

Abstract
The MESA erythrocyte cytoskeleton binding (MEC) motif is a 13-amino acid sequence found in 14 exported Plasmodium falciparum proteins. First identified in the P. falciparum Mature-parasite-infected Erythrocyte Surface Antigen (MESA), the MEC motif is sufficient to target proteins to the infected red blood cell cytoskeleton. To identify host cell targets, purified MESA MEC motif was incubated with a soluble extract from uninfected erythrocytes, precipitated and subjected to mass spectrometry. The most abundant co-purifying protein was erythrocyte ankyrin (ANK1). A direct interaction between the MEC motif and ANK1 was independently verified using co-purification experiments, the split-luciferase assay, and the yeast two-hybrid assay. A systematic mutational analysis of the core MEC motif demonstrated a critical role for the conserved aspartic acid residue at the C-terminus of the MEC motif for binding to both erythrocyte inside-out vesicles and to ANK1. Using a panel of ANK1 constructs, the MEC motif binding site was localized to the ZU5C domain, which has no known function. The MEC motif had no impact on erythrocyte deformability when introduced into uninfected erythrocyte ghosts, suggesting the MEC motif’s primary function is to target exported proteins to the cytoskeleton. Finally, we show that PF3D7_0402100 (PFD0095c) binds to ANK1 and band 4.1, likely through its MEC and PHIST motifs, respectively. In conclusion, we have provided multiple lines of evidence that the MEC motif binds to erythrocyte ANK1.
Keywords: Plasmodium, exported protein, MESA, PF3D7_0500800, PF3D7_0402100, ankyrin, band 4.1, host-pathogen interaction
1. Introduction
During the intra-erythrocytic stage, Plasmodium falciparum secretes hundreds of proteins into the infected red blood cell (iRBC) [1–4]. Some of these proteins interact with the RBC membrane or cytoskeletal proteins and are involved in the extensive remodeling of iRBC membrane properties, including membrane deformability [5, 6], permeability [7] and adhesiveness to the endothelium [5, 8, 9]. Although the molecular bases for these changes are not yet fully understood, Plasmodium exported proteins that interact with RBC cytoskeletal proteins are thought to remodel the iRBC membrane partly through the disruption of the spectrin-actin cytoskeleton network (horizontal interactions) or the protein-protein interaction linkages that tether the cytoskeleton to the overlying membrane bilayer (vertical interactions) [9–13].
Horizontal interactions in the erythrocyte cytoskeleton consist of head to head interacting heterotetramers of laterally aligned anti-parallel α- and β-spectrin repeats [14, 15]. These spectrin tetramers are then crosslinked into a lattice at their tails via the formation of junctional complexes consisting of F-actin, 4.1R (band 4.1, EPB41) and several accessory actin-binding proteins [16, 17]. This organization provides a mechanically stable spectrin skeleton that runs along the inside of the RBC membrane bilayer. Two distinct vertical protein interactions link the spectrin skeleton to the lipid bilayer [15]. One of these is mediated by ankyrin (ANK1) and couples the spectrin skeleton to the integral membrane proteins band 3 and Rh associated glycoprotein (RhAG) [16]. The second is mediated by 4.1R and connects the spectrin junctional complex to the transmembrane protein glycophorin C [18]. Linking of the spectrin-actin skeleton to the overlying membrane bilayer maintains membrane cohesion and prevents shear stress-induced membrane fragmentation in circulation [16, 19]. Manual rupture or mutations that weaken the spectrin-ANK1-band 3 bridge induce spontaneous membrane vesiculation and fragmentation [20–22], pointing to its central role in the maintenance of membrane cohesion. On the other hand, disrupting the spectrin-protein 4.1-glycophorin C bridge did not reveal any obvious impact on membrane mechanical properties [20, 21].
Interactions of Plasmodium exported proteins with erythrocyte cytoskeletal proteins play an important role in development, survival, viability and pathogenicity of the malaria parasite [23–26]. One of the first identified P. falciparum exported proteins was the Mature-parasite-infected Erythrocyte Surface Antigen (MESA) [27]. Although initially thought to be a surface antigen, subsequent studies showed that it was in fact localized beneath the host cell membrane and associated with the erythrocyte cytoskeleton [28]. Detailed mapping experiments with deletion constructs and competitive binding experiments with overlapping peptides revealed the 19 amino acid sequence DHLYSIRNYIECLRNAPYI as the minimal region responsible for binding to the cytoskeleton [29]; this element was sufficient to direct GFP to the erythrocyte cytoskeleton [30]. Co-immunoprecipitation experiments identified 4.1R as a binding partner of MESA [31]. Subsequent studies indicated that MESA binds to the 4.1R 30-kDa domain at a site that overlaps with p55 binding domain, suggesting that the two proteins compete for interaction with 4.1R [32].
Using iterative BLASTP searches, we discovered sequences closely related to the MESA cytoskeleton targeting sequence in 13 exported P. falciparum proteins that otherwise shared little sequence similarity [33]. Multiple sequence alignments suggested the minimal conserved sequence element from MESA consisted of a 13 amino acid sequence element (NYxxC[L/I]xxAPYID), the MESA Erythrocyte Cytoskeleton (MEC) binding motif. The C-terminal aspartic acid residue is conserved in all MEC motifs, but had been overlooked due to its location within the restriction site used for cloning [33]. Constructs containing MEC motifs from PF3D7_0500800/PFE0040c (MESA), PF3D7_0937000/PFI1790w, PF3D7_1038800/PF10_0378, PF3D7_0114000/PFA0675w, PF3D7_0220400/PFB0925w, PF3D7_0402100/PFD0095c and PF3D7_0631100/PFF1510w specifically interacted with inside out vesicles (IOVs) prepared from normal human RBCs [33]. As with MESA, the molecular functions of the predicted MEC motif proteins are poorly understood. However, gene knockout experiments suggested MEC motif proteins may be essential for parasite growth (PF3D7_0402100/PFD0095c and PF3D7_1102200/PF11_0034), cytoadherence (PF3D7_1039100/PF10_0381) or alteration of RBC membrane deformability (PF3D7_1401600/PF14_0018) [5].
In this report, we provide evidence that the MEC motif specifically interacts with the erythrocyte protein ANK1. In multiple independent experiments we find that the MESA MEC motif binds to a region of ANK1 adjacent to, but distinct from the spectrin-binding site. Amino acid substitutions revealed that the conserved aspartic acid residue at the C-terminus of the MEC motif is required for binding to ANK1 and inside-out vesicles. Binding of the MESA MEC motif to ANK1 has no apparent effect on RBC deformability, suggesting the primary function of the MEC motif is to recruit exported P. falciparum proteins to the iRBC cytoskeleton.
2. Material and methods
2.1. Constructs and deletion mutants.
Generation of the fragments encoding the MEC domains from MESA (PF3D7_0500800/PFE0040c), PF3D7_1038800 (PF10_0378), PF3D7_0114000 (PFA0675w), and the control MESA-MF4 clone [29] have been described previously [33]. Full length ORFs and gene fragments of erythroid Ankyrin-1 (ANK1) (variant 2) and protein 4.180KDa (4.1R, variant 2) were amplified from human bone marrow cDNA by PCR and cloned in frame with the fusion tags in the following plasmids: the bacterial expression vectors pGEX-6p-1, pMal-C4e, or a modified version of pMal-C4e in which a hexahistidine tag was inserted for addition to the 3’ end of the gene to be expressed; the wheat germ expression vectors p424-BYDV-UTR-3XFlag and p424-BYDV-UTR-SBP, which were derived from pF3K WG (BYDV) Flexi® vector (Promega); the split-luciferase vectors p424-BYDV-NFLuc and p424-BYDV-CFLuc; and the yeast two-hybrid plasmids pOAD2, pOAD102, and pOBD2. Additional details are provided in the results section and figure legends. Mutations in the 13 amino acid that constitute the conserved core of the MEC motif from MESA (PFE0040c) [33] were generated by overlap primer extension PCR. Mutant MESA MEC motif fragments were cloned into pMal-C4e in frame with the maltose binding protein (MBP) for bacteria expression, the split-luciferase vector p424-BYDV-NFLuc, or the yeast two-hybrid plasmid pOAD2. All clones were verified by sequencing.
2.2. Bacterial expression and purification of GST and MBP fusion proteins.
Expression and purification of MBP fusion proteins was as described [33]. For expression and purification of glutathione S-transferase (GST) - 4.1R-F5 fusion protein, the MBP fusion protein purification protocol was followed with the following modifications. Cleared bacterial supernatants were incubated for 1 h at room temperature with pre-washed glutathione-agarose beads (ThermoFisher), transferred to a gravity flow chromatography column and washed 3 times with cold PBS (1.05 mM KH2PO4, 154 mM NaCl, 5.48 mM Na2HPO4). To elute bound fusion proteins, 500 μl of 25 mM reduced glutathione (GSH) in cold PBS was applied to the column and incubated for 10 minutes at RT. The flow through was collected from multiple consecutive elutions and subjected to SDS-PAGE followed by staining with Coomassie Blue to evaluate purity. Fractions of interest were pooled and dialyzed overnight at 4° C against PBS supplemented with 10% glycerol. Purified and dialyzed fusion proteins were stored at −20° C in PBS plus 50% glycerol. Protein concentrations were determined as described previously [33]. For some experiments, MBP-His and MBP-MESA were further purified by size exclusion chromatography on an ÄKTAprime plus liquid chromatography system with a HiLoad 16/600 Superdex 200 column (GE Healthcare). Proteins were loaded onto the pre-equilibrated column (PBS, pH 7.4) and eluted at flow rate of 1 mL/min. Samples containing MBP-His and MBP-MESA were collected, subjected to SDS-PAGE and visualized with InstantBlue protein stain (Expedeon).
2.3. In vitro translation of parasite and RBC proteins.
In vitro transcription and translation of all p424-BYDV expression vector-based constructs was carried out using the TNT® SP6 High-Yield Wheat Germ Protein Expression System (Promega) as described [33]. To normalize the amount of protein used in each experiment, equal volumes of in vitro translation reactions were subjected to SDS-PAGE followed by western blot analysis with anti-FLAG antibody. The bands were then quantified using the Image J software package (NIH) [34]. Dilution ratios to normalize sample concentrations were obtained by dividing the densitometry value of all bands by that of the weakest band. Samples were then diluted with translation reaction buffer and kept on ice until use in subsequent experiments.
