

Summary
The RET proto-oncogene encodes receptor tyrosine kinase, expressed primarily in tissues of neural crest origin. De-regulation of RET signaling is implicated in several human cancers. Recent phosphatome interactome analysis identified PTPRA interacting with the neurotrophic factor (GDNF)-dependent RET-Ras-MAPK signaling-axis. Here, by identifying comprehensive interactomes of PTPRA and RET, we reveal their close physical and functional association. The PTPRA directly interacts with RET, and using the phosphoproteomic approach, we identify RET as a direct dephosphorylation substrate of PTPRA both in vivo and in vitro. The protein phosphatase domain-1 is indispensable for the PTPRA inhibitory role on RET activity and downstream Ras-MAPK signaling, whereas domain-2 has only minor effect. Furthermore, PTPRA also regulates the RET oncogenic mutant variant MEN2A activity and invasion capacity, whereas the MEN2B is insensitive to PTPRA. In sum, we discern PTPRA as a novel regulator of RET signaling in both health and cancer.
Subject Areas: Biological Sciences, Molecular Biology, Cancer
Graphical Abstract
Highlights
-
•
PTPRA inhibits ligand (GDNF-GFRα1)-mediated RET activity on Ras-MAPK signaling axis
-
•
PTPRA dephosphorylate RET on key functional phosphotyrosine sites
-
•
PTPRA catalytic (PTPase) domain 1 regulates RET-driven signaling
-
•
PTPRA suppresses RET oncogenic mutant MEN2A in both Ras-MAPK and cell invasion models
Biological Sciences; Molecular Biology; Cancer
Introduction
Protein tyrosine phosphorylation is a prime eukaryotic regulatory step for intracellular signal transduction and is maintained by opposing activities of protein tyrosine kinases (PTKs) and phosphatases (PTPs). Strikingly, the number of PTPs (107) encoded by the human genome roughly matches that of PTKs (90), indicating that PTPs might also have equivalent functional complexity and specificity as their kinase counterparts. However, unlike PTKs, biological circuitry and activity control mechanisms of many PTPs are still undefined. Recently, through global systematic interactome analysis of human protein phosphatases, we have demonstrated that GDNF (glial cell line-derived neurotrophic factor) and GRB2 (growth factor receptor-bound protein 2) form a complex with the protein tyrosine phosphatase receptor-type A (PTPRA) (Yadav et al., 2017). To our interest, GDNF acts as a key homodimeric neurotrophic factor family ligand, which in conjunction with GDNF α-receptors (GFRα1-4; glycosylphosphatidylinositol-anchored proteins) incite RET (REarranged during Transfection) receptor tyrosine kinase via dimerization to prompt Ras-MAPK (mitogen-activated protein kinase) cascade and other signaling pathways (Airaksinen and Saarma, 2002, Arighi et al., 2005). Notably, only a few protein phosphatases such as PTPRF (Leukocyte common antigen-related; LAR), PTN6 (Src homology region 2 domain-containing phosphatase-1; SHP1), and PTN11 (Src homology region 2 domain-containing phosphatase-2; SHP2) have been suggested to balance the phosphorylation and oncogenic activity of RET (Hennige et al., 2001, Perrinjaquet et al., 2010, Qiao et al., 2001).
Similar to PTPRF, PTPRA is a membrane-bound “receptor-type” protein tyrosine phosphatase, with a highly glycosylated ectodomain, a single membrane-spanning region, and two intracellular catalytic phosphatase domains (PTPase; membrane-proximal D1 and -distal D2) (Daum et al., 1994, Wang and Pallen, 1991, Wu et al., 1997). Although ubiquitously expressed, it is particularly abundant in the brain (neurons and glial cells) and insulin target tissues, where it induces cell differentiation, migration, activation of voltage-gated potassium channels, and insulin secretion (Chen et al., 2009, Imbrici et al., 2000, Kaplan et al., 1990, Kapp et al., 2003, Norris et al., 1997, Petrone et al., 2003). Nevertheless, several previous studies have disclosed its critical role as a main positive regulator of Src family kinases (Fyn, FAK, and Src) in cell growth and oncogenic transformation (Huang et al., 2011, Tremper-Wells et al., 2010, Zheng et al., 2002). Conforming with this, PTPRA has been suggested to play a dual role in regulating EGFR kinase signaling via Src dephosphorylation and activation (Yao et al., 2017). Despite these findings, relatively few signaling pathways (cell adhesion- and integrin-mediated processes) and cellular targets (or substrates) have been suggested that could unravel the PTPRA-mediated regulation of cellular signaling (Bodrikov et al., 2005, Truffi et al., 2014, Yao et al., 2017).
