

Abstract
Synthetic polymers are employed to create highly defined microenvironments with controlled biochemical and biophysical properties for cell culture and tissue engineering. Chemical modification is required to input biological or chemical ligands, which often changes the fundamental structural properties of the material. Here, we report on a simple modular biomaterial design strategy that employs functional cyclodextrin nanobeads threaded onto poly(ethylene glycol) polymer necklaces to form multifunctional hydrogels. Nanobeads with desired chemical or biological functionalities can be simply threaded onto the PEG chains to form hydrogels, creating an accessible platform for users. We describe the design and synthesis of these multifunctional hydrogels, elucidate structure-property relationships, and demonstrate applications ranging from stem cell culture and differentiation to tissue engineering.
Keywords: Functional biomaterials, Poly(ethylene glycol), α-cyclodextrin, Tissue Engineering, Hydrogels
Graphical Abstract
Poly(ethylene glycol) is employed to create synthetic hydrogel microenvironments for cells but the ether backbone lacks sites for functionalization. Here, we apply supramolecular chemistry to create modular hydrogels using α-cyclodextrins modified with biological and chemical functional groups with independently controlled crosslinking densities designed to direct stem cell functions.
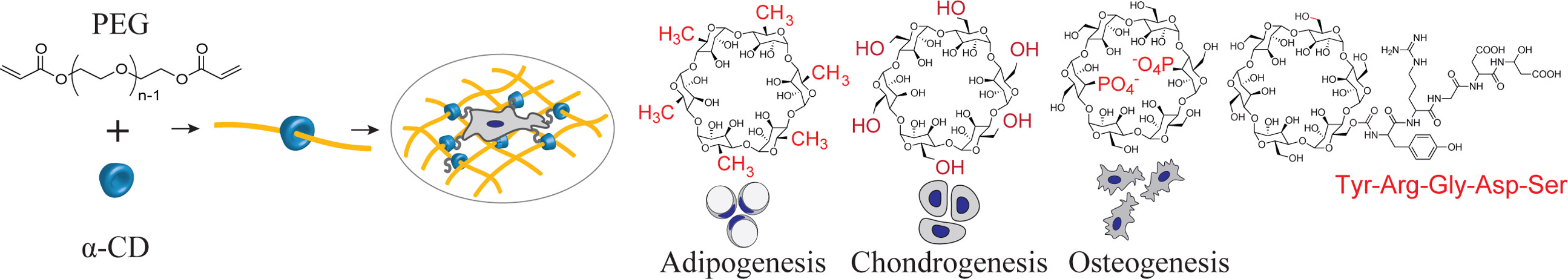
1. Introduction
Biomaterials are fundamental components of medical devices, such as heart valves and stents, sutures, prosthetics, and tissue engineering scaffolds[1]. The functionalization of materials is key to modulation of their properties, and plays an important role at chemical and biological interfaces to target design requirements for applications in applied science, engineering and technology. Hydrogels are a class of biomaterials composed of crosslinked polymer networks[2,3] employed in drug delivery[4], microfabrication of complex devices[5], and medical devices, such as contact lenses[6]. In recent years, hydrogels have become popular vehicles for culturing cells in a 3D environment[2,3]. For this application, hydrogels are often decorated with biological signals to modulate cell functions, understand cell-extracellular matrix (ECM) relationships and direct tissue regeneration[7]. However, the biological outcomes are dependent on many nanoscale cell-ECM interactions[8]; and mimicking the cell environment is challenging due to lack of multifunctional materials with precisely defined microenvironments that can be easily modulated at the nanoscale level[8–12].
Both synthetic and biologically derived materials have been employed to create hydrogels for cell culture and tissue engineering applications. Biologically derived materials such as hyaluronic acid (HA), alginate and collagens, can be employed to create highly compatible hydrogels that support cell-material interactions, proliferation and tissue growth. Unfortunately, biological materials have complexities in biological and chemical substitutions that make creating highly controlled environments challenging (e.g., cells interact with HA via CD44 receptors[13]). For this reason, researchers have explored synthetic materials as highly controlled artificial microenvironments. Poly(ethylene glycol), PEG, in particular is widely used as a synthetic vehicle for cell culture and tissue engineering[14–17]. PEG is considered to be highly compatible with cells and does not significantly absorb proteins or cells, providing a blank slate to start incorporating ligands or chemical functional groups[17]. Current strategies for creating synthetic scaffolds that mimic specific characteristics of the native ECM require covalent conjugation of multivalent ligands to the main chain of polymers, which requires chemical expertise and can result in spatial mismatching, and limited mobility and accessibility[17–22]. In particular, PEG is a polyether that does not have modifiable functional groups on the backbone so only the end groups can be manipulated, altering the overall hydrogel network structure[17]. In this work, we synthesized a cytocompatible PEG hydrogel system with modular mobile functionalities that does not require chemical modification of the PEG backbone, to create synthetic microenvironments.
