

Abstract
Autophagy contributes to cellular quality control and energetics through lysosomal breakdown and recycling of essential cellular components. Chaperone-mediated autophagy (CMA) adds to these autophagic functions the ability to timely and selectively degrade single tagged proteins to terminate their cellular function and, in this way, participate in the regulation of multiple cellular processes.
Many cancer cells upregulate CMA for pro-tumorigenic and pro-survival purposes. However, growing evidence supports a physiological role for CMA against malignant transformation. Understanding the mechanisms behind this functional switch of CMA from anti-oncogenic to pro-oncogenic is fundamental for targeting CMA in cancer treatment. Here, we summarize the current understanding of the CMA functions in cancer biology and discuss the basis for its context-dependent dual role in oncogenesis.
Keywords: chaperones, lysosomes, metabolism, oncogenes, protein degradation, tumorigenesis
CMA: the “newbie” in cancer biology
Autophagy degrades intracellular proteins and organelles in lysosomes and was, for decades, considered a non-selective process for cellular turnover [1]; however, nowadays, it is well-accepted that autophagy selectively discriminates components to be degraded (cargo) from those that should be spared. Out of the three main types of autophagy co-existing in most mammalian cells (Fig.1) [2], selective degradation was first described for chaperone-mediated autophagy (CMA), the main focus of this review. Contrary to macroautophagy and microautophagy, where cargo is sequestered in vesicles for lysosomal delivery [2], proteins degraded via CMA, are recognized one-by-one by a cytosolic chaperone that brings them to lysosomes [3]. Selective macroautophagy was demonstrated after the discovery of cargo receptors and adaptors that bring the macroautophagy machinery to the material to be degraded [4]. Selectivity adds regulatory capabilities to the known functions of autophagy in cellular quality control and energetics.
Figure 1. CMA in the context of mammalian autophagic pathways.

Macroautophagy starts with sequestration of cargo by a limiting membrane that seals into a double-membrane vesicle named autophagosome. Guided by microtubules autophagosome fuses with lysosomes, where degradation occurs. Chaperone-mediated autophagy (CMA) degrades proteins selected by the chaperone Hsc70 that binds a KFERQ-like sequence. Internalization of substrate proteins into lysosomes occurs upon multimerization of LAMP2A. Microautophagy entraps cytosolic cargo in small vesicles formed by invagination of the lysosomal membrane. In endosomal microautophagy KFERQ-proteins recognized by hsc70 are targeted for degradation in late endosomes through invaginations in the membrane,
Both, maintenance and regulatory function of macroautophagy have proven important in its complex role in cancer biology. Macroautophagy inhibits or promotes cancer cell proliferation and tumorigenesis in a context-dependent manner [5, 6]. Activation of macroautophagy provides recycled essential components required to maintain anabolic processes in many cancers. However, in others, degradation by macroautophagy is blocked to favor anabolic over catabolic processes and thus sustain rapid cell division and proliferation [5]. Activation of macroautophagy in many cancer cells when subjected to anti-oncogenic interventions confers them resistance through mechanisms still under intensive investigation [5, 6]. For example, recent studies have shown that cancer cells can avoid activation of apoptosis in response to chemotherapy drugs by modulating cellular levels of FOXO3a through macroautophagy degradation [7].
Only recently a possible relation of CMA with cancer biology has been considered. The first report, 8 years ago, showing CMA dependence for growth and survival of many cancer cell types [8], triggered the growing number of studies in support of this pro-oncogenic function of CMA in cancer cells and set the basis for the recent interest in targeting CMA in cancer. However, as for macroautophagy, additional consideration needs to be given before CMA can be used as a therapeutic target. In contrast with the pro-oncogenic functions of CMA in cancer cells, studies in untransformed cells support that one of the physiological functions of CMA in these cells is to protect them against malignant transformation. Here, we review this intricate role of CMA in cancer and the different mechanisms described behind this dual function.
CMA: the basics
Recent reviews had summarized in detail the current understanding of the steps, components and physiology of CMA [3]. Here and in Box 1, we highlight the main characteristics that distinguish CMA from other autophagies.
BOX 1. CMA REGULATION
Substrate targeting:
Binding of hsc70 to substrates occurs when the KFERQ-like motif is exposed and accessible (i.e. partial protein unfolding, protein dissociation from protein complexes or membranes). In some instances, only four of the five amino acids of the targeting motif are constitutively present in the protein, and posttranslational modifications such as phosphorylation or acetylation can provide the missing charge and complete the motif [10]. The same chaperone, hsc70, and targeting motif, are shared by CMA and endosomal microautophagy, a selective form of microautophagy [78]. It is likely that the conformation of the substrate protein along with hsc70 cochaperones participate in cargo triage between both autophagic pathways. Cargo is delivered to late endosomes in endosomal microautophagy and to secondary lysosomes in the case of CMA [3, 78].
Lysosomal regulation of CMA:
Dynamics of LAMP2A at the lysosomal membrane are tightly regulated by two pairs of proteins at the lysosomal surface: i) monomers of the intermediate filament protein glial fibrillary acidic protein (GFAP) and its GTP-binding partner elongation factor 1 alpha (EF1α) determine the stability of the LAMP2A translocation complex through a series of events that require GTP hydrolysis and GFAP phosphorylation [13]; ii) a second set of lysosome associated proteins that regulate CMA activity are the kinases mTORC2 and AKT1 and the phosphatase PHLPP1 [17]. Phosphorylation of lysosome-associated AKT1 by mTORC2 represses CMA activation by reducing LAMP2A assembly into the translocation complex, in part through phosphorylation of GFAP. Contrary to mTORC1, the negative regulator of macroautophagy, that shuttles in and out of lysosomes, mTORC2 is stably associated with the membrane of CMA-dedicated lysosomes, making thus necessary the recruitment of the phosphatase PHLPP1 to counteract the inhibitory effect of mTORC2 by actively dephosphorylating AKT1 [17].
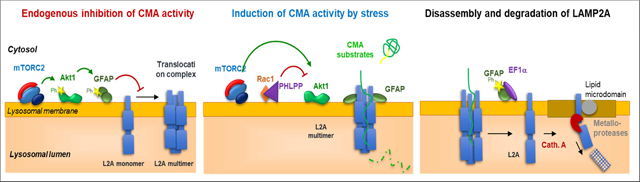
Extra-lysosomal regulation of CMA:
Nuclear signaling through the retinoic acid receptor alpha (RARa) has been shown to be inhibitory on the CMA-related transcriptional program [76]. Thus, chemical inhibition of RARa enhances transcription of LAMP2A and also other CMA components such as Rab11, necessary for LAMP2A trafficking from Golgi to lysosomes [79]. Transcription of LAMP2A can be directly initiated by the transcription factor NFAT1 [32] and by NRF2 [80], in both instances in close relation with the oxidative cellular conditions. Lastly, mitochondria can also upregulate CMA activity through release of small peptides such as humanin that stabilizes binding of substrates during their unfolding at the lysosomal membrane [15].