In addition to the matching cellular localization, the RET and PTPRA tissue expression also coincide. RET is distinctly expressed in neural tissues (brain and enteric nervous system) and in the developing kidney, where it instigates axonal guidance, neuronal survival, and ureteric bud morphogenesis (Pachnis et al., 1993). Its malfunctioning is described in neuroendocrine tumors and diseases (neurocristopathies) such as renal cell carcinoma, multiple endocrine neoplasia type 2 (MEN2), neuroblastoma, and Parkinson's disease (Drinkut et al., 2016, Mulligan, 2014, Schedl, 2007). For example, in MEN2 syndrome, RET carries numerous gain-of-function mutations in extracellular (C634W; MEN2A) and catalytic (M918T; MEN2B) domains, which leads to aberrant kinase activation and, eventually, to pheochromocytomas and medullary thyroid carcinomas (Eng and Mulligan, 1997, Santoro et al., 2004). Importantly, GDNF-GFRα1-activated RET is autophosphorylated at discrete intracellular tyrosine-sites, Y981, Y1015, Y1062, and Y1096 (Y1096 found only in RET51 isoform), which provide docking sites for downstream adaptors or effectors (Src, SHC, GRB2, Enigma, and DOK proteins) and coordinate four key signaling routes: Ras-MAPK, PI3K-AKT, Src, and PLC-γ pathways (Amoresano et al., 2005, Besset et al., 2000, Coulpier et al., 2002, Goodman et al., 2014, Melillo et al., 2001). Among these sites, autophosphorylation of Y1062 is critical for the initiation of Ras-MAPK (GRB2-SOS complex) and PI3K-AKT (GRB2-GAB1 complex) relays in response to GDNF-GFRα1 co-complex during neuronal survival and proliferation (Besset et al., 2000, Coulpier et al., 2002, Kawamoto et al., 2004).
To decipher the GDNF-GFRα1-mediated RET-Ras-MAPK signaling-axis regulation by PTPRA, we mapped in-depth molecular interactions and mechanisms involved. Additionally, we focused on the plausible anti-cancer function of PTPRA and, especially, on its role in the regulation of the two oncogenic RET mutants MEN2A (C634W) and MEN2B (M918T) in cancer cessation. The possibility to modulate the oncogenic effects of MEN2A or MEN2B via regulating PTPRA activity would be intriguing and could likely offer novel therapeutic avenues for treating MEN2-type tumors.
Results and Discussion
Structural-Functional Coherence of PTPRA and RET Complexes
To expand the analysis of the functional cross talk between PTPRA and the GDNF-induced RET signaling, we applied both affinity purification-mass spectrometry (AP-MS) and proximity-dependent biotin identification (BioID) interaction proteomic approaches, now, using GDNF, RET, and GRB2 as bait proteins (Liu et al., 2018, Yadav et al., 2017). The AP-MS allows purification of the intact protein complexes and estimation of their stoichiometry, whereas the BioID enables capturing of extremely transient and close-by interactions (Figure 1A). Hence, for analysis, GDNF, RET, and GRB2 were subcloned into StrepIII-HA and BioID vectors, their corresponding stable, transient, and close-proximity interactors were purified using Strep-Tactin resin, and the interactors were identified with liquid chromatography-mass spectrometry (LC-MS) (Figure 1A) (Liu et al., 2018, Yadav et al., 2017).
Figure 1.

