

Abstract
Diabetic cardiomyopathy (DCM) is a chronic complication of diabetes mellitus, characterized by abnormalities of myocardial structure and function. Researches on the models of type 1 and type 2 diabetes mellitus as well as the application of genetic engineering technology help in understanding the molecular mechanism of DCM. DCM has multiple hallmarks, including hyperglycemia, insulin resistance, increased free radical production, lipid peroxidation, mitochondrial dysfunction, endothelial dysfunction, and cell death. Essentially, cell death is considered to be the terminal pathway of cardiomyocytes during DCM. Morphologically, cell death can be classified into four different forms: apoptosis, autophagy, necrosis, and entosis. Apoptosis, as type I cell death, is the fastest form of cell death and mainly occurs depending on the caspase proteolytic cascade. Autophagy, as type II cell death, is a degradation process to remove damaged proteins, dysfunctional organelles and commences by the formation of autophagosome. Necrosis is type III cell death, which contains a great diversity of cell death processes, such as necroptosis and pyroptosis. Entosis is type IV cell death, displaying “cell-in-cell” cytological features and requires the engulfing cells to execute. There are also some other types of cell death such as ferroptosis, parthanatos, netotic cell death, lysosomal dependent cell death, alkaliptosis or oxeiptosis, which are possibly involved in DCM. Drugs or compounds targeting the signals involved in cell death have been used in clinics or experiments to treat DCM. This review briefly summarizes the mechanisms and implications of cell death in DCM, which is beneficial to improve the understanding of cell death in DCM and may propose novel and ideal strategies in future.
Keywords: diabetic cardiomyopathy, cell death, apoptosis, autophagy, necrosis, entosis
Introduction
Diabetic cardiomyopathy (DCM) is manifested as specific abnormalities of myocardial structure and function in diabetic patients without hypertension or coronary artery disease in clinic. DCM is regarded as the one of the most common complications of diabetes to increase the risk of heart failure (Paolillo et al., 2019). Hyperglycemia, insulin resistance, fatty acids, oxidative stress, inflammation, myocardial fibrosis and hypertrophy, mitochondrial dysfunction, endoplasmic reticulum stress, and endothelial dysfunction are possible molecular foundations for DCM (Wang et al., 2014; Cao et al., 2015; Chen et al., 2018; Li et al., 2018a). Reactive oxygen species (ROS) produced by fatty-acid oxidation or nicotinamide adenine dinucleotide phosphate (NADPH) oxidase induce cell death or tissue damage (Parim et al., 2019). Essentially, cell death is considered to be the terminal pathway of cardiomyocytes during DCM. Experimental models of DCM show that metabolic dysfunction, cardiac structural, or functional abnormalities are similar to the pathology of human DCM. Streptozotocin (STZ) is the most common agent to induce type 1 diabetes mellitus (T1DM). Zucker diabetic fatty rat, leptin receptor deficiency mice (db/db), and leptin deficiency (ob/ob) mice are useful models to study type 2 DM (T2DM). Recently, echocardiography, computed tomography, magnetic resonance imaging, and single-photon emission computed tomography are used for the diagnosis of DCM (Adingupu et al., 2019; Parim et al., 2019; Tan et al., 2019). In vitro, DCM is often imitated in rat embryonic heart derived H9C2 cells, neonatal cardiomyocytes or cardiac fibroblasts with high glucose (HG) or advance glycation end product (AGE) stimulation (Sun et al., 2019a; Tang et al., 2019b; Wang et al., 2019a).
A large number of studies have shown several distinct types of cardiomyocyte death. Morphologically, cell death can be classified into four different forms. (1) Type I or apoptosis, featuring with cytoplasmic shrinkage, nuclear pyknosis, karyorrhexis, DNA fragmentation, and plasma membrane blebbing, and eventually apoptotic body formation. (2) Type II, or autophagy, showing extensive cytoplasmic vacuolization to form autophagosome, phagocytosis and subsequent lysosomal degradation. (3) Type III or necrosis, manifesting with distinctive morphology, is different from type I and type II cell death. Its morphological changes contain organelle and plasma membrane rupture, finalizing with disposal of cell corpses absent of evident phagocytic and lysosomal involvement. (4) Type IV or entosis, displaying “cell-in-cell” cytological features in which cell winners engulf and kill cell losers (Martins et al., 2017; Tang et al., 2019a) (Figure 1). Biopsy showed that the apoptosis of a diabetic heart was 85 times higher than that of the nondiabetic heart, indicating that cardiomyocytes in diabetes were sensitive to suffer cell death (Cai and Kang, 2003). To understand the detailed mechanism of cell death in cardiomyocytes is beneficial to propose novel and ideal strategies for DCM.
Figure 1.

Classification of cell death. Morphologically, cell death is classified into four different forms: type I or apoptosis, type II or autophagy, type III or necrosis, and type IV or entosis.
Mechanisms and Implications of Cell Death in DCM
Apoptosis in DCM
Apoptosis is also called programmed cell death (PCD), which is the fastest form of cell death. Proteolytic cascade induced by caspases is the key biochemical characteristic of apoptosis. Caspases with an inactive proenzyme form are widely expressed in cardiomyocytes. Immediately after activation, a protease cascade is initiated by other procaspases depending on their proteolytic activity. This proteolytic cascade amplifies apoptotic pathways and finally results in rapid and irreversible cell death (Huang et al., 2019). There are two main pathways, including extrinsic apoptotic pathway and intrinsic apoptotic pathway, involved in the mechanisms of apoptosis. The extrinsic apoptotic pathway is also known as death receptor pathway, during which multiple death ligands, such as tumor necrosis factor-α (TNF-α) and Fas, bind to their homologous receptors to trigger cell death. Then, caspase 8 and caspase 9 are initiated to subsequently activate executioner caspases (caspase 3, caspase 6, and caspase 7), and finally result in apoptosis. The intrinsic pathway is known as mitochondrial pathway. During intrinsic pathway, a group of proteins called the complex permeability transition pore (CPTP) form a megapore and cover the inner and outer membranes of the mitochondria. Mitochondrial proteins such as cytochrome C (Cyto C), high temperature requirement protein A2 (HtrA2)/Omi, and second mitochondria-derived activator of caspases/direct inhibitors of apoptosis-binding protein with low pI (Smac/Diablo) are released into the cytosol. Then, Cyto C combines with apoptosis protease activating factor-1 (Apaf-1) to form a complex named “apoptosome”, which serves as a platform for the cleavage and activation of downstream caspases (D'Arcy, 2019). Activation of caspases ultimately leads to cell dismantling (Figure 2).
Figure 2.

Mechanism of apoptosis in DCM. Both extrinsic apoptotic pathway and intrinsic apoptotic pathway are involved in DCM. Extrinsic pathway: long-term hyperglycemia triggers TNF-α to bind to TNF-α receptor (TNFR), initiates caspase 8 and subsequent caspase 3 activation, and finally results in apoptosis in the cardiomyocytes. Intrinsic pathway: Cyto C released into the cytosol combines with Apaf-1 to form a complex serving as a platform for caspase 9 and caspase 3 activation. Apoptosis induces cardiomyocyte cell loss to ultimately promote DCM.