2.4. RBC ghosts and IOVs preparation.
RBC ghosts were prepared from normal human RBCs obtained from anonymous donors (Bioreclamation, LLC) with slight modifications [33]. Briefly, 5 - 30 mL of packed and washed RBCs were diluted 1:10 with ice-cold lysis buffer (5 mM phosphate buffer, pH 8.0, protease inhibitor cocktail, and 0.1 mM EDTA), mixed several times by inversion and incubated on ice for 10 min. The lysed cells were collected by centrifugation at 48,000 x g for 10 min. After removing ~95% of the supernatant, the previous step was repeated to ensure all cells were lysed, at which point the loose pellet was transferred to a fresh tube while leaving behind the intact RBCs. This process was repeated until the pellet had a creamy white appearance and the supernatant appeared free of hemoglobin (typically 2-3 repetitions). This stock of white RBC ghosts was either used immediately for the preparation of IOVs as described in [33] or extraction of RBC cytoskeletal proteins for co-affinity purification/protein-protein interaction assays. Erythrocyte ghost cells and IOVs were stored at 4° C for a maximum of 1 week.
2.5. Extraction of cytoskeletal proteins from RBC ghost cells.
RBC cytoskeletal proteins were extracted from white RBC ghosts using lithium 3, 5-diiodosalicylate (LIS, Sigma) as described in [33] with slight modifications. Five ml of packed white RBC ghosts were mixed with 50 ml of 20 mM LIS in 5 mM phosphate buffer, pH 8.0 supplemented with protease inhibitor cocktail (Roche), incubated on ice for 1 h with occasional gentle mixing, and cleared by centrifugation at 48,000 x g for 30 minutes. The supernatant was concentrated to 10 ml using a Centricon Plus-70 column (Millipore). Triton X-100 was added to a final concentration of 0.5% and the sample dialyzed twice against 1X PBS to remove LIS. Alternatively, cytoskeletal proteins were extracted by incubating RBC ghost cells with 1 M KCl solution with 0.5 M sodium phosphate (pH 8.0) plus PI cocktail on ice for 2 h with occasional mixing. Insoluble material was collected by centrifugation at 210,000 X g for 30 min at 4° C and the supernatant was dialyzed against PBS overnight. The resulting dialyzed RBC cytoskeletal protein extracts were used immediately in co-affinity purification assays.
2.6. Co-affinity purification assays
Co-affinity purification assays were performed using purified recombinant MBP-, GST-, or SBP-tagged proteins. Bait proteins were incubated with pre-washed affinity beads (amylose, GST, or streptavidin) in 500 μl of binding buffer A (1X PBS, plus protease inhibitor [PI] cocktail and 0.5% or 1% Triton X-100) for 2 h at 4° C with constant mixing. Beads were collected by brief, low-speed centrifugation and washed once with chilled binding buffer A. Prey protein was added in a total volume of 500 μL binding buffer A and the mixture incubated at 4° C for 6-8 h with constant mixing. Beads were washed three times with cold wash buffer A (1X PBS, 0.5 or 1% Triton X-100, and 0.25 mM KCl). For each wash, beads were mixed for 5 min at 4° C before centrifugation to ensure thorough removal of non-specific binding proteins.
2.7. IOV binding assays.
IOV binding assays were performed as described in [33] and Shakya et al. (submitted). Additional details can be found in individual figure legends.
2.8. Mass spectrometry.
Purified MBP MESA MEC motif proteins (10 μg) were pre-bound to amylose resin and mixed with 500 μg of dialyzed, LIS-extracted RBC cytoskeleton proteins. Co-affinity purification was performed as described above. Bound complexes were eluted by adding 100 μL of 3X SDS sample loading buffer supplemented with DTT to a final concentration of 1 mM followed by boiling for 5 minutes. 50 μL of cleared supernatant was then resolved on 10% SDS-polyacrylamide gel (SDS-PAGE) following standard protein Tris-glycine electrophoresis protocols. Resolved protein bands were visualized after staining with Coomassie Brilliant Blue (CBB). Bands were excised and prepared for liquid chromatography-tandem mass spectrometry (LC-MS/MS) as described in [35] and Shakya et al (submitted). Erythrocyte cytoskeleton proteins that bound to purified MBP-PF3D7_0402100 were identified as described in Shakya et al (submitted).
2.9. Yeast two-hybrid assays.
A fragment of MESA containing amino acids 80-140 and a series of N- and C-terminal deletion constructs derived from this fragment were cloned in frame with the GAL4 activation domain (AD) in pOAD102 by in vivo homologous recombination in the yeast strain BK100 (MATa ura3-52 ade2-101 trp1-901 leu2-3,112 his3-200 gal4Δ gal80Δ GAL2-ADE2 LYS2∷GAL1-HIS3 met2∷GAL7-lacZ) [36, 37]. Similarly, gene fragments from ANK1 and 4.1R were PCR amplified and cloned in frame with the GAL4 DNA binding domain (DBD) in pOBD2 by homologous recombination in the yeast strain R2HMet (MATa ura3-52 ade2-101 trp1-901 leu2-3,112 his3-200 met2Δ∷hisG gal4Δ gal80Δ) [36, 37]. Yeast strains expressing AD and DBD fusion proteins were mated to introduce both plasmids into the same cell. Diploid cells were selected by growth on synthetic defined (SD) medium deficient in tryptophan and leucine (-TL). For yeast two-hybrid assays, diploid cells were grown in synthetic defined liquid medium lacking tryptophan and leucine (SD–TL), adjusted to an OD600 = 1.0, serially diluted five-fold, and plated on synthetic defined medium lacking tryptophan, leucine, uracil, and histidine, and containing 3-amino-1,2,4-triazole (SD–TLUH + 3-AT) [36, 37]. 3-AT is a competitive inhibitor of the His3 reporter enzyme; higher concentrations of 3-AT represent more stringent selection conditions. To confirm the expression of AD fusion proteins in the diploid strains, the yeast lysates were prepared as described [38], followed by SDS-PAGE and western blotting with anti-GAL4AD antibody.
2.9. Split-luciferase assays.
Split-luciferase assays were performed as previously described ([35, 39] and Shakya et al. submitted). Kinetic split-luciferase assays were performed by adding N-FLuc-ankyrin to samples containing C-FLuc-MESA MEC construct and luciferase substrate in binding buffer. Luciferase activity was monitored at approximately 1 min intervals for at least 15 min. Statistical significance was determined by one-way ANOVA with multiple comparisons to wild type MESA MEC motif using Graphpad Prism 6 software.
2.10. Microsphere filtration assays.
Microsphere filtration (microsphiltration) was performed as described [35, 40].
3. Results
3.1. The MESA MEC motif binds ANK1.
We previously discovered an amino acid motif similar to the cytoskeleton binding element of the P. falciparum mature parasite-infected erythrocyte surface antigen (MESA) in 13 putative exported P. falciparum proteins [27–29, 33]. Co-precipitation experiments suggested this sequence binds to erythrocyte band 4.1 (4.1R) in infected cells and in assays using purified proteins [31, 32]. We found that 4.1R co-purified with the MEC motifs from three P. falciparum proteins when the MEC motifs were expressed in E. coli and mixed with lysates from erythrocyte ghosts (Fig. 1). However, we also observed that other erythrocyte cytoskeletal proteins, in particular ankyrin and spectrin, co-purified with MEC motif proteins at levels above the negative controls in the same assay (Fig. 1). Since both ankyrin and 4.1R bind to spectrin, it was possible that one or more of these proteins purified with the MEC motifs via bridging interactions with other erythrocyte cytoskeletal proteins. In an effort to confirm a direct interaction between the MEC motif and 4.1R, we attempted co-precipitations with MEC motif proteins and the minimal 4.1R fragment that bound to MESA (fragment F5, [32]), as well as larger fragments that spanned the length of 4.1R (Supplementary Fig. 1). Although the 4.1R F5 fragment bound to MEC motifs from some P. falciparum proteins, it did not co-purify with the MESA MEC motif in any format.
Fig.1.
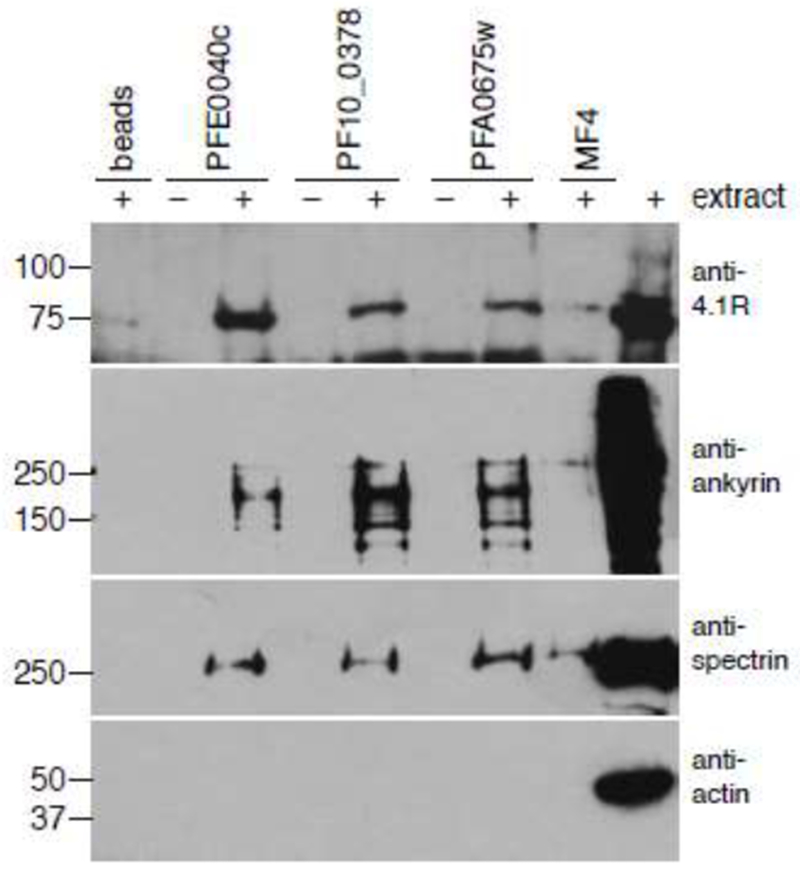
Erythrocyte cytoskeleton proteins co-precipitated with MEC motif constructs. E. coli-expressed and purified MBP-tagged MEC motif constructs from P. falciparum proteins MESA (PFE0040c), PF10_0378, and PFA0675w were incubated with extracts from erythrocyte membranes, precipitated on amylose beads, and subjected to western blotting with antibodies that recognize human 4.1R, ankyrin, spectrin, and actin. MBP-tagged MF4, a fragment from MESA that does not bind to erythrocyte inside out vesicles, was included as a negative control. Molecular weight markers are shown to the left of each blot.
