

SUMMARY
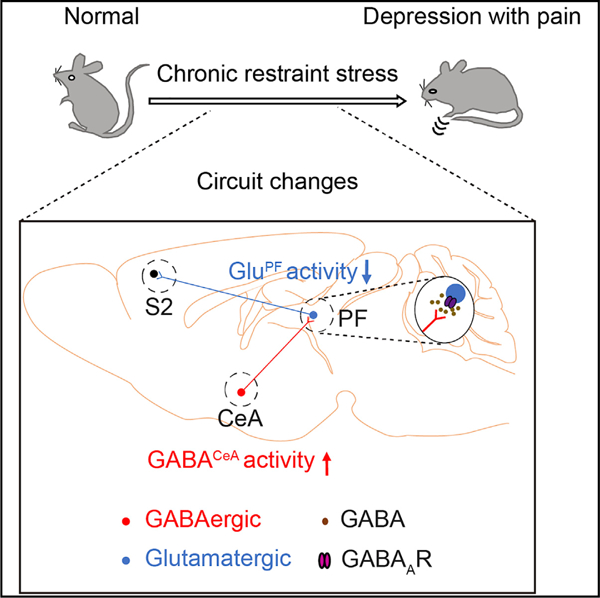
While comorbid pain in depression (CP) occurs at a high rate worldwide, the neural connections underlying the core symptoms of CP have yet to be elucidated. Here, we define a pathway whereby GABAergic neurons from the central nucleus of the amygdala (GABACeA) project to glutamatergic neurons in the parafascicular nucleus (GluPF). These GluPF neurons relay directly to neurons in the second somatosensory cortex (S2), a well-known area involved in pain signal processing. Enhanced inhibition of the GABACeA→GluPF→S2 pathway is found in mice exhibiting CP symptoms. Reversing this pathway using chemogenetic or optogenetic approaches alleviates CP symptoms. Together, the current study demonstrates the putative importance of the GABACeA→GluPF→S2 pathway in controlling at least some aspects of CP.
Graphical Abstract
In Brief
Zhu et al. identify that GABAergic neurons from the central nucleus of the amygdala (GABACeA) project to glutamate neurons in the parafascicular nucleus (GluPF) and uncover the role of this pathway in regulation of pain symptoms in depression via connecting with the second somatosensory cortex.
INTRODUCTION
Recently, chronic pain has emerged as the leading cause of years lived with disability, while major depressive disorder is the fifth (GBD 2016 Disease and Injury Incidence and Prevalence Collaborators, 2017). Pain symptoms occur with an estimated prevalence of up to 65% in patients with major depression, which is much higher than that observed in non-depressed cohorts (Bair et al., 2003; Robinson et al., 2009; Rudy et al., 1988). Furthermore, comorbid pain in depression (CP) symptoms can worsen depressed mood and are associated with poor responses to both depression and pain treatment (Bushnell et al., 2013; Tracey and Mantyh, 2007), creating a cycle of depression and pain that is often difficult to break (Chapman and Gavrin, 1999). The proper treatment for CP represents a major challenge in the field, as the underlying mechanisms of CP are poorly understood.
Chronic pain varies in etiology, with different conditions exhibiting distinct profiles of pain symptomatology (Basbaum et al., 2009; Todd, 2010; Woolf and Ma, 2007). Alterations in serotonergic (Morita et al., 2015) and noradrenergic (Hermans et al., 2011) systems are considered the common pathological roots of depression and chronic pain, and they share a clinical pattern of persistence beyond the precipitant. However, approximately one-third of depression patients do not respond well to treatment by targeting these systems (Hieronymus et al., 2016). Notably, the acute use of serotonin-selective serotonin reuptake inhibitors (SSRIs) produces early adverse events in some patients, which may exacerbate pain symptoms (Hieronymus et al., 2016). Together, the current evidence indicates that the neuroanatomical and molecular substrates that underlie CP may be different from those that underlie other various somatic pain syndromes (Bair et al., 2003), which could be beyond the serotonergic and noradrenergic systems.
Many brain regions, such as the amygdala (Zhou et al., 2019), anterior cingulate cortex (ACC) (Barthas et al., 2015), prefrontal cortex (PFC) (Moda-Sava et al., 2019), insular cortex (IC) (Gehrlach et al., 2019), and the core of the nucleus accumbens (NAc) (Descalzi et al., 2017), are involved in the central mechanisms of depression. However, little is known about whether, or how, depression may influence the brain neural system that would modify the central mechanisms of chronic pain. In addition, the brain regions involved in chronic pain closely mirror those involved in depression regulation, such as the amygdala, ACC (Bliss et al., 2016), IC (Zhuo, 2008), and PFC (Wang et al., 2015). Therefore, chronic pain and depressive symptoms appear to be associated. However, the mechanism, especially with regard to how the two are linked on the level of neuroanatomical and molecular substrates, is unknown. For example, it is unclear whether CP results from maladaptive changes in depression-related brain circuits.
The amygdaloid complex is well known to be relevant to fear learning, anxiety, reward, and pain (Duvarci and Pare, 2014; Li et al., 2013; Tovote et al., 2015; Tye et al., 2011; Zhuo, 2008). Even the PFC (Huang et al., 2019) and IC (Berret et al., 2019) have been reported to interact with the amygdala in processing cognition, emotion, and pain. This raises the possibility that the amygdala could be an important site for processing the depressive symptoms associated with pain (Senn et al., 2014). The central nucleus of the amygdala (CeA), referred to as the ‘‘nociceptive amygdala,’’ serves as the main output nucleus for amygdala functions (Janak and Tye, 2015). Highly processed, polymodal information reaches the CeA indirectly from the thalamus and cortical areas or direct nociceptive inputs from the spinal cord and brainstem. Imaging studies have shown significant alterations in the activity of the CeA in chronic pain. Furthermore, the CeA forms widespread connections with forebrain areas (Penzo et al., 2015) and the brainstem (Tovote et al., 2016), which has been implicated in mediating fear and mood disorders. These studies suggest that the CeA is a probable convergent point of chronic pain and depression. However, the cause-and-effect relationship between the adaptation of CeA circuits and the pathology of CP is unknown.
The thalamus is a key hub that regulates pain signal transmission (Vartiainen et al., 2016) and processing of depressive emotions (Greicius et al., 2007). It shares anatomical connections with other brain areas, such as the medial PFC (Ährlund-Richter et al., 2019), superior colliculus (SC), brainstem, basal ganglia (Assous et al., 2017; Bieler et al., 2018), and amygdala (LeDoux et al., 1990). The precise organization and the function of the CeA-PF circuit remains unknown. Based on evidence linking the amygdala with both pain and depression, the current study sought to examine the pathological causes of the CP by defining the neurocircuitry, architectural rules, and plasticity of the CeA.
Combining viral tracing, optogenetic and electrophysiological methods, we dissected the functional organization of the CeA→thalamus pathway and explored this adaptation of this pathway in CP mice. Collectively, we describe an inhibition GABACeA→GluPF→S2 pathway, the alteration of which is both necessary and sufficient for manifesting CP.
RESULTS
An Inhibitory GABACeA→GluPF Pathway
First, the H129-G4 virus, used as an anterograde polysynaptic tracer (Zeng et al., 2017), was stereotaxically delivered into the CeA of a wild-type mouse (Figures 1A and 1B, left). Thirty-six hours later, the GFP+ signal was observed in many regions, such as S2 (Bushnell et al., 2013; Navratilova and Porreca, 2014), zona incerta (ZI) (Masri et al., 2009), and ventrolateral periaqueductal gray (vlPAG) (Tovote et al., 2016), which are involved in the pathophysiology of pain (Figure S1). Interestingly, GFP-labeled neurons were identified only in the PF of the thalamus (Figure 1B, right), which were predominantly co-localized with the glutamate antibody (Figure 1C). These results suggest that GluPF neurons may receive CeA projections.
Figure 1. Dissection of the GABACeA→GluPF Pathway.
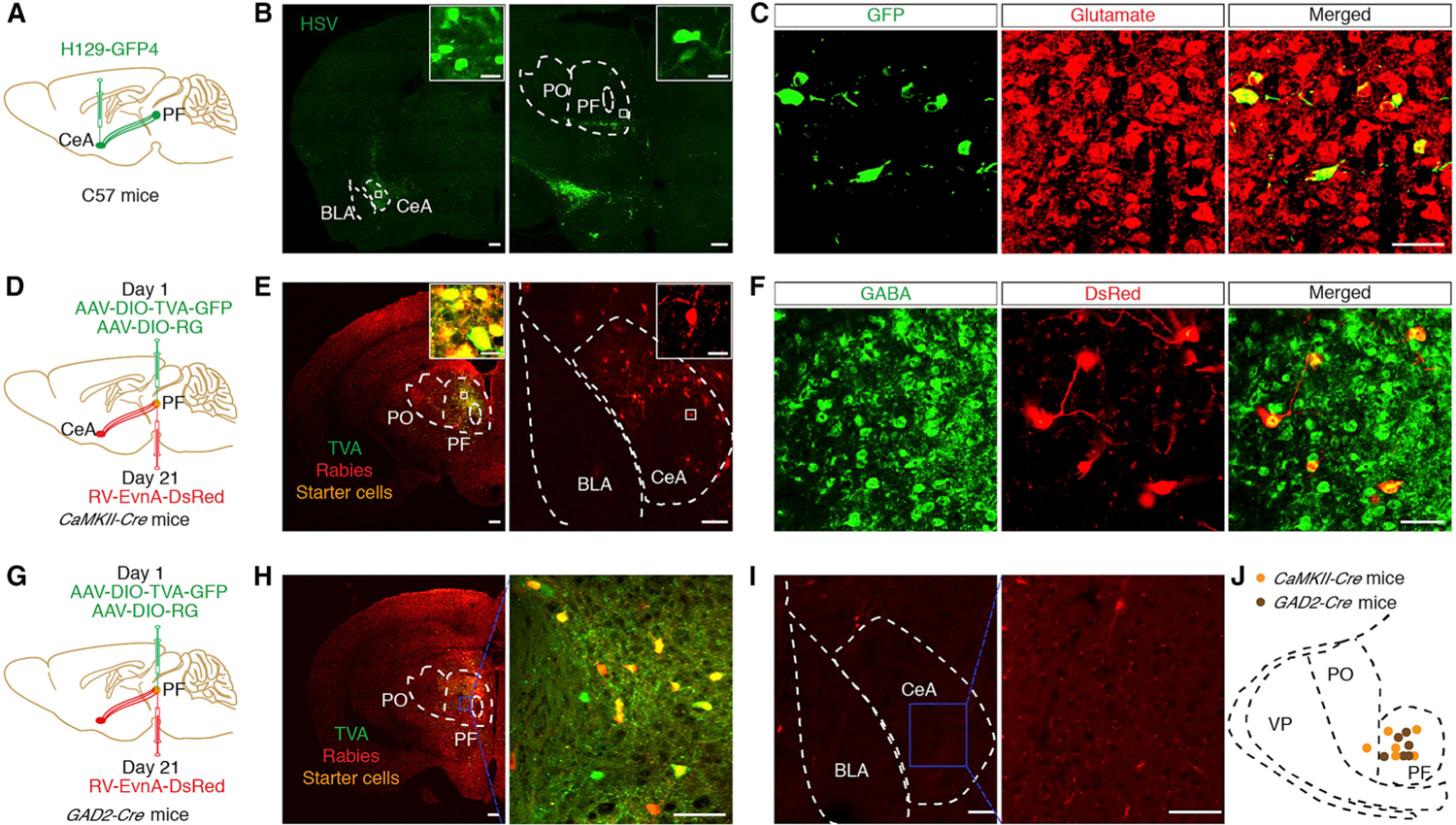
(A) Schematic of trans-polysynaptic H129-G4 virus tracing strategy.
(B) Typical images of H129-G4 injection sites within the CeA (left) and viral expression in the PF (right), which were spliced with a confocal microscope. Scale bar, 200 μm. The white boxes depicting the area shown in the boxes of the CeA or PF. Scale bars, 20 μm.
(C) Herpes simplex virus (HSV)-labeled neurons within the PF traced from the CeA (seen in B, right) were co-localized with glutamate immunofluorescence. Scale bar, 50 μm.
(D) Schematic of the Cre-dependent retrograde trans-monosynaptic rabies virus tracing strategy.