CMA degrades proteins targeted to lysosomes by a cytosolic chaperone, the heat shock-cognate chaperone of 70 kDa, hsc70, that recognizes and binds a pentapeptide motif (KFERQ-like) in the substrate protein [9, 10] (Fig.1). At the lysosome, the substrate protein binds the cytosolic tail of the lysosome-associated membrane protein type 2A (LAMP2A), one of the three splice variants of the lamp2 gene [11]. Substrate binding triggers assembly of LAMP2A into a protein complex that mediates translocation of the substrate into lysosomes [12, 13] with the help of a hsc70 resident in the lysosomal lumen (Fig.1).
Chaperones participate in multiple CMA steps. Besides substrate targeting, hsc70 at the lysosome surface facilitates substrate unfolding and disassembly of the translocation complex after substrate internalization [12]. Luminal resident hsc70 completes translocation of the substrates [14]. Hsp90 is present on both sides of the lysosomal membrane: Cytosol-facing hsp90 binds substrate proteins during the unfolding step that precedes translocation to prevent unwanted interactions [15], whereas hsp90 in the luminal part of the lysosomal membrane, binds and stabilizes LAMP2A as it transitions from monomer to multimer [12].
LAMP2A and luminal hsc70 content determine rates of CMA, but since binding occurs first, LAMP2A levels are limiting [16]. Consequently, LAMP2A abundance is often used as an indirect indicator of CMA status, and interventions changing lysosomal levels of this receptor are used to modulate CMA activity [11, 16]. Often, CMA activation does not require de novo synthesis of LAMP2A, because its lysosomal abundance can be modified by changing its stability, organization and dynamics at the lysosomal membrane [12]. These events are tightly regulated by lysosome-associated proteins such as the GFAP/EF1α pair [13] and the mTORC2/AKT1/PHLPP1 axis [17] (Box 1). Beyond this local regulation, signaling from nucleus, plasma membrane and other organelles also provide inputs on CMA (Box 1).
Physiologic functions of CMA
Although reconstitution of CMA in vitro using isolated lysosomes [18] allowed molecular dissection and identification of lysosomal regulators of this pathway, measurement of CMA in intact cells is now possible using novel fluorescent reporters [19]. One of such reporters (KFERQ-Dendra) has been recently used to generate a transgenic mouse model where CMA can be tracked in whole tissues in vivo [20]. This model and tissue-specific CMA incompetent LAMP2A knock-out mice have expanded the physiological functions attributed to CMA.
CMA contributes to cellular quality control [21–24] and its inhibition turns cells sensitive to stressors that disrupt proteostasis (i.e. oxidative stress [23], protein-damaging chemicals [25]) and leads to accumulation of oxidized and aggregated proteins [21–24].
Although the first stimulus known to activate CMA was removal of nutrients [26] a role for CMA in cellular energetics has only recently been demonstrated. Activation of CMA during starvation was proposed to contribute amino acids to maintain essential proteins’ synthesis. However, the main contribution of CMA to cellular energetics is through selective degradation of fully functional key enzymes in lipid and glucose metabolism that terminates their function and reduces flux through these metabolic pathways [27]. CMA degradation of glycolytic and lipogenic enzymes and of lipid droplet-associated proteins stops hepatic glycolysis and lipogenesis and facilitates lipolysis during starvation [27, 28].
This ability of CMA to selectively degrade still functional proteins is the bases of the physiological regulatory functions nowadays attributed to this pathway [3]. Besides metabolic pathways, CMA contributes to cell cycle re-entry after DNA repair through timely degradation of Chk1. In absence of CMA, nuclear levels of phospho-Chk1 remain elevated, thus disrupting the DNA repair machinery [29]. CMA also modulates HIF1α, another regulator of the cell cycle, under hypoxic conditions [30, 31], and controls transcriptional programs, by directly degrading transcription factors and their activators and inhibitors. For example, in CD4+ T cells, CMA degradation of Itch and RCAN1, negative regulators of TCR signaling, is required to attain full T cell activation. Consequently, CMA-incompetent T cells show defective responses to immunization and pathogen infection [32].
Anti-oncogenic function of CMA
Dysfunction of CMA was first described in aging, coincidentally the highest risk factor of many types of cancer. Age-dependent CMA decline occurs in almost all mammalian cell types and tissues [22, 33], in many instances due to lower stability of LAMP2A in old cells’ lysosomes because of changes in their membrane lipid composition [33, 34]. The consequences of this systemic decline of CMA and its contribution to aging are being investigated by reproducing CMA blockage in young mice. For example, mice with selective liver CMA inhibition develop a marked metabolic phenotype [27] and display higher incidence of spontaneous hepatic tumors with age [24]. These findings support a physiological anti-oncogenic function for CMA, at least in part, by preventing intracellular changes that favor transformation and by directly modulating the cellular content of oncogenic proteins (Fig.2).
Figure 2. (Key figure). Anti-oncogenic function of CMA turns pro-oncogenic in cancer cells.

Studies in vitro and in vivo support an anti-oncogenic function for CMA in normal cells (1) through a variety of mechanism (blue box). This could explain why conditions in which CMA activity is reduced (2) such as aging or disease favor malignant transformation. Right after transformation CMA activity is upregulated (3) and remains constitutively active in most tumor cells (4). High CMA activity in cancer cells sustains different pro-oncogenic functions (yellow box).
CMA prevents intracellular changes that favor malignant transformation.
The most studied changes, so far, are those that contribute to tumors in CMA-defective livers [24]. Higher rates of malignant transformation in this context can be, in part, secondary to the underlying hepatic steatosis [27]. Liver-specific LAMP2AKO mice also show higher predisposition to fibrosis in response to pro-oxidant insults [24] mimicking, to some extent, the progression in patients of non-alcoholic fatty liver to fibrosis and hepatocellular carcinoma (HCC). CMA is activated in response to lipid challenges [28] and LAMP2A upregulation also occurs in experimental mouse models of non-alcoholic steatohepatitis [35]. However, abnormally elevated intracellular lipids are inhibitory to CMA [36] and have been shown to reduce LAMP2A expression in different models of steatosis [37, 38]. This vicious circle, whereby reduced CMA promotes steatosis that in turn further inhibits CMA may perpetuate the metabolic derangement and progression to fibrosis and HCC. The basis for the pro-fibrotic nature of CMA-deficient livers is unknown. Upregulation of macroautophagy in stellate cells contributes to liver fibrosis [39]. Stellate cells show lower basal CMA activity than hepatocytes but similar inducible activity in response to stress [20]. Further studies will determine if stress-induced CMA upregulation in stellate cells contributes to the pro-fibrotic phenotype of hepatocyte-specific LAMP2AKO mice.