Affinity Purification and Proximity-Dependent Labeling Mass Spectrometry Reveal the Overlap between PTPRA and RET Interactomes
(A) The stable Flp-In-T-REx 293 cells are generated for expression of StrepIII-HA- (AP-MS) and MAC- (BioID) tagged bait proteins (PTPRA, RET, GDNF, and GRB2). The bait expression is induced with tetracycline (AP-MS and BioID baits), and extra biotin is added for BioID baits. Next, the bait complexes are single-step affinity purified and the interacting proteins are identified with LC-MS/MS (n = 4 replicates).
(B) The AP-MS and BioID derived composite protein interaction map of PTPRA and RET as well as GDNF and GRB2 baits (hexagons). Various prey proteins (circles) are grouped based on their biological functions and color coded. Uniprot entry names are used for protein nomenclature. Key: bait-prey interactions (green = BioID; red = AP-MS; and black = both).
Interestingly, the PTPRA and RET interactomes shared many core proteins that operate in various cell growth events (Figure 1B and Table S1). For example, Ras-MAPK pathway commencing cell-surface receptors (IGF1R, TGFR1, EGFR, and ERBB2), intracellular bona fide docking proteins (SOS1, SOS2, Src, SHB, SHC1, GAB1, GRB2, and FRS2), and other regulators (MARK3, CRK, and MERL) were identified not only in complex with RET but also with PTPRA (Figure 1B). More so, through extended AP-MS analysis of GRB2, as well as of GDNF, we confirmed GDNF-RET, GRB2-PTPRA, and GRB2-SHC1-SOS1-SOS2 associations (Table S1). Consistent with these results, previous studies have described that GDNF-activated RET promotes Ras-MAPK activation, which is essential for development of nervous system (enteric and brain), spermatogenesis, and kidney during embryogenesis (Costantini and Shakya, 2006, Li et al., 2006, Soba et al., 2015, Xiao et al., 2015). Noticeably, we have also retrieved PTPRA interaction with other receptor tyrosine kinases (RTKs) EGFR and ERBB2 (Figure 1B and Table S1), where PTPRA was earlier shown to dephosphorylate EGFR and subject a positive effect on downstream Ras-MAPK signaling through Src activation (Yao et al., 2017). Therefore, we sought to check the specificity of the derived PTPRA and RET interactomes by comparing them with that of EGFR and ERBB2 along with IGF1R kinase, related to RET in neural oncogenesis (Denardo et al., 2013). For this purpose, both AP-MS and BioID approaches were applied to draw the proteomes of these RTKs: EGFR (219 interactions), ERBB2 (111 interactions), and IGF1R (209 interactions) (Salokas et al., Unpublished Data). Upon comparison, PTPRA exhibited <15% common interactions with these RTKs, whereas with RET it shared nearly 49% interactions (Figure S1A and Table S1). Nevertheless, within the Ras-MAPK module, ∼7.3 (average) interactions reoccurred in EGFR, ERBB2, and IGF1R proteomes (Figure S1B). Intriguingly, RET interactome also differed substantially from these RTKs with a similarity of about 11% with EGFR, 5% with ERBB2, and 10% with IGF1R, suggesting their characteristic signaling complexes (Figure S1C). Hence, this comparative analysis data not only verified the distinctiveness of our PTPRA and RET interactomes but also indicated cross talk of PTPRA in regulating RET-mediated Ras-MAPK signaling, by virtue of their converging interaction frameworks (PTPRA-EGFR-FRS2-GRB2-GAB1 and RET-FRS2-GRB2-GAB1-SHC1) (Figure 1B and Table S1).
Additionally, overlap of the PTPRA and RET interactomes was also detected with a large cohort of proteins such as UCN5(B-C), NRP1, BASP1, CERS2, VANG2, MARK2, and TULP3, linked to varied aspects of neuronal development, axon guidance, pathfinding, neuronal polarization, and pattern (axis) formation (Figure 1B and Table S1) (Chen et al., 2006, Imgrund et al., 2009, Larrivee et al., 2007, Mosevitsky, 2005, Norman et al., 2009, Poliak et al., 2015, Telley et al., 2016, Torban et al., 2004). Although many of these biological functions are linked to RET, much less is known about PTPRA in regulating these processes. Moreover, the gene expression pattern of PTPRA shows its high levels in the neuroendocrine tissues of the nervous system, kidney, thyroid, and pituitary gland, along with the tumors derived from these tissues (Figure S2). These include astrocytoma, glioblastoma (GBM), oligodendroglioma, mixed glioma, nephroblastoma, and thyroid carcinoma, further implicating the importance of PTPRA in neural development (Figure S2). Therefore, collectively, as our proteomic results point toward physical and functional interaction between PTPRA and RET, we set out to study if RET activity would be PTPRA regulated and if RET would be a direct substrate of PTPRA.