(E) Left: typical images of injection sites and viral expression within the PF of CaMKII-Cre mice. Starter cells (yellow) co-expressing AAV-DIO-TVA-GFP, AAV-DIO-RVG (green), and rabies RV-EnvA-∆G-DsRed (red). Scale bars, 200 μm. Right: DsRed-labeled neurons within the CeA. Scale bars, 100 μm. The white boxes depict the area shown in the boxes of the PF or CeA. Scale bars, 20 μm.
(F) DsRed-labeled neurons within the CeA traced from the PF in (E, right) were co-localized with the GABA immunofluorescence. Scale bar, 100 μm.
(G) Schematic of the cell-type-specific retrograde trans-monosynaptic rabies virus tracing strategy.
(H) Typical images of injection sites and viral expression within the PF of GAD2-Cre mice (left, scale bar, 200 μm). The blue box (right, Sclar bar, 100 μm) depicting the area shown in the PF (left). Starter cells (yellow) coexpressing AAV-DIO-TVA-GFP, AAV-DIO-RVG (green), and rabies RV-EnvA-∆G-DsRed (red).
(I) DsRed-labeled neurons within the amygdala traced from the PF (left, scale bar, 100 μm). The blue box (right, scale bar, 50 μm) depicts the area shown in the CeA (left).
(J) Infusion sites with AAV-helper and RV virus in CaMKII-Cre mice and GAD2-Cre mice.
See also Figures S1 and S2.
This finding is interesting because the thalamus is a well-known ‘‘pain nucleus’’ in mammals (Tracey and Mantyh, 2007). To characterize the CeA-GluPF organization, a cell-type-specific retrograde trans-monosynaptic tracing system was employed. Cre-dependent helper viruses (AAV-EF1α-DIO-TVA-GFP and AAV-EF1α-DIO-RVG) were injected into the PF of Ca2+/calmodulin-dependent protein kinase II (CaMKII, an enzyme in glutamatergic neurons)-Cre mice (Figure 1D). After 3 weeks, the rabies virus (RV) (EnvA-pseudotyped RV-∆G-DsRed) was injected into the same site (Figure 1E, left). The presence of these helper viruses facilitated monosynaptic retrograde RV spread (Wickersham et al., 2007). In addition to multiple inputs to the PF, such as the PFC, bed nucleus of the stria terminalis (BNST), and lateral hypothalamus (LH; Figure S2), we also identified intensely DsRed-labeled neurons in the CeA (Figure 1E, right). The DsRed signal was co-localized with the GABA antibody (Figure 1F), which is consistent with previous research suggesting that the CeA consists of 95% GABA neurons (Janak and Tye, 2015; Martin-Fernandez et al., 2017). By contrast, the same tracing strategy conducted in glutamic acid decarboxylase 2 (GAD2, a GABA synthetic enzyme)-Cre mice resulted in no DsRed+ neurons detected in the CeA (Figures 1G–1J). These findings suggest a GABACeA→GluPF pathway.
To examine the functional connections of the GABACeA→GluPF pathway, an adeno-associated virus expressing Cre-dependent channelrhodopsin-2 (AAV-DIO-ChR2-mCherry) was infused into the CeA of GAD2-Cre mice (Figure 2A). We observed mCherry+ (GABA) cell bodies in the CeA (Figures 2B and 2C) and numerous mCherry+ fibers in the PF (Figure 2D) and other regions, such as ZI and vlPAG but not S2 (Figure S3). By whole-cell recording in brain slices, brief light stimulation of ChR2-containing GABACeA terminals in the PF reliably elicited inhibitory postsynaptic currents (IPSCs, 121.75 ± 13.26 pA) in GluPF neurons, which were eliminated by the GABA receptor antagonist bicuculline (12.98 ± 10.05 pA; Figure 2E). Post hoc immunostaining showed that the recorded neurons labeled with neuronbiotin-488 in pipette solution were glutamatergic (Figure 2F). In addition, in vivo electrophysiology recordings were performed in the PF of freely moving mice. AAV-DIO-ChR2-mCherry viruses were injected into the CeA in GAD2-Cre mice. After 3 weeks, multi-tetrode recording and optic fibers were implanted into the PF (Figure 2G). Multiple channel recordings of the firing rate of the PF before, during, and after light photostimulation (Figures 2H and 2I) were conducted. We found that blue light decreased the firing rate in PF neurons. To confirm the CeA-PFGlu projection, we injected anterograde monosynaptic AAV2/1-Cre virus into the CeA of Ai14 (Cre-dependent tdTomato reporter) mice (Zingg et al., 2017) (Figures 2J and 2K). After 3 weeks, we observed tdTomato-expressing neurons in the PF (Figure 2L), which were co-localized with the glutamate antibody (Figure 2M). These data confirm that GABACeA neurons send afferents to the PF that synapse on GluPF neurons exerting inhibition.
Figure 2. Identification of the Functional Connection of the GABACeA→GluPF Pathway.
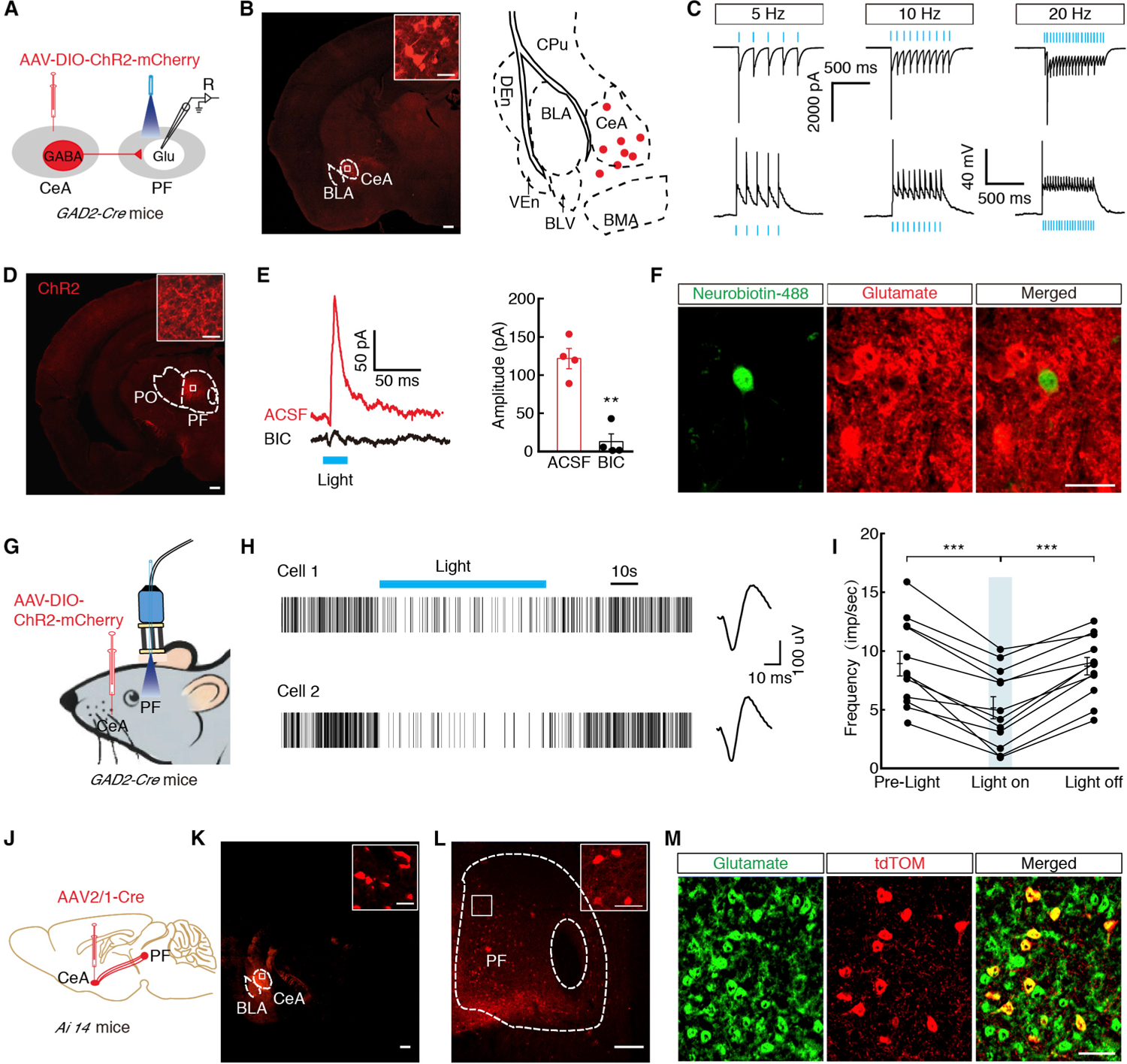
(A) Schematic of CeA injection of AAV-DIO-ChR2-mCherry in GAD2-Cre mice and recording configuration in acute slices.
(B) Typical images (left) and infusion sites (right) of an AAV-DIO-ChR2-mCherry viral expression in the CeA of GAD2-Cre mice (scale bar, 200 μm). The white boxes depicting the area shown in the box of the CeA (scale bar, 20 μm).
(C) Sample traces of action potentials evoked by 473 nm light recorded from CeA mCherry+ neurons in acute brain slices.
(D) The images showed ChR2-containning GABACeA terminals in the PF, which were spliced with a confocal microscope. Scale bar, 200 μm. The white boxes depicting the area shown in the box of the PF. Scale bar, 20 μm.
(E) Representative traces (left) and summarized data (right) of light-evoked currents (473 nm, 10 Hz, 20 ms, blue bar) before and after bicuculline (BIC, 10 μM, n = 4 neurons).
(F) Representative images of neurobiotin-488 (green), anti-glutamate immunostaining (red), merged (yellow) showing glutamate neurons by post hoc staining after patch in (E). Scale bar, 20 μm.
(G) Schematic of in vivo recording of freely moving GAD2-Cre mice.
(H and I) Representative traces (H) and summarized data (I) showed the firing rate of the PF neurons in the GAD2-Cre mice before, during, and after light photostimulation with multiple channel recordings (n = 14 neurons).
(J) Schematic of trans-monosynaptic AAV2/1-Cre virus tracing strategy.
(K) Typical images of an AAV2/1-Cre injection site and viral expression in the CeA of Ai14 mice (scale bar, 200 μm). The white boxes depicting the area shown in the box of the CeA (scale bar, 20 μm).
(L) Typical images of viral expression in the PF. Scale bar, 200 μm. The white boxes depicting the area shown in the boxes of the PF. Scale bar, 20 μm.
(M) tdTomato-labeled neurons within the PF traced from the CeA (seen in L) were co-localized with glutamate immunofluorescence. Scale bar, 50 μm.
All of the data are presented as mean ± SEM. **p < 0.01, ***p < 0.001. Paired t test for (E). One-way repeated-measures ANOVA with Bonferroni post hoc analysis is for (I). See also Figure S3.
An Enhanced Inhibition of the GABACeA→GluPF Pathway in CP
To determine the role of the GABACeA→GluPF pathway in CP, we used the chronic restraint stress (CRS)-induced depression model in mice (Nestler and Hyman, 2010) (Figure 3A). After 3 weeks of CRS (CRS 3W), the mice displayed multiple depressive-like behaviors in routine assays: forced-swim test (i.e., increased immobility), tail suspension test (i.e., increased immobility), open-field test (i.e., reduced time and entries into the center zone), and elevated plus maze test (i.e., reduced open arms time and entries; Figure 3B; Figures S4A and S4B). The total travel distance was unchanged, suggesting that locomotor ability was not affected by CRS 3W (Figure S4C). Of note, after 1 and 2 weeks of CRS (CRS 1W and CRS 2W, respectively) mice did not display pain sensitization or depressive-like behaviors (Figures S4D–S4H). However, at 3 weeks, CRS mice displayed significant pain sensitization (Figure 3C). Furthermore, traditional analgesic agents, which were effective in Complete Freund’s adjuvant (CFA)-induced inflammatory (Figures 3D, 3E, and 3H) and spared nerve injury (SNI)-induced neuropathic pain (Figures 3F, 3G, and 3I), had no effect on CRS-induced pain sensitization (Figure 3J).
Figure 3. Pharmacological Effects on CRS-, Inflammatory-, and Neural Injury-Induced Pain.