Because protein-protein interaction assays have high false-negative rates [41], we sought an unbiased approach to identify the predominant erythrocyte cytoskeleton proteins that bind to the MEC motif. The MESA MEC motif was expressed in E. coli as a fusion to MBP, purified, dialyzed with PBS and incubated with an extract prepared from erythrocyte ghosts. MBP-MESA MEC and associated proteins were precipitated with amylose beads, washed to remove non-specifically associated proteins and subjected to SDS-PAGE. Proteins that specifically co-purified with MBP MESA MEC were excised from the gel and identified by mass spectrometry (Fig. 2A). In all six gel slices, the predominant human protein was ankyrin (ANK1, Table 1).
Fig. 2.
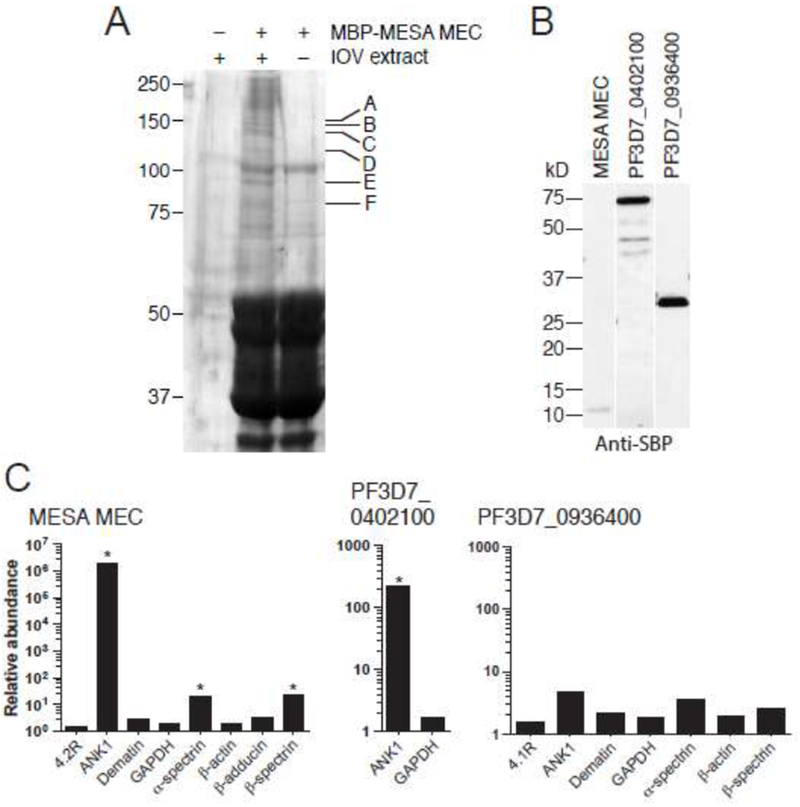
A. Co-affinity purification of erythrocyte cytoskeletal proteins with the MESA MEC motif. MBP-MESA was expressed in E. coli, purified, incubated with LIS-extracted erythrocyte ghosts, and purified again on amylose beads. Eluted proteins were subjected to SDS-PAGE and visualized by Coomassie blue staining. Bands found specifically in the MBP-MESA plus erythrocyte extract sample (indicated by letters) were excised from the gel, digested with trypsin, and subjected to mass spectrometry (see Table 1 for protein identification).
B. Expression of SBP-tagged P. falciparum proteins. PF3D7_0402100, and PF3D7_0402100. C-terminal SBP-tagged MESA MEC motif, PF3D7_0402100, (minus the region N-terminal to the predicted export motif) and PF3D7_0936400 (does not bind to IOVs) were in vitro translated in WGE, subjected to SDS-PAGE followed by western blotting with anti-SBP antibody.
C. Co-purification of erythrocyte cytoskeletal proteins with SBP-tagged P. falciparum proteins. In vitro translated SBP-tagged proteins were incubated with a soluble erythrocyte cytoskeletal extract and purified using streptavidin-coated beads. Co-purifying proteins were identified by LC-MS/MS. The plot shows the relative abundance of erythrocyte proteins that co-purified compared to the negative control (SBP-tagged GFP), which was set at 1 (* SAINT score >0.9).
Table 1.
Erythrocyte proteins that co-purified with MBP-MESA MEC.
| Band | Protein | Accession | Number of peptides |
|---|---|---|---|
| A | Ankyrin-1 (ANK1) | P16157 | 6 |
| B | Ankyrin-1 (ANK1) | P16157 | 13 |
| Spectrin beta chain, erythrocyte (SPTB) | P11277 | 2 | |
| C | Ankyrin-1 (ANK1) | P16157 | 12 |
| D | Ankyrin-1 (ANK1) | P16157 | 4 |
| Kininogen-1 (KNG1) | P01042 | 1 | |
| E | Ankyrin-1 (ANK1) | P16157 | 2 |
| Pro-opiomelanocortin (POMC) | P01189 | 1 | |
| Kininogen-1 (KNG1) | P01042 | 1 | |
| F | Ankyrin-1 (ANK1) | P16157 | 5 |
| Kininogen-1 (KNG1) | P01042 | 1 | |
| Pro-opiomelanocortin (POMC) | P01189 | 2 |
To obtain additional support for an interaction with ANK1, we expressed the MESA MEC motif and PF3D7_0402100 (full-length minus the region N-terminal to the PEXEL motif) as fusions to the streptavidin binding peptide (SBP) in wheat germ extracts. The MEC motif from PF3D7_0402100 diverges from others family members in that it has H and F instead of N rather than Y at positions 1 and 11, respectively, of the core motif. The PF3D7_0402100 MEC motif also appeared to bind less tightly to IOVs [33]. As a negative control, we included SBP-tagged PF3D7_0936400 (full-length minus the region N-terminal to the PEXEL motif), which does not bind to erythrocyte IOVs (Shakya et al, submitted, and data not shown).
SBP-tagged proteins were incubated with a soluble cytoskeleton extract, which was prepared by incubating erythrocyte ghost cells with 1 M KCL followed by dialysis with PBS. After washing the beads, the co-purifying proteins were digested with trypsin and submitted to liquid chromatography-tandem mass spectrometry (LC-MS/MS) for identification. ANK1 was the only significantly enriched cellular protein that co-purified with SBP-MESA MEC and SBP-PF3D7_0402100 (Fig. 2C). In contrast, no cellular proteins were significantly enriched in the SBP-PF3D7_0936400 pull-down. Thus, erythrocyte-expressed ANK1 co-purified with the isolated MESA MEC motif and with a nearly full-length protein containing a MEC motif in multiple independent experiments.
3.2. Confirmation of the interaction between the MEC motif and ANK1 in independent assays.
To confirm that ankyrin bound directly to the MESA MEC motif, we tested the interaction in three independent assays. First, we used the yeast two-hybrid assay, a genetic approach in which binding of a pair of proteins in yeast reconstitutes the Gal4 transcription factor, which in turn stimulates expression of a reporter gene. Data from a previous screen for host cell proteins that interacted with P. falciparum proteins suggested that a MESA fragment containing the MEC motif interacted with ankyrin 2 and 3 [42]. Here we tested the equivalent fragment of ANK1 for interaction with a MESA construct encompassing the MEC motif (amino acids 80-143, Fig. 3A). The MESA MEC construct interacted strongly with ankyrin (Fig. 3B), but did not interact with a 4.1 R construct that included the domain responsible for binding to the MESA in co-precipitation experiments [32]. To determine if the MEC motif was sufficient for binding to ankyrin, we tested a 19-residue fragment spanning the MEC motif (amino acids 100-118) and two constructs containing the N- or C-terminal flanking sequences. The 19-amino acid MEC motif construct bound to ankyrin in the yeast two-hybrid assay, though not as well as MESA fragment 80 – 143 (Fig. 3C, compare growth on media containing 1 mM and 10 mM 3-AT, a competitive inhibitor of the His3 reporter protein). Neither the N- nor C-terminal flanking sequences interacted with ankyrin. To determine if the flanking sequences contributed to binding, a series of N- and C-terminal truncations were constructed. C-terminal truncations had no impact on the interaction between the MESA MEC motif and ankyrin, whereas deletion of 15 or 20 amino acids from the N-terminal region reduced binding (Fig. 3D). However, the effect could only be observed when high concentrations of 3-AT were included in the growth medium, suggesting the impact of the N-terminal flanking sequence was modest. Thus, the MEC motif is sufficient for binding to ankyrin, but residues between 88 and 100 contribute to the interaction, at least in the context of the yeast two-hybrid assay. It is not clear if these residues are directly involved in binding or if they are needed as part of linker between the Gal4 binding domain and the MEC motif.
Fig. 3.
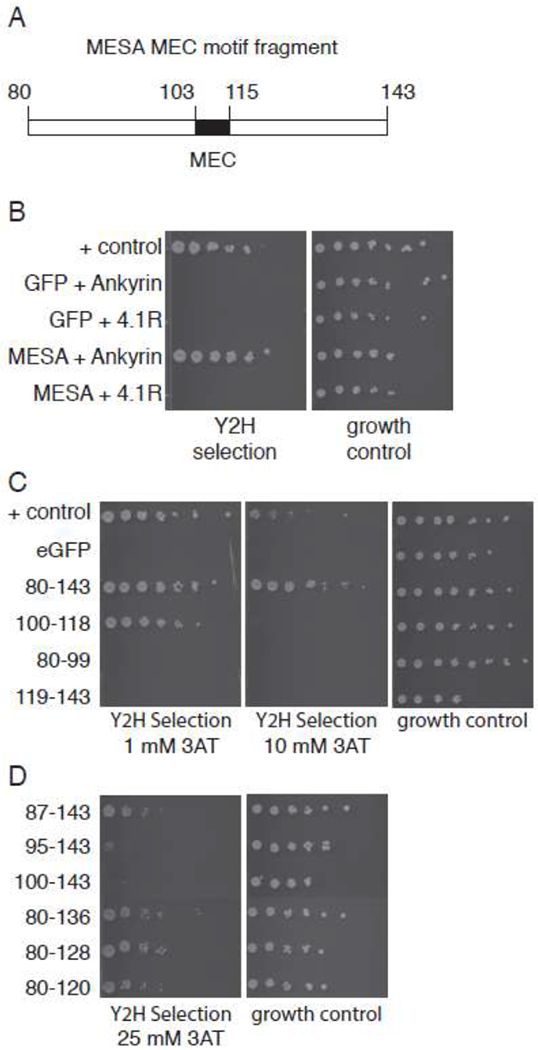
The MESA MEC motif interacts with erythrocyte ankyrin in the yeast two-hybrid assay.