Decreased protein quality control and subsequent lower resistance to stress in LAMP2AKO mice [24] may also favor malignant transformation and liver tumorigenesis (Fig.2). In fact, both steatosis and reduced quality control have been shown to promote oncogenic transformation and liver tumors in LAMP2AKO mice display areas of eosinophilic foci (proteinaceous aggregates), lipidosis, and inflammation [24].
Furthermore, because CMA is part of the cellular response to genotoxic insults, through the above-mentioned role in DNA repair [29], it is likely that increased genomic instability in absence of CMA also favors malignant transformation [24].
CMA modulates oncogenic cellular load and pro-oncogenic signaling.
Recent studies support that the antioncogenic capacity of CMA is, at least in part, due to its ability to reduce levels of pro-oncogenic proteins. CMA degradation of the protooncogene protein MDM2 [40] and of the tumor-associated TCTP (upon acetylation) [41] were the first hints of a tumor suppressive function for CMA. However, the first direct proof that regulation of oncogene levels by CMA prevents malignant transformation was described for c-MYC [42]. CMA inhibition was shown sufficient to increase c-MYC-induced transformation in cultured fibroblasts [42]. The acute nature of these experiments does not allow for changes in metabolism, proteostasis or genotoxicity, thus permitting to attribute the increase in transformation to the higher c-MYC levels in CMA-incompetent cells. Interestingly, in this context, c-MYC is not directly degraded by CMA. Instead, CMA promotes c-MYC proteasomal degradation by degrading CIP2A, the phosphatase that regulates the site-specific phosphorylation of c-MYC that controls its proteolysis through the ubiquitin-proteasome system [42].
Recent studies also support an anti-oncogenic role for CMA in cells of the tumor micro-environment (Fig.2). For example, because p65, central component of nuclear factor-kappa B, is a CMA substrate, reduced CMA activity in epithelial cells augments NF-κB signaling, contributing to progression of epithelial-mesenchymal transition and tumorigenesis [43].
Immuno-oncogenic response and CMA.
Although still a poorly explored area, CMA facilitates immunogenic apoptosis - a type of cell death caused by some cytostatic agents - by mediating exposure of surface-exposed calreticulin [44]. Immunogenic apoptosis of cancer cells can induce an effective antitumor immune response through activation of dendritic cells, and consequent activation of specific T cell response [45].
Interestingly, there are already examples of cancer cells modulating CMA in host cells to actively suppress the anti-oncogenic response. Thus, glioblastoma cells induce cancer immunotolerance by actively deregulating CMA in pericytes -perivascular stromal cells that promote an immunological defense through secretion of inflammatory molecules [46]. In this case, tumor-induced aberrant upregulation of CMA in pericytes elicits an anti-inflammatory phenotype that prevents T cell activation for tumor clearance. Normalizing pericyte CMA could be an effective anti-glioblastoma intervention since experimental blockage of CMA in pericytes during glioblastoma-pericyte interaction was sufficient to promote tumor cell death [46].
The multiplicity of mechanisms behind the anti-oncogenic function of CMA is in line with the increasing number of cell-type specific CMA functions described, and it highlights that conditions when CMA declines (i.e. extreme dietary challenges, disease and in aging) has an associated increased risk of malignant transformation (Fig.2).
Changes and regulation of CMA in cancer cells
The first connection established between CMA and cancer was with its pro-survival and pro-tumorigenic activity in cancer cells [8]. Contrary to macroautophagy, that is upregulated or downregulated in cancer cells depending on the type and stage of cancer [5], CMA was found consistently upregulated in multiple cancer cell lines and primary tumors (including HCC, melanoma, lung, stomach, colon, uterus, ovary and breast cancers) [8]. Subsequent studies confirmed that, with still few exceptions (mostly acute myeloid leukemia (AML) [47]), constitutive upregulation of CMA seems a common feature of most cancer cells. Levels of LAMP2A, used as surrogate for CMA activity in human tumors, positively correlate with tumor size and recurrence in HCC patients [48] and have been proposed of prognostic value in gastric [49] and breast cancer patients [50]. Even in cancer cells with LAMP2A levels similar to their non-transformed counterpart cells, blockage of CMA pronouncedly reduces tumor cell viability [48], suggesting that basal CMA may be enough to support their pro-oncogenic activities, or that other CMA components may be responsible for its upregulation in these cells.
Studies in cultured fibroblasts support that CMA upregulation is cell-intrinsic and occurs shortly upon transformation [8] (Fig. 2). However, the mechanisms of CMA activation in cancer cells are, for the most part, unknown (Box 3). Intrinsic characteristics of the tumor microenvironment such as lack of nutrients, hypoxia and high ROS content are well-established CMA activators [23, 31, 51–53]. Some cancer cells upregulate CMA through the same mechanisms as non-transformed cells [54, 55], whereas other bypass standard CMA regulatory pathways [17], utilize new ones or change the function of known CMA regulators [56] (Box 3).
BOX 3. CMA-REGULATED PROTEOME IN CANCER CELLS
Cancer cells utilize selective degradation of proteins by CMA to remodel the proteome for their advantage through different mechanisms:
CMA degradation of undesired proteins in cancer cells can be attained by
Global CMA upregulation: to eliminate anti-proliferative proteins such as RND3 [49], tumor suppressors such as MST1 [73], PED [82] and mutant p53 [83], or pro-apoptotic proteins such as BBC3/PUMA [84].
CMA targeting through posttranslational modifications or cancer-associated mutations: Deacetylation of the tumor-suppressor MST1 in breast cancer cells triggers its degradation by CMA [73]. In the case of p53, whereas CMA promotes wild type p53 degradation by the proteasome in cancer cells [8], in specific cancer conditions (macroautophagy inhibition and imposed metabolic stress), p53 mutant variants undergo degradation in lysosomes by CMA [83].
Modulation of protein stability by CMA: CMA can regulate intracellular protein content by mechanisms other than their direct lysosomal degradation. Thus, in addition to promoting p53 proteasomal degradation [8], CMA can also modulate p53 expression in cancer cells [68]. CMA modulates proteasomal degradation of c-Myc to keep its levels low [42]. Whether CMA may contribute instead to stabilize c-Myc in cancer cells requires future investigation. These findings suggest that the proteome regulated by CMA expands probably beyond the already large group of proteins that contain the KFERQ motif. Interestingly, this indirect regulation applies even to proteins that contain the KFERQ motif such as c-Myc.