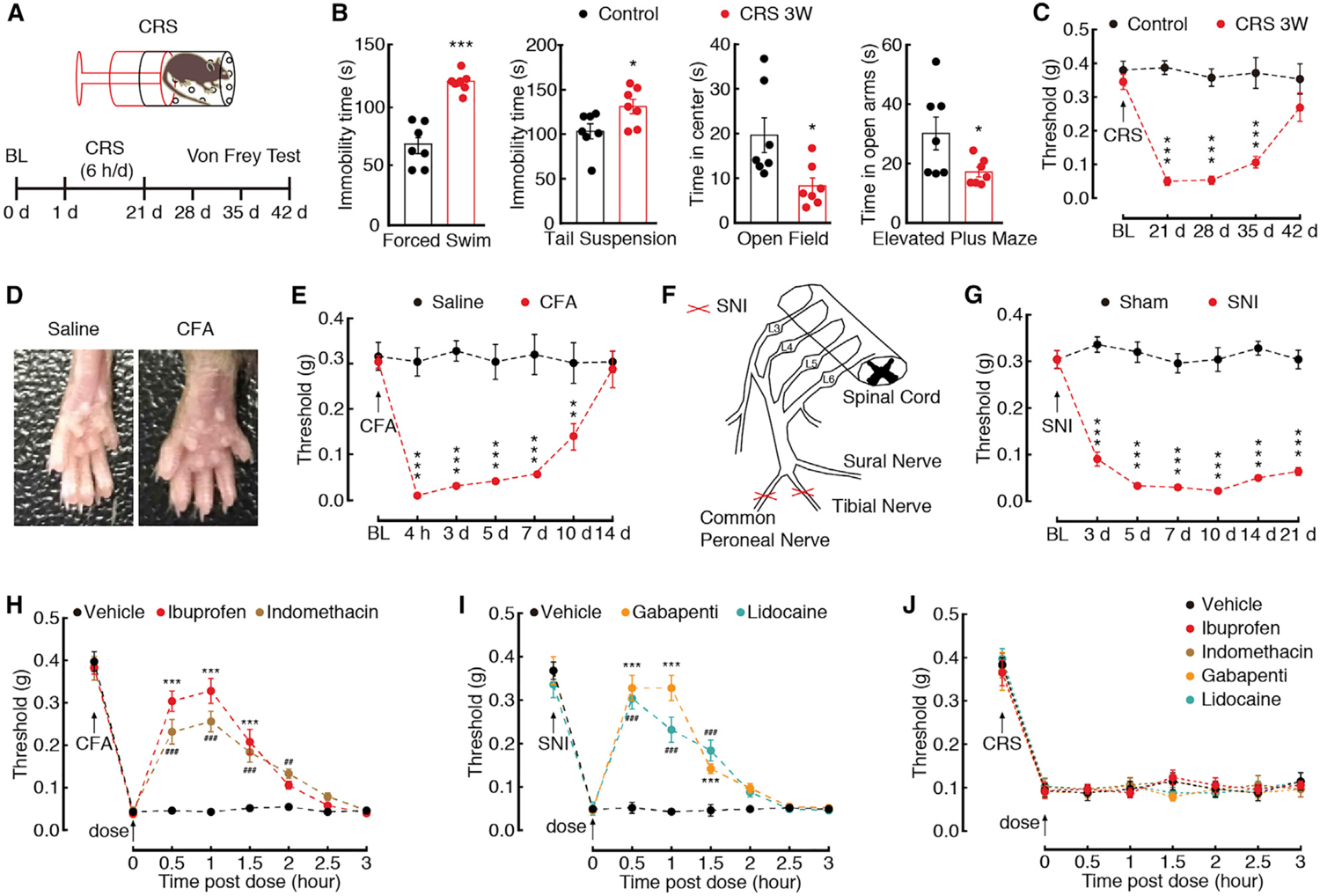
(A) An outline of the experimental procedure for mice with CRS treatment and behavioral tests.
(B) Behavioral tests in forced swim, tail suspension, open field, and elevated plus maze (n = 7 mice per group).
(C) Time course of CRS-induced changes in pain threshold by Von Frey tests (n = 7 mice per group).
(D) The mouse left hind paw at day 3 after saline or CFA injection.
(E) CFA-induced changes in pain threshold (n = 5 mice per group).
(F) Schematic of the animal model of SNI.
(G) SNI-induced changes in pain threshold (n = 5 mice per group).
(H) Effects of ibuprofen (10 mg/kg, i.p.) or indomethacin (10 mg/kg, i.p.) on pain thresholds at day 3 after CFA treatment (n = 5 mice per group). *Preference comparison within the ibuprofen group; #preference comparison within the indomethacin group.
(I) Effects of gabapentin (10 mg/kg, i.p.) or lidocaine (10 mg/kg, i.p.) on pain thresholds at day 7 after SNI (n = 5 mice per group). *Preference comparison within the gabapentin group;, #preference comparison within the lidocaine group.
(J) Effects of ibuprofen, indomethacin, gabapentin, or lidocaine on pain thresholds of CRS 3W mice (n = 5 mice per group).
All data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. ## p < 0.01, ### p < 0.001. Unpaired t test for (B); two-way repeated-measures ANOVA with Bonferroni post hoc analysis for (C), (E), and (G)–(J). See also Figure S4.
Next, we investigated the activity of PF-projecting GABACeA neurons in CRS 3W mice. To visualize GABA neurons, GAD2-Cre mice were used to cross with Ai14 mice to produce transgenic mice with red tdTomato-expressing GABA neurons (GAD2-tdT; Figure S5A). Whole-cell recordings were performed in acute brain slices on PF-projecting GABACeA neurons by PF infusion of the retrograde tracer CTB-488 (Figures 4A and 4B). We found an increase in the spike number (Figure 4C) and a decrease in rheobase (Figure 4D) in CeA CTB-488+ GAD2-tdT neurons but not in CTB-488− GAD2-tdT neurons (Figures S5B–S5D), from CRS 3W mice. However, these phenotypes were not observed in CRS 2W (Figures S5E–S5G), CFA 3D (Figures S5H–S5J), or SNI 7D mice (Figures S5K–S5M).
Figure 4. Enhanced Inhibition of the GABACeA→GluPF Pathway in CP.
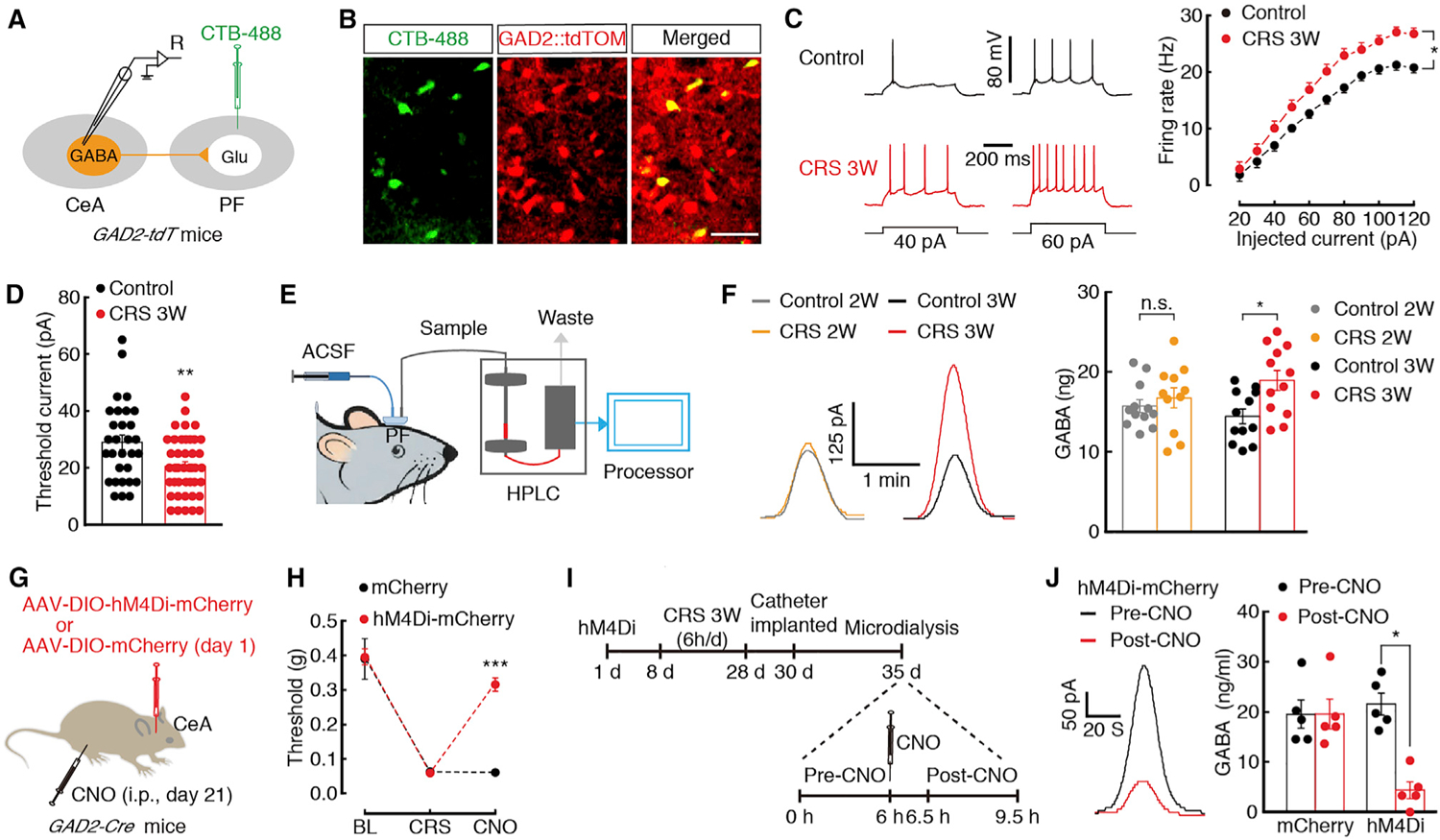
(A) Schematic of CTB-488 injection and recording configuration in acute slices.
(B) Representative images of GABACeA neurons (yellow) labeled by CTB-488 (green) injected in the PF of GAD2-tdTOM mice (neurons with red tdTOM). Scale bar, 50 μm.
(C and D) Sample traces and statistical data for firing rate (C) and rheobase (D) recorded from PF-projecting GABACeA neurons (yellow cells in B) in mice treated with control (n = 30 neurons) or CRS 3W (n = 42 neurons).
(E) Schematic of microdialysis-HPLC detection in freely moving mice with probe implanted in the PF.
(F) Sample traces (left) and summarized data (right) of GABA signal in the PF from the indicated groups (n = 11–12 mice per group).
(G) Schematic of chemogenetics in GAD2-Cre mice.
(H) Behavioral effects of the chemogenetic inhibition of GABACeA neurons on pain threshold at 30 min after CNO intraperitoneal injection (i.p., n = 5 mice per group).
(I) Timeline of microdialysis-HPLC detection.
(J) Sample traces (left) and summarized data (right) of GABA signal in the PF before and after CNO injection (n = 5 mice per group).
All of the data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Unpaired t test for (D); paired t test for (J); two-way Repeated-measures ANOVA with Bonferroni post hoc analysis for (C) and (H); one-way ANOVA with Bonferroni post hoc analysis for (F). See also Figures S5 and S6.
Based on this result, we subsequently investigated whether GABA levels were increased in the PF. Using in vivo microdialysis and high-performance liquid chromatography (HPLC) measurements by implanting a microdialysis probe into the PF (Cheng et al., 2009) (Figure 4E), we found that the concentration of GABA in the PF was increased in the CRS 3W mice (18.94 ± ng) but not CRS 2W mice (16.75 ± 1.67 ng) relative to control mice (14.44 ± 0.82 ng; Figure 4F; Figure S6A). These results suggest enhanced PF-projecting GABACeA neuronal activity in CP states.
Necessary and Sufficient Role of the GABACeA→GluPF Pathway for CP
Given the increased GABACeA→GluPF inhibitory input in the presence of CRS, we infused Cre-dependent expression of chemogenetic inhibitory hM4Di in the CeA and intraperitoneally injected its ligand clozapine-N-oxide (CNO), to selectively inhibit GABACeA neurons of GAD2-Cre mice (Figure 4G; Figures S6E–S6G). We found that the CRS-induced pain sensitization (Figure 4H) and depressive-like behaviors (Figure S6I) were alleviated and that the GABA concentration in the PF decreased (mCherry, 21.57 ± 2.18 ng; hM4Di, 4.37 ± 1.68 ng) after chemogenetic inhibition of GABACeA neurons (Figures 4I and 4J; Figure S6J). Chemogenetic inhibition of GABACeA neurons did not affect distance traveled in the open-field test (Figure S6H).