PTPRA Inhibits the RET-Ras-MAPK Signaling Pathway
The activation of RET by GDNF-GFRα1 complex is the first event in the activation of the Ras-MAPK signaling and acts as a catalyst for the downstream relay. Therefore, to determine the effect of PTPRA on this pathway, we developed a RET-Ras-MAPK activation detection system in HEK293 cells, which included StrepIII-HA-tagged RET (RET9), Elk1-Gal4-binding domain (GBD) effector (pGBD-Elk1), and Gal4-activation domain (GAD) containing Firefly luciferase (pGAD-FR-Luc) reporter along with Renilla luciferase (phR-Luc) control reporter (Figure 2A). Upon activation by GDNF-GFRα1, the reporter system shows >5-fold pathway activity induction. Since HEK293 cells exhibit undetectable levels of endogenously expressed PTPRA (https://amp.pharm.mssm.edu › Harmonizome and Geiger et al., 2012), this system was then used for assessing the outcomes of transfecting increasing amounts of PTPRA (0, 10, 25, and 50 ng), alongside GFP control, on the RET-Ras-MAPK pathway activity in the presence or absence of soluble GDNF-GFRα1 (100–500 ng/mL; 24 h) ligand complex (Figure 2B). The maximal inhibition on the ligand-activated pathway (orange bar) was achieved with transfection of 50 ng PTPRA (∼2-fold, p = 0.0001) (Figure 2B). Notably, the basal pathway activity (blue bar) was also restricted to a similar extent (∼1.5-fold) (Figure 2B). The detected inhibitory role of PTPRA in RET-Ras-MAPK cascade was further verified using MG87RET reporter fibroblast cells stably expressing RET (Eketjall et al., 1999). Even under steady RET levels, the PTPRA expression moderated (1.9- to 2.4-fold) the MAPK activation to nearly comparable extents (Figure 2C). This not only validated the attained HEK293 luciferase-reporter assay results but also affirmed the reliability and usability of the RET-Ras-MAPK activation detection system in transiently transfected HEK293 cells.
Figure 2.

RET-Ras-MAPK Dual-Luciferase Reporter System
(A) HEK293 cells in 96-well plate format are transiently transfected with RET (StrepIII-HA C-terminal tag) and Gal4-binding domain-containing Elk1 (pGBD-Elk1) together with Firefly Gal4-activation domain-containing luciferase (pGAD-FR-Luc), transfection control Renilla luciferase (hR-Luc), PTPRA (StrepIII-HA), and GFP control (StrepIII-HA) expression vectors. GDNF (100 ng/mL) and GFRα1 (500 ng/mL) co-complex is added for 24 h to induce the RET-Ras-MAPK pathway activation. Then, the cells are rapidly lysed and the dual-luciferase signal is measured with a luminometer. The Firefly luciferase values are normalized by the transfection control Renilla luciferase signal values to obtain relative luciferase signal (RLU).
(B and C) (B) HEK293 and (C) MG87RET cells expressing RET, transiently (B) or stably (C), show decreased RET-Ras-MAPK pathway activity with increasing amounts (0, 10, 25, 50 ng) of PTPRA alongside GFP control (50, 25, 40, 0 ng) in absence (blue bars) and presence (orange bars) of GDNF-GFRα1 ligands. The bar graph is representative of two independent experiments, where all bars indicate average ratios (n = 5 replicates) of Firefly to Renilla luciferase counts (relative luciferase unit, RLU) and the error bars designate one standard deviation. The statistical significance is obtained by a two-tailed Student's t test (***p < 0.0001 and **p < 0.001).
