

Abstract
Purpose of the Review:
Acute kidney injury (AKI) remains a major unmet medical need and associates with high morbidity, mortality, and healthcare costs. Among survivors, long-term outcomes of AKI can include development of chronic kidney disease (CKD) or progression of pre-existing CKD. In this review, we focus on ongoing efforts by the AKI community to understand the human AKI to CKD continuum, with an emphasis on the cellular stress responses that underlie AKI and the maladaptive responses that persist in the acute to chronic phase. The emphasis is on work that has been published in the past year in this rapidly expanding field.
Recent Findings:
Recent studies in preclinical models highlight the importance of mitochondrial dysfunction, cell death and inflammation in the underlying pathogenesis of AKI. These pathogenic mechanisms can resolve with adaptive kidney repair but persist in maladaptive repair that leads to progressive chronic disease. The complexity and interconnections of these pathways involve cross-talk between the tubular epithelium, endothelium and interstitial compartments.
Summary:
Approaches which lessen or counteract these cellular responses represent novel strategies to prevent AKI and stop or slow down the progression to CKD.
Keywords: acute renal failure, senescence, cellular stress, maladaptive repair, AKI to CKD transition
INTRODUCTION
Despite the combined efforts of the acute kidney injury (AKI) community to harmonize clinical definition and staging, advance novel renal biomarkers for clinical use, improve clinical trial design and methodology, as well as advance our understanding of the pathophysiology of AKI, no pharmacological agent exists for prevention or treatment of acute tubular injury. Since AKI is defined by a change in creatinine or decrease in urine output, it can be caused by a number of tubular and vascular-interstitial as well as glomerular etiologies. In this short review, we will focus on processes that affect predominantly the tubular/interstitial compartments of the kidney. AKI impacts approximately 13.3 million patients per year worldwide [1] and associates with high morbidity, healthcare costs, and mortality---approximately 1.7 million deaths per year globally [1]. Moreover, among survivors, long-term outcomes of AKI can include the development of chronic kidney disease (CKD) and end-stage renal disease (ESRD) or acceleration of pre-existing CKD to ESRD [2, 3].
The AKI community has focused on clinical translation in an attempt to develop effective therapies. The National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK) recently proposed an overarching research strategy to overcome translational barriers in AKI which integrates the collective efforts of the AKI community [4–7] in order to facilitate the development and testing of therapies that could meaningfully improve clinical outcomes [8*]. Using a reverse translational medicine approach starting with the patient first, a path forward has been defined which includes: 1) a better understanding of human AKI, achieved by applying ‘omic technologies (i.e. transcriptomics, proteomics, genomics, epigenetics, metabolomics) to kidney biopsy tissue combined with analyses of blood and urine; 2) application of this information to expansion of efforts to identify AKI endophenotypes that then could be used for patient stratification in clinical trials to optimize response to treatment; and 3) integration of knowledge on human AKI with refined new and existing animal models which would include disease stages and comorbidities common in AKI patients.
There has been progress in these areas. The Kidney Precision Medicine Project has been funded by the NIDDK to obtain and interrogate kidney biopsy samples from patients with AKI (and CKD) in order to determine disease subgroups, generate a kidney tissue atlas to catalog the various kidney cell types and elucidate biological pathways that are drivers of disease in order to identify promising drug targets [9]. ‘Omic technologies are being used to probe kidney biopsy tissue [10*, 11–13], even though there remain practical issues to safely obtaining kidney tissue from AKI patients at a time in the disease process when tissue interrogation will be most informative [14, 15]. Wu et al 2018 [12**] are the first to use single cell RNA sequencing of human kidney biopsy samples from healthy adult and renal transplant patients to simultaneously measure expression of thousands of genes in thousands of cells to allow molecular characterization of cellular responses. Single nucleus RNA sequencing may offer additional advantages such as detection of rare or fragile kidney cell types, and the ability to use archival frozen tissue and analysis of inflamed fibrotic kidney [10*]. These technologies may help redefine AKI at the cellular and molecular level [16]. Endophenotypes of AKI representing two common, but disparate clinical syndromes—AKI due to volume depletion and AKI due to intrinsic kidney injury—have very different transcriptional programs [17**]. Lastly, a concerted effort is underway to have preclinical models recapitulate human AKI. Comorbidities, such as aging or pre-existing CKD, are being incorporated into existing rodent models [18] and large animals are being used with greater frequency to probe pathophysiology and to test novel therapies for proof-of-concept [19, 20]. Recently, the human kidney organoid 3D culture system, which consists of multiple cell types with the characteristics of podocytes, proximal tubules, loops of Henle and distal convoluted tubule in a contiguous arrangement resembling nephrons in vivo [21, 22**], has been manipulated to model kidney injury to probe pathophysiology of different cell types simultaneously and to evaluate potential therapies [23**].
Our understanding of the pathogenesis of AKI continues to advance. Herewith the goal of identifying novel therapeutics for prevention and treatment. In this article, we highlight recent findings from studies in vitro and in vivo designed to better understand the human AKI to CKD continuum, with an emphasis on the cellular stress responses that underlie AKI, primarily mitochondrial dysfunction, cell death and inflammation as well as consequent adaptive and maladaptive repair. Depending on the severity of the injury or the presence of pre-existing CKD, the inability of the repairing kidney to launch adaptive responses allows maladaptive repair to persist leading to the onset or progression of a chronic phase of injury, which is characterized by persistent injury, inflammation and fibrosis. The contribution of these cellular stress responses to chronic disease will be discussed.