Selective preservation of desired proteins in cancer cells by preventing their degradation through CMA is also attained through
Global CMA downregulation: In those few cancer types where CMA activity is reduced compared with non-transformed cells, failure to degrade specific cytosolic proteins by CMA may contribute to cancer progression. Thus, for example, in pediatric acute myeloid leukemia, AF1Q that fuses to translocation patterns and contributes to leukemogenesis, has been validated as a bona fide CMA substrate that accumulates when CMA activity is inhibited [85].
Selective exclusion from CMA degradation: Hexokinase-II, whose activity is critical for cancer cell resistance to paclitaxel and previously validated as CMA substrate in non-transformed cells, becomes resistant to CMA degradation in breast cancer through its phosphorylation on Thr473 [62]. A similar change in CMA vulnerability has been described for 17beta-hydroxysteroid dehydrogenase type 4 (HSD17B4) that undergoes normally regulated degradation by CMA upon acetylation at lysine 669 (K669), but that when mutated in this lysine in breast cancer persists at high levels promoting cancer cell migration and invasion [66]
Microenvironment cells can also contribute to activation of CMA in tumor cells. In response to cytostatic agents, HCC-resident macrophages secrete interleukin-17 that increases levels of LAMP2A in tumor cells and likely their CMA activity thus conferring them resistance [57].
CMA functions in cancer cells
Although presently subject of intensive investigation, here we summarize examples of some of described pro-oncogenic functions of CMA in cancer cells (Fig. 2). A current limitation is that many studies lack functional CMA analysis and rely instead on levels of LAMP2A or other CMA components. However, it is encouraging that studies where CMA activity has been measured, seem to confirm the proposed pro-oncogenic activity of CMA in cancer cells.
Energy homeostasis:
Cancer cells depend often on anaerobic glycolysis for energy production even in the presence of oxygen [58] (known as the Warburg effect) [59]. Functional CMA is required to sustain the Warburg effect in lung cancer cells and in melanoma [8]. Contrary to the described increase in glycolytic enzymes in control cells upon CMA blockage [27], in lung cancer and melanoma cells, CMA inhibition stabilizes p53 which in turn reduces transcription of glycolytic enzymes [60]. Poor energetics upon CMA blockage are the main reason for reduced proliferation and cell death in these cells, since restoration of ATP levels is sufficient to reinitiate their proliferation [8].
Glycolytic enzymes are also regulated by CMA in other types of cancer but instead through their direct degradation by this pathway. CMA degradation of acetylated pyruvate kinase 2 leads to accumulation of glycolytic intermediates with proliferating signal properties [61]. Hexokinase-II, required for tumorigenesis, undergoes CMA degradation depending on glucose availability in non-AML cells [55]. Hexokinase-II depletion through CMA upregulation has been proposed as effective strategy to induce a metabolic crisis in these cells and their subsequent cell death. However, upregulation of CMA may not be enough to promote hexokinase II degradation since phosphorylation of this enzyme at Thr473, frequent in breast cancer, prevents its CMA degradation [62]. Inhibition of the kinase that phosphorylates Hexokinase-II shows potential in reducing cancer cells growth [62]. Studies in HCC cells and xenografts also support a role for CMA in energetic maintenance in these cells since blockage of CMA made them particularly sensitive to starvation [48].
Resistance to stress:
CMA upregulation in breast cancer cells contributes to their survival during oxidative stress since LAMP2A inhibition leads to their apoptosis [63]. CMA upregulation protects against ER stress in some types of lung cancers expressing misfolded N-CoR. This misfolded protein is required for activation of oncogenic survival pathways but its rapid removal by CMA is required to reduce the ER stress associated with its expression [47]. CMA may also serve as a cancer cell survival response to tumor-associated hypoxia. Hypoxia increases transcription of CMA genes in different types of cancer cells which in turn could contribute to regulate the adaptative response to hypoxia through CMA degradation of hypoxia-inducible factor-1 alpha (HIF-1α) [31, 64].
Proliferative potential, invasive and migratory properties:
Besides energetic regulation, CMA can sustain cancer cell proliferation through regulation of cell cycle. CMA degradation of the cell cycle-related protein RND3/RhoE and of HIF-1α (in this case induced by cyclin-dependent kinase 2) were shown necessary for rapid proliferation of gastric cancer cells [49] and cervical carcinoma and HCC [65], respectively.
CMA may contribute to tumor metastasis since blockage of CMA in lung cancer cells markedly decreases their metastatic potential by reducing migration and resistance to anoikis [8]. Similar positive correlation between CMA activity and metastasis was noticed in breast cancer [50]. The molecular mechanisms behind the CMA-dependence of metastasis remain unknown, but at least in cancer cells, CMA degradation of the multifunctional protein HSD17B4 have been proposed to modulate their invasive and migratory properties [66] and CMA downregulation of Atg5-dependent macroautophagy to promote metastasis [50].
Resistance to anti-oncogenic interventions:
CMA is a common protective mechanism against different anti-oncogenic interventions. CMA can selectively remove proteins damaged during the treatment such as oxidized proteins generated by pro-oxidant agents [63] or by photodynamic therapy [67]. In other instances, CMA confers resistance to the treatment through cell-type specific mechanisms. For example, CMA prevents apoptosis and contributes to resistance to oxaliplatin in HCC by degrading the apoptosis trigger cyclin D1 [57] or to irradiation by degrading HMGB1 [68]. Resistance to 5-fluorouracil in colorectal cancer has been linked to active degradation of the acetyltransferase p300/CBP by CMA [69]. Upregulation of LAMP2A by exosomes released by hepatitis B virus-associated cancer have been shown to reduce cell death induced by oxaliplatin, but direct CMA upregulation and the specific substrate behind apoptosis protection in this context has not yet been identified [70] (Fig. 3).
Figure 3. CMA targeting in cancer prevention and treatment.

Before malignant transformation maintaining fully functional CMA should help preserving its anti-oncogenic function. ??? denotes that experimental evidence is lacking to support value of targeting CMA in the pre-malignant lesion (before or at the moment that CMA gets upregulated), but systemic activation of CMA may help boosting the T cell mediated immune-oncogenic response. Once the tumor is formed, inhibition of CMA in tumor cells and tumor-associated macrophages has been shown to reduce tumor growth and to induced tumor death both alone or in combination with other anti-oncogenic interventions to prevent cancer cell resistance. Active upregulation of CMA has also shown effective in inducing cancer cell death in specific conditions combining inhibition of macroautophagy and metabolic stress. Boxes summarize the main cellular processes affected by the CMA-targeting intervention at each stage. (t): transcription; (d): degradation.