PTPRA Impacts Downstream RET-Ras-MAPK Signaling
We then followed up the ramification of PTPRA expression on subsequent signaling target MAPKs (ERK1 and ERK2). To do so, HEK293-MSR cell lysates, containing RET (StrepIII-HA-tagged) and increasing amounts of wild-type PTPRA (V5-tagged), were prepared in the absence and presence (15 min stimulation) of GDNF-GFRα1 ligands. The immunoblotting with site-specific p44/42 MAPK (T202/Y204) antibody shows that the phosphorylation of endogenous ERKs (1 and 2) was readily induced by the ligand-activated RET and expression of PTPRA potentiated their phosphorylation, which concluded the PTPRA-facilitated RET-Ras-MAPK inhibition (Figure 3A).
Figure 3.

PTPRA Impedes Downstream MAPK (ERK1 and ERK2) Activation and Requires GRB2 for Regulating RET-Ras-MAPK Signaling
(A) HEK293-MSR are co-transfected with RET (StrepIII-HA-tagged) and increasing amounts of V5-tagged PTPRA (0, 0.5, and 1.5 μg) construct. After 15 min stimulation with GDNF-GFRα1 ligands (100–500 ng/mL), the cell lysates were immunoblotted with p44/42 MAPK (T202/Y204) antibody to detect endogenous ERK1 and ERK2 phosphorylation status. Both ERK1 and 2 were dephosphorylated by PTPRA. RET and PTPRA protein expression are determined by anti-HA and anti-V5 antibodies, respectively. Tubulin is used as a loading control.
(B) Either StrepIII-HA-tagged wild-type (WT) PTPRA or Y789F mutant along with V5-tagged GRB2 expression vectors are transiently co-transfected in HEK293-MSR cells. The GRB2 co-immunoprecipitate with WT PTPRA only. The immunoprecipitated (IP) proteins, as well as cell lysates, were immunoblotted with anti-V5 and anti-HA antibodies to detect GRB2 and PTPRA proteins, respectively. The phosphorylation of ERK1 and ERK2 proteins from cell lysate was detected with p44/42 MAPK (T202/Y204) antibody. Tubulin is used as a loading control.
(C) PTPRA Y789F mutant displays significantly decreased phosphatase activity in comparison with WT PTPRA. However, the YF mutant still inhibited RET activity in the Ras-MAPK luciferase-reporter assays, shown in the absence (blue bars) and presence (orange bars) of GDNF-GFRα1 stimulation (100–500 ng/mL; 24 h). The data represent average ratios (n = 5 replicates) of the relative luciferase unit (RLU), and the error bars indicate one standard deviation. The statistical significance is obtained by a two-tailed Student's t test (***p < 0.0001).
Furthermore, from PTPRA and RET complex analysis, we have observed their strong interaction with GRB2, an adaptor signaling protein (Table S1). Previously, GRB2, in complex with other docking (adaptor) proteins including Src kinase, has been reported to bind RET and harmonize the Ras-MAPK cascade (Alberti et al., 1998, Ohiwa et al., 1997), whereas PTPRA could sequester the SH2 domain of not only GRB2 but also of Src through its phosphorylated Y789 site, thereby, linking PTPRA to other key components of cellular signaling (Den Hertog et al., 1994, Denhertog and Hunter, 1996, Zheng et al., 2000). Apparently, in our proteomic analysis, we spotted RET-Src interaction, but with low overall interaction strength, whereas PTPRA-Src interaction was not retrieved (Table S1). Hence, we tested the role of GRB2 in PTPRA-induced RET-Ras-MAPK down-regulation by creating PTPRA Y789F (tyrosine; Y to phenylalanine; F) mutation (Figure 4A). We carried out co-immunoprecipitation of either wild-type PTPRA or Y789F mutant (StrepIII-HA-tagged) with GRB2 (V5-tagged). As shown in Figure 3B, the PTPRA-GRB2 interaction was fully abolished and slightly higher phosphorylation of MAPKs (ERK1 and ERK2) was obtained with Y789F mutant. Then, in Ras-MAPK luciferase assays, PTPRA Y789F mutant competently reduced (∼1.63-fold; p = 2.68 × 10−5) the activated RET-Ras-MAPK reporter signal, despite being less active (∼1.51-fold; p = 4.95 × 10−4) than wild-type PTPRA, confirming that Y789 phosphorylation not only is essential for PTPRA-GRB2 interaction but also has direct influence on RET signaling (Figure 3C).
