

Abstract
Chloramphenicol acetyltransferases (CATs) were among the first antibiotic resistance enzymes identified and have long been studied as model enzymes for examining plasmid‐mediated antibiotic resistance. These enzymes acetylate the antibiotic chloramphenicol, which renders it incapable of inhibiting bacterial protein synthesis. CATs can be classified into different types: Type A CATs are known to be important for antibiotic resistance to chloramphenicol and fusidic acid. Type B CATs are often called xenobiotic acetyltransferases and adopt a similar structural fold to streptogramin acetyltransferases, which are known to be critical for streptogramin antibiotic resistance. Type C CATs have recently been identified and can also acetylate chloramphenicol, but their roles in antibiotic resistance are largely unknown. Here, we structurally and kinetically characterized three Vibrio CAT proteins from a nonpathogenic species (Aliivibrio fisheri) and two important human pathogens (Vibrio cholerae and Vibrio vulnificus). We found all three proteins, including one in a superintegron (V. cholerae), acetylated chloramphenicol, but did not acetylate aminoglycosides or dalfopristin. We also determined the 3D crystal structures of these CATs alone and in complex with crystal violet and taurocholate. These compounds are known inhibitors of Type A CATs, but have not been explored in Type B and Type C CATs. Based on sequence, structure, and kinetic analysis, we concluded that the V. cholerae and V. vulnificus CATs belong to the Type B class and the A. fisheri CAT belongs to the Type C class. Ultimately, our results provide a framework for studying the evolution of antibiotic resistance gene acquisition and chloramphenicol acetylation in Vibrio and other species.
Keywords: Aliivibrio fischeri, antibiotic resistance, biochemical education, chloramphenicol acetyltransferase, functional characterization, gene annotation, integron, Vibrio cholerae, Vibrio vulnificus, xenobiotic acetyltransferase
Abbreviations
- AcCoA
acetyl coenzyme A
- CAT
chloramphenicol acetyltransferase
- Cm
chloramphenicol
- CSGID
Center for Structural Genomics of Infectious Diseases
- DTNB
5,5′‐dithio‐bis‐[2‐nitrobenzoic acid]
- GNATs
Gcn5‐related N‐acetyltransferases
- IMAC
immobilized metal affinity chromatography
- IPTG
isopropyl‐β‐d‐thiogalactoside
- LB
Luria‐Bertani
- LβH
left‐handed β‐helix
- SAT
streptogramin acetyltransferase
- TCEP
tris[2‐carboxyethyl]phosphine
- XAT
xenobiotic acetyltransferase
1. INTRODUCTION
One strategy bacteria use for survival is to interfere with the mode of action of antibiotics by modifying them with some sort of functional group. The genes encoding enzymes that modify antibiotics can be transferred to different species, which increases the likelihood that other bacteria will become resistant to the same types of drugs and allow them to survive under diverse environmental pressures. Many types of antibiotic resistance enzymes perform these modifications and have been identified, including chloramphenicol acetyltransferases (CATs). These enzymes acetylate the antibiotic chloramphenicol (Cm) at the 3′‐hydroxyl position using the acetyl donor acetyl coenzyme A (AcCoA). CATs are known to be important for bacterial antibiotic resistance to Cm because they acetylate the antibiotic and thereby render it unable to bind to the 50S subunit of the bacterial ribosome to inhibit protein translation.1 They have also been widely studied as model systems for examining the evolution, acquisition, and expression of bacterial antibiotic resistance genes.
Since their discovery, CAT classification, gene designation, and nomenclature have been somewhat confusing in the literature. In general, CATs are divided into two distinct classes: the classical/native CATs (Type A) and the novel CATs (Type B).1 A third type of CAT (Type C) has been recently designated by Zhang et al.2 Shaw initially divided the native CATs into two families: constitutive (designated by roman numerals) and inducible (designated by capital letters).3 Type B CATs have also been called xenobiotic acetyltransferases (XATs) in the literature, but there is debate as to whether they should continue to maintain this nomenclature since no alternative substrates other than Cm have been identified.4, 5 It is not currently understood whether the XATs that acetylate Cm actually have different native substrates that have yet to be identified. Regardless, Type B CATs confer low resistance to Cm and are not structural or sequence homologs of classical (Type A) CATs.4 Streptogramin acetyltransferases (SATs), such as virginiamycin acetyltransferases VatA and VatD, were originally grouped collectively with the XAT designation but have since been renamed based on the substrates they acetylate. Although they are structurally related to Type B CATs, they do not acetylate Cm. It is worth noting that streptogramin antibiotics are structurally unrelated to Cm. It is not currently known whether Type C CATs are structurally related to the other CATs, but they do acetylate Cm.2
Cm was introduced to human and veterinary medicine in the early 1950s and many pathogens have developed resistance to this drug. However, some organisms apparently never exposed to Cm have resistance genes due to their acquisition in mobile genetic elements or integrons.1 To explore this concept further in Vibrio species, we selected computationally annotated CAT proteins from two pathogens (Vibrio cholerae (VcCAT) and Vibrio vulnificus (VvCAT)) and one symbiont (Vibrio (Aliivibrio) fischerii (AfCAT)) for functional and structural characterization. The rationale for selecting these specific proteins for study was that they were targeted by the Center for Structural Genomics of Infectious Diseases (CSGID) for structure determination, and relatively little was known about their functions. Previous studies did show that the catB9 gene from V. cholerae (VcCAT) provided resistance toward Cm,6 but to our knowledge the enzyme had not been functionally characterized.
In an age where antibiotic resistance threatens to alter life as we know it, the youth of the next generation will be forced to identify strategies to combat this ever‐increasing problem. Thus, it is vital that students have opportunities to learn about this issue and develop critical thinking and problem‐solving skills so they are prepared to contribute to future solutions. Therefore, we designed an undergraduate laboratory course that incorporated genuine research around the topic of elucidating protein function for enzymes annotated as CATs from three different Vibrio species. Students explored the structures and functions of the selected Vibrio CATs in a culminating biochemistry/biophysical laboratory course as a way to engage them in authentic research on the topic of antibiotic resistance. The goals of this course were to (a) improve student scientific literacy on antibiotic resistance, (b) enhance student critical thinking, problem solving, and laboratory skills prior to graduation, and (c) determine whether the three Vibrio proteins were indeed CATs as annotated.
2. RESULTS
2.1. Sequence comparison of Vibrio proteins
We performed a pairwise sequence alignment between the VcCAT, VvCAT, and AfCAT proteins and other characterized Type B and C CATs and SATs and generated a simple phylogenetic tree. Specifically, we compared our Vibrio protein sequences with those of CatB7 from Pseudomonas aeruginosa (Uniprot ID P26841; locus PA0706), CatB from Elizabethkingia anophelis (Uniprot ID X5KVH4), CatC from Vibrio parahaemolyticus (NCBI Accession WP 025635165), VatA from Staphylococcus aureus (Uniprot ID P26839; plasmid pIP630), and VatD from Enterococcus faecium (Uniprot ID P50870) (Figure 1a). We found the VcCAT and VvCAT proteins were 76% identical and were between 62 and 64% identical to CatB7 of P. aeruginosa, which was previously classified as a Type B CAT. The AfCAT protein was 54% identical to VcCAT and VvCAT proteins, and showed 67% identity to the previously characterized V. parahaemolyticus Type C protein. VcCAT, VvCAT, and AfCAT showed 42% identity to VatA and 40–42% identity to VatD. VcCAT, VvCAT, and AfCAT showed 14–19% identity with Type A CATs (data not shown). Therefore, the VcCAT and VvCAT proteins most closely resemble Type B CATs, whereas the AfCAT most closely resembles Type C. There are two regions that contributed to the predominant sequence differences in the Type B, C, and SAT proteins. The first is an extended N‐terminus of SAT proteins (Figure 1a, orange box) and the second is an insertion in Type C CATs that is not found in either Type B CATs or SATs (Figure 1a, green box). When we examined the phylogenetic tree, we found the VcCAT and VvCAT proteins clustered with other Type B CATs and AfCAT clustered with Type C CATs. Interestingly, the Type C CATs were more closely related to SATs than Type B CATs (Figure 1b).
Figure 1.