There is only one report showing that reduction of CMA, instead of upregulation, confers resistance to azacytidine treatment in the context of AML [71]. Interestingly, low levels of LAMP2A makes these cells more sensitive to lysosomal and autophagy inhibitors offering the potential of combinatorial therapies [71] (Fig. 3).
CMA-modulated proteome in cancer cells:
The dependence of many cancer cells on CMA suggests that selective degradation of a fraction of the proteome by CMA contributes to cancer properties. CMA changes in cancer cells are both quantitative and qualitative, as the sub-proteome degraded by CMA is different in untransformed and transformed cells (Box 3). Cancer cells preferentially target inhibitors of transformation or of tumor growth to CMA [49], but selectively shield proteins that facilitate growth and survival from CMA degradation [72]. Modifications in these proteins often determine their different fate in cancer and normal cells [73] (Box 3).
An elegant proteomic study of proteins degraded in a LAMP2A-dependent manner has identified the subset of proteins degraded by CMA in breast cancer cells upon blockage of macroautophagy and inducing metabolic stress [74]. Besides already known CMA substrates, this study unveiled a role for CMA in regulating protein translation through selective degradation of EIF4A1, EIF4H, and DDX3X [74]. Future studies in cancer cells with preserved macroautophagy should help in elucidating if regulation of translation is a constitutive function of CMA or only elicited upon the double insult.
Changes in the tumor microenvironment:
Recent studies highlight that CMA in cells within the tumor microenvironment contributes to tumorigenesis. Besides the above-described glioblastoma-dependent increase of CMA in pericytes to ablate their immune anti-oncogenic response [46], increased LAMP2A levels in tumor-associated macrophages have been shown to be required for breast cancer cell progression. In fact, levels of macrophage LAMP2A inversely correlate with cancer prognosis [75]. Induction of CMA in macrophages promotes their activation to aid in tumor growth [75]. The specific mechanisms whereby tumor cells modulate CMA in the surrounding cells are not known, but direct connections through microtube-like projections [46] and tumor-generated exosomes [70] are attractive possibilities.
CMA as a new target in cancer therapeutics
Despite numerous connections between CMA and cancer biology, different factors have limited the therapeutic translatability of these findings. An important impediment is the absence of selective chemical modulators of CMA, due in part to lack of exclusive and easily “druggable” CMA components. Most CMA effectors and regulators (i.e. chaperones, signaling elements) are involved in many other cellular processes. LAMP2A, the most unique CMA component, is a difficult target due to its high homology (almost 85% identity) with the other spliced variants of the LAMP2 gene involved in other cellular functions. A first-in-class type of selective CMA activators have been recently developed that target only the CMA-related transcriptional program under the retinoic acid receptor alpha signaling [76]. However, no selective inhibitors of CMA have been generated yet.
A limitation to therapeutically target macroautophagy in cancer, has been its opposite roles depending on the type of cancer and the context. Although this is less of a problem in CMA, due to its predominant pro-oncogenic function in most cancer cells, there are still two important challenges: 1) the dual role of CMA as anti-oncogenic mechanism in healthy cells but pro-tumorigenic in transformed cells and 2) the differences observed in CMA regulation in cancer cells when compared to non-transformed cells. The key events that mediate the switch from low to high CMA during transformation are unknown, making even more complicated the search for efficient CMA modulators in the context of cancer.
Preventive interventions:
As reflected in Fig 3, considering the physiological anti-oncogenic role of CMA, there is general agreement that preventive interventions should aim at restoring CMA activity to that in healthy conditions. For example, genetic prevention of the age-dependent decrease of CMA in mouse liver has proven effective in slowing-down hepatic proteotoxicity and accumulation of intracellular damage [22]. It is anticipated that the positive effect of this intervention on liver homeostasis will also prevent age-related oncogenic transformation in this organ and exert similar protection when applied systemically. However, because the possible tissue-dependence of the anti-oncogenic function of CMA has not been tested, future studies are needed to determine the value of systemic CMA restoration. In fact, studies with the novel selective CMA activators are underway, but in addition, the tight connection between CMA and nutrition, makes nutrient interventions an attractive alternative to preserve CMA activity and protect against malignant transformation in old organisms.
Therapeutic interventions:
Proof-of-concept for the therapeutic value of targeting CMA has been obtained for many different types of cancers through genetic modulation of LAMP2A. Knock-down of LAMP2A in multiple cancer cell lines have proven to reduce their proliferative capability, increase their susceptibility to stressors and attenuate their tumorigenic properties when used in xenografts [8, 49, 50, 63, 66–68, 70]. Direct injection of shRNA against LAMP2A in already formed xenograft tumors induced tumor regression and reduced metastasis [8]. The role of CMA in resistance to anti-oncogenic interventions, points towards a possible value of CMA blockage in combinatorial treatments (Fig. 3). Upregulation of CMA above its already high constitutive levels in cancer cells, has been shown effective to induce cancer cell death upon blockage of macroautophagy and induction of metabolic stress [55]. Under these conditions, depletion of key glycolytic enzymes by CMA results in metabolic crisis and subsequent cell death [55].
Despite this mounting genetic evidence in support of the value of modulating CMA with therapeutic purposes in cancer, the number of drugs targeting CMA is still extremely low and we still lack selective inhibitors of CMA. In fact, the only reports of chemical inhibition of CMA have used agents that inhibit the proteolytic activity of lysosomes and thus disrupt all forms of autophagy and not only CMA. On this respect, it is interesting that recent clinical anti-cancer interventions designed to interfere with macroautophagy utilize agents such as hydroxychloroquine, that inhibits intralysosomal proteolysis by raising the lysosomal pH. Although degradation of CMA substrates once internalized in lysosomes is not limiting for this process, a sustained increase in lysosomal pH contributes to destabilize the luminal form of hsc70 that is essential for substrate translocation, and consequently should result in inhibition of CMA. Future studies are needed to determine the contribution of CMA blockage to beneficial effects observed in trials using hydroxychloroquine (alone or in combination with other drugs).
Upregulation of CMA to induce metabolic crisis in macroautophagy-inhibited AML cells has been recently attempted with new small molecules; although their selectivity for CMA and ability to activate this pathway outside these specific conditions is still under investigation [55]. The recently developed selective chemical CMA activators could be of potential use in this context, but only if CMA in cancer cells is still under RARa inhibition (Fig. 3).
As the CMA-regulated subproteome in cancer cells becomes better characterized, an alternative approach could be interventions that specifically modulate targeting of the key CMA substrates responsible of its pro-oncogenic effects. A recent study in AML has attempted rerouting mutant p53-R248 to CMA by inhibition of hsp90 which promotes instead binding of the mutant protein to hsc70 upon conditions of metabolic stress [77]. However, experimental evidence of lysosomal degradation by CMA under these conditions is still missing. Inhibitors of hsp90 have been also used to promote degradation of the pro-tumorigenic proteins IGF-1R by CMA. [56]
Concluding Remarks and Future Perspectives
Although still a relatively young field, the interplay between CMA and carcinogenesis is currently subject of intensive investigation driven, for the most part, by the potential of targeting this autophagic pathway with therapeutic purposes in cancer.