We created a modular system to build synthetic matrices based on supramolecular chemistry of PEG[23] and α-cyclodextrin (α-CD)[24,25]. α-CD is a six-member oligosaccharide doughnut ring structure (i.e., bead) with an inner cavity and an outside diameter of ~1.5 nm[24]. The α-CD beads physically thread onto the PEG chains to resemble a molecular necklace[25]. α-CD bears many hydroxyl groups that can be easily modified to create a variety of functionalities[26] that also provide sites for conjugation of bioactive agents or ligands onto these rings[27–29]. The α-CD nanobeads are also highly mobile, and slide freely along and rotate around the polymer chain[29–31]. Furthermore, α-CD rings conjugated with biological ligands can be dethreaded from the PEG chains when end-capped with user-specified cleavable linkages, providing temporal control on hydrogel properties[32,33]. Post-threading, the PEG “necklaces” decorated with the modular α-CD are crosslinked to form hydrogels with user-controlled material properties. Because the α-CD molecules are threaded onto the PEG necklace, they do not take part in the hydrogel crosslinking process and can be independently manipulated and functionalized irrespective of the hydrogel network structure. Ultimately, the hydrogel can be decorated with chemical functional groups or biological signals, such as cell adhesion peptides via the nanobeads to create highly controlled microenvironments for cells (Figures 1a-d). The simple modular design strategy that utilizes functional molecular necklaces to synthesize hydrogels with independently tunable physical and chemical properties requires minimal user chemistry, promoting accessibility to a wider scientific community. This strategy also provides a key advantage for PEG polyether chains by incorporating functional arms along the macromer that can be chemically manipulated. In this work, we synthesized an array of synthetic modular multifunctional hydrogels to control stem cell differentiation and selectively stimulate lineage-specific tissue production.
Figure 1. Design of a molecular-necklace system to create tunable, multifunctional hydrogels with independent control of mechanics, cell adhesion properties, and chemical functionality.

a, Alpha-cyclodextrin (α-CD), with its nanobead-like structure, forms an inclusion complex with poly(ethylene glycol)-diacrylate (PEGDA) (R = hydroxyl or other functional groups). After threading α-CD onto polymer chains, PEGDA is crosslinked to form a hydrogel. b, The mechanical properties of α-CD-PEG hydrogel can be varied independent of α-CD by manipulating the cross-linking density of PEG, which is directly related to the stiffness of the hydrogel. c, The α-CD can be substituted with cell adhesion peptides before threading and hydrogel formation. The concentration of cell integrin-binding peptide conjugated to α-CD can be varied independent of the cross-linking density. d, The chemical functionality of α-CD can be varied (i.e., hydrophobic, hydrophilic or charged groups) to create specific microenvironments.
2. Results and Discussion
The first application of the molecular necklace hydrogels was to evaluate stem cell response to mechanical and cell adhesion cues. To achieve this, α-CD was chemically modified with the adhesion peptide, YRGDS (Supplementary Figure S1a) as confirmed by Matrix-Assisted Laser Desorption/Ionization (MALDI) time of flight (TOF) mass spectrometry analysis (Figure 2a). The substituted α-CD-YRGDS nanobeads were plugged onto the PEG chains in aqueous solution by formation of inclusion complexes (Figure S1b) that were then crosslinked to form hydrogels. The chemistry of the hydrogels was characterized by X-ray photoelectron spectroscopy (XPS) (Figure 2b) and Fourier transform infrared spectroscopy (FTIR) (Figure 2c) to validate material structure. To confirm the biological functionality of the molecular necklace, cells were cultured on 2D hydrogel surfaces (Figure 2d) with α-CD-YRGDS concentration varying from 0.1% to 2.0% (w/v) while maintaining a constant hydrogel crosslinking density, or PEGDA concentration (10%, w/v). We could change the concentration of cell-adhesion sites by plugging in varying amounts of nanobeads, which are physically threaded onto the PEGDA chains and not a part of the hydrogel crosslinking. Cells spread more and adhered better to the hydrogel surfaces (Figure 2e) decorated with nanobeads substituted with cell adhesion peptides compared to those on the hydrogels containing only α-CD at similar concentrations. This confirmed the successful functionalization and retention of biological activity of the adhesion peptides attached to the α-CD-decorated hydrogel.
Figure 2. Synthesis and characterization of cell-interactive molecular necklace, α-CD-YRGDS-PEG hydrogels.

a, MALDI-TOF spectrum to confirm peptide conjugation, and synthesis of α-CD-YRGDS (mol wt ~1595 Da) (see in Supplementary Fig. S1a). b, XPS spectrum for α-CD-YRGDS to determine the presence of nitrogen with peptide conjugation to α-CD. c, FTIR spectra for α-CD-YRGDS threading onto PEGDA chains. The spectrum for PEGDA provides a baseline before threading, followed by the α-CD-YRGDS alone, mixed with PEGDA, and α-CD-YRGDS-PEGDA after threading. A peak at 1750 cm−1 arises for the amide stretching of the peptide conjugated to α-CD, and a broader peak at ~3550 cm−1 is for the hydroxyl groups of α-CD and YRGDS. Threading efficiency was ~20% as determined by the ninhydrin assay on rigorously washed and dried hydrogels (see in Supplementary Fig. S1b-d). d, PEGDA (10% w/v) was threaded with α-CD-YRGDS nanobeads and cross-linked to form hydrogels, which were then seeded with MSCs. e, MSCs cultured on the surface of the α-CD-YRGDS-PEGDA hydrogels decorated with varying numbers of α-CD-YRGDS nanobeads (0.1%, 0.5%, and 2.0%, w/v) had significantly greater cell areas and complex actin structures compared to the respective α-CD and PEGDA controls after 4 days of culture (TCP = tissue culture plate; F-actin staining with Texas Red®-X phalloidin).
Next, we evaluated the stem cell response to the multifunctional hydrogels with independently modulated mechanical properties and cell adhesion peptide concentration. Hydrogels with three PEGDA concentrations (5 %, 10 %, and 15 %, defined as soft, medium, and stiff in Figure 3a), or crosslinking densities and hence mechanical properties (shear modulus) were synthesized with varying concentrations of α-CD-YRGDS nanobeads (0 %, 0.5 %, 2.0 %, and 10 %, defined as none, low, moderate, and high in Figure 3a). The modulus of the hydrogels ranged from ~0.5 to ~10 and ~33 kPa, and there was no significant difference in the moduli with varying α-CD nanobead concentration (Figure 3a). As expected, the swelling ratio was inversely proportional to the crosslinking density such that the less crosslinked, softer gels contained more water (Figure 3b). Consequently, an array of hydrogels was generated with independently varied mechanical and cell adhesion properties. Figure 3c pictures the cell morphology on soft and stiff hydrogel substrates with varying concentration of cell adhesion molecule, α-CD-YRGDS. Cells on control PEG hydrogels retained a spherical morphology regardless of mechanical properties. As α-CD-YRGDS concentration increased, cell interactions with the material changed; however, the response varied depending on the mechanical properties. On the soft hydrogels containing adhesion peptides, the cell area and elongation increased significantly with α-CD-YRGDS concentration. Over time, the cells aggregated, particularly as the α-CD-YRGDS concentration increased. On the stiffer hydrogels, extensive cell spreading was also observed at higher adhesive peptide levels but there was less aggregation compared to softer substrates. Most importantly, by manipulating the cell adhesivity of the soft hydrogel, the cell response and morphology appeared qualitatively similar to the stiffer hydrogel substrates (Figure 3c). These changes in cell morphology were quantified and confirmed by evaluating the cell inverse shape ratio and cell perimeter (Figures 4a & b). Quantitative analysis confirmed the morphological observations that there were significant differences in the cells cultured on soft and stiff substrates with minimal cell adhesion, but the incorporation of more cell adhesion peptides reduced the differential response to mechanical properties. Ultimately, the cellular response to the material varied not only with the mechanical properties, but also with the cell adhesivity of the hydrogel, which was easily controlled via modular input into the molecular necklace hydrogel.
Figure 3. α-CDYRGDS-PEG hydrogels with independently tunable mechanical and cell adhesion properties.

An array of hydrogels was synthesized with independently varied cross-linking densities (% of PEGDA, necklace) and cell adhesions (α-CD-YRGDS, nanobeads). a, The shear storage moduli of the hydrogels varied only with the crosslinking density and did not change with the incorporation of α-CD nanobeads. The PEGDA 15% (w/v) hydrogels with shear storage modulus ~30 kPa are defined as stiff substrates, while PEGDA 10% (w/v) with shear storage modulus ~7–10 kPa as moderately stiff, and PEGDA 5% (w/v) with shear storage modulus ~0.5 kPa as soft substrates. b, The amount of water absorbed into the hydrogels (swelling ratio) also varied with respect to the crosslinking density, and it did not change with α-CD nanobeads. c, MSCs cultured on the surfaces of stiff and soft hydrogels with varying concentrations of cell adhesive nanobeads have different morphological and organization characteristics (F-actin staining with Texas Red®-X phalloidin at 16 h). Cells were able to spread on the soft surfaces and aggregated with increasing adhesion compared to cells seeded on the stiffer substrates that had longer cell extensions in isolation (little aggregation). Data collected throughout the study of compression modulus and swelling ratio are presented as a mean ± standard deviation of three or more data samples (*p ≤ 0.05).
Figure 4. Stem cell response to materials with independently controlled stiffness and adhesion.

The morphological changes of cells cultured on surfaces with independently controlled stiffness and adhesion were quantified over time in terms of inverse shape ratio (perimeter2/4π*Area) and projected perimeter. Inverse shape ratio characterizes cell spreading and deviation from non-adherent circular shape. Cells were characterized on: a, low adhesion (α-CD-YRGDS) and b, high adhesion (α-CD-YRGDS) content surfaces with variable cross-linking density or mechanical properties. Larger differences in both cell shape parameters developed when the hydrogels were decorated with higher concentrations of α-CD-YRGDS nanobeads. Cell differentiation was related to cell shape, and gene expression was evaluated after 21 days of culture for markers related to c, adipogenesis, d, myogenesis, e, chondrogenesis, and f, osteogenesis. Data collected throughout the study of cell morphology is presented as a mean ± standard deviation of multiple data samples (n = 15–20), except for PEGDA samples.
We observed that changes in stem cell morphology correlate with tissue-specific differentiation in the molecular necklace hydrogels. For example, mesenchymal stem cells (MSCs) permitted to spread over a large micropatterned area undergo osteogenic differentiation while those constrained to small areas prefer the adipogenic lineage[34,35]. Furthermore, cells restricted to a small attachment area cannot form geometrically separated adhesions that otherwise act as anchors to increase tension in the cytoskeleton, whereas cells allowed to spread over a larger area can form numerous geometrically opposed focal adhesion complexes and can generate significant cytoskeletal tension[34]. The mechanical and cell adhesive properties of a material may also play a role in determining if cells aggregate with each other or interact with the material in a singular fashion, which plays a role in directing cell differentiation[36–39]. The cell adhesion characteristics of the hydrogel (i.e., the quantity of cell adhesive α-CD nanobeads) regulated the differentiation profile of stem cells in response to mechanical stimuli. Adipogenic specific markers, such as fatty acid-binding protein (FABP) and CCAAT/enhancer-binding proteins (CEBPA) were expressed on the softer substrate irrespective of the concentrations of cell adhesion peptides (Figure 4c). Myogenic markers desmin and myogenic factor 5 (MYF5) were upregulated on hydrogels with intermediate mechanical properties, though only in the case of low levels of adhesion peptide (Figure 4d). Adipogenic markers, both FABP and CEBPA, along with two myogenic markers MYF5 and desmin, were upregulated on hydrogel substrates with medium stiffness at lower adhesion level. This suggests that hydrogel substrates with medium stiffness and low cell adhesivity promote upregulation of both adipogenic and myogenic markers. Stiffer substrates induced both chondrogenesis and osteogenesis differentiation pathways with either low or high (Figures 4e & 4f) cell adhesion characteristics. However, the highest cell adhesion concentrations induced significantly greater expression of tissue-specific genes compared to the low-adhesion environment. Hydrogel mechanical properties influence differentiation of stem cells; however, the interface between the cells and the biomaterial, in the form of cell adhesion, played a critical role in how the cells responded to the mechanical properties of the substrate and the level of differentiated gene expression. Hence, a synergy between the mechanics and cell-adhesive signals in biomaterials exists in dictating stem cell response. The modulation of transcription factors for lineage specific differentiation of MSCs has also been shown in earlier studies to be dependent on the interplay of substrate stiffness and composition of covalently bound tissue-specific ECM proteins, such as collagen, laminin and fibronectin[40].
Small molecules and even specific chemical functionalities impact stem cell differentiation and tissue development[41–44]. We demonstrated the utility of the molecular necklace modular methodology to create specific chemical environments for tissue development independent of the mechanics and cell-adhesion properties of the material. Development of three musculoskeletal tissues (cartilage, fat, and bone)[41–44] was directed and stimulated with specific α-CD chemical functionalities that were selected based on the charge and hydrophobicity. Cells were encapsulated in the 3D PEG hydrogels with varying nanobead composition and concentration (10% PEG, w/v), and cultured in appropriate differentiation conditions (Supplementary Figure S2a). In the case of α-CD-OH nanobeads in the hydrogel network, bone marrow-derived MSCs increased expression of the cartilage specific matrix molecule type II collagen compared to a standard PEG hydrogel (Figure 5a). More cartilage extracellular matrix was present throughout the α-CD-OH containing hydrogel as evidenced by staining with safranin-O for negatively charged proteoglycans.
Figure 5.

Chemical functionality of molecular necklace directs differentiation of stem cells in 3D hydrogels. Stem cells encapsulated in hydrogels with α-CD nanobeads functionalized with polar, hydrophobic, or charged chemical groups were cultured for 3 weeks to evaluate differentiation. a, α-CD (1%, w/v) with hydroxyl groups stimulated chondrogenesis of MSCs compared to control PEG hydrogels with increased early expression of Sox9 transcription factor and (matrix molecules or ECM proteins) aggrecan and type II collagen. Increased cartilage matrix production is visible by safranin-O histological staining. b, The chemical structure of hydrophobic α-CD-CH3 was confirmed by MALDI-TOF spectrometry. Human adipose-derived stem cells (hADSCs) encapsulated in the α-CD-CH3/PEG hydrogels increased expression of adipose-related genes several-fold. Genes include fatty acid-binding protein (FABP), lipoprotein lipase (LPL) and CEBPA (also see Supplementary Figure S2b) and produced more lipid droplets visualized by Oil red O staining, compared to control α-CD hydrogels. c, Cells cultured in hydrogels with α-CD-PO4− functionalized nanobeads (the chemical structure confirmed by MALDI-TOF) produced more mRNA for osteogenic genes COL X, OCN, and OPN compared to control hydrogels (also see Supplementary Figure S2e). Morphologically, mineralization characterized by alizarin red staining also increased in α-CD-PO4−/PEG hydrogels compared to controls.
Hydrophobic-substituted, or α-CD-CH3 nanobeads were investigated for adipogenic differentiation. The MALDI spectrum of α-CD-CH3 confirmed the presence of the hydrophobic functional groups (Figure 5b). Quantitative gene expression for adipose-related markers FABP, lipoprotein lipase (LPL), and CEBPA significantly increased in stem cells encapsulated in the presence of the hydrophobic α-CD-CH3 (1%, w/v) nanobeads in the hydrogels compared to control PEG and α-CD-OH-loaded (both, 1% and 5%, w/v) hydrogels (Figure 5b, Supplementary Figures S2b & S2c). Oil red O staining correlated with gene expression results; the hydrophobic decorated hydrogels producing the greatest amount of lipids (Figure 5b). Finally, nanobeads with phosphate charged groups that are known to enhance bone differentiation and mineralization[43] were threaded onto the PEG chains. Again, phosphate functionalization of the α-CD nanobead was confirmed by MALDI analysis (Figure 5c). Cells cultured in osteogenic conditions significantly increased gene expression for the hypertrophic matrix molecule type X collagen (COL X) as well as for the bone matrix molecules osteocalcin (OCN) and osteopontin (OPN) compared to control PEG and α-CD-OH-loaded (Figure 5c, Supplementary Figure S2d–f) hydrogels. Mineralization, as characterized by alizarin red staining, also increased compared to control hydrogels (Figure 5c, Supplementary Figure S2d). Changing hydrogel physical properties have an impact on tissue production; however, the hydrogels containing different chemical functionalities had swelling ratios (water content) similar to control hydrogels (Supplementary Figure S2a), suggesting similar cross-linking densities and mechanical properties irrespective of threading. Ultimately, the alcohol, hydrophobic methyl group, and phosphate-substituted α-CD-nanobeads stimulated chondrogenic, adipogenic, and osteogenic differentiation, respectively.
The application of α-CD-nanobeads holds numerous advantages in creating a synthetic microenvironment for stem cells. This mobile, nanostructural feature of the molecular necklace can be utilized for modulating receptor-mediated cellular functions by facilitating the interactions of bioactive agent- or ligand-conjugated α-CDs in hydrogels with receptor sites of proteins on the plasma cell membranes (e.g., cell-adhesion via integrin binding to ECM proteins)[19,20,28]. α-CD has primarily been employed as a nanoscale drug delivery vehicle[24,28,29,45] and as a crosslinking agent to create hydrogels for fibroblast culture[22,32,46], cartilage[27,33] and bone[47,48] tissue engineering. However, critical features such as the modular design, cytocompatibility[45], ease of modification[26], enhanced cell-material interactions[19,20,28], and controlled presentation of biological ligands via user-controlled dethreading[30–33], enable application of this powerful hydrogel tool for directed stem cell differentiation and tissue production as demonstrated in the present studies.
3. Conclusions
In summary, we present synthesis and application of a modular molecular necklace with decorated α-CD nanobeads to create multifunctional PEG hydrogels. Functional α-CD nanobeads plugged onto the PEG chains were used to form hydrogels with highly controlled biophysical and biochemical properties to probe stem cell function and stimulate development of specific tissues. This modular design enables independent control of mechanics, cell adhesivity, and chemistry of the hydrogels without modifying the base PEG network structure. The simplicity of the system will further allow researchers in multiple disciplines such as physics, biology, and engineering to create highly controlled and variable synthetic environments. The system allows precise control of the spacing and presentation of integrin binding and growth factor binding ligands on the same α-CD with control and variable spacing while the rotational freedom of the α-CD supports optimal engagement of the ligand-receptor interactions. This spatially dynamic presentation of biochemical cues, adaptive to the contractile or focal adhesion assembly/disassembly processes[49], is unavailable in any other 3D biomaterial design.
4. Experimental
Preparation of 2D and 3D hydrogels.
PEGDA (Mw ~ 3400 Da, pdi 1.1, SunBio) was dissolved in PBS (pH 7.4) at different concentrations, 5%, 10% and 15% (w/v). This solution was added either directly to α-CD (Sigma-Aldrich) and α-CD-derivatives (α-CDPO4− and α-CDCH3 purchased from Cyclodextrin-Shop, Netherlands) or to their saturated solutions in PBS with a final concentration of 0.5%, 2% and 10% (w/v). The solution was properly mixed on a shaker overnight. A photoinitiator, Irgacure® 2959 (Ciba®) (1.0%, w/v) in 70% (v/v) ethanol was dissolved and added to the PEGDA solution with a final concentration of (0.05%, w/v). The pre-gel solution was exposed to ultraviolet (UV) light (wavelength~365 nm) for 5 min in either Corning® Transwell® inserts (diameter 6.5 mm or 24 mm, 3.0 μm polyester membrane) for 2D hydrogel substrates or caps of Eppendorf tubes (0.5 mL) for 3D hydrogels. In 3D hydrogels, cells (~2 million) were added to the pre-gel solution (100 μL). For 2D hydrogels, cells were seeded onto the substrates with a cell density of 20,000 cells/cm2. For most of the 2D studies, a base hydrogel substrate with appropriate concentration and volume was made, followed by another hydrogel layer of PEGDA/α-CD (derivatives). As an example, 250 μL of PEGDA (10%, w/v) solution was added to a 24 mm diameter Corning® Transwell® insert and polymerized under UV for 3 min. This hydrogel layer was added with 30 μL of PEGDA/α-CD (10%/0.5%, w/v) to cover the substrate uniformly, followed by UV polymerization for 4 min. The 2D hydrogel was kept in PBS (pH 7.4) overnight to remove unthreaded α-CD before culturing the cells with expansion media.
Synthesis, characterization, mechanical testing and optical microscopy.
α-CDYRGDS was synthesized in a two-step reaction. First, activated α-CD was isolated by reacting α-CD with N, N’-carbonyldiimidazole in dimethylformamide (DMF). Second, YRGDS (Biomatik corporation) was conjugated to activated α-CD in DMF followed by multiple precipitations in acetone. The product was analyzed by MALDI-TOF spectrometry (Voyager DE™-STR, AppliedBiosystems®). Threading efficiency of α-CD-YRGDS onto PEGDA chains was determined by the ninhydrin assay[50]. The hydrogels were rigorously washed with deionized water and lyophilized. A known amount of the dried hydrogel was hydrolyzed overnight at 115 °C with 6 N HCl, followed by neutralization with NaOH. The absorbance at 574 nm (DU® 500 UV/Visible Spectrophotometer, Beckman Coulter) of these aliquots were recorded and compared to PEGDA/α-CD-YRGDS pre-gel standard solution. The nitrogen content in PEGDA/α-CD-YRGDS hydrogels was determined by XPS (PHI 5400 XPS). FTIR-Attenuated Total Reflectance (ATR) spectroscopy (Bruker Vector 22 with a Pike MIRacle ATR attachment) was performed on dried hydrogel surfaces. The shear storage moduli of the gels were measured using a parallel plate rheometer (RFS 3, Rheometric Scientific) in the strain-controlled mode within linear viscoelastic region at frequency 1 Hz. The swelling ratio was determined by ratio of wet weight to the dried weight of the hydrogel. The images were recorded from Nikon Digital Eclipse DXM1200 and Zeiss Axio optical microscopes, and processing and analysis were performed by ImageJ ver 4.0 (NIH, MD).
Histochemistry.
Harvested constructs were fixed for 24 h in 4% paraformaldehyde at 4 °C and then stored in 70% ethanol until processing. The constructs were then dehydrated in a sequential series of ethanol solutions (i.e., 80%, 95% and 100%) and 100% xylene, and embedded in paraffin at 60 °C overnight. The paraffin block was sliced into 5 μm sections and mounted onto microscope slides, and incubated on a 40 °C plate for at least 1 h. Prior to staining, samples were de-waxed and rehydrated immediately before staining. Safranin-O staining was used for detecting proteoglycan content. For calcium deposition staining, fixed sections were incubated for 30 min at room temperature in a 40 mM alizarin red solution (pH 4.1), then washed with PBS to remove unincorporated dye. For lipid droplet staining, samples were rinsed with PBS and embedded in O.C.T. The frozen block was sliced into 10 μm by cryosectioning and then stained with Oil Red O solution (in 60% isopropanol) for 45 min, followed by repeated washing with isopropanol.
F-actin staining.
The samples were rinsed thrice with PBS, fixed with 4% paraformaldehyde for 10 min, and treated with 0.1% TritonX-100 for 5 min at room temperature. After rinsing twice with PBS, 2.5% v/v Texas Red®-X phalloidin (Invitrogen™) and 4 μM Hoechst 33258 solution were added and kept for 30 min. After washing with PBS three times, images were taken with Nikon DXM1200 or Zeiss Axio optical microscopes. The images were merged and analyzed using ImageJ (NIH, MD).
Supplementary Material
Acknowledgements
The authors gratefully acknowledge the Maryland Stem Cell Research Foundation for a Postdoctoral research fellowship (A.S.), NSF-CMMI 0948053, the Jules Stein Chair and NIH-NIAMS for financial support. We also thank Dr. Qiongyu Guo and Prof. Denise Montell of Johns Hopkins University for their valuable suggestions and comments of this manuscript. We also acknowledge the Johns Hopkins A.B. Mass Spectrometry/Proteomics Facility and the Johns Hopkins Department of Chemistry Instrumentation Facilities for providing access to the MALDI-TOF, FTIR, and XPS instruments.
Footnotes
Supporting Information
The supporting information is available from the Wiley Online Library or from the authors.
Contributor Information
Anirudha Singh, 400 N. Broadway, Robert H. & Clarice Smith Building, Wilmer Eye Institute & Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD-21231, USA..
Jianan Zhan, 400 N. Broadway, Robert H. & Clarice Smith Building, Wilmer Eye Institute & Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD-21231, USA..
Zhaoyang Ye, The State Key Laboratory of Bioreactor Engineering, School of Bioengineering, East China University of Science and Technology, Shanghai-200237, China..
Jennifer H. Elisseeff, Email: jhe@jhu.edu, 400 N. Broadway, Robert H. & Clarice Smith Building, Wilmer Eye Institute & Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD-21231, USA..
References:
- [1].Langer R, Tirrell DA, Nature 2004, 428, 487. [DOI] [PubMed] [Google Scholar]
- [2].Lee KY, Mooney DJ, Chem. Rev 2001, 101, 1869. [DOI] [PubMed] [Google Scholar]
- [3].Hoffman AS, Adv. Drug Deliv. Rev 2002, 54, 3. [DOI] [PubMed] [Google Scholar]
- [4].Langer R, Peppas NA, AICHE J. 2003, 49, 2990. [Google Scholar]
- [5].Weibel DB, Diluzio WR, Whitesides GM, Nat. Rev. Microbiol 2007, 5, 209. [DOI] [PubMed] [Google Scholar]
- [6].Kopecek J, J. Polym. Sci. Part A: Polym. Chem 2009, 47, 5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lutolf MP, Hubbell JA, Nat. Biotechnol 2005, 23, 47. [DOI] [PubMed] [Google Scholar]
- [8].Wheeldon I, Farhadi A, Bick AG, Jabbari E, Khademhosseini A, Nanotechnology 2011, 22, 212001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ferreira L, Karp JM, Nobre L, Langer R, Cell Stem Cell 2008, 3, 136–146. [DOI] [PubMed] [Google Scholar]
- [10].Keung AJ, Kumar S, Schaffer DV, Annu. Rev. Cell. Dev. Bio 2010, 26, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].von der Mark K, Park J, Bauer S, Schmuki P, Cell Tissue Res. 2010, 339, 131. [DOI] [PubMed] [Google Scholar]
- [12].Shekaran A, Garcia AJ, Biochim. Biophys. Acta 2011, 1810, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B, Cell 1990, 61, 1303. [DOI] [PubMed] [Google Scholar]
- [14].Peppas NA, Keys KB, Torres-Lugo M, Lowman AM, J. Control Release 1999, 62, 81. [DOI] [PubMed] [Google Scholar]
- [15].Lin CC, Anseth KS, Pharmaceut. Res 2009, 26, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hwang NS, Varghese S, Li H, Elisseeff J, Cell Tissue Res. 2011, 344, 499. [DOI] [PubMed] [Google Scholar]
