

SUMMARY
Piezo1 ion channels are activated by mechanical stimuli and mediate the sensing of blood flow. Although cryo-electron microscopy (cryo-EM) structures have revealed the overall architecture of Piezo1, the precise domains involved in activation and subsequent inactivation have remained elusive. Here, we perform a targeted chimeric screen between Piezo1 and the closely related isoform Piezo2 and use electrophysiology to characterize their inactivation kinetics during mechanical stimulation. We identify three small subdomains within the extracellular cap that individually can confer the distinct kinetics of inactivation of Piezo2 onto Piezo1. We further show by cysteine crosslinking that conformational flexibility of these subdomains is required for mechanical activation to occur and that electrostatic interactions functionally couple the cap to the extensive blades, which have been proposed to function as sensors of membrane curvature and tension. This study provides a demonstration of internal gating motions involved in mechanotransduction by Piezo1.
Graphical Abstract
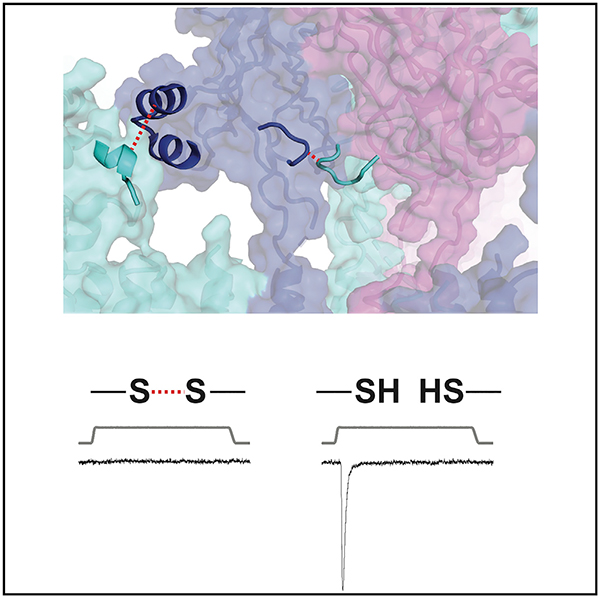
In Brief
Lewis and Grandl combine a chimeric screen and cysteine crosslinking to identify small subdomains of the cap of mechanically activated Piezo1 ion channels that must have conformational flexibility for mechanical gating. They further show that electrostatic interactions couple one of these domains to the channel blade.
INTRODUCTION
Piezos are non-selective cation channels that open in response to mechanical forces (Wu et al., 2017a). In vertebrates, there are two isoforms that together mediate a wide variety of mechanotransduction processes. For example, Piezo1 is expressed in the vascular system, where it senses shear stress and vascular flow in order to regulate blood pressure, arterial remodeling, and blood cell volume (Cahalan et al., 2015; Retailleau et al., 2015; Wang et al., 2016; Zeng et al., 2018), as well as in neural stem cells, where it senses cellular traction forces and local environment to control differentiation (Blumenthal et al., 2014; Del Mármol et al., 2018; Ellefsen et al., 2019; Pathak et al., 2014). In contrast, Piezo2 is highly expressed in sensory tissue, including Merkel cells, where it initiates the organism’s detection of light touch, as well as neurons of the nodose ganglia, where it senses airway stretch to regulate respiration (Ikeda and Gu, 2014; Nonomura et al., 2017; Woo et al., 2014).
Partial cryo-electron microscopy (cryo-EM) structures for Piezo1 and Piezo2 revealed that both isoforms share the same general architecture: both channels are homotrimers with large curved blades (36 transmembrane domains per subunit) that may confer mechanosensitivity, two pore helices per trimer (inner pore helix [IH] and outer pore helix [OH]), and a large extracellular cap that connects to the pore helices in a domain-swapped manner (Guo and MacKinnon, 2017; Saotome et al., 2018; Wang et al., 2019; Zhao et al., 2018). Ion permeation likely occurs through a central pore lined by the IH from each subunit and an intracellular C-terminal domain (CTD), which forms the narrowest constriction; in all existing structures, this constriction site is too narrow to allow permeation of cations, and thus, the structures are interpreted to be in closed states (Saotome et al., 2018; Taberner et al., 2019; Wang et al., 2019; Zhao et al., 2018; Zheng et al., 2019a). The extracellular site of entry for ions has not been experimentally defined but could occur through lateral fenestrations between the cap and the blades or, alternatively, through the center of the cap itself.
A key feature of Piezos is that, following mechanical activation, currents decay substantially, even in the presence of continued stimulation, owing to the presence of at least two kinetically distinct inactivated states (Coste et al., 2010, 2012; Lewis et al., 2017). Inactivation modulates mechanical sensitivity and endows Piezos with the ability to function as frequency filters of repetitive stimuli, though the physiological implications of these findings have yet to be explored (Lewis et al., 2017; Lewis and Grandl, 2015). Regulation of inactivation occurs through many mechanisms, including alternative splicing, pH, temperature, divalent ions, osmotic swelling, co-expression of other membrane proteins, membrane lipid composition, and G-protein-coupled pathways (Anderson et al., 2018; Bae et al., 2015; Dubin et al., 2012; Eijkelkamp et al., 2013; Gottlieb and Sachs, 2012; Jia et al., 2013; Romero et al., 2019; Szczot et al., 2017; Zheng et al., 2019b). Moreover, mutations in both Piezo1 and Piezo2 have been identified in human patients diagnosed with xerocytosis and distal arthrogryposis that cause a gain-of-function phenotype by slowing inactivation, indicating that normal physiology requires inactivation to be intact (Albuisson et al., 2013; Bae et al., 2013; Coste et al., 2013; Zarychanski et al., 2012).
Although the physiological importance of inactivation is well established, our understanding of its mechanism and structural basis remains incomplete. Consistent with their disparate roles in physiology, Piezo1 and Piezo2 also have distinct inactivation kinetics, as Piezo1 inactivates relatively slowly as compared to Piezo2 (Coste et al., 2010, 2012; Wu et al., 2017b). The isoform-specific rate of inactivation can be conferred by the cap, and mechanical pulling specifically on the cap with a force normal to the membrane slows inactivation, together providing evidence for a critical role of this domain in inactivation (Wu et al., 2016, 2017b). Other domains must contribute to inactivation as well, however; most notably, a single lysine residue (K2479) in the adjacent IH confers the voltage dependence of inactivation, and nearby leucine and valine residues (L2475 and V2476) constitute a hydrophobic constriction site that may serve as an inactivation gate (Figure 1A; Wu et al., 2017b; Zheng et al., 2019a).
Figure 1. A Chimeric Screening Strategy Reveals Subdomains of Piezo1 Required for Slow Inactivation Kinetics.
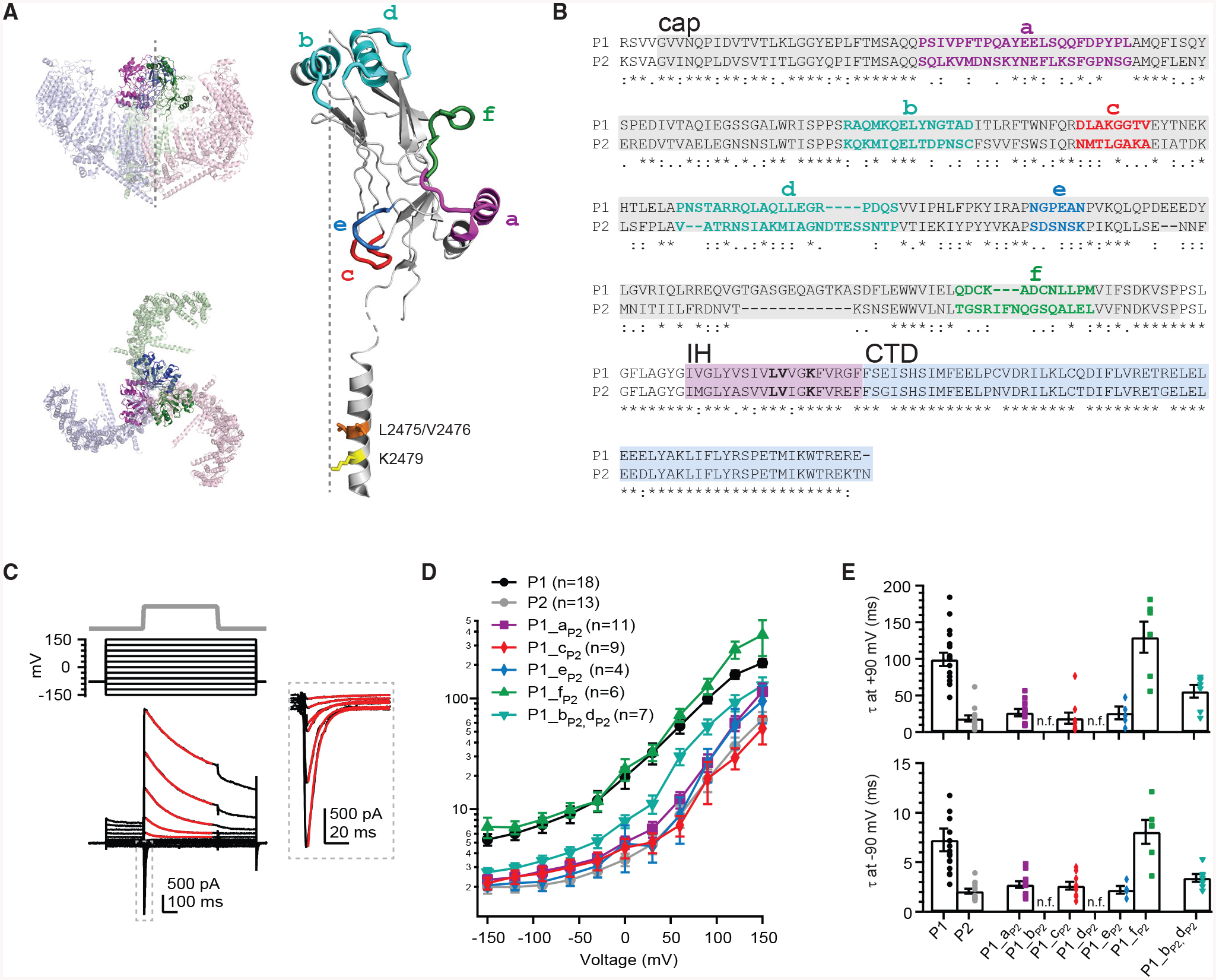
(A) Left: structural model of full-length trimeric mouse Piezo1 (PDB: 6B3R) viewed from the side (top) and from the top (bottom). Right: ribbon model of one monomer of the C-terminal extracellular domain (cap) and inner helix (IH) with subdomains containing distinct secondary structure highlighted according to the naming and color scheme in (B) is shown. Highlighted residues L2475/V2476 form a previously identified constriction site posited to be involved in inactivation (Zheng et al., 2019a), and K2479 confers voltage dependence of inactivation (Wu et al., 2017b).
(B) Alignment of the cap (gray), IH (pink), and C-terminal domain (CTD) (blue) of mouse Piezo1 (wtP1; residues 2,214–2,457) and mouse Piezo2 (wtP2; residues 2,497–2,731). Poorly conserved regions of the cap with distinct secondary structure are colored and correspond to structural subdomains in (A). Residues L2475, V2476, and K2479 are highlighted in bold lettering.
(C) Mechanical indentation protocol (8 mm; top), voltage protocol (middle), and representative whole-cell current (bottom) from a HEK293t-P1Ko cell transiently transfected with wtP1. Red lines indicate single exponential fits to the current decay; inset shows current responses at negative voltages at higher magnification.
(D) Mean inactivation time constants from single exponential fits to currents from 4–18 individual cells transfected with P1, P2, and chimeric channels, as a function of voltage.
(E) Mean inactivation time constants at +90 mV and −90 mV for P1, P2, and chimeric constructs. Each symbol represents one individual cell. Constructs P1_bP2 and P1_dP2 were non-functional (n.f.).
All data are mean ± SEM. See also Figure S1.
These studies leave open the question of whether the cap acts as a static structural unit that indirectly modulates a distal inactivation gate or, alternatively, whether structural rearrangements within the cap itself directly mediate inactivation. All available Piezo1 cryo-EM structures revealed an identical fold for the cap and also share notable similarity to a previously determined crystal structure of the cap of C. elegans Piezo (Kamajaya et al., 2014). Remarkably, comparing distinct Piezo1 structures and cryo-EM particle classes suggests a conformational rotation of the cap by ~15° in relation to the blades (Ge et al., 2015; Saotome et al., 2018; Zhao et al., 2018). Moreover, in Piezo2, the cap is similarly rotated and the putative gate in the IH dilated with respect to Piezo1 (Wang et al., 2019). Together, these observations suggest that the cap may play a dynamic role in gating.
Here, we use a chimeric strategy to identify small and distinct subdomains of the cap that confer isoform-specific inactivation kinetics, as well as others that do not contribute. We further find that conformational flexibility of these domains is required for mechanical gating. Finally, we demonstrate that stabilizing previously predicted electrostatic interactions between the cap and the blades inhibits mechanical gating. Together, our findings reveal that inactivation kinetics and mechanical gating in Piezo1 rely on small subdomains within the cap.
RESULTS
A Chimeric Screen Reveals Cap Subdomains Sufficient for Conferring Fast Inactivation Kinetics to Piezo1
Previously, Zheng et al. (2019a) identified two potential inactivation gates within the IH and CTD. Our lab demonstrated that the C-terminal extracellular domains (cap) of both mouse Piezo1 and mouse Piezo2 were sufficient for conferring their characteristic slow and fast inactivation kinetics, respectively (Wu et al., 2017b). Both studies suggested to us that the structural determinants of inactivation are likely to be found within these domains of the channel.
By comparing sequences of mouse Piezo1 and Piezo2, we noticed that the IH and CTD are remarkably well conserved (75% identity and 95% similarity); this includes two sets of residues previously identified as potential inactivation gates (Zheng et al., 2019a). In contrast, the cap is substantially less conserved (47% identity and 57% similarity). Further, by comparing the sequence alignment to the cap structure (PDB: 6B3R; Guo and MacKinnon, 2017), we identified six cap subdomains with relatively poor sequence conservation that also contain distinct secondary structure (Figures 1A and 1B). Together, these observations formed our hypothesis that some of these subdomains within the cap account for the distinct inactivation kinetics of Piezo1 and Piezo2, and in contrast, others are not important.
In order to test this hypothesis, we used a chimeric screening strategy in which we individually replaced each of the six subdomains from mouse wild-type Piezo1 (wtP1) with the corresponding sequence from mouse wild-type Piezo2 (wtP2) and assessed chimera function using whole-cell electrophysiology. Specifically, we transiently transfected HEK293-P1ko cells lacking endogenous Piezo1 (and thus mechanically activated currents; Dubin et al., 2017) with cDNA for chimeric constructs or wild-type channels as controls and stimulated cells mechanically with a blunt glass probe for 500 ms at voltages from −150 mV to +150 mV (Δ30 mV). We then fit the decay of evoked currents with single exponential functions (τ) in order to quantify the kinetics of entry into inactivated states (Figure 1C).
As previously described (Coste et al., 2010; Wu et al., 2017b), both wtP1 and wtP2 inactivation kinetics were steeply voltage dependent (30- to 40-fold slower at +150 mV as compared to −150 mV) and, across all voltages, kinetics were 3- to 5-fold faster in wtP2 as compared to wtP1 (at −90 mV: wtP1 τ = 7.2 ± 1.1 ms, n = 18, wtP2 τ = 2.1 ± 0.2 ms, n = 13; at +90 mV: wtP1 τ = 99.3 ± 9.2 ms, wtP2 τ = 18.6 ± 4.3 ms; Figures 1C–1E and S1A).
Remarkably, three chimeric constructs (P1-aP2, P1-cP2, and P1-eP2) exhibited inactivation kinetics that were drastically accelerated and statistically indistinguishable from that of wtP2 (at −90 mV: wtP2 τ = 2.1 ± 0.2 ms, P1-aP2 τ = 2.8 ± 0.3 ms, P1-cP2 τ = 2.6 ± 0.4 ms, and P1-eP2 τ = 2.2 ± 0.4 ms; p > 0.05 for each chimera versus wtP2, Student’s t test; at +90 mV: wtP2 τ = 18.6 ± 4.3 ms, P1-aP2 τ = 26.4 ± 4.9 ms, P1-cP2 τ = 18.9 ± 7.7 ms, and P1-eP2 τ = 26.3 ± 8.8 ms; p > 0.05 for each chimera versus wtP2, Student’s t test, n = 4–11; Figures 1D, 1E, and S1B–S1D). We conclude from this result that, despite their small sizes, subdomains a, c, and e, all of which are located at the base of the cap, are each individually sufficient for conferring the fast inactivation kinetics of Piezo2 onto Piezo1.
In contrast, one chimera (P1-fP2) had inactivation kinetics that were unchanged and statistically indistinguishable from that of wtP1 (at −90 mV: wtP1 τ = 7.2 ± 1.1 ms, P1-fP2 τ = 8.1 ± 1.1 ms, p > 0.05; at +90 mV: wtP1 τ = 99 ± 9 ms, P1-fP2 τ = 130 ± 21 ms, p > 0.05; n = 6; Student’s t test; Figures 1D, 1E, and S1E). This contrasting result provides proof of concept for our initial hypothesis that some, but not all, cap subdomains are sufficient for conferring fast inactivation kinetics onto Piezo1. The functional separation of this particular domain is also reflected in its structure, as subdomain f is the only domain that does not contact other domains of the protein but instead protrudes into the extracellular space.
Two chimeras (P1-bP2 and P1-dP2) were non-functional (data not shown). Both subdomains are located at the top of the cap, and because the cryo-EM structures predict extensive contacts between them (Guo and MacKinnon, 2017; Saotome et al., 2018; Zhao et al., 2018), we designed a chimeric construct in which both subdomains were replaced in tandem. Indeed, this resulting chimera (P1-bP2+dP2) was functional and had inactivation kinetics that were accelerated, but not fully to wtP2-like rates (at −90 mV: wtP2 τ = 2.1 ± 0.2 ms, P1-bP2+dP2 τ = 3.4 ± 0.4 ms, p < 0.05 versus wtP2; at +90 mV, wtP2 τ = 18.6 ± 4.3 ms, P1-bP2+dP2 τ = 55.6 ± 8.6 ms, p < 0.05 versus wtP2; Student’s t test; n = 7; Figures 1D, 1E, and S1F). We conclude that one or both of these subdomains can modulate inactivation kinetics but that other domains must contribute as well. In summary, this targeted screen singled out three distinct and substantially smaller subdomains that are each individually sufficient to fully accelerate Piezo1 inactivation kinetics to Piezo2 levels.
Conformational Flexibility of Distinct Cap Subdomains Is Required for Mechanical Gating
We reasoned that the subdomains we identified could contribute to inactivation via several mechanisms. The entire cap may be a rigid structural unit that interacts with the rest of the protein to block the pore, without state-dependent changes in the cap itself. Alternatively, conformational changes within the cap may either directly or allosterically modulate an inactivation gate. In the case of the latter, we hypothesized that restricting conformational changes within the cap may alter channel gating and would thus allow us to distinguish between both mechanisms.
In order to constrain conformational flexibility, we introduced double cysteine mutations in wtP1 within the cap subdomains our screen identified and in residues predicted to be nearby, with the goal of crosslinking them covalently. Notably, some pairs of residues resided within the same monomer and thus probed intrasubunit interactions, whereas others were in adjacent monomers and probed intersubunit interactions.
The partial cryo-EM structure of Piezo1 (PDB: 6B3R) predicts two potential double cysteine bonds, and the full Piezo1 sequence contains a total of 57 cysteine residues, thus potentially rendering gating of wtP1 sensitive to disruption of disulfide bonds by the reducing agent dithiothreitol (DTT). We therefore first tested whether DTT affects wtP1 function. More specifically, we developed a protocol where we locally perfused cells with buffer with or without 10 mM DTT in order to inhibit or allow cysteine crosslinking, respectively. Cells were then mechanically stimulated at 10-s intervals at −80 mV and +80 mV so that we would capture any voltage-dependent effects of DTT on both activation and subsequent inactivation (see STAR Methods; Figures 2A–2D).
Figure 2. Cysteine Crosslinking of Two Subdomains at Base of Piezo1 Cap Prohibits Channel Gating.
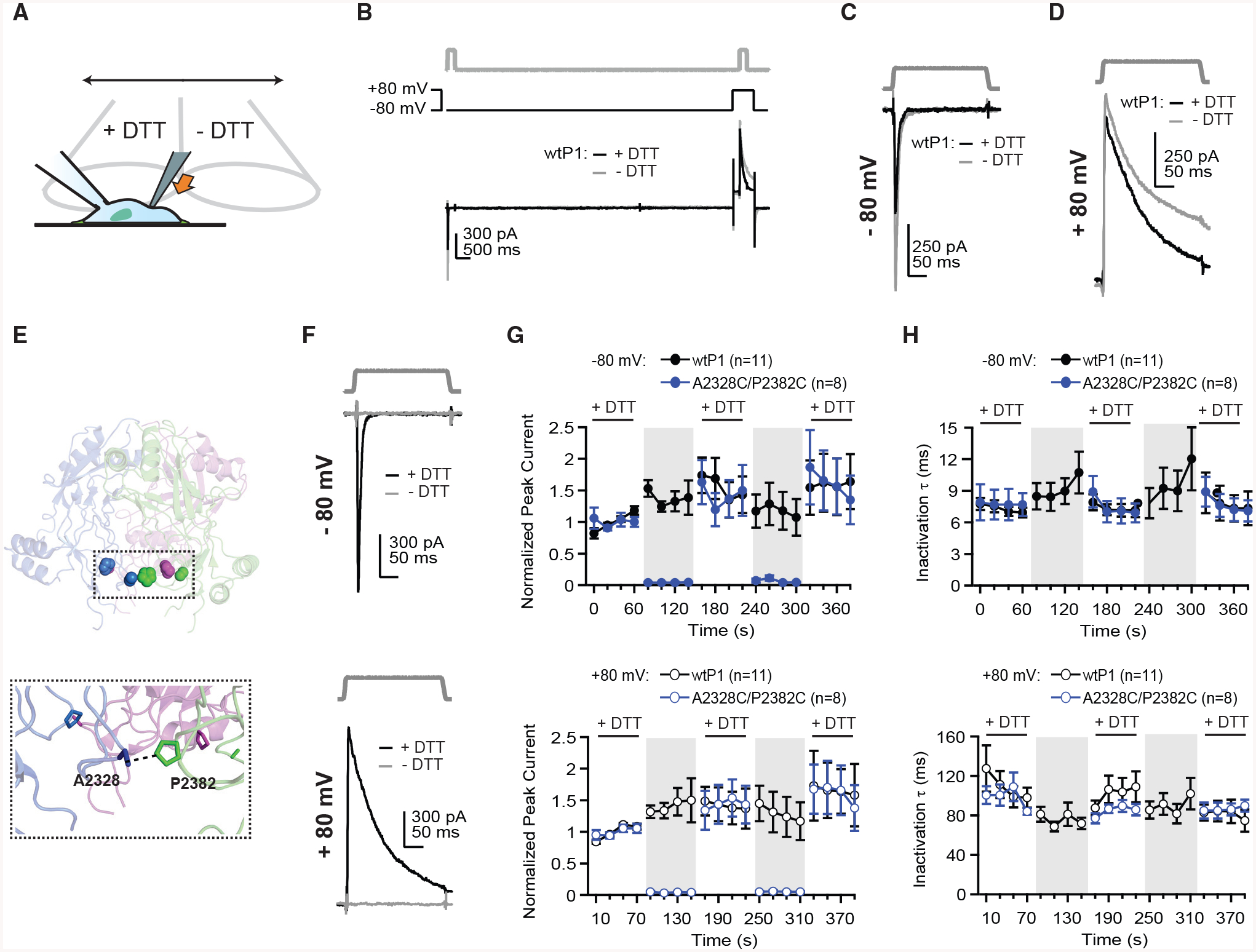
(A) Schematic depicting whole-cell recording setup with mechanical indentation stimulation and gravity perfusion.
(B) Indentation stimulus protocol (5 μm, top), voltage protocol (middle), and representative whole-cell current (bottom) from a HEK293t-P1Ko cell transiently transfected with wtP1 in with (black) and without (gray) 10 mM DTT in the bath.
(C and D) Magnification of indentation stimulus protocol and currents from (B) at −80 mV (C) and +80 mV (D).
(E) Structural model of Piezo1 cap highlighting cysteine pair A2328C and P2382C. Colors indicate three subunits of Piezo1.
(F) Indentation stimulus protocol (5 μm) and representative currents from a cell transfected with A2328C/P2382C at −80 mV (top) and +80 mV (bottom) with (black) and without (gray) 10 mM DTT in the bath.
(G) Mean peak current from 8–11 individual cells, normalized to the average of the first four peak currents in DTT, for wtP1 and A2328C/P2382C, at −80 mV (top) and +80 mV (bottom).
(H) Mean inactivation time constant (τ) from 8–11 individual cells for wtP1 and A2328C/P2382C at −80 mV (top) and +80 mV (bottom).
All data are mean ± SEM. See also Figures S2 and S3.
In these experiments, wtP1 current amplitudes increased slightly in control buffer (at −80 mV: 137% ± 12% of the initial current in DTT; at +80 mV: 141% ± 17%; Figure 2G; n = 11). There are several possible explanations for this increase. First, irreversible changes in the cell structure upon repeated stimulation (20 total stimuli) could alter the effective membrane tension evoked by a given stimulus. Second, guanosine triphosphate (GTP)-dependent run up over the long duration of the 6-min experiment could increase current amplitudes (Jia et al., 2013). In either case, this is unlikely to be a DTT-specific effect, as currents further rose upon reapplication of DTT (at −80 mV: 166% ± 30%; at +80 mV: 142% ± 25%). Inactivation kinetics of wtP1 were also unaffected by DTT (at −80 mV: τ = 7.3 ± 0.5 ms during first four stimuli in DTT, τ = 9.2 ± 1.4 ms during next four stimuli in control, and τ = 7.4 ± 0.6 ms during four stimuli upon return to DTT; at +80 mV: τ = 109.0 ± 14.0 ms in DTT, τ = 75.6 ± 6.7 ms in control, and τ = 101.7 ± 12.7 ms in DTT; Figure 2H). DTT also had no effect on mechanosensitivity or single-channel conductance of wtP1 (Figures S2A–S2E). The lack of effect of DTT on wtP1, as well as the observation that there are no unpaired cysteines in the cap domain, together suggested that any effects we would observe of DTT on channel gating are occurring due to disruption of newly introduced disulfide bonds. We then proceeded with testing three double cysteine mutant constructs using the exact same protocol.
First, we probed crosslinking an alanine residue in subdomain c, a loop at the base of the cap (Figure 1), and a proline in nearby subdomain e, which are predicted to be 6.2 Å apart (Cα-Cα; PDB: 6B3R) and are located in different subunits (Figure 2E). This construct (A2328C/P2382C) retained normal current amplitudes in the presence of DTT (Figures S2F and S2G), but strikingly, currents were completely abolished in the presence of control buffer. In fact, residual current amplitudes were indistinguishable from noise (at −80 mV: 4% ± 1% of the current during the first DTT wash; at +80 mV: 4% ± 1%; n = 8; Figures 2F, 2G, S2F, and S2G; STAR Methods). This reduction of current was complete within 10 s, as the current remaining during the first stimulus in control buffer was indistinguishable from the average of all subsequent stimuli (at −80 mV: 4% ± 1%; at +80 mV: 5% ± 2%). The effect was reversible and repeatable: current amplitudes returned to original levels upon reapplication of DTT (at −80 mV: first stimulus in DTT = 163% ± 35%, mean of all four stimuli = 142% ± 42%; at +80 mV: first stimulus in DTT = 134% ± 31%, mean of all four = 144% ± 29%) and were again abolished by a second washout of DTT. Inactivation kinetics, which could only be measured in DTT, were indistinguishable from wild-type (during first DTT application at −80 mV: τ = 7.8 ± 1.4 ms; at +80 mV: τ = 98.6 ± 8.2 ms; Figure 2H). These data suggest that these two residues sample conformations that allow them to form a disulfide linkage. More importantly, restricting the conformational flexibility of subdomains c and e via intersubunit crosslinking of A2328 and P2382 stabilizes a non-conducting state of the channel.
Second, we tested the effect of crosslinking a glycine residue G2330 in subdomain c and valine residue V2453 in the linker that leads from the cap to the inner pore helix; these residues are located in the same subunit and are predicted to be 6.1 Å apart (Cα-Cα) (Figure S3A). Reducing conditions had no effect on this construct (G2330C/V2453C), as current amplitudes remained constant in control buffer (at −80 mV: 104% ± 11% of the first DTT application; at +80 mV: 98% ± 8%; n = 9; Figures S3B and S3C). Inactivation kinetics were also unaffected by DTT, although time constants were faster in both conditions compared to those observed in wtP1 (during first DTT application at −80 mV: τ = 4.2 ± 0.3 ms; at +80 mV: τ = 29.6 ± 2.7 ms; Figure S3D), consistent with our previous observation that subdomain c contributes to inactivation kinetics (Figure 1).
Third, we probed the effect of crosslinking two residues in the α helices we identified at the top of the cap that modulate inactivation kinetics, subdomains b and d. Two residues, K2305 in subdomain b and E2359 in subdomain d, are predicted to be 8.4 Å apart (Cα-Cα), and their side chains could potentially make an intrasubunit salt bridge interaction (Figure S3E). Current amplitudes for this construct (K2305C/E2359C) were unaffected by wash out of DTT (at −80 mV: 94% ± 7% of the first DTT application; at +80 mV, 104% ± 9%, n = 6; Figures S3F and S3G). Similarly, inactivation kinetics were unaffected by the presence of DTT and were identical to wtP1 (at −80 mV in DTT: 5.6 ± 0.5 ms; at +80 mV in DTT: 71.3 ± 11.0 ms; Figure S3H). These results could indicate that these two sets of residues (G2330/V2453 and K2305/E2359) do not interact closely enough to form a disulfide bond. Alternatively, it is possible that crosslinking these residues does not affect channel function. Together, this series of experiments demonstrates that intersubunit conformational flexibility between specific subdomains (c and e) at the base of the cap is required for mechanical activation.
Crosslinking between the Cap and Blade Residues Inhibits Mechanical Gating
Subdomain a, which consists of two anti-parallel α helices, stood out from our screen because previous structural data from the MacKinnon lab resolved a series of negatively charged residues in this domain that likely contact positively charged residues on the blades, which they predicted to form salt bridges and stabilize the channel in a curved conformation (Guo and MacKinnon, 2017). Direct experimental evidence from the same lab suggests that this curvature induces a membrane “footprint,” and a theoretical framework provides a rationale for this footprint being essential for the ability of Piezo1 to sense changes in membrane tension (i.e., open in response to membrane flattening; Guo and MacKinnon, 2017; Haselwandter and MacKinnon, 2018; Lin et al., 2019). Moreover, Piezo1 structures from the labs of Ward and Xiao suggest that the cap may adopt at least two rotational conformations with respect to the blades (Saotome et al., 2018; Wang et al., 2019; Zhao et al., 2018), which may underlie mechanical gating. However, the Ward and Xiao structures did not resolve the putative cap-blade salt bridges and thus do not provide any detailed clues about their potential role in promoting a rotation of the cap and/or mechanical gating. We therefore set out to systematically probe the mechanical coupling between subdomain a and the blades by again mutating pairs of charged residues to cysteines and probing for functional effects of crosslinking.
First, we attempted to crosslink R1762 and E2257, which is the closest pair of charged residues and are located 9.2 Å apart (Figure 3A). Current amplitudes for this construct (R1762C/E2257C) remained normal in DTT, but strikingly, in the absence of DTT, currents were abolished (at −80 mV in control: 5.5% ± 0.9% of the original current in DTT; at +80 mV in control, 5.9% ± 1.0% of the original current in DTT; n = 8; Figures 3B and 3C). In DTT, inactivation kinetics were similar to wtP1, with a trend toward slightly slower values at positive potentials (at −80 mV: 8.6 ± 0.9 ms; at +80 mV: 143 ± 19 ms; Figure 3D). These data demonstrate that both residues can crosslink and further suggest that stabilizing this intersubunit interaction promotes a non-conducting conformation, which is consistent with the closed pore observed in the Piezo1 structure in which this salt bridge is prominent (Guo and MacKinnon, 2017).
Figure 3. Cysteine Crosslinking between a Glutamate Residue in the Piezo1 Cap and Two Arginine Residues in the Blade Prevents Channel Opening.
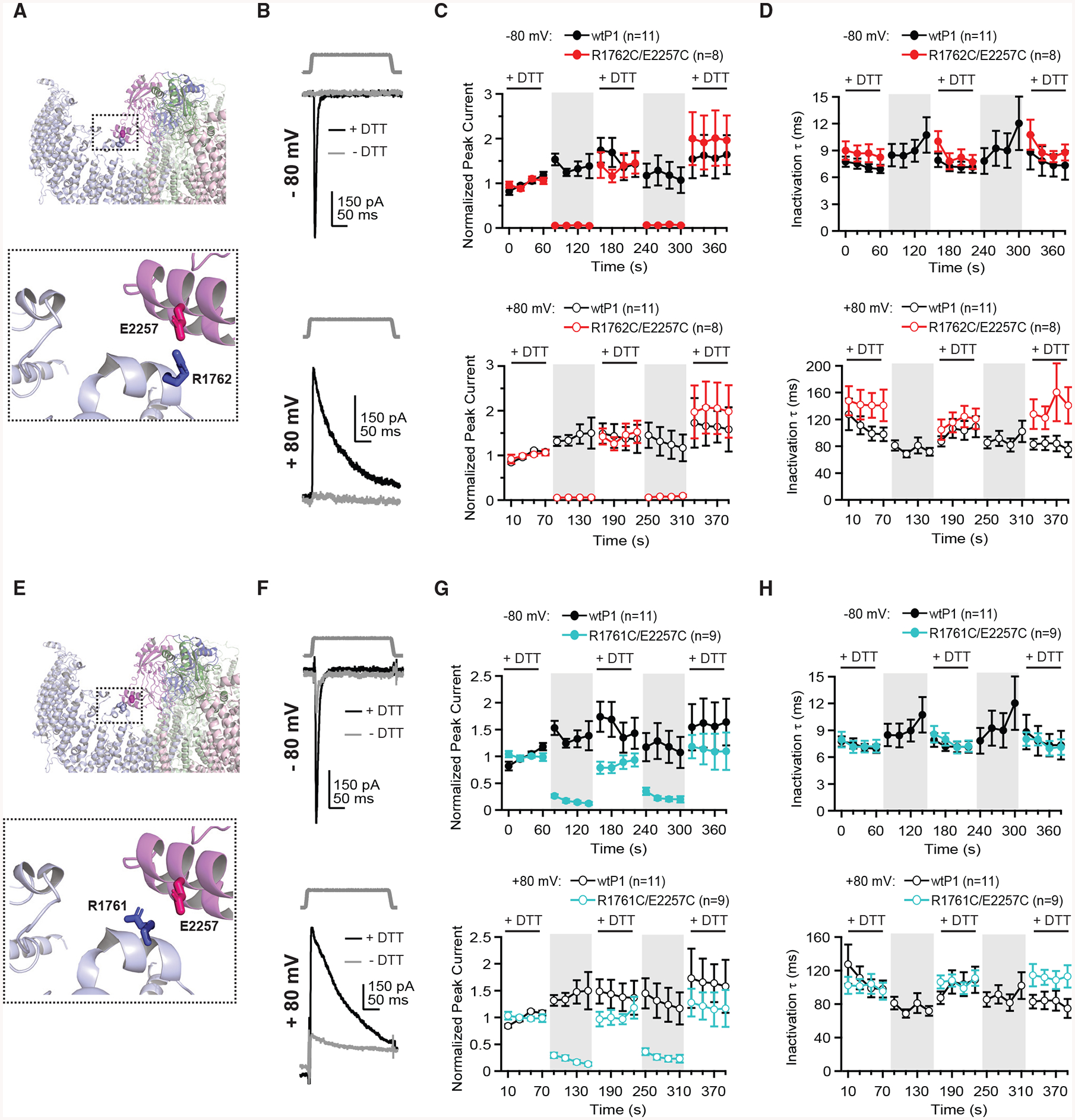
(A) Structural model of Piezo1 cap highlighting cysteine pair R1762C and E2257C. Colors indicate three subunits of Piezo1.
(B) Indentation stimulus protocol (3 μm) and representative currents from a cell transfected with R1762C/E2257C at −80 mV (top) and +80 mV (bottom) with (black) and without (gray) 10 mM DTT in the bath.
(C) Mean peak current from 8–11 individual cells, normalized to the average of the first four peak currents in DTT, for wtP1 and R1762C/E2257C, at −80 mV (top) and +80 mV (bottom).
(D) Mean inactivation time constant (τ) from 8–11 individual cells for wtP1 and R1762C/E2257C at −80 mV (top) and +80 mV (bottom).
(E) Structural model of Piezo1 cap highlighting cysteine pair R1761C and E2257C. Colors indicate three subunits of Piezo1.
(F) Indentation stimulus protocol (6 mm) and representative currents from a cell transfected with R1761C/E2257C at −80 mV (top) and +80 mV (bottom) with (black) and without (gray) 10 mM DTT in the bath.
(G) Mean peak current from 9–11 individual cells, normalized to the average of the first four peak currents in DTT, for wtP1 and R1761C/E2257C, at −80 mV (top) and +80 mV (bottom).
(H) Mean inactivation time constant (τ) from 9–11 individual cells for wtP1 and R1761C/E2257C at −80 mV (top) and +80 mV (bottom).
All data are mean ± SEM. See also Figures S2, S4, and S5.
Second, we tested whether E2257 could crosslink with the adjacent arginine residue on the blades, R1761, which is located at a slightly further distance of 10.2 Å (R1761C/E2257C; Figure 3E). Interestingly, although current amplitudes for this construct were greatly reduced upon removal of DTT, they were not abolished (Figures 3F and 3G). Additionally, unlike the instantaneous loss of current in control wash for R1762C/E2257C, currents at −80 mV for R1761C/E2257C decreased gradually, from 26.3% ± 3.0% at the beginning of the control wash to 12.2% ± 2.8% at the end of the control wash (+80 mV: from 29.5% ± 5.0% to 13.4% ± 3.0%; n = 9). Inactivation kinetics in DTT were again unchanged compared to wtP1 (at 80 mV: 7.4 ± 0.7 ms; at +80 mV: 100.8 ± 9.3 ms; Figure 3H).
One explanation for the attenuated currents is that the cysteine residues were not oxidized, thus preventing crosslinking. To test for this, we repeated our indentation assay but added 10 mM H2O2 to our control solution to promote oxidation and subsequent formation of disulfide bonds. We found that H2O2 had no additional effect beyond that of control buffer for R1761C/E2257C. Currents at −80 mV decreased from 35.8% ± 11.0% at the beginning of the H2O2 wash to 25.4% ± 7.4% at the end of the wash. H2O2 had no effect on current amplitudes or inactivation kinetics in wtP1 (Figures S4A–S4F). Therefore, the reason for attenuated, but not abolished, currents for R1761C/E2257C was not because the local environment did not allow for full oxidation of the cysteine residues involved. Thus far, we concluded from these experiments that conformational flexibility between the cap and blade, which may well be a rotation of the cap, allows residue E2257 to crosslink readily not only to R1762 but also to a lesser extent to R1761.
Third, we tested the ability of E2257 to crosslink with the third positive charge in the cluster, R1269, which is located even more distally at 15.7 Å (R1269/E2257C; Figure S5A) and thus probes for an even greater degree of conformational flexibility between the blade and the cap. Current amplitudes remained high in the presence and absence of DTT (R1269C/E2257C: at −80 mV in control, 108% ± 23% of original current in DTT; at +80 mV, 101% ± 15%; n = 9; Figures S5B–S5D).
Finally, we tested potential crosslinking of R1269 and D2264, which are similarly distant at 15.1 Å (R1269C/D2264C; Figure S5E). Again, currents remained stable throughout experiments (D2264C/R1269C: at −80 mV in control, 100% ± 10% of the original current in DTT; at +80 mV, 98% ± 10%; n = 8; Figures S5G and S5H). It is possible that crosslinking can occur between R1269C and E2257C or D2264C but has no effect on channel function. However, given the complete absence of current when other residues in these subdomains are crosslinked, the more likely explanation is that crosslinking does not occur between R1269C and E2257C or D2264C, which is consistent with the further distance between these residues.
Altogether, this set of experiments suggests that, during gating, conformational flexibility allows residue E2257 to interact with both R1762 and R1761, but not R1269, with the interaction between E2257 and R1762 being the most probable. More importantly, covalently linking the blade to the cap, which restricts conformational flexibility in this region, prevents channels from opening in response to mechanical force.
Destabilizing Electrostatic Interactions between Cap and Blades Specifically Promotes an Open State
Although our experiments revealed that crosslinking the cap to the blade can inhibit mechanical gating, they did not reveal the precise role the naturally occurring salt bridges present in wild-type channels play in mechanical gating. We therefore hypothesized that destabilizing the cap-blade interaction, as might happen upon membrane flattening due to tension and subsequent blade extension (Lin et al., 2019), may in return promote an open state of the channel. To test this hypothesis, we mutated all three negative charges in the putative interaction site of the cap to positively charged lysines (E2257K/E2258K/D2264K), thereby eliminating potential salt bridge interactions between the cap and blades (Figure 4A). We then used three different stimulation protocols to assess how the kinetics of inactivation, deactivation, and recovery from inactivation were affected:
Figure 4. Disruption of an Electrostatic Interaction between the Blade and Cap of Piezo1 Promotes an Open State.
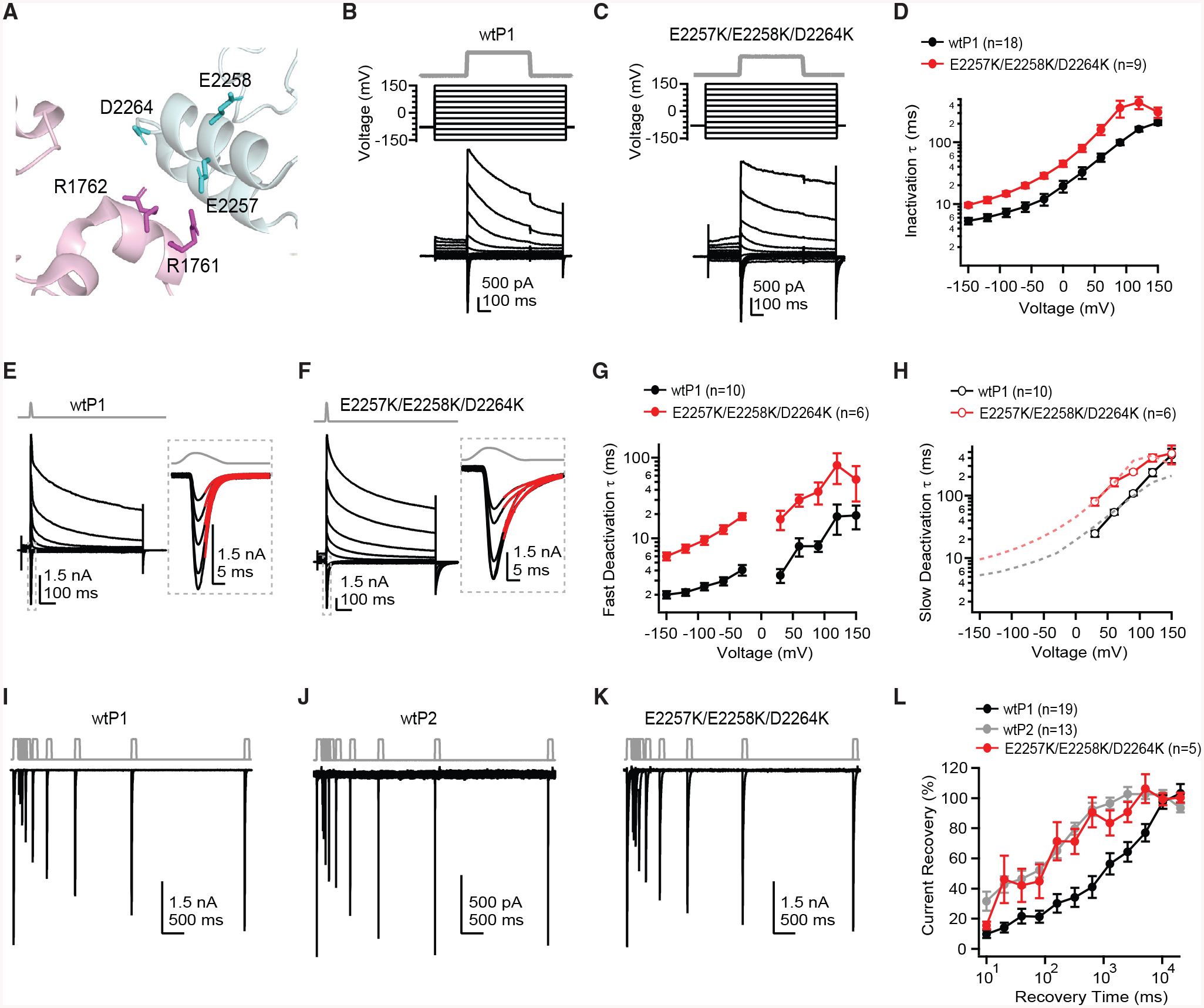
(A) Structural model of Piezo1 (PDB: 6B3R) highlighting the region of a monomer of the blade (pink) and a different monomer of the cap (blue) with predicted salt bridge interactions and interacting residues shown as sticks.
(B and C) Indentation protocol (B, 8 μm; C, 11 μm) designed to measure inactivation (top), voltage protocol (middle), and representative whole-cell current (bottom) from a HEK293t-P1Ko cell transiently transfected with wtP1 (B) and from a cell transfected with E2257K/E2258K/D2264K (C).
(D) Mean inactivation time constants from single exponential fits to currents from 9–18 individual cells transfected with wtP1 (black circles) or E2257K/E2258K/D2264K (red squares), as a function of voltage.
(E and F) Indentation protocol (E, 5 μm; F, 7 μm) designed to measure deactivation and representative whole-cell current from a HEK293t-P1ko cell transiently transfected with wtP1 and from a cell transfected with E2257K/E2258K/D2264K. Insets show negative voltages at higher magnification.
(G) Mean fast deactivation time constants from single (−150 to −30 mV) or double (+30 to +150 mV) exponential fits to currents from 6–10 individual cells transfected with wtP1 (black circles) or E2257K/E2258K/D2264K (red squares), as a function of voltage.
(H) Mean slow deactivation time constants from single (−150 to −30 mV) or double (+30 to +150 mV) exponential fits to currents from 6–10 individual cells transfected with wtP1 or E2257K/E2258K/D2264K, as a function of voltage. Dashed lines are inactivation time constants (as in D) for wtP1 (black) and E2257K/E2258K/D2264K (red), showing the overlap with the slow component of deactivation that emerges at positive potentials.
(I–K) Two-step indentation recovery protocol (I, 3 μm; J, 3 μm; K, 9 μm) and representative whole-cell currents from HEK293t-P1ko cells transiently transfected with wtP1, wtP2, and E2257K/E2258K/D2264K.
(L) Mean current recovery from 5–19 individual cells, calculated as peak current during the second pulse as a percentage of peak current during the first pulse.
All data are mean ± SEM.
Inactivation kinetics, tested with a 500-ms mechanical stimulus, were markedly slower for the charge-reversal construct as compared to wtP1 (at −90 mV: wtP1 τ = 7.2 ± 1.1 ms, n = 18; E2257K/E2258K/D2264K τ = 14.7 ± 1.3 ms, n = 9, p = 0.001; at +90 mV: wtP1 τ = 99.3 ± 9.2 ms, E2257K/E2258K/D2264K τ = 361 ± 111 ms; p = 0.002; Figures 4B–4D).
Deactivation kinetics were tested by mechanically stimulating cells very briefly (2–4 ms) and measuring the time constant of current decay after removal of the stimulus (Figures 4E–4H; see STAR Methods). Like inactivation, deactivation is strongly voltage dependent, with slower kinetics at positive potentials as compared to negative potentials. The kinetics are more complicated to extract, however, as at all negative potentials, decays are well fit with a single exponential, whereas at positive potentials, decays are bi-exponential. The fast component at positive potentials likely reflects channels closing (deactivation). The slower component likely reflects channels entering inactivated, rather than closed states, as the slow time constant is indistinguishable from that of the inactivation time constant measured with the former protocol (Figures 1D and 4H). We therefore compared the single time constant at negative potentials and the fast time constant at positive potentials and found that deactivation was dramatically slowed for E2257K/E2258K/D2264K across the voltage range tested (at −90 mV: wtP1 τ = 2.5 ± 0.3 ms, n = 10; E2257K/E2258K/D2264K τ = 9.5 ± 1.1 ms, n = 6; p < 5e−7; at +90 mV: wtP1 fast τ = 108.1 ± 7.7 ms; E2257K/E2258K/D2264K fast τ = 239.3 ± 14.3 ms, p < 5e−4).
To quantify recovery from inactivation, we used a two-step protocol in which first a 100-ms mechanical stimulus was applied to open channels and allow them to transition into inactivated states; the stimulus was then removed for varying time intervals (10–20,480 ms) before application of a second 100-ms mechanical stimulus to assay the fraction of channels that had recovered from inactivation (Lewis et al., 2017). We first tested the kinetics of recovery from inactivation in both wtP1 and wtP2, as no study has yet directly compared the two isoforms in this respect. Although recovery for both isoforms was complete after ~10 s (at 10.24 s: wtP1 = 97% ± 4%, n = 19; wtP1 = 102% ± 3%, n = 13), wtP2 recovered to a greater extent at all shorter time intervals (for example, at 320 ms: wtP1 = 34% ± 6%, wtP2 = 80% ± 4%; Figures 4I–4L). Remarkably, recovery of E2257K/E2258K/D2264K was qualitatively more similar to wtP2 than to its parent construct, wtP1 (at 320 ms, 71% ± 9%; n = 5). In summary, the slowed inactivation and deactivation, together with the accelerated recovery from inactivation, demonstrate that disrupting the charge interactions between the cap and the blade both stabilizes the open state of Piezo1 and destabilizes the inactivated state(s). In other words, the naturally occurring electrostatic coupling between the cap and the blades promotes Piezo1 closing upon mechanical activation.
DISCUSSION
Our targeted screen successfully identified three distinct subdomains (a, c, and e) of the Piezo2 cap that, individually, are each sufficient for conferring fast inactivation kinetics onto Piezo1. The subdomains are 23, 8, and 6 residues in length, which substantially narrows down the determinants of inactivation kinetics in a channel comprising 2,500 residues per subunit.
A common feature of the three subdomains we identified is that they are all located at the base of the cap. In contrast, other subdomains that have a more modest effect on kinetics despite their large size (b and d; 34 total residues), or no effect at all (f; 12 residues), are all located in the middle or top of the cap. Together, this result highlights the base of the cap as a locus that determines inactivation kinetics and further implicates it as a potential structural target to manipulate inactivation pharmacologically.
The base of the cap is further interesting because of its multiple predicted interactions with the blades. These include the previously identified negatively charged residues in subdomain a that face opposing positive countercharges on the blades in the Piezo1 structure revealed by the MacKinnon lab (Guo and MacKinnon, 2017). Additionally, all available cryo-EM structures of Piezo1 and Piezo2 show electron densities that fill the space between cap and the blades and that likely correspond to yet unresolved extracellular domains of the blades. The lack of resolution in these regions suggests they may be flexible. Thus, subdomains a, c, and e likely not only contact the blades and/or the top of the pore helices extensively but may also couple to them allosterically.
The base of the cap is also a location suited for directly controlling access of ions to the central permeation pathway. The structure of the cap is consistent with a potential permeation pathway either down the central axis of the cap or through lateral fenestrations between the cap and blades (Zhao et al., 2018), although previous cysteine-modifying experiments from our lab were unable to provide direct evidence for the central pathway (Wu et al., 2017b). Subdomains a, c, and e may therefore act as gates, either separately or in concert with the previously identified constrictions in the inner pore helix (Zheng et al., 2019a).
Of course, it is possible that our screen missed additional structures within the cap that confer inactivation kinetics. In fact, our finding that three structurally distinct subdomains are each sufficient for conferring fast inactivation suggests additional domains may exist and further argues that inactivation of Piezo1 may be a complex allosteric mechanism in which structural rearrangements in the cap and elsewhere modulate a distal gate. The concept of multiple gates has precedence in many ion channel families. For example, Shaker potassium channels contain structurally distinct inactivation gates corresponding to N-type (pore obstruction) and C-type (selectivity filter rearrangement) inactivation mechanisms (Armstrong and Hollingworth, 2018; Hoshi et al., 1990). Previous work from our lab likewise suggests multiple loci of inactivation may exist for Piezo1, as we found evidence for at least two kinetically distinct inactivated states (Lewis et al., 2017). The inner pore helix likely serves as one locus of inactivation, as it not only contains a residue that confers the voltage dependence of inactivation but also contains a hydrophobic constriction site that may serve as a bona fide inactivation gate (Wu et al., 2017b; Zheng et al., 2019a). Consistent with these ideas, disease-causing mutations that slow inactivation kinetics are spread throughout Piezo1, located in the blades, beam, anchor, IH, and cap, all of which could allosterically couple to multiple gates (Glogowska et al., 2017).
A completely unexpected outcome from our work is that restricting conformational flexibility of two subdomains contained entirely within the cap (subdomains c and e) via intersubunit crosslinking inhibits mechanical activation altogether. This result demonstrates that motion within the cap, and more specifically, conformational flexibility between subunits, is required for opening of Piezo1. That is, the cap is not merely a static domain, but rather conformational changes within it are absolutely required for mechanical gating to occur.
We also found evidence that electrostatic intersubunit blade-cap interactions that were previously predicted from a cryo-EM structure (Guo and MacKinnon, 2017) play an essential role in normal Piezo function. Specifically, crosslinking subdomain a of the cap with the blades prohibits channel opening, whereas preventing the putative salt bridge interactions between the two domains by reversing the charges on the residues in the cap not only slows both inactivation and deactivation but also accelerates recovery from inactivation, thus promoting the open state. This finding resonates with a recently proposed mechanism by which changing curvature of the blades may provide the major driving force for mechanical gating (Haselwandter and MacKinnon, 2018; Lin et al., 2019) and the interpretation that the previously captured structures are in closed or inactivated conformations. As a consequence, our construct that stabilizes the open state (E2257K/E2258K/D2264K) may be useful in future structural studies aimed at capturing an open state.
We have shown that restricting intersubunit conformational flexibility both within the cap and between the cap and blades prevents channel opening. Exactly which domain(s) requires conformational flexibility is less clear, however, because crosslinking two domains with each other necessarily restricts the motion of both. Also, it is currently unclear whether crosslinking inhibits the transition to the open state (activation), stabilizes an inactivated state, or directly prevents ion permeation. Elucidating the complete gating mechanism of the channel will require decoupling of the two (or more) inactivation gates, as well as identifying the structural correlates of both activation and inactivation. Currently, structural information provides little insight into the functional consequence of motion in the observed constriction sites in the CTD and IH, which have been proposed to be sites of both activation and inactivation (Saotome et al., 2018; Wang et al., 2019; Zheng et al., 2019a). Further, specific manipulations of single gating transitions have not yet been reported. For example, depolarization stabilizes the open state of Piezo1 and slows both inactivation and deactivation indiscriminately, as does our construct (E2257K/E2258K/D2264K) that stabilizes the open state.
The functional studies presented here thus begin to extend our mechanistic understanding of Piezo1 activation and inactivation beyond what had been obtained from structural data: none of the three Piezo1 cryo-EM structures alone or by comparison suggest internal conformational flexibility of the cap. This may be due to insufficient spatial resolution or due to the fact that all structures captured the same functional state. Our study therefore predicts that future structures of distinct functional states will reveal differences in at least one of the cap subdomains we identified.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Jörg Grandl (grandl@neuro.duke.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HEK293t-P1Ko cells (Piezo1 knockout human embryonic kidney cells; (Dubin et al., 2017)), a gift of Ardem Patapoutian, were cultured at 37°C and 5% CO2 in DMEM-HG (Life Technologies) supplemented with 10% FBS (Clontech), 50 U/mL penicillin, and 50 mg/mL streptomycin (Life Technologies). The cell line was not authenticated.
METHOD DETAILS
Cloning and Cell Culture
Mouse Piezo1-pIRES-EGFP in pcDNA3.1(+) was obtained from Ardem Patapoutian and previously described (Coste et al., 2012). Mouse Piezo2 was synthesized to be codon optimized for expression in human cells by Genewiz and ligated into pcDNA3.1(+) between restriction sites Kpn1 and Not1 (Lewis et al., 2017). Piezo1-Piezo2 chimeras were created using the Q5 Site-Directed Mutagenesis kit (New England Biosciences) following the manufacturer’s protocol for large insertions using non-overlapping primers. Point-mutations in Piezo1 were created using the Quikchange Lightning Multi Site-Directed Mutagenesis kit (Agilent Technologies). All constructs were sequence-verified (Genewiz).
40–48 hours before recording, cells were transiently transfected in 6-well plates (Piezo1 and Piezo1-based constructs: 3 μg; Piezo2 and Piezo2-based constructs: 2.5 μg + 0.5 μg GFP) with Fugene 6 (Promega) according to the manufacturer’s protocol in the presence of 10 μM ruthenium red. Transfected cells were reseeded from 6-well plates onto poly-L-lysine and laminin-coated coverslips 18–24 hours before recording.
Electrophysiology
Electrophysiological recordings were performed at room temperature using an EPC10 amplifier and Patchmaster software (HEKA Elektronik, Lambrecht, Germany). Data were sampled at 5 kHz (cell-attached) or 10 kHz (whole-cell) and filtered at 2.9 kHz.
For whole-cell experiments, borosilicate glass pipettes (1.5 OD, 0.85 ID; Sutter Instrument Company, Novato, CA) had a resistance of 2–5 MΩ when filled with pipette buffer solution (in mM: 133 CsCl, 10 HEPES, 5 EGTA, 1 MgCl2, 1 CaCl2, 4 MgATP, 0.4 Na2GTP, pH 7.3 with CsOH. Internal solution was allowed to dialyze for at least five minutes before recording to promote GTP-mediated run-up of currents (Jia et al., 2013). Series resistance was compensated by 40%–70%. The control bath solution for all whole-cell experiments was (in mM) 130 NaCl, 3 KCl, 1 MgCl2, 10 HEPES, 2.5 CaCl2, 10 glucose (pH 7.3 with NaOH).
For crosslinking experiments, cells were perfused by placing custom-built pipes made from silica tubing (ID: 320 μm; Trajan Scientific) within ~100 μm of the cell and solution was allowed to flow via gravity (~0.5 mL/min). Fresh solutions of 10 mM dithiothreitol (DTT) or 10 mM hydrogen peroxide (H2O2) were prepared from frozen stocks every hour and kept on ice. For crosslinking experiments, all cells were initially patched and stimulus intensity was chosen in the presence of DTT. Voltages were not corrected for a liquid junction potential.
For cell-attached experiments, pipettes had a resistance of 1.5–4 MΩ when filled with pipette buffer solution (in mM: 130 NaCl, 5 KCl, 10 HEPES, 10 TEACl, 1 CaCl2, 1 MgCl2, pH 7.3 with NaOH). The cell-attached bath solution used to zero the membrane potential was (in mM): 140 KCl, 10 HEPES, 1 MgCl2, 10 glucose, pH 7.3 with KOH. Patches were held at −80 mV except where described otherwise.
Mechanical Stimulation
In cell-attached patches, negative pressure was applied through the patch pipette with an amplifier-controlled high-speed pressure clamp system (HSPC-1; ALA Scientific Instruments, Farmingdale, NY).
For whole-cell experiments, cells were indented with a fire-polished glass pipette (tip diameter ~3–5 μm) by an amplifier-controlled piezo-electric driver (E625 LVPZT Controller/Amplifier; Physik Instrumente) operated in closed-loop mode. The probe was initially positioned ~2–4 μm from the cell and advanced at 0.5 μm/ms in 1 μm increments at an 80° angle. The step increment was stopped after eliciting a current of > 200 pA and the last indentation depth maintained for subsequent protocols (mean indentation depth: 6.5 μm, where 0 μm is the last step before touching the cell). For both stimulus paradigms, the intersweep interval was 10 s to allow for recovery from inactivation (Lewis et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were analyzed using Igor Pro 8.02 (WaveMetrics). Whole-cell electrophysiology recordings were analyzed for cells with a seal resistance of > 500 MΩ and a series resistance of < 12 MΩ. Cells with maximum whole-cell currents less than 200 pA were excluded from the analysis. Baseline currents before mechanical stimulation were subtracted offline. Current amplitudes were quantified as the peak current during the stimulus (with no smoothing). To quantify the effects of DTT on channel function, we further normalized each peak current amplitude to the average of the first four peak current amplitudes in DTT and then calculated the average of four stimulus responses in each condition. Because the peak-to-peak variation due to noise in whole-cell currents was ~25 pA, peak currents in the absence of DTT that were smaller than this value (~5% of a 500 pA peak current in DTT) were indistinguishable from noise, and these constructs were deemed non-functional in the absence of DTT (see Figure S2).
The time constant of inactivation (τ) was obtained by fitting a single exponential function between the peak current and the stimulus offset (~500 ms): I = I0 + A * exp(−(t−t0)/τ)
In a subset of cells, steady-state current at positive potentials was decreased by DTT, but this effect was inconsistent (5 of 9 wild-type cells) and we chose not to analyze it further. The time constant of deactivation (τ) was obtained by visually identifying the inflection point in the current (where current transitions from inactivating to deactivating; typically within 2–3 ms following the beginning of the stimulus offset) and fitting a single exponential function (negative potentials; same equation as inactivation) or double exponential function (positive potentials) for 500 ms following the inflection point:
Single channel amplitudes were measured by performing a Gaussian multi-peak analysis on current amplitude histograms. Slope conductances were fit for each individual cell from single channel amplitudes at −60 mV, −80 mV, and −100 mV (3–5 openings per voltage).
For two-pulse recovery experiments, because Piezo current amplitudes occasionally ran up or down during the long duration of the experiment due to GTP-dialysis (Jia et al., 2013), any sweep in which the test pulse amplitude deviated by more than 30% from that of the following sweep was discarded from analysis.
All structural figures were prepared using Pymol and Cα-Cα distances were measured using the Pymol distance wizard and PDB: 6B3R (Guo and MacKinnon, 2017).
All data are reported as mean ± SEM. Statistical analyses were performed using unpaired Student’s t test. Significance thresholds were set as p < 0.05.
DATA AND CODE AVAILABILITY
The published article includes all datasets generated or analyzed during this study. All data are available from the Lead Contact upon reasonable request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental Models: Cell Lines | ||
| HEK-p1ko cells (Piezo1 knockout human embryonic kidney cells) | Dubin et al., 2017 | N/A |
| Recombinant DNA | ||
| Mouse Piezo1-pIRES-EGFP | Coste et al., 2010 | N/A |
| Mouse Piezo2 in pcDNA3.1(+) | Lewis et al., 2017 | N/A |
| P1-aP2 | This paper | N/A |
| P1-cP2 | This paper | N/A |
| P1-eP2 | This paper | N/A |
| P1-fP2 | This paper | N/A |
| P1-bP2, dP2 | This paper | N/A |
| P1- A2328C/P2382C | This paper | N/A |
| P1- G2330C/V2453C | This paper | N/A |
| P1- K2305C/E2359C | This paper | N/A |
| P1- R1762C/E2257C | This paper | N/A |
| P1- R1761C/E2257C | This paper | N/A |
| P1- R1269C/E2257C | This paper | N/A |
| P1- R1269C/D2264C | This paper | N/A |
| Other | ||
| High Speed Pressure Clamp (HSPC-1) | ALA Scientific | N/A |
| Piezoelectric driver (E625 LVPZT Controller/Amplifier) | Physik Instrumente | N/A |
Highlights.
Piezo1 inactivation kinetics are conferred by small subdomains within the cap
Restricting intersubunit conformational flexibility inhibitsPiezo1 gating
An electrostatic interaction couples the Piezo1 cap to the blade to control gating
ACKNOWLEDGMENTS
This study was supported by NIH F32NS094088 (A.H.L.) and Duke University. We thank Malcolm McDonald for pilot experiments and Jason Wu, Son Le, and all members of the Grandl lab for thoughtful comments on the study.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.040.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albuisson J, Murthy SE, Bandell M, Coste B, Louis-Dit-Picard H, Mathur J, Fénéant-Thibault M, Tertian G, de Jaureguiberry JP, Syfuss PY, et al. (2013). Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat. Commun 4, 1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EO, Schneider ER, Matson JD, Gracheva EO, and Bagriantsev SN (2018). TMEM150C/Tentonin3 is a regulator of mechano-gated ion channels. Cell Rep. 23, 701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, and Hollingworth S (2018). A perspective on Na and K channel inactivation. J. Gen. Physiol 150, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae C, Gnanasambandam R, Nicolai C, Sachs F, and Gottlieb PA (2013). Xerocytosis is caused by mutations that alter the kinetics of the mechanosensitive channel PIEZO1. Proc. Natl. Acad. Sci. USA 110, E1162–E1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae C, Sachs F, and Gottlieb PA (2015). Protonation of the human PIEZO1 ion channel stabilizes inactivation. J. Biol. Chem 290, 5167–5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal NR, Hermanson O, Heimrich B, and Shastri VP (2014). Stochastic nanoroughness modulates neuron-astrocyte interactions and function via mechanosensing cation channels. Proc. Natl. Acad. Sci. USA 111, 16124–16129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan SM, Lukacs V, Ranade SS, Chien S, Bandell M, and Patapoutian A (2015). Piezo1 links mechanical forces to red blood cell volume. eLife 4, e07370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, and Patapoutian A (2010). Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Xiao B, Santos JS, Syeda R, Grandl J, Spencer KS, Kim SE, Schmidt M, Mathur J, Dubin AE, et al. (2012). Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 483, 176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Houge G, Murray MF, Stitziel N, Bandell M, Giovanni MA, Philippakis A, Hoischen A, Riemer G, Steen U, et al. (2013). Gain-of-function mutations in the mechanically activated ion channel PIEZO2 cause a subtype of distal arthrogryposis. Proc. Natl. Acad. Sci. USA 110, 4667–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Mármol JI, Touhara KK, Croft G, and MacKinnon R (2018). Piezo1 forms a slowly-inactivating mechanosensory channel in mouse embryonic stem cells. eLife 7, e33149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin AE, Schmidt M, Mathur J, Petrus MJ, Xiao B, Coste B, and Patapoutian A (2012). Inflammatory signals enhance piezo2-mediated mechanosensitive currents. Cell Rep. 2, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin AE, Murthy S, Lewis AH, Brosse L, Cahalan SM, Grandl J, Coste B, and Patapoutian A (2017). Endogenous Piezo1 can confound mechanically activated channel identification and characterization. Neuron 94, 266–270.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N, Linley JE, Torres JM, Bee L, Dickenson AH, Gringhuis M, Minett MS, Hong GS, Lee E, Oh U, et al. (2013). A role for Piezo2 in EPAC1-dependent mechanical allodynia. Nat. Commun 4, 1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellefsen KL, Holt JR, Chang AC, Nourse JL, Arulmoli J, Mekhdjian AH, Abuwarda H, Tombola F, Flanagan LA, Dunn AR, et al. (2019). Myosin-II mediated traction forces evoke localized Piezo1-dependent Ca2+ flickers. Commun. Biol 2, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Li W, Zhao Q, Li N, Chen M, Zhi P, Li R, Gao N, Xiao B, and Yang M (2015). Architecture of the mammalian mechanosensitive Piezo1 channel. Nature 527, 64–69. [DOI] [PubMed] [Google Scholar]
- Glogowska E, Schneider ER, Maksimova Y, Schulz VP, Lezon-Geyda K, Wu J, Radhakrishnan K, Keel SB, Mahoney D, Freidmann AM, et al. (2017). Novel mechanisms of PIEZO1 dysfunction in hereditary xerocytosis. Blood 130, 1845–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb PA, and Sachs F (2012). Piezo1: properties of a cation selective mechanical channel. Channels (Austin) 6, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YR, and MacKinnon R (2017). Structure-based membrane dome mechanism for Piezo mechanosensitivity. eLife 6, e33660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselwandter CA, and MacKinnon R (2018). Piezo’s membrane footprint and its contribution to mechanosensitivity. eLife 7, e41968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, and Aldrich RW (1990). Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250, 533–538. [DOI] [PubMed] [Google Scholar]
- Ikeda R, and Gu JG (2014). Piezo2 channel conductance and localization domains in Merkel cells of rat whisker hair follicles. Neurosci. Lett 583, 210–215. [DOI] [PubMed] [Google Scholar]
- Jia Z, Ikeda R, Ling J, and Gu JG (2013). GTP-dependent run-up of Piezo2-type mechanically activated currents in rat dorsal root ganglion neurons. Mol. Brain 6, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamajaya A, Kaiser JT, Lee J, Reid M, and Rees DC (2014). The structure of a conserved piezo channel domain reveals a topologically distinct b sandwich fold. Structure 22, 1520–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH, and Grandl J (2015). Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. eLife 4, e12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH, Cui AF, McDonald MF, and Grandl J (2017). Transduction of repetitive mechanical stimuli by Piezo1 and Piezo2 ion channels. Cell Rep. 19, 2572–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-C, Guo YR, Miyagi A, Levring J, MacKinnon R, and Scheuring S (2019). Force-induced conformational changes in PIEZO1. Nature 573, 230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonomura K, Woo SH, Chang RB, Gillich A, Qiu Z, Francisco AG, Ranade SS, Liberles SD, and Patapoutian A (2017). Piezo2 senses airway stretch and mediates lung inflation-induced apnoea. Nature 541, 176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak MM, Nourse JL, Tran T, Hwe J, Arulmoli J, Le DT, Bernardis E, Flanagan LA, and Tombola F (2014). Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc. Natl. Acad. Sci. USA 111, 16148–16153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retailleau K, Duprat F, Arhatte M, Ranade SS, Peyronnet R, Martins JR, Jodar M, Moro C, Offermanns S, Feng Y, et al. (2015). Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell Rep. 13, 1161–1171. [DOI] [PubMed] [Google Scholar]
- Romero LO, Massey AE, Mata-Daboin AD, Sierra-Valdez FJ, Chauhan SC, Cordero-Morales JF, and Vásquez V (2019). Dietary fatty acids fine-tune Piezo1 mechanical response. Nat. Commun 10, 1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome K, Murthy SE, Kefauver JM, Whitwam T, Patapoutian A, and Ward AB (2018). Structure of the mechanically activated ion channel Piezo1. Nature 554, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczot M, Pogorzala LA, Solinski HJ, Young L, Yee P, Le Pichon CE, Chesler AT, and Hoon MA (2017). Cell-type-specific splicing of Piezo2 regulates mechanotransduction. Cell Rep. 221, 2760–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberner FJ, Prato V, Schaefer I, Schrenk-Siemens K, Heppenstall PA, and Lechner SG (2019). Structure-guided examination of the mechanogating mechanism of PIEZO2. Proc. Natl. Acad. Sci. USA 116, 14260–14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, and Offermanns S (2016). Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Invest 126, 4527–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhou H, Zhang M, Liu W, Deng T, Zhao Q, Li Y, Lei J, Li X, and Xiao B (2019). Structure and mechanogating of the mammalian tactile channel PIEZO2. Nature 573, 225–229. [DOI] [PubMed] [Google Scholar]
- Woo SH, Ranade S, Weyer AD, Dubin AE, Baba Y, Qiu Z, Petrus M, Miyamoto T, Reddy K, Lumpkin EA, et al. (2014). Piezo2 is required for Merkel-cell mechanotransduction. Nature 509, 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Goyal R, and Grandl J (2016). Localized force application reveals mechanically sensitive domains of Piezo1. Nat. Commun 7, 12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lewis AH, and Grandl J (2017a). Touch, tension, and transduction - the function and regulation of piezo ion channels. Trends Biochem. Sci 42, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Young M, Lewis AH, Martfeld AN, Kalmeta B, and Grandl J (2017b). Inactivation of mechanically activated Piezo1 ion channels is determined by the C-terminal extracellular domain and the inner pore helix. Cell Rep. 21, 2357–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarychanski R, Schulz VP, Houston BL, Maksimova Y, Houston DS, Smith B, Rinehart J, and Gallagher PG (2012). Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood 120, 1908–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng WZ, Marshall KL, Min S, Daou I, Chapleau MW, Abboud FM, Liberles SD, and Patapoutian A (2018). PIEZOs mediate neuronal sensing of blood pressure and the baroreceptor reflex. Science 362, 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Zhou H, Chi S, Wang Y, Wang J, Geng J, Wu K, Liu W, Zhang T, Dong MQ, et al. (2018). Structure and mechanogating mechanism of the Piezo1 channel. Nature 554, 487–492. [DOI] [PubMed] [Google Scholar]
- Zheng W, Gracheva EO, and Bagriantsev SN (2019a). A hydrophobic gate in the inner pore helix is the major determinant of inactivation in mechanosensitive Piezo channels. eLife 8, e44003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Nikolaev YA, Gracheva EO, and Bagriantsev SN (2019b). Piezo2 integrates mechanical and thermal cues in vertebrate mechanoreceptors. Proc. Natl. Acad. Sci. USA 116, 17547–17555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. All data are available from the Lead Contact upon reasonable request.