In addition, reduced pain sensitization effects (Figure 5E) but not locomotion (Figure 5D) or depressive-like behaviors (Figures S7A–S7D) were obtained by injecting Cre-dependent AAV carrying eNpHR (AAV-DIO-eNpHR3.0-EYFP) into the CeA and optical inhibition of eNpHR-containing GABACeA terminals in the PF of GAD2-Cre mice (Figures 5A–5C). It should be noted that the optogenetic inhibition had no effect on CFA-induced inflammatory and SNI-induced pain sensitization (Figure 5F). In naive mice, optical activation of GABACeA terminals in the PF through CeA injection of Cre-dependent AAV-DIO-ChR2-mCherry in GAD2-Cre mice caused the animals to display pain sensitization and did not affect distance traveled (Figures S7E–S7G). In addition, brief (5 min) light stimulation of ChR2-containing GABACeA terminals in the PF (Figure S7H) elicited a significant decrease in the spike number of the GluPF neurons, and this effect could last 15 min after light withdrawal (Figures S7I and S7K). Meanwhile, the spike number of the GluPF neurons did not show a change after GAD2-Cre mice with CeA infusion of AAV-DIO-mCherry (Figures S7I and S7J). These results suggest that the GABACeA→PF pathway is necessary and sufficient for the development of CP symptoms.
Figure 5. Necessary and Sufficient Role of the GABACeA→GluPF Pathway for CP.
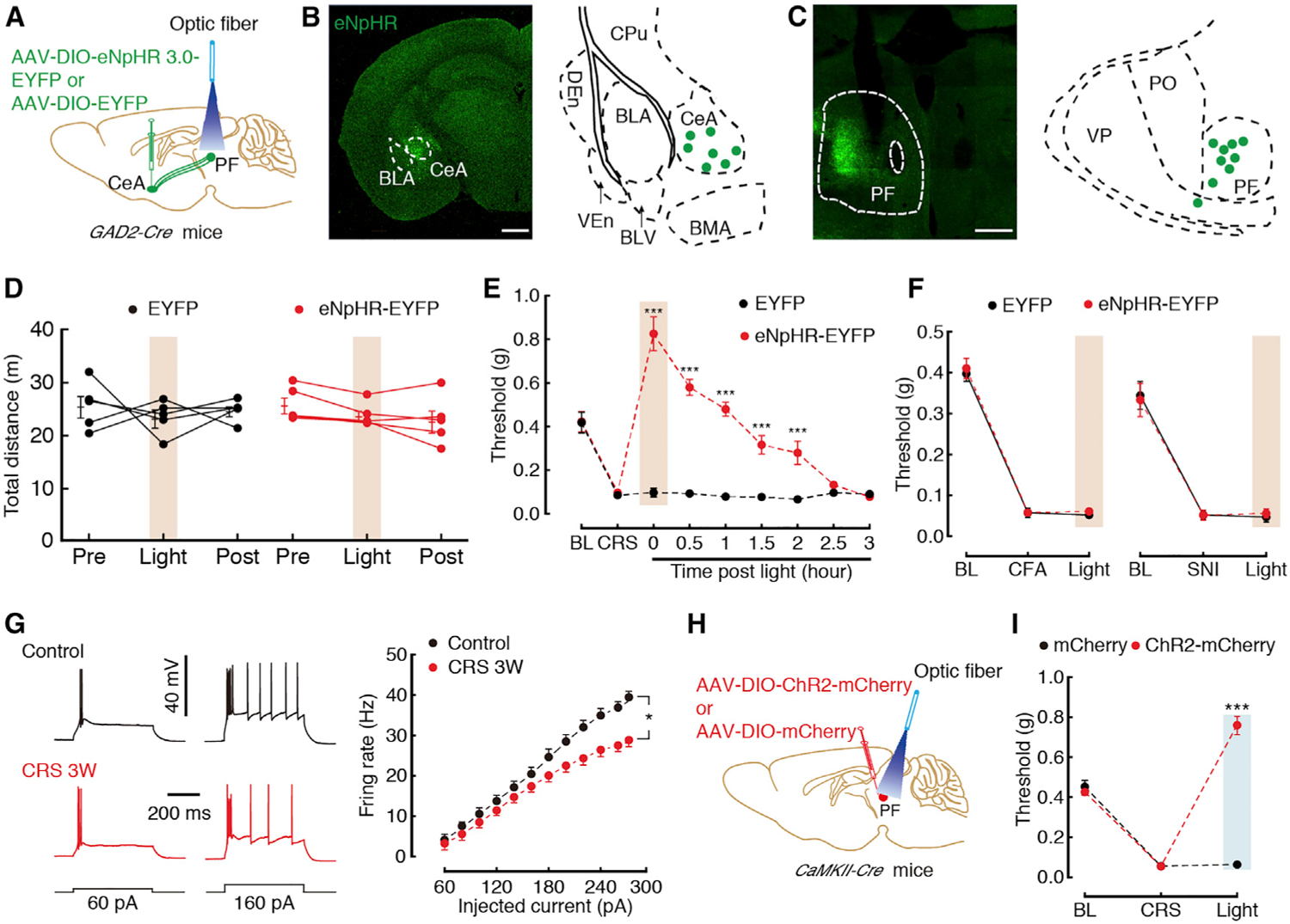
(A) Schematic of optogenetic experiments in GAD2-Cre mice.
(B) Typical image (left) and injection sites (right) within the CeA. Scale bar, 200 μm.
(C) Typical image (left) that were spliced with a confocal microscope, and optic fiber sites (right) within the PF. Scale bar, 200 μm.
(D) Locomotion of the open-field test before, with, and after light photostimulation in the EYFP (n = 5) or eNpHR3.0-EYFP mice (n = 5).
(E) Effects of optogenetic inhibition of GABACeA terminals in the PF on pain threshold in CRS 3W mice (n = 5 mice per group).
(F) Effects of optogenetic inhibition of GABACeA terminals in the PF on pain threshold in CFA (at day 3) or SNI (at day 7) mice (n = 5–6 mice per group).
(G) Sample traces (left) and statistical data (right) of action potential firing recorded from GluPF neurons in CaMKII-tdT mice treated with CRS 3W (n = 56 neurons) or control (n = 33 neurons).
(H) Schematic of optogenetic experiments in CaMKII-Cre mice.
(I) Behavioral effects of optogenetic inhibition of GluPF neurons on pain threshold in CaMKII-Cre mice treated with CRS 3W (n = 5 mice per group).
All of the data are presented as mean ± SEM. *p < 0.05, ***p < 0.001. Two-way repeated-measures ANOVA with Bonferroni post hoc analysis for (D)–(G) and (I). See also Figures S7–S9.
If GluPF neurons are inhibited by GABACeA inputs, the increase of the inputs under CRS conditions should cause inhibitory effects. We next investigated GluPF neuronal activity by whole-cell recordings performed in visualized glutamate neurons in slices from CaMKII-tdT mice, which were generated by CaMKII-Cre mice crossed with Ai14 mice (Figure S8A). A decrease in the spike number in PF CaMKII-tdT neurons from CRS 3W mice (Figure 5G) but not CRS 2W (Figure S8B) was observed when compared with control mice. In addition, optical activation of GluPF neurons through PF injection of Cre-dependent AAV-DIO-ChR2-mCherry in CaMKII-Cre mice relieved CRS-induced pain sensitization (Figures 5H and 5I; Figures S8C and S8D), while, in naive mice, chemogenetic inhibition of GluPF neurons induced severe pain sensitization (Figures S8E–S8J). Furthermore, we investigated the neuronal excitability of GABA neurons in the PF (GABAPF) via whole-cell recordings in visualized GABA neurons in slices from GAD2-tdT mice (Figure S9A). We found that the firing rate (Figure S9B) and rheobase (Figure S9C) were unchanged in CRS 3W mice when compared to control mice. In addition, optogenetic or chemogenetic inhibition of GABAPF neurons in GAD2-Cre mice could not relieve CRS-induced pain sensitization (Figures S9D–S9K). These data suggest that the occurrence of CP is likely caused by an enhanced inhibitory process that involves direct GABACeA-mediated inhibition of GluPF neurons.
The S2 Is the Output of the GABACeA→GluPF Pathway for CP
We subsequently aimed to identify the GABACeA→GluPF output circuitry that may mediate CP behavior. Following injection of AAV-DIO-ChR2-mCherry into the PF of CaMKII-Cre mice (Figures 6A and 6B), we observed an extremely large number of the mCherry+ fibers in the S2 (Figure 6C), a key brain region purportedly involved in the pathophysiology of pain, in addition to other brain regions (Figure S10A). Optical activation of GluPF terminals in the S2 elicited excitatory postsynaptic currents (EPSCs) in brain slices (−140.25 ± 13.08 pA), which were blocked by the glutamate receptor antagonist KOH 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX, 10 μM; −10.24 ± 0.78 pA; Figure 6D). In addition, in vivo electrophysiology recordings were performed in the S2 of freely moving mice. AAV-DIO-ChR2-mCherry viruses were injected into the PF of CaMKII-Cre mice. After 3 weeks, multi-tetrode recording and optic fiber were implanted into the S2 (Figure 6E). Multiple channel recordings of the firing rate of the S2 before, during, and after light photostimulation were conducted (Figures 6F and 6G). We found that blue light increased the firing rate in the S2 neurons. Furthermore, optical stimulation of these fibers in the S2 reduced CRS-induced pain sensitization (Figures 6H–6J) but did not affect locomotion (Figures S10B–S10D). These results suggest that the GABACeA→GluPF outputs to the S2 are part of the inhibitory circuit that underlies CP behavior.
Figure 6. The GABACeA→GluPF→S2 Pathway Controls CP-like Behavior.
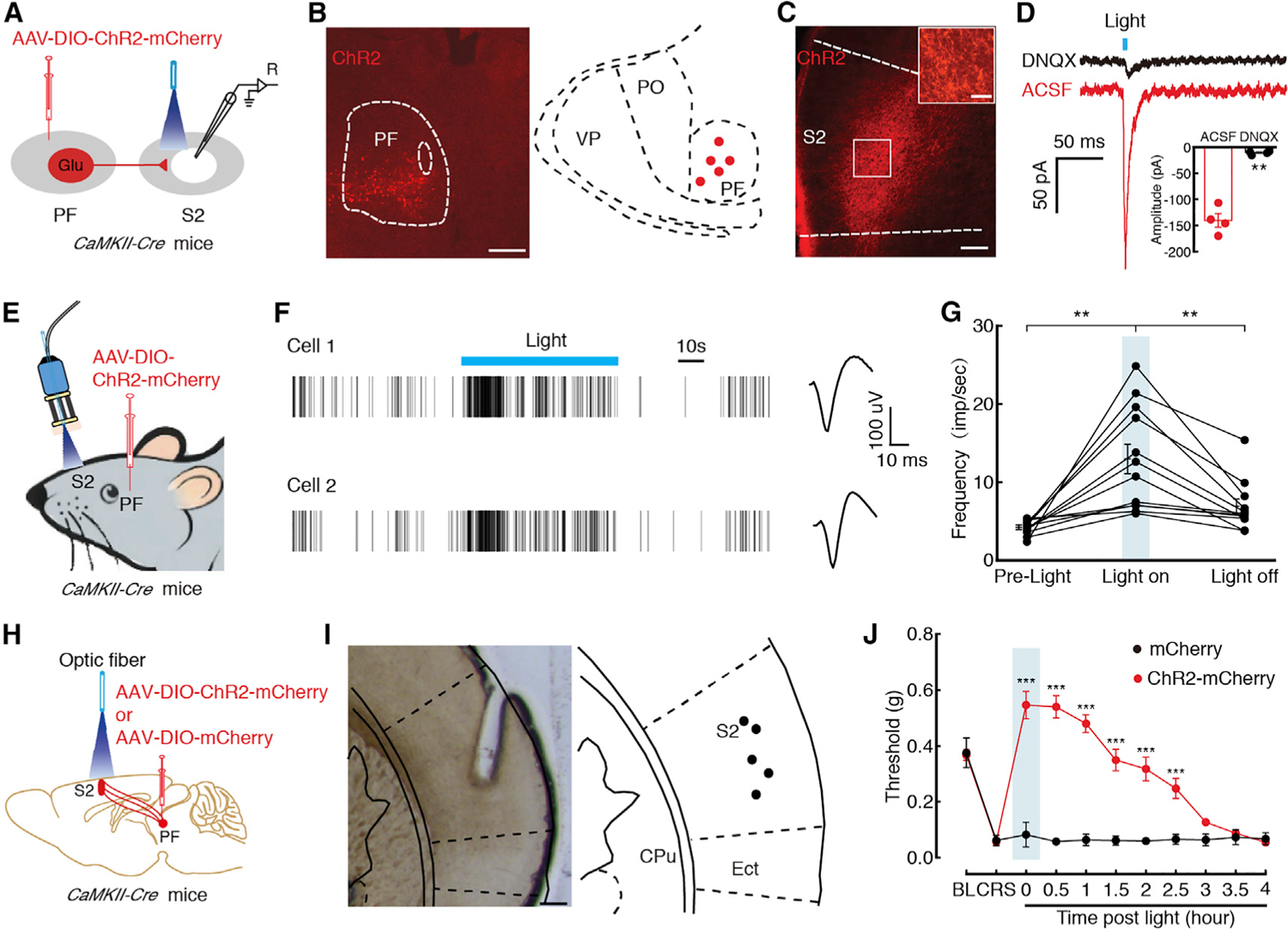
(A) Schematic of viral infection and whole-cell recording configuration in acute slices.