A. MESA MEC fragment for yeast two-hybrid experiments. Numbers indicate amino acid positions in full length MESA.
B. Yeast two-hybrid assays. Diploid yeast expressing GFP or MESA MEC motif as fusions to the Gal4 activation domain (AD) and ankyrin or 4.1R as fusions to the Gal4 DNA binding domain (BD) were plated on yeast two-hybrid selection medium and medium lacking tryptophan and leucine (to show equal numbers of cells). Positive control cells express PFE1350c and PFC0255c as fusions to Gal4 AD and DBD.
C. MESA MEC motif N- and C-terminal flanking regions do not bind to ankyrin. Subfragments of the MESA MEC motif construct in (A) containing the MEC motif or the N- or C-terminal flanking regions were tested for binding to ankyrin in the yeast two-hybrid assay as in part B, except that the 3-AT concentrations were 1 and 10 mM in the left and middle panels, respectively. Higher 3-AT concentrations are more stringent.
D. Binding of MESA MEC motif truncation mutants to ankyrin. Six N- and C-terminal deletions of the MESA MEC motif construct were tested for binding to ankyrin in the yeast two-hybrid assay. Five-fold serial dilutions of diploid yeast were plated on yeast two-hybrid selection medium containing 25 mM 3-AT or medium lacking tryptophan and leucine.
To provide additional independent support for the interaction, we employed the split-luciferase assay. In this protein complementation assay, the N- and C-terminal fragments of firefly luciferase are fused to a pair of proteins. If the proteins interact, the luciferase fragments assemble to form a functional enzyme. The MESA MEC motif and full-length PF3D7_0402100 (minus the region N-terminal to the predicted PEXEL motif) were tested for binding to a panel of 42 fragments from 22 RBC cytoskeletal proteins in the split-luciferase assay; Fig. 4 shows the relative luciferase activity from 14 constructs that interacted with at least one exported P. falciparum protein and were therefore functional in the split-luciferase assay (Shakya et al, submitted). MESA yielded significantly higher luciferase levels relative to negative controls when paired with ANK1 fragment 3 (amino acids 823 - 1375) (Fig. 4A). Consistent with this result, PF3D7_0402100 interacted with ANK1 fragment 3, but also bound to ANK1 fragments 1 and 2 (Fig. 4B). An N-terminal fragment of 4.1R interacted with PF3D7_0402100, but not the MESA MEC, suggesting that the binding site for PF3D7_0402100 may reside in a different domain. Indeed, the PHIST domain has been reported to bind to 4.1 R (Shakya et al, submitted, and [35, 43]).
Fig. 4.
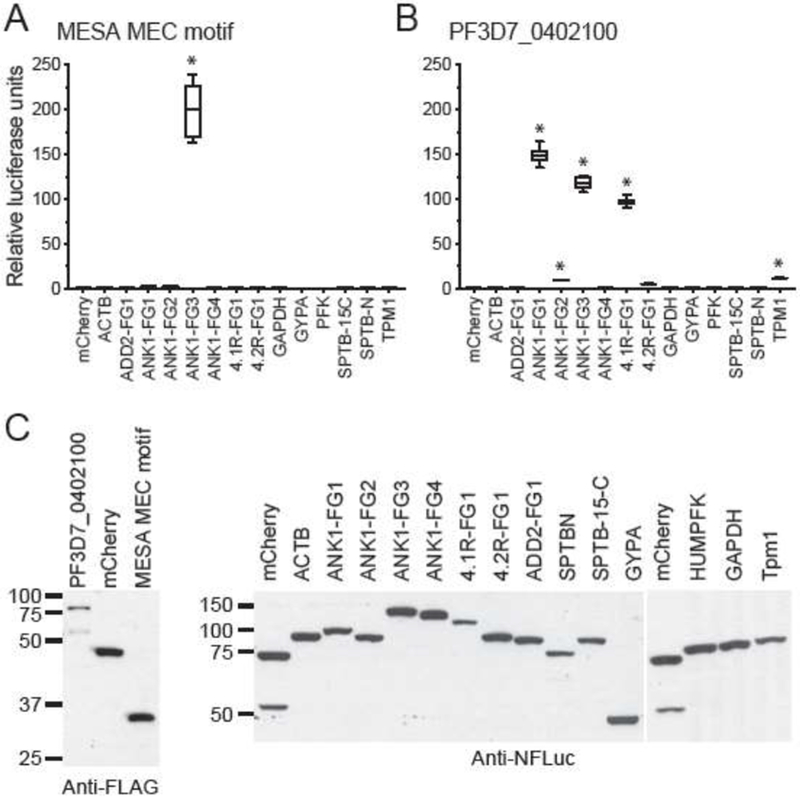
MEC motif proteins bind to erythrocyte ankyrin in the split-luciferase assay.
A. The MESA MEC motif binds to erythrocyte ankyrin. Proteins were in vitro translated in wheat germ extracts as fusions to N- and C-terminal fragments of firefly luciferase (N-FLuc and C-FLuc, respectively), mixed, and assayed for luciferase activity. Average relative luciferase activity (RLA) (± SEM) was calculated from duplicate readings from three independent replicates (six readings total) and was normalized to MESA-MEC motif plus N-FLuc-mCherry, which was set to 1. * indicates P value ≤ 0.001, as determined by one-way ANOVA with multiple comparisons to N-FLuc-mCherry using Graphpad Prism 6 software. ACTB, β-actin; ANK1, Ankyrin 1; 4.1R, Band 4.1; 4.2R, Band 4.2; GYPA, glycophorin A; GAPDH, glyceraldehyde phosphate dehydrogenase; SPTB-N, N-terminal fragment of β-spectrin; SPTB 15-C, β-spectrin fragment spanning spectrin repeat 15th to the C-terminus; ADD2, β-adducin; PFK, phosphofructokinase; TPM1, Tropomyosin 1.
B. PF3D7_0402100 binds to ANK1, 4.1R and TPM1 in the split-luciferase assay. Average RLA of C-FLuc-PF3D7_0402100 was calculated as above except that values were normalized to PF3D7_0402100 plus N-FLuc-mCherry. (* P ≤ 0.001).
C. Western blots showing the relative amounts of N-FLuc and C-FLuc fusion proteins used in panels A and B.
As a final approach to evaluate the MEC motif-ANK1 interaction, we performed co-precipitation assays in three formats. First, MEC motifs from four proteins were expressed in E. coli as MBP fusions, purified, dialyzed and mixed with four 3X-FLAG epitope-tagged ankyrin fragments that were in vitro translated in wheat germ extracts (Fig. 5A and B). Complexes were precipitated with amylose beads and washed. Consistent with the split-luciferase and yeast two-hybrid data, all four MEC motifs specifically co-precipitated ANK1 fragment 3 (FG-3), but not fragments spanning the ankyrin repeats or the C-terminus (Fig. 5C). MBP and MBP-MESA MF4, which do not bind to erythrocyte inside-out vesicles (IOVs), bound weakly or not at all to any ankyrin fragment. Blotting with anti-MBP antisera confirmed that each MBP protein was efficiently precipitated.
Fig. 5.
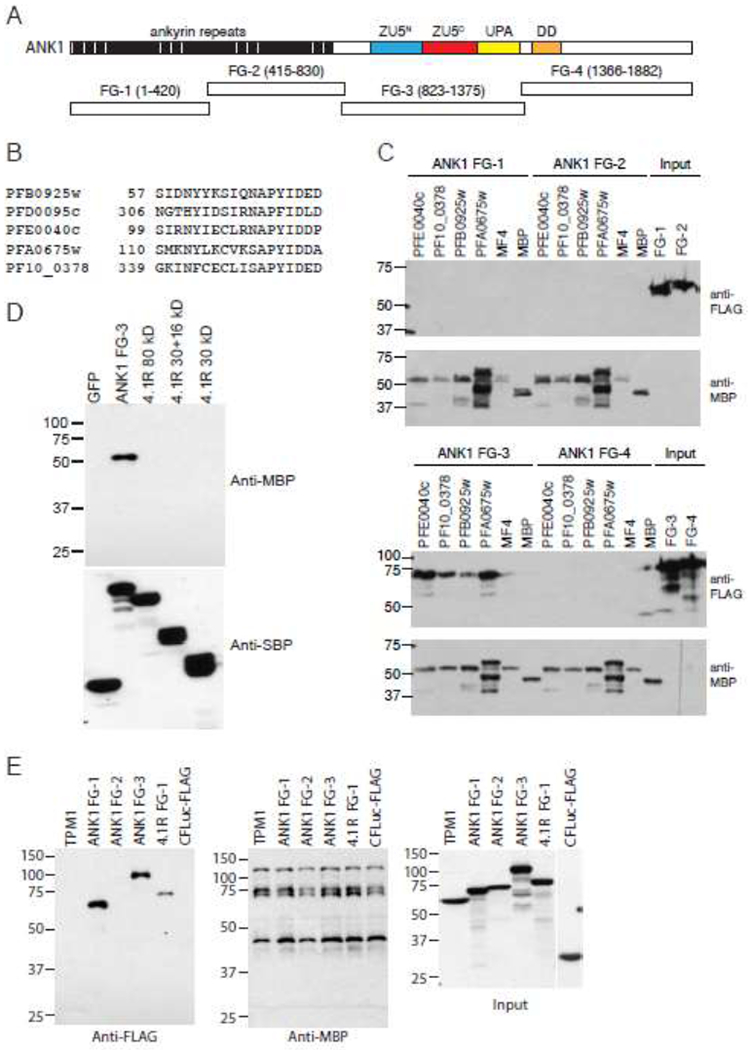
The MEC motif co-precipitates with erythrocyte ankyrin in pull-down assays.
A. Fragments used to map the MEC motif binding site on ankyrin. Numbers in parentheses indicate the amino acid start and end points of the fragments. Cyan and red boxes represent ZU5 domains (domain present in ZO-1 and Unc5-like netrin receptors); yellow box, UPA domain (domain conserved in UNC5, PIDD, and Ankyrins); orange box, death domain (DD).
B. Alignment of the MEC motifs from MESA (PF3D7_0500800/PFE0040c), PF3D7_1038800 (PF10_0378), PF3D7_0220400(PFB0925w), PF3D7_0114000 (PFA0675w) and PF3D7_0402100 (PFD0095c).