Figure 4.

The Activity of PTPase Domain 1 (D1) Is Indispensable for PTPRA-Mediated Negative Regulation of RET
(A) Schematic representation of wild-type (WT) PTPRA and the domain deletion (DD) mutants. The WT PTPRA is a 793 amino acid (aa) long protein. The ΔD1 (1–231/492–793 aa) and ΔD2 (1–523/782–793 aa) correspond to deletions of PTPase I and PTPase II domains, respectively. The catalytic cysteine-to-serine inactivation mutations C433S and C723S in PTPas domain D1 and D2, respectively, along with C-terminal Y789F mutation are indicted as vertical red bars. N, N-terminal; ECD, extracellular domain; TM, transmembrane region; JM, juxtamembrane region; D1, protein tyrosine phosphatase domain 1 (PTPase I); L, interdomain linker region; D2, protein tyrosine phosphatase domain 2 (PTPase II); C, C-terminal tail. See also Figure S3.
(B) HEK293-MSR cells are transiently transfected with either StrepIII-HA-tagged wild-type (WT) PTPRA or DD mutants (ΔD1and ΔD2) along with V5-tagged RET expression vectors. The RET co-precipitates with all the tested PTPRA constructs. The immunoprecipitated (IP) proteins, as well as cell lysates, were immunoblotted with anti-V5 and anti-HA antibodies to detect RET and PTPRA proteins, respectively. Tubulin is used as a loading control. See also Figure S3.
(C) PTPRA D1 domain deletion mutant ΔD1 displays significant loss of phosphatase activity, whereas both WT and ΔD2 constructs potently inhibit the RET activity in the Ras-MAPK luciferase-reporter assays. The RET-Ras-MAPK pathway activity is shown in the absence (blue bars) and presence (orange bars) of GDNF-GFRα1 co-complex stimulation (100-500 ng/mL; 24 h). The bar graph is representative of two independent experiments, where all bars indicate average ratios (n = 5 replicates) of Firefly to Renilla luciferase counts (relative luciferase unit, RLU) and the error bars designate one standard deviation. The statistical significance is obtained by a two-tailed Student's t test (***p < 0.0001 and **p < 0.001).
(D) HEK293-MSR cells are transiently transfected with either StrepIII-HA-tagged wild-type (WT) PTPRA or CS mutants (C433S and C723S) along with V5-tagged RET expression vectors. The RET co-immunoprecipitates with all the PTPRA constructs. The immunoprecipitated (IP) proteins, as well as cell lysates, were immunoblotted with anti-V5 and anti-HA antibodies to detect RET and PTPRA proteins, respectively. The amount of immunoprecipitated tyrosine-phosphorylated RET was detected with a general anti-phosphotyrosine (pTyr) antibody. Tubulin is used as a loading control. See also Figure S3.
(E) PTPRA C433S mutant shows a significant decrease in RET activity in the Ras-MAPK luciferase-reporter assays, whereas C723S did not. The effect of CS mutation is measured in the absence (blue bars) and presence (orange bars) of GDNF-GFRα1 ligand co-complex (100–500 ng/mL; 24 h). The bar graph is representative of two independent experiments, where all bars indicate average ratios (n = 5 replicates) of Firefly to Renilla luciferase counts (relative luciferase unit, RLU) and the error bars designate one standard deviation. The statistical significance is obtained by a two-tailed Student's t test (***p < 0.0001).