(B) Typical image (left) and injection sites (right) with AAV-DIO-ChR2-mCherry of PF in CaMKII-Cre mice. Scale bar, 200 μm.
(C) Representative images of mCherry signals in the S2 at day 21 after PF infusion of AAV-DIO-ChR2-mCherry in CaMKII-Cre mice. Scale bar, 100 μm. The white boxes depicting the area shown in the box of the S2. Scale bar, 20 μm.
(D) Representative current traces (left) and summarized data (right, n = 4 neurons) evoked through photostimulation of ChR2-containing GluPF terminals in the S2 before and after DNQX (10 μM).
(E) Schematic of in vivo recording of freely moving CaMKII-Cre mice.
(F and G) Representative traces (F) and summarized data (G) showed the firing rate of the S2 neurons in the CaMKII-Cre mice before, with, and after light photostimulation with multiple channel recordings (n = 12 neurons).
(H) Schematic of optogenetic experiments in CaMKII-Cre mice.
(I) Typical image (left) and optic fiber sites (right) within the S2. Scale bar,100 μm.
(J) Behavioral effects of optical activation of GluPF terminals in the S2 on pain threshold (n = 5 mice per group).
All of the data are presented as mean ± SEM. **p < 0.01; ***p < 0.001. Paired t test for (D); one-way repeated-measures ANOVA with Bonferroni post hoc analysis for (G); two-way repeated-measures ANOVA with Bonferroni post hoc analysis for (J). See also Figure S10.
To characterize the connectivity of the GABACeA→GluPF→S2 pathway, we took advantage of an efficient combination strategy that permits direct visualization of these projections (Figure 7A). We initially injected retro-AAV (AAV2/2Retro-CMV-bGl-Cre-EGFP) into the S2 that allowed retrograde virus transport to the soma of GluPF neurons, allowing these neurons to express Cre-GFP (Tervo et al., 2016) (Figure 7B, left). Subsequently, the Cre-dependent retrograde trans-monosynaptic tracing system was used to infect GluPF neurons that expressed Cre-GFP (Figure 7B, middle; Figures S11A and S11B). We visualized numerous DsRed+ cell bodies in the CeA (Figure 7B, right), which were identified as GABA neurons by immunofluorescence staining (Figure 7C).
Figure 7. The GABACeA→GluPF→S2 Pathway Were Identified.
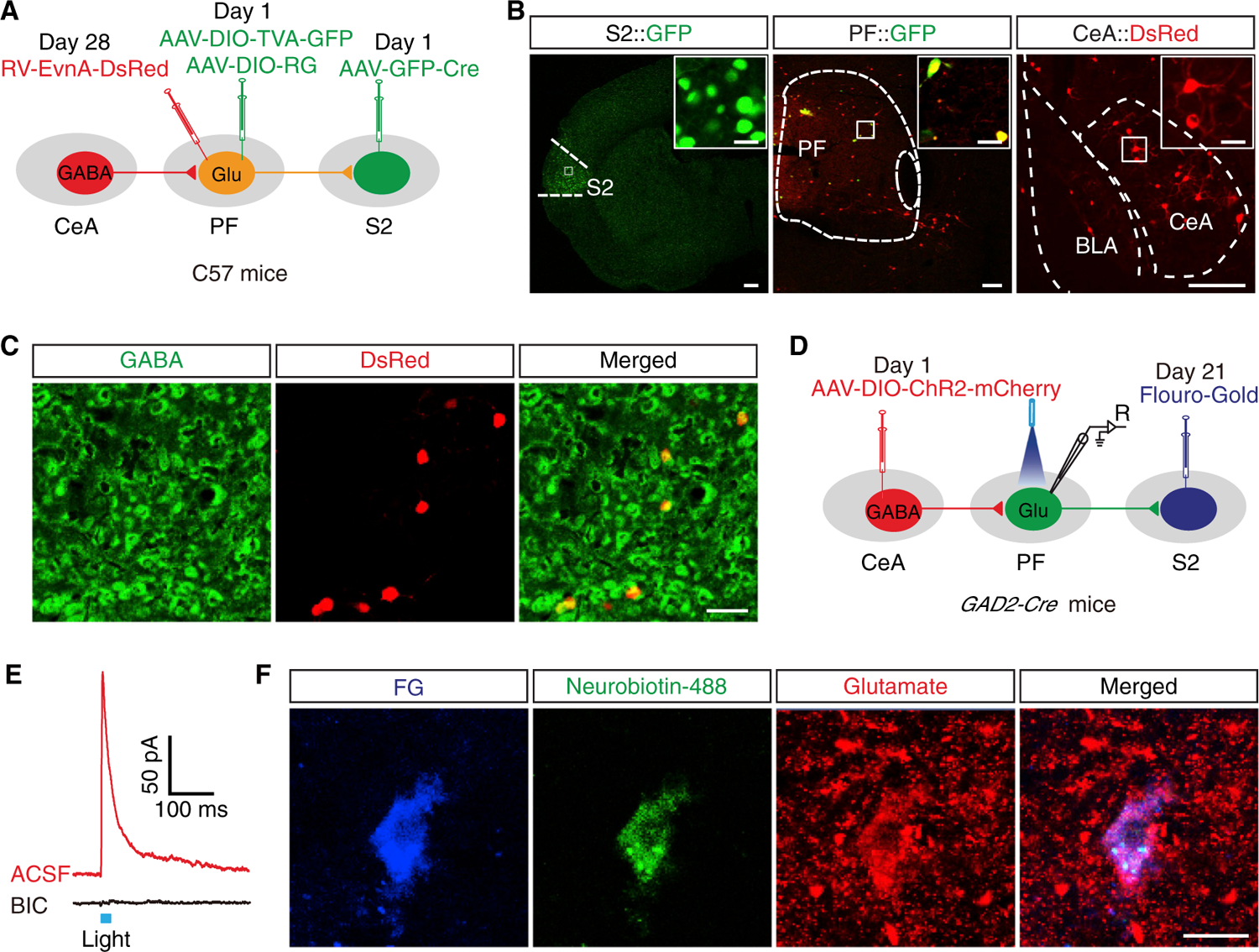
(A) Schematic of viral injection for triple retrograde tracing.
(B) Left: images of an AAV-GFP-Cre injection site in the S2, scale bar, 200 μm; Middle: images of AAV-GFP-Cre and AAV helper co-expression in the PF, scale bar, 200 μm; Right: images of RV-labeled neurons in the CeA, scale bar, 100 μm. The white boxes depicting the area shown in the boxes of the S2, PF, and CeA. Scale bars, 20 μm.
(C) DsRed signals traced from the PF in (B, right) were co-localized with GABA immunofluorescence in the CeA. Scale bar, 100 μm.
(D) Schematic of viral and FG injection and whole-cell recording configuration in acute slices from GAD2-Cre mice.
(E) Representative current traces evoked through photostimulation (blue bar) before and after bath application bicuculline (BIC, 10 μM).
(F) Representative images of FG (blue), neurobiotin-488 (green), anti-glutamate immunostaining (red), and merged showing glutamate neurons by post hoc staining after patch in (E). Scale bar, 20 μm.
See also Figures S11 and S12.
To examine the functional synaptic connectivity of the GABACeA→GluPF→S2 pathway, we injected Fluoro-Gold (FG) into the S2 to label the S2-projecting GluPF neurons and injected Cre-dependent AAV-DIO-ChR2-mCherry into the CeA of GAD2-Cre mice (Figure 7D). Similar to the findings in GluPF neurons with GABACeA inputs (see Figure 2E), photostimulation of GABACeA fibers in the PF elicited an IPSC in FG+ neurons, which was reversed by bicuculline (Figure 7E). Post hoc immunofluorescence by intracellularly labeling with neuronbiotin-488 confirmed the recorded FG+ neurons were glutamate neurons (Figure 7F). In addition, following injection of FG into the S2 and H129-GFP4 into the CeA (Figures S11C and S11D), total overlapping of GFP+ and FG+ signals in the PF was observed (Figure S11E), which were identified as glutamate neurons (Figure S11F). These results demonstrate a direct functional connectivity of GABACeA→GluPF→S2 pathway (Figure S12).
DISCUSSION
This study defines a GABACeA→GluPF→S2 pathway through which CP is generated. Central to this process is a circuit mechanism involving increased inhibition of CeA GABAergic neurons on S2-projecting PF glutamatergic neurons under depression conditions to prime pain behavior.
The major problem of why certain depression treatments are ineffective for CP patients is far from understood (Bair et al., 2003). For instance, the SSRI paroxetine has been shown to be effective for treating depression but not effective for CP (Marks et al., 2009). Consistent with this notion, we demonstrate that typical analgesic agents for inflammatory and neuropathic pain had no effect on the symptoms of CP in animals, implicating that a pathway is necessary for the occurrence of CP. In marked contrast, the inhibition of the GABACeA→GluPF→S2 pathway relieved the symptoms of CP animals, indicating that the GABACeA→GluPF output to the S2 is likely critical for CP treatment. This also suggests that typical analgesic agents are likely inappropriate for treating CP patients. In addition, data supporting pain-reducing effects of selectively serotonergic medications are more limited, presumably due to the role of serotonin in both inhibiting and enhancing pain via descending pathways. In fact, the exact targets for these antidepressants remain elusive. For their analgesic effects, one possible mechanism is the activation of the descending modulatory pathway from the rostral ventromedial medulla and periaqueductal gray to the dorsal horn neurons, via inhibition of serotonin and norepinephrine reuptake. These results raise the possibility that selective activation of this pathway is a possible intervention for CP.
The advantage of studying the neural circuit of a disorder seems to reveal convergent points of drug action and pathologically relevant behavioral consequences. Although the neural circuits underlying major depression (Knowland et al., 2017) and chronic pain (Cheng et al., 2017; Liu et al., 2018) have been extensively investigated, the mechanism underlying CP remains unknown. Based on our present findings, we would like to propose a hypothesis for the neural circuit responsible CP, in which depression enhances the inhibition of the GABACeA→GluPF pathway, thus decreasing excitatory outputs to the S2, and resulting in the symptoms observed in CP. Of note, CP occurred in a state-dependent manner, accompanied by a gradual increase of GABA in the PF and a decrease of GluPF neuronal activity at CRS 3W but not at CRS 2W. Thus, it appears that the duration of stress is important for the emergence of pain symptoms. This complex behavioral outcome can likely be attributed to the dynamic network activity during progressive stress. Adaptation of the GABACeA→GluPF pathway at CRS 3W primes the development of CP. In addition, we found that reversing the dysfunction of the GABACeA→GluPF pathway alleviated the symptoms of CP but not those of inflammation- or neural-injury-induced pain, suggesting that this pathway is likely specifically recruited under chronic stress conditions.
