

Abstract
Most breast cancer (BC) risk-associated single-nucleotide polymorphisms (raSNPs) identified in genome-wide association studies (GWAS) are believed to cis-regulate the expression of genes. We hypothesise that cis-regulatory variants contributing to disease risk may be affecting microRNA (miRNA) genes and/or miRNA binding. To test this, we adapted two miRNA-binding prediction algorithms—TargetScan and miRanda—to perform allele-specific queries, and integrated differential allelic expression (DAE) and expression quantitative trait loci (eQTL) data, to query 150 genome-wide significant () raSNPs, plus proxies. We found that no raSNP mapped to a miRNA gene, suggesting that altered miRNA targeting is an unlikely mechanism involved in BC risk. Also, 11.5% (6 out of 52) raSNPs located in 3′-untranslated regions of putative miRNA target genes were predicted to alter miRNA::mRNA (messenger RNA) pair binding stability in five candidate target genes. Of these, we propose RNF115, at locus 1q21.1, as a strong novel target gene associated with BC risk, and reinforce the role of miRNA-mediated cis-regulation at locus 19p13.11. We believe that integrating allele-specific querying in miRNA-binding prediction, and data supporting cis-regulation of expression, improves the identification of candidate target genes in BC risk, as well as in other common cancers and complex diseases.
Subject terms: Risk factors, Data mining
Introduction
In the past 10 years, genome-wide association studies (GWAS) identified hundreds of common low-penetrance variants to be associated with breast cancer (BC) risk.1 Most of these risk-associated single-nucleotide polymorphisms (raSNPs) are located in non-coding regions,2 often with no established, or easily perceived, biological function. Rather than altering protein sequence, and consequently protein function or structure, it seems that most raSNPs, or those in linkage disequilibrium (LD) with them, may act in cis to regulate the expression levels of target genes located distally and proximally.3–5 The biological effect of raSNPs has so far been detected by expression quantitative trait loci analysis (eQTL),3,6–8 but also, although less frequently, through the analysis of differential allelic expression (DAE).9,10 A few functional studies for BC raSNPs have confirmed this cis-regulatory role, but have mainly focused on their potential to alter transcription factor binding sites.3,6–8 Nevertheless, genetic variation can modulate gene expression by several other mechanisms, such as microRNA-mediated regulation.
MicroRNA (miRNAs) are small non-coding RNA (ncRNA) molecules that bind messenger RNA (mRNA) complementary sequences and generally direct post-transcriptional silencing in the 3′-untranslated region (UTR) of target genes.11 There is strong, albeit episodic, evidence of SNPs within miRNA genes and mRNA binding sites affecting the susceptibility to some cancers,12,13 including BC.14–16 However, hitherto, the systematic analysis of BC risk loci via miRNA regulation is still lacking.
Here, we set out to evaluate the effect of common genetic variants associated with BC susceptibility on miRNA-regulatory mechanisms. Our initial list of raSNPs was established by selecting the 150 most significant () BC raSNPs from published GWAS (retrieved on 13 February 2017), along with their proxies in high LD. Next, we filtered these by genomic location, keeping those in or near miRNA genes and/or in protein-coding genes (PCGs; potential miRNA target sequences). Finally, we modified existing prediction tools to perform allele-specific miRNA target prediction analysis. We used both miRNAs and putative mRNA target genes, expressed in normal breast tissue, and also cis-regulated genes as supported by DAE and eQTL data in normal breast tissue.
Here we present a systematic miRNA pathway-based study from published BC GWAS, using allele-differential prediction analysis, further improved by integration of DAE and eQTL data from normal breast tissue.
Results
Some BC risk variants locate to the 3′-UTR of PCGs, but none to miRNA genes
To evaluate the contribution to BC risk of genetic variation modelling miRNA::mRNA binding, we first assessed how many GWAS SNPs and their proxies were located in either miRNA genes or 3′-UTRs of PCGs. We identified 2749 raSNPs, resulting from 150 BC GWAS SNPs (Supplementary Table S1) and their proxies, of which almost one-third (805 raSNPs) were solely annotated to “gene deserts” (585 raSNPs) or intergenic regions (220 raSNPs). The remainder 1944 raSNPs were located in either ncRNA genes or PCGs (see Supplementary Fig. S1), in a total of 161 unique Ensembl gene IDs, correspondent to 129 HGNC (HUGO Gene Nomenclature Committee) symbols.
Next, we assessed how many would change the miRNA gene sequence, thus affecting their biogenesis or target genes. Interestingly, none of the raSNPs mapped to miRNA genes, even after the LD threshold was lowered to when defining proxy SNPs (results not shown). This suggests that altered miRNA biogenesis or altered seed region sequence are unlikely mechanisms associated with BC risk. However, 13 SNPs were annotated as downstream or upstream variants of miRNA genes (Supplementary Table S2), raising the possibility of them being regulating the expression of the miRNA itself. However, we did not pursue this hypothesis further due to unavailability of DAE or eQTL data for these particular miRNA genes.
The vast majority of the raSNPs located within PCGs were in non-coding regions (1881 out of 1915, 98%) (see Supplementary Fig. S1), consistent with previous reports.17 SNPs located at the 3′-UTR of the mRNA sequence of PCGs could potentially modify, create or destroy miRNA-binding sites, and we found 52 raSNPs (1.9% of total queried, 2.7% of total in PCGs), at 16 risk loci, with at least one annotation at the 3′-UTR of PCGs.
Development and validation of allele-specific miRNA target prediction analysis
raSNPs located at 3′-UTR of PCGs were then evaluated for their potential to generate allelic-differential miRNA binding (Fig. 1). To do so, we started by looking at existing miRNA target prediction algorithms, but none could straightforwardly perform SNP allele queries in an automatic way (see Supplementary Table S3 for a systematic review). We, therefore, modified the input of two prediction algorithms, TargetScan18 and miRanda,19 to account for SNP alleles queries and indirectly implemented them in R.
Fig. 1. Schematic overview of the bioinformatics pipeline used for the prediction of allele-specific miRNA-binding sites in breast cancer risk variants identified in GWAS.
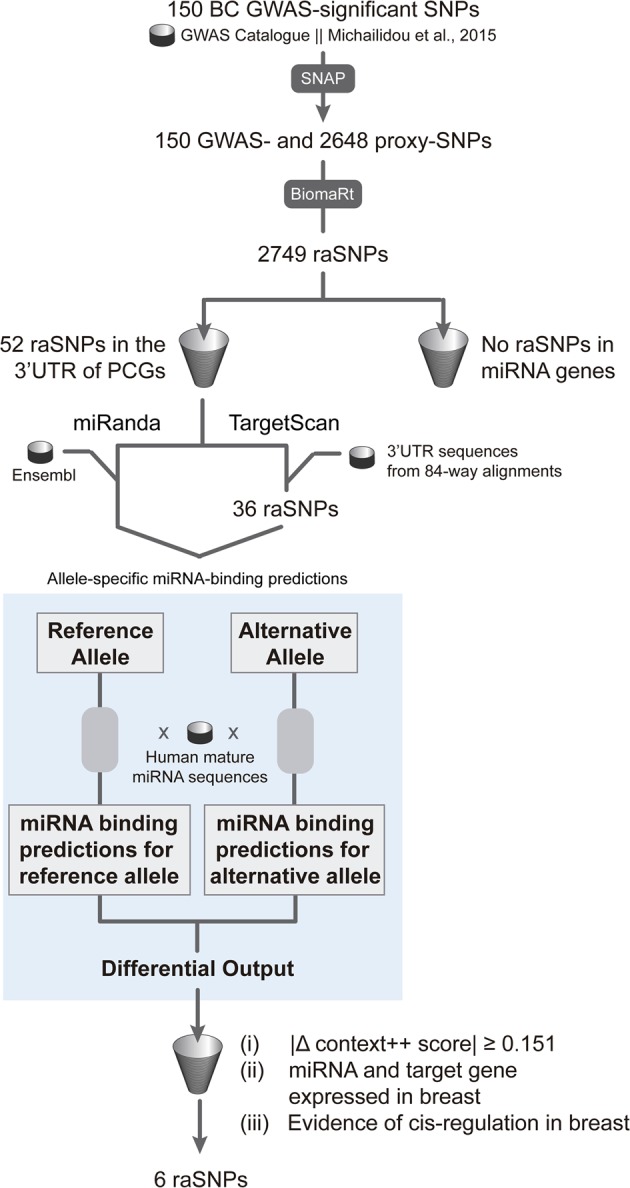
Genome-wide significant () SNPs associated with breast cancer risk in published GWAS were retrieved from the GWAS Catalog and from a GWAS meta-analysis.49 Proxies in high linkage disequilibrium () were obtained through SNAP v2.2, using data from the pilot release of the 1000 Genomes Project for the CEU population. The biomaRt R package (v2.34.2) was used to retrieve genomic annotations from the Ensembl database v92. Risk-associated SNPs (raSNPs) were filtered for their location either in the 3′-UTR of protein-coding genes (PCGs) or in miRNA genes. Next, allele-specific miRNA-binding predictions were performed by modifying the input of two prediction algorithms—TargetScan and miRanda. First, each raSNP allele (reference and alternative) located at the 3′-UTR of PCGs was independently evaluated for putative miRNA binding through the algorithms. Then, allele-specific miRNA-binding predictions for each SNP were obtained by comparing each output file of corresponding SNP alleles and extracting their differences in miRNA binding. miRNA-binding predictions common to both algorithms were filtered for: (i) context++ score absolute difference ( context++ score) ; (ii) miRNA expression () in adjacent-normal breast tissue from the miRmine database and PCG expression () in normal breast from the GTEx Project; and (iii) evidence of PCG cis-regulation in normal breast tissue from in-house differential allelic expression analysis and eQTL data from the GTEx Project.
To validate this approach of differential allelic miRNA-binding querying, we ran the novel pipeline on seven SNPs that had previous functional validation supporting allele-specific miRNA binding. All seven SNPs were predicted to have allele-preferential binding of the corresponding miRNAs in at least one of the algorithms (Table 1), according to previous reports.13,16,20–24 One of the SNPs previously published, and functionally validated, is rs11540855 in the ABHD8 gene, which is in high LD () with rs8170, a variant associated with risk to BC in BRCA1 mutation carriers.25,26 Li et al.20 showed that the G allele of rs11540855 was preferentially bound by the hsa-miR-4707-3p agomir in the BC cell line MCF116, leading to both decreased luciferase activity and ABHD8 protein levels. This is consistent with our prediction, using TargetScan, of hsa-miR-4707-3p specifically binding to the G allele of rs11540855 (context++ score = −0.225 for the G allele and no predicted binding for the allele A). The miRanda algorithm only predicted a minor difference in the maximum absolute minimum free energy (MFE) (−25.95 kcal/mol for the G allele and −23.93 kcal/mol for the A allele), although corroborating the preferential binding to the G allele (Table 1).
Table 1.
Previously functionally validated SNPs affecting miRNA binding evaluated by TargetScan and miRanda.
| SNP | Locus | Gene | Transcripta | miRNAb | Allelesc | Ref. allele | Alt. allele | Associated disease | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Context ++d | MFEe | Context ++d | MFEe | |||||||
| rs4245739[f21 | 1q32 | MDM4 | ENST00000367182.3 | hsa-miR-191-5p | A/C | — | — | −0.309 | — | Ovarian cancer |
| rs793013 | 1q42.3 | TOMM20 | ENST00000366607.4 | hsa-miR-4273-5p | A/G | −0.151 | — | — | — | Colorectal cancer |
| rs3559256722 | 3q28 | TP63 | ENST00000264731.3 | hsa-miR-140-5p | C/T | −0.234 | — | — | — | Bladder cancer |
| rs107173816 | 4q32.3 | PALLD | ENST00000261509.6 | hsa-miR-96-5p | G/C | — | — | −0.12 | — | Breast cancer |
| hsa-miR-182-5p | — | — | −0.298 | — | ||||||
| ENST00000335742.7 | hsa-miR-96-5p | — | — | −0.126 | — | |||||
| hsa-miR-182-5p | — | — | −0.317 | — | ||||||
| rs1272020823 | 8p22 | FGF20 | ENST00000180166.5 | hsa-miR-433-3p | G/A | −0.201 | — | — | — | Parkinson’s disease |
| rs422524 | 11q23.3 | APOC3 | ENST00000227667.3 | hsa-miR-4271 | G/T | — | −16.69 | −0.172 | −21.07 | Coronary heart disease |
| rs11540855f20 | 19p13.11 | ABDH8 | ENST00000247706.3 | hsa-miR-4707-3p | A/G | — | −23.93 | −0.225 | −25.95 | — |
aTranscript ID of the 3′-UTR sequence used in TargetScan’s miRNA-binding predictions
bmiRNA ID from miRbase v21
cReference allele/alternative allele(s)
dObtained from TargetScan v7.1
eMinimum free energy (MFE, from miRanda v3.3a) for the corresponding prediction obtained in TargetScan
fSNP associated in GWAS with breast cancer risk, all others are proxies GWAS-significant variants in the corresponding loci
Another previously published prediction was that of the preferential binding of the hsa-miR-191-5p to the alternative allele (C allele) of rs4245739, located in the 3′-UTR of the MDM4 gene, in ovarian cancer cell lines.21 This result was also concordant with our differential allelic miRNA-binding predictions (context++ score of 0.309 for allele C vs. no binding for allele A; Table 1).
The results from the validation step, presented in Table 1, suggest that functional allelic differences are easier to identify using the TargetScan algorithm. Additionally, they provided a guide to establish a biological significance threshold for the prediction scores. We set this threshold at the weakest binding prediction for any of the validated loci, corresponding to the lowest TargetScan context++ score of −0.151 obtained for rs7930 in TOMM20.13 This was later used to set the list of variants with stronger potential to affect miRNA binding.
Five per cent of the tested raSNPs are predicted to alter miRNA binding
To assess how many of the 52 raSNPs located in the 3′-UTR of PCGs (Supplementary Table S4) were likely to alter the miRNA::mRNA pairing stability, the analysis pipeline was applied and the difference of scores obtained for each pair of alleles was calculated. Sixteen of these raSNPs could not be analysed by TargetScan, as they were 3′-UTR variants of nonsense-mediated decay transcripts, which are excluded by this tool (Supplementary Table S4). Of the 36 raSNPs analysed for differential miRNA binding by TargetScan, allele-specific context++ scores were generated, which revealed that a total of 311 unique miRNAs had potential targets altered by raSNPs, with an average of nine miRNAs per raSNP (Supplementary Dataset 1). As for miRanda analysis, all 52 raSNPs generated maximum absolute MFE differences for a total of 2227 unique miRNAs (average 43 miRNAs per SNP; Supplementary Dataset 2). Together, both algorithms commonly predicted a total of 160 combinations of gene–SNP–allele–miRNA. These were then filtered for the established TargetScan context++ score threshold, and evidence of both putative target mRNA (GTEx Project) and miRNA expression (miRmine database) in normal breast tissue, resulting in ten common predictions for seven raSNPs at six PCGs.
To identify candidate regulatory SNPs (rSNPs), we further filtered the resulting seven raSNPs based on previous evidence of cis-regulation of the target gene in breast tissue. For this purpose, we used the requirement of DAE of the target gene in normal breast tissue.27,28 Furthermore, we used normal breast tissue eQTL data29 as supporting evidence of cis-regulation. Overall, six BC raSNPs located in the 3′-UTRs of five PCGs were predicted to modify miRNA::mRNA pair binding stability in an allele-specific manner, with supporting evidence of cis-regulation of the putative target gene. These raSNPs correspond to five initial GWAS-significant associations in four BC risk loci (5% of the initial 83 BC GWAS loci) (Table 2). These variants were rs17354678 (in RNF115, at 1q21.1 locus), rs1019806 and rs6884232 (in ATG10, at 5q14.1-2 locus), rs3734805 (in CCDC170, at 6q25.1 locus), rs4808616 (in ABHD8, at 19p13.11 locus) and rs2385088 (in ISYNA1, at 19p13.11 locus).
Table 2.
BC risk loci with putative allele-specific miRNA binding.
| GWAS SNP | Locus | Candidate rSNP | LDa | Gene | Transcriptb | Allelesc | miRNAd | Ref. allele | Alt. allele | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D′ | Context++e | MFEf | Context++e | MFEf | ||||||||
| rs1240513249 | 1q21.1 | rs17354678 | 0.85 | 0.96 | RNF115 | ENST00000369291.5 | T/C | hsa-miR-486-5p | −0.033 | −21.85 | −0.212 | −24.63 |
| rs770792149 | 5q14.2 | rs1019806 | 1 | 1 | ATG10 | ENST00000282185.3 | A/G | hsa-miR-3138 | −0.22 | −20.18 | −0.069 | −20.12 |
| rs6884232 | 1 | 1 | ATG10 | ENST00000282185.3 | A/G | hsa-miR-21-3p | — | — | −0.169 | −18.95 | ||
| rs12662670 49 | 6q25.1 | rs3734805 | 0.89 | 1 | CCDC170 | ENST00000239374.7 | A/C | hsa-miR-766-5p | — | −17.04 | −0.151 | −21.72 |
| rs4808075 34 | 19p13.11 | rs4808616 | 1 | 1 | ABHD8 | ENST00000247706.3 | C/A | hsa-miR-7705 | −0.155 | −17.11 | — | — |
| rs4808801 33 | 19p13.11 | rs2385088 | 0.96 | 1 | ISYNA1 | ENST00000338128.8 | A/G | hsa-miR-6842-5p | — | −24.45 | −0.279 | −26.8 |
Common miRNA-binding predictions found using TargetScan and miRanda algorithms. Results are filtered for (i) context++ score absolute fold change between matching SNP alleles, (ii) miRNA expression with , using the miRmine dataset (SRX513286), and (iii) gene expression with from the GTEx Project. Only genes with evidence of cis-regulation are shown. Results are ranked for decreasing context++ score
aLinkage disequilibrium with GWAS SNP (1000 Genomes Project Pilot, CEU population)
bTranscript ID of the 3′-UTR sequence used in TargetScan’s miRNA-binding predictions
cReference allele alternative allele(s)
dmiRNA ID from miRbase v21
eContext++ score from TargetScan v7.1
fMaximum absolute minimum free energy (Max |MFE|, from miRanda v3.3a)
RNF115 is a novel strong candidate target gene for BC risk
Following the canonical mechanism of action of miRNAs, we based our next analysis on the premise that the allele with preferential binding prediction would be the least expressed.
For CCDC170, the proposed rSNP rs3734805 is in very weak LD () with all the DAE variants analysed (see Supplementary Fig. S2), and it is not an eQTL for the expression of any gene according to GTEx. Therefore, we could not establish direct association between the rSNP alleles and preferential allelic expression.
As for the two candidate rSNPs in ATG10 (5q14.1-2 locus), both are reported eQTLs for ATG10 expression using GTEx dataset (data not shown), with the alternative alleles associated with lower expression. This is concordant with the DAE data for rs1428940 (see Supplementary Fig. S2), in high LD with these variants ( and 0.93, respectively). However, the predictions for allelic-preferential binding of miRNAs at rs6884232 is discordant with this evidence, as it is the reference allele which is predicted to have preferential binding. As for rs1019806, only the TargetScan prediction points to a concordant allelic difference in binding, while miRanda predicts almost no allelic difference.
For the pair hsa-miR-6842-5p::ISYNA1, the G allele of rs2385088 was predicted to bind preferentially, but this was also the preferentially expressed allele in normal breast (see Supplementary Fig. S2). Moreover, the DAE data was concordant with the role of ISYNA1 as a reported tumour suppressor gene,30–32 with protective G allele of the GWAS-significant variant rs4808801 (odds ratio (OR) for G allele = 0.93, 95% confidence interval (CI) = [0.91–0.95] )33 in high LD with the preferentially expressed G allele of rs2385088 (, being G the alternative allele for both variants).
Regarding locus 19p13.11, a novel variant in the 3′-UTR of ABHD8’s only expressed transcript ENST00000247706.3 (see Supplementary Fig. S3), rs4808616, was predicted to have allelic-specific binding of hsa-miR-7705 to the reference C allele. The DAE measured at this variant indicates this allele is the less expressed (Fig. 2a). rs4808616 is in complete LD with rs4808075, associated with cancer pleiotropy (OR for alternative C allele = 1.1, ),34 suggesting that risk may be caused via increasing expression of ABHD8.
Fig. 2. Breast cancer risk loci with strong predictions for allelic-differential binding of miRNA.

a BC risk locus 19p13.11 with candidate target gene ABHD8 and b BC risk locus 1q21.1 with candidate target gene RNF115. Both figures display a top panel with the genomic organisation of RefSeq genes (NCBI Release 109), with the candidate target genes in red and displaying the transcript predicted to have differential binding of miRNAs. The middle panel displays the genomic organisation of the tested raSNPs (GWAS-significant variants in green, plus proxy SNPs in high LD in black) in each locus, identified and tested in this study. raSNPs located in the 3′-UTR of candidate target genes with the strongest predictions for allelic-differential binding of miRNAs (rSNP candidates) are indicated in cyan. raSNPs in red have weaker predictions for allelic-differential binding of miRNAs. The third panel shows the DAE data for heterozygous individuals tested for the SNP indicated immediately below. The alleles are indicated for each SNP in the order of the AE ratio calculated (i.e. A/G corresponds to the ratio of allele A by allele G). Dashed horizontal lines indicate the threshold for DAE set at 1.5-fold difference between alleles ().
The last candidate rSNP, rs17354678, locates to the 3′-UTR of RNF115 and was predicted to have more stable pair binding of hsa-miR-486-5p::RNF115 in the presence of the alternative C allele, for the only protein-coding transcript ENST00000369291.5 (see Supplementary Fig. S3). rs17354678 is in high LD with variants for which DAE was detected, namely rs12402867 () and rs17352469 (), both of which displayed preferential expression of the reference alleles (G and A, respectively) (Fig. 2b). This was congruent with the prediction of preferential binding of the alternative C allele of rs17354678. This variant is also in high LD () with the BC GWAS-significant SNP rs12405132 (OR for reference C allele = 1.03, 95% CI = [1.01–1.05], ).35 Therefore, these results suggest that risk may be conferred by higher expression of RNF115.
Discussion
GWASes have established the importance of normal non-coding genetic variation in common diseases, including BC. Currently, the challenges are to identify the true causal variant in risk-associated loci, as well as to determine the mechanism by which they act and the target genes they control. Our study focused on understanding the extent by which miRNA-mediated cis-regulation contributes to BC risk.
We found that none of the tested raSNPs from BC GWAS mapped to miRNA genes, suggesting that altered miRNA biogenesis, or targeting, are unlikely mechanisms to be involved in BC risk. However, this result could be due to the low representation of miRNA SNPs in the commercial genotyping arrays. On a biological perspective, however, miRNAs can bind hundreds of different mRNA targets36 across the genome, and variants in both the precursor elements, as well as the mature miRNA sequence, may drive changes in transcription in a much more widespread and significant manner, than those on target sequences.37 Thus, we can hypothesise that SNPs in miRNA genes would have larger effect sizes than those observed in GWAS, explaining their under-representation in these studies.
Next, we found 13 BC raSNPs located upstream and downstream of miRNA genes, which could be regulating miRNA transcription; however, most were also intronic variants of PCGs with evidence of cis-regulation from DAE data in normal breast tissue. This suggests that these raSNPs are more likely regulating PCG expression instead. Also, since the DAE data was generated using PCG exon-centric SNP microarrays, and as such did not cover the great majority of miRNA genes, we cannot exclude the possibility that our result is biased against miRNA gene cis-regulation. Further studies will be needed to evaluate whether raSNPs are located in regulatory regions affecting miRNA-gene expression levels.
We also found 52 raSNPs located at the 3′-UTR of PCGs, where miRNAs are generally known to bind,11 supporting a possible role for miRNA-mediated cis-regulation in BC predisposition. However, co-expression of both miRNAs and target sequences in the same tissue is imperative to validate such findings. While no comprehensive datasets are currently available to quantify the impact of miRNA expression on the putative target gene expression levels, we performed a qualitative filter for miRNA expression in adjacent-normal breast tissue,38 as well as for the target-gene expression in breast mammary tissue.39
GWAS follow-up studies have mostly used eQTL mapping in normal tissue, to identify cis-acting variants and prioritise candidate cis-rSNPs for functional analysis.40–42 However, cis-regulatory signals can be masked in eQTL studies by trans-acting factors or environmental effects.43 Direct assessment of cis-regulation requires allele-specific approaches, such as DAE studies, where the effect of trans-regulation is eliminated when comparing the relative expression of two alleles in an heterozygous individual, within the same cellular context.44 Here we combined both DAE and eQTL to filter our results, and we predict that six 3′-UTR raSNPs have the potential to alter miRNA-binding stability in five genes with evidence of cis-regulation in normal breast.
Our strongest result was obtained for raSNP rs17354678, mapping to the 3′-UTR of the transcript ENST00000369291.5 of the RNF115 gene, for which the reference allele was predicted to decrease the binding of hsa-miR-6842-5p in normal breast tissue. According to the DAE data, this allele is congruently associated with higher expression of RNF115, and is in high LD with the risk variant for BC, rs12405132,35 supporting that risk might be conferred by upregulation of RNF115. RNF115 encodes for the three ubiquitin ligase RING finger protein 115, which has been reported as upregulated in BC, particularly in oestrogen receptor α-positive tumours.45 RNF115 has also been proposed to promote proliferation possibly through downregulation of the expression of the tumour suppressor p21.46 Our data further support RNF115’s role as an oncogene, as the predicted preferential binding of hsa-miR-486-5p and consequent lower expression of RNF115, is associated with protection against BC.
In addition, we also found a strong evidence that the reference allele of rs4808616, located at the 3′-UTR of the transcript ENST00000247706.3 of the ABHD8 gene could promote a binding site for hsa-miR-7705 in normal breast tissue. Expression and LD analysis with the BC risk variant support a role for miRNA-mediated regulation and decreased expression of ABHD8 in BC risk. This prediction is in accordance to what has been functionally validated for other candidate rSNP rs1154085520 in ABHD8. Previously, rs4808616 had been also functionally studied for mechanisms underlying pleiotropic risk to breast and ovarian cancer.47 The authors found evidence of allelic expression and identified multiple risk alleles, which they associated with increased ABHD8 promoter activity. For rs4808616, in particular, the authors identified a link between the risk allele and higher expression of ABHD8 through inclusion in a putative regulatory elements, but did not test for miRNA-mediated mechanisms. Nevertheless, all data, ours and from others, support that increased expression of ABHD8, which encodes for a poorly studied lipase,48 is linked to higher risk to BC. Furthermore, our study adds evidence for altered miRNA-binding through cis-regulatory variation as a mechanism of risk in this locus.
The remaining predictions for rs1019806 and rs6884232 (in ATG10), rs3734805 (in CCDC170) and rs2385088 (in ISYNA1), albeit supported by DAE evidence for the corresponding genes, were not directly explained by the preferential allelic expression pattern observed in normal breast tissue. Given that gene expression regulation is a complex trait in itself, with multiple possible cis-regulatory variants acting on the same gene, via different possible mechanisms, and with different allelic effects, we should not rule these candidates out. Particularly, ATG10 and CCDC170 are large genes, with many potential cis-regulatory variants in LD with the GWAS variants.
In this study, we proposed a systematic post-GWAS framework focused on miRNA regulation, integrating DAE and eQTL data from normal tissue, to prioritise candidate rSNPs in already known risk loci. Although searching for altered transcription factor binding has been a popular approach following GWASes, other mechanisms, or even more than one at the same time, may be at play at susceptibility loci. Thus, it is important to look at the whole cis-regulation context when searching for the causal rSNP(s). Here, we showed that five genes, at four BC risk loci, have putative altered miRNA binding and that these genes have evidence of cis-regulation in normal breast tissue, supporting a functional role. In the future, it will be important to validate such findings through in vitro and in vivo assays.
Finally, our study provides a quick, powerful and systematic way of assessing the allelic-differential miRNA-mediated cis-regulation. As other common cancer GWASes have similar genomic distribution of risk variants to BC, it will be interesting to determine whether similar findings of putative altered miRNA regulation is also present in other common cancers.
Methods
BC risk loci dataset
GWAS-significant raSNPs for BC were retrieved from the NHGRI-EBI Catalog of published GWAS,2 available at www.ebi.ac.uk/gwas (accessed on 13 February 2017), using and the catalogue traits “Breast cancer”, “Breast cancer (male)”, “Breast cancer (early onset)”, “Breast Cancer in BRCA1 mutation carriers”, “Breast cancer in BRCA2 mutation carriers”, “Breast cancer (oestrogen receptor negative, progesterone-receptor negative, and human epidermal growth factor-receptor negative)”, “Cancer” and “Cancer (pleiotropy)”. Furthermore, we retrieved the 15 SNPs previously identified by Michailidou et al.49 in a BC GWAS meta-analysis, as well as those mentioned in their Supplementary Table 3, with a , which included variants found associated by previous candidate gene association studies.
Proxy SNP query
Proxy SNPs were identified using the SNP Annotation and Proxy Search 50 online tool (version 2.2), available at archive.broadinstitute.org/mpg/snap/ldsearch.php, using genotype data from the pilot release of the 1000 Genomes Project51 for the CEU population (Utah residents with Northern and Western European ancestry), with a distance limit of 500 kb and an LD threshold of .
Retrieval of SNP annotations
We used the getBM function from the biomaRt R package v2.34.252 to retrieve the genomic annotations, alleles and variation consequence (Ensembl release 9253) of each SNP, as well as its molecular position (according to Ensembl release 75.54) SNPs flagged by Ensembl53,55 for containing errors or inconsistencies in their annotation were automatically excluded from further analysis.
Allele-specific miRNA-binding predictions
raSNPs were filtered for the Sequence Ontology term “3_prime_UTR_variant” as a variant consequence.56 For each allele of 3′-UTR-located SNPs, miRNA::mRNA interactions were searched using the default settings of the predictive algorithm TargetScan (release 7.1)18 and custom settings for the miRanda (v3.3a) algorithm,19,57 as described below. The R code used to perform both analyses is available at https://github.com/maialab/postgwas-miRNA.
TargetScan predicts biological targets of miRNAs by searching for the presence of conserved canonical sites in 3′-UTRs that match the “seed” region (2–7 nucleotides of the mature miRNA) of each miRNA.18 The matches are made to human 3′-UTRs from Gencode v19 (Ensembl 75) and their orthologues, as defined by UCSC whole-genome alignments (hg19).18 For each site, a context++ score is calculated; the lower the score, the higher the probability of effective target repression.18 TargetScan source code, and accompanying datasets, were downloaded from http://www.targetscan.org/cgi-bin/targetscan/data_download.vert71.cgi, and run over the two alleles of each raSNP. Briefly, for each SNP located within a specific human 3′-UTR multiple sequence alignment (as provided by TargetScan), independent text files containing either the reference or the alternative allele were generated according to source code instructions for the reference allele. We excluded from further analysis SNPs annotated as 3′-UTR variants, but not located within the available 3′-UTR sequence alignments (Supplementary Table S4). Default instructions available for context++ score calculation were followed. For each miRNA-binding prediction, context++ score differences between correspondent SNP alleles were calculated.
miRanda detects potential miRNA target sites in genomic sequences by carrying a dynamic programming local alignment between query miRNA sequences and target mRNA sequences.19 For each detected complementary match between a miRNA and a potential target gene two measures are calculated: (i) a score S based on sequence complementary and (ii) the MFE of the optimal miRNA–mRNA interaction. High S and low MFE values indicate potential target sites.19 We used the getBM function of the R package biomaRt 52 to retrieve a target-sequence centred on each allele and flanked by 25 nucleotides on either side, based on annotation in the Ensembl release 92.53 miRNA mature sequences were retrieved from miRbase database (release 21, ftp://mirbase.org/pub/mirbase/21/) and filtered for Human.58 miRanda’s software (v3.3a) was obtained from MicroRNA.org, a comprehensive resource of miRNA target predictions and expression profiles,59 at www.microrna.org. A cut-off of S 80 and an MFE ≤ −16 kcal/mol was used as previously described14 to select for miRNA binding. Maximum absolute MFE differences between matching SNP alleles for each SNP::miRNA pair were calculated.
miRNA and miRNA target gene expression
miRNA expression data, previously generated by miRNA-sequencing from pooled adjacent-normal breast tissue samples from eight BC patients,60 was obtained from the miRmine Database,38 available under the Sequence Read Archive ID SRX513286 (http://guanlab.ccmb.med.umich.edu/mirmine, accessed on 22 January 2017). miRNAs with expression values >1 read per million were considered as expressed.61
Gene and transcript expression levels for 290 breast mammary tissue samples were obtained from the GTEx Portal (v7) at www.gtexportal.org (accessed on 12 December 2017).39 Genes with median expression levels >1 transcript per million were considered as expressed.62,63
Defining cis-regulation of gene expression
Cis-rSNPs act on regulatory elements, including promoters, enhancers and miRNA-binding sites, by modifying the binding affinity of trans-acting factors and thus specifically affecting gene expression in an allelic manner. This gives rise to unequal expression of transcribed alleles of the gene, a common feature in the human genome.64,65 Comparison of the relative expression of the two alleles in a heterozygous individual by DAE analysis is therefore a direct indicator of rSNPs acting in cis. Furthermore, measure of mRNA transcripts and association of their expression levels with genetic variants in eQTL studies can also indicate the presence of cis-rSNPs.
DAE analysis was performed with data from 64 normal breast tissue samples from healthy controls, collected from women submitted to reduction mastectomy, for reasons not related to cancer, at Addenbrooke’s Hospital in Cambridge, United Kingdom (under Addenbrooke’s Hospital Local Research Ethics Committee approval, REC reference 06/Q0108/221). DNA and total RNA were extracted as previously described.66 Briefly, DNA and cDNA samples derived from total RNA from a given individual were run on Illumina Infinium Exon510S-Duo arrays, data were filtered and normalised as described previously.28 The raw data is deposited in the Gene Expression Omnibus under accession number GSE35023.
After normalisation, quality control (QC) was carried for allelic expression and genotyping as follows: (1) SNPs with average log 2 RNA signal intensity values lower than 9.5 were excluded, to avoid low-intensity-related false positives; (2) to verify allelic discrimination at RNA level a two-samples Student’s t test was applied to compare RNA log ratios between heterozygous (AB) and homozygous groups (AA and BB), and only SNPs with p values for all comparisons were further analysed; (3) QC for the genotyping analysis was carried by excluding SNPs with call rate , Hardy–Weinberg equilibrium p value and less than five heterozygotes; finally, (5) SNPs with multiple genomic mapping entries, flagged as suspected in dbSNP149 GRCh38p7 and located in sexual chromosomes were also excluded.
The following equation was used for normalisation of allelic expression, to cancel DNA dosage effects:
| 1 |
DAE was inferred when (1.5-fold or greater allelic difference) for at least 10% of the heterozygotes and a minimum of three samples for a given SNP. Genes with at least one SNP with DAE were considered to show evidence of being cis-regulated.
cis-eQTL data (evaluated for ±1 Mb around the transcriptional start site of each gene) for 251 breast mammary tissue samples from GTEx’s v7 release was obtained from the GTEx Portal on 12 December 2017.29 Genes whose expression levels were associated with at least one significant cis-eQTL, at a false discovery rate of 0.05, were selected.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank Unidade de Apoio à Investigação (UAIC) at Universidade do Algarve (UAlg), in particular Mr. Vitor Morais, and the Informatics Services of UAlg. We would also like to thank Dr. Suet-Feung Chin and Dr. Mae Goldgraben from University of Cambridge for valuable discussions. This work was supported by national Portuguese funding through FCT – Fundação para a Ciência e a Tecnologia and CRESC ALGARVE 2020, institutional support CBMR-UID/BIM/04773/2013, POCI-01-0145-FEDER-022184 “GenomePT”, the contract DL 57/2016/CP1361/CT0042 (J.M.X.) and individual postdoctoral fellowship SFRH/BPD/99502/2014 (J.M.X.). The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/under REA grant agreement no. 303745 (A.-T.M.), and a Maratona da Saúde Award (A.-T.M.).
Author contributions
A.J.-F. performed the computational work, and drafted the manuscript. J.M.X. and R.M. performed the DAE analysis, critically revised the manuscript and supervised the computational work. J.G.L. conducted the computational analysis regarding SNPs located near miRNA genes. A.-T.M. conceived and directed the study, secured funding and drafted the manuscript. All authors read and approved the final manuscript.
Data availability
All datasets analysed in this study were obtained from publicly available databases and websites. SNPs associated with BC risk, as well as their proxies, were obtained from the GWAS Catalog website at www.ebi.ac.uk/gwas and from Broad Institute’s SNAP (v2.2) website at archive.broadinstitute.org/mpg/snap, respectively. SNP data was obtained from the Ensembl database (version 92 and 75) available at www.ensembl.org. Mature miRNA sequences were retrieved from miRbase (release 21) at www.mirbase.org. The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS. Gene and transcript expression, and eQTL data for breast tissue from the GTEx Project (v7) were retrieved from the GTEx Portal at www.gtexportal.org. Other detailed results are available in Supplementary Information and Supplementary Datasets 1 and 2.
Code availability
The Perl code used to perform TargetScan’s miRNA-binding predictions was obtained from the TargetScan (v7.1) website at www.targetscan.org/vert_71. miRanda’s software (v3.3a) was retrieved from MicroRNA.org at 34.236.212.39/microrna/home.do. Code used to perform the analysis and generate this paper is available on GitHub at https://github.com/maialab/postgwas-miRNA. Further code may be made available upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41525-019-0112-9.
References
- 1.MacArthur J, et al. The new nhgri-ebi catalog of published genome-wide association studies (gwas catalog) Nucleic Acids Res. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer KB, et al. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freedman ML, et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 2011;43:513–518. doi: 10.1038/ng.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.French JD, et al. Functional variants at the 11q13 risk locus for breast cancer regulate cyclin d1 expression through long-range enhancers. Am. J. Hum. Genet. 2013;92:489–503. doi: 10.1016/j.ajhg.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glubb DM, et al. Fine-scale mapping of the 5q11.2 breast cancer locus reveals at least three independent risk variants regulating map3k1. Am. J. Hum. Genet. 2015;96:5–20. doi: 10.1016/j.ajhg.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghoussaini M, et al. Evidence that the 5p12 variant rs10941679 confers susceptibility to estrogen-receptor-positive breast cancer through fgf10 and mrps30 regulation. Am. J. Hum. Genet. 2016;99:903–911. doi: 10.1016/j.ajhg.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maia A-T, et al. Effects of BRCA2 cis-regulation in normal breast and cancer risk amongst BRCA2 mutation carriers. Breast Cancer Res. 2012;14:R63. doi: 10.1186/bcr3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamdi Y, et al. Association of breast cancer risk with genetic variants showing differential allelic expression: Identification of a novel breast cancer susceptibility locus at 4q21. Oncotarget. 2016;7:80140–80163. doi: 10.18632/oncotarget.12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 12.Chin LJ, et al. A snp in a let-7 microrna complementary site in the kras 3’ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–8540. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee A-r, Park J, Jung KJ, Jee SH, Kim-Yoon S. Genetic variation rs7930 in the miR-4273-5p target site is associated with a risk of colorectal cancer. OncoTargets Ther. 2016;9:6885–6895. doi: 10.2147/OTT.S108787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicoloso MS, et al. Single-nucleotide polymorphisms inside microrna target sites influence tumor susceptibility. Cancer Res. 2010;70:2789–2798. doi: 10.1158/0008-5472.CAN-09-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brewster BL, et al. Identification of fifteen novel germline variants in the brca1 3’utr reveals a variant in a breast cancer case that introduces a functional mir-103 target site. Hum. Mutat. 2012;33:1665–1675. doi: 10.1002/humu.22159. [DOI] [PubMed] [Google Scholar]
- 16.Gilam A, et al. Local microRNA delivery targets Palladin and prevents metastatic breast cancer. Nat. Commun. 2016;7:12868. doi: 10.1038/ncomms12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal V, Bell GW, Nam J-W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:1–38. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enright AJ, et al. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li MJ, et al. Exploring genetic associations with ceRNA regulation in the human genome. Nucleic Acids Res. 2017;45:5653–5665. doi: 10.1093/nar/gkx331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wynendaele J, et al. An illegitimate microRNA target site within the 3’ UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010;70:9641–9. doi: 10.1158/0008-5472.CAN-10-0527. [DOI] [PubMed] [Google Scholar]
- 22.Wang M, et al. A functional variant in TP63 at 3q28 associated with bladder cancer risk by creating an miR-140-5p binding site. Int. J. Cancer. 2016;139:65–74. doi: 10.1002/ijc.29978. [DOI] [PubMed] [Google Scholar]
- 23.Wang G, et al. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am. J. Hum. Genet. 2008;82:283–9. doi: 10.1016/j.ajhg.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu S-l, Cui G-l, Huang J, Jiang J-g, Wang D-w. An APOC3 3aUTR variant associated with plasma triglycerides levels and coronary heart disease by creating a functional miR-4271 binding site. Scientific Rep. 2016;6:32700. doi: 10.1038/srep32700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Couch FJ, et al. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet. 2013;9:e1003212. doi: 10.1371/journal.pgen.1003212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antoniou AC, et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat. Genet. 2010;42:885–892. doi: 10.1038/ng.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xavier J, et al. Abstract A31: integrative differential allelic expression analysis efficiently reveals the biology underlying risk to breast cancer. Mol. Cancer Res. 2016;14:A31–A31. [Google Scholar]
- 28.Liu R, et al. Allele-specific expression analysis methods for high-density SNP microarray data. Bioinformatics. 2012;28:1102–8. doi: 10.1093/bioinformatics/bts089. [DOI] [PubMed] [Google Scholar]
- 29.Consortium TG, et al. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kassie F, et al. Combinations of N-acetyl-S-(N-2-phenethylthiocarbamoyl)-l-cysteine and myo-inositol inhibit tobacco carcinogen-induced lung adenocarcinoma in mice. Cancer Prev. Res. 2008;1:285–97. doi: 10.1158/1940-6207.CAPR-08-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wattenberg LW, Estensen RD. Chemopreventive effects of myo-inositol and dexamethasone on benzo[a]pyrene and 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone-induced pulmonary carcinogenesis in female a/j mice. Cancer Res. 1996;56:5132–5. [PubMed] [Google Scholar]
- 32.Koguchi T, Tanikawa C, Mori J, Kojima Y, Matsuda K. Regulation of myo-inositol biosynthesis by p53-ISYNA1 pathway. Int. J. Oncol. 2016;48:2415–24. doi: 10.3892/ijo.2016.3456. [DOI] [PubMed] [Google Scholar]
- 33.Michailidou K, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013;45:353–61, 361e1–2. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fehringer G, et al. Cross-cancer genome-wide analysis of lung, ovary, breast, prostate, and colorectal cancer reveals novel pleiotropic associations. Cancer Res. 2016;76:5103–14. doi: 10.1158/0008-5472.CAN-15-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michailidou K, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:1–24. doi: 10.1038/nature24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 37.Sun G, et al. SNPs in human miRNA genes affect biogenesis and function. RNA. 2009;15:1640–51. doi: 10.1261/rna.1560209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panwar B, Omenn GS, Guan Y. miRmine: a database of human miRNA expression profiles. Bioinformatics. 2017;33:1554–1560. doi: 10.1093/bioinformatics/btx019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mele M, et al. The human transcriptome across tissues and individuals. Science. 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gamazon ER, et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet. 2018;50:956–967. doi: 10.1038/s41588-018-0154-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lonsdale J, et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Q, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152:633–641. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pastinen T, Ge B, Hudson TJ. Influence of human genome polymorphism on gene expression. Hum. Mol. Genet. 2006;15:R9–16. doi: 10.1093/hmg/ddl044. [DOI] [PubMed] [Google Scholar]
- 44.Pastinen T, Hudson TJ. Cis-acting regulatory variation in the human genome. Science. 2004;306:647–50. doi: 10.1126/science.1101659. [DOI] [PubMed] [Google Scholar]
- 45.Burger AM, et al. A novel RING-type ubiquitin ligase breast cancer-associated gene 2 correlates with outcome in invasive breast cancer. Cancer Res. 2005;65:10401–10412. doi: 10.1158/0008-5472.CAN-05-2103. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z, et al. RNF115/BCA2 E3 ubiquitin ligase promotes breast cancer cell proliferation through targeting p21Waf1/Cip1 for ubiquitin-mediated degradation. Neoplasia (New York, NY) 2013;15:1028–1035. doi: 10.1593/neo.13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawrenson K, et al. Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast-ovarian cancer susceptibility locus. Nat. Commun. 2016;7:12675–22. doi: 10.1038/ncomms12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lord CC, Thomas G, Brown JM. Mammalian alpha beta hydrolase domain (ABHD) proteins: lipid metabolizing enzymes at the interface of cell signaling and energy metabolism. Biochim. Biophys. Acta. 2013;1831:792–802. doi: 10.1016/j.bbalip.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michailidou K, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015;47:373–380. doi: 10.1038/ng.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson AD, et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Consortium TGP, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Durinck S, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 53.Zerbino DR, et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flicek P, et al. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Y, et al. Ensembl variation resources. BMC Genomics. 2010;11:293. doi: 10.1186/1471-2164-11-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McLaren W, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.John B, et al. Human microRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kozomara A, Griffiths-Jones S. MiRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu J, et al. Different miRNA expression profiles between human breast cancer tumors and serum. Front. Genet. 2014;5:1–7. doi: 10.3389/fgene.2014.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gong J, et al. Comprehensive analysis of human small RNA sequencing data provides insights into expression profiles and miRNA editing. RNA Biol. 2014;11:1375–1385. doi: 10.1080/15476286.2014.996465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wagner GP, Kin K, Lynch VJ. A model based criterion for gene expression calls using RNA-seq data. Theory Biosci. 2013;132:159–164. doi: 10.1007/s12064-013-0178-3. [DOI] [PubMed] [Google Scholar]
- 64.Lo HS, et al. Allelic variation in gene expression is common in the human genome. Genome Res. 2003;13:1855–62. doi: 10.1101/gr.885403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morley M, et al. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maia A, et al. Extent of differential allelic expression of candidate breast cancer genes is similar in blood and breast. Breast Cancer Res. 2009;11:R88. doi: 10.1186/bcr2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets analysed in this study were obtained from publicly available databases and websites. SNPs associated with BC risk, as well as their proxies, were obtained from the GWAS Catalog website at www.ebi.ac.uk/gwas and from Broad Institute’s SNAP (v2.2) website at archive.broadinstitute.org/mpg/snap, respectively. SNP data was obtained from the Ensembl database (version 92 and 75) available at www.ensembl.org. Mature miRNA sequences were retrieved from miRbase (release 21) at www.mirbase.org. The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS. Gene and transcript expression, and eQTL data for breast tissue from the GTEx Project (v7) were retrieved from the GTEx Portal at www.gtexportal.org. Other detailed results are available in Supplementary Information and Supplementary Datasets 1 and 2.
The Perl code used to perform TargetScan’s miRNA-binding predictions was obtained from the TargetScan (v7.1) website at www.targetscan.org/vert_71. miRanda’s software (v3.3a) was retrieved from MicroRNA.org at 34.236.212.39/microrna/home.do. Code used to perform the analysis and generate this paper is available on GitHub at https://github.com/maialab/postgwas-miRNA. Further code may be made available upon request.