, 4419–4425. [Google Scholar]
- (608).Qiao S; Pindi S; Spigener PT; Jiang B; Li G Asymmetric Synthesis of Homoallylic Amines via 1,2-Addition of Grignard Reagent to Aliphatic N-Phosphonyl Hemiaminal. Tetrahedron Lett. 2016, 57, 619–622. [Google Scholar]
- (609).Kattuboina A; Kaur P; Nguyen T; Li G Chiral N-Phosphonyl Imine Chemistry: Asymmetric 1,2-Additions of Allylmagnesium Bromides. Tetrahedron Lett. 2008, 49, 3722–3724. [Google Scholar]
- (610).Chang Z-Y; Coates RM Enantioselective Synthesis of Primary Amines via Grignard Additions to Stereogenic N-(α-Phenyl-β-(benzyloxy)ethyl)nitrones. J. Org. Chem 1990, 55, 3475–3483. [Google Scholar]
- (611).Zhu Q; Cheng L; Lu Y Asymmetric Organocatalytic Michael Addition of Ketones to Vinylsulfone. Chem. Commun 2008, 6315–6317. [DOI] [PubMed] [Google Scholar]
- (612).Ritter RH; Cohen T Carbenoid Anion Behavior of Dilithio Derivatives of Thioacetal Alcohols. Stereochemistry and Mechanism of Ring Closures by Oxyanion-Facilitated CH Bond Insertion. J. Am. Chem. Soc 1986, 108, 3718–3725. [Google Scholar]