All evidence gathered so far support that defense against oncogenesis is one of the physiological functions of CMA. Consequently, interventions to preserve or restore normal CMA activity could become effective in cancer prevention. In contrast, future studies in an even broader range of cancer types to the ones already studied are needed to consolidate the proposed pro-oncogenic effect in cancer cells and implement blockage or further abnormal upregulation of this pathway in cancer therapeutics. The fact that reduced CMA has already been identified in some cancer cells calls for some caution before generalizing that CMA upregulation is a universal feature of cancer, but fortunately does not diminish its possible therapeutic potential since the reduced levels of CMA in those cells seem to still make them more vulnerable to some cytostatic agents.
Future efforts, summarized in the Outstanding Questions box should be directed to understand the mechanisms that mediate the change in CMA from non-transformed to transformed cells as it can provide clues on specific players on cancer CMA that could be targeted without affecting the host CMA. In addition, before clinical implementation becomes possible, we need to develop effective methods to measure CMA in humans and increase the number of the still very limited selective modulators of CMA.
BOX 2. CMA REGULATION IN CANCER CELLS
Mechanisms by which cancer cells modulate CMA fall under the following categories:
Mechanisms shared with non-transformed cells:
During starvation, non-transformed cells upregulate CMA by reducing lysosomal turnover of LAMP2A to increase its lysosomal abundance [16]. Similarly, the higher content of LAMP2A in colorectal cancer with reduced sorting nexin 10 (SNX10) has been attributed to reduced LAMP2A degradation in absence of SNX10 [54].
Mechanisms that bypass pathways that regulate CMA in non-transformed cells:
The inhibitory effect of the TORC2/AKT1 signal pathway on CMA is no longer observed in a variety of cancer cells (i.e. HC-2, MCF12, HEK, N2a) that display elevated CMA activity independently of their differently mTORC2/AKT1 signaling activity [17]. A second CMA regulatory mechanism evaded by many cancer cells is the coordinated activity of this pathway with that of other autophagic processes. In un-transformed cells [21, 34] and rodent tissues, [27, 32] crosstalk between macroautophagy and CMA mediates activation of one of these pathways upon blockage of the other. Upregulation of macroautophagy in response to CMA blockage in liver maintains protein quality control, although the differences in timing of activation between both autophagic pathways makes it not possible to compensate for regulatory functions of CMA (i.e. on metabolic pathways or on DNA repair [24, 27]). In most cancer cells, CMA is constitutively activated independently of the status of macroautophagy [8].
There are however examples of cancer cells where autophagic crosstalk is preserved. For example, in non-acute myeloid leukemia cells blockage of macroautophagy upregulates CMA thus accelerating turnover of mutant p53. [55]. Reduced macroautophagy and higher levels of CMA markers have also been described in cirrhotic human livers with HCC, leading to propose that upregulated CMA may contribute to survival of cancer cells defective in macroautophagy; however additional direct experimental support for this attractive hypothesis and to identify if the cross-talk regulators are conserved are still missing [81].
CMA regulators with opposite effect in cancer and non-transformed cells:
Inhibition of hsp90, reduces CMA activity in normal cells as this chaperone stabilizes LAMP2A at the lysosomal membrane [12]. However, hsp90 inhibition in cancer cells promotes degradation of specific substrates through CMA such as oxidized proteins [53] or the insulin-like growth factor 1 in pancreatic cancer [56]. Downregulation of IGF-1R, should in turn further upregulate CMA by reducing levels of the CMA repressor p-AKT1 [17].
BOX: TRENDS HIGHLIGHTS
Chaperone-mediated autophagy (CMA) is a component of the proteostasis network for protein quality control, but it has additional regulatory roles by terminating the function of key cellular proteins through their timely and selective degradation.
Cells with reduced CMA have higher facility for malignant transformation in support of a physiological anti-oncogenic function of CMA.
Multiple types of cancer cells and tumors upregulate CMA, and blockage of CMA in those cells reduces their tumorigenic capabilities.
Cancer cells depend on CMA for a variety of pro-oncogenic functions such as sustained glycolytic activity, resistance to stressors and maintenance of high oncogene load.
The mechanisms behind the switch between the anti-oncogenic function of CMA in untransformed cells and its pro-oncogenic function in cancer cells remain poorly understood.
BOX: OUTSTANDING QUESTIONS
Is CMA upregulation a universal characteristic of most cancer cells? Despite CMA upregulation in majority of cancer cells analyzed so far, deficient CMA has been recently noted in some types of leukemia.
What triggers CMA upregulation upon malignant transformation?
Is CMA regulated through similar mechanisms in non-transformed and cancer cells? If cancer-specific regulators of CMA are identified they could become effective targets in cancer therapeutics.
How do tumor cells modulate CMA of cells in the tumor-microenvironment?
Is it possible to modulate the selective degradation of only some CMA substrates in cancer therapeutics? Preventing CMA degradation of anti-oncogenic proteins in cancer cells could be attained through disruption of their CMA targeting motif through posttranslational modifications or by blocking peptides.
Are levels of LAMP2A always a good surrogate marker for CMA activity? Other CMA components could become limiting (i.e. lysosomal hsc70) and dynamics rather than only levels of LAMP2A are important for CMA. Because the literature on CMA in cancer is still populated by a large number of studies based on 1) correlative data without directly measuring CMA activity and 2) the modulation of CMA with non-selective agents that affect primarily other cellular processes such as 6-aminonicotinamide, it is not clear how many of the earlier observations will be proven true once functional studies are performed in the same context.
Does therapeutic inhibition of lysosomal proteolysis through pH neutralization wok in part by inhibiting CMA? Dozen of ongoing clinical trials using hydroxychloroquine as one of the anti-oncogenic agents, offer a unique opportunity to gain a better understanding of the contribution of CMA upregulation to disease progression and the potential of targeting this autophagic pathway in cancer.