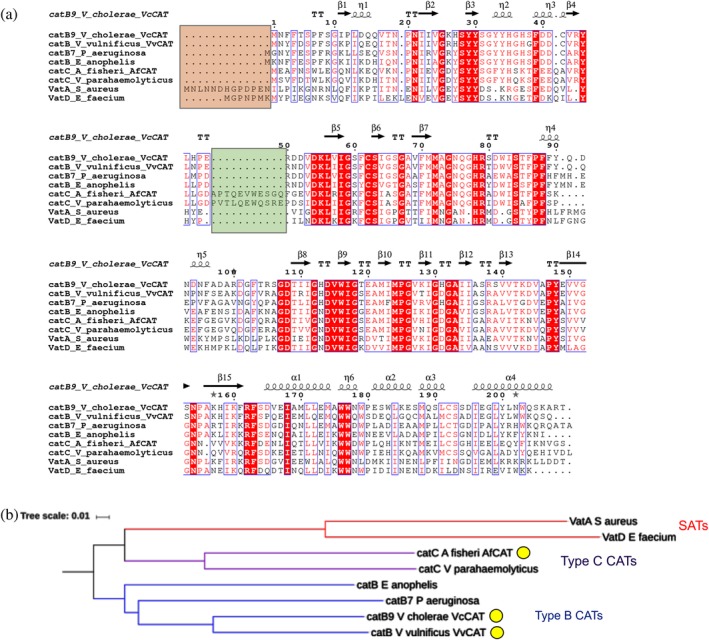
Pair‐wise sequence alignment and phylogenetic tree of CAT enzymes. (a) Sequence alignment comparing Vibrio proteins in this study to other known Type B and C CATs and SATs. The orange box highlights the differences in N‐terminus of SATs compared with Type B and C CATs. The green box highlights the alpha helical insertion of Type C CAT proteins not found in other CATs or SATs. (b) Phylogenetic tree of Type B and C CATs and SATs. Red nodes correspond to SATs, purple nodes correspond to Type C CATs, and blue nodes correspond to Type B CATs. The same sequences were used for the sequence alignment and phylogenetic tree. CATs discussed in this work are highlighted in a yellow circle. Clustal Omega, ESPRIPT, iTOL, and Microsoft PowerPoint were used to create the figure images (see the Materials and Methods section for more details)
2.2. Overall structure and domain organization of Vibrio proteins
To learn whether the VcCAT, VvCAT, and AfCAT proteins adopt a similar structural fold to other CATs with known structures, we crystallized them and compared their structures to other CATs, XATs, and SATs previously determined and deposited into the Protein Data Bank (PDB). We determined a total of seven structures of VcCAT, VvCAT, and AfCAT proteins (Table 1). Four structures of VcCAT were determined in the presence and absence of different ligands: complex with MPD ([4S]‐2‐methyl‐2,4‐pentanediol) (PDB ID: 3EEV), with 1,2‐ethanediol and MPD (PDB ID: 6PUA), with AcCoA and citrate (PDB ID: 6U9C), and with crystal violet (PDB ID: 6PUB). One structure of VvCAT with 1,2‐ethanediol (PDB ID: 6PU9) was determined, and two structures of AfCAT in the presence of MES buffer and acetate (PDB ID: 5UX9) and taurocholate, formic acid, glycerol, and acetate (PDB ID: 6PXA) were also determined.
Table 1.
Data collection and refinement statistics
| Data collection | VcCAT | VcCAT | VcCAT‐CV | VcCAT‐AcCoA | VvCAT | AfCAT | AfCAT‐TCH |
|---|---|---|---|---|---|---|---|
| Wavelength (Å) | 0.9793 | 0.9792 | 0.9792 | 0.9792 | 0.9792 | 0.9792 | 0.9789 |
| Resolution range (Å) (highest resolution) | 32.7–2.60 (2.64–2.60) | 33.6–2.00 (2.03–2.00) | 32.6–2.43 (2.47–2.43) | 47.0–2.20 (2.24–2.20) | 40.8–1.70 (1.73–1.70) | 2.70–47.0 (2.70–2.75) |
1.82–47.0 (1.82–1.84) |
| Space group | P3121 | P3121 | P6322 | P3121 | P31 | P63 | P1 |
| Unit cell dimensions | |||||||
| a, b, c (Å) | 99.97, 99.97, 127.4 | 100.1, 100.1, 128.0 | 83.26, 83.26, 161.8 | 101.2, 101.2, 126.2 | 71.46, 71.46, 108.6 | 147.4, 147.4, 101.2 | 43.52, 121.4, 146.2 |
| α, β, γ (°) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 89.40, 89.90, 87.68 |
| # molecules in ASU | 3 | 3 | 1 | 3 | 3 | 4 | 12 |
| Unique reflectionsa | 22,994 (1,129) | 50,618 (2,510) | 13,039 (621) | 37,660 (1,509) | 65,032 (2,229) | 34,158 (1,724) | 257,772 (9,761) |
| Multiplicitya | 9.7 (9.1) | 7.6 (6.3) | 12.0 (10.1) | 10.2 (9.7) | 3.9 (3.0) | 4.2 (4.1) | 3.4 (3.2) |
| Completenessa (%) | 99.8 (99.6) | 99.9 (100) | 98.9 (99.0) | 97.9 (80.2) | 95.6 (65.8) | 99.1 (99.4) | 96.3 (92.0) |
| I/σ(I)2 | 21.2 (2.5) | 37.2 (2.2) | 20.5 (2.6) | 20.4 (2.5) | 19.4 (3.2) | 21.6 (2.5) | 25.3 (1.4) |
| Wilson B‐factor (Å2) | 48.7 | 37.4 | 40.2 | 48.7 | 24.7 | 38.1 | 33.5 |
| R‐mergea | 0.103 (0.660) | 0.065 (0.969) | 0.132 (0.992) | 0.110 (0.667) | 0.099 (0.245) | 0.055 (0.779) | 0.084 (0.718) |
| Phasing | MR (1XAT) | MR (3EEV) | MR (3EEV) | MR (3EEV) | MR (3EEV) | ||
| Resolution (Å) | 32.7–2.60 | 33.56–2.0 | 32.64–2.43 | 47.0–2.20 | 40.8–1.70 | 47.0–2.70 | 47.0–1.82 |
| CullR_ano (%) | 0.87 | ||||||
| FOM before DM | 0.19 | ||||||
| Correlation coefficientb | 0.56 | 0.62 | 0.57 | 0.71 | 0.58 | 0.47 | |
| Refinement | |||||||
| Resolution (Å) | 32.7–2.61 | 33.6–2.0 | 32.6–2.43 | 47.0–2.20 | 40.8–1.70 | 47.0–2.70 | 47.0–1.82 |
| cc1/2 (highest resolution shell) | n/a | 0.746 | 0.722 | 0.925 | 0.949 | ||
| No. reflections (work/test) | 20,580/1,177 | 48,059/2,356 | 12,436/593 | 37,576/1,753 | 61,659/3,144 | 32,088/1,625 | 244,504/12,605 |
| R work/R free | 0.175/0.242 | 0.163/0.188 | 0.211/0.255 | 0.206/0.251 | 0.194/0.241 | 0.200/0.240 | 0.197/0.238 |
| No. of atoms | |||||||
| Protein | 4,926 | 5,008 | 1,672 | 4,951 | 4,850 | 6,547 | 20,749 |
| Water/others | 218/24 | 356/111 | 83/73 | 36/168 | 311/13 | 86/277 | 776/531 |
| Average B‐factor (Å2) | 44.6 | 44.8 | 44.6 | 70.9 | 39.9 | 46.07 | 50.89 |
| Protein main chain/side chain | 44.0/44.9 | 43.1/45.5 | 42.2/43.5 | 67.4/70.1 | 36.8/41.6 | 44.47/46.87 | 48.28/53.38 |
| Waters/others | 45.5/50.0 | 46.4/75.2 | 43.0/83.2 | 57.8/126.8 | 41.3/46.4 | 35.17/59.81 | 47.14/64.30 |
| RMS deviation | |||||||
| Bond length (Å) | 0.017 | 0.007 | 0.001 | 0.007 | 0.006 | 0.003 | 0.009 |
| Bond angle (°) | 1.61 | 0.769 | 0.404 | 0.916 | 0.764 | 0.568 | 1.010 |
| Ramachandran plot (%) | |||||||
| Favored regions | 95.58 | 98.2 | 95.2 | 96.4 | 96.4 | 94.61 | 96.45 |
| Allowed regions | 3.27 | 1.32 | 4.8 | 3.09 | 3.09 | 5.27 | 3.50 |
| Outliers | 1.15 | 0.48 | 0.0 | 0.51 | 0.51 | 0.12 | 0.05 |
| Clashscore | 12.0 | 1.90 | 2.99 | 4.86 | 4.85 | 5.78 | 4.03 |
| PDB ID | 3EEV | 6PUA | 6PUB | 6U9C | 6PU9 | 5UX9 | 6PXA |
Note: Not including three N‐terminal vector‐derived residues, SNA.
ASU = asymmetric unit; DM = density modification; FOM = figure of merit; PDB = Protein Data bank; RMS = root mean square.
Statistics for the last resolution bin in parentheses.
Molecular replacement.
All three Vibrio proteins crystallized as homotrimers (Figure 2a), and we found by analytical size‐exclusion chromatography that indeed all three proteins eluted as trimers in solution (Figure S1). Each monomer in the trimer is comprised of three domains that protrude from the left‐handed β‐helix (LβH) central core. This core resembles a triangular prism and is constructed from a series of hexapeptide repeats where every sixth residue is either an isoleucine or small aliphatic amino acid. The three domains attached to the LβH central core include the following: (a) a loop between the first and second turns of the LβH that is unstructured in the VcCAT and VvCAT structures (residues 43–54 in the VcCAT structure; Figure 2b,c) but is longer and has alpha helical character in the AfCAT structure (Figure 2d), (b) a long loop that we term the anchor domain that protrudes between the second and third turns of the LβH and helps to stabilize the trimer (residues 71–108 in the VcCAT structure; Figure 2b), and (c) a C‐terminal alpha helical domain (residues 163–209 in the VcCAT structure; Figure 2b) that is connected at the base of the LβH and interacts with anchor domain of the adjacent monomer of the trimer (Figure 2e). A network of interactions between a C‐terminal alpha helical domain of one monomer and the extended anchor domain of a second monomer stabilize the trimer.
Figure 2.
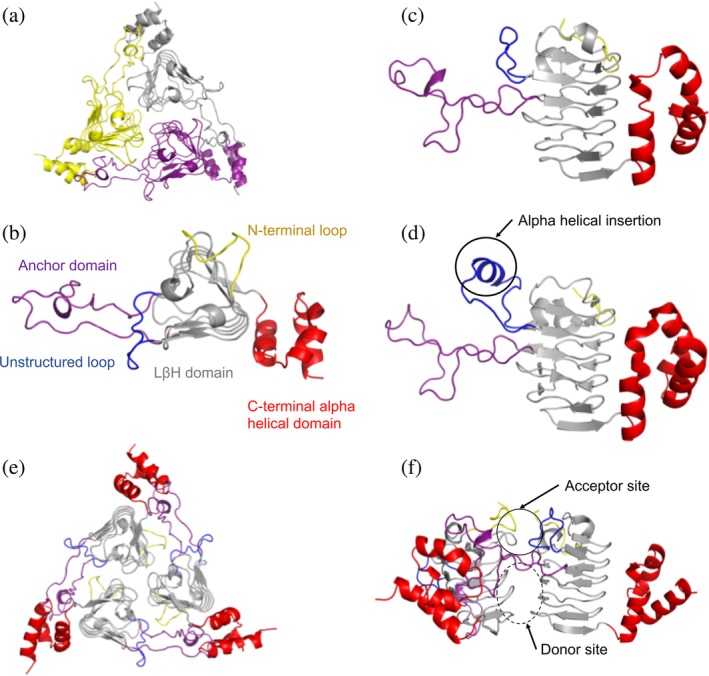
Structure and domain organization of VcCAT, VvCAT, and AfCAT proteins. (a) All three proteins were crystallized as homotrimers. A representative VcCAT structure (PDB ID: 6PUA) is shown with each monomer of the trimer highlighted in different colors. (b) Top view of domains found in each protein monomer; VcCAT structure is used as the representative. The first 10 residues of the N‐terminus are highlighted in yellow, the left‐handed beta‐helix (LBH) core is shown in gray, an unstructured loop (residues 43–54) is highlighted in blue, the anchor domain (residues 71–108) is highlighted in purple, and the C‐terminal alpha helical domain (residues 163–209) is in red. (c) Side view of domains of the VcCAT and VvCAT monomers. Colors are the same as panel B. (d) Side view of domains of the AfCAT (PDB ID: 5UX9) protein. Colors are the same as panel B. The alpha helical insertion unique to the AfCAT protein is circled. (e) Homotrimer of the VcCAT protein with domains highlighted as indicated in Panel B. (f). The interface of two monomers of the VcCAT protein that create the active site. The acceptor site and donor site are circled. All figures were prepared using PyMOL and Microsoft PowerPoint
The active site of each protein contains an AcCoA donor site and an acetyl acceptor site (Figure 2f). The acceptor site of each protein is located between monomers of the trimer in a pocket fashioned from (a) a single face of the triangular prism and a loop created by the first 10–12 N‐terminal residues of one monomer, and (b) a large portion of the extended arm of the anchor domain and the unstructured loop of the LβH of the adjacent monomer. The AcCoA binding site is located beneath the anchor domain of the acceptor site and is at the interface of two LβH between two monomers. Despite multiple attempts, we were unable to crystallize any of the three Vibrio proteins in the presence of Cm or desulfocoenzyme A. We did, however, obtain one structure of the VcCAT in the presence of AcCoA (PDB ID: 6U9C), but a portion of the pantothenate of AcCoA is partially disordered. AcCoA was bound in a similar location to desulfocoenzyme A from a homologous structure of a Type B CAT/XAT from P. aeruginosa (PDB ID: 2XAT).
2.3. Structural comparison of Vibrio proteins to other CATs and SATs
Type A CATs that have been structurally characterized have a different fold from Type B CATs and SATs. Prior to our work, the only structurally characterized Type B CATs were from P. aeruginosa (PDB IDs: 1XAT, 2XAT7) and E. anophelis (PDB ID: 6MFK). Known SAT structures include VatA from S. aureus (PDB IDs: 4MYO, 4HUR, 4HUS8) and VatD from E. faecium (PDB IDs: 1MR7, 1MR9, 1MRL then;9 3DHO; 1KHR, 1KK6, 1KK5, 1KK45). SAT structures typically have a longer N‐terminal loop compared with Type B CATs (Figure 1a). When we compared our structures of the Vibrio proteins with CATs and SATs from the PDB, we found the VcCAT and VvCAT proteins aligned with both these types of proteins, but had a lower RMSD value when compared with that of the P. aeruginosa Type B CAT structures. We did not observe any major structural differences between the VcCAT and VvCAT proteins. The AfCAT structure had an insertion (residues 51–61) not previously observed in other Type B CAT or SAT structures (Figures 1a and 2d). The region that varied was in a comparable location to the unstructured loop of the VcCAT and VvCAT proteins. In the case of the AfCAT protein, the insertion adopted an alpha helical secondary structure. This structural difference along with the sequence and kinetic comparison (as described later) to other CAT proteins led us to the conclusion that the AfCAT protein most closely resembles a Type C CAT and is therefore the first structural representative of this class of proteins.
2.4. Crystal violet and taurocholate bind to VcCAT and AfCAT
It has been previously shown that E. coli Type I CAT enzymes mediate resistance to the antibiotic fusidic acid by binding it competitively with Cm in its acceptor site, but Type II and III CATs do not.10 It was also shown that the Type I CATs bind steroidal antibiotics like bile salts10, 11 and triphenylmethane dyes are competitive inhibitors of Cm and crystal violet in Enterococcus CATs.12, 13 To our knowledge, it is not known whether Type B and C CATs are capable of exhibiting the same type of behavior toward fusidic acid, bile salts, and triphenylmethane dyes. Therefore, we screened all three proteins for crystals in the presence of a variety of compounds (see the Materials and Methods section). None of the crystals we obtain showed fusidic acid bound, but we were able to determine the structures of VcCAT with crystal violet (PDB ID: 6PUB) and AfCAT with taurocholate (PDB ID: 6PXA). We found crystal violet bound at the interface between monomers of the trimer (Figure 3a) in a pocket created by the first 10 N‐terminal residues of the VcCAT protein where a portion of the N‐terminal loop (residues 7–10) are pulled into the Cm acceptor site (Figure 3b). This loop appears to be drawn inward due to Pro7 and Phe8 in a similar location to the aromatic ring of Cm in the 2XAT structure (Figure 3b). Crystal violet binding is stabilized by hydrophobic interactions with aromatic side chains of Phe4 (through a partial ring staking), Phe8, Tyr34, Met1 and Pro7 (Figure 3a). His47 from the neighboring protein chain also provides a longer range weak interaction. The side chains of His47 and Glu49 from the neighboring protein chain are displaced to make a space for crystal violet (Figure 3a).
Figure 3.
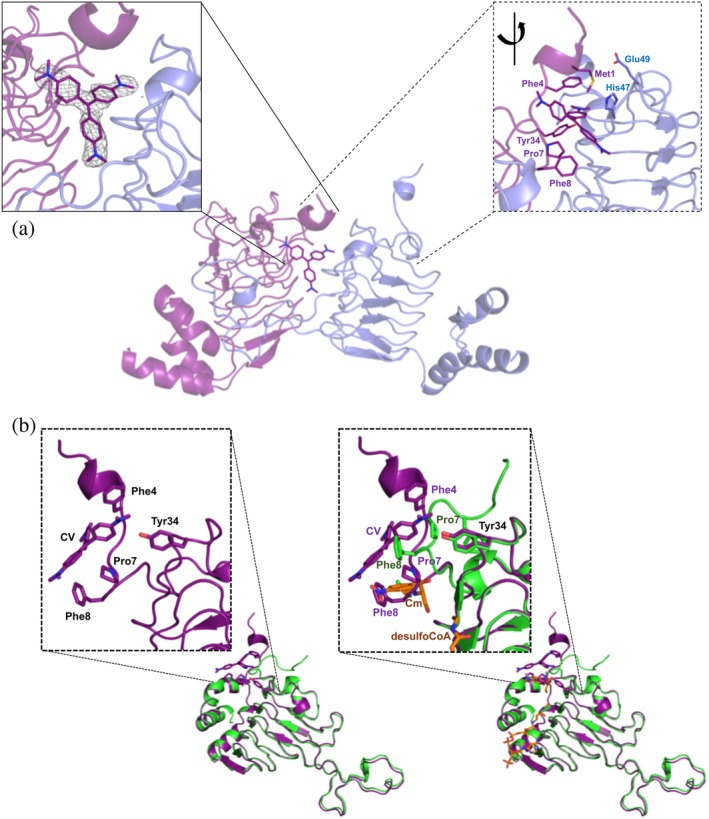
VcCAT structure in the presence of crystal violet. (a) Crystal violet binding site at dimer interface (PDB ID: 6PUB). The VcCAT structure is a trimer, but only two monomers are shown for clarity (one monomer is colored purple and the other is colored slate blue). Crystal violet is shown as purple sticks with its nitrogens in dark blue. Two views of the crystal violet binding site are shown: The left box with solid lines shows the F o weighted omit map in grey mesh surrounding the crystal violet molecule drawn at a contour level of 1.2σ. The right box with dashed lines shows a rotated view of the dimer interface with key residues of the binding site labeled and shown as sticks. (b) Comparison of VcCAT (PDB ID: 6PUA) (green) and VcCAT‐CV (PDB ID: 6PUB) (purple) structures. The ligand binding mode of crystal violet (CV) in the 6PUB structure is shown in the left panel, and the right panel shows the overlay of 6PUB and 6PUA structures. Chloramphenicol (Cm) and desulfocoenzyme A (desulfoCoA) are shown in orange and are modeled from the structure of the xenobiotic acetyltransferase from Pseudomonas aeruginosa (PDB ID: 2XAT). The secondary structures of each crystal structure were defined using Stride (http://webclu.bio.wzw.tum.de/stride/) and altered manually in PyMOL. All figures were prepared using PyMOL and Microsoft PowerPoint
The two AfCAT crystal structures showed either MES buffer (PDB ID: 5UX9) or taurocholate (PDB ID: 6PXA) bound to the acceptor site of the AfCAT protein at the interface between monomers of the trimer (Figure 4). The sulfonic acid moieties of the MES buffer molecules were stabilized by H‐bond interactions with Ser101 on one side of the pocket and Asp111 on the opposite side of the pocket (Figure 4a). Some of the monomers of the structure had two molecules of MES bound, whereas only one MES molecule was observed in other monomers. Binding taurocholate in the acceptor site caused a few local conformational adjustments in the N‐terminal residues and the anchor domain. Upon binding taurocholate, the packing of the three LβH domains at their N‐terminal ends became more tightened and caused a larger tilting of each LβH domain toward the threefold axis of the trimer. The sulfonic acid of taurocholate was H‐bonded to Ser101 like that of one of the MES buffer molecules in the AfCAT structure and the cyclopentanophenanthrene portion of the molecule was loosely packed in a rather shallow hydrophobic pocket (Figure 4b). This pocket was lined by residues Trp7 and Leu8 from the N‐terminus of one monomer, and Leu48 and Phe62 from the small alpha‐helical loop (unstructured loop in VvCAT and VcCAT proteins) and Phe113 from the anchor domain of a second monomer (Figure 4b). A total of three H‐bond interactions were observed between taurocholate and the protein or other ligands in the structure: (a) an H‐bond between O12H of the steroid and the backbone oxygen of Leu8, (b) an H‐bond between to the N24H of the taurine moiety and O2 of a glycerol molecule from the cryoprotectant, and (c) H‐bonds between the side chain of Ser101 and O1S and O2S of the sulfonic acid of taurocholate (Figure 4b).
Figure 4.
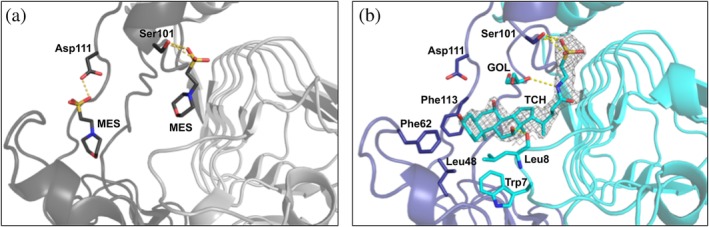
AfCAT structures in the presence of MES and taurocholate. (a) Zoomed view of the structure of AfCAT (PDB ID: 5UX9) in complex with two molecules of MES buffer. The acceptor site is located between two monomers, and residues of the AfCAT protein that form H‐bonds with MES buffer molecules are shown in dark gray sticks. H‐bonds are indicated with yellow dashed lines. The ribbon diagrams of two monomers are shown in dark gray and light gray, respectively. (b) AfCAT‐TCH (PDB ID: 6PXA) structure in complex with taurocholate (TCH) and glycerol (GOL). Ribbon diagrams of two monomers of the trimer are shown in purple and cyan, respectively. Key residues that line the acceptor site of the AfCAT structure are represented as sticks and colored according to the monomer in which they lie. H‐bonds between TCH, GOL, and the AfCAT protein are indicated with yellow dashed lines. The weighted 2F o–F c map in gray mesh surrounds the taurocholate molecule and is drawn at a contour level of 1σ. The secondary structures of each crystal structure were defined using Stride (http://webclu.bio.wzw.tum.de/stride/) and altered manually in PyMOL. All figures were prepared using PyMOL and Microsoft PowerPoint
2.5. Kinetic characterization of vibrio CAT proteins
There are three main types of antibiotics that can be O‐acetylated by various acetyltransferases: Cm, aminoglycosides, and streptogramins. Since the Vibrio proteins were annotated as CATs and it was likely they would perform O‐acetylation, we performed an initial substrate acetylation screening assay toward the three types of antibiotics. We screened activity toward Cm, tobramycin, gentamycin, streptomycin, kanamycin B, and dalfopristin and found Cm was the only substrate for these enzymes. Therefore, we used Cm to further characterize them.
Prior studies with Gcn5‐related N‐acetyltransferases (GNATs) have shown that in many cases the polyhistidine affinity tag affects enzymatic activity,14 but we did not know if the same would be true for CATs, which are not GNATs. Therefore, we compared the activity of all three Vibrio proteins with the tag retained and cleaved from the protein (Table 2, Figure 5). In general, we found the catalytic efficiency increased for all three proteins when the tag was removed: 1.2‐fold for VcCAT, 1.8‐fold for AfCAT, and 2‐fold for VvCAT. The main driver for this increase was the higher apparent affinity for Cm for AfCAT and the increased turnover for the VcCAT and VvCAT proteins (Table 2). Regardless of whether the tag was retained or removed, all three proteins showed a relatively poor apparent affinity for Cm under the described reaction conditions.
Table 2.
Steady‐state kinetic parameters for tagged and cleaved enzymes toward chloramphenicol (Cm)
| Enzyme | K m for Cm (mM) | k cat (s−1) | k cat/K m (M−1 s−1) |
|---|---|---|---|
| Tagged protein | |||
| VcCAT | 0.764 ± 0.037 | 63 | 8.25 × 104 |
| VvCAT | 1.02 ± 0.03 | 43 | 4.22 × 104 |
| AfCAT | 0.803 ± 0.064 | 21 | 2.62 × 104 |
| Cleaved protein | |||
| VcCAT | 0.939 ± 0.056 | 95 | 1.01 × 105 |
| VvCAT | 0.913 ± 0.059 | 78 | 8.54 × 104 |
| AfCAT | 0.659 ± 0.082 | 31 | 4.70 × 104 |
Figure 5.