MITOCHONDRIAL DYSFUNCTION
Mitochondria have been implicated in the pathobiology of AKI for many decades but there has been renewed focus on this cellular organelle in the pathogenesis of AKI and consequent CKD [24, 25]. Mitochondrial fragmentation has been implicated in cell death in kidney as well as other organs. Prevention of mitochondrial fragmentation by proximal tubule–specific deletion of dynamin-related protein 1 (DRP1), a mitochondrial fission protein that constricts and cleaves mitochondria leading to fragmentation, prevented renal ischemia-reperfusion injury and promoted epithelial recovery [26]. Moreover, deletion of DRP1 in proximal tubules after ischemia-reperfusion attenuated progressive kidney injury and fibrosis [26], suggesting that proximal tubule DRP1 increases kidney susceptibility to AKI and its activation has long term consequences in maladaptive repair. It has been also recently reported [27] that microRNA-668 is upregulated by hypoxia-inducible factor (HIF-1) and serves to reduce mitochondrial fragmentation, tissue injury and renal apoptosis. miR-668 negatively regulates MTP18, a protein involved in fission of mitochondria.
In contrast to mitochondrial fission, mitochondrial biogenesis has been linked to organ protection, including the kidney. PGC-1α is the primary regulator of mitochondrial biogenesis [28] and its expression and activation are protective in models of renal ischemia reperfusion injury and nephrotoxicity [29, 30**]. PGC-1α also protects from Notch-induced kidney fibrosis by restoring mitochondrial morphology, and improving fatty acid oxidation, which is the preferred substrate for kidney energy production, and ameliorating kidney fibrosis [31].
PGC-1α is renoprotective likely because it drives the biosynthesis of nicotinamide adenine dinucleotide (NAD), which plays an essential role in energy metabolism and adaptive stress responses [32]. The depletion of NAD is a fundamental feature of aging and associates with age related renal, cardiac and neurodegenerative diseases [32, 33]. NAD biosynthesis is impaired in preclinical models of AKI and in patients at risk to develop AKI [34**] due to reduced levels of the enzyme quinolinate phosphoribosyltransferase (QPRT), which leads to elevated urinary levels of the precursors quinolinate and tryptophan [34**]. Increased ratios of urinary quinolinate to tryptophan (uQ/T) has been proposed to be a potential new biomarker to identify patients at risk of AKI and for predicting adverse clinical outcomes [34**]. Moreover, augmenting renal NAD metabolism with oral nicotinamide (a precursor of NAD) has been reported to prevent human AKI [34**], as it did in mouse models [30*], providing a potential new therapy for AKI prevention. Further support for targeting the NAD pathway comes from a recent study demonstrating that inhibition of α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD), an enzyme that controls cellular NAD levels, is protective against AKI in both the cisplatin and renal-ischemia reperfusion models. Genetic and pharmacological inhibition of ACMSD increases NAD synthesis, thereby enhancing sirtuin activity and mitochondrial function to mediate protection [35**].
Targeting isocitrate dehydrogenase 2, a key enzyme in the citric acid cycle required for maintaining mitochondrial redox balance, may also have clinical utility. When deleted, mitochondrial damage, oxidative stress, and cell death are worse following renal ischemia reperfusion [36], whereas delivery of this enzyme by gene transfer attenuates renal injury, and increases mitochondrial membrane potential, respiratory capacity and intracellular ATP levels, mimicking the effects of ischemic preconditioning [37]. Targeting microRNA-709 (miR-709), which impairs mitochondrial biogenesis by negatively regulating the mitochondrial transcription factor A (TFAM), may also be beneficial. Not only does the expression of miR-709 correlate with the severity of kidney injury in patients with AKI, but antagonizing miR-709 attenuates mitochondrial dysfunction and kidney injury in the cisplatin model [38].
Recently, a mechanism for the coordinated regulation of renal mitochondrial metabolism and the reabsorptive functions of the kidney has been uncovered involving the master oxidative nuclear receptor, estrogen-related receptor gamma (ERRy) in epigenomics programming [39*]. ERRy directly binds to and activates transcription of a large number of genes important for mitochondrial metabolism and fatty acid oxidation in neurons, heart and skeletal muscle cells [40, 41]. In addition to directly regulating oxidative phosphorylation and fatty acid oxidation in the kidney, ERRy cooperates with hepatocyte nuclear factor 1 beta (HNF1β) to activate the expression of renal reabsorption genes, and in this way links energy metabolism to tubular reabsorption [39*].
CELL DEATH PATHWAYS
Besides the well-established role of mitochondria in cellular metabolism, regulation of cell death is another major function of these organelles. In addition to apoptosis, recent evidence points to regulated necrosis and its pathways---necroptosis, ferroptosis, mitochondrial permeability transition-—as mediators of AKI and its consequent progressive CKD [42]. While tubular cell death is an important feature of many forms of AKI, endothelial injury and vascular rarefaction are also key features. In a recent manuscript [43*] the authors found that in the absence of the pro-apoptotic enzyme, caspase-3, ischemia of the mouse kidney resulted in higher tubular injury scores, upregulation of the necroptosis marker receptor-interacting serine/threonine protein kinase 3 (RIPK3 ) but reduced microvascular loss [43*] . Three weeks after injury the caspase-3 deleted mice had less vascular rarefaction and less evidence for fibrosis. This important study supports the key role of microvascular injury and vascular rarefaction as perhaps being more critical than tubular injury in defining the long-term consequences of the injury [43*]. It also highlights that under the experimental conditions used in this study, the tubular epithelium can be re-directed to necroptosis when apoptosis is blocked, whereas the endothelium appears to engage only the apoptotic pathway for cell death.
Various modes of cell death among cellular compartments highlight the complexity and the potential for cross-talk among cell death pathways in AKI. Necroptosis is mediated through the necrosome, a signaling complex containing RIPK1, RIPK3, and mixed lineage kinase domain-like (MLKL) protein. Phosphorylation of MLKL by RIPK3 directly disrupts the plasma membrane bilayer. Release of danger-associated molecular patterns (DAMPs) and activation of NOD-like receptors (NLRs), including the NLRP3 inflammasome, trigger inflammation. Inhibition of these components is protective in models of acute tubular injury both in vitro and in vivo [44], whereas activation contributes to persistence of renal injury [45] and progression of AKI to CKD [46]. Less is known about ferroptosis, an iron-dependent cell death pathway in which lipid peroxidation ruptures the plasma membrane. Glutathione peroxidase 4 (GPX4) prevents ferroptosis and lipid peroxidation because genetic depletion of GPX4 in renal tubules results in lethal tubular necrosis [47]. Moreover, in experimental AKI, when ferroptosis or necroptosis is compromised, there is compensation of one regulated necrosis pathway for another [48], although this is not well understood. Ferroptosis appears to be the main pathway of regulated necrosis in folic-acid induced nephropathy [49]. There are other forms of regulated necrosis that have been implicated in AKI including mitochondrial permeability transition-mediated regulated necrosis, parthanatos and pyroptosis but less is known about the relative importance and regulation of these processes in AKI [42].
INFLAMMATION
A hallmark of regulated necrosis is necroinflammation, in which kidney injury and inflammation are reciprocally enhanced in an auto-amplification loop [50, 51]. Necroinflammation can be initiated by a few necrotic cells that activate the innate immune system, which subsequently leads to necrosis of more cells, triggering more inflammation in a process that may lead to organ failure. In renal injury, damaged, surviving cells also release pro-inflammatory cytokines and chemokines [52] which, in combination with resident dendritic cells and macrophages, as well as the innate immune response [53–55] of infiltrating neutrophils, monocytes and lymphocytes, intensify the inflammatory milieu, rendering inflammation a key pathophysiologic component of AKI.
Recent attention has focused on the contribution of members of the IL-1 superfamily of cytokines in both the acute and chronic phases of AKI. IL-36, a newly named member of the IL-1 superfamily, has been reported to mediate renal ischemia-reperfusion injury. Knock-out of the IL-36 receptor leads to reduced loss in renal function, cytokine expression, and tubular injury, presumably through diminished activation of NF-κB and Erk phosphorylation [56]. In the unilateral ureteral obstruction and folic acid-induced AKI models, overexpression of IL-36α associates with renal dysfunction and the progression of tubulointerstitial inflammation [57, 58]. Furthermore, in AKI patients, IL-36α is elevated in urine and biopsy samples [56]. Thus, inhibition of this pathway may serve as a potential new therapeutic approach. The data are less clear with IL-33, another member of the IL-1 superfamily, which can exacerbate [58, 59] or prevent [60, 61] renal injury, depending on the timing of administration. In response to renal ischemia-reperfusion, IL-33 is immediately released from the microvascular endothelium, where it is constitutively expressed, to promote invariant natural killer T cell (iNKT) recruitment and cytokine production, resulting in neutrophil infiltration and renal injury [58]. Mice lacking IL-33 or its specific receptor, ST2, are protected [58]. In agreement with these data, IL-33 treatment exacerbates AKI following cisplatin administration [59] and worsens renal fibrosis following renal ischemia reperfusion [58]. In contrast, pretreatment with recombinant IL-33 before induction of renal insult prevents tissue injury and mortality [60] by inducing expansion of kidney-resident type 2 innate lymphoid cells [61], suggesting a protective role of the IL-33-ILC2 axis. Clearly, further research is required to clarify the role of IL-33 signaling in the pathogenesis of AKI.
In addition to a potential role for members of the IL-1 superfamily in the injury phase, a novel mechanism has been uncovered for IL-1β in the initiation and progression of tubulointerstitial fibrosis [23**]. Drawing on the identification of metabolic gene signatures associated with CKD progression and tubulointerstitial fibrosis from a cohort of patient kidney biopsies, a combination of in vitro and in vivo models was used to demonstrate that IL-1β activates kidney resident stromal cells, inducing a shift to glycolytic metabolism which supports stromal cell proliferation and deposition of fibrotic matrix, through a mechanism that involves stabilization of the oncoprotein MYC. IL-1β driven MYC accumulation is impaired by the interleukin-1 receptor-associated kinase 4 (IRAK4) small molecule inhibitor, BIIB-IRAK4i, and the MYC inhibitor, (+)-JQ1, reducing tubular injury and abrogating fibrosis. Corroborating data from the human kidney organoid 3D culture system [22**], used for the first time to model results from in vitro human kidney stromal cells and in vivo mouse models of kidney injury, further support that inhibiting the IL-1β pathway may be a new therapeutic approach to limit fibrosis in the progression of AKI to CKD.
CELL SENESCENCE AND REPAIR
Targeting senescence of tubular epithelial cells following renal injury may provide another opportunity to limit fibrosis following AKI [52, 62*, 63**]. Senescent tubular epithelial cells have survived the renal insult but, because of DNA damage and perhaps other stimuli, are in cell cycle arrest and thus cannot engage proliferative pathways to restore the tubular epithelial lining. Senescent cells contribute in an important way to fibrosis. By acquiring the senescence-associated secretory phenotype (SASP), they are responsible for the secretion of growth factors and pro-inflammatory cytokines and chemokines [64], and thus contribute to a microenvironment that promotes fibrosis and inflammation [65]. New data now demonstrate that cyclin G1 (CG1), an atypical cyclin, promotes G2-M arrest in proximal tubular cells, forming target of rapamycin (TOR)-autophagy spatial coupling compartments (TASCCs), which promote profibrotic secretion [63**], similar to the SASP. Prevention of TASCC formation, either in vitro or in vivo, prevents kidney fibrosis whereas deletion of CG1 reduces G2-M arrested cells and TASCC formation.
Wnt9a also has been linked to renal fibrosis and cellular senescence [62*]. In biopsy samples from CKD patients, there is a direct correlation in expression of Wnt9a with renal fibrosis and the cellular senescence marker, p16INK4a. Additionally, overexpression of Wnt9a in the renal ischemia reperfusion model exacerbates fibrosis and tubular senescence, whereas knockdown of Wnt9a suppresses these effects. In vitro, Wnt9a induces epithelial senescence by upregulating the senescence markers, p16INK4a, p19ARF, p53 and p21, while decreasing phosphorylation of retinoblastoma protein. Moreover, Wnt9a induces senescent tubular cells to produce TGF-β1, providing a direct link of Wnt9a senescing cells to interstitial fibrosis.
As indicated previously in this review, endothelial dysfunction likely contributes to tubulointerstitial fibrosis via vascular rarefaction. In addition, the endothelial secretome, derived from dysfunctional endothelial cells, has a profibrogenic signature. The secretome contains ligands of the Wnt-β-catenin and Notch pathways, as well as proteolytic fragments of the glycocalyx which normally lines the vascular endothelium luminal surface and is a crucial regulator of endothelial function and homeostasis. Components of the endothelial secretome activate fibroblasts to form myofibroblasts [66, 67*]. Restoration of the endothelial glycocalyx by sulodexide [68] or liposomal nanocarriers of preassembled glycocalyx restores endothelial homeostasis and in this way may dampen fibrogenic signaling.
In adaptive repair there is the potential reversibility of intracellular signaling pathways that can trigger inflammation and mediate fibrosis [69] related at least in part to the proliferation of non-senescent tubular epithelial cells that have survived the insult as a mechanism to replace lost epithelia, without evidence of a stem cell compartment that plays a significant role [70]. By contrast, when the injury is severe or occurs in the setting of chronic disease, the kidney undergoes maladaptive repair involving a number of known (some of which are described previously in the review) and likely unknown signaling pathways and cell-cell interactions whose relative importance is not well delineated. For example, there is activation of the epidermal growth factor receptor (EGFR)-Akt signaling pathway which plays an essential role in mediating epithelial cell regeneration following AKI. Activation of Yes associated protein (YAP), a downstream effector of the Hippo signaling pathway, leads to cell cycle progression and cell migration [71], processes that are essential for epithelial cell regeneration, whereas deletion of transcriptional coactivator with PDZ binding domain (TAZ), has no effect [71]. In contrast, activation of YAP/TAZ signaling in Gli1+ fibroblasts transform fibroblasts into myofibroblasts which in turn leads to kidney fibrosis [72]. The maladaptive repair has been prevented in part by small molecule stimulators of tubular epithelial cell proliferation [73] and exosomes derived from tubular epithelial cells which improve renal function, inflammation, fibrosis and rarefaction when administered after renal injury [74].
New attention is also being focused on the kidney lymph node (KLN) in the pathogenesis of AKI, particularly the role of fibroblastic reticular cells (FRCs) in the initiation and repair phases of injury following renal ischemia reperfusion. FRCs maintain the integrity of high endothelial venules and produce interconnected fibers of extracellular matrix, allowing the movement of antigens and immune cells within the lymph node [75*]. In response to renal ischemia-reperfusion injury, activation of FRCs drives T cell activation and structural changes in the KLN. When FRCs are depleted, T cell activation is reduced and renal injury is ameliorated. In contrast, repetitive bouts of renal ischemia reperfusion associates with senescence of FRCs, fibrosis of the KLN and renal scarring, which are ameliorated by exogenous administration of FRCs.
CONCLUSION
With a renewed emphasis on understanding human AKI to improve clinical translation, coupled with new technologies that will allow interrogation of patient samples at the cellular, biochemical and molecular levels, and continued insight obtained from “traditional” and newer animal models reflecting more co-morbidities, a greater understanding of human AKI is continually emerging. This undoubtedly will lead to new targets for the translational pipeline. Refined preclinical animal models reflecting the human condition will be used to test novel therapeutic agents. In this review, we have highlighted recent findings relating to the AKI to CKD continuum in the context of organizational initiatives all focused on translating pathophysiological insight, informed by animal and human tissue analyses, to patient therapies. The interconnection and cross-talk among pathways of mitochondrial dysfunction, cell death, inflammation and senescence interacting across vascular, cellular, and interstitial compartments is complex and incompletely understood but offers promise of new therapeutic approaches (Figure 1). Indeed, several potential agents are in clinical trials (www.Clinicaltrials.gov) for the prevention and treatment of AKI, building on lessons learned from recent preclinical and clinical studies. The future appears promising for the AKI patient.
Figure 1.
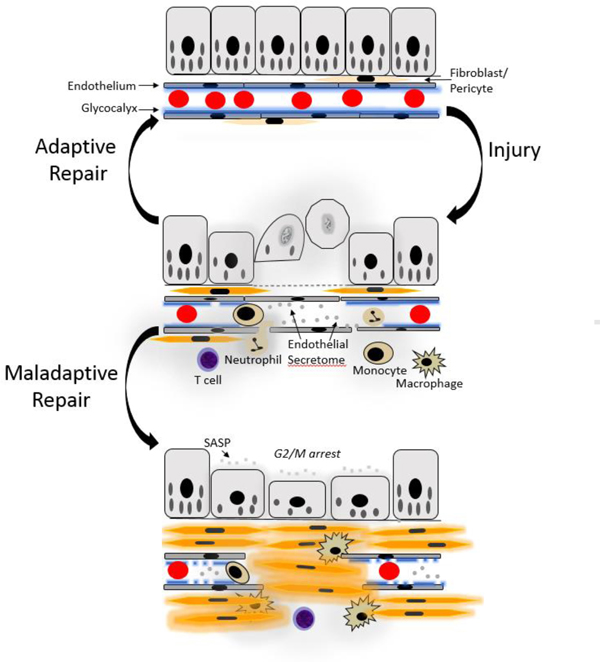
Recent advances in the acute kidney injury to chronic kidney disease continuum address the complex interactions among epithelial, interstitial and vascular compartments. In response to renal injury, mitochondrial fragmentation and loss [26] are linked to activation of cell death pathways, including apoptosis and necroptosis. There is endothelial dysfunction and damage to the glycocalyx [68] which can lead to rarefaction, a long-term consequence. Cytokines and chemokines, containing the newly described members of the IL-1 superfamily, IL-33 and IL-36 [56–61], exacerbate the pro-inflammatory milieu created by injured and dying cells as well as infiltrating immune cells. Adaptive responses lead to kidney repair. Dedifferentiation and proliferation of injured epithelial cells due to activation of gene programs and intracellular signaling pathways, such as YAP/TAZ signaling [71], promote kidney recovery. Mitochondrial biogenesis mediated by PGC-1alpha restores energy metabolism by driving the biosynthesis of NAD [30**, 32, 34**, 35**]. Restoration of the glycocalyx promotes vascular homeostasis [68]. With severe and repeated insults, the reparative process becomes maladaptive. Wnt9a accelerates senescence of tubular epithelial cells, which release cytokines and chemokines, such as TGF-β1, to promote fibrosis [62*]. Cyclin G1 and TASCC regulate G2-M arrest of kidney epithelial cells and fibrotic maladaptive repair [63**]. Components of the endothelial secretome released from damaged vascular cells also are pro-fibrogenic, activating fibroblasts to form myofibroblasts [66, 67*], as does YAP/TAZ signaling in Gli1+ fibroblasts [72]. Moreover, inflammation drives fibrosis. IL-1β activates kidney stromal cells to proliferate and deposit fibrotic matrix through a mechanism that involves stabilization of the oncoprotein MYC [23**]. The complex cross-talk between cellular compartments requires further study.
KEY FINDINGS:
Mitochondrial dysfunction and altered bioenergetics are linked to cell death, persistent nflammation and fibrosis in AKI.
The inability of the injured kidney to successfully launch adaptive responses allows maladaptive repair to persist, ushering in chronic disease.
Complex interactions among epithelial, vascular and interstitial compartments that contribute to AKI underlie adaptive and maladaptive responses and provide opportunities for therapeutic intervention.
There is a growing focus on human tissue to better understand human AKI and the factors driving maladaptive vs adaptive repair.
New therapies are critically needed to prevent and treat the growing population of patients with AKI worldwide
ACKNOWLEDGEMENTS
FINANCIAL SUPPORT AND SPONSORSHIP
This work was supported by NIH grants R37DK039773 and R01DKD072381 (J.V.B.)
CONFLICTS OF INTEREST
AZ is employed by Akebia Therapeutics, Inc. JVB is co-inventor on KIM-1 patents assigned to Partners Healthcare. He is co-founder of Goldfinch Bio. He has grant support from Astellas and has received grant support from Boehringer Ingelheim. He is a consultant for Boehringer Ingelheim, Cadent, Aldeyra and an advisor with equity in Medibeacon Inc, Rubius, Theravance, Sensor Kinesis, Goldilocks, Thrasos and Sentien.
REFERENCES AND RECOMMENDED READING
- 1.Mehta RL, Cerda J, Burdmann EA, et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): A human rights case for nephrology. Lancet 2015; 385:2616–2643. [DOI] [PubMed] [Google Scholar]
- 2.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 2014; 371:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishani A, Xue JL, Himmelfarb J, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 2009; 20:223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Caestecker M, Humphreys BD, Liu KD, et al. Bridging translation by improving preclinical study design in AKI. J Am Soc Nephrol 2015; 26:2905–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basile DP, Bonventre JV, Mehta R, et al. Progression after AKI: Understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol 2016; 27:687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humphreys BD, Cantaluppi V, Portilla D, et al. Targeting endogenous repair pathways after AKI. J Am Soc Nephrol 2016; 27:990–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matejovic M, Ince C, Chawla LS, et al. Renal hemodynamics in AKI: In search of new treatment targets. J Am Soc Nephrol 2016; 27:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *8.Zuk A, Palevsky PM, Fried L, et al. Overcoming translational barriers in acute kidney injury: A report from an NIDDK workshop. Clin J Am Soc Nephrol 2018; 13:1113–1123.This report lays out an overarching strategy for the AKI field in an attempt to address the challenges with successful clinical translation. Understanding human AKI is key and a path forward to achieve this is discussed.
- 9.Norton JM, Ketchum CJ, Narva AS, Star RA, Rodgers GP. Complementary initiatives from the niddk to advance kidney health. Clin J Am Soc Nephrol 2017; 12:1544–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *10.Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 2018.This report compares single-nucleus and single-cell RNA sequencing using kidney tissue from mouse models to highlight potential advantages of the former. These technologies will be used to build the first comprehensive single-cell atlas of healthy and diseased kidneys.
- 11.Malone AF, Wu H, Humphreys BD. Bringing renal biopsy interpretation into the molecular age with single-cell rna sequencing. Semin Nephrol 2018; 38:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **12.Wu H, Malone AF, Donnelly EL, et al. Single-cell transcriptomics of a human kidney allograft biopsy specimen defines a diverse inflammatory response. J Am Soc Nephrol 2018; 29:2069–2080.This report is the first to describe the cutting edge technology of single cell sequencing from biopsy samples of human kidney allografts to molecularly characterize a heterogeneous tissue and to demonstrate feasibility of use.
- 13.Marx D, Metzger J, Pejchinovski M, et al. Proteomics and metabolomics for AKI diagnosis. Semin Nephrol 2018; 38:63–87. [DOI] [PubMed] [Google Scholar]
- 14.Moledina DG, Luciano RL, Kukova L, et al. Kidney biopsy-related complications in hospitalized patients with acute kidney disease. Clin J Am Soc Nephrol 2018; 13:1633–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weisbord SD. Kidney biopsy in hospitalized patients with acute kidney disease: Is there an increased risk? Clin J Am Soc Nephrol 2018; 13:1617–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiryluk K, Bomback AS, Cheng YL, et al. Precision medicine for acute kidney injury (AKI): Redefining AKI by agnostic kidney tissue interrogation and genetics. Semin Nephrol 2018; 38:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **17.Xu K, Rosenstiel P, Paragas N, et al. Unique transcriptional programs identify subtypes of AKI. J Am Soc Nephrol 2017; 28:1729–1740.This is the first report to identify subtypes of AKI based on unique transcriptional programs, differentiating AKI patients with intrinsic renal injury from those with volume depletion.
- 18.Skrypnyk NI, Siskind LJ, Faubel S, de Caestecker MP. Bridging translation for acute kidney injury with better preclinical modeling of human disease. Am J Physiol Renal Physiol 2016; 310:F972–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Post EH, Su F, Taccone FS, et al. The effects of fenoldopam on renal function and metabolism in an ovine model of septic shock. Shock 2016; 45:385–392. [DOI] [PubMed] [Google Scholar]
- 20.O’Kane D, Gibson L, May CN, et al. Zinc preconditioning protects against renal ischemia reperfusion injury in a preclinical sheep large animal model. BioMetals 2018. 31:821–834. [DOI] [PubMed] [Google Scholar]
- 21.Morizane R, Bonventre JV. Kidney organoids: A translational journey. Trends Mol Med 2017; 23:246–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **22.Morizane R, Bonventre JV. Generation of nephron progenitor cells and kidney organoids from human pluripotent stem cells. Nat Protoc 2017; 12:195–207.This report provides detailed methodology to differentiate human pluripotent stem cells into nephron progenitor cells and kidney organoids for use in studies of human kidney development, kidney diseases, nephrotoxicity and kidney regeneration.
- **23.Lemos DR, McMurdo M, Karaca G, et al. Interleukin-1beta activates a MYC-dependent metabolic switch in kidney stromal cells necessary for progressive tubulointerstitial fibrosis. J Am Soc Nephrol 2018; 29:1690–1705.Excellent well-designed series of experiments that uses in vitro and in vivo approaches, as well as human kidney organoids, to demonstrate that in kidney stromal cells, the IL-1β pathway stabilizes MYC, shifting cellular metabolism to glycolysis to drive fibrosis. Inhibition of MYC or the IL-1β pathway prevents fibrosis and injury.
- 24.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol 2017; 13:629–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morigi M, Perico L, Benigni A. Sirtuins in renal health and disease. J Am Soc Nephrol 2018; 29:1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perry HM, Huang L, Wilson RJ, et al. Dynamin-related protein 1 deficiency promotes recovery from AKI. J Am Soc Nephrol 2018; 29:194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei Q, Sun H, Song S, et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J Clin Invest 2018; 128:5448–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanchis-Gomar F, Garcia-Gimenez JL, Gomez-Cabrera MC, Pallardo FV. Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr Pharm Des 2014; 20:5619–5633. [DOI] [PubMed] [Google Scholar]
- 29.Funk JA, Schnellmann RG. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1α activation following ischemia-reperfusion injury. Toxicol Appl Pharmacol 2013; 273:345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **30.Tran MT, Zsengeller ZK, Berg AH, et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016; 531:528–532.Excellent series of studies demonstrating that the mitochondrial regulator, PGC1α, is a crucial determinant of renal recovery. PGC1α regulates NAD (nicotinamide adenine dinucleotide) biosynthesis, which links oxidative metabolism to kidney protection.
- 31.Han SH, Wu MY, Nam BY, et al. PGC-1alpha protects from notch-induced kidney fibrosis development. J Am Soc Nephrol 2017; 28:3312–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang EF, Lautrup S, Hou Y, Croteau DL, Mattson MP, Bohr VA. NAD+ in aging: Molecular mechanisms and translational implications. Trends Mol Med 2017; 23:899–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hershberger KA, Martin AS, Hirschey MD. Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol 2017; 13:213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **34.Poyan Mehr A, Tran MT, Ralto KM, et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat Med 2018; 24:1351–1359.Excellent example of bench to bedside translation. Impaired NAD+ biosynthesis in murine AKI and patients predisposed to AKI is due to reduced levels of the enzyme, quinolinate phosphoribosyltransferase, leading to elevated levels in urine of the ratio of quinolinate to tryptophan, a potential new biomarker for AKI. Oral nicotinamide augments renal NAD+ metabolism and prevents AKI.
- **35.Katsyuba E, Mottis A, Zietak M, et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 2018; 563:354–359.Further support that enhancing NAD biosynthesis is protective in preclinical models of AKI, including liver injury. Genetic and pharmacologic inhibition of the enzyme, α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD), which controls NAD levels, enhances sirtuin activity and mitochondrial function.
- 36.Han SJ, Jang HS, Noh MR, et al. Mitochondrial NADP(+)-dependent isocitrate dehydrogenase deficiency exacerbates mitochondrial and cell damage after kidney ischemia-reperfusion injury. J Am Soc Nephrol 2017; 28:1200–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolb AL, Corridon PR, Zhang S, et al. Exogenous gene transmission of isocitrate dehydrogenase-2 mimics ischemic preconditioning protection. J Am Soc Nephrol 2018; 29:1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Y, Ni J, Chen S, et al. MicroRNA-709 mediates acute tubular injury through effects on mitochondrial function. J Am Soc Nephrol 2018; 29:449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *39.Zhao J, Lupino K, Wilkins BJ, et al. Genomic integration of ERRgamma-HNF1beta regulates renal bioenergetics and prevents chronic kidney disease. Proc Natl Acad Sci U S A 2018; 115:E4910-E4919.This report identifies ERRgamma in the regulation of renal bioenergetics. With HNF1β, ERRgamma simultaneously coordinates kidney reabsorptive functions. ERRgamma thus links bioenergetics to tubular function.
- 40.Fan W, He N, Lin CS, et al. Errgamma promotes angiogenesis, mitochondrial biogenesis, and oxidative remodeling in PGC1alpha/beta-deficient muscle. Cell Rep 2018; 22:2521–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang T, McDonald C, Petrenko NB, et al. Estrogen-related receptor alpha (ERRalpha) and ERRgamma are essential coordinators of cardiac metabolism and function. Mol Cell Biol 2015; 35:1281–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kers J, Leemans JC, Linkermann A. An overview of pathways of regulated necrosis in acute kidney injury. Semin Nephrol 2016; 36:139–152. [DOI] [PubMed] [Google Scholar]
- *43.Yang B, Lan S, Dieude M, et al. Caspase-3 is a pivotal regulator of microvascular rarefaction and renal fibrosis after ischemia-reperfusion injury. J Am Soc Nephrol 2018; 29:1900–1916.This study is the first to report that microvascular endothelial cells undergo apoptosis whereas tubular epithelial cells can undergo either apoptosis or necroptosis. The data highlight inherent differences in cell death pathways among tissue compartments and the potential for complex cross-talk between the various cell types.
- 44.Liu W, Chen B, Wang Y, et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc Natl Acad Sci U S A 2018; 115:E1475-E1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin-Sanchez D, Fontecha-Barriuso M, Carrasco S, et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc Natl Acad Sci U S A 2018; 115:4182–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen H, Fang Y, Wu J, et al. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis 2018; 9:878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator GPX4 triggers acute renal failure in mice. Nat Cell Biol 2014; 16:1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muller T, Dewitz C, Schmitz J, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci 2017; 74:3631–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin-Sanchez D, Ruiz-Andres O, Poveda J, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol 2017; 28:218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mulay SR, Kumar SV, Lech M, Desai J, Anders HJ. How kidney cell death induces renal necroinflammation. Semin Nephrol 2016; 36:162–173. [DOI] [PubMed] [Google Scholar]
- 51.Mulay SR, Linkermann A, Anders HJ. Necroinflammation in kidney disease. J Am Soc Nephrol 2016; 27:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 2010; 16:535–543, 531p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med 2016; 67:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonavia A, Singbartl K. A review of the role of immune cells in acute kidney injury. Pediatric Nephrology 2018; 33:1629–1639. [DOI] [PubMed] [Google Scholar]
- 55.Lee SA, Noel S, Sadasivam M, Hamad ARA, Rabb H. Role of immune cells in acute kidney injury and repair. Nephron 2017; 137:282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishikawa H, Taniguchi Y, Matsumoto T, et al. Knockout of the interleukin-36 receptor protects against renal ischemia-reperfusion injury by reduction of proinflammatory cytokines. Kidney Int 2018; 93:599–614. [DOI] [PubMed] [Google Scholar]
- 57.Ichii O, Kimura J, Okamura T, et al. IL-36alpha regulates tubulointerstitial inflammation in the mouse kidney. Front Immunol 2017; 8:1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferhat M, Robin A, Giraud S, et al. Endogenous IL-33 contributes to kidney ischemia-reperfusion injury as an alarmin. J Am Soc Nephrol 2018; 29:1272–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akcay A, Nguyen Q, He Z, et al. IL-33 exacerbates acute kidney injury. J Am Soc Nephrol 2011; 22:2057–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stremska ME, Jose S, Sabapathy V, et al. IL233, a novel IL-2 and IL-33 hybrid cytokine, ameliorates renal injury. J Am Soc Nephrol 2017; 28:2681–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao Q, Wang Y, Niu Z, et al. Potentiating tissue-resident type 2 innate lymphoid cells by IL-33 to prevent renal ischemia-reperfusion injury. J Am Soc Nephrol 2018; 29:961–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *62.Luo C, Zhou S, Zhou Z, et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J Am Soc Nephrol 2018; 29:1238–1256.This report demonstrates that expression of Wnt9a by tubular epithelial cells drives cellular senescence and production of TGF-β1, which in turn accelerates renal fibrosis through fibroblast activation.
- **63.Canaud G, Brooks CR, Kishi S, et al. Cyclin G1 and TASCC regulate kidney epithelial cell G2-M arrest and fibrotic maladaptive repair. Sci Transl Med 2019; 11. doi: 10.1126/scitranslmed.aav4754This important study is the first to describe the in vivo presence and role that the TASCC plays in fibrotic maladaptive repair following AKI using an arsenal of various in vitro and in vivo approaches. It is the first study to demonstrate the TASCC in human CKD.
- 64.Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol Med 2010; 16:238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clements ME, Chaber CJ, Ledbetter SR, Zuk A. Increased cellular senescence and vascular rarefaction exacerbate the progression of kidney fibrosis in aged mice following transient ischemic injury. PLoS One 2013; 8:e70464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lipphardt M, Dihazi H, Muller GA, Goligorsky MS. Fibrogenic secretome of sirtuin 1-deficient endothelial cells: Wnt, notch and glycocalyx rheostat. Front Physiol 2018; 9:1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *67.Lipphardt M, Song JW, Matsumoto K, et al. The third path of tubulointerstitial fibrosis: Aberrant endothelial secretome. Kidney Int 2017; 92:558–568.This review focuses on endothelial-fibroblast communication and the putative role of the endothelial secretome derived from dysfunctional endothelial cells in reprogramming (myo)fibroblasts.
- 68.Song JW, Zullo JA, Liveris D, Dragovich M, Zhang XF, Goligorsky MS. Therapeutic restoration of endothelial glycocalyx in sepsis. J Pharmacol Exp Ther 2017; 361:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grgic I, Campanholle G, Bijol V, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 2012; 82:172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Humphreys BD, Valerius MT, Kobayashi A, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008; 2:284–291. [DOI] [PubMed] [Google Scholar]
- 71.Chen J, You H, Li Y, Xu Y, He Q, Harris RC. EGFreceptor-dependent YAP activation is important for renal recovery from AKI. J Am Soc Nephrol 2018; 29:2372–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liang M, Yu M, Xia R, et al. YAP/TAZ deletion in Gli(+) cell-derived myofibroblasts attenuates fibrosis. J Am Soc Nephrol 2017; 28:3278–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Monteiro MB, Ramm S, Chandrasekaran V, et al. A high-throughput screen identifies DYRK1a inhibitor ID-8 that stimulates human kidney tubular epithelial cell proliferation. J Am Soc Nephrol 2018; 29:2820–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dominguez JH, Liu Y, Gao H, Dominguez JM 2nd, Xie D, Kelly KJ. Renal tubular cell-derived extracellular vesicles accelerate the recovery of established renal ischemia reperfusion injury. J Am Soc Nephrol 2017; 28:3533–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *75.Maarouf OH, Uehara M, Kasinath V, et al. Repetitive ischemic injuries to the kidneys result in lymph node fibrosis and impaired healing. JCI Insight 2018; 3:1–17.Interesting study which details the potential role of the kidney lymph node and resident fibroblastic reticular cells in the pathogenesis of AKI. This is a relatively new area of study, of which little is known, but these intriguing findings point to the potential of fibroblastic reticular cells as a novel cell therapy for AKI.