A great number of studies have demonstrated that long-term hyperglycemia in diabetic patients induces cardiomyocyte apoptosis (Joubert et al., 2019). Cardiomyocyte apoptosis leads to cell loss to decrease cardiac contractile function and to ultimately promote cardiac remodeling (Hu et al., 2017). It has been proven that astragalus polysaccharides (APS) protected HG induced-H9C2 cell apoptosis by reducing Cyto C release and inhibiting caspase activity. That is to say APS has the potential ability to attenuate DCM through suppressing both extrinsic and intrinsic apoptotic pathways (Sun et al., 2017). One study reported that nicorandil decreased TdT-mediated DUTP nick end labeling (TUNEL)-positive cells, B-cell lymphoma-2 (bcl-2)-associated X (bax) as well as cleaved caspase 3 expression, while it increased bcl-2 expression in the heart of diabetic rats. Moreover, 5-HD, the competitive antagonist of nicorandil, increased apoptosis but reduced the phosphorylation of phosphatidylinositol 3-kinase (P13K), protein kinase B (AKT), endothelial nitric oxide synthase (eNOS), and mammalian target of rapamycin (mTOR) in H9C2 cardiomyocyte with high glucose stimulation. These data demonstrated that nicorandil alleviated hyperglycemia-caused cardiomyocyte apoptosis (Wang et al., 2019c). Li et al. found that long noncoding RNAs H19/MicroRNA-675 axis was involved in HG-induced cardiomyocyte apoptosis by downregulation of voltage-dependent anion channel 1 (VDAC1), which was required for the mitochondria-mediated apoptosis (Li et al., 2016). Recent studies also demonstrated that miR-186-5p overexpression suppressed apoptosis in HG-treated cardiomyocytes (Liu et al., 2019). Chronic and severe endoplasmic reticulum (ER) stress also resulted in cell apoptosis. APS provided cardioprotective effects on DCM by inhibiting cardiomyocytes apoptosis via suppressing protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 (ATF6)-related pathway of ER stress (Sun et al., 2019c). Matrine induced caspase 3 and caspase 9 down-regulation along with bcl-2 and p53 up-regulation. In addition, matrine administration inhibited the transforming growth factor beta (TGF-β) induced PERK signal pathway activation, which was involved in ER stress-induced apoptosis (Hou et al., 2019). Together, these events suggest that apoptosis promotes cardiomyocyte damage during DCM through multiple upstream signal pathways. Thus, compounds or molecules inhibiting apoptosis may serve as potential therapeutic agents for DCM.
Autophagy in DCM
Autophagy is initially described as a degradation process to eliminate damaged proteins and dysfunctional organelles. Autophagy is usually induced by nutrient deprivation, hypoxia, oxidative stress, genotoxic stress, or high glucose. Autophagy commences by autophagosome formation, a double-membrane vesicle swallowing up cytoplasmic materials (Meng et al., 2019). Autophagy is tightly regulated by autophagy-related (ATG) proteins, which is encoded by a family of highly conserved genes. Multiple signaling pathways including adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK), mTOR, unc-51-like kinase 1 (ULK1), PI3K/AKT, GTPases, calcium, and protein synthesis are involved in autophagy (Yang et al., 2005; Li et al., 2017; Bootman et al., 2018; Li et al., 2018b; Wang et al., 2019b).
The real role of autophagy on DCM is quite conflicting. Various studies have demonstrated the cardioprotective effect of autophagy in the heart of diabetic animals (Bhattacharya et al., 2018). In a study that explored the effect of aldehyde dehydrogenase 2 (ALDH2) on diabetes-induced myocardial dysfunction, there was decreased ratio of microtubule-associated protein 1 light chain 3-II (LC3II)-to-LC3I, reduced autophagy related 7 (ATG7) expression but increased sequestosome 1 (p62) levels, suggesting depressed autophagy in diabetes. ALDH2 overexpression or its agonist Alda-1 improved autophagy to reverse diabetes or high glucose-induced dysfunctions via AMPK and forkhead box O3a (FOXO3a) (Guo et al., 2015). Sirtuin3 (Sirt3) deficiency decreased LC3 puncta, autophagosomes, and LC3II level, via acetylated-Foxo3A enhancement and parkin inhibition, indicating that Sirt3 deficiency exacerbates diabetic cardiac dysfunction (Yu et al., 2017). Activating cannabinoid receptor 2 (CB2) produced a cardioprotective effect in DCM as well as cardiomyocytes under HG challenge through inducing AMPK-mTOR-p70S6K signaling-mediated autophagy (Wu et al., 2018a). Surprisingly, another study suggested that diminished autophagy gave adaptive response to limit diabetic cardiac injury in type 1 diabetic mice (Xu et al., 2013). Consistently, diabetes-associated myocardial mitochondrial dysfunction was associated with enhanced autophagy in the myocardium of diabetic Goto-Kakizaki rats (Liu et al., 2014). Hence, determining whether autophagy is adaptive or maladaptive is critical for the execution of efficient therapeutic treatment for DCM.
Necrosis in DCM
Necrotic cell death contains a great diversity of cell death processes. Necrosis may occur in the case of extensive damage such as high temperature and mechanical stress, leading to cell integrity destruction (Marunouchi and Tanonaka, 2015). In this case, necrosis is passive and does not require any specific signaling pathways. Another type of necrosis, called secondary necrosis, occurs at the late stage of apoptosis or autophagy when dead cells can't be removed by phagocytosis. Secondary necrosis is regarded as independent of initial signal events such as apoptosis or autophagy (Wu et al., 2018b). However, necrosis can also be a result of signaling cascade. Early study showed that diabetes increased necrosis by fourfold in cardiomyocytes (Frustaci et al., 2000).
Necroptosis is the best-characterized form of programmed necrosis, showing features of both apoptosis and necrosis (Tang et al., 2019a). The pathway of necroptosis activation after high glucose stimulation is mediated by ligand to death receptors such as tumor necrosis factor receptor 1 (TNFR1) and Fas receptors. Damage-associated molecular patterns (DAMPs), nucleotide-binding, and oligomerization domain (NOD)-like receptors (NLRs) ripoptosome, and protein kinase R (PKR) complexes also trigger necroptosis. TNFR1-mediated necroptosis is the most thoroughly studied pathway to activate necroptosis. After TNF-α binds to TNFR1, there are three possible divergent functions including cell survival, apoptosis, or necroptosis via different signaling complexes. Complex I is prosurvival and Complex IIa is proapoptotic. Complex IIb formation results in necroptosis (Galluzzi et al., 2018). Receptor-interacting protein 3 (RIP3) is regarded as a critical regulator of necroptosis. RIP3 regulates necroptosis on a RIP1-dependent manner in diabetes. Once recruited by RIP1 after HG exposure, RIP3 is activated by auto-phosphorylation to promote mixed lineage kinase domain like protein (MLKL) phosphorylation. Then, MLKL oligomerizes and translocates to the cell membrane, interacts with phosphatidylinositol lipids and cadiolipin to result in membrane permeabilization (Gupta et al., 2018). Calmodulin-dependent protein kinase II (CaMKII) is a newly found RIP3 substrate to induce necroptosis. CaMKII is abundant in the myocardium and is inactivated under normal conditions. CaMKII phosphorylation by intracellular Ca2+ or RIP1 facilitates necroptosis (Nomura et al., 2014). Meanwhile, HG also increases ROS to activate CaMKII by oxidation (Feng and Anderson, 2017). Altogether, the above pathological changes trigger the opening of mitochondrial permeability transition pore (mPTP), which is involved in the final pathway of necroptosis (Figure 3). Some research found that knockdown of cyclophilin D (CypD), a protein that increases mPTP opening probability, protected against RIP3-induced cardiomyocyte necrosis (Zhang et al., 2016). RIP3 up-regulation increased phosphoglycerate mutase 5 (PGAM5) expression, enhanced CypD phosphorylation, and finally resulted in mPTP opening. Excessive opening of the mPTP also amplified the death signal and ultimately led to necroptosis in the endothelial cells (Zhou et al., 2018a).
Figure 3.

Mechanism of necroptosis in DCM. Once recruited by RIP1 after HG exposure, RIP3 is activated by auto-phosphorylation to promote the recruitment and activation of mixed lineage kinase domain like protein (MLKL). RIP3 also phosphorylates CaMKII to induce mPTP opening. High glucose also increases ROS to activate CaMKII by oxidation and finally triggers mPTP opening, which is a final pathway of necroptosis during DCM.
It was observed that when H9C2 cells were exposed to high glucose, cell viability, the activity of ALDH2 mRNA or protein expression were suppressed, while ROS levels as well as RIP1, RIP3, and MLKL expression were enhanced. Necrostain-1 (Nec-1, a specific inhibitor of necroptosis) or Alda-1 (the ALDH2 activator) attenuated HG-induced down-regulation of ALDH2 and enhancement of RIP1, RIP3 and MLKL. In general, ALDH2 activation protected against HG-induced H9C2 cell injury, partly by inhibiting necroptosis (Fang et al., 2018). Another study showed that HG markedly increased the expression of Toll-like receptor 4 (TLR4), which was attenuated by NAC, a ROS scavenger. HG also significantly increased the expression of RIP3, which was ameliorated by TAK-242 (an inhibitor of TLR4) or Nec-1. In addition, NAC mitigated HG-induced RIP3 expression up-regulation. More importantly, DZ (a mitochondrial KATP channel opener) and Pin (a non-selective KATP channel opener) reduced the increased levels of TLR4 and RIP3 induced by HG. Taken together, the opening of KATP channels inhibited necroptosis through ROS-TLR4 pathway to protect H9C2 cells against HG-induced damage (Liang et al., 2017). Our recent research revealed that inhibitor 1 of protein phosphatase 1 (I1PP1) over-expression alleviated CaMKIIδ alternative splicing disorder, suppressed ROS overproduction, inhibited CaMKII oxidation, suppressed necroptosis, and ultimately alleviated high glucose-induced cardiomyocyte injury (Sun et al., 2019a).
Pyroptosis is another novel form of programmed necrosis (Robinson et al., 2019). It is responsible for cellular lysis and extracellular release of proinflammatory cytokines such as interleukin-1β (IL-1β) and interleukin-18 (IL-18) by HG stimulation (Zeng et al., 2019). There are two different pathways including the canonical pathway and noncanonical pathway in pyroptosis. In the canonical pathway, cytoplasmic multiprotein complexes, named inflammasome, consist of the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family (including NLRP3, NLRP1, NLRC4, NLRP9 and NLRP6), the pyrin and HIN domain (PYHIN) protein families (absent in melanoma 2, AIM2), and pyrin proteins. Distinct inflammasomes are recognized by pathogen-associated molecular patterns (PAMPs) or DAMPs to activate caspase 1, leading to pyroptosis (Lee et al., 2019). In the noncanonical pathway, bacterial lipopolysaccharide (LPS) is delivered to the cytosol to activate caspase 11. Activated caspase 11 directly induces pyroptosis. The cleaved caspase 11 also activates the Gasdermin-D (GSDMD) to form a membrane pore. Meanwhile, the NLRP3 inflammasome is activated by the N-terminal fragment of GSDMD (GSDMD-N) to trigger pyroptosis via the canonical pathway (Zhao et al., 2019).
Emerging evidence has verified that pyroptosis is involved in the progression of DCM. Jeyabal et al. found that NLRP3, caspase 1, and ELAV-like RNA binding protein 1 (ELAVL1, also called Hu-Antigen, HuR) expression in both human diabetic heart and HG-exposed human ventricular cardiomyocytes were augmented. Further study identified HuR as a direct target of miR-9. MiR-9 mimic transfection reduced HuR, caspase 1, and IL-1β expression to inhibit HG-induced pyroptosis in human cardiomyocytes. This study highlighted that targeting miR-9/HuR would play a therapeutic role in cell death during DCM (Jeyabal et al., 2016). Recently, Wang et al. found that absent in melanoma 2 (AIM2) inflammasome, caspase-1, IL-1β, and GSDMD were elevated in the myocardium of diabetes mellitus. ROS was also increased in H9C2 cells with HG stimulation. AIM2-shRNA reduced caspase 1 and IL-1β expression, suppressed GSDMD-N level and attenuated HG-induced pyroptosis. Furthermore, AIM2 level dropped significantly if ROS was inhibited. This study indicated that AIM2 inhibition is beneficial to attenuate diabetic cardiomyopathy via alleviating GSDMD-N-related pyroptosis (Wang et al., 2019d) (Figure 4). These findings confirmed the distinctive role of pyroptosis in DCM. To inhibit the pyroptosis signaling pathways is to capably expand the potential therapeutic targets for DCM treatment.
Figure 4.
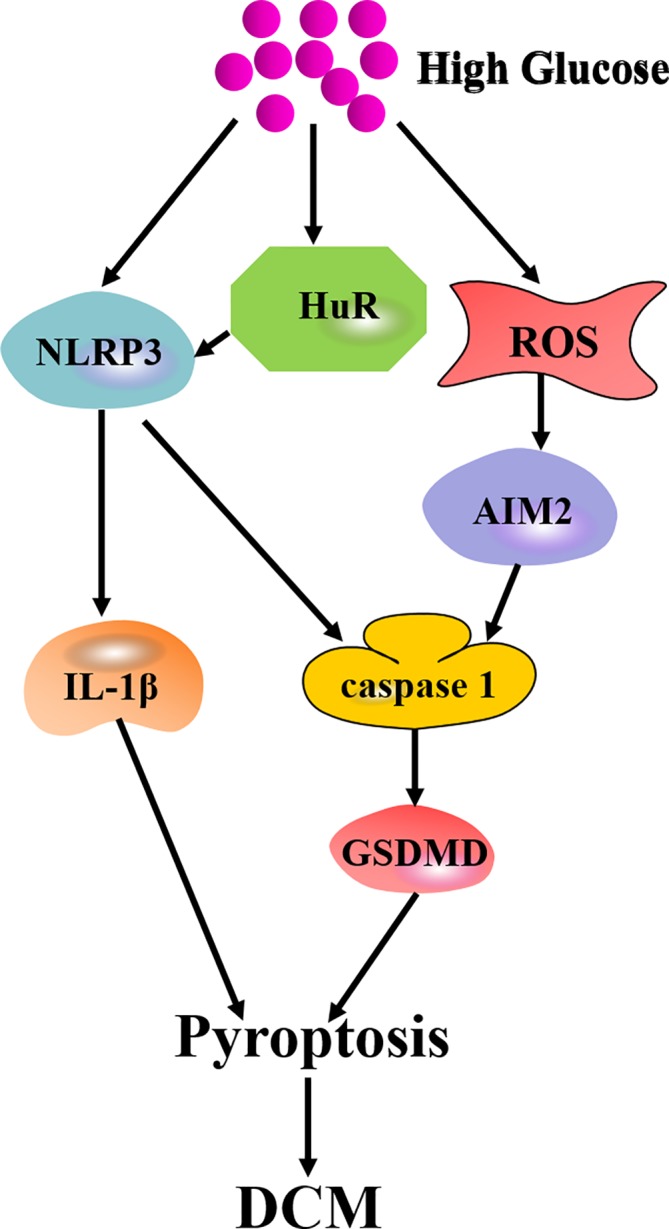
Mechanism of pyroptosis in DCM. High glucose augments expression of NLRP3 and HuR to activate IL-1β or caspase 1/GSDMD-mediated pyroptosis. High glucose also elevates ROS production to increase AIM2 expression and ultimately mediates pyroptosis through caspase 1/GSDMD pathway.
Other Types of Cell Death in DCM
Entosis seems fundamentally different from most typical forms of cell death due to the requirement of engulfing cells to execute. Entosis is mediated by cellular engulfment via E-cadherin, α-catenin, Rho family of GTPases, and rho-associated kinase (ROCK). Entosis is identified as an essential mechanism of embryo implantation (Li et al., 2015), which plays pro- and antitumorigenic roles in cancer. However, no data regarding the role of entosis in cardiovascular diseases has been found until now, and the possible mechanisms were incompletely defined.
Ferroptosis is an iron- and lipotoxicity-dependent form of regulated cell death (RCD). It is characterized by small mitochondria with reduced crista and condensed or ruptured outer membrane (Baba et al., 2018). Acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and arachidonate lipoxygenase (ALOXs) pathways mediate the oxidation of polyunsaturated fatty acids, which is necessary for the lipid toxicity of ferroptosis (Dixon et al., 2015; Chu et al., 2019; Xiao et al., 2019). ACSL4 up-regulation is the marker of ferroptosis (Doll et al., 2017). Decreasing antioxidant glutathione or inhibiting glutathione peroxidases inevitably enhanced ROS formation by erastin, a cell-permeable ferroptosis activator (Yang et al., 2014). Ferroptosis has been reported to be regulated during drug therapy; whether it participates in the pathology of DCM is unclear. In consideration of ROS formation promoting ferroptosis, there is a serious possibility that ferroptosis is involved in DCM.
Parthanatos is a poly ADP-ribose (PAR) polymerase-1 (PARP-1)-dependent type of cell death, which is activated by oxidative stress-induced DNA damage (Wang et al., 2019e). Neither apoptotic body formation nor DNA fragmentation is observed during parthanatos. Parthanatos does not produce cell swelling or lysosomal degradation. Energy exhaustion and apoptosis-inducing factor (AIF) release from the mitochondria mediated by PAR or calpain are possibly the main mechanisms of parthanatos. Parthanatos has been reported to be involved in cardiovascular diseases, renal diseases, diabetes, and neurodegeneration (Linkermann, 2016; Barany et al., 2017; Kam et al., 2018; Aizawa et al., 2019; Li et al., 2019d). PARP inhibitor L-2286 prevented cardiac remodeling, improved systolic function, and delayed the development of heart failure (Bartha et al., 2009). Thus, PARP inhibition may have the potential ability to attenuate DCM.
Netotic cell death is a form of RCD driven by neutrophil extracellular traps (NETs), which is regulated by NADPH oxidase-mediated ROS production and histone citrullination. NET formation and release, or NETosis relies on ROS production, autophagy, granular enzyme release, and translocation from the cytosol to the nucleus. Histone citrullination eventually leads to chromatin depletion, nuclear membrane destruction, and chromatin fiber release (Hemmers et al., 2011; Mitroulis et al., 2011; Tang et al., 2019a). Efforts could be endeavored to explore the mechanisms of NETosis underlying DCM, thereby providing new therapeutic policies for DCM.
Lysosomal-dependent cell death (LCD), also known as lysosomal cell death, is a form of regulated cell death mediated by intralysosomal components or iron translocation resulting from lysosomal membrane permeabilization (LMP) to amplify or initiate cell death during apoptosis, autophagy, and ferroptosis (Wang et al., 2018). Although the relation between LCD and DCM was easy to be deduced, further studies are still indispensable.
Alkaliptosis is driven by intracellular alkalization. It is known that down-regulation of inhibitor of nuclear factor kappa B kinase subunit beta (IKBKB) and nuclear factor-κB (NF-κB) induces alkaliptosis (Song et al., 2018). The pathological significance of alkaliptosis in human diseases remains complicated, and the main signals of allkaliptosis need to be illuminated in the future.
Oxeiptosis is a novel caspase-independent RCD induced by ROS, which is mediated through Kelch-like ECH-associated protein 1 (KEAP1)/PGAM5/apoptosis-inducing factor 1 mitochondrial (AIFM1) pathway (Scaturro and Pichlmair, 2018). Since ROS accumulation links to multiple physiological and pathological processes, it is very likely that oxeiptosis promotes the development of several diseases including DCM.
Different types of cell death are mediated by distinct but overlapping central pathways. One study found that autophagy was inhibited but apoptosis was amplified in diabetic heart (Xing et al., 2019). Elevated bax/bcl-2 ratio, increased cleaved caspase 3 level, augmented NLRP3, cleaved caspase 1, IL-1β, and cleaved GSDMD expression along with increased ROS production were detected in the myocardium of db/db mice, indicating the involvement of apoptosis and pyroptosis in DCM (Xue et al., 2019). Altogether, several types of cell death may coexist in the occurrence and development of DCM. Moreover, apoptosis, necrosis, necroptosis, pyroptosis, autophagy, or ferroptosis might act simultaneously during DCM. Inhibition of one kind of cell death may possibly promote other types of cell death and cause compensation, which eventually increases the complexity of pathogenesis about DCM and enhance difficulty of treatment for DCM.
Conclusion and Prospective
Current therapeutic strategies for DCM involve insulin and insulin-secreting agents, oral antihyperglycemic medication, β-blockers, angiotensin converting enzyme inhibitors, angiotensin II receptor antagonists, calciumion channel antagonists, and hydroxymethylglutaryl CoA reductase inhibitors. However, the above synthetic drugs have many adverse effects including gastrointestinal disturbances, hepatotoxicity, abdominal pain, genitourinary tract infection, and so on (Parim et al., 2019). As cell death is a determinant pathological fate in DCM, the executors of cell death signal pathways are obviously potential therapeutic targets for DCM. Many drugs or compounds are being used in or under clinical trials. Take some examples: Metformin, a common antidiabetic drug, activates AMPK and improves autophagy via inhibiting the mTOR pathway and alleviating pyroptosis (Yang et al., 2019). Crocin, a carotenoid extracted from saffron, improves the deteriorated cardiac function in diabetic animals by inhibiting apoptosis and normalizing autophagy (Feidantsis et al., 2018). Protein phosphatase 2A (PP2A) is a central cardiac phosphatase that regulates the functions of diverse myocytes through target molecules. Okadaic acid (OA), an inhibitor of PP2A, inhibited apoptosis of experimental diabetic mellitus-related cardiomyopathy (Guan et al., 2019). These researches will improve the understanding of cell death in DCM and set the stage for novel therapies in future. Several drug or compounds targeting the signals in cell death have been used in clinics or experiments for DCM (Table 1).
Table 1.
Drugs or compounds against cell death in diabetic cardiomyopathy.
| Drugs or compounds | Targets of cell death type | Effects | References |
|---|---|---|---|
| Astragalus Polysaccharides | apoptosis | oxidative stress↓, antioxidant capacity↑ |
(Sun et al., 2019b) |
| Catalpol | apoptosis | bax/bcl-2↓, cleaved caspase 3↓ |
(Zou et al., 2019b) |
| Crocim | autophagy | LC3II/LCI↑ | (Feidantsis et al., 2018) |
| apoptosis | bax/bcl-2↓, cleaved caspase 3↓ |
||
| Curcumin | apoptosis | cleaved caspase 3↓, cytochrome C↓, bcl-2↑ | (Yao et al., 2018) |
| autophagy | LC3II/LC3I↑, p62↓ | ||
| Dexmedetomidine | autophagy | ATG7↓ | (Oh et al., 2019) |
| Dihydromyricetin | autophagy | LC3II/LC3I↑, p62↓, ATG7↓ | (Wu et al., 2017) |
| apoptosis | bax/bcl-2↓, cleaved caspase3↓, cleaved caspase 9↓ |
||
| 1,25-Dihydroxyvitamn-D3 | autophagy | LC3II/LC3I↑ | (Wei et al., 2017) |
| Empagliflozin | apoptosis | bax/bcl-2↓, cleaved caspase 3↓ |
(Xue et al., 2019) |
| pyroptosis | NLRP3↓, caspase 1↓, IL-1β↓, cleaved GSDMD↓, full-length GSDMD↑ |
||
| GYY413 | apoptosis | bax/bcl-2↓, caspase 3↓ | (Li et al., 2019b) |
| Helix B surface peptide | autophagy | LC3II/LC3I↑, p62↓ | (Lin et al., 2017) |
| Metformin | pyroptosis | NLRP3↓, caspase 1↓, IL-1β↓, GSDMD-N↓ | (Zou et al., 2019a) |
| NaHS | necroptosis | RIP3↓, cleaved caspase 3↓ |
(Liang et al., 2016) |
| Nicorandil | apoptosis | bax/bcl-2↓, cleaved caspase 3↓ |
(Wang et al., 2019c) |
| Notoginsenoside R1 | apoptosis | mitochondrial membrane depolarization↓, ROS↓ | (Zhang et al., 2018) |
| Okadaic acid | apoptosis | bax/bcl-2↓, cleaved caspase 3↓ |
(Ren et al., 2016) |
| Palbociclib | apoptosis | bax/bcl-2↓, caspase 3↓ | (Wang et al., 2019f) |
| Piceatonnol | apoptosis | bax/bcl-2↓, caspase 3↓ | (Li et al., 2019a) |
| Resveratrol | autophagy | LC3II/LC3I↑, p62↓ | (Xu et al., 2018) |
| apoptosis | cleaved caspase 3↓ | ||
| Sitagliptin | autophagy | LC3II/LC3I↑, p62↓ | (Zhou et al., 2018b) |
| Sophocarpine | apoptosis | bax/bcl-2↓, cleaved caspse3↓, cleaved caspase 9↓ |
(Zou et al., 2019a) |
| Vaspin | autophagy | LC3II/LC3I↑, p62↓ | (Li et al., 2019c) |
Taken together, cell death is the terminal pathway of cardiomyocytes during DCM. Although upstream signal regulation seems to attenuate cell death effectively, it may produce a series of nonspecific effects. Targeting cell death directly may be more practicable than its upstream signaling pathway.
Author Contributions
YC researched documents and wrote the manuscript. YH, XL, IA, and WZ revised the manuscript. GM designed the study and reviewed the manuscript. All authors approved the final version of the manuscript.
Funding
The work was funded by grants (81770279, 81670243 and 81873470) from the National Natural Science Foundation of China, a major project of Natural Science Research in Jiangsu Higher Education Institutions (18KJA310005), the Six Talent Peaks Project in Jiangsu Province (2018-WSN-062), grants from the China Postdoctoral Science Foundation (2017M610342 and 2019T120449), a Jiangsu Planned Project for Postdoctoral Research Funds (1701050A), a Research and Innovation Project of Graduate Students in Jiangsu Province (KYCX18_2401), a Science and Technology Project of Taicang City (TC2019KJFZ02), and the Nantong University Cooperative Innovation Program of Small Molecular Compound R&D (NTU2016-1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Adingupu D. D., Gopel S. O., Gronros J., Behrendt M., Sotak M., Miliotis T., et al. (2019). SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob(-/-) mice. Cardiovasc. Diabetol. 18, 16. 10.1186/s12933-019-0820-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizawa S., Brar G., Tsukamoto H. (2019). Cell death and liver disease. Gut. Liver. 14, 20–29. 10.5009/gnl18486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba Y., Higa J. K., Shimada B. K., Horiuchi K. M., Suhara T., Kobayashi M., et al. (2018). Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 314, H659–H668. 10.1152/ajpheart.00452.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barany T., Simon A., Szabo G., Benko R., Mezei Z., Molnar L., et al. (2017). Oxidative stress-related parthanatos of circulating mononuclear leukocytes in heart failure. Oxid. Med. Cell. Longev. 2017, 1249614. 10.1155/2017/1249614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartha E., Solti I., Kereskai L., Lantos J., Plozer E., Magyar K., et al. (2009). PARP inhibition delays transition of hypertensive cardiopathy to heart failure in spontaneously hypertensive rats. Cardiovasc. Res. 83, 501–510. 10.1093/cvr/cvp144 [DOI] [PubMed] [Google Scholar]
- Bhattacharya D., Mukhopadhyay M., Bhattacharyya M., Karmakar P. (2018). Is autophagy associated with diabetes mellitus and its complications? A review. EXCLI. J. 17, 709–720. 10.17179/excli2018-1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootman M. D., Chehab T., Bultynck G., Parys J. B., Rietdorf K. (2018). The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium. 70, 32–46. 10.1016/j.ceca.2017.08.005 [DOI] [PubMed] [Google Scholar]
- Cai L., Kang Y. J. (2003). Cell death and diabetic cardiomyopathy. Cardiovasc. Toxicol. 3, 219–228. 10.1385/CT:3:3:219 [DOI] [PubMed] [Google Scholar]
- Cao H., Chen T., Shi Y. (2015). Glycation of human serum albumin in diabetes: impacts on the structure and function. Curr. Med. Chem. 22, 4–13. 10.2174/0929867321666140912155738 [DOI] [PubMed] [Google Scholar]
- Chen X. F., Li X. L., Yang M., Song Y., Zhang Y. (2018). Osteoprotective effects of salidroside in ovariectomized mice and diabetic mice. Eur. J. Pharmacol. 819, 281–288. 10.1016/j.ejphar.2017.12.025 [DOI] [PubMed] [Google Scholar]
- Chu B., Kon N., Chen D., Li T., Liu T., Jiang L., et al. (2019). ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 21, 579–591. 10.1038/s41556-019-0305-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcy M. S. (2019). Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 43, 582–592. 10.1002/cbin.11137 [DOI] [PubMed] [Google Scholar]
- Dixon S. J., Winter G. E., Musavi L. S., Lee E. D., Snijder B., Rebsamen M., et al. (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609. 10.1021/acschembio.5b00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S., Proneth B., Tyurina Y. Y., Panzilius E., Kobayashi S., Ingold I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. 10.1038/nchembio.2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang T., Cao R., Wang W., Ye H., Shen L., Li Z., et al. (2018). Alterations in necroptosis during ALDH2mediated protection against high glucoseinduced H9c2 cardiac cell injury. Mol. Med. Rep. 18, 2807–2815. 10.3892/mmr.2018.9269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feidantsis K., Mellidis K., Galatou E., Sinakos Z., Lazou A. (2018). Treatment with crocin improves cardiac dysfunction by normalizing autophagy and inhibiting apoptosis in STZ-induced diabetic cardiomyopathy. Nutr. Metab. Cardiovasc. Dis. 28, 952–961. 10.1016/j.numecd.2018.06.005 [DOI] [PubMed] [Google Scholar]
- Feng N., Anderson M. E. (2017). CaMKII is a nodal signal for multiple programmed cell death pathways in heart. J. Mol. Cell. Cardiol. 103, 102–109. 10.1016/j.yjmcc.2016.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frustaci A., Kajstura J., Chimenti C., Jakoniuk I., Leri A., Maseri A., et al. (2000). Myocardial cell death in human diabetes. Circ. Res. 87, 1123–1132. 10.1161/01.RES.87.12.1123 [DOI] [PubMed] [Google Scholar]
- Galluzzi L., Vitale I., Aaronson S. A., Abrams J. M., Adam D., Agostinis P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541. 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Zhou L., Zhang Y., Tian H., Li A., Han X. (2019). Effects of PP2A/Nrf2 on experimental diabetes mellitus-related cardiomyopathy by regulation of autophagy and apoptosis through ROS dependent pathway. Cell. Signal. 62, 109339. 10.1016/j.cellsig.2019.06.004 [DOI] [PubMed] [Google Scholar]
- Guo Y., Yu W., Sun D., Wang J., Li C., Zhang R., et al. (2015). A novel protective mechanism for mitochondrial aldehyde dehydrogenase (ALDH2) in type i diabetes-induced cardiac dysfunction: role of AMPK-regulated autophagy. Biochim. Biophys. Acta 1852, 319–331. 10.1016/j.bbadis.2014.05.017 [DOI] [PubMed] [Google Scholar]
- Gupta K., Phan N., Wang Q., Liu B. (2018). Necroptosis in cardiovascular disease - a new therapeutic target. J. Mol. Cell. Cardiol. 118, 26–35. 10.1016/j.yjmcc.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmers S., Teijaro J. R., Arandjelovic S., Mowen K. A. (2011). PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PloS One 6, e22043. 10.1371/journal.pone.0022043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H., Zhang Q., Dong H., Ge Z. (2019). Matrine improves diabetic cardiomyopathy through TGF-beta-induced protein kinase RNA-like endoplasmic reticulum kinase signaling pathway. J. Cell. Biochem. 120, 13573–13582. 10.1002/jcb.28632 [DOI] [PubMed] [Google Scholar]
- Hu X., Bai T., Xu Z., Liu Q., Zheng Y., Cai L. (2017). Pathophysiological fundamentals of diabetic cardiomyopathy. Compr. Physiol. 7, 693–711. 10.1002/cphy.c160021 [DOI] [PubMed] [Google Scholar]
- Huang M. L., Chiang S., Kalinowski D. S., Bae D. H., Sahni S., Richardson D. R. (2019). The role of the antioxidant response in mitochondrial dysfunction in degenerative diseases: cross-talk between antioxidant defense, autophagy, and apoptosis. Oxid. Med. Cell. Longev. 2019, 6392763. 10.1155/2019/6392763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyabal P., Thandavarayan R. A., Joladarashi D., Suresh Babu S., Krishnamurthy S., Bhimaraj A., et al. (2016). MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem. Biophys. Res. Commun. 471, 423–429. 10.1016/j.bbrc.2016.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert M., Manrique A., Cariou B., Prieur X. (2019). Diabetes-related cardiomyopathy: the sweet story of glucose overload from epidemiology to cellular pathways. Diabetes Metab. 45, 238–247. 10.1016/j.diabet.2018.07.003 [DOI] [PubMed] [Google Scholar]
- Kam T. I., Mao X., Park H., Chou S. C., Karuppagounder S. S., Umanah G. E., et al. (2018). Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson's disease. Science 362, eaat8407. 10.1126/science.aat8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C., Do H. T. T., Her J., Kim Y., Seo D., Rhee I. (2019). Inflammasome as a promising therapeutic target for cancer. Life Sci. 231, 116593. 10.1016/j.lfs.2019.116593 [DOI] [PubMed] [Google Scholar]
- Li Y., Sun X., Dey S. K. (2015). Entosis allows timely elimination of the luminal epithelial barrier for embryo implantation. Cell Rep. 11, 358–365. 10.1016/j.celrep.2015.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Wang H., Yao B., Xu W., Chen J., Zhou X. (2016). lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci. Rep. 6, 36340. 10.1038/srep36340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Song Y., Liu L., Hou N., An X., Zhan D., et al. (2017). miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 24, 1205–1213. 10.1038/cdd.2015.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G. X., Jiao X. H., Cheng X. B. (2018. a). Correlations between blood uric acid and the incidence and progression of type 2 diabetes nephropathy. Eur. Rev. Med. Pharmacol. Sci. 22, 506–511. 10.26355/eurrev_201801_14202 [DOI] [PubMed] [Google Scholar]
- Li Y., Wang Y., Zou M., Chen C., Chen Y., Xue R., et al. (2018. b). AMPK blunts chronic heart failure by inhibiting autophagy. Biosci. Rep. 38 BSR20170982 10.1042/BSR20170982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Shi Y., Wang X., Li P., Zhang S., Wu T., et al. (2019. a). Piceatannol alleviates inflammation and oxidative stress via modulation of the Nrf2/HO-1 and NF-kappaB pathways in diabetic cardiomyopathy. Chem. Biol. Interact. 310, 108754. 10.1016/j.cbi.2019.108754 [DOI] [PubMed] [Google Scholar]
- Li J., Yuan Y. Q., Zhang L., Zhang H., Zhang S. W., Zhang Y., et al. (2019. b). Exogenous hydrogen sulfide protects against high glucose-induced apoptosis and oxidative stress by inhibiting the STAT3/HIF-1alpha pathway in H9c2 cardiomyocytes. Exp. Ther. Med. 18, 3948–3958. 10.3892/etm.2019.8036 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li X., Ke X., Li Z., Li B. (2019. c). Vaspin prevents myocardial injury in rats model of diabetic cardiomyopathy by enhancing autophagy and inhibiting inflammation. Biochem. Biophys. Res. Commun. 514, 1–8. 10.1016/j.bbrc.2019.04.110 [DOI] [PubMed] [Google Scholar]
- Li Y., Yang Y., Zhao Y., Zhang J., Liu B., Jiao S., et al. (2019. d). Astragaloside IV reduces neuronal apoptosis and parthanatos in ischemic injury by preserving mitochondrial hexokinase-II. Free Radic. Biol. Med. 131, 251–263. 10.1016/j.freeradbiomed.2018.11.033 [DOI] [PubMed] [Google Scholar]
- Liang W., Chen J., Mo L., Ke X., Zhang W., Zheng D., et al. (2016). ATP-sensitive K(+) channels contribute to the protective effects of exogenous hydrogen sulfide against high glucose-induced injury in H9c2 cardiac cells. Int. J. Mol. Med. 37, 763–772. 10.3892/ijmm.2016.2467 [DOI] [PubMed] [Google Scholar]
- Liang W., Chen M., Zheng D., Li J., Song M., Zhang W., et al. (2017). The opening of ATP-Sensitive K+ channels protects H9c2 cardiac cells against the high glucose-induced injury and inflammation by Inhibiting the ROS-TLR4-necroptosis pathway. Cell. Physiol. Biochem. 41, 1020–1034. 10.1159/000461391 [DOI] [PubMed] [Google Scholar]
- Lin C., Zhang M., Zhang Y., Yang K., Hu J., Si R., et al. (2017). Helix B surface peptide attenuates diabetic cardiomyopathy via AMPK-dependent autophagy. Biochem. Biophys. Res. Commun. 482, 665–671. 10.1016/j.bbrc.2016.11.091 [DOI] [PubMed] [Google Scholar]
- Linkermann A. (2016). Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int. 89, 46–57. 10.1016/j.kint.2015.10.008 [DOI] [PubMed] [Google Scholar]
- Liu J., Tang Y., Feng Z., Liu J., Liu J., Long J. (2014). (-)-Epigallocatechin-3-gallate attenuated myocardial mitochondrial dysfunction and autophagy in diabetic Goto-Kakizaki rats. Free Radic. Res. 48, 898–906. 10.3109/10715762.2014.920955 [DOI] [PubMed] [Google Scholar]
- Liu Y., Zheng W., Pan Y., Hu J. (2019). Low expression of miR-186-5p regulates cell apoptosis by targeting toll-like receptor 3 in high glucose-induced cardiomyocytes. J. Cell. Biochem. 120, 9532–9538. 10.1002/jcb.28229 [DOI] [PubMed] [Google Scholar]
- Martins I., Raza S. Q., Voisin L., Dakhli H., Law F., De Jong D., et al. (2017). Entosis: The emerging face of non-cell-autonomous type IV programmed death. Biomed. J. 40, 133–140. 10.1016/j.bj.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marunouchi T., Tanonaka K. (2015). Cell death in the cardiac myocyte. Biol. Pharm. Bull. 38, 1094–1097. 10.1248/bpb.b15-00288 [DOI] [PubMed] [Google Scholar]
- Meng T., Lin S., Zhuang H., Huang H., He Z., Hu Y., et al. (2019). Recent progress in the role of autophagy in neurological diseases. Cell Stress 3, 141–161. 10.15698/cst2019.05.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitroulis I., Kambas K., Chrysanthopoulou A., Skendros P., Apostolidou E., Kourtzelis I., et al. (2011). Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PloS One 6, e29318. 10.1371/journal.pone.0029318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M., Ueno A., Saga K., Fukuzawa M., Kaneda Y. (2014). Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 74, 1056–1066. 10.1158/0008-5472.CAN-13-1283 [DOI] [PubMed] [Google Scholar]
- Oh J. E., Jun J. H., Hwang H. J., Shin E. J., Oh Y. J., Choi Y. S. (2019). Dexmedetomidine restores autophagy and cardiac dysfunction in rats with streptozotocin-induced diabetes mellitus. Acta Diabetol. 56, 105–114. 10.1007/s00592-018-1225-9 [DOI] [PubMed] [Google Scholar]
- Paolillo S., Marsico F., Prastaro M., Renga F., Esposito L., De Martino F., et al. (2019). Diabetic cardiomyopathy: definition, diagnosis, and therapeutic implications. Heart Fail. Clin. 15, 341–347. 10.1016/j.hfc.2019.02.003 [DOI] [PubMed] [Google Scholar]
- Parim B., Sathibabu Uddandrao V. V., Saravanan G. (2019). Diabetic cardiomyopathy: molecular mechanisms, detrimental effects of conventional treatment, and beneficial effects of natural therapy. Heart Fail. Rev. 24, 279–299. 10.1007/s10741-018-9749-1 [DOI] [PubMed] [Google Scholar]
- Ren X. M., Zuo G. F., Wu W., Luo J., Ye P., Chen S. L., et al. (2016). Atorvastatin alleviates experimental diabetic cardiomyopathy by regulating the GSK-3beta-PP2Ac-NF-kappaB signaling axis. PloS One 11, e0166740. 10.1371/journal.pone.0166740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson N., Ganesan R., Hegedus C., Kovacs K., Kufer T. A., Virag L. (2019). Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 26, 101239. 10.1016/j.redox.2019.101239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaturro P., Pichlmair A. (2018). Oxeiptosis-a cell death pathway to mitigate damage caused by radicals. Cell Death Differ. 25, 1191–1193. 10.1038/s41418-018-0134-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X., Zhu S., Xie Y., Liu J., Sun L., Zeng D., et al. (2018). JTC801 induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology 154, 1480–1493. 10.1053/j.gastro.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Yang S., Dai M., Jia X., Wang Q., Zhang Z., et al. (2017). The effect of Astragalus polysaccharides on attenuation of diabetic cardiomyopathy through inhibiting the extrinsic and intrinsic apoptotic pathways in high glucose -stimulated H9C2 cells. BMC Complement. Altern. Med. 17, 310. 10.1186/s12906-017-1828-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Chen Y., Luo H., Xu M., Meng G., Zhang W. (2019. a). Ca(2+)/calmodulin-dependent protein kinase II regulation by inhibitor 1 of protein phosphatase 1 alleviates necroptosis in high glucose-induced cardiomyocytes injury. Biochem. Pharmacol. 163, 194–205. 10.1016/j.bcp.2019.02.022 [DOI] [PubMed] [Google Scholar]
- Sun Q., Wu X., Wang H., Chen W., Zhao X., Yang Y., et al. (2019. b). Protective effects of astragalus polysaccharides on oxidative stress in high glucose-induced Or SOD2-Silenced H9C2 cells based On PCR Array Analysis. Diabetes Metab. Syndr. Obes. 12, 2209–2220. 10.2147/DMSO.S228351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Yang S., An N., Wang G., Xu Q., Liu J., et al. (2019. c). Astragalus polysaccharides inhibits cardiomyocyte apoptosis during diabetic cardiomyopathy via the endoplasmic reticulum stress pathway. J. Ethnopharmacol. 238, 111857. 10.1016/j.jep.2019.111857 [DOI] [PubMed] [Google Scholar]
- Tan X., Hu L., Shu Z., Chen L., Li X., Du M., et al. (2019). Role of CCR2 in the development of streptozotocin-treated diabetic cardiomyopathy. Diabetes 68, 2063–2073. 10.2337/db18-1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Kang R., Berghe T. V., Vandenabeele P., Kroemer G. (2019. a). The molecular machinery of regulated cell death. Cell Res. 29, 347–364. 10.1038/s41422-019-0164-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S. G., Liu X. Y., Wang S. P., Wang H. H., Jovanovic A., Tan W. (2019. b). Trimetazidine prevents diabetic cardiomyopathy by inhibiting Nox2/TRPC3-induced oxidative stress. J. Pharmacol. Sci. 139, 311–318. 10.1016/j.jphs.2019.01.016 [DOI] [PubMed] [Google Scholar]
- Wang J., Liu H., Li N., Zhang Q., Zhang H. (2014). The protective effect of fucoidan in rats with streptozotocin-induced diabetic nephropathy. Mar. Drugs 12, 3292–3306. 10.3390/md12063292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Gomez-Sintes R., Boya P. (2018). Lysosomal membrane permeabilization and cell death. Traffic 19, 918–931. 10.1111/tra.12613 [DOI] [PubMed] [Google Scholar]
- Wang J., Tang Z., Zhang Y., Qiu C., Zhu L., Zhao N., et al. (2019. a). Matrine alleviates AGEs- induced cardiac dysfunctions by attenuating calcium overload via reducing ryanodine receptor 2 activity. Eur. J. Pharmacol. 842, 118–124. 10.1016/j.ejphar.2018.10.010 [DOI] [PubMed] [Google Scholar]
- Wang L., Yuan D., Zheng J., Wu X., Wang J., Liu X., et al. (2019. b). Chikusetsu saponin IVa attenuates isoprenaline-induced myocardial fibrosis in mice through activation autophagy mediated by AMPK/mTOR/ULK1 signaling. Phytomedicine 58, 152764. 10.1016/j.phymed.2018.11.024 [DOI] [PubMed] [Google Scholar]
- Wang X., Pan J., Liu D., Zhang M., Li X., Tian J., et al. (2019. c). Nicorandil alleviates apoptosis in diabetic cardiomyopathy through PI3K/Akt pathway. J. Cell. Mol. Med. 23, 5349–5359. 10.1111/jcmm.14413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Pan J., Liu H., Zhang M., Liu D., Lu L., et al. (2019. d). AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci. 221, 249–258. 10.1016/j.lfs.2019.02.035 [DOI] [PubMed] [Google Scholar]
- Wang Y., Luo W., Wang Y. (2019. e). PARP-1 and its associated nucleases in DNA damage response. DNA Repair (Amst) 81, 102651. 10.1016/j.dnarep.2019.102651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Li J., Wang Y., Liu Q. (2019. f). Palbociclib improves cardiac dysfunction in diabetic cardiomyopathy by regulating Rb phosphorylation. Am. J. Transl. Res. 11, 3481–3489. [PMC free article] [PubMed] [Google Scholar]
- Wei H., Qu H., Wang H., Ji B., Ding Y., Liu D., et al. (2017). 1,25-Dihydroxyvitamin-D3 prevents the development of diabetic cardiomyopathy in type 1 diabetic rats by enhancing autophagy via inhibiting the beta-catenin/TCF4/GSK-3beta/mTOR pathway. J. Steroid. Biochem. Mol. Biol. 168, 71–90. 10.1016/j.jsbmb.2017.02.007 [DOI] [PubMed] [Google Scholar]
- Wu B., Lin J., Luo J., Han D., Fan M., Guo T., et al. (2017). Dihydromyricetin protects against diabetic cardiomyopathy in streptozotocin-induced diabetic mice. Biomed. Res. Int. 2017, 3764370. 10.1155/2017/3764370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A., Hu P., Lin J., Xia W., Zhang R. (2018. a). Activating Cannabinoid receptor 2 protects against diabetic cardiomyopathy through autophagy induction. Front. Pharmacol. 9, 1292. 10.3389/fphar.2018.01292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M. Y., Yiang G. T., Liao W. T., Tsai A. P., Cheng Y. L., Cheng P. W., et al. (2018. b). Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol. Biochem. 46, 1650–1667. 10.1159/000489241 [DOI] [PubMed] [Google Scholar]
- Xiao F. J., Zhang D., Wu Y., Jia Q. H., Zhang L., Li Y. X., et al. (2019). miRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem. Biophys. Res. Commun. 515, 448–454. 10.1016/j.bbrc.2019.05.147 [DOI] [PubMed] [Google Scholar]
- Xing R., Liu D., Cheng X., Tian X., Yan C., Han Y. (2019). MiR-207 inhibits autophagy and promotes apoptosis of cardiomyocytes by directly targeting LAMP2 in type 2 diabetic cardiomyopathy. Biochem. Biophys. Res. Commun. 520, 27–34. 10.1016/j.bbrc.2019.09.092 [DOI] [PubMed] [Google Scholar]
- Xu X., Kobayashi S., Chen K., Timm D., Volden P., Huang Y., et al. (2013). Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J. Biol. Chem. 288, 18077–18092. 10.1074/jbc.M113.474650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K., Liu X. F., Ke Z. Q., Yao Q., Guo S., Liu C. (2018). Resveratrol modulates apoptosis and autophagy induced by high glucose and palmitate in cardiac cells. Cell Physiol. Biochem. 46, 2031–2040. 10.1159/000489442 [DOI] [PubMed] [Google Scholar]
- Xue M., Li T., Wang Y., Chang Y., Cheng Y., Lu Y., et al. (2019). Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin. Sci. (Lond) 133, 1705–1720. 10.1042/CS20190585 [DOI] [PubMed] [Google Scholar]
- Yang Y. P., Liang Z. Q., Gu Z. L., Qin Z. H. (2005). Molecular mechanism and regulation of autophagy. Acta Pharmacol. Sin. 26, 1421–1434. 10.1111/j.1745-7254.2005.00235.x [DOI] [PubMed] [Google Scholar]
- Yang W. S., SriRamaratnam R., Welsch M. E., Shimada K., Skouta R., Viswanathan V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F., Qin Y., Wang Y., Meng S., Xian H., Che H., et al. (2019). Metformin inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int. J. Biol. Sci. 15, 1010–1019. 10.7150/ijbs.29680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q., Ke Z. Q., Guo S., Yang X. S., Zhang F. X., Liu X. F., et al. (2018). Curcumin protects against diabetic cardiomyopathy by promoting autophagy and alleviating apoptosis. J. Mol. Cell. Cardiol. 124, 26–34. 10.1016/j.yjmcc.2018.10.004 [DOI] [PubMed] [Google Scholar]
- Yu W., Gao B., Li N., Wang J., Qiu C., Zhang G., et al. (2017). Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochim. Biophys. Acta Mol. Basis. Dis. 1863, 1973–1983. 10.1016/j.bbadis.2016.10.021 [DOI] [PubMed] [Google Scholar]
- Zeng C., Wang R., Tan H. (2019). Role of pyroptosis in cardiovascular diseases and its therapeutic implications. Int. J. Biol. Sci. 15, 1345–1357. 10.7150/ijbs.33568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Zhang Y., Cui M., Jin L., Wang Y., Lv F., et al. (2016). CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 22, 175–182. 10.1038/nm.4017 [DOI] [PubMed] [Google Scholar]
- Zhang B., Zhang J., Zhang C., Zhang X., Ye J., Kuang S., et al. (2018). Notoginsenoside R1 protects against diabetic cardiomyopathy through activating estrogen receptor alpha and its downstream signaling. Front. Pharmacol. 9, 1227. 10.3389/fphar.2018.01227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z., Guo L., Shi W., Zuo W. (2019). Role of pyroptosis in cardiovascular disease. Cell Prolif. 52, e12563. 10.1111/cpr.12563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Li D., Zhu P., Ma Q., Toan S., Wang J., et al. (2018. a). Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia-reperfusion injury. J. Pineal. Res. 65, e12503. 10.1111/jpi.12503 [DOI] [PubMed] [Google Scholar]
- Zhou Y., Wang H., Man F., Guo Z., Xu J., Yan W., et al. (2018. b). Sitagliptin protects cardiac function by reducing nitroxidative stress and promoting autophagy in Zucker Diabetic Fatty (ZDF) rats. Cardiovasc. Drugs Ther. 32, 541–552. 10.1007/s10557-018-6831-9 [DOI] [PubMed] [Google Scholar]
- Zou F., Wang L., Liu H., Wang W., Hu L., Xiong X., et al. (2019. a). Sophocarpine suppresses NF-kappaB-mediated inflammation both in vitro and in vivo and inhibits diabetic cardiomyopathy. Front. Pharmacol. 10, 1219. 10.3389/fphar.2019.01219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou G., Zhong W., Wu F., Wang X., Liu L. (2019. b). Catalpol attenuates cardiomyocyte apoptosis in diabetic cardiomyopathy via Neat1/miR-140-5p/HDAC4 axis. Biochimie 165, 90–99. 10.1016/j.biochi.2019.05.005 [DOI] [PubMed] [Google Scholar]