C. Co-purification of FLAG-tagged erythrocyte ankyrin with MBP-tagged MEC motifs. MEC motifs from MESA (PF3D7_0500800/PFE0040c), PF3D7_1038800 (PF10_0378), PF3D7_0220400 (PFB0925w), and PF3D7_0114000 (PFA0675w), and the negative control MESA fragment MF4 were expressed in E. coli as fusions to MBP, purified, and dialyzed with PBS. 3X-FLAG-tagged ankyrin fragments were in vitro translated in wheat germ extracts. MBP- and 3X-FLAG tagged proteins were mixed, incubated at 4°C, precipitated on amylose beads, and subjected to SDS-PAGE and western blotting with anti-FLAG (upper panels) and anti-MBP antisera (lower panels). The two right lanes on the upper blots show the amount of FLAG-tagged ankyrin proteins used in each co-purification.
D. Co-purification of MBP-MESA with SBP-tagged erythrocyte ankyrin. MBP-MESA MEC motif was expressed in E. coli, purified and dialyzed with PBS. SBP-tagged ankyrin fragment 3 and 4.1 R were in vitro translated in wheat germ extracts, mixed with MBP-MESA MEC, incubated at 4°C, precipitated on streptavidin beads, and subjected to SDS-PAGE and western blotting with anti-MBP and anti-SBP antisera.
E. Co-purification of FLAG-tagged erythrocyte ankyrin and 4.1R with MBP-PF3D7_0402100. MBP-His-tagged PF3D7_0402100 was incubated with 3X-FLAG/C-FLuc tagged human erythrocyte proteins, purified with amylose resin, and subjected to SDS-PAGE and western blotting with anti-FLAG (left panel) and anti-MBP (middle panel) antibodies. 3XFLAG/C-FLuc with no insert was included as a negative control. Right panel shows input amounts of 3X-FLAG/C-FLuc tagged human erythrocyte proteins. Molecular weight markers (sizes in kDa) are indicated at left.
We next performed co-precipitation assays using SBP-tagged ANK1 and 4.1 R as baits. SBP-tagged proteins were expressed in wheat germ extracts and incubated with MBP-MESA MEC that was expressed and purified from E. coli. MBP-MESA MEC co-precipitated with ANK1 FG-3 but not GFP or three 4.1R constructs (full length, 30+16 kDa, and 30 kDa domains) (Fig. 5D). Blotting with anti-SBP antisera confirmed that each SBP-tagged protein was efficiently precipitated. To ensure that MBP-MESA did not nonspecifically co-purify due to aggregation, we applied MBP-His and MBP-MESA MEC to a size exclusion column and used protein from the peak fraction at 50 kDa, corresponding to soluble monomer, for the pull-down experiment. Once again, MBP-MESA MEC, but not MBP-His, co-purified specifically SBP-ANK1 (Supplementary Fig. 2). Neither MBP-tagged protein co-precipitated with GFP or 4.1 R constructs.
Finally, we performed co-purification experiments with PF3D7_0402100. MBP-PF3D7_0402100 was expressed and purified from E. coli and incubated with in vitro translated erythrocyte cytosketon proteins. PF3D7_0402100 co-precipitated ANK1 fragments 1 and 3, and an N-terminal fragment of 4.1 R (Fig 5E). Tropomyosin and ANK1 FG-2, which yielded a relatively weak signal in the split-luciferase assay, did not co-purify with PF3D7_0402100. Blotting with anti-MBP antisera confirmed that MBP-PF3D7_0402100 was efficiently precipitated in each sample. Thus, in multiple, independent assays with diverse read outs, ANK1 specifically interacted with the MEC motif.
3.3. The ZU5C domain contains the MEC motif binding site on ANK1.
Ankyrin fragment FG-3 contains the ZU5N, ZU5C, and UPA (UNC5, PIDD, and ankyrins) domains. ZU5N binds to spectrin [44, 45], but the functions of ZU5C and UPA are not known. To determine if the MESA MEC and spectrin binding sites overlapped, we tested the ability of MBP-MESA MEC to co-precipitate subfragments of ankyrin FG-3. We verified that similar amounts of each ankyrin fragment were included in each sample and that MBP-MESA MEC was efficiently purified. Ankyrin fragment D6, and to a lesser extent D7, co-purified with the MESA MEC motif (Fig. 6B). D6 is the only fragment that contains a complete U5C domain.
Fig. 6.
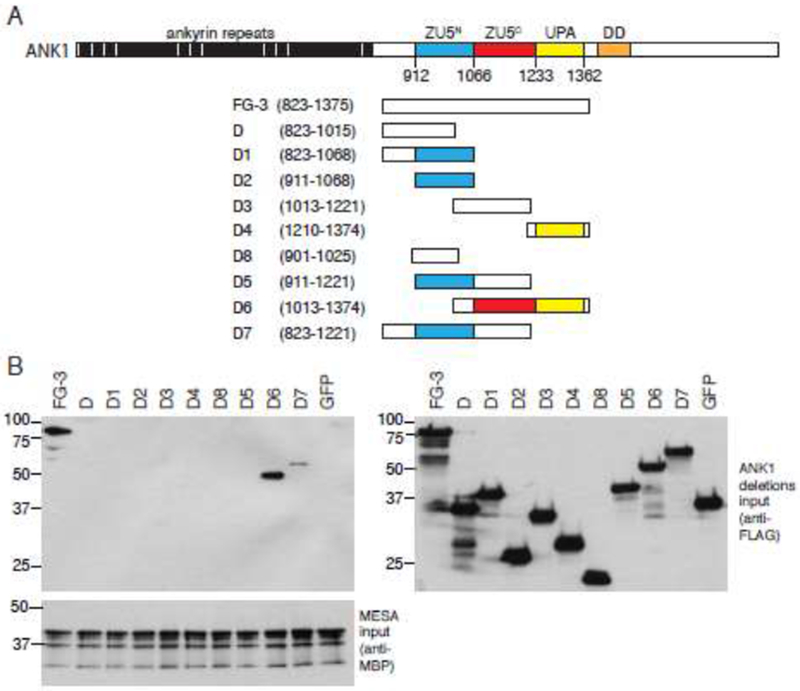
The MEC motif binding site is requires the ankyrin ZU5C domain.
A. Deletion fragments used to map the MEC motif binding site on ankyrin. Numbers in parentheses indicate the amino acid start and end points of the fragments.
B. The MESA MEC motif binds to an ankyrin fragment containing the ZU5C domain. Nine partially overlapping truncation mutants spanning ankyrin FG3 were in vitro translated in wheat germ extracts as fusions to three copies of the FLAG epitope tag and subjected to co-purification experiments with MBP-MESA as described for Fig. 5C. Left panels show immunoblots of copurified proteins using anti-FLAG (upper) and anti-MBP antisera (lower). An equivalent amount of each 3X-FLAG-tagged ankyrin fragment used in the co-precipitations was subjected to SDS-PAGE and immunoblotting with anti-FLAG antisera (right panel). Molecular weight markers are shown to the left of each blot. Co-purifications were performed three times with similar results; representative blots are shown.
3.4. Identification of MEC motif residues required for binding to ANK1.
To further analyze the binding of the MEC motif to ankyrin, each residue in the 13-amino acid conserved core motif from MESA was changed to alanine, with the exception of A110, which was changed to arginine. MEC motif mutants were introduced in the context of the 80 – 143 fragment, expressed in E. coli as fusions to MBP, purified, and tested for binding to IOVs (Fig. 7). Single mutations at positions 103, 110 and 114, and double or triple mutations in amino acids 110-114 eliminated binding to IOVs, whereas mutation of N102 reduced binding. The remaining mutations either had no effect or slightly increased binding.
Fig. 7.
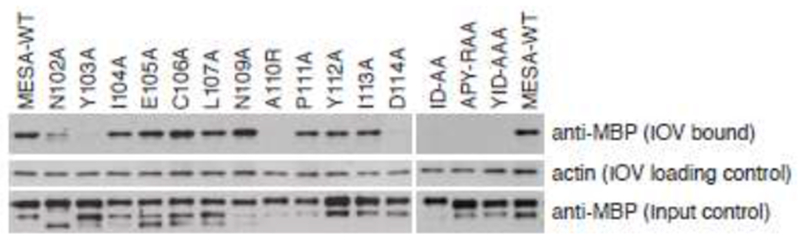
Amino acid substitutions in the MESA MEC motif disrupt binding to IOVs. Twelve single and three double or triple amino acid substitutions were generated in the MESA MEC motif by PCR-mediated mutagenesis. All mutations converted the wildtype amino acid to alanine except at position 110, in which alanine was converted to arginine. Substitution mutants were expressed in E. coli as fusions to MBP, purified on amylose beads, dialyzed with PBS, and incubated with erythrocyte IOVs. Proteins that co-precipitated with IOVs were separated on SDS-PAGE and subjected to immunoblotting with antisera that recognized MBP (top) and actin (middle; to demonstrate equal amounts of IOVs were present). The amount of MBP fusion protein used in each co-precipitation is shown in the bottom panel.
To examine the impact of the mutations on binding to ankyrin, the mutant MEC motif fragments were cloned into the split-luciferase expression plasmid and translated in vitro in wheat germ extracts. Binding to ankyrin was assessed in a kinetic split-luciferase assay in which the reaction was initiated by addition of the ankyrin fragment. The increase in luminescence was measured over time and used as an estimate of the rate of association. Similar to the IOV-binding results, single mutations at residues 102, 110 and 114, as well as the double and triple mutations reduced the binding to ankyrin (Fig. 8A, Supplementary Fig. 3). Surprisingly, however, Y103A increased the association of the MESA MEC motif with ankyrin. To determine if this was an effect of the fusion to the luciferase fragment or a difference in the ability of the MESA MEC motif to bind to IOVs versus ankyrin, the split-luciferase-MEC motif mutants were assayed for their binding to IOVs (Fig. 8B). IOV-binding closely paralleled luciferase activity, indicating that the differences in the binding properties of the Y103A mutant was most likely due to the fusion tag.
Fig. 8.
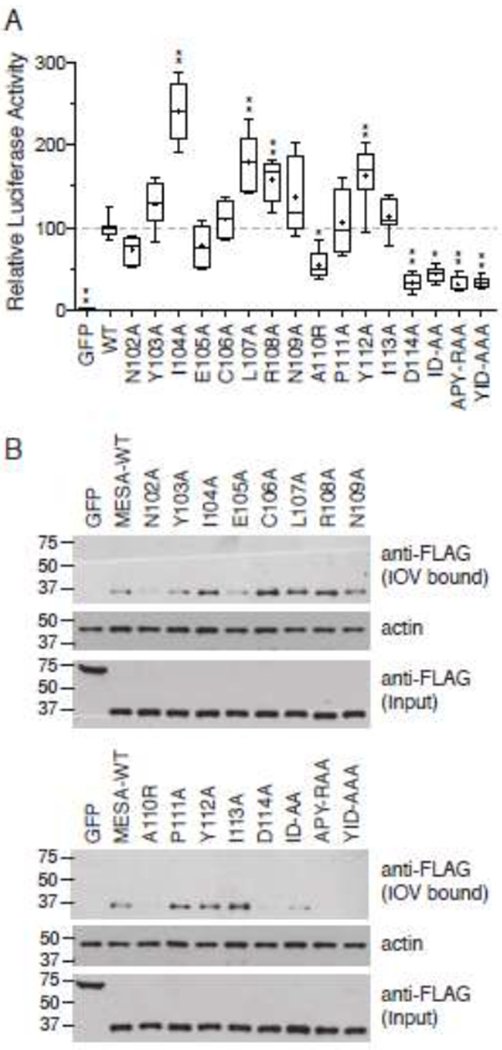
Amino acid substitutions in the MESA MEC motif disrupt binding to ankyrin.
A. Split-luciferase assay. The 12 single amino acid substitutions described in Fig. 6 were in vitro translated in wheat germ extracts as fusions to C-Fluc and tested for their interaction with N-FLuc-tagged ankyrin fragment 3 in the split-luciferase assay. Assays were initiated by addition of N-FLuc-ankyrin to samples containing C-FLuc-MESA MEC construct and luciferase substrate in binding buffer. The graph shows the mean luciferase activity (± S.D.) derived by subtracting the initial luciferase value from the third measurement (see Supplementary Fig. 3 for relative luciferase activity versus time for each sample) and normalizing the data to wild type MESA MEC, which was set to 100 (n = 8, from three independent experiments). Statistical significance was determined by one-way ANOVA with multiple comparisons to wild type MESA MEC motif using Graphpad Prism 6 software. * p value ≤ 0.01, ** p value ≤ 0.001, *** p value ≤ 0.0001.
B. IOV binding. C-FLuc-MESA MEC motif mutants were tested for binding to erythrocyte IOVs as described in the legend to Fig. 6. Upper blot shows a western blot analysis of IOV-bound 3X-FLAG and C-FLuc-tagged MESA MEC mutants; middle blot, erythrocyte actin, which serves as a loading control for the amount of IOVs present in each sample; lower blot, amount of C-FLuc construct added to each IOV-binding reaction.
3.5. The MESA MEC motif does not affect RBC rigidity.
Since ANK1 is a key component of the RBC cytoskeleton, we assessed the impact of the MESA MEC on erythrocyte rigidity (Fig. 9). Purified MBP-MESA was loaded into erythrocyte ghosts at a concentration (30 μM) predicted to exceed the amount of ankyrin and 4.1R. Protease protection assays confirmed loading of the MBP fusion proteins into ghosts (Fig. 9C). To demonstrate association of MBP-MESA with the erythrocyte cytoskeleton, ghost cells were lysed in a hypotonic buffer and the insoluble membrane fraction was collected by centrifugation. Membranes were incubated with 7.5 mM Tris HCI (pH 8.0), 6 M urea, 100 μM sodium carbonate (pH 11.5) or 2% SDS + 1% Triton X-100 [44, 45]. MBP-MESA was associated with the membrane fraction and could be partially solubilized with 6M urea and alkaline sodium carbonate, suggesting it was peripherally associated with the membrane (Supplementary Fig. 4).
Fig. 9. The MESA MEC motif does not alter erythrocyte rigidity.
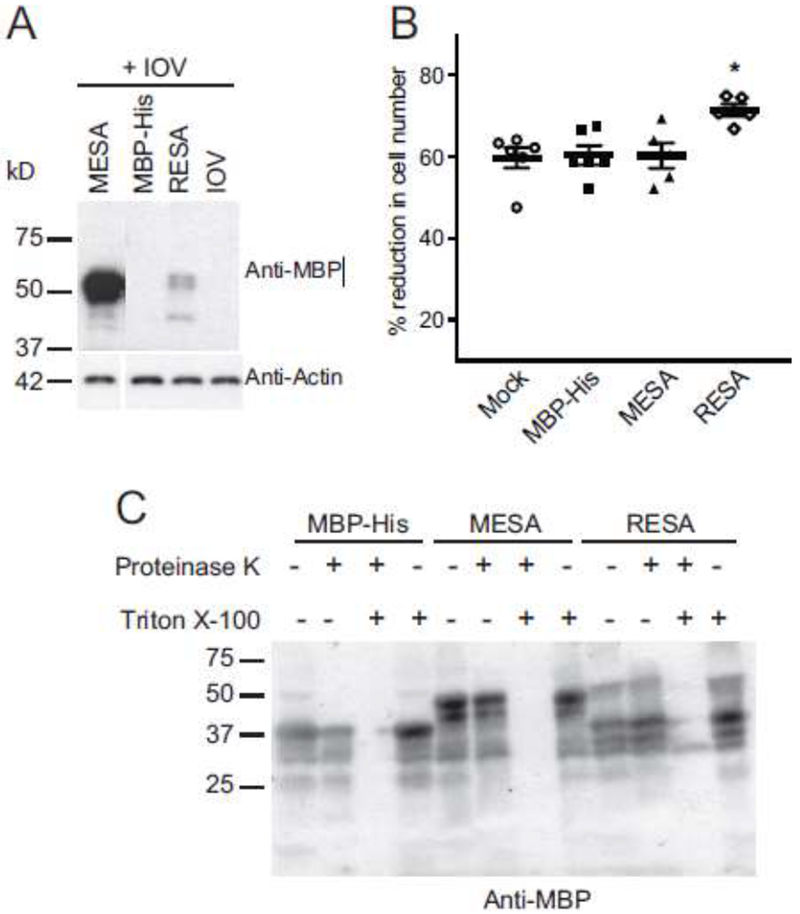
A. MBP-MESA binds to IOVs. Purified MBP-MESA, MBP-RESA, and MBP-His were incubated with erythrocyte IOVs and washed with binding buffer. IOVs and co-precipitating proteins were lysed with SDS sample buffer, separated by SDS-PAGE and analyzed by immunoblotting with anti-MBP antibody. Molecular weight markers (sizes in kDa) are shown at left.
B. MESA does not affect migration of erythrocyte ghosts through a microsphere filtration column. Erythrocytes ghost cells were loaded with purified MBP-MESA, MBP-RESA, or MBP-His (30 μM each) and passed through a microsphere matrix. Ghost cells in pre- and post-column samples were counted with a hemocytometer. Graphs show the percent reduction in cell numbers in the post-column samples. Statistical significance was calculated with the Mann-Whitney U test (*P<0.05).
C. Proteinase protection assay. Erythrocyte ghosts were purified MBP-MESA, MBP-RESA, or MBP-His and mock-treated or incubated with Proteinase K (100 μg/ml), 0.5% Triton X-100 or both. Samples were subjected to SDS-PAGE followed by western blotting with an anti-MBP antibody.
Loaded ghost cell were applied to a microsphere filtration column [40]. This column consists of metal microspheres of two sizes that pack together to create passages similar to inter-endothelial splenic slits [40]. To migrate through these openings, RBCs must distort their cell membranes. The efficiency with which RBCs pass through the microsphere filtration column is therefore an indication of RBC rigidity. Erythrocyte ghosts loaded with MBP-MESA MEC motif passed through the microsphere matrix at the same rate as negative control cell that were mock treated or loaded with MBP-His (Fig. 9B). In contrast, ghost cells loaded with MBP-RESA were retained in the column at a significantly higher rate, confirming previous results with RESA and demonstrating the assay functioned as expected [46, 47].
4. Discussion
Here we provide multiple lines of evidence that the MESA MEC motif binds to ANK1. First, the MESA MEC motif co-precipitated native, erythrocyte ANK1 from soluble extracts derived from erythrocyte ghost cells. Full length MEC motif proteins PF3D7_0402100 and PF3D7_1401600 also co-precipitated ANK1 from soluble extracts obtained from erythrocyte ghosts (Fig. 2 and Shakya et al, submitted). Using E. coli expressed and in vitro translated proteins, the MESA MEC motif co-precipitated with ANK1 in pair-wise co-purification experiments. The MESA MEC motif interacted strongly with ANK1 in the yeast two-hybrid assay, a genetic approach performed in Saccharomyces cerevisiae. In the split-luciferase assay, an in vitro protein complementation assay, the relative luciferase level with the MESA MEC motif and ANK1 was significantly higher than negative controls, and is one of the most active pairs we have found in this assay. Finally, mutations in the MEC motif that disrupted binding to ANK1 also disrupted binding to IOVs, suggesting that ANK1 is a valid target for the MEC motif.
The MEC motif binding site was localized to ankyrin fragment 3 (Fig. 4). This region includes two tandemly repeated copies of the ZU5 domain (ZU5N and ZU5C, also referred to as ZU5A and ZU5B) and a UPA domain [48, 49]. ZU5N and ZU5C have the same overall fold but little amino acid homology and several key differences. ZU5N contains a positively charged patch that binds to spectrin. This patch is missing in ZU5C, and as a result ZU5C does not bind to spectrin [48, 49]. ZU5N, but not ZU5C, makes extensive contacts with UPA, resulting in a cloverleaf structure with ZU5C protruding out from the ZU5N-UPA axis [48]. ZU5C has an additional alpha helix near the C-terminus of the domain, which covers a hydrophobic beta sheet [48, 49]. As such, this helix has been proposed to contribute to the stability of the domain [48]. ZU5C also has a positive patch near β-sheet 7 that is absent in ZU5N but is conserved in other ZU5C domains [48]. Despite the presence of potential binding sites, no function has been described for ZU5C.
Based on the mapping experiment in Fig. 5, the MEC motif binding site is C-terminal to the spectrin binding site and requires an intact ZU5C domain. Only fragment D6, which contained a complete ZU5C domain, bound efficiently to the MESA MEC motif (Fig. 6). Fragments that included ZU5N or UPA alone (D1, D2 and D4) did not co-purify. Fragments with truncated ZU5C domains bound weakly (D7) or not at all (D3 and D5). These truncated ZU5C constructs lacked amino acids 1222 – 1233, which comprise the ZU5c-specific α-helix thought to be required for ZU5C stability [48]. The absence of this helix could therefore cause misfolding of the ZU5C domain in constructs D3 and D5 and indirectly prevent binding, though we cannot rule out the possibility that the α-helix is directly involved in binding. Alternatively, the MEC motif may interact with ZU5C via the unique positively charged region, which has been proposed to serve as a binding site for negatively charged proteins [48].