Molecular Determinants of PTPRA-RET Interaction
To investigate the mechanism of PTPRA imposed constraints on RET-Ras-MAPK signaling, we tested how stable or transient the probable direct interaction between PTPRA and RET is and which PTPRA domains are required for its functions. Therefore, we constructed PTPRA membrane-proximal and membrane-distal PTPase D1 and D2 domain deletion (DD; ΔD1 and ΔD2) mutants, respectively (Figure 4A). In particular, both DD mutants retain the juxtamembrane region (JM) involved in protein-protein interactions, as well as the C-terminal tail containing GRB2- and Src-binding sites (Figure 4A). After validating the analogous intracellular localization of these DD mutants with that of wild-type PTPRA, we performed co-immunoprecipitation from HEK293-MSR cells co-expressing either wild-type or DD mutants of PTPRA (StrepIII-HA-tagged) together with RET (RET9; V5-tagged) (Figures 4B and S3A). As presented in Figure 4B, the RET co-precipitated with wild-type PTPRA as well as both DD mutants, which indicated that RET interacts with this phosphatase and might represent its physiological target. Noticeably, RET was predominantly recognized as a 150-kDa band (endoplasmic reticulum [ER] glycosylated species) in HEK293-MSR cells, which was explained previously by inefficient delivery of RET9 (1,072 amino acids) isoform from ER and Golgi to plasma membrane relative to another isoform RET51 (1,114 amino acids) in these cells (Iwamoto et al., 1993, Richardson et al., 2012, Takahashi et al., 1991).
Next, we examined the effect of PTPRA DD mutants (ΔD1 and ΔD2) in the RET-Ras-MAPK reporter assay. In this assay, the ΔD2 PTPRA mutant showed activity equivalent to the wild-type phosphatase post GDNF-GFRα1 stimulation, whereas the ΔD1 mutant failed to suppress the RET activity (Figure 4C). This demonstrates that the D1 domain is the main contributor toward RET inhibition, as well as highlights that the PTPRA membrane-proximal D1 domain contains most of the phosphatase catalytic activity toward RET.
To further understand the molecular details of PTPRA-RET interaction, we utilized PTPRA substrate-trapping (or phosphatase-dead) CS mutants (catalytic cysteine-to-serine): C433S (in D1 domain) and C723S (in D2 domain) (Figures 4A and S3A) (Den Hertog et al., 1994). The co-immunoprecipitation from HEK293-MSR cells, co-transfected with either wild-type or CS mutants of PTPRA (StrepIII-HA-tagged) and RET (V5-tagged), shows that both CS mutants could bind to (de)phosphorylated RET with no significant difference with wild-type PTPRA (Figure 4D). Also, the PTPRA interaction is greatly reduced with kinase-dead RET (K758R) mutants, suggesting that autophosphorylation of RET mediates this interaction (Figure S3B). However, in the luciferase assays, only C433S mutant leads to a greater (∼2.7-fold; p = 7.33 × 10−7) dampening of reporter signal in contrast to C723S, indicating that C433S acts as a true substrate-tapping mutant and D1 domain catalytic activity is indeed required for PTPRA function (Figure 4E).
Taken together, although the D2 domain appeared to be non-essential for the RET-Ras-MAPK repression, it could be required for other possible PTPRA-substrate interactions or might play, if any, regulatory role.
PTPRA Instigates Direct Dephosphorylation of RET and Downstream RET Substrates
Furthermore, we explored the direct consequences of PTPRA phosphatase on tyrosine phosphorylation of RET kinase. Through in vitro phosphatase assay, where recombinant RET protein was incubated with either active PTPRA or recombinant GFP (control), we observed a significant decrease in RET tyrosine phosphorylation using general anti-phosphotyrosine (pTyr) antibody (Figure 5A). Moreover, LC-MS/MS-based phosphopeptide quantification (MaxQuant) of these samples showed the PTPRA-regulated dynamics of specific RET pTyr-sites (RET + GFP versus RET + PTPRA) (Figure 5A). Several conventional (known) RET pTyr-sites spanning juxtamembrane region, catalytic domain, and C-terminal tail such as Y752, Y826, Y981, and Y1015 underwent notable phosphorylation changes (54%, 95%, 17%, and 81% dephosphorylation, respectively), whereas Y687, Y900, Y905, Y1029, Y1062, Y1090, and Y1096 were completely dephosphorylated by PTPRA (Figure 5A). Interestingly, two of these phosphosites, Y1062 and Y1096, are shown to be critical for RET-Ras-MAPK activation (marked with an asterisk in Figure 5A) