

Abstract
Intrinsically disordered proteins play vital roles in biology, and their dysfunction contributes to many major disease states. These proteins remain challenging targets for rational ligand discovery or drug design because they are highly dynamic and fluctuate through a diverse set of conformations, frustrating structure-based approaches. To meet this challenge, we have developed protocols to efficiently identify active small molecule ligands of disordered proteins. Our approach utilizes enhanced sampling molecular dynamics and conformational analysis approaches optimized for disordered targets, coupled with computational docking and machine learning-based screens of compound libraries. By applying this protocol to an amyloid-forming segment of microtubule-associated protein tau, we successfully identified novel, chemically diverse tau ligands, including an inhibitor that delays the aggregation reaction in vitro without affecting the amount of aggregate formed at steady state. Our results indicate that we have expanded the toolkit of protein aggregation inhibitors into new areas of chemical space, and demonstrate the feasibility of our ligand discovery strategy.
Keywords: intrinsically disordered protein, amyloid, protein aggregation, protein-ligand interactions, drug discovery
Graphical Abstract
Introduction
Current approaches to rational drug design and discovery are frustrated by the highly dynamic nature of many protein targets relevant to major human diseases. Specifically, conventional approaches rely on the twin assumptions that a protein populates a single, well-defined structure, and that a small molecule ligand compatible with that structure can bind specifically to the target so as to interrupt pathological activity or restore normal function. However, a significant fraction (estimated to be 30–50%) of human proteins are completely or partially unstructured.1 These intrinsically disordered proteins (IDPs) rapidly fluctuate between an ensemble of structures instead of adopting a single well-defined conformation, and cannot be characterized using classical structural biology methods such as X-ray crystallography.2–4 Consequently, it has been challenging to rationally develop new therapies for diseases linked to IDP function and dysfunction.
IDPs and intrinsically disordered regions (IDRs) are involved in many widespread and severe diseases for several reasons.5,6 First, IDPs tend to play important roles in cell signaling pathways. Due to their remarkable structural plasticity, IDPs can bind transiently yet specifically to a variety of different partners, serving as hubs for the flow of information in the cell. Aberrant signaling (due to mutation, toxins, or stress- or age-related post-translational modification) can drastically impair cellular function, accounting for the fact that most cancers involve the dysfunction of particular IDPs, including the tumor suppressors p53 and BRCA1.7,8
Second, the unique physical and chemical properties of IDPs enable them to play vital roles as tunable structural components of subcellular macromolecular assemblies such as non-membrane-bound organelles (NMBOs), the nuclear pore complex, and cytoskeletal architecture.9–11 For instance, NMBOs such as stress granules and P-bodies form by reversible co-aggregation or co-acervation of specific proteins and RNA resulting in liquid-liquid phase separation and droplet formation within the cytoplasm.9,12 These droplets appear to perform important metabolic and physiological functions, with their formation being switchable and regulated. Defects in particular IDPs can cause disorders such as amyotrophic lateral sclerosis (TDP-43, FUS etc.) and hypertrophic cardiomyopathy (troponin-C).13,14
Third, many IDPs are key players in degenerative amyloid disorders such as Alzheimer’s disease (AD), type II diabetes, and Parkinson’s disease.15,16 In these conditions, particular IDPs convert from soluble, monomeric native states into a heterogeneous and dynamic ensemble of toxic intermediates and then into highly ordered β-sheet-rich amyloid aggregates. Amyloid toxicity frequently involves the remodeling and disruption of membranes by partially structured monomeric or oligomeric species.
There is therefore an urgent need for drug discovery strategies targeting IDPs, exemplified by the focus of this study: microtubule-associated protein tau. In its physiological role, tau stabilizes microtubules, regulates axonal trafficking in neurons, and is involved in neurite outgrowth.17 Tau remains predominantly disordered even when bound to microtubules or tubulin, outside of the residues that interact with these partners.11,18,19 Amyloid formation by tau (often accompanied by hyperphosphorylation and oxidative modifications) is a hallmark of a range of neurodegenerative disorders, including Alzheimer’s disease (AD) and chronic traumatic encephalopathy, that are collectively called tauopathies.20 AD is also characterized by the aggregation of the IDP amyloid-β into senile plaques; conversely, tau neurofibrillary tangles are frequently observed in synucleinopathies such as Parkinson’s disease (PD) and dementia with Lewy bodies. Membrane interactions are central to the pathological activity of tau: tau causes membrane leakage, disrupts vesicles, and forms lipid-protein co-aggregates.21–23 Tau occurs in six different isoforms generated by alternative splicing, up to 441 residues long. The central microtubule binding region (MTBR) of tau, consisting of three or four repeats, is responsible both for interacting with tubulin, and for pathological self-association and cytotoxicity.21,24,25 Two hexapeptide repeats dubbed PHF6 and PHF6* are important drivers of aggregation in vitro, and (along with surrounding portions of the MTBR) form the structured core of tau fibrils.26–28 We focus our studies on the tau4RD construct (residues 244–372, comprising all four repeats of the MTBR), which recapitulates tau amyloidogenesis and membrane interactions.23,29 Tau is a particularly intriguing model system because it displays the full gamut of IDP behavior: disorder is important to its native function, and it remains largely unstructured even when bound to microtubules;11,19 its conformational ensemble is very sensitive to environment and binding partners;30,31 it forms amyloid aggregates and causes membrane leakage;21,25 it forms lipid-protein co-aggregates,22,23 and also undergoes liquid-liquid phase transitions.32
The discovery and design of drug-like small molecules capable of ameliorating aggregation by tau and similar IDPs has been a long-standing goal.3,33 Despite substantial effort, our toolkit of amyloid-modulating compounds had until recently been limited to relatively confined regions of chemical space: a handful of dyes and their analogs, polyphenols like tannic acid and epigallocatechin gallate (EGCG), and other natural products such as curcumin.34–41 This generation of active compounds has generally been discovered serendipitously or by high-throughput screening, and has yielded valuable insight into the conformation and aggregation mechanisms of diverse IDPs.
Although IDPs are undeniably challenging targets, there has been exciting recent progress towards the goal of rationally discovering or designing active ligands. Advances in spectroscopic techniques including NMR, single-molecule Förster resonance energy transfer (FRET), time-resolved FRET, tryptophan triplet state quenching, and electron paramagnetic resonance have all provided invaluable insights into IDP conformation and dynamics.4,30,42–45 Simultaneously, the development of enhanced sampling simulation techniques such as replica exchange molecular dynamics (REMD) and metadynamics, as well as optimized Monte Carlo methods, have generated unprecedented molecular insight into the conformational ensembles sampled by IDPs.30,46–50 Tau and tau4RD have been modeled using Monte Carlo and molecular dynamics approaches.51–53 Several groups have been able to leverage this conformational insight into monomeric, oligomeric and bound states of IDPs to generate new, biologically active ligands for a variety of disordered targets.3,54–59 For example, novel ligands have been discovered or designed for amyloid states of tau and other IDPs,58,59 for membrane-bound dimers of islet amyloid polypeptide (IAPP or amylin),56 and to disrupt p53/MDM binding.60 Broad-spectrum amyloid modulators have been developed based on oligothiophene,61 oligoquinolone,62,63 (D,L)-peptide64,65 and molecular tweezer66 architectures. Moreover, monomeric disordered states of targets such as c-Myc, amyloid-β and α-synuclein have been successfully targeted by identifying transiently sampled, druggable structures.54,55,57 In these works, experimentally constrained or filtered simulations were used to define an ensemble of target states, which were then subjected to computational screening followed by experimental validation.
Here, we have been able to improve on these efforts using new enhanced sampling simulation protocols for IDPs, methods to identify preferentially sampled local structures within a larger disordered ensemble, and a combination of conventional computational docking and machine learning (ML)-based screening. Using this approach (schematized in Figure 1), examining thousands of compounds computationally and just ten in vitro, we have identified one novel compound that inhibits tau aggregation as well as a second that binds to fibrils without altering the extent of aggregation.
Figure 1:
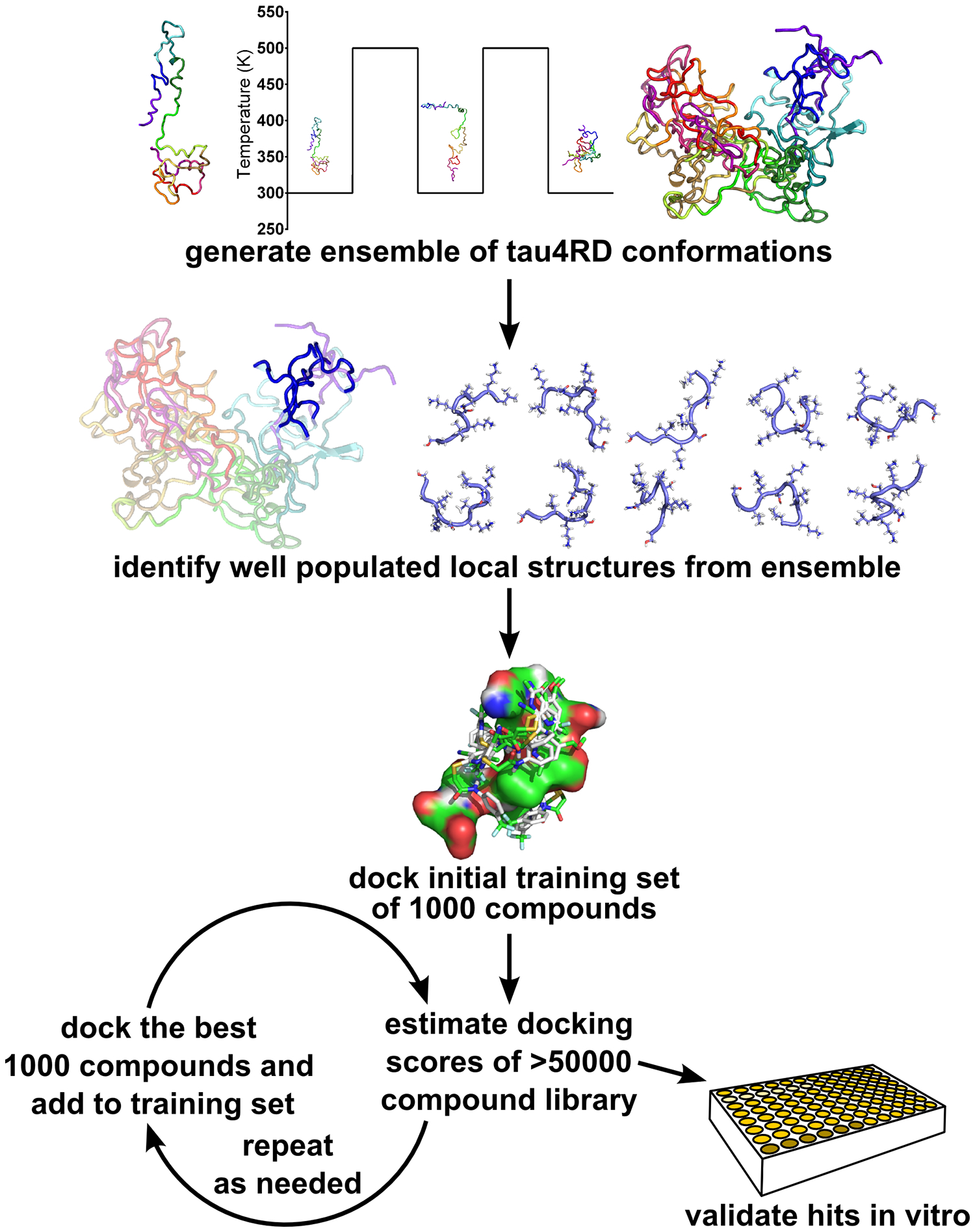
Schematic of our combined computational and experimental strategy to discover novel ligands for IDPs (see text for details).
Experimental Procedures
Molecular Dynamics Simulations
Molecular dynamics simulations including ReSA-MD were done using GROMACS 4.6.567 with the Amber99SB forcefield. Ten starting states for the MTBR were generated, one using a conformation from FRET-restrained Monte Carlo simulations,30 and the remaining nine using randomized Ramachandran accessible phi-psi angles and correcting for clashes. Systems were solvated using the tip4p-ew water model, with ten chlorine atoms were added to neutralize charge. Starting structures were energy minimized using a steepest-descent algorithm, then allowed to relax for 0.25 ps with a 0.05 fs timestep before starting ReSA simulations. ReSA was performed using PME electrostatic calculations, NVT ensemble with v-rescale thermostat, and a timestep of 5 fs. Simulations were stopped every 2 ns, then restarted, alternating the reference temperature between 300 and 500K each time.
Clustering and Small Molecule Docking
Simulation data from the 300K simulations was taken and used for clustering analysis. Clusters were determined for all 25 segments of the peptide using all-atom RMSD values. Various clustering algorithms and criteria were examined, and the GROMOS algorithm68 using an all-atom RMSD cutoff of 0.2 nm was determined to give the best representative structures based on structure homology and population of the clusters. Median structures from each of the 10 most populated clusters of each segment were converted to receptor models for docking in AutoDock Vina.69 Structures of the ChemBridge CNS collection were obtained from PubChem and parameters were calculated using OpenBabel.70 Ligand models were built using AutoDock’s ligand preparation utility. CACTVS fingerprints71 were obtained from PubChem. Ligand docking was done using AutoDock Vina, with a search area encompassing the space of a cube extending 2 nm beyond the receptor model in each dimension.
Protein Expression
Plasmids containing the His-tagged tau4RD or full-length tau2N4R genes with a TEV cleavage site were received as a gift from the Rhoades lab at the University of Pennsylvania. Plasmid was transfected into two E. coli lines: DH-5α for long-term stocks, and BL-21 for protein expression. To express protein, BL21 (DE3) cells were incubated in 1 L of Luria-Bertani (LB) medium in baffled flasks. Cells were grown in at 37°C, 220 RPM in a New Brunswick Innova 44/44R incubator until the OD reached 0.6–0.7. IPTG was then added to a final concentration of 0.4 mM, the temperature was dropped to 16°C and the culture was allowed to grow overnight. The following day, cells were harvested by centrifugation and resuspended in lysis buffer: 50 mM Tris, 500 mM NaCl, and 10 mM imidazole, pH 8 @ 4°C. Halt Protease inhibitor cocktail (Life Technologies; 300 μL), 0.1 mg/mL DNAse, 0.1 mg/mL RNAse, and 300 μL of a saturated solution of PMSF in ethanol was added per 1 L of growth before lysis via French press. The lysate was then centrifuged at 2800 g for 45 minutes. The supernatant was passed through 0.8 and 0.4 μm filters, before being loaded onto a 5 mL Ni-NTA agarose column. The protein was eluted by increasing the concentration of imidazole in the solvent from 10 to 250 mM. The eluent was buffer exchanged back into 10 μM imidazole buffer, while incubating with 1mM dithiothreitol and 250 μL of TEV protease at 2.8 mg/mL overnight at 4°C. This solution was then run over the Ni-NTA column and the flowthrough was collected and concentrated to 1–2 mL using a 3 kilodalton molecular weight cutoff centrifugal filter. The concentrate was fractionated using a 25 mL S200 extended gel-filtration column with buffer containing 20 mM Tris HCL, 50 mM NaCl, 1 mM TCEP pH 7.4 as the mobile phase. Purity was confirmed by SDS-PAGE. Protein was concentrated, aliquoted and flash frozen before being stored at −80°C. Concentrations of protein were obtained using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific).
Preparation and Storage of Selected Compounds
Selected compounds were identified from the ChemBridge CNS small molecule collection, and dry samples were obtained through Hit2Lead (ChemBridge, San Diego, CA). Samples were dissolved in DMSO to 1 mM stock solutions and stored at −80°C. The identity of active compounds was confirmed with Quantitative time of flight mass spectroscopy using an Agilent Technologies 6520 Q-TOF (Figure S6). Dynamic light scattering (DLS) was used to confirm that at experimental conditions, artifacts due to compound insolubility72 were unlikely to contribute to observed activity (Figure S7). DLS assays of compound aggregation were performed using a DynaPro NanoStar Laser Photometer and accompanying software. DMSO control and compound samples were incubated overnight at room temperature or at 37°C, and then immediately assayed.
Aggregation Assays
All fibrillization reactions were carried out at 37°C at pH 7.4, in buffer containing 20 mM Tris HCL, 50 mM NaCl, 1 mM TCEP, and 50 μM thioflavin T (ThT). 100 μL aliquots of tau4RD and ThT at twice the desired final concentration were prepared in black-walled polystyrene 96-well plates (Corning Life Sciences, Tewksbury, MA). Equal volumes of unfractionated ~3KDa heparin sodium (Acros Organics, Morris Plains, NJ) at twice the desired final concentration were added to the wells to initiate the reactions. Final concentrations of protein and heparin were 5 μM and 3 μM respectively. Reaction progress was monitored by increase in Thioflavin T fluorescence with λex = 440 nm and λem = 485 nm in a BioTek Synergy HTX (BioTek, Winooski, VT) plate reader. Reads were taken every 5 minutes after 1 minute of agitation (for tau4RD), or every 10 minutes with continuous agitation (for full-length tau). Intrinsic fluorescence from compounds was in all cases ≤ 5% of the background, which was corrected for by baseline subtraction. Experiments were performed with at least 3 technical replicates.
Tyrosine Fluorescence Assays
Tau4RD contains a single native tyrosine, which provides an intrinsic spectroscopic probe of concentration. Tyrosine fluorescence assays were performed to measure tau4RD concentration in solution using an Agilent Cary Eclipse spectrophotometer. Samples were excited at 276 nm, and fluorescence was measured from 300–310 nm. A standard curve was constructed using buffer-matched samples of tau4RD, and used to estimate soluble tau levels. Samples were incubated under the same condition as the aggregation assays listed above, save that duplicate samples were made without ThT. Initial concentrations were measured immediately after the addition of heparin. Additionally, after 3 hours of incubation, samples were centrifuged (30 minutes at 21,100 g) at 4°C, which was sufficient to separate large aggregates and bona fide fibrils from monomer and small, soluble oligomers. After centrifugation the intrinsic tyrosine fluorescence of tau in the supernatant was measured. Each experiment was run with 4 technical replicates, and the results averaged.
Gel Densitometry
Aggregation reactions were allowed to proceed for 3 hours and spun down as described above. 15 μL of the supernatant was boiled with 3 μL of loading dye and then run on an Invitrogen Bolt 12% Bis-Tris 15 well gel, along with T=0 controls and standard samples of 1, 3 and 5 μM tau4RD. Gels were scanned with a Li-Cor Odyssey CLx gel-scanner, and band intensity was analyzed using Fiji.73
Results
Our approach is depicted schematically in Figure 1. We began by performing enhanced sampling molecular dynamics on tau4RD to sample its native conformational ensemble. We then used clustering methods to identify preferentially sampled, locally structured states within this ensemble. These preferentially sampled structures were then used in computational screening procedures to identify promising compounds from a library of ~5×104 small molecules. ML techniques were used to refine and guide molecular docking calculations. Finally, we assayed ten selected compounds in terms of their effects on tau4RD amyloid formation in vitro.
While replica exchange molecular dynamics (REMD) simulations are physically rigorous and are useful for smaller IDPs, they are impractical for an IDP as large as tau4RD. (For example, using explicit solvent, approximately 200 replicas would be required to ensure an exchange probability of 0.01.74) To overcome this limitation, we developed and implemented an alternative technique we term Repeated Simulated Annealing (ReSA). In ReSA, a single MD trajectory is periodically (every 2 ns) exchanged from a low temperature (300 K) to a high temperature (500 K) and then returned to 300K. Upon raising the temperature, the target protein rapidly samples conformational space. When the temperature is lowered, the protein dynamics slow down and the simulation samples the local conformational space of the protein (Figure S1A). We ran ten 100ns simulations from 10 distinct starting positions, then sampled the 300 K portions of the ReSA trajectory every 50 ps (thereby omitting most structures sampled as tau4RD relaxed from 500 K to 300 K), resulting in 500 ns of usable conformational data. As expected, ReSA samples a broader conformational space in a given time period than conventional MD (Figure S1B). Moreover, ReSA more closely recapitulates the radius of gyration (Rg) derived from SM-FRET30 and small-angle X-ray scattering75 than conventional MD does (Figure S2D). NMR chemical shifts calculated from the ReSA ensemble are also in good agreement with experimental measurements (Figure S2). Both the predicted chemical shifts and the contact map (Figure S3A) are very similar to those obtained by extensive (4.8 μs) REMD simulations.53
After generating a library of conformations using ReSA, we used a local clustering approach to identify potential docking targets. One of the challenges in identifying targets from an IDP conformational ensemble is determining which conformations are the most frequently sampled in solution. More popular conformations are desirable as targets: the more frequently sampled a conformation is, the greater the chance it encounters a ligand. However, identifying well-sampled conformations for dynamic protein targets is not straightforward. Conformational fluctuations in distant, unrelated parts of the protein can cause the clustering algorithm to classify two conformations as distinct and relatively uninteresting even if they both displayed the same local structural motif. Because our conformational library displayed few long-range contacts, we expect local structures to be relevant targets (Figure S3). Attempts to generate clusters based on global conformational similarity (e.g., Cα RMSD for the entire tau4RD sequence) would overlook bona fide, preferentially sampled local structures. Instead, we performed clustering on overlapping segments of tau4RD extracted from the larger ReSA ensemble. We tested segment lengths of 5, 10 and 20 residues, and found that ten-residue segments (Figure 2) provided the best compromise between cluster resolution and cluster population. This approach allowed us to model the conformations of tau4RD in the context of the complete molecule, yet also identify relevant local structures. After experimentation with different clustering methods and all-atom RMSD cutoff values, we clustered our structures using the GROMOS geometric clustering method68 using a RMSD cutoff of 0.2 nm. This allowed for the identification of well-resolved preferentially sampled structures for each of the 10-residue segments (Figure S4).
Figure 2:
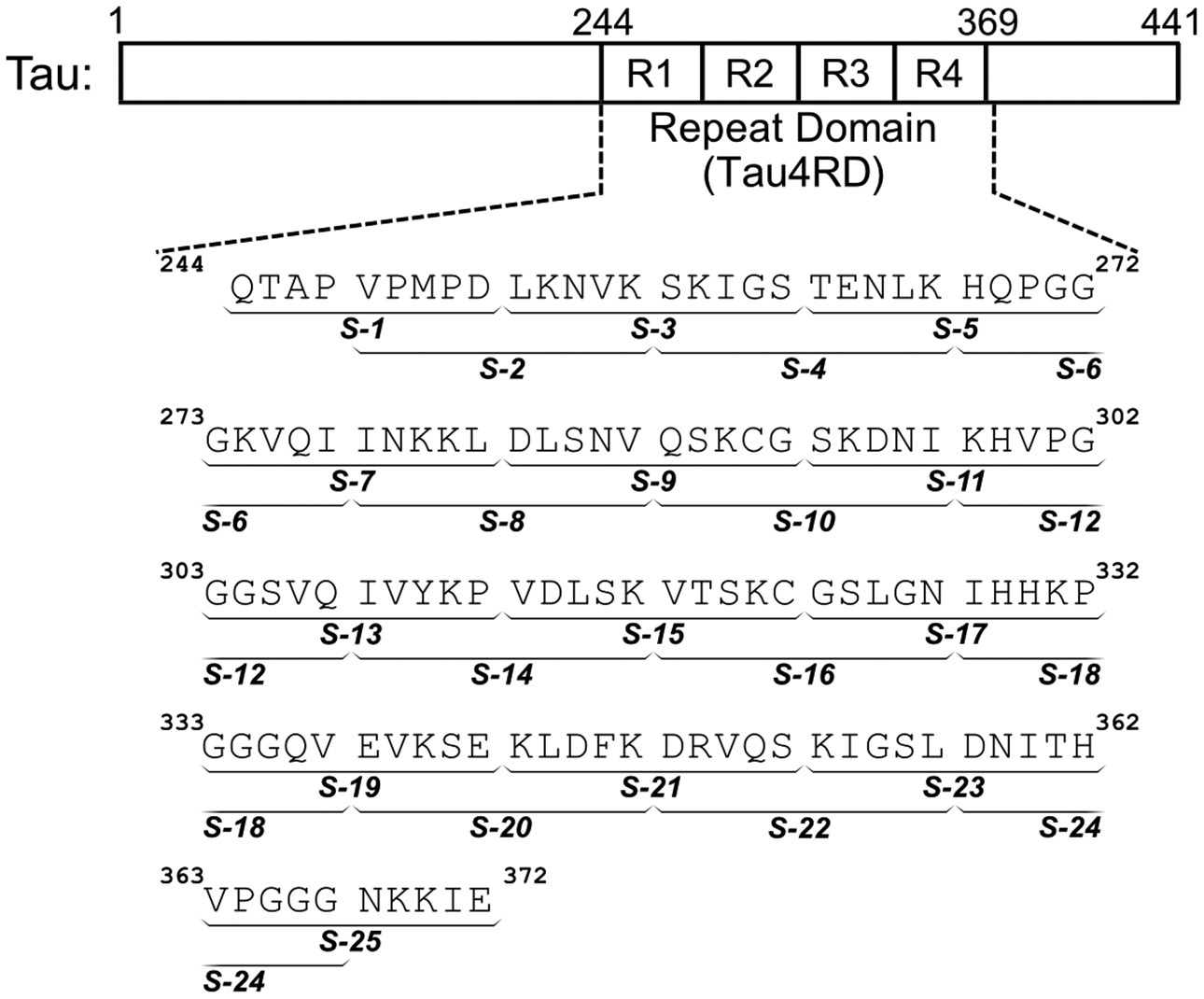
Sequence of tau4RD and segments used for local clustering. Tau4RD spans the 4-repeat microtubule-binding domain of the IDP tau, and models the full length protein’s capacity for aggregation and membrane interaction. To identify well-populated local structures within the conformational ensemble of tau4RD, the sequence was divided into 25 overlapping segments (denoted S-1 through S-25) and each segment was clustered independently.
We used these structures as docking targets for virtual screening of small molecules. Each of the 10 most populated clusters for each of the 25 segments was used as a target for computational docking. Each cluster was representative of 0.5% to 4% of the conformational library. A pilot set of 1000 compounds of the ChemBridge CNS collection was docked against the 250 targets using AutoDock Vina. These results showed that compounds preferentially bound to particular regions of the tau4RD (Figure 3A), suggesting that these segments are essentially the most “druggable” portions of the protein. We also found that similar docking results are obtained when targets were generated from either half of the ReSA 300K dataset (Figure S4C), which suggests that the ReSA conformational ensemble has converged sufficiently for our purpose of ligand discovery.
Figure 3:
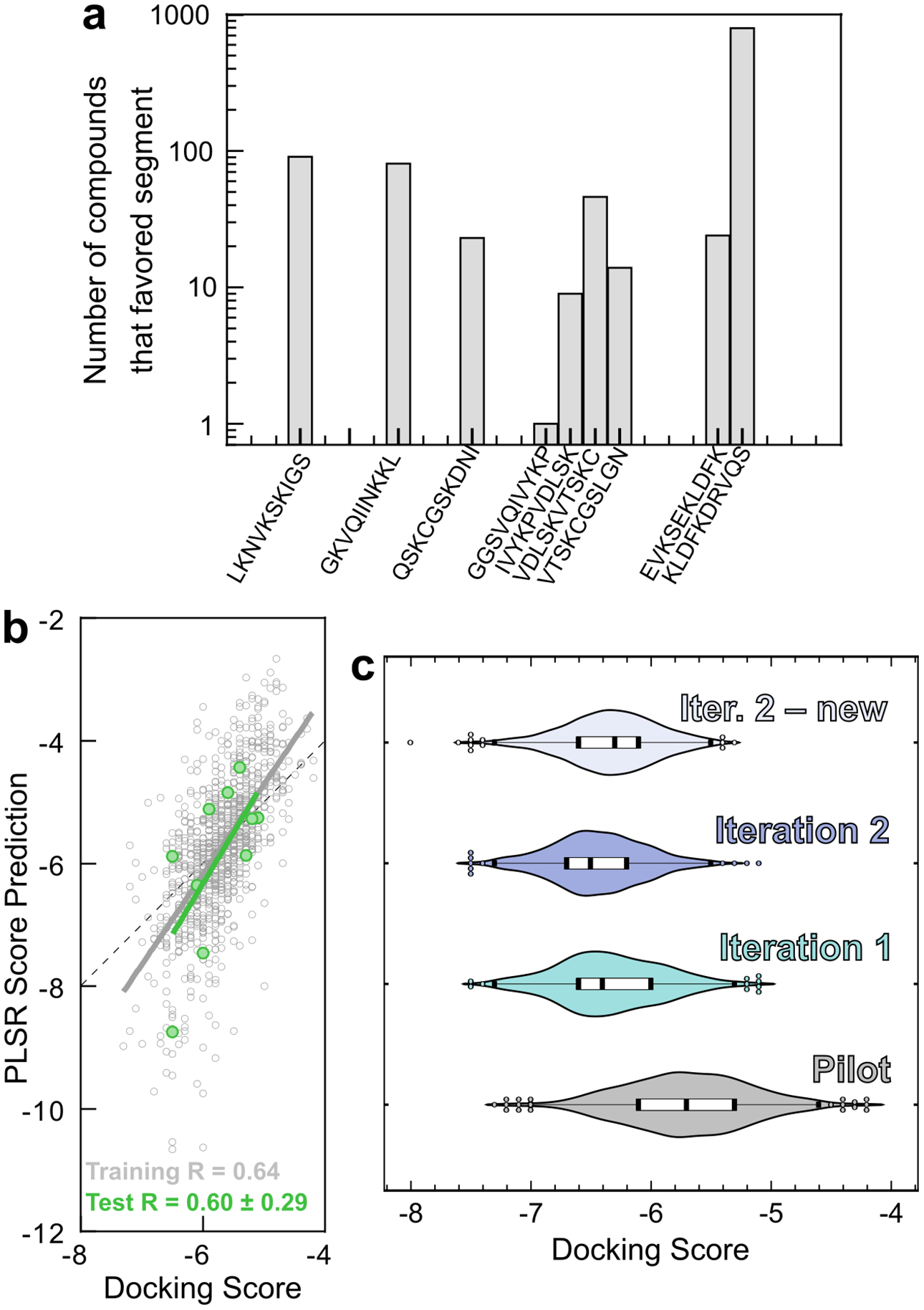
Discovery of potentially active compounds through iterative machine learning. a) A pilot set of 1000 compounds randomly chosen from the CNS library was docked against the 10 most populated clusters from each of the 25 segments. The histogram of each compound’s most favored target illustrates that some segments of tau4RD are better able to accommodate small molecule binding. b) PLSR trained on 990 compounds from the pilot set was able to predict the docking scores of the remaining 10 compounds with reasonable accuracy. Pearson correlation coefficients are shown at the bottom of the panel. c) PLSR was used to select compounds from the diverse set of small molecules in the CNS library. Iteration 1 yielded 1000 compounds based on the results and fingerprints of the pilot set. Iteration 2, based on the docking scores and fingerprints of the 2000 compounds from the pilot set and Iteration 1, yielded primarily compounds that had already been examined and docked in Iteration 1. When these were excluded from the library, the next 1000 compounds (“Iter. 2 – new”) displayed worse docking scores on average. This indicated that the PLSR search had largely converged.
It would be computationally prohibitive to dock larger (> 104 compound) libraries to all 250 targets. Therefore, we used ML to mine large compound collections and prioritize small molecules for more detailed computational screening. ML techniques can robustly and efficiently predict diverse aspects of chemical function from simplified descriptions of a compound’s structure.56,76–79 We used CACTVS chemical fingerprints80 to quantify the presence of chemical features of the compounds in the CNS collection, then used partial least squares regression (PLSR) analysis to correlate the presence and absence of chemical features with docking scores. A CACTVS fingerprint is an 882-bit binary string, with a 0 or 1 at each position corresponding to the absence or presence of a particular chemical feature in a compound of interest. PLSR is a versatile technique capable of identifying the relationships between two matrices, one of which is dependent in some way on the other.76 PLSR simultaneously decomposes those matrices to identify patterns of covariance to build a regression matrix that, when multiplied by the independent matrix, approximates the dependent matrix. In our case, the independent matrix F consists of the CACTVS fingerprints for each compound in a training set, while the dependent matrix S consists of docking scores for each compound in that training set to each target in a panel of ReSA-derived clusters. PLSR yields a prediction matrix P such that F⋅P≈S. If a vector f consists of the fingerprint of a new compound not in the training set, then the vector s=f⋅P will contain the predicted docking score of that compound for each target in the panel. PLSR thus approximates the results of multiple, computationally intensive docking calculations with a single matrix multiplication step.
We validated our PLSR predictions using a subsampling approach. Briefly, 10 randomly selected molecules were withheld from the training set, and the model was trained with the remaining 990 (Figure 3B). We then compared the predicted scores of the 10 test compounds with docking scores generated by Autodock Vina. We repeated this subsampling ten times, each with a different set of 10 test compounds. The Pearson (Spearman rank-order) correlations between PLSR predictions and docking scores were 0.60 ± 0.29 (0.58 ± 0.20), as compared to 0.64 (0.67) for the training sets. This indicated that PLSR was able to predict the docking scores of unknown compounds with reasonable accuracy.
We then applied our validated PLSR protocol to predict docking scores for all 50,000 compounds in the ChemBridge CNS library, and selected the top 1000 molecules for another round of docking. For computational efficiency, all subsequent docking calculations were performed only with the segments of tau4RD identified to be druggable (Figure 3A). The docking results of these 1000 compounds were used to update the PLSR training set, and the PLSR predictions were repeated. The 1000 compounds predicted to have the best docking scores were again selected for docking, and this process was repeated, at which point the proportion of new compounds (i.e., not identified in previous rounds) dropped to ~40% and the docking scores of the best compounds stayed approximately constant. At this stage, we considered the ML screen to have converged, and ranked all compounds in terms of their docking score for any segment of tau4RD (Figure 3C). High-scoring compounds tended to be hydrophobic (mean clogP of the top 1% of compounds was 3.8, as compared to 3.1 for the entire compound library), and to feature heteroatomic conjugated systems. Selected docked poses are shown in Figure S5.
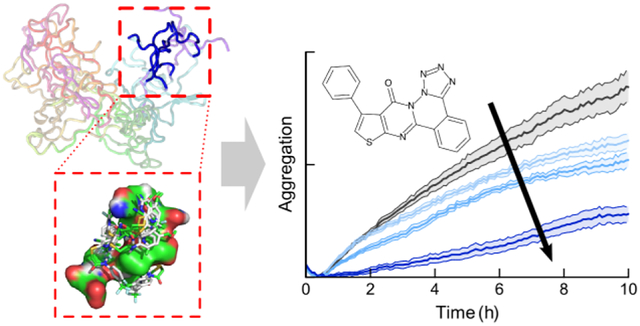
We selected 10 compounds (see Table S1) for experimental testing based on docking score, targeted segment, and chemical structure, so as to prioritize high-affinity ligands that bound different regions of tau4RD and sampled diverse regions of chemical space. These 10 compounds were tested for their effects on the pathological aggregation of tau4RD using a kinetic assay that relies on the enhancement of ThT fluorescence when bound to amyloid structure. We compared the aggregation of tau4RD in the presence and absence of each compound and found that 1 and 6 markedly altered the observed trajectory (Figure 4). Compound 1 delayed and decreased the ThT fluorescence in a dose-dependent manner, and compound 6 increased the magnitude of ThT fluorescence in a dose dependent manner (In contrast, 0 of 10 compounds randomly selected from the ChemBridge CNS library showed any activity). To corroborate the results of the ThT assay, we centrifuged aggregation reactions after 3 hours and measured the amount of soluble tau4RD remaining in the supernatant using intrinsic tyrosine fluorescence. Compound 1 consistently decreased the amount of aggregated protein (confirmed by gel densitometry, Figure S8), while 6 had no effect. These results suggest that 1 is a bona fide aggregation inhibitor, while 6’s effects on the ThT fluorescence are likely photophysical in origin. Since this signal results from fibril-bound ThT, the enhancement of fluorescence suggests that 6 does interact with tau4RD fibrils without affecting the rate or extent of aggregation. In contrast, 1 is a fairly potent aggregation inhibitor, capable of delaying aggregation substoichiometrically. However, despite this effect on kinetics, neither 1 nor 6 appears to affects the total amount of aggregate formed once the reaction has plateaued (see 96 h data in Figure S9).
Figure 4:
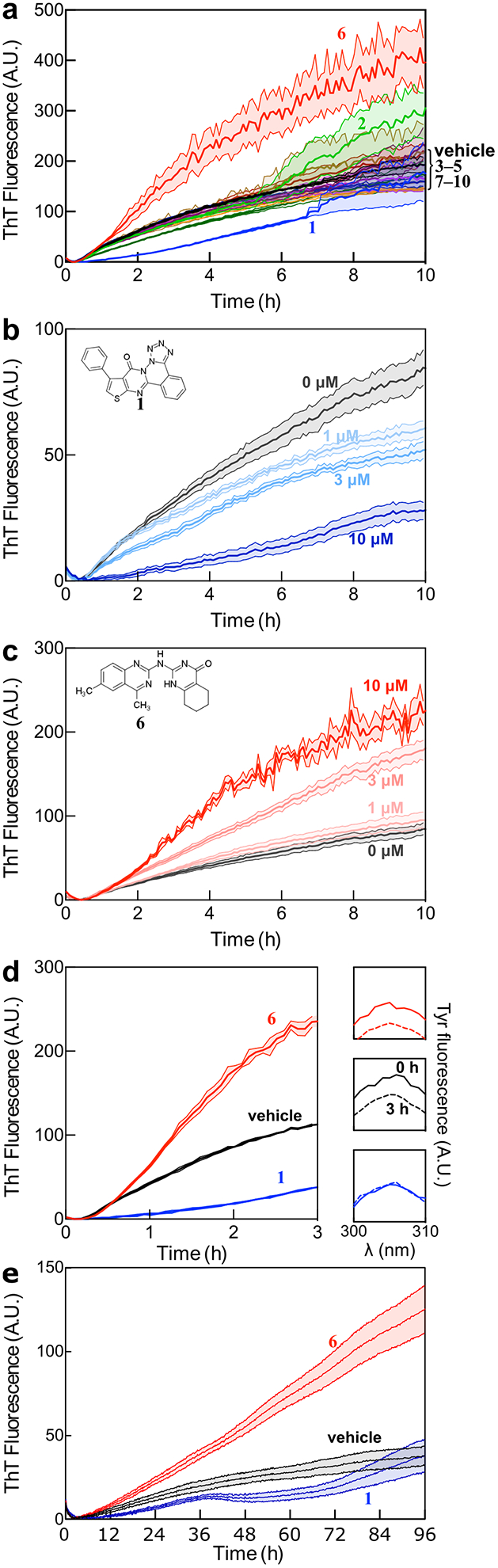
Experimental validation of selected compounds. a) ThT-monitored tau4RD fibril formation kinetics in the presence of 10 compounds selected based on computational screening. Compounds 1 and 6 consistently altered the observed fluorescence kinetics relative to vehicle control. Traces show mean ± SEM of 3 technical replicates, and are representative of three independent experiments. b) Compound 1 extends the lag phase and decreases ThT fluorescence intensity in a dose-dependent manner. c) Compound 6 increases ThT fluorescence in a dose-dependent manner, but does not dramatically affect the lag phase. For B and C, traces show mean ± SEM of 4 technical replicates, and are representative of three independent experiments. d) Measurement of soluble tau content after 3 hours of aggregation indicates that 1 but not 6 affects the extent of aggregation. The main panel shows ThT-monitored aggregation, while the panels on the right show the amount of tau remaining in solution after centrifugation, measured by intrinsic tyrosine fluorescence. Treatment with Compound 1 (blue) shows that soluble tau4RD content is unchanged over the course of the experiment. In contrast, treatments with either Compound 6 (red) or vehicle (black) show a similar loss of fluorescence intensity. e) Compounds 1 and 6 exhibit similar effects on ThT-monitored aggregation of full-length tau, with 1 delaying aggregation and 6 appearing to raise the final level of fluorescence. For D and E, traces show mean ± SEM of 4 or 5 technical replicates respectively, and are representative of at least three independent experiments. Concentration of compounds was set at 10 μM unless otherwise indicated and the concentration of DMSO for all samples was 1%.
The tau4RD construct has been extensively studied in vitro, in part because it forms fibrils much more readily than full-length tau. In order to determine whether 1 and 6 retain their activity towards biologically relevant forms of tau, we studied the aggregation of full-length (2N4R) tau in the presence and absence of each compound. The response of full-length tau to these modulators mirrored that of tau4RD, albeit on much slower timescales: 1 delayed the onset of aggregation, while 6 increased ThT fluorescence intensity without seeming to affect the kinetics of aggregation in any systematic way. This suggests that 1 and 6 could be useful reagents in cellular and animal models of tau pathology, whether studying full-length tau or more experimentally tractable constructs.
Importantly, both 1 and 6 are chemically quite distinct from any other compound known to affect amyloid formation by tau or any other protein. This suggests that our approach has indeed enabled us to explore new regions of chemical space for tau aggregation inhibitors.
Discussion
The role of computation in ligand discovery is to reduce the amount of in vitro testing needed to identify active compounds. High throughput screening for IDP ligands is often challenging because binding or other functional outcomes can be difficult to detect. Rates of success for these undirected screening are as low as 0.04%.81 As such, computational approaches are appealing in that much of the selection process can be shifted to the relatively cheap and quick in silico methods. Recent attempts have aimed to identify targets (often using computational methods guided by in vitro measurements), and then use computational docking approaches to identify a limited number of promising compounds. For example, Yu et al. targeted the IDP c-Myc based on models built from molecular dynamics biased with a known inhibitor. Their computational screening identified 273 compounds from which they identified 7 novel ligands.57 Vendruscolo et al. used clustering methods to identify the most populated global conformations in REMD simulations of α-synuclein, and then used fragment mapping to identify binding hotspots. From computational docking to these hotspots, the authors identified 89 compounds for in vitro characterization, and reported one biologically active compound capable of rescuing dopaminergic cells in a Parkinson’s cell model.55
The process we described here distinguishes itself in three major ways. Firstly, appropriate and efficient enhanced sampling molecular dynamics methods explored conformational space and generated a valuable conformational library. Secondly, the segmented clustering approach allowed the identification of well-populated local conformations that serve as appealing ligand targets. Thirdly, machine learning techniques identified and utilized chemical characteristics that focused screening attempts and drastically increased the efficiency of computational screening. Our approach identified two novel tau ligands with promising activity – 1 as an aggregation inhibitor, and 6 as a fibril-binding ligand – out of just ten compounds characterized in vitro. This suggests that our methodology successfully identified promising targets within a dynamic conformational ensemble and used this information to select compounds likely to interact with an IDP target. Future studies will examine the biological activities of 1, 6 and related compounds in cellular and in vivo assays of tau pathology. Importantly, while the compounds identified here are novel and interesting, they will require substantial further development to become lead compounds in drug development efforts. Bioavailability and other pharmacokinetic/pharmacodynamic parameters will need to be optimized, and so will selectivity for tau over other amyloid-forming proteins. Nevertheless, we believe that the development of 1 and 6 represents an exciting advance in the collective effort to improve diagnosis and treatment of AD and other tauopathies. We anticipate that screening larger compound collections using the methods described here will yield multiple, distinct modulators and probes of tau aggregation.
The work presented here provides a framework that can easily be applied to other disordered protein targets. ReSA is easily adaptable to work on other systems, and does not have extensive computational requirements beyond those of canonical molecular dynamics. We stress that ReSA is not likely to generate a complete, physically rigorous landscape of an IDP conformational ensemble, but nevertheless is evidently able to model an IDP at sufficient resolution and accuracy for rational ligand discovery. The segmentation and clustering approach is also well suited for other larger IDPs, although the parameters for clustering will need to be adjusted to accommodate for the flexibility and local structural diversity of the system being examined. Our hope is that this work provides valuable tools to the research community that aid in the development of ligands capable of modulating IDP function or dysfunction, so as to gain insight into the mechanisms of toxicity, and to advance the eventual development of therapeutics and diagnostics for IDP-mediated pathologies.
Supplementary Material
Acknowledgements
We thank Hannah Baughman for helpful discussions and assistance with NMR chemical shift comparisons, and gratefully acknowledge Prof. Elizabeth Rhoades (University of Pennsylvania) for the gift of tau4RD plasmids.
Funding Sources
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number T32-GM007750 (to D.W.B.), by the University of Washington Royalty Research Fund (to A.N.) and by the Brady Fund for Natural Products Research (to A.N.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- DLS
dynamic light scattering
- FRET
Förster resonance energy transfer
- IDP
intrinsically disordered protein
- IDR
intrinsically disordered region
- ML
machine learning
- MTBR
microtubule-binding region
- PLSR
partial least-squares regression
- REMD
replica exchange molecular dynamics
- ReSA
repeated simulated annealing
- RMSD
root-mean-square deviation
- ThT
thioflavin T
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. It comprises a PDF file with further analysis of ReSA and conventional MD simulations, and details on all ten compounds screened in vitro.
References
- (1).Uversky VN, Oldfield CJ, and Dunker AK (2008) Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys 37, 215–246. [DOI] [PubMed] [Google Scholar]
- (2).Cheng Y, LeGall T, Oldfield CJ, Mueller JP, Van Y-YJ, Romero P, Cortese MS, Uversky VN, and Dunker AK (2006) Rational drug design via intrinsically disordered protein. Trends Biotechnol 24, 435–42. [DOI] [PubMed] [Google Scholar]
- (3).Ambadipudi S, and Zweckstetter M (2016) Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov 11, 65–77. [DOI] [PubMed] [Google Scholar]
- (4).Nath A, and Rhoades E (2013) A flash in the pan: dissecting dynamic amyloid intermediates using fluorescence. FEBS Lett 587, 1096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dunker AK, Cortese MS, Romero P, Iakoucheva LM, and Uversky VN (2005) Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J 272, 5129–5148. [DOI] [PubMed] [Google Scholar]
- (6).Cortese MS, Uversky VN, and Dunker AK (2008) Intrinsic disorder in scaffold proteins: getting more from less. Prog Biophys Mol Biol 98, 85–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Uversky VN (2016) p53 proteoforms and intrinsic disorder: An illustration of the protein structure-function continuum concept. Int. J. Mol. Sci 17, 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mark W-Y, Liao JCC, Lu Y, Ayed A, Laister R, Szymczyna B, Chakrabartty A, and Arrowsmith CH (2005) Characterization of segments from the central region of BRCA1: An intrinsically disordered scaffold for multiple protein–protein and protein–DNA interactions? J. Mol. Biol 345, 275–287. [DOI] [PubMed] [Google Scholar]
- (9).Weber SC, and Brangwynne CP (2012) Getting RNA and protein in phase. Cell 149, 1188–1191. [DOI] [PubMed] [Google Scholar]
- (10).Lemke EA (2016) The multiple faces of disordered nucleoporins. J. Mol. Biol 428, 2011–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Melo AM, Coraor J, Alpha-Cobb G, Elbaum-Garfinkle S, Nath A, and Rhoades E (2016) A functional role for intrinsic disorder in the tau-tubulin complex. Proc. Natl. Acad. Sci. U. S. A 113, 14336–14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Stroberg W, and Schnell S (2017) On the origin of non-membrane-bound organelles, and their physiological function. J. Theor. Biol 434, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mackenzie IR, Rademakers R, and Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9, 995–1007. [DOI] [PubMed] [Google Scholar]
- (14).Metskas LA, and Rhoades E (2016) Order-disorder transitions in the cardiac troponin complex. J. Mol. Biol 428, 2965–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Eisenberg D, and Jucker M (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Knowles TPJ, Vendruscolo M, and Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol 15, 384–396. [DOI] [PubMed] [Google Scholar]
- (17).Drubin DG, and Kirschner MW (1986) Tau protein function in living cells. J. Cell Biol 103, 2739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, Mandelkow E, and Zweckstetter M (2015) Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. U. S. A 112, 7501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Santarella RA, Skiniotis G, Goldie KN, Tittmann P, Gross H, Mandelkow E-M, Mandelkow E, and Hoenger A (2004) Surface-decoration of microtubules by human tau. J. Mol. Biol 339, 539–553. [DOI] [PubMed] [Google Scholar]
- (20).Lee G, and Leugers CJ (2012) Tau and tauopathies. Prog. Mol. Biol. Transl. Sci 107, 263–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Flach K, Hilbrich I, Schiffmann A, Gärtner U, Krüger M, Leonhardt M, Waschipky H, Wick L, Arendt T, and Holzer M (2012) Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem 287, 43223–43233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Elbaum-Garfinkle S, Ramlall T, and Rhoades E (2010) The role of the lipid bilayer in tau aggregation. Biophys. J 98, 2722–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ait-Bouziad N, Lv G, Mahul-Mellier A-L, Xiao S, Zorludemir G, Eliezer D, Walz T, and Lashuel HA (2017) Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun 8, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Barghorn S, Biernat J, and Mandelkow E (2005) Purification of recombinant tau protein and preparation of Alzheimer-paired helical filaments in vitro. Methods Mol. Biol 299, 35–51. [DOI] [PubMed] [Google Scholar]
- (25).Künze G, Barré P, Scheidt HA, Thomas L, Eliezer D, and Huster D (2012) Binding of the three-repeat domain of tau to phospholipid membranes induces an aggregated-like state of the protein. Biochim. Biophys. Acta - Biomembr 1818, 2302–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ganguly P, Do TD, Larini L, Lapointe NE, Sercel AJ, Shade MF, Feinstein SC, Bowers MT, and Shea JE (2015) Tau assembly: The dominant role of PHF6 (VQIVYK) in microtubule binding region repeat R3. J. Phys. Chem. B 119, 4582–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow E-M, Mandelkow E, and Kirschner MW Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306 VQIVYK 311) forming β structure [DOI] [PMC free article] [PubMed]
- (28).Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Anthony Crowther R, Ghetti B, Goedert M, and Scheres SHW (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Baughman HER, Clouser AF, Klevit RE, and Nath A (2018) HspB1 and Hsc70 chaperones engage distinct tau species and have different inhibitory effects on amyloid formation. J. Biol. Chem 293, 2687–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nath A, Sammalkorpi M, DeWitt DC, Trexler AJ, Elbaum-Garfinkle S, O’Hern CS, and Rhoades E (2012) The conformational ensembles of α-synuclein and tau: combining single-molecule FRET and simulations. Biophys. J 103, 1940–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Elbaum-Garfinkle S, and Rhoades E (2012) Identification of an aggregation-prone structure of tau. J. Am. Chem. Soc 134, 16607–16613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ambadipudi S, Biernat J, Riedel D, Mandelkow E, and Zweckstetter M (2017) Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein tau. Nat. Commun 8, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Campos HC, da Rocha MD, Viegas FPD, Nicastro PC, Fossaluzza PC, Fraga CAM, Barreiro EJ, and Viegas C (2011) The role of natural products in the discovery of new drug candidates for the treatment of neurodegenerative disorders I: Parkinson’s disease. CNS Neurol. Disord. Drug Targets 10, 239–50. [DOI] [PubMed] [Google Scholar]
- (34).Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S, Goedert M, and Hasegawa M (2006) Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry 45, 6085–94. [DOI] [PubMed] [Google Scholar]
- (35).Lakey-Beitia J, Berrocal R, Rao KS, and Durant AA (2015) Polyphenols as therapeutic molecules in Alzheimer’s disease through modulating amyloid pathways. Mol. Neurobiol 51, 466–479. [DOI] [PubMed] [Google Scholar]
- (36).Alavez S, Vantipalli MC, Zucker DJS, Klang IM, and Lithgow GJ (2011) Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 472, 226–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ono K, and Yamada M (2006) Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J. Neurochem 97, 105–15. [DOI] [PubMed] [Google Scholar]
- (38).Hauber I, Hohenberg H, Holstermann B, Hunstein W, and Hauber J (2009) The main green tea polyphenol epigallocatechin-3-gallate counteracts semen-mediated enhancement of HIV infection. Proc Natl Acad Sci U S A 106, 9033–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Akoury E, Gajda M, Pickhardt M, Biernat J, Soraya P, Griesinger C, Mandelkow E, and Zweckstetter M (2013) Inhibition of tau filament formation by conformational modulation. J. Am. Chem. Soc 135, 2853–2862. [DOI] [PubMed] [Google Scholar]
- (40).Ballatore C, Brunden KR, Piscitelli F, James MJ, Crowe A, Yao Y, Hyde E, Trojanowski JQ, Lee VM-Y, and Smith AB (2010) Discovery of brain-penetrant, orally bioavailable aminothienopyridazine inhibitors of tau aggregation. J. Med. Chem 53, 3739–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Akoury E, Pickhardt M, Gajda M, Biernat J, Mandelkow E, and Zweckstetter M (2013) Mechanistic basis of phenothiazine-driven inhibition of tau aggregation. Angew. Chemie Int. Ed 52, 3511–3515. [DOI] [PubMed] [Google Scholar]
- (42).Salmon L, Nodet G, Ozenne V, Yin G, Jensen MR, Zweckstetter M, and Blackledge M (2010) NMR characterization of long-range order in intrinsically disordered proteins. J. Am. Chem. Soc 132, 8407–8418. [DOI] [PubMed] [Google Scholar]
- (43).Grupi A, and Haas E (2011) Time resolved FRET detection of subtle temperature induced conformational biases in ensembles of α-synuclein molecules. J. Mol. Biol 411, 234–247. [DOI] [PubMed] [Google Scholar]
- (44).Drescher M (2011) EPR in protein science. Top. Curr. Chem 321, 91–119. [DOI] [PubMed] [Google Scholar]
- (45).Soranno A, Longhi R, Bellini T, and Buscaglia M (2009) Kinetics of contact formation and end-to-end distance distributions of swollen disordered peptides. Biophys. J 96, 1515–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ferrie JJ, Haney CM, Yoon J, Pan B, Lin Y-C, Fakhraai Z, Rhoades E, Nath A, and Petersson EJ (2018) Using a FRET library with multiple probe pairs to drive Monte Carlo simulations of α-synuclein. Biophys. J 114, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Scheraga HA, Khalili M, and Liwo A (2007) Protein-folding dynamics: overview of molecular simulation techniques. Annu. Rev. Phys. Chem 58, 57–83. [DOI] [PubMed] [Google Scholar]
- (48).Leone V, Marinelli F, Carloni P, and Parrinello M (2010) Targeting biomolecular flexibility with metadynamics. Curr. Opin. Struct. Biol 20, 148–54. [DOI] [PubMed] [Google Scholar]
- (49).Vitalis A, and Pappu RV (2009) ABSINTH: a new continuum solvation model for simulations of polypeptides in aqueous solutions. J. Comput. Chem 30, 673–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Vitalis A, and Pappu RV (2009) Methods for Monte Carlo simulations of biomacromolecules. Annu. Rep. Comput. Chem 5, 49–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Fisher CK, Huang A, and Stultz CM (2010) Modeling intrinsically disordered proteins with Bayesian statistics. J. Am. Chem. Soc 132, 14919–14927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Ozenne V, Schneider R, Yao M, Huang J, Salmon L, Zweckstetter M, Jensen MR, and Blackledge M (2012) Mapping the potential energy landscape of intrinsically disordered proteins at amino acid resolution. J. Am. Chem. Soc 134, 15138–15148. [DOI] [PubMed] [Google Scholar]
- (53).Luo Y, Ma B, Nussinov R, and Wei G (2014) Structural insight into tau protein’s paradox of intrinsically disordered behavior, self-acetylation activity, and aggregation. J. Phys. Chem. Lett 5, 3026–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhu M, De Simone A, Schenk D, Toth G, Dobson CM, and Vendruscolo M (2013) Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J. Chem. Phys 139, 035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Tóth G, Gardai SJ, Zago W, Bertoncini CW, Cremades N, Roy SL, Tambe MA, Rochet JC, Galvagnion C, Skibinski G, Finkbeiner S, Bova M, Regnstrom K, Chiou SS, Johnston J, Callaway K, Anderson JP, Jobling MF, Buell AK, Yednock TA, Knowles TP, Vendruscolo M, Christodoulou J, Dobson CM, Schenk D, and McConlogue L (2014) Targeting the intrinsically disordered structural ensemble of α-synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLoS One 9, e87133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Nath A, Schlamadinger DE, Rhoades E, and Miranker AD (2015) Structure-based small molecule modulation of a pre-amyloid state: Pharmacological enhancement of IAPP membrane-binding and toxicity. Biochemistry 54, 3555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yu C, Niu X, Jin F, Liu Z, Jin C, and Lai L (2016) Structure-based inhibitor design for the intrinsically disordered protein c-myc. Sci. Rep 6, 22298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Münch J, Baker D, and Eisenberg D (2011) Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 475, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Seidler PM, Boyer DR, Rodriguez JA, Sawaya MR, Cascio D, Murray K, Gonen T, and Eisenberg DS (2017) Structure-based inhibitors of tau aggregation. Nat. Chem 10, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Yu X, Narayanan S, Vazquez A, and Carpizo DR (2014) Small molecule compounds targeting the p53 pathway: are we finally making progress? 19, 1055–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Civitelli L, Sandin L, Nelson E, Khattak SI, Brorsson A-C, and Kågedal K (2016) The luminescent oligothiophene p-FTAA converts toxic Aβ 1–42 species into nontoxic amyloid fibers with altered properties. J. Biol. Chem 291, 9233–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Kumar S, and Miranker AD (2013) A foldamer approach to targeting membrane bound helical states of islet amyloid polypeptide. Chem. Commun. (Camb). 49, 4749–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kumar S, Brown MA, Nath A, and Miranker AD (2014) Folded small molecule manipulation of islet amyloid polypeptide. Chem. Biol 21, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Kellock J, Hopping G, Caughey B, and Daggett V (2016) Peptides composed of alternating L- and D-amino acids inhibit amyloidogenesis in three distinct amyloid systems independent of sequence. J. Mol. Biol 428, 2317–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Bleem A, Francisco R, Bryers JD, and Daggett V (2017) Designed α-sheet peptides suppress amyloid formation in Staphylococcus aureus biofilms. NPJ Biofilms Microbiomes 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Sinha S, Lopes DHJ, Du Z, Pang ES, Shanmugam A, Lomakin A, Talbiersky P, Tennstaedt A, McDaniel K, Bakshi R, Kuo P-Y, Ehrmann M, Benedek GB, Loo JA, Klärner F-G, Schrader T, Wang C, and Bitan G (2011) Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J. Am. Chem. Soc 133, 16958–16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Hess B, Kutzner C, van der Spoel D, and Lindahl E (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 4, 435–447. [DOI] [PubMed] [Google Scholar]
- (68).Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, and Mark AE (1999) Peptide folding: When simulation meets experiment. Angew. Chemie Int. Ed 38, 236–240. [Google Scholar]
- (69).Trott O, and Olson AJ (2010) AutoDock Vina. J. Comput. Chem 31, 445–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, and Hutchison GR (2011) Open Babel: an open chemical toolbox. J. Cheminform 3, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang J, Yu B, Zhang J, and Bryant SH (2016) PubChem substance and compound databases. Nucleic Acids Res 44, D1202–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Shoichet BK (2006) Screening in a spirit haunted world. Drug Discov. Today 11, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, and Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Patriksson A, and van der Spoel D (2008) A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys 10, 2073. [DOI] [PubMed] [Google Scholar]
- (75).Mylonas E, Hascher A, Bernadó P, Blackledge M, Mandelkow E, and Svergun DI (2008) Domain conformation of tau protein studied by solution small-angle X-ray scattering. Biochemistry 47, 10345–53. [DOI] [PubMed] [Google Scholar]
- (76).Abdi H (2003) Partial Least Squares (PLS) regression, in Encyclopedia of Social Science Research Methods (Lewis-Beck M, Bryman A, and Futing T, Eds.), pp 792–795. Sage, Thousand Oaks, CA. [Google Scholar]
- (77).Plewczynski D, Spieser SAH, and Koch U (2009) Performance of machine learning methods for ligand-based virtual screening. Comb. Chem. High Throughput Screen 12, 358–68. [DOI] [PubMed] [Google Scholar]
- (78).Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, and Duchesnay É (2011) Scikit-learn: Machine learning in Python. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
- (79).Nath A, Zientek MA, Burke BJ, Jiang Y, and Atkins WM (2010) Quantifying and predicting the promiscuity and isoform specificity of small-molecule cytochrome P450 inhibitors. Drug Metab. Dispos 38, 2195–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Ihlenfeldt WD, Takahashi Y, Abe H, and Sasaki S (1994) Computation and management of chemical properties in CACTVS: An extensible networked approach toward modularity and compatibility. J. Chem. Inf. Comput. Sci 34, 109–116. [Google Scholar]
- (81).Bulic B, Pickhardt M, Mandelkow EM, and Mandelkow E (2010) Tau protein and tau aggregation inhibitors. Neuropharmacology 59, 276–289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.