Mutagenesis of each amino acid in the core conserved MEC motif revealed a critical role for aspartic acid at the end of the motif. Changing D to A individually or in combination with adjacent amino acids had the greatest impact on binding to ankyrin. This absolutely conserved aspartic acid residue may facilitate binding to the unique positively charged patch on ZU5C. Mutating alanine 110 to arginine also disrupted binding, but this drastic change may have impacted the overall structure. The amino terminal asparagine had a small but consistent effect on binding to IOVs and to ANK1. Mutating the adjacent tyrosine had no effect on binding to IOVs or to ANK1 when expressed as a fusion to 3X-FLAG and C-FLuc, but disrupted binding to IOVs when expressed as a fusion to MBP-His. Several MEC motif mutants appeared to bind better to ANK1 than wildtype, suggesting it may be possible to develop reagents with higher affinity for this site that may be able to block the MEC motif-ANK1 interaction. Some mutations behaved differently in the kinetic split-luciferase assay than in the IOV-binding experiments. For example, mutations at positions 107 and 108 increased the rate of association with ANK1, but had little effect on equilibrium binding to IOVs.
Binding of the MESA MEC motif to ANK1 had no effect on erythrocyte rigidity in the microsphere filtration assay (Fig. 9). This is consistent with data from MESA gene deletion strains, which did not reveal any obvious phenotypes [27, 28, 31]. In addition, the MEC motif binding site, ZU5C, has no known function. ZU5C does not bind directly to spectrin, nor does the presence of ZU5C affect the binding of ZU5N to spectrin based on both structural modelling and experimental data [48, 49]. Thus, MEC motif proteins are unlikely to compete with spectrin for binding to ankyrin. This suggests the MEC motif is most likely a mechanism to recruit exported proteins to the erythrocyte cytoskeleton but by itself does not affect cytoskeletal properties. Rather, interactions mediated by other domains in MEC motif proteins are responsible for the biochemical effects of the proteins. For example, PF3D7_1401600 binds to ANK1 D2 domain and to 4.1R via its PHIST domain and affected the rigidity of ghost cells (Shakya et al, submitted). Since the MEC motif by itself did not affect the rigidity of RBC ghost cells, the effect of PF3D7_1401600 on passage through the microsphere filtration column must be due to other interactions with the erythrocyte cytoskeleton, presumably those mediated by the PHIST domain.
Intriguingly, PF3D7_0402100 binds to similar set of erythrocyte cytoskeletal proteins to PF3D7_1401600 (Fig. 4 and 5, and Shakya et al, submitted). Both PF3D7_0402100 and PF3D7_1401600 bind to ANK1 fragments 1 and 3, and to the 4.1R membrane binding domain. Both have PHIST domains that precede their MEC motifs, although the overall amino acid similarity is low. However, PF3D7_0402100 bound to 4.1R to a greater extent in the split-luciferase assay than PF3D7_1401600 and also interacted with tropomyosin, while PF3D7_1401600 did not. Although PF3D7_0402100 (PFD0095c) was initially thought to be essential because it could not be deleted from the P. falciparum genome [5, 33], a large-scale piggyBac transposition screen has found that PF3D7_0402100 can be disrupted [50]. The ability to generate PF3D7_0402100-deficient strains will enable careful analyses of any phenotypes related to erythrocyte membrane properties in the future.
In contrast to ANK1, we were not able to recapitulate the interaction between MESA and 4.1R. In five formats (four pair-wise assays and complex purification from a soluble erythrocyte cytoskeleton extract) with different sources of 4.1R (full length 4.1R, fragments corresponding to defined domains and to the minimum region required for binding to MESA, using protein extracted from RBCs, expressed in E. coli or in vitro translated in WGE), the MESA MEC construct failed to co-precipitate with 4.1R at levels above the background from the negative controls. Similarly, the MESA MEC did not interact with 4.1R in the yeast two-hybrid assay. Only the split-luciferase assay yielded evidence of a direct interaction with 4.1R, but that was inconsistent. In Fig. 4, The combination of the MESA MEC motif and the membrane binding domain of 4.1R, which included the reported MESA binding site [32], was inactive, whereas the combination of ankyrin and MESA increased relative luciferase activity 200-fold. However, 4.1R fragment F5, the smallest fragment reported to interact with 4.1R [32], resulted in a statistically significant increase in luciferase activity relative to GFP and mCherry negative controls, though at a level 7 times lower than ankyrin (Supplementary Fig. 5). This same construct also caused a significant increase in luciferase activity when paired with PF3D7_0308600 (PFC0365w), a negative control not known or predicted to interact with MESA, suggesting F5 may be somewhat promiscuous.
The first report of 4.1R as a binding partner for MESA utilized co-immunoprecipitation experiments with an anti-MESA antibody that recognized the hexapeptide repeat sequence GESKET [31]. P. falciparum-infected cells labelled with 32P orthophosphate were lysed and immunoprecipitated with anti-MESA antibodies. Based on the presence of a radiolabeled band at 80 kDa and the pattern of fragments generated by proteolysis, this protein was determined to be 4.1R [31]. In a reciprocal experiment, MESA was co-immunoprecipitated with 4.1R antisera [29]. However, the technologies available at the time of these studies did not allow other non-radiolabeled RBC proteins to be identified, so other components such as ANK1 and spectrin were potentially missed. Co-precipitation experiments such as these typically purify complexes and are unable to definitively distinguish direct binding partners. Indeed, in our co-purification experiments followed by western blotting (Fig. 1), 4.1 R, ankyrin and spectrin were detected above binding to the negative controls, likely through bridging interactions that link these proteins in the cytoskeleton.
Although we were unable to reliably detect the MESA MEC-4.1R interaction, it is important to note that all protein-protein interaction assays have false-negatives and false-positives. A systematic analysis of five different protein interaction assays using well validated interacting proteins found that each method had a false-negative rate of 65 – 75 % [41]. Thus, we cannot rule out the possibility that the lack MESA MEC motif interaction with 4.1 R was a false negative in each of our assays. Furthermore, and in contrast to MESA, MEC motifs from other proteins, in particular PF3D7_1038800 (PF10_0378) and PF3D7_0114000 (PFA0675w), co-purified with 4.1 R fragment F5, which was previously identified as the smallest region of 4.1 R that bound to MESA (Supplementary Fig. 1) [32]. MEC motif variants may have different binding properties or greater propensities to bind either ANK1, 4.1R or both. Alternatively, MESA may interact with 4.1 R via sequences other than the MEC motif. Amino acids 850 – 1147 from MESA targeted GFP to the erythrocyte membrane, most likely through the actions of two lysine-rich elements [51]. Similar lysine-rich sequences from GARP were sufficient to direct GFP to the periphery of infected red blood cells [51]. The molecular target of these sequences is not known.
In conclusion, we have provided strong evidence from multiple independent assays that the MEC motifs present in exported P. falciparum proteins binds to erythrocyte ANK1, specifically to the ZU5C domain. This interaction does not affect erythrocyte rigidity. Rather the interaction appears to be a means to target exported proteins to a specific location in the infected RBC.
Supplementary Material
Acknowledgements
We thank Dr. H. Choi (National University of Singapore) for help with the SAINT analysis. The authors acknowledge the use of the facilities of the Bindley Bioscience Center, a core facility of the NIH-funded Indiana Clinical and Translational Sciences Institute, and the national scientific user facility at the Environmental Molecular Sciences Laboratory, sponsored by the U.S. DOE OBER.
This work was supported by the National Institutes of Health [grant numbers R01GM092829 and R01AI114814] and the Indiana Clinical and Translational Sciences Institute, funded in part by grant #UL1 TR001108 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. B. S. was supported in part by a fellowship from the Purdue Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- [1].Hiller NL, Bhattacharjee S, van Ooij C, Liolios K, Harrison T, Lopez-Estrano C, Haldar K, A host-targeting signal in virulence proteins reveals a secretome in malarial infection, Science 306(5703) (2004) 1934–7. [DOI] [PubMed] [Google Scholar]
- [2].Marti M, Good RT, Rug M, Knuepfer E, Cowman AF, Targeting malaria virulence and remodeling proteins to the host erythrocyte, Science 306(5703) (2004) 1930–3. [DOI] [PubMed] [Google Scholar]
- [3].Sargeant TJ, Marti M, Caler E, Carlton JM, Simpson K, Speed TP, Cowman AF, Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites, Genome Biol 7(2) (2006) R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].van Ooij C, Tamez P, Bhattacharjee S, Hiller NL, Harrison T, Liolios K, Kooij T, Ramesar J, Balu B, Adams J, Waters AP, Janse CJ, Haldar K, The malaria secretome: from algorithms to essential function in blood stage infection, PLoS Pathog 4(6) (2008) e1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Maier AG, Rug M, O’Neill MT, Brown M, Chakravorty S, Szestak T, Chesson J, Wu Y, Hughes K, Coppel RL, Newbold C, Beeson JG, Craig A, Crabb BS, Cowman AF, Exported proteins required for virulence and rigidity of Plasmodium falciparum-infected human erythrocytes, Cell 134(1) (2008) 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Suwanarusk R, Cooke BM, Dondorp AM, Silamut K, Sattabongkot J, White NJ, Udomsangpetch R, The deformability of red blood cells parasitized by Plasmodium falciparum and P. vivax, J Infect Dis 189(2) (2004) 190–4. [DOI] [PubMed] [Google Scholar]
- [7].Desai SA, Bezrukov SM, Zimmerberg J, A voltage-dependent channel involved in nutrient uptake by red blood cells infected with the malaria parasite, Nature 406(6799) (2000) 1001–5. [DOI] [PubMed] [Google Scholar]