The CeA forms widespread connections with forebrain areas and the hypothalamus, to regulate emotional behavior (Pessoa, 2008; Roozendaal et al., 2009). This is confirmed by our findings that the chemogenetic inhibition of GABACeA neurons reduced CRS-induced depressive-like behaviors. Although the CeA is part of a descending endogenous pain control system that includes circuits in the brainstem (e.g., periaqueductal gray [Tovote et al., 2016]) and spinal cord (Fields, 2004), how the CeA integrates affective information to increase pain processing through the ascending pain pathway is unknown. Our results show that the PF is a direct target of the CeA that contributes to CP. Moreover, we identified that the S2 receives glutamatergic inputs from the GABACeA→GluPF pathway to control the symptoms of CP. As the S2 is the final path for processing polymodal sensory information integration (Keysers et al., 2010; Milligan and Watkins, 2009; Navratilova and Porreca, 2014), cell-type-specific regulation of the tone of the GABACeA→GluPF pathway could be one of the specific rules for the development of CP. Interestingly, inhibiting GABACeA neurons relieved both the pain and depressive symptoms, while inhibiting GABACeA terminals in the PF only relieved pain symptoms. These data suggest that CRS-induced pain sensitization and depressive-like behaviors are regulated by the CeA through different individual pathways. Although GABACeA neuronal activity likely contributes to multiple behavioral outputs, including fear, anxiety, and depression (Li et al., 2013), the specific projection of this type of neuron to the PF could be relatively more important for CP in the current animal models. The significance of these findings with respect to treatment-resistant CP is that manipulations that target this neural circuitry would have dual effects on the symptoms of depression and chronic pain.
As pharmacological options for the treatment of CP remain quite limited, these findings raise the possibility of developing optimal treatments that involve the use of dual-acting drugs or non-drug approaches, such as deep-brain stimulation (Mayberg et al., 2005) or transcranial magnetic stimulation (Pascual-Leone et al., 1996), which target the converging pathway.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhi Zhang (zhizhang@ustc.edu.cn).
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
In all experiments, C57BL/6J, GAD2-Cre, CaMKII-Cre and Ai14 (RCL-tdT) male mice (purchased from Charles River or Jackson Laboratories) at 8–10 weeks of age were used. Unless cannula surgery was performed, the mice were housed five per cage in a colony with ad libitum access to water and food. They were maintained under a 12-hour light/dark cycle (lights on from 7:00 a.m. to 7:00 p.m.) at a stable temperature (23–25°C). The animal protocols were approved by the Care Committee of the University of Science and Technology of China. A total of 350 mice were recruited for behavioral and viral tracing experiments in the current study, with 85 mice excluded due to missed targets, including the injection of viruses, or the placement of optic/microdialysis fibers. The viral tracing and behavioral data of these animals were excluded from further analyses.
Animal models
CRS
Mice were periodically constrained from moving by placing them in a 50 mL syringe for 6 h, every day, for 3 weeks. Holes were drilled in the ends of the syringes to allow the mice to breathe (see Figure 3A). During the restraint period, control mice were allowed to freely move around in the cage without water or food provided. Syringes were thoroughly washed every day after the restraint period. To exclude the effect of acute stress, mice were allowed to rest one day after CRS before the anatomical and electrophysiological experiments were conducted.
CFA
Complete Freund’s adjuvant (CFA, 25 ul) or saline was injected into the left hind paw under brief isoflurane anesthesia for inflammatory pain model and control.
SNI
The SNI surgery was performed under anesthesia with isoflurane. The skin and muscle of the left thigh were incised to explore the sciatic nerve, consisting of the sural, common peroneal, and tibial nerves. After exploration, nonabsorbent 4–0 chromic gut were used to ligate the common peroneal and tibial nerves, and then the nerves were transected, and about 2 mm sections from the dot were removed. The skin was stitched and disinfected with iodophor (see Figure 3F). For the sham mice, the procedure was the same as for the experimental group except that the nerves were left intact.
METHODS DETAILS
Virus and trace injection
Mice were fixed in a stereotactic frame (RWD, Shenzhen, China) under a combination of xylazine (10 mg/kg) and ketamine (100 mg/kg) anesthesia. A heating pad was used to maintain the core body temperature of the animals at 36°C. A volume of 100–300 nL virus (depending on the expression strength and viral titer) was injected using calibrated glass microelectrodes connected to an infusion pump (micro 4, WPI, USA) at a rate of 30 nl/min. The coordinates were defined as dorsal-ventral (DV) from the brain surface, anterior-posterior (AP) from bregma and medio-lateral (ML) from the midline (in mm).
For polysynaptic anterograde tracing, HSV129-GFP4 (H129-G4, 1×109PFU/ml, 300 nl) were injected into the CeA (AP: −1.0 mm, ML: −2.64 mm, DV: −4.30 mm) of C57BL/6J mice. After 36 hours, mice that had been anesthetized with pentobarbital (20 mg/kg, i.p.) were transcardially perfused, and brain slices were prepared (40 μm) for tracing GFP or co-staining with glutamate antibodies. For monosynaptic anterograde tracing, AAV2/1-hSyn-Cre-WPRE-hGH (AAV2/1-Cre, AAV2/1, 1.23 × 1013 vg/ml, 200 nl; Taitool, Shanghai, China) were delivered into the CeA to allow the virus to spread antegradely to the PF soma to express Cre of Ai14 mice.
For retrograde monosynaptic tracing, helper viruses that contained rAAV-Ef1α-DIO-RVG-WPRE-pA (AAV-DIO-RVG, AAV2/9, 2 × 1012 vg/ml) and rAAV-Ef1α-DIO-EGFP-2a-TVA-WPRE-pA (AAV-DIO-TVA-GFP, AAV2/9, 2 × 1012 vg/ml; 1:2, 200 nl) were co-injected into the PF (AP: −2.10 mm, ML: −0.77 mm, DV: −3.20 mm) of CaMKII-Cre mice. After three weeks, the rabies virus RV-EnvA-∆G-dsRed (2 × 108 IFU/ml, 300 nl) was injected into the same site of the PF. For GABACeA→GluPF→S2 triple tracing, the AAV2/2Retro-CMV-bGl-Cre-EGFP virus (AAV-Cre-GFP, 1.83 × 1013 vg/ml, 100 nl; Taitool, Shanghai, China) was first injected into the S2 (AP: −0.50mm, ML: −4.50mm, DV: −0.90mm with a 20° angle) to allow the virus to spread retrogradely to the PF soma to express Cre-GFP; simultaneously, the Cre-dependent mixed helper virus was infused into the ipsilateral PF. The RV was injected after three weeks. Mice that had been anesthetized with pentobarbital (20 mg/kg, i.p.) were transcardially perfused 7 days after the last injection, and brain slices were prepared (40 μm) for tracing DsRed signal or co-staining with GABA antibody. The retrograde tracer cholera toxin B subunit conjugated to CTB-488 (0.1% mg/mL, 100 nl, Thermolife) was injected into the PF of GAD2-tdT mice for 7 days to label PF projecting GABACeA neurons for visualized electrophysiological recordings. Retrograde tracer Fluoro-Gold (0.1% mg/mL, 100 nl, Santa Cruz Biotechnology) was injected into the S2 to label S2 projecting PF neurons.
The Cre-dependent virus rAAV-Ef1α-DIO-hChR2 (H134R)-mCherry-WPRE-pA (AAV-DIO-ChR2-mCherry, AAV2/9, 1.63 × 1013 vg/ ml, 200 nl) was delivered into the CeA of GAD2-Cre mice or into the PF of CaMKII-Cre mice. After three weeks, the expression of mCherry was detected in the whole brain, and optogenetic manipulation was performed. In some experiments, rAAV-Ef1α-DIO-eNpHR3.0-EYFP-WPRE-pA (AAV-DIO-eNpHR3.0-EYFP, AAV2/9, 1.18 × 1013 vg/ml) were used for optogenetic manipulation. The rAAV-Ef1α-DIO-hM4D(Gi)-mCherry-WPRE-pA (AAV-DIO-hM4Di-mCherry, AAV2/9, 3.69 × 1013 vg/ml) viruses were used for chemogenetic manipulations 3 weeks after viral injection, with intraperitoneal injection of CNO (5 mg/kg, Sigma-Aldrich, USA) 30 min before behavior test. The rAAV-Ef1α-DIO-mCherry-WPRE-pA (AAV2/8, 8.93 × 1012 vg/ml) and rAAV-DIO-EYFP-WPRE-pA (AAV2/9, 1.95 × 1012 vg/ml) viruses were used as the controls. Unless otherwise stated, all viruses were packaged by BrainVTA (Wuhan, China). All mice were transcardially perfused with 0.9% saline followed by ice-cold phosphate buffer (0.1 M) that contained 4% paraformaldehyde. Images of the signal expression were acquired with a confocal microscope (LSM 710, ZEISS, Germany). Animals with missed injections were excluded.
Optogenetic manipulations in vivo
An optical fiber cannula was initially implanted into areas of interest, which included the PF and S2, in the brain of an anesthetized mouse that had been immobilized in a stereotaxic apparatus. The implant was secured to the animal’s skull with dental cement. Chronically implantable fibers (diameter, 200 μm, Newdoon, Hangzhou) were connected to a laser generator using optic fiber sleeves. The delivery of a 5-min pulse of blue light (473 nm, 2–5 mW, 15-ms pulses, 20 Hz) or yellow light (594 nm, 5–8 mW, constant) was controlled by a Master-8 pulse stimulator (A.M.P.I., Jerusalem, Israel). The same stimulus protocol was applied to the mice in the control group. Optical activation of CeA GABA neurons induced obvious freezing behavior (Video S1) and reduced movement during light on (Figures S6B and S6C), which is consistent with previous studies (Janak and Tye, 2015; Li et al., 2013). Locomotion would recover during 5-min light off (Figure S6D). Thus, it is inappropriate to analyze depressive-like behavior during this time point, because of the deficiency in locomotion during freezing. To avoid the effect of locomotion and unify the behavioral paradigm, we performed behavioral experimentation after light stimulation withdrawal. The location of the fibers was examined in all mice at the conclusion of the experiments, and data obtained from mice in which the fibers were located outside of the desired brain region were discarded.
Brain slice electrophysiology
Brain slice preparation
Acute brain slices were prepared as previously described (Zhang et al., 2011). Mice were deeply anesthetized with pentobarbital sodium (2%, w/v, i.p.) and intracardially perfused with ~20 mL ice-cold oxygenated modified ice-cold oxygenated N-Methyl-D-glucamine (NMDG) artificial cerebrospinal fluid (ACSF) that contained (in mM) 93 NMDG, 2.5 KCl, 0.5 CaCl2, 20 HEPES, 1.2 NaH2PO4, 30 NaHCO3, 10 MgSO4, 25 glucose, 5 Na-ascorbate, 3 Na-pyruvate, 2 thiourea and 3 glutathione (pH: 7.3–7.4, osmolarity: 300–305 mOsm/kg). Coronal slices (300 μm) that contained the CeA, PF or S2 were sectioned with a vibrating microtome (VT1200s, Leica, Germany) and were initially incubated in HEPES ACSF that contained (in mM) 2.5 KCl, 92 NaCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 3 glutathione (GSH), 2 CaCl2 and 2 MgSO4 (pH 7.3–7.4, osmolarity 300–305 mOsm/kg) for at least 1 hour at 25°C. The brain slices were transferred to a slice chamber (Warner Instruments, USA) with ACSF perfusion at 2.5–3 ml/min at 32°C for electrophysiological recording; the temperature of the ACSF was maintained by an in-line solution heater (TC-344B, Warner Instruments, USA).
Whole-cell patch-clamp recordings
Neurons were visualized using a 40 × water immersion objective on an upright microscope (BX51WI, Olympus, Japan) equipped with interference contrast (IR/DIC) and an infrared camera connected to the video monitor. Whole-cell patch-clamp recordings were obtained from visually identified CeA, PF or S2 cells. Patch pipettes (3–5 MΩ) were pulled from borosilicate glass capillaries (VitalSense Scientific Instruments Co., Ltd., Wuhan, China) with an outer diameter of 1.5 mm on a four-stage horizontal puller (P1000, Sutter Instruments, USA). The signals were acquired via a Multiclamp 700B amplifier, low-pass filtered at 2.8 kHz, digitized at 10 kHz and analyzed with Clampfit 10.7 software (Molecular Devices, Sunnyvale, CA, USA). The data were collected from the neurons with the series resistance < 30 MΩ and input resistance > 100 MΩ. In some experiments, the 0.5% neurobiocytin 488 was included in the intracellular solution, together with immunofluorescence staining, for cell type identification. The current-evoked firing was recorded in current-clamp mode (I = 0 pA). The threshold current for firing was defined as the minimum strength of current injection required to elicit at least one or two spikes. For recording the intrinsic membrane properties, the pipettes (5–7 MΩ) were filled with potassium gluconate-based internal solution resistance containing (in mM): 130 K-gluconate, 2 MgCl2, 5 KCl, 0.6 EGTA, 10 HEPES, 2 Mg-ATP and 0.3 Na-GTP (osmolality: 285–290 mOsm/kg, pH: 7.2). Measurement of the intrinsic membrane properties were conducted under current-clamp mode and data were collected from neurons with a resting membrane potential negative than –50 mV and an overshot of the action potentials. The threshold current of the action potential was defined as the minimum current to elicit an action potential.
Light-evoked response
Optical stimulation was delivered using a laser (Shanghai Fiblaser Technology Co., Ltd. China) through an optical fiber 200 μm in diameter positioned 0.2 mm from the surface of the brain slice. To test the functional characteristics of AAV-DIO-ChR2-mCherry, fluorescently labeled neurons that expressed ChR2 in GAD2-Cre or CaMKII-Cre mice 3–4 weeks after virus injection were visualized and stimulated with a blue (473 nm, 5–10 mV) laser light using 5-Hz, 10-Hz, and 20-Hz stimulation protocols with a pulse width of 15 ms. In some experiments, the function of eNpHR3.0 was assessed by applying sustained yellow (594 nm, 5–10 mV, 200 ms) laser light stimulation.
In vivo electrophysiology recording
For chronic extracellular recordings, a custom-made four movable tetrode array was implanted into the PF (AP: −2.10 mm, ML: −0.77 mm, DV: −3.00 mm) and the S2 (AP: −0.50mm, ML: −4.00mm, DV: −1.2mm). Each tetrode was made of four twisted fine platinum/iridium wires (12.5 μm diameter, California Fine Wire). The optrode was constructed by surrounding an optical fiber (200 μm, Newdoon) with tetrodes. The screw-based microdrive scaffolds for lowering the electrodes were cemented onto the skull. The mice were allowed to recover for at least 3 days before recordings were made. Recording electrodes were attached to a 16-channel headstage, and neuronal signals were amplified, filtered at a bandwidth of 300–5000 Hz, and stored using Neurostudio software (Jiangsu Brain Medical Technology, China). Neuronal firings in the PF or S2 were recorded in the absence and presence of blue light stimulation (60 s). Spike sorting was performed with a sorting method involving a T-Dis E-M algorithm built in Offline Sorter 4 (Plexon, USA). The firing rates of sorted units were calculated using Neuroexplorer 4 (Nex Technologies, USA).
Immunohistochemistry and imaging
The mice were deeply anesthetized with pentobarbital sodium (50 mg/kg, i.p.) and sequentially perfused with saline and 4% (w/v) paraformaldehyde (PFA). The brains were subsequently removed and post-fixed in 4% PFA at 4°C overnight. After cryoprotection of the brains with 30% (w/v) sucrose, coronal sections (40 μm) were cut on a cryostat (Leica CM1860) and used for immunofluorescence. The sections were incubated in 0.3% (v/v) Triton X-100 for 0.5 h, blocked with 10% donkey serum for 1 h at room temperature, and incubated with primary antibodies, including anti-GABA (1:500, rabbit, Sigma), anti-Glutamate (1:500, rabbit, Sigma), at 4°C for 24 h, followed by the corresponding fluorophore-conjugated secondary antibodies for 2h at room temperature. Fluorescence signals were visualized using Zeiss LSM710 microscope.
For post hoc immunofluorescence, slices were collected after whole-cell recording, in which the patched neurons were labeled with neuronbictin-488, and were immersed into 4% PFA for post-fixation in 4°C for 1 week. Then, slices were washed in 0.01M PBS for 3 × 30 minutes, followed by incubation overnight with blocking solution [10% (v/v) donkey serum, 1.0% (v/v) Triton X-100 dissolved in 0.01M PBS]. Then, slices were incubated with primary antibody diluted in blocking solution at 4°C for three days, including anti-Glutamate (1:100, rabbit, Sigma). After washing in 0.01M PBS, slices were immersed in Alexa 594- or Alexa 647-conjugated secondary antibody (1:100, Invitrogen) diluted in 0.01M PBS at RT for 8 hours, then washed in 0.01M PBS for 3 × 30 minutes, mounted on the glass slides. Images were captured via confocal microscopy.
Assessment of depressive-like behaviors
Mice were transported to the behavioral testing room in a holding cabinet to habituate at least 2 hours prior to testing. The behavior of the animals during testing was recorded using a video tracking system and was subsequently analyzed offline. Dim light (~20 lux) was used in the room to minimize the anxiety of the animals.
Forced swim test
A mouse was placed in a transparent Plexiglas cylinder (30 cm high and 12 cm in diameter) that contained fresh water (24 ± 1°C) up to a height of 25cm from the bottom for 6 min. The animal’s behavior was videotaped from the side, and the duration of immobility of each mouse during the last 5 min of recording was measured offline. Despair behavior was defined as floating or remaining motionless such that the overall immobility time was two s.d. greater than that of the control.
Tail suspension test
Mice were individually suspended about 50 cm above the surface of a table using adhesive tape that was placed approximately 1 cm from the tip of the tail. Each mouse was tested only once for 6 min; the test was videotaped from the side, and the immobility time of the animal was measured in the last 5 min. Mice were considered immobile without initiated movements; immobility was considered to include passive swaying.
Open field test
Mice were placed in one corner of an open field apparatus that consisted of a square area (25 cm × 25 cm) and a marginal area (50 cm × 50 cm × 60 cm); the mice were allowed to freely explore their surroundings. The animals’ movement trajectories were recorded for 5 min using EthoVision XT software, which records the number of entries into and the amount of time spent in the central area. The area was cleaned with 75% ethanol after each test to remove olfactory cues from the apparatus.
Elevated plus maze test
The elevated plus maze consisted of a central platform (6 × 6 cm), two closed arms (30 × 6 × 20 cm) and two opposing open arms (30 × 6 cm), which was placed 100 cm above the floor. A mouse was placed in the central platform facing a closed arm and was allowed to explore the maze during 5 min session. The time spent in the open arms and the number of entries into the open arms were analyzed using EthoVision XT software. Seventy-five percent ethanol was used to clean the area between tests.
Von Frey filament test
Mice were individually placed in transparent plastic chamber on a wire mesh grid to allow for calibrated Von Frey hairs insertion on the surface of hind paw. Before testing, mice were allowed to acclimate to the testing environment for 30 min. We tested the withdrawal threshold (g) of the planta when stimulating the paw’s surface with the calibrated Von Frey hairs. The mean threshold was calculated from five applications. Pain thresholds were measured every 5 min or daily by the paw-withdrawal test on a freely moving animal with the Hargreaves apparatus (IITC Life Science Inc., Woodland Hills, CA, USA) for thermal hyperalgesia. Pain thresholds were tested immediately after 1 min of light stimulation with optogenetic manipulations.
In vivo microdialysis-HPLC
A microdialysis probe (CMA7, CMA, USA), connected to a syringe infusion pump (CMA402, CMA, USA) via polyethylene tubing, was initially implanted into the right PF of deeply anesthetized mice. The tissue was perfused with normal Ringer’s solution (145 mM NaCl, 3 mM KCl and 1.3 mM CaCl2) via the pump at a rate of 0.5 ml/min, and the dialysate was collected in a 15 mL quantitative loop through the probe in freely moving mice. Then the dialysate was derived using ophthaldialdehyde (OPA) solution (37 mM OPA, 50 mM Na2SO3, 90 mM H3BO3, and 5% MeOH) for 3 min. After that, dialysate was automatically loaded to the mobile phase (0.1 M NaH2PO4 and 10% MeOH, pH = 3.2) and separated on a 1 mm × 50 mm column (ALF-105, Antec, Netherlands) with a 3 μm particle size at a rate of 0.35 ml/min. Detection was conducted using an Alexys online analysis system (Antec Leyden) that consisted of a DECADE II electrochemical detector and VT-3 electrochemical flow cells. The data were analyzed using Clarity software (ANTEC, Netherlands) based on standard samples.
For chemogenetic-microdialysis experiments, samples were collected for 3 hours before CNO intraperitoneal injection. Then, sample was collected at 1, 2, and 3 hours after CNO injection. The mean threshold of GABA concentration after CNO was calculated from three applications.
QUANTIFICATION AND STATISTICAL ANALYSIS
We conducted simple statistical comparisons using Student’s t test. ANOVA (one-way and two-way) and post hoc analyses were used to statistically analyze the data from the experimental groups with multiple comparisons. All data are expressed as the mean ± SEM, and significance levels are indicated as * p < 0.05, ** p < 0.01 and *** p < 0.001. OriginPro 2017 software (Origin Lab Corporation, USA) and GraphPad Prism 5 (Graph Pad Software, Inc., USA) were used for the statistical analyses and graphing. Offline analysis of the data obtained from electrophysiological recordings was conducted using Clampfit software version 10.7 (Axon Instruments, Inc., USA) and MiniAnalysis software version 6.03 (Synaptosoft Inc., USA). All statistical data are presented in Table S1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-Glutamate antibody | Sigma-Aldrich | Cat#G6642 |
| Rabbit Anti-GABA antibody | Sigma-Aldrich | Cat#A2052 |
| Alexa 488-conjugated secondary antibody | Invitrogen | Cat#A21206 |
| Alexa 594-conjugated secondary antibody | Invitrogen | Cat#A21207 |
| Alexa 647-conjugated secondary antibody | Invitrogen | Cat#A31573 |
| Bacterial and Virus Strains | ||
| AAV2/1-hSyn-Cre-WPRE-hGH | Taitool | Cat#S0292 |
| rAAV-Ef1α-DIO-RVG-WPRE-pA | BrainVTA | Cat#PT-0023 |
| rAAV-Ef1α-DIO-EGFP-2a-TVA-WPRE-pA | BrainVTA | Cat#PT-0062 |
| RV-EnvA-∆G-dsRed | BrainVTA | Cat#R01002 |
| AAV2/2-Retro-CMV-bGl-Cre-EGFP | Taitool | Cat#S0231 |
| rAAV-Ef1α-DIO-hChR2 (H134R)-mCherry-WPRE-pA | BrainVTA | Cat#PT-0002 |
| rAAV-Ef1α-DIO-eNpHR3.0-EYFP-WPRE-pA | BrainVTA | Cat#PT-0006 |
| rAAV-Ef1α-DIO-hM4D(Gi)-mCherry-WPRE-pA | BrainVTA | Cat#PT-0043 |
| rAAV-Ef1α-DIO-mCherry-WPRE-pA | BrainVTA | Cat#PT-0013 |
| rAAV-DIO-EYFP-WPRE-pA | BrainVTA | Cat#PT-0012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DNQX | Sigma-Aldrich | Cat#D0540; CAS:2379–57-9 |
| bicuculline | Sigma-Aldrich | Cat#O7639; CAS:722456–08-8 |
| CTB-488, Alexa Fluor 488 conjugate | Thermolife | Cat#C22841; Lot:1797975 |
| Fluoro-Gold | Santa Cruz | CAS: 223769–64-0 |
| Clozapine N-oxide | Sigma-Aldrich | Cat#H0832; CAS:34233–69-7 |
| Ibuprofen | MedChemExpress | HY-78131; CAS: 15687–27-1 |
| Indomethacin | Sigma-Aldrich | I7378; CAS: 53–86-1 |
| Gabapentin | MedChemExpress | HY-A0057; CAS: 60142–96-3 |
| Lidocaine | Sigma-Aldrich | L5738; CAS: 21306–56-9 |
| Experimental Models: Organisms/Strains | ||
| Mouse: wild type C57BL/6J | Charles River | N/A |
| Mouse: CaMKII-Cre: B6.Cg-Tg(Camk2a-cre)T29–1stl/J | Jackson Laboratory | 005359 |
| Mouse: GAD2-Cre:B6J.Cg-Gad2tm2(cre)Zjh/J | Jackson Laboratory | 028867 |
| Mouse: Ai 14: B6;129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratory | 007914 |
| Software and Algorithms | ||
| OriginPro 2017 | Origin Lab | https://www.originlab.com/index.aspx?go=Products/Origin |
| Illustrator CS6 | Adobe | https://www.adobe.com/products/illustrator.html |
| GraphPad Prism 5 | GraphPad Software | https://www.graphpad.com/scientific-software/ |
| ZEN | Zeiss | https://www.zeiss.com/microscopy/int/products/microscope-software/zen-lite.html |
| Other | ||
| Optogenetic fibers | Newdoon | N/A |
| Microdialysis probes | CMA | N/A |
Highlights.
GABAergic neurons from the CeA project to glutamatergic neurons in the PF
Enhanced inhibition of the GABACeA→GluPF pathway in CRS mice with pain
Optical inhibition of the GABACeA→GluPF pathway relieves pain symptoms in CRS mice
ACKNOWLEDGMENTS
We thank Xiang Yu and Yupeng Yang for providing Ai14 mice. We thank Fu-Qiang Xu, Tian Xue, and Ben-Sheng Qiu for technical support. All data are available in the main text or the STAR Methods. Support for this study was provided by the National Natural Science Foundation of China (grants 81870877, 91732303, and 91849119), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant XDB02010000), and the NIH (R01-DE025943).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.003.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
This study did not generate/analyze [datasets/code].
REFERENCES
- Ährlund-Richter S, Xuan Y, van Lunteren JA, Kim H, Ortiz C, Pollak Dorocic I, Meletis K, and Carlén M. (2019). A whole-brain atlas of monosynaptic input targeting four different cell types in the medial prefrontal cortex of the mouse. Nat. Neurosci 22, 657–668. [DOI] [PubMed] [Google Scholar]
- Assous M, Kaminer J, Shah F, Garg A, Koós T, and Tepper JM. (2017). Differential processing of thalamic information via distinct striatal interneuron circuits. Nat. Commun 8, 15860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bair MJ, Robinson RL, Katon W, and Kroenke K. (2003). Depression and pain comorbidity: a literature review. Arch. Intern. Med 163, 2433–2445. [DOI] [PubMed] [Google Scholar]
- Barthas F, Sellmeijer J, Hugel S, Waltisperger E, Barrot M, and Yalcin I. (2015). The anterior cingulate cortex is a critical hub for pain-induced depression. Biol. Psychiatry 77, 236–245. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, and Julius D. (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berret E, Kintscher M, Palchaudhuri S, Tang W, Osypenko D, Kochubey O, and Schneggenburger R. (2019). Insular cortex processes aversive somatosensory information and is crucial for threat learning. Science 364, Published online May 31, 2019 10.1126/science.aaw0474. [DOI] [PubMed] [Google Scholar]
- Bieler M, Xu X, Marquardt A, and Hanganu-Opatz IL. (2018). Multisensory integration in rodent tactile but not visual thalamus. Sci. Rep 8, 15684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, Kaang BK, and Zhuo M. (2016). Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat. Rev. Neurosci 17, 485–496. [DOI] [PubMed] [Google Scholar]
- Bushnell MC, Ceko M, and Low LA. (2013). Cognitive and emotional control of pain and its disruption in chronic pain. Nat. Rev. Neurosci 14, 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman CR, and Gavrin J. (1999). Suffering: the contributions of persistent pain. Lancet 353, 2233–2237. [DOI] [PubMed] [Google Scholar]
- Cheng GW, Hsu KC, Lee CF, Wu HL, and Huang YL. (2009). On-line microdialysis coupled with liquid chromatography for biomedical analysis. J. Chromatogr. Sci 47, 624–630. [DOI] [PubMed] [Google Scholar]
- Cheng L, Duan B, Huang T, Zhang Y, Chen Y, Britz O, Garcia-Campmany L, Ren X, Vong L, Lowell BB, et al. (2017). Identification of spinal circuits involved in touch-evoked dynamic mechanical pain. Nat. Neurosci 20, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descalzi G, Mitsi V, Purushothaman I, Gaspari S, Avrampou K, Loh YE, Shen L, and Zachariou V. (2017). Neuropathic pain promotes adaptive changes in gene expression in brain networks involved in stress and depression. Sci. Signal 10, Published online March 21, 2017 10.1126/scisignal.aaj1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators (2017). Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1211–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvarci S, and Pare D. (2014). Amygdala microcircuits controlling learned fear. Neuron 82, 966–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields H. (2004). State-dependent opioid control of pain. Nat. Rev. Neurosci 5, 565–575. [DOI] [PubMed] [Google Scholar]
- Gehrlach DA, Dolensek N, Klein AS, Roy Chowdhury R, Matthys A, Junghänel M, Gaitanos TN, Podgornik A, Black TD, Reddy Vaka N, et al. (2019). Aversive state processing in the posterior insular cortex. Nat. Neurosci 22, 1424–1437. [DOI] [PubMed] [Google Scholar]
- Greicius MD, Flores BH, Menon V, Glover GH, Solvason HB, Kenna H, Reiss AL, and Schatzberg AF. (2007). Resting-state functional connectivity in major depression: abnormally increased contributions from subgenual cingulate cortex and thalamus. Biol. Psychiatry 62, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans EJ, van Marle HJ, Ossewaarde L, Henckens MJ, Qin S, van Kesteren MT, Schoots VC, Cousijn H, Rijpkema M, Oostenveld R, and Fernández G. (2011). Stress-related noradrenergic activity prompts largescale neural network reconfiguration. Science 334, 1151–1153. [DOI] [PubMed] [Google Scholar]
- Hieronymus F, Emilsson JF, Nilsson S, and Eriksson E. (2016). Consistent superiority of selective serotonin reuptake inhibitors over placebo in reducing depressed mood in patients with major depression. Mol. Psychiatry 21, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Gadotti VM, Chen L, Souza IA, Huang S, Wang D, Ramakrishnan C, Deisseroth K, Zhang Z, and Zamponi GW. (2019). A neuronal circuit for activating descending modulation of neuropathic pain. Nat. Neurosci 22, 1659–1668. [DOI] [PubMed] [Google Scholar]
- Janak PH, and Tye KM. (2015). From circuits to behaviour in the amygdala. Nature 517, 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keysers C, Kaas JH, and Gazzola V. (2010). Somatosensation in social perception. Nat. Rev. Neurosci 11, 417–428. [DOI] [PubMed] [Google Scholar]
- Knowland D, Lilascharoen V, Pacia CP, Shin S, Wang EH, and Lim BK. (2017). Distinct Ventral Pallidal Neural Populations Mediate Separate Symptoms of Depression. Cell 170, 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Farb C, and Ruggiero DA. (1990). Topographic organization of neurons in the acoustic thalamus that project to the amygdala. J. Neurosci 10, 1043–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, and Li B. (2013). Experience-dependent modification of a central amygdala fear circuit. Nat. Neurosci 16, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Latremoliere A, Li X, Zhang Z, Chen M, Wang X, Fang C, Zhu J, Alexandre C, Gao Z, et al. (2018). Touch and tactile neuropathic pain sensitivity are set by corticospinal projections. Nature 561, 547–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks DM, Shah MJ, Patkar AA, Masand PS, Park GY, and Pae CU. (2009). Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr. Neuropharmacol 7, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fernandez M, Jamison S, Robin LM, Zhao Z, Martin ED, Aguilar J, Benneyworth MA, Marsicano G, and Araque A. (2017). Synapse-specific astrocyte gating of amygdala-related behavior. Nat. Neurosci 20, 1540–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri R, Quiton RL, Lucas JM, Murray PD, Thompson SM, and Keller A. (2009). Zona incerta: a role in central pain. J. Neurophysiol 102, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Schwalb JM, and Kennedy SH. (2005). Deep brain stimulation for treatment-resistant depression. Neuron 45, 651–660. [DOI] [PubMed] [Google Scholar]
- Milligan ED, and Watkins LR. (2009). Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci 10, 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, Witztum J, Shaver DC, Rosenthal DL, Alway EJ, et al. (2019). Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364, Published online April 12, 2019 10.1126/science.aat8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T, McClain SP, Batia LM, Pellegrino M, Wilson SR, Kienzler MA, Lyman K, Olsen AS, Wong JF, Stucky CL, et al. (2015). HTR7 Mediates Serotonergic Acute and Chronic Itch. Neuron 87, 124–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova E, and Porreca F. (2014). Reward and motivation in pain and pain relief. Nat. Neurosci 17, 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, and Hyman SE. (2010). Animal models of neuropsychiatric disorders. Nat. Neurosci 13, 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Leone A, Rubio B, Pallardó F, and Catalá MD. (1996). Rapid-rate transcranial magnetic stimulation of left dorsolateral prefrontal cortex in drugresistant depression. Lancet 348, 233–237. [DOI] [PubMed] [Google Scholar]
- Penzo MA, Robert V, Tucciarone J, De Bundel D, Wang M, Van Aelst L, Darvas M, Parada LF, Palmiter RD, He M, et al. (2015). The paraventricular thalamus controls a central amygdala fear circuit. Nature 519, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessoa L. (2008). On the relationship between emotion and cognition. Nat. Rev. Neurosci 9, 148–158. [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Edwards SE, Iyengar S, Bymaster F, Clark M, and Katon W. (2009). Depression and pain. Front. Biosci 14, 5031–5051. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, and Chattarji S. (2009). Stress, memory and the amygdala. Nat. Rev. Neurosci 10, 423–433. [DOI] [PubMed] [Google Scholar]
- Rudy TE, Kerns RD, and Turk DC. (1988). Chronic pain and depression: toward a cognitive-behavioral mediation model. Pain 35, 129–140. [DOI] [PubMed] [Google Scholar]
- Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Gründemann J, Fadok JP, Müller C, Letzkus JJ, and Lüthi A. (2014). Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 81, 428–437. [DOI] [PubMed] [Google Scholar]
- Tervo DG, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron 92, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ. (2010). Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci 11, 823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, and Lüthi A. (2015). Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci 16, 317–331. [DOI] [PubMed] [Google Scholar]
- Tovote P, Esposito MS, Botta P, Chaudun F, Fadok JP, Markovic M, Wolff SB, Ramakrishnan C, Fenno L, Deisseroth K, et al. (2016). Midbrain circuits for defensive behaviour. Nature 534, 206–212. [DOI] [PubMed] [Google Scholar]
- Tracey I, and Mantyh PW. (2007). The cerebral signature for pain perception and its modulation. Neuron 55, 377–391. [DOI] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, and Deisseroth K. (2011). Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature 471, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen N, Perchet C, Magnin M, Creac’h C, Convers P, Nighoghossian N, Mauguiére F, Peyron R, and Garcia-Larrea L. (2016). Thalamic pain: anatomical and physiological indices of prediction. Brain 139, 708–722. [DOI] [PubMed] [Google Scholar]
- Wang GQ, Cen C, Li C, Cao S, Wang N, Zhou Z, Liu XM, Xu Y, Tian NX, Zhang Y, et al. (2015). Deactivation of excitatory neurons in the prelimbic cortex via Cdk5 promotes pain sensation and anxiety. Nat. Commun 6, 7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR, Lyon DC, Barnard RJ, Mori T, Finke S, Conzelmann KK, Young JA, and Callaway EM. (2007). Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 53, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, and Ma Q. (2007). Nociceptors–noxious stimulus detectors. Neuron 55, 353–364. [DOI] [PubMed] [Google Scholar]
- Zeng WB, Jiang HF, Gang YD, Song YG, Shen ZZ, Yang H, Dong X, Tian YL, Ni RJ, Liu Y, et al. (2017). Anterograde monosynaptic transneuronal tracers derived from herpes simplex virus 1 strain H129. Mol. Neurodegener 12, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Cai YQ, Zou F, Bie B, and Pan ZZ. (2011). Epigenetic suppression of GAD65 expression mediates persistent pain. Nat. Med 17, 1448–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Jin Y, Meng Q, Zhu X, Bai T, Tian Y, Mao Y, Wang L, Xie W, Zhong H, et al. (2019). A neural circuit for comorbid depressive symptoms in chronic pain. Nat. Neurosci 22, 1649–1658. [DOI] [PubMed] [Google Scholar]
- Zhuo M. (2008). Cortical excitation and chronic pain. Trends Neurosci 31, 199–207. [DOI] [PubMed] [Google Scholar]
- Zingg B, Chou XL, Zhang ZG, Mesik L, Liang F, Tao HW, and Zhang LI. (2017). AAV-Mediated Anterograde Transsynaptic Tagging: Mapping Corticocollicular Input-Defined Neural Pathways for Defense Behaviors. Neuron 93, 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.