GLOSSARY
- Anoikis
form of programed cell death (apoptosis) that occurs in tumors cells when they detach from the surrounding extracellular matrix
- Eosinophilic foci
small to large protein aggregates sometimes considered putative preneoplastic lesions in the liver. Based upon their phenotypic appearance they are classified as basophilic, eosinophilic, clear cell, vacuolated, and mixed foci
- Hepatic steatosis
pathological accumulation of fat deposits (mostly triglycerides) within hepatocytes. Hepatic steatosis or fatty liver can originate from excessive alcohol consumption, but is also frequently associated with obesity, insulin resistance and dyslipidemia in non-alcoholic patients (termed non-alcoholic fatty liver disease (NAFLD))
- Immunogenic apoptosis
also called immunogenic cell death (ICD), is type of apoptosis induced by a specific set of chemotherapeutic drugs or ionizing irradiation and photodynamic therapy. Contrary to conventional autophagy that does not elicit an immune response, cells undergoing immunogenic apoptosis release pro-inflammatory molecules that promote their elimination by phagocytes
- Leukemogenesis
induction and development of any of the blood cancers commonly referred to as leukemia
- Lipidosis
accumulation of fats or lipids in any tissue. Hepatic lipidosis, probably the most common of this lipid storage disorders, results from massive fatty degeneration of the liver as result of accumulation of triglycerides
- Pentapeptide motif KFERQ
specific combination of amino acids in a protein sequence recognized by hsc70. Binding of the chaperone is based on the physical properties of the amino acids and requires always a Q flanking either side of the motif, a positively (K or R) and a negatively (E or D) charged amino acid, a hydrophobic residue (f, V, L or I) and an additional positive or hydrophobic amino acid
- Proteome
protein composition of a cell or tissue at a given time. It includes all proteins expressed within a cell (cellular proteome) or organism (complete proteome)
- Proteotoxicity
cellular consequence of the intracellular accumulation of proteins in an abnormal conformation, often as inclusions (aggregates) or oligomeric toxic species. Failure to timely eliminate these proteins often results in cell death and it is the basis of diseases known as proteinopathies or protein conformational disorders. Cells with low rates of division are usually more susceptible to proteotoxicity since they cannot dilute their protein content through division
- Translocation complex (for CMA)
a 700 kDa protein complex at the lysosomal membrane that mediates internalization of single proteins into the lumen. LAMP2A, main component of the complex, multimerizes in a step-wise manner that includes the formation of an intermediate LAMP2A homotrimer. Glial fibrillary acidic protein (GFAP) also forms part of this complex and contributes to its stabilization
- Warburg effect
common characteristic of the metabolism of cancer cells that refers to their ability to sustain anaerobic glycolysis even when exposed to aerobic conditions. The lower energetic efficiency of anaerobic glycolysis when compared to oxidative phosphorylation (preferred by most un-transformed cells) forces cancer cells to internalize large amount of glucose. This metabolic preference results in large production of lactate as well as additional metabolites that have been shown to have proliferation stimulating properties and contribute to tumor growth
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levine B and Klionsky DJ (2017) Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc Natl Acad Sci U S A 114 (2), 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N. et al. (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15 (3), 1101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaushik S and Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19, 365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stolz A. et al. (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16 (6), 495–501. [DOI] [PubMed] [Google Scholar]
- 5.White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12 (6), 401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang X. et al. (2015) Autophagy in cellular metabolism and cancer. J Clin Invest 125 (1), 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzwalter BE. et al. (2018) Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev Cell 44 (5), 555–565 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kon M. et al. (2011) Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med 3 (109), 109ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dice JF (1990) Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 15, 305–309. [DOI] [PubMed] [Google Scholar]
- 10.Kirchner P. et al. (2019) Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLOS Biology 17 (5), e3000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuervo AM and Dice JF (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503. [DOI] [PubMed] [Google Scholar]
- 12.Bandyopadhyay U. et al. (2008) The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28 (18), 5747–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bandyopadhyay U. et al. (2010) Identification of regulators of chaperone-mediated autophagy. Mol Cell 39 (4), 535–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuervo AM. et al. (1997) A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem 272 (9), 5606–15. [DOI] [PubMed] [Google Scholar]
- 15.Gong Z. et al. (2018) Humanin is an endogenous activator of chaperone-mediated autophagy. J Cell Biol 217 (2), 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuervo AM and Dice JF (2000) Regulation of lamp2a levels in the lysosomal membrane. Traffic 1, 570–83. [DOI] [PubMed] [Google Scholar]
- 17.Arias E. et al. (2015) Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone- Mediated Autophagy. Mol Cell 59 (2), 270–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arias E (2017) Methods to Study Chaperone-Mediated Autophagy. Methods Enzymol 588, 283–305. [DOI] [PubMed] [Google Scholar]
- 19.Koga H. et al. (2011) A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun 2, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong S. et al. (2019) A novel mouse model for spatio-temporal analysis of chaperone-mediated autophagy in vivo. submitted. [Google Scholar]
- 21.Massey AC. et al. (2006) Consequences of the selective blockage of chaperone-mediated autophagy. Proc Nat Acad Sci USA 103 (15), 5805–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C and Cuervo AM (2008) Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 14 (9), 959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiffin R. et al. (2004) Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 15 (11), 4829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider JL. et al. (2015) Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 14 (2), 249–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cuervo AM. et al. (1999) Direct lysosomal uptake of alpha 2-microglobulin contributes to chemically induced nephropathy. Kidney international 55 (2), 529–45. [DOI] [PubMed] [Google Scholar]
- 26.Backer J and Dice J (1986) Covalent linkage of ribonuclease S-peptide to microinjected proteins causes their intracellular degradation to be enhanced by serum withdrawal. Proc Nat Acad Sci USA 83, 5830–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider JL. et al. (2014) Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 20 (3), 417–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaushik S and Cuervo AM (2015) Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 17 (6), 759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park C. et al. (2015) Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun 6, 6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira JV. et al. (2013) STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 9 (9). [DOI] [PubMed] [Google Scholar]
- 31.Hubbi ME. et al. (2013) Chaperone-mediated Autophagy Targets Hypoxia-inducible Factor-1alpha (HIF-1alpha) for Lysosomal Degradation. J Biol Chem 288 (15), 10703–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valdor R. et al. (2014) Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol 15 (11), 1046–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuervo AM and Dice JF (2000) Age-related decline in chaperone-mediated autophagy. J Biol Chem 275 (40), 31505–13. [DOI] [PubMed] [Google Scholar]
- 34.Kiffin R. et al. (2007) Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci 120 (Pt 5), 782–91. [DOI] [PubMed] [Google Scholar]
- 35.Das S. et al. (2013) Purinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 305 (12), G950–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Navarro JA. et al. (2012) Inhibitory effect of dietary lipids on chaperone- mediated autophagy. Proc Natl Acad Sci U S A 109 (12), E705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai Y. et al. (2016) The Detrimental Role Played by Lipocalin-2 in Alcoholic Fatty Liver in Mice. Am J Pathol 186 (9), 2417–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma S. et al. (2011) GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS One 6 (9), e25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hernandez-Gea V. et al. (2012) Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142 (4), 938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu TL. et al. (2010) Hispolon promotes MDM2 downregulation through chaperone-mediated autophagy. Biochem Biophys Res Commun 398 (1), 26–31. [DOI] [PubMed] [Google Scholar]
- 41.Bonhoure A. et al. (2017) Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur J Cell Biol 96 (2), 83–98. [DOI] [PubMed] [Google Scholar]
- 42.Gomes LR. et al. (2017) Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 13 (5), 928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang J. et al. (2017) Impaired p65 degradation by decreased chaperone-mediated autophagy activity facilitates epithelial-to-mesenchymal transition. Oncogenesis 6 (10), e387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garg AD. et al. (2013) Calreticulin surface exposure is abrogated in cells lacking, chaperone-mediated autophagy-essential gene, LAMP2A. Cell Death Dis 4, e826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spisek R and Dhodapkar MV (2007) Towards a better way to die with chemotherapy: role of heat shock protein exposure on dying tumor cells. Cell Cycle 6 (16), 1962–5. [DOI] [PubMed] [Google Scholar]
- 46.Valdor R. et al. (2019) Glioblastoma ablates pericytes anti-tumor immune function through aberrant upregulation of chaperone-mediated autophagy. . Proc Nat Acad Sci USA E-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ali AB. et al. (2011) Role of chaperone mediated autophagy (CMA) in the degradation of misfolded N-CoR protein in non-small cell lung cancer (NSCLC) cells. PLoS One 6 (9), e25268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding ZB. et al. (2016) Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int J Oncol 49 (6), 2367–2376. [DOI] [PubMed] [Google Scholar]
- 49.Zhou J. et al. (2016) Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 12 (3), 515–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han Q. et al. (2017) Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci Rep 7 (1), 4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dice JF (1982) Altered degradation of proteins microinjected into senescent human fibroblasts. J Biol Chem 257 (24), 14624–7. [PubMed] [Google Scholar]
- 52.Cuervo AM. et al. (1995) Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Pathol 269 (5 Pt 1), C1200–8. [DOI] [PubMed] [Google Scholar]
- 53.Finn PF and Dice JF (2005) Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem 280 (27), 25864–70. [DOI] [PubMed] [Google Scholar]
- 54.Zhang S. et al. (2018) Sorting nexin 10 acts as a tumor suppressor in tumorigenesis and progression of colorectal cancer through regulating chaperone mediated autophagy degradation of p21(Cip1/WAF1). Cancer Lett 419, 116–127. [DOI] [PubMed] [Google Scholar]
- 55.Xia HG. et al. (2015) Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol 210 (5), 705–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xue N. et al. (2019) Chaperone-mediated autophagy degradation of IGF-1Rbeta induced by NVP-AUY922 in pancreatic cancer. Cell Mol Life Sci E-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo B. et al. (2017) M2 tumor-associated macrophages produce interleukin-17 to suppress oxaliplatin-induced apoptosis in hepatocellular carcinoma. Oncotarget 8 (27), 44465–44476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bartrons R and Caro J (2007) Hypoxia, glucose metabolism and the Warburg’s effect. J Bioenerg Biomembr 39 (3), 223–9. [DOI] [PubMed] [Google Scholar]
- 59.Warburg O (1956) On the origin of cancer cells. Science 123 (3191), 309–14. [DOI] [PubMed] [Google Scholar]
- 60.Vousden KH and Ryan KM (2009) p53 and metabolism. Nat Rev Cancer 9 (10), 691–700. [DOI] [PubMed] [Google Scholar]
- 61.Lv L. et al. (2011) Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 42 (6), 719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang T. et al. (2018) PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene 37 (45), 5997–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saha T (2012) LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 8 (11), 1643–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferreira JV. et al. (2015) K63 linked ubiquitin chain formation is a signal for HIF1A degradation by Chaperone-Mediated Autophagy. Sci Rep 5, 10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hubbi ME. et al. (2014) Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1alpha to promote cell-cycle progression. Proc Natl Acad Sci U S A 111 (32), E3325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y. et al. (2017) Acetylation targets HSD17B4 for degradation via the CMA pathway in response to estrone. Autophagy 13 (3), 538–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dewaele M. et al. (2011) Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J Cell Mol Med 15 (6), 1402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu JH. et al. (2017) CMA down-regulates p53 expression through degradation of HMGB1 protein to inhibit irradiation-triggered apoptosis in hepatocellular carcinoma. World J Gastroenterol 23 (13), 2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Du C. et al. (2017) 5-Fluorouracil targets histone acetyltransferases p300/CBP in the treatment of colorectal cancer. Cancer Lett 400, 183–193. [DOI] [PubMed] [Google Scholar]
- 70.Liu DX. et al. (2019) Exosomes derived from HBV-associated liver cancer promote chemoresistance by upregulating chaperone-mediated autophagy. Oncol Lett 17 (1), 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dubois A. et al. (2019) LAMP2 expression dictates azacytidine response and prognosis in MDS/AML. Leukemia 33 (6), 1501–1513. [DOI] [PubMed] [Google Scholar]
- 72.Suzuki J. et al. (2017) Chaperone-mediated autophagy promotes lung cancer cell survival through selective stabilization of the pro-survival protein, MCL1. Biochem Biophys Res Commun 482 (4), 1334–1340. [DOI] [PubMed] [Google Scholar]
- 73.Li L. et al. (2016) Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene 35 (31), 4048–57. [DOI] [PubMed] [Google Scholar]
- 74.Hao Y. et al. (2019) Targetome analysis of chaperone-mediated autophagy in cancer cells. Autophagy 15 (9), 1558–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang R. et al. (2019) Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. EBioMedicine 40, 118–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anguiano J. et al. (2013) Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol 9 (6), 374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Allende-Vega N and Villalba M (2019) Metabolic stress controls mutant p53 R248Q stability in acute myeloid leukemia cells. Sci Rep 9 (1), 5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sahu R. et al. (2011) Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20 (1), 131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
References unique to Boxes
- 79.Zhang J. et al. (2017) Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J Biol Chem 291 (25), 10328–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pajares M. et al. (2018) Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 14 (8), 1310–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chava S. et al. (2017) Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 8 (25), 40019–40036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quintavalle C. et al. (2014) Phosphorylation-regulated degradation of the tumorsuppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J Cell Physiol 229 (10), 1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vakifahmetoglu-Norberg H. et al. (2013) Chaperone-mediated autophagy degrades mutant p53. Genes Dev 27 (15), 1718–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xie W. et al. (2015) Chaperone-mediated autophagy prevents apoptosis by degrading BBC3/PUMA. Autophagy 11 (9), 1623–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li P. et al. (2014) Degradation of AF1Q by chaperone-mediated autophagy. Exp Cell Res 327 (1), 48–56. [DOI] [PubMed] [Google Scholar]