- [8].Magowan C, Wollish W, Anderson L, Leech J, Cytoadherence by Plasmodium falciparum-infected erythrocytes is correlated with the expression of a family of variable proteins on infected erythrocytes, J Exp Med 168(4) (1988) 1307–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Maier AG, Cooke BM, Cowman AF, Tilley L, Malaria parasite proteins that remodel the host erythrocyte, Nat Rev Microbiol 7(5) (2009) 341–54. [DOI] [PubMed] [Google Scholar]
- [10].Boddey JA, Cowman AF, Plasmodium nesting: remaking the erythrocyte from the inside out, Annu Rev Microbiol 67 (2013) 243–69. [DOI] [PubMed] [Google Scholar]
- [11].Lavazec C, Molecular mechanisms of deformability of Plasmodium-infected erythrocytes, Curr Opin Microbiol 40 (2017) 138–144. [DOI] [PubMed] [Google Scholar]
- [12].Mohandas N, An X, Malaria and human red blood cells, Med Microbiol Immunol 201(4) (2012) 593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Proellocks NI, Coppel RL, Mohandas N, Cooke BM, Malaria parasite proteins and their role in alteration of the structure and function of red blood cells, Adv Parasitol 91 (2016) 1–86. [DOI] [PubMed] [Google Scholar]
- [14].Fowler VM, The human erythrocyte plasma membrane: a Rosetta Stone for decoding membrane-cytoskeleton structure, Curr Top Membr 72 (2013) 39–88. [DOI] [PubMed] [Google Scholar]
- [15].Lux S.E.t., Anatomy of the red cell membrane skeleton: unanswered questions, Blood 127(2) (2016) 187–99. [DOI] [PubMed] [Google Scholar]
- [16].Baines AJ, The spectrin-ankyrin-4.1-adducin membrane skeleton: adapting eukaryotic cells to the demands of animal life, Protoplasma 244(1-4) (2010) 99–131. [DOI] [PubMed] [Google Scholar]
- [17].Mohandas N, Gallagher PG, Red cell membrane: past, present, and future, Blood 112(10) (2008) 3939–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Salomao M, Zhang X, Yang Y, Lee S, Hartwig JH, Chasis JA, Mohandas N, An X, Protein 4.1R-dependent multiprotein complex: new insights into the structural organization of the red blood cell membrane, Proc Natl Acad Sci U S A 105(23) (2008) 8026–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mohandas N, Lie-Injo LE, Friedman M, Mak JW, Rigid membranes of Malayan ovalocytes: a likely genetic barrier against malaria, Blood 63(6) (1984) 1385–92. [PubMed] [Google Scholar]
- [20].Anong WA, Franco T, Chu H, Weis TL, Devlin EE, Bodine DM, An X, Mohandas N, Low PS, Adducin forms a bridge between the erythrocyte membrane and its cytoskeleton and regulates membrane cohesion, Blood 114(9) (2009) 1904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Anong WA, Weis TL, Low PS, Rate of rupture and reattachment of the band 3-ankyrin bridge on the human erythrocyte membrane, J Biol Chem 281(31) (2006) 22360–6. [DOI] [PubMed] [Google Scholar]
- [22].Van Dort HM, Knowles DW, Chasis JA, Lee G, Mohandas N, Low PS, Analysis of integral membrane protein contributions to the deformability and stability of the human erythrocyte membrane, J Biol Chem 276(50) (2001) 46968–74. [DOI] [PubMed] [Google Scholar]
- [23].Aikawa M, Iseki M, Barnwell JW, Taylor D, Oo MM, Howard RJ, The pathology of human cerebral malaria, Am J Trop Med Hyg 43(2 Pt 2) (1990) 30–7. [DOI] [PubMed] [Google Scholar]
- [24].Dondorp AM, Angus BJ, Hardeman MR, Chotivanich KT, Silamut K, Ruangveerayuth R, Kager PA, White NJ, Vreeken J, Prognostic significance of reduced red blood cell deformability in severe falciparum malaria, Am J Trop Med Hyg 57(5) (1997) 507–11. [DOI] [PubMed] [Google Scholar]
- [25].Dondorp AM, Kager PA, Vreeken J, White NJ, Abnormal blood flow and red blood cell deformability in severe malaria, Parasitol Today 16(6) (2000) 228–32. [DOI] [PubMed] [Google Scholar]
- [26].Pongponratn E, Riganti M, Punpoowong B, Aikawa M, Microvascular sequestration of parasitized erythrocytes in human falciparum malaria: a pathological study, Am J Trop Med Hyg 44(2) (1991) 168–75. [DOI] [PubMed] [Google Scholar]
- [27].Coppel RL, Culvenor JG, Bianco AE, Crewther PE, Stahl HD, Brown GV, Anders RF, Kemp DJ, Variable antigen associated with the surface of erythrocytes infected with mature stages of Plasmodium falciparum, Mol Biochem Parasitol 20(3) (1986) 265–77. [DOI] [PubMed] [Google Scholar]
- [28].Coppel RL, Lustigman S, Murray L, Anders RF, MESA is a Plasmodium falciparum phosphoprotein associated with the erythrocyte membrane skeleton, Mol Biochem Parasitol 31(3) (1988) 223–31. [DOI] [PubMed] [Google Scholar]
- [29].Bennett BJ, Mohandas N, Coppel RL, Defining the minimal domain of the Plasmodium falciparum protein MESA involved in the interaction with the red cell membrane skeletal protein 4.1, J Biol Chem 272(24) (1997) 15299–306. [DOI] [PubMed] [Google Scholar]
- [30].Black CG, Proellocks NI, Kats LM, Cooke BM, Mohandas N, Coppel RL, In vivo studies support the role of trafficking and cytoskeletal-binding motifs in the interaction of MESA with the membrane skeleton of Plasmodium falciparum-infected red blood cells, Mol Biochem Parasitol 160(2) (2008) 143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lustigman S, Anders RF, Brown GV, Coppel RL, The mature-parasite-infected erythrocyte surface antigen (MESA) of Plasmodium falciparum associates with the erythrocyte membrane skeletal protein, band 4.1, Mol Biochem Parasitol 38(2) (1990) 261–70. [DOI] [PubMed] [Google Scholar]
- [32].Waller KL, Nunomura W, An X, Cooke BM, Mohandas N, Coppel RL, Mature parasite-infected erythrocyte surface antigen (MESA) of Plasmodium falciparum binds to the 30-kDa domain of protein 4.1 in malaria-infected red blood cells, Blood 102(5) (2003) 1911–4. [DOI] [PubMed] [Google Scholar]
- [33].Kilili GK, LaCount DJ, An erythrocyte cytoskeleton-binding motif in exported Plasmodium falciparum proteins, Eukaryot Cell 10(11) (2011) 1439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Abramoff MD, Magelhaes PJ, Ram SJ, Image Processing with ImageJ, Biophotonics International 11(7) (2004) 36–42. [Google Scholar]
- [35].Shakya B, Penn WD, Nakayasu ES, LaCount DJ, The Plasmodium falciparum exported protein PF3D7_0402000 binds to erythrocyte ankyrin and band 4.1, Mol Biochem Parasitol 216 (2017) 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].LaCount DJ, Interactome mapping in malaria parasites: challenges and opportunities, Methods Mol Biol 812 (2012) 121–45. [DOI] [PubMed] [Google Scholar]
- [37].LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C, Fields S, Hughes RE, A protein interaction network of the malaria parasite Plasmodium falciparum, Nature 438(7064) (2005) 103–7. [DOI] [PubMed] [Google Scholar]
- [38].Kushnirov VV, Rapid and reliable protein extraction from yeast, Yeast 16 (2000) 857–860. [DOI] [PubMed] [Google Scholar]
- [39].Brown HF, Wang L, Khadka S, Fields S, Lacount DJ, A densely overlapping gene fragmentation approach improves yeast two-hybrid screens for Plasmodium falciparum proteins, Mol Biochem Parasitol 178(1-2) (2011) 56–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lavazec C, Deplaine G, Safeukui I, Perrot S, Milon G, Mercereau-Puijalon O, David PH, Buffet P, Microsphiltration: a microsphere matrix to explore erythrocyte deformability, Methods Mol Biol 923 (2013) 291–7. [DOI] [PubMed] [Google Scholar]
- [41].Braun P, Tasan M, Dreze M, Barrios-Rodiles M, Lemmens I, Yu H, Sahalie JM, Murray RR, Roncari L, de Smet AS, Venkatesan K, Rual JF, Vandenhaute J, Cusick ME, Pawson T, Hill DE, Tavernier J, Wrana JL, Roth FP, Vidal M, An experimentally derived confidence score for binary protein-protein interactions, Nat Methods 6(1) (2009) 91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vignali M, McKinlay A, Lacount DJ, Chettier R, Bell R, Sahasrabudhe S, Hughes RE, Fields S, Interaction of an atypical Plasmodium falciparum ETRAMP with human apolipoproteins, Malar J 7(1) (2008) 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Parish LA, Mai DW, Jones ML, Kitson EL, Rayner JC, A member of the Plasmodium falciparum PHIST family binds to the erythrocyte cytoskeleton component band 4.1, Malar J 12 (2013) 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mohler PJ, Yoon W, Bennett V, Ankyrin-B targets beta2-spectrin to an intracellular compartment in neonatal cardiomyocytes, J Biol Chem 279(38) (2004) 40185–93. [DOI] [PubMed] [Google Scholar]
- [45].Ipsaro JJ, Mondragon A, Structural basis for spectrin recognition by ankyrin, Blood 115(20) (2010) 4093–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pei X, Guo X, Coppel R, Bhattacharjee S, Haldar K, Gratzer W, Mohandas N, An X, The ring-infected erythrocyte surface antigen (RESA) of Plasmodium falciparum stabilizes spectrin tetramers and suppresses further invasion, Blood (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Diez-Silva M, Park Y, Huang S, Bow H, Mercereau-Puijalon O, Deplaine G, Lavazec C, Perrot S, Bonnefoy S, Feld MS, Han J, Dao M, Suresh S, Pf155/RESA protein influences the dynamic microcirculatory behavior of ring-stage Plasmodium falciparum infected red blood cells, Scientific reports 2 (2012) 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang C, Yu C, Ye F, Wei Z, Zhang M, Structure of the ZU5-ZU5-UPA-DD tandem of ankyrin-B reveals interaction surfaces necessary for ankyrin function, Proc Natl Acad Sci U S A 109(13) (2012) 4822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yasunaga M, Ipsaro JJ, Mondragon A, Structurally similar but functionally diverse ZU5 domains in human erythrocyte ankyrin, J Mol Biol 417(4) (2012) 336–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, Udenze K, Bronner IF, Casandra D, Mayho M, Brown J, Li S, Swanson J, Rayner JC, Jiang RHY, Adams JH, Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis, Science 360(6388) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Davies HM, Thalassinos K, Osborne AR, Expansion of lysine-rich repeats in Plasmodium proteins generates novel localization sequences that target the periphery of the host erythrocyte, J Biol Chem 291(50) (2016) 26188–26207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brown GR, Hem V, Katz KS, Ovetsky M, Wallin C, Ermolaeva O, Tolstoy I, Tatusova T, Pruitt KD, Maglott DR, Murphy TD, Gene: a gene-centered information resource at NCBI, Nucleic Acids Res 43(Database issue) (2015) D36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nilsson S, Angeletti D, Wahlgren M, Chen Q, Moll K, Plasmodium falciparum antigen 332 is a resident peripheral membrane protein of Maurer’s clefts, PLoS One 7(11) (2012) e46980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Proellocks NI, Herrmann S, Buckingham DW, Hanssen E, Hodges EK, Elsworth B, Morahan BJ, Coppel RL, Cooke BM, A lysine-rich membrane-associated PHISTb protein involved in alteration of the cytoadhesive properties of Plasmodium falciparum-infected red blood cells, FASEB J (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.