- [17].Zhu JM, Biomaterials 2010, 31, 4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cairo CW, Gestwicki JE, Kanai M, Kiessling LL, J. Am. Chem. Soc 2002, 124, 1615. [DOI] [PubMed] [Google Scholar]
- [19].Nelson A, Belitsky JM, Vidal S, Joiner CS, Baum LG, Stoddart JF, J. Am. Chem. Soc 2004, 126, 11914. [DOI] [PubMed] [Google Scholar]
- [20].Hyun H, Yui N, Macromol. Biosci 2011, 11, 765. [DOI] [PubMed] [Google Scholar]
- [21].Yui N, Ooya T. Chem. Eur. J 2006, 12, 6730. [DOI] [PubMed] [Google Scholar]
- [22].Seo J-H, Kakinoki S, Inoue Y, Yamaoka T, Ishihara K, Yui N, Soft matter 2012, 8, 5477. [Google Scholar]
- [23].Salinas CN, Anseth KS, J. Tiss. Eng. Regen. Med 2008, 2, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].van de Manakker F, Vermonden T, van Nostrum CF, Hennink WE, Biomacromolecules 2009, 10, 3157. [DOI] [PubMed] [Google Scholar]
- [25].Harada A, Li J, Kamachi M, Nature 1992, 356, 325. [Google Scholar]
- [26].Khan AR, Forgo P, Stine KJ, D’Souza VT, Chem. Rev 1998, 98, 1977. [DOI] [PubMed] [Google Scholar]
- [27].Tachaboonyakiat W, Furubayashi T, Katoh M, Ooya T, Yui N, J. Biomater. Sci.-Polym. Ed 2004, 15, 1389. [DOI] [PubMed] [Google Scholar]
- [28].Li JJ, Zhao F, Li J, Adv. Biochem. Eng. Biotechnol 2011, 125, 207. [DOI] [PubMed] [Google Scholar]
- [29].Loethen S, Kim J-M, Thompson DH, Polym. Rev 2007, 47, 383. [Google Scholar]
- [30].Ceccato M, LoNostro P, Baglioni P, Langmuir 1997, 13, 2436. [Google Scholar]
- [31].Onagi H, Blake CJ, Easton CJ, Lincoln SF, Chem. Eur. J 2003, 9, 5978. [DOI] [PubMed] [Google Scholar]
- [32].Watanabe J, Ooya T, Nitta KH, Park KD, Kim YH, Yui N, Biomaterials 2002, 23, 4041. [DOI] [PubMed] [Google Scholar]
- [33].Lee WK, Ichi T, Ooya T, Yamamoto T, Katoh M, Yui N, J. Biomed. Mater. Res. Part A 2003, 67A, 1087. [DOI] [PubMed] [Google Scholar]
- [34].Kilian KA, Bugarija B, Lahn BT, Mrksich M, Proc. Natl. Acad. Sci. USA 2010, 107, 4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS, Dev. Cell 2004, 6, 483. [DOI] [PubMed] [Google Scholar]
- [36].Reilly GC, Engler AJ, J. Biomech. 2010, 43, 55. [DOI] [PubMed] [Google Scholar]
- [37].Engler A, Bacakova L, Newman C, Hategan A, Griffin M, Discher D, Biophys. J 2004, 86, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Nat. Mater 2010, 9, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Griffith LG, Naughton G, Science 2002, 295, 1009. [DOI] [PubMed] [Google Scholar]
- [40].Rowlands AS, George PA, Cooper-White JJ, Am. J. Physiol. Cell Physiol 2008, 295, C1037. [DOI] [PubMed] [Google Scholar]
- [41].Keselowsky BG, Collard DM, Garcia AJ, Biomaterials 2004, 25, 5947. [DOI] [PubMed] [Google Scholar]
- [42].Ayala R, Zhang C, Yang D, Hwang Y, Aung A, Shroff SS, Arce FT, Lal R, Arya G, Varghese S, Biomaterials 2011, 32, 3700. [DOI] [PubMed] [Google Scholar]
- [43].Curran JM, Chen R, Hunt JA, Biomaterials 2006, 27, 4783. [DOI] [PubMed] [Google Scholar]
- [44].Benoit DSW, Schwartz MP, Durney AR, Anseth KS, Nat. Mater 2008, 7, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Davis ME, Brewster ME, 2004, 3, 1023. [DOI] [PubMed] [Google Scholar]
- [46].Tran NQ, Joung YK, Lih E, Park KM, Park KD, Macromol. Res 2011, 19(3), 300. [Google Scholar]
- [47].Hein CD, Liu X-M, Chen F, Cullen DM, Wang D, Macromol. Biosci 2010, 10, 1544. [DOI] [PubMed] [Google Scholar]
- [48].Fujimoto M, Isobe M, Yamaguchi S, Amagasa T, Watanabe A, Ooya T, Yui N, J. Biomater. Sci.-Polym. Ed 2005, 16, 1611. [DOI] [PubMed] [Google Scholar]
- [49].Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA, Nature 2010, 466, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Starcher B, Anal. Biochem 2001, 292, 125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.