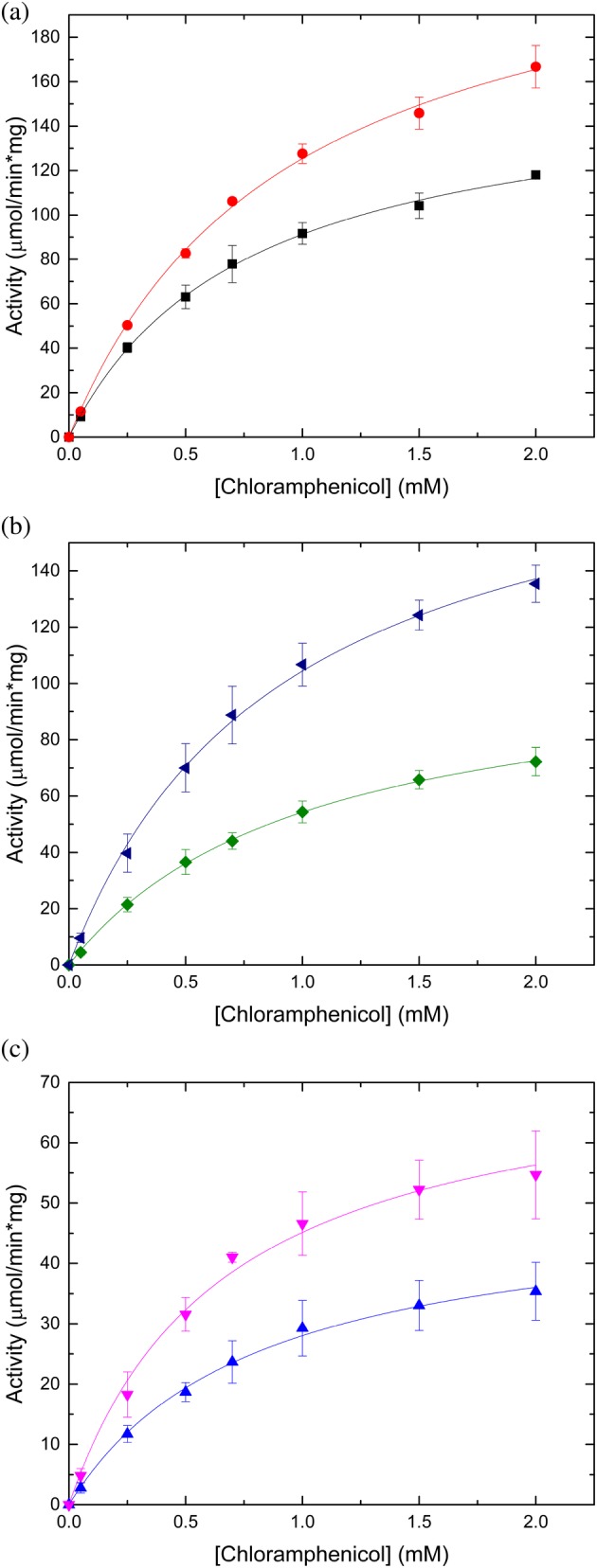
Substrate saturation curves of VcCAT, VvCAT, and AfCAT toward chloramphenicol. All three proteins were kinetically characterized in the presence and absence of the N‐terminal polyhistidine affinity tag; see the Materials and Methods section for more details. (a) VcCAT in the presence (black squares) and absence of tag (red circles). (b). VvCAT in the presence (green diamonds) and absence of tag (blue triangles). (c) AfCAT in the presence (blue triangles) and absence of tag (pink triangles). Figures were prepared using Origin 2016 software
Next, we compared the kinetic parameters of the cleaved Vibrio proteins to previously characterized CATs in the literature. We found all three proteins showed a similar apparent affinity for Cm (939, 913, and 659 μM for VcCAT, VvCAT, and AfCAT, respectively) to that of CatB7 from P. aeruginosa (812 μM), which is at least one order of magnitude lower apparent affinity compared to Type A CATs (Table 3). Since k cat was not reported for many of the previously characterized CATs, we could only compare our results for these parameters with three other CATs: CatIII (Type A) from S. aureus, CAT (Type A) from V. anguillarum, and CatC (Type C) from V. parahaemolyticus. The turnover number for VcCAT, VvCAT, and AfCAT was one order of magnitude lower than the CatIII (Type A) S. aureus enzyme, but one order of magnitude greater than the CatC (Type C) V. parahaemolyticus enzyme under the described reaction conditions (Table 3). Both Type C CATs, CatC from V. parahaemolyticus and AfCAT, showed very similar catalytic efficiencies. We also found the VcCAT exhibited the highest catalytic efficiency of all Type B and Type C enzymes assayed to date, but the catalytic efficiency of VvCAT was only 1.2‐fold lower than VcCAT. In general, we observed the following trend in catalytic efficiency toward Cm across the three types of CATs: Type A CATs show the highest catalytic efficiencies toward Cm (~106) followed by Type B (~105), and Type C CATs exhibit the lowest catalytic efficiencies (~104). Based on our kinetic characterization, we found VcCAT and VvCAT kinetically resembled Type B CATs and AfCAT resembled Type C CATs. Since all three proteins do acetylate Cm, we chose to retain the nomenclature of CAT rather than XAT.
Table 3.
Comparison of kinetic parameters of CATs toward chloramphenicol (Cm)
| Gene | Organism | K m for Cm (μM) | k cat (s−1) | k cat/K m (M−1 s−1) | Reference |
|---|---|---|---|---|---|
| Type A CAT | |||||
| cat | Agrobacterium tumefaciens | 20.5 | 39 | ||
| cat | Clostridium perfringens | 21.5 | 39 | ||
| cat | Diplococcus pneumoniae | 10.0 | 39 | ||
| catI | Escherichia coli | 11.5 | 39 | ||
| catII | Escherichia coli | 18.0 | 39 | ||
| catIII | Escherichia coli | 16.3 | 39 | ||
| catII | Haemophilus parainfluenzae | 17.5 | 39 | ||
| cat | Proteus mirabilis | 31.0 | 39 | ||
| catI | Pseudomonas aeruginosa | 94 | 4 | ||
| catA | Staphylococcus aureus | 2.6 | 40 | ||
| catB | Staphylococcus aureus | 2.7 | 40 | ||
| catC | Staphylococcus aureus | 2.5 | 39 | ||
| catD | Staphylococcus aureus | 2.7 | 40 | ||
| catIII | Staphylococcus aureus | 162 | 209 | 1.29 × 106 | 41 |
| cat | Streptomyces acrimycini | 16.6 | 39 | ||
| cat | Streptococcus agalactiae | 9.3 | 39 | ||
| cat | Vibrio anguillarum | 34.5 | 26 | 7.54 × 105 | 42 |
| Type B CATa | |||||
| catB1 | Pseudomonas aeruginosa | 140 | 4 | ||
| catB3 | Pseudomonas aeruginosa | 156 | 4 | ||
| catB5 | Pseudomonas aeruginosa | 136 | 4 | ||
| catB7 | Pseudomonas aeruginosa | 812 | 4 | ||
| catB9 | VCA0300 (VcCAT) Vibrio cholerae | 939 | 95 | 1.01 × 105 | This work |
| catB | VV20610 (VvCAT) Vibrio vulnificus | 913 | 78 | 8.54 × 104 | This work |
| Type C CAT | |||||
| catC | VFA0790 (AfCAT) Aliivibrio fisherii | 659 | 31 | 4.70 × 104 | This work |
| catC | Vibrio parahaemolyticus VPA‐67 | 145 | 5.6 | 3.86 × 104 | 2 |
Type B CATs are sometimes referred to as xenobiotic acetyltransferases (XATs) in the literature.
3. DISCUSSION
Multiple mechanisms for bacterial antibiotic drug resistance have been identified, but the one relevant to our study includes the acquisition of genes for antibiotic resistance or altered metabolism by integrons. Gram‐negative bacteria use integrons to pick up and express genes found in mobile genetic elements.15 There are two types of integrons: mobile integrons and sedentary chromosomal integrons, including superintegrons.16 Mobile integrons have been associated with antibiotic resistance, whereas superintegrons are chromosomal, are not mobile, and have a very large number of gene cassettes that can include antibiotic resistance determinants (reviewed in Reference 15). While mobile integrons have been shown to have a direct role in antibiotic resistance, less is known about the roles of antibiotic genes found in superintegrons since it appears they may only be functionally expressed under specific pressures or environmental conditions.
Under antibiotic resistance pressure, Rowe‐Magnus et al. have shown that multiple resistance integrons in a conjugative plasmid can recruit genes from superintegrons in V. cholerae, and these genes can then be passed to clinical pathogenic strains.6 This was demonstrated with a phenotypically silent V. cholerae CAT (VCA0300; catB9) gene found in a superintegron—the same gene that encodes the VcCAT protein we characterized in this study. The acquisition of this gene rendered E. coli resistant to Cm compared with strains that lacked the gene, but its distance from the promoter was a critical determinant for whether it conferred resistance (i.e. the further the gene was from the promoter, the more sensitive the bacterium was to Cm). Therefore, catB9 encodes a functional CAT that enables bacteria to be resistant to Cm as long as it is close to the promoter. Baharoglu et al. also showed that during plasmid conjugation in V. cholerae an SOS response is induced, which turns on the expression of an integrase and rearranges placement of the catB9 gene in the superintegron closer to the promoter. Thus, VcCAT becomes functional and directly affects V. cholerae resistance to Cm.17 This is important because it shows that phenotypically silent genes are innately functional, but are only awakened under specific environmental conditions or genetic placement, which could provide an evolutionary advantage to pathogens that acquire these genes. Moreover, it shows that SOS induction as a result of conjugation can confer bacterial antibiotic resistance through the recombination of antibiotic resistance genes at specific locations in cassettes and in genetic mobile elements.
In addition to V. cholerae, other Vibrio species contain superintegrons (e.g., V. parahaemolyticus, V. vulnificus,18 A. fisheri 19); however, the VvCAT and AfCAT genes in our study are not found in superintegrons and appear to be part of their core genome. It has been suggested that many proteins important for virulence and antibiotic resistance in P. aeruginosa are part of its core genome,20 and while we currently do not know whether these cat genes confer Cm resistance to V. vulnificus and A. fisheri, they likely have important functions. Interestingly, the catB9 gene was not found in the V. cholerae El Tor O1 genome prior to the seventh cholera pandemic,6, 21 and its acquisition does not appear to be related to the clinical use of Cm since it is not used to treat cholera. We have shown that the VvCAT protein shares significant sequence, structural, and kinetic similarity to the VcCAT protein, which makes it tempting to speculate that the catB9 gene may have been acquired from another bacterium like V. vulnificus in the environment. Therefore, further studies are needed to discern the path and cause of gene acquisition.
On the surface, it is puzzling why a marine bacterium like V. vulnificus or A. fisheri would have a protein that acetylates Cm when it is likely to have never encountered this antibiotic that is produced by a soil bacterium. Moreover, V. cholerae and V. vulnificus are human pathogens, but A. fisheri is not and yet still retains a protein capable of Cm acetylation. Based on our kinetic characterization, the VcCAT, VvCAT, and AfCAT enzymes exhibit a decrease in catalytic efficiency of up to two orders of magnitude toward Cm compared with Type A CATs, which presents an alternative hypothesis that their native substrates are not Cm. Indeed, some proteins such as β‐lactamases and aminoglycoside acetyltransferases are thought to have native housekeeping functions, but have been shown to contribute to antibiotic resistance due to inherent promiscuity in their active sites (reviewed in Reference 20). It is therefore likely that the Vibrio proteins we characterized have functions beyond Cm acetylation and warrant further studies. Other known LβH fold proteins such as LpxA, LpxD, and GlmU have been implicated in a variety of bacterial cellular processes, including modifying cell wall polysaccharides and lipids, and are clearly important players in the life of a bacterium. These proteins, however, exhibit longer LβHs than the CATs we structurally characterized but provide evidence that molecules other than antibiotics can bind to this type of structural fold.
Our structural studies of these proteins from a variety of Vibrio species have expanded our knowledge of structures of Type B and C CATs and provide a framework for determining their native functions or delving deeper into their ability to modify Cm. Indeed, our structure of AfCAT in the presence of taurocholic acid may provide insight to the type of ligands that can bind to this protein and aid in elucidating a better substrate than Cm. Moreover, similar to how Type I CATs aid in fusidic acid resistance by binding it in their active sites10 and inducible CATs increase their copy numbers,3 AfCAT may sequester taurocholate similarly if the enzyme is overproduced under specific environmental conditions. Although there is still much to be learned about these proteins, our results provide a strong foundation for further exploration into the roles of these proteins in Vibrio species and beyond.
Many undergraduate educators have implemented course‐based undergraduate research experiences in their curriculum as a mechanism for increasing persistence in science, developing critical thinking and problem‐solving skills, and providing more inclusive research experiences for more students than are typically admitted to research laboratories.22, 23, 24 Here, we exposed undergraduate students to an authentic research experience where they were partners in the discovery process and dissemination of results on proteins important in antibiotic resistance. To accomplish this, we formed a collaboration between scientists from the University of Chicago CSGID, Argonne National Laboratory Structural Biology Center (SBC) and undergraduate students at San Francisco State University (SFSU) to explore the structural and biochemical characterization of these proteins.
The workflow of the one‐semester course was as follows. CSGID provided purified protein to SFSU students so they could focus on assay design, kinetic characterization, and structural comparisons. Students performed literature searches on CATs and XATs to determine what they do and why they are important. They then learned what reactions are catalyzed by these enzymes and used this information to search the literature for possible enzyme assays they could use to test if the uncharacterized proteins acetylate chloramphenicol. Based on this information, they determined what solutions they needed to make and learned to design an experiment. Students performed multiple sequence alignments to formulate a hypothesis about their proteins based on sequence and performed structural comparisons of their proteins with others found in the PDB. They also collected kinetic data and compared their results to the literature. At the end of the course, students presented their conclusions regarding how these proteins should be functionally annotated. Finally, they presented their results and conclusions via: (a) poster presentations at the annual student project showcase at SFSU, (b) video conference presentations to scientists at CSGID, and (c) a written draft manuscript. After the semester concluded, some students volunteered to perform additional experiments over the summer and edit the draft manuscript with the instructor for publication.
Although the impact of this pedagogical strategy remains to be seen, the instructor observed an increase in the breadth and depth of student knowledge on topics of antibiotic resistance, protein functional annotation, protein structure, and essential skills of critical thinking, teamwork, and problem solving. However, proper pedagogical research would need to be performed to determine whether there are statistically significant increases in student learning compared to a control course. Regardless, the idea of involving undergraduate students in research on antibiotic resistance is critical since they will certainly be part of finding solutions to this critical global health issue in the future.
4. MATERIALS AND METHODS
4.1. Cloning
The following three genes on chromosome II of each Vibrio species were selected for study: (a) V. cholerae O1 biovar El Tor str. N16961 locus tag VCA0300, Accession NP_232696, UniProtID Q9KMN1; (b) Aliivibrio fischeri ES114 locus tag VF_A0790, Accession YP_206748, UniProtID Q5DZD6; and (c) V. vulnificus CMCP6 locus tag WP_011081553, Accession WP_011081553, UniProtID A0A1V8MQW9 (misannotated as an N‐acetylglutamate synthase). The gene from V. cholerae was cloned into the pMCSG7 vector (N‐terminal polyhistidine tag and TEV protease recognition site: MHHHHHHSSGVDLGTENLYFQ/SNA)25 and genes from A. fischeri and V. vulnificus were cloned into the pMCSG53 vector (N‐terminal polyhistidine tag and TEV protease recognition site: MHHHHHHSSGVDLGTENLYFQ/SNA)26 using ligation‐independent cloning as described earlier.25 Both vectors are ampicillin resistant, and after TEV cleavage the three amino acids SNA remain at the N‐terminus of the protein. All three clones were transformed into E. coli BL21(DE3) Gold (Strategene) cells. In cells containing the pMCSG7 vector, an extra kanamycin‐resistant plasmid encoding three rare tRNAs27 was also introduced. This additional plasmid was not necessary for cells containing the pMCSG53 vector because it contains these rare tRNAs on the same plasmid.26 The single monomer molecular weights of each protein are VcCAT (23.5 kDa), AfCAT (24.4 kDa), and VvCAT (23.4 kDa).
4.2. Sequence alignment and phylogenetic tree
We performed a sequence alignment using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/), which also generated a basic phylogenetic tree using sequences mentioned in the results section. ESPRIPT (http://espript.ibcp.fr/ESPript/ESPript/) was used to generate the final figure and indicate the secondary structural elements of the VcCAT 6PUA structure in relation to the sequence. iTOL (https://itol.embl.de/) was used to generate the figure for the phylogenetic tree.
4.3. Protein expression and purification
Cells were grown over several days in Luria‐Bertani** (LB) medium with 150 μg/ml of ampicillin at 37°C; 30 μg/mL of kanamycin was also added to cultures that contained the V. cholerae clone. On the first day, a 1 ml starter culture was inoculated and grew overnight. The following day, 100 μl of this culture was used to inoculate 50 ml of the overnight culture. 25 ml of this culture was added to 940 ml of LB medium with appropriate antibiotics in 2 L plastic bottles and shaken at 180 rpm until the OD600nm reached 0.8. Cells were then cooled for 50 min in a 4°C incubator. Next, 10 mM K2HPO4 was added to each culture for 10 min, and then protein expression was induced with 0.5 mM IPTG (isopropyl‐β‐d‐thiogalactoside; Sigma Aldrich) and shaken overnight at 18°C. Cells were harvested by centrifugation at 7,000 g in a Sorvall Evolution RC centrifuge, and pellets were resuspended in 5 ml of lysis buffer per 1 g of cells (50 mM HEPES pH 8.0, 500 mM NaCl, 20 mM imidazole, 10 mM beta‐mercaptoethanol, 5% [v/v] glycerol, and protease inhibitor cocktail [cOmplete ULTRA EDTA‐free, Sigma]). Lysozyme (from chicken egg white, lyophilized powder, Sigma Aldrich) was then added at a concentration of 1 mg/ml and cells were frozen at −80°C. The cells were then thawed and sonicated and subjected to centrifugation at 28,000 g for 50 min. The supernatant was filtered through a 0.45 μm filter (Millex Durapore, Millipore) prior to purification with an AKTA Express System (GE Healthcare). Proteins were purified using nickel immobilized metal affinity chromatography (IMAC) with a 5 ml HiTrap chelating HP column charged with Ni2+ as described earlier.28 The protein was then subjected to polyhistidine tag cleavage using TEV protease as described earlier,28 and the cleaved protein was purified using subtractive IMAC since TEV has a noncleavable polyhistidine tag.28, 29 We performed buffer exchange and protein concentration using an Amicon Ultra‐15 Centrifugal Filter Device (Millipore). All proteins were exchanged into and stored in crystallization buffer (20 mM HEPES pH 8.0, 150 mM NaCl, and 1.5 mM TCEP (tris[2‐carboxyethyl]phosphine; Amresco Inc). Aliquots of both tagged and cleaved proteins were frozen in liquid nitrogen and stored at −80°C until ready to use for enzyme kinetics.
4.4. Analytical size exclusion chromatography
The oligomeric states of the proteins were determined by size‐exclusion chromatography on a Dionex HPLC (Thermo Scientific™) using an SRT SEC‐150 column (7.8 x 250 mm; Sepax Technologies) in 20 mM HEPES, pH 8.0, 150 mM NaCl, and 1.5 mM TCEP. A 5‐μl sample injection volume was used and the flow rate was 1.2 ml/min. The column was calibrated with ribonuclease A (13.7 kDa), carbonic anhydrase (29 kDa), ovalbumin (44 kDa), albumin (66 kDa), and aldolase (158 kDa) as standards. The separation was carried out at 22°C at a flow rate of 1.2 ml/min. The calibration curve of K av = (V e–V o)/(V t–V o) was used, where V e is the elution volume for the protein, V o is the column void volume, and V t is the total bed volume. The results were compared with the predictions from the PISA website (https://www.ebi.ac.uk/pdbe/pisa/pistart.html).30
4.5. Protein crystallization
Crystallization experiments were performed using the sitting drop vapor‐diffusion method in 96‐well CrystalQuick plates (Greiner, Bio‐One) with a total volume of 135 μl of well solution and protein mixed with crystallization solution in a 1:1 ratio (0.4 μl of each); the final protein concentration in the crystallization droplets was 6–8 mg/ml and plates were stored at 16°C. A Mosquito (TTP Labtech) robot was used to set the following crystallization screens: INDEX, crystal screen (Hampton Research), PEGsII Suite (Qiagen), MCSG1, and MCSG4 (Anatrace). For co‐crystallization, ligands were used at a 10‐fold to 20‐fold molar excess over the protein concentration. The ligands tried were as follows: crystal violet and methyl green (Allied Chemicals), fusidic acid, glycodeoxycholate, taurocholate, cholesterol, Ellman's reagent or 5,5′‐dithio‐bis‐[2‐nitrobenzoic acid] (DTNB), N‐acetylneuraminic acid, coenzyme A, β,γ‐imidoadenosine 5′‐triphosphate lithium salt hydrate (AMP‐PNP), acetyl coenzyme A sodium salt (all from Sigma Aldrich), desulfo‐coenzyme A (Jena Bioscience), Cm, thiamphenicol, and chloramphenicol succinate (all from Sigma Aldrich). Crystals suitable for structure determination appeared in 2–7 days and were soaked in cryoprotecting solution (the same crystallization condition as with added glycerol or ethylene glycol to prevent ice formation during freezing) and flash‐frozen in liquid nitrogen before data collection. All proteins used for structure determination had the tag removed prior to crystallization trials. High‐resolution diffracting crystals grew under the conditions as follows: VcCAT (PDB ID: 3EEV) was co‐crystallized with 16 mM AcCoA and 10 mM Cm. The best crystals appeared in Crystal Screen H7 (0.2 M ammonium phosphate monobasic, 0.1 M Tris pH 8.5, 50% (v/v) (±)‐2‐methyl‐2,4‐pentanediol). VcCAT (PDB ID: 6PUA) was also cocrystallized with 20 mM AcCoA and 10 mM MgCl2 in Crystal Screen H7 (50% v/v MPD). No additional cryoprotectant was needed for this condition. VcCAT (PDB ID: 6PUB) was cocrystallized with 15 mM crystal violet, 20 mM AcCoA, and 10 mM MgCl2 in Crystal Screen F11 (1.6 M ammonium sulfate, 0.1 M MES monohydrate pH 6.5, 10% (v/v) 1,4‐dioxane). 10% ethylene glycol was added as a cryoprotectant. VcCAT (PDB ID: 6U9C) was crystallized in PEGs II F1 (0.1 M trisodium citrate, 20% (w/v) PEG 4000, 20% (w/v) isopropanol), and crystals were soaked for 4 days in mother liquor supplemented with 50 mM AcCoA and 5 mM Cm. Crystals were cryoprotected with 10% ethelene glycol. AfCAT (PDB ID: 5UX9) was crystallized in MCSG1 A11 (0.2 M MgCl2, 0.1 M MES:NaOH pH 6.5, and 10% [w/v] PEG 4000). AfCAT (PDB ID: 6PXA) was cocrystallized with 8 mM sodium taurocholic hydrate pH 6.0 under PEGs II C8 condition (0.2 M sodium acetate, 0.1 M Tris pH 8.5, 16% [w/v] PEG 4000), and 15% glycerol was added for cryoprotection. VvCAT (PDB ID: 6PU9) was cocrystallized with 20 mM AcCoA and 10 mM MgCl2 in Crystal Screen D1 (0.1 M sodium acetate trihydrate pH 4.6, 8% (w/v) PEG 4000) and cryoprotected with 25% ethylene glycol.
4.6. Data collection and structure determination
All data sets were collected at the Structural Biology Center ID‐19 or BM‐19 beamlines at the Advanced Photon Sources (Argonne National Laboratory) at 100 K. Diffraction images were integrated and scaled using HKL3000,31 and the structures were determined by molecular replacement with MOLREP and REFMAC in the HKL3000 software package. The VcCAT (PDB ID: 3EEV) and AfCAT (PDB ID: 5UX9) structures previously deposited into the PDB as part of structural genomics efforts were used as the search models for the remaining structures described in this article. The initial models were manually adjusted using COOT32 and where then iteratively refined using COOT, PHENIX33 and/or REFMAC.34 In the case of VvCAT, the molecular replacement structure was rebuilt to contain the proper sequence by BUCCANEER35 and refined initially by REFMAC. The structure was then moved to PHENIX to continue refinement. Throughout the refinement for all structures, the same 5% of reflections were not included in the refinement for R free calculations when using both REFMAC and PHENIX. X‐ray coordinates were deposited into the PDB using accession codes 3EEV, 6PUA, 6PUB, and 6U9C for VcCAT, 5UX9, and 6PXA for AfCAT, and 6PU9 for VvCAT (see Table 1 for data collection and refinement statistics for all structures).
4.7. Enzyme kinetics assay
A buffer exchange was performed with all proteins using 0.5 ml Zeba spin desalting columns (Thermo Scientific) since TCEP reacts with DTNB in the enzyme kinetics assay.36 Proteins were eluted into 100 mM Tris–HCl at pH 8.0 and 150 mM NaCl, and their concentrations were determined using the absorbance of the protein at 280 nm, their extinction coefficients, and Beer's law. Extinction coefficients for each protein were calculated with ProtParam (https://web.expasy.org/protparam/)37 as 50,420, 48,930, and 51,910 M−1 cm−1 for VcCAT, AfCAT, and VvCAT, respectively. We used a discontinuous steady‐state enzyme kinetic assay8, 38 and measured the absorbance of TNB2− at 412 nm with the following modifications. Each reaction contained 100 mM Tris–HCl at pH 8.0, 150 mM NaCl, 2% EtOH, 0.5 mM AcCoA (trilithium salt from Sigma), and 1 mM acceptor substrate. Initially, we screened the enzymes for their ability to acetylate a variety of potential substrates including chloramphenicol, aminoglycoside antibiotics (tobramycin, gentamycin, streptomycin, kanamycin B), and the streptogramin antibiotic dalfopristin. Only chloramphenicol was a substrate; therefore, we performed substrate saturation curves using chloramphenicol (concentration varied between 0 and 2 mM). Reactions were initiated with enzyme and were allowed to proceed for 10 min at 37°C and then terminated with guanidine HCl and reacted with DTNB as described earlier.38 The following concentration of each enzyme (based on the monomer MW) was used for steady‐state kinetics: 10.7 nM tagged VcCAT, 5.1 nM cleaved VcCAT, 31.5 nM tagged AfCAT, 19.7 nM cleaved AfCAT, 13.4 nM tagged VvCAT, and 5.9 nM cleaved VvCAT. Each reaction was performed in duplicate, and the average of two replicates was used to fit data. Kinetic parameters for each enzyme were determined using the Michaelis–Menten equation fitted with Origin 2016 software.
Supporting information
Figure S1 Chromatograms for analytical size exclusion chromatography of VcCAT, VvCAT, and AfCAT. Each protein elution profile was compared to a set of protein standards described in Materials and Methods. The VcCAT, VvCAT, and AfCAT are shown as gray, orange, and blue lines, respectively. The VcCAT protein eluted at 5.77 ml, VvCAT eluted at 5.74 ml, and AfCAT eluted at 5.76 ml, which corresponded to 92 kDa, 96 kDA, and 94 kDa, respectively.
ACKNOWLEDGMENTS
We sincerely thank Dr. Andrzej Joachimiak for his expertise and willingness to participate in video conference calls with SFSU students and providing resources for enzyme kinetics assays. Additionally, we are most grateful to Dr. Didier Mazel for help determining whether genes that encode VvCAT and AfCAT were found in superintegrons. We also thank Michael Endres, Robert Jedrzejczak, and JCVI for their help cloning the genes in this study into expression vectors. Additionally, we thank Professor Sean McFarland at the SFSU School of Art for his help creating anaglyphs for student poster presentations. Dalfopristin was a gift from Sanofi‐Aventis and was solubilized as described.8 Funding for this project was provided in part by federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract Nos. HHSN272201200026C and HHSN272201700060C. The use of SBC beamlines at the Advanced Photon Source is supported by the U.S. Department of Energy (DOE) Office of Science and operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357. Additional funding for this project was provided by the National Science Foundation (Grant CHE‐1708863 to MLK).
Alcala A, Ramirez G, Solis A, et al. Structural and functional characterization of three Type B and C chloramphenicol acetyltransferases from Vibrio species. Protein Science. 2020;29:695–710. 10.1002/pro.3793
Ashley Alcala, Guadalupe Ramirez, and Allan Solis contributed equally to this study.
Funding information Department of Energy, Grant/Award Number: DE‐AC02‐06CH11357; National Institute of Allergy and Infectious Diseases, Grant/Award Numbers: HHSN272201200026C, HHSN272201700060C; National Science Foundation, Grant/Award Number: CHE‐1708863
Contributor Information
Natalia Maltseva, Email: nmaltseva@anl.gov.
Misty L. Kuhn, Email: mkuhn@sfsu.edu.
REFERENCES
- 1. Schwarz S, Kehrenberg C, Doublet B, Cloeckaert A. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol Rev. 2004;28:519–542. [DOI] [PubMed] [Google Scholar]
- 2. Zhang G, Sun K, Ai G, et al. A novel family of intrinsic chloramphenicol acetyltransferase CATC in Vibrio parahaemolyticus: Naturally occurring variants reveal diverse resistance levels against chloramphenicol. Intl J Antimicrob Agents. 2019;54:75–79. [DOI] [PubMed] [Google Scholar]
- 3. Shaw WV. Chloramphenicol acetyltransferase: Enzymology and molecular biology. CRC Crit Rev Biochem. 1983;14:1–46. [DOI] [PubMed] [Google Scholar]
- 4. White PA, Stokes HW, Bunny KL, Hall RM. Characterisation of a chloramphenicol acetyltransferase determinant found in the chromosome of Pseudomonas aeruginosa . FEMS Microbiol Lett. 1999;175:27–35. [DOI] [PubMed] [Google Scholar]
- 5. Sugantino M, Roderick SL. Crystal structure of vat(D): An acetyltransferase that inactivates streptogramin group A antibiotics. Biochemistry. 2002;41:2209–2216. [DOI] [PubMed] [Google Scholar]
- 6. Rowe‐Magnus DA, Guerout A‐M, Mazel D. Bacterial resistance evolution by recruitment of super‐integron gene cassettes. Mol Microbiol. 2002;43:1657–1669. [DOI] [PubMed] [Google Scholar]
- 7. Beaman TW, Sugantino M, Roderick SL. Structure of the hexapeptide xenobiotic acetyltransferase from Pseudomonas aeruginosa . Biochemistry. 1998;37:6689–6696. [DOI] [PubMed] [Google Scholar]
- 8. Stogios PJ, Kuhn ML, Evdokimova E, Courvalin P, Anderson WF, Savchenko A. Potential for reduction of streptogramin a resistance revealed by structural analysis of acetyltransferase VatA. Antimicrob Agents Chemother. 2014;58:7083–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kehoe LE, Snidwongse J, Courvalin P, Rafferty JB, Murray IA. Structural basis of Synercid® (quinupristin‐dalfopristin) resistance in gram‐positive bacterial pathogens. J Biol Chem. 2003;278:29963–29970. [DOI] [PubMed] [Google Scholar]
- 10. Bennett AD, Shaw WV. Resistance to fusidic acid in Escherichia coli mediated by the type I variant of chloramphenicol acetyltransferase. A plasmid‐encoded mechanism involving antibiotic binding. Biochem J. 1983;215:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Proctor GN, McKell J, Rownd RH. Chloramphenicol acetyltransferase may confer resistance to fusidic acid by sequestering the drug. J Bacteriol. 1983;155:937–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Proctor GN, Rownd RH. Rosanilins: Indicator dyes for chloramphenicol‐resistant enterobacteria containing chloramphenicol acetyltransferase. J Bacteriol. 1982;150:1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanaka H, Izaki K, Takahashi H. Some properties of chloramphenicol acetyltransferase, with particular reference to the mechanism of inhibition by basic triphenylmethane dyes. J Biochem. 1974;76:1009–1019. [PubMed] [Google Scholar]
- 14. Majorek KA, Kuhn ML, Chruszcz M, Anderson WF, Minor W. Double trouble‐buffer selection and His‐tag presence may be responsible for nonreproducibility of biomedical experiments. Protein Sci. 2014;23:1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mazel D. Integrons: Agents of bacterial evolution. Nat Rev Microbiol. 2006;4:608–620. [DOI] [PubMed] [Google Scholar]
- 16. Cury J, Jové T, Touchon M, Néron B, Rocha EP. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 2016;44:4539–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baharoglu Z, Bikard D, Mazel D. Conjugative DNA transfer induces the bacterial SOS response and promotes antibiotic resistance development through integron activation. PLoS Genet. 2010;6:e1001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen CY, Wu KM, Chang YC, et al. Comparative genome analysis of Vibrio vulnificus, a marine pathogen. Genome Res. 2003;13:2577–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ruby EG, Urbanowski M, Campbell J, et al. Complete genome sequence of Vibrio fischeri: A symbiotic bacterium with pathogenic congeners. Proc Natl Acad Sci U S A. 2005;102:3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martínez J. Ecology and Evolution of Chromosomal Gene Transfer between Environmental Microorganisms and Pathogens. Microbiol Spectrum. 2018;6(1):MTBP‐0006‐2016. 10.1128/microbiolspec.MTBP-0006-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dziejman M, Balon E, Boyd D, Fraser CM, Heidelberg JF, Mekalanos JJ. Comparative genomic analysis of Vibrio cholerae: Genes that correlate with cholera endemic and pandemic disease. Proc Natl Acad Sci U S A. 2002;99:1556–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bangera G, Brownell SE. Course‐based undergraduate research experiences can make scientific research more inclusive. CBE Life Sci Educ. 2014;13:602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Auchincloss LC, Laursen SL, Branchaw JL, et al. Assessment of course‐based undergraduate research experiences: A meeting report. CBE Life Sci Educ. 2014;13:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brownell SE, Hekmat‐Scafe DS, Singla V, et al. A high‐enrollment course‐based undergraduate research experience improves student conceptions of scientific thinking and ability to interpret data. CBE Life Sci Educ. 2015;14:ar21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high‐throughput, ligation‐independent cloning encoding a tobacco etch virus protease cleavage site. Protein Express Purif. 2002;25:8–15. [DOI] [PubMed] [Google Scholar]
- 26. Eschenfeldt WH, Makowska‐Grzyska M, Stols L, Donnelly MI, Jedrzejczak R, Joachimiak A. New LIC vectors for production of proteins from genes containing rare codons. J Struct Funct Genomics. 2013;14:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dieckman L, Gu M, Stols L, Donnelly MI, Collart FR. High throughput methods for gene cloning and expression. Protein Express Purif. 2002;25:1–7. [DOI] [PubMed] [Google Scholar]
- 28. Makowska‐Grzyska M, Kim Y, Maltseva N, et al. Protein production for structural genomics using E. coli expression. Methods Mol Biol. 2014;1140:89–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kapust RB, Waugh DS. Controlled intracellular processing of fusion proteins by TEV protease. Protein Express Purif. 2000;19:312–318. [DOI] [PubMed] [Google Scholar]
- 30. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. [DOI] [PubMed] [Google Scholar]
- 31. Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL −3000: The integration of data reduction and structure solution—From diffraction images to an initial model in minutes. Acta Cryst. 2006;D62:859–866. [DOI] [PubMed] [Google Scholar]
- 32. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 33. Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: A comprehensive Python‐based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murshudov GN, Skubák P, Lebedev AA, et al. REFMAC 5 for the refinement of macromolecular crystal structures. Acta Cryst. 2011;D67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cowtan K. The buccaneer software for automated model building. 1. Tracing protein chains. Acta Cryst. 2006;D62:1002–1011. [DOI] [PubMed] [Google Scholar]
- 36. Han JC, Han GY. A procedure for quantitative determination of tris(2‐carboxyethyl)phosphine, an odorless reducing agent more stable and effective than dithiothreitol. Analyt Biochem. 1994;220:5–10. [DOI] [PubMed] [Google Scholar]
- 37. Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server The proteomics protocols handbook. Totowa, NJ: Humana Press, 2005; p. 571–607. [Google Scholar]
- 38. Kuhn ML, Majorek KA, Minor W, Anderson WF. Broad‐substrate screen as a tool to identify substrates for bacterial Gcn5‐related N‐acetyltransferases with unknown substrate specificity. Protein Sci. 2013;22:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zaidenzaig Y, Fitton JE, Packman LC, Shaw WV. Characterization and comparison of chloramphenicol acetyltransferase variants. Eur J Biochem. 1979;100:609–618. [DOI] [PubMed] [Google Scholar]
- 40. Fitton JE, Shaw WV. Comparison of chloramphenicol acetyltransferase variants in staphylococci. Purification, inhibitor studies and N‐terminal sequences. Biochem J. 1979;177:575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kobayashi J, Furukawa M, Ohshiro T, Suzuki H. Thermoadaptation‐directed evolution of chloramphenicol acetyltransferase in an error‐prone thermophile using improved procedures. Appl Microbiol Biotech. 2015;99:5563–5572. [DOI] [PubMed] [Google Scholar]
- 42. Masuyoshi S, Okubo T, Inoue M, Mitsuhashi S. Purification and some properties of a chloramphenicol acetyltransferase mediated by plasmids from vibrio anguillarum. J Biochem. 1988;104:131–135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Chromatograms for analytical size exclusion chromatography of VcCAT, VvCAT, and AfCAT. Each protein elution profile was compared to a set of protein standards described in Materials and Methods. The VcCAT, VvCAT, and AfCAT are shown as gray, orange, and blue lines, respectively. The VcCAT protein eluted at 5.77 ml, VvCAT eluted at 5.74 ml, and AfCAT eluted at 5.76 ml, which corresponded to 92 kDa, 96 kDA, and 94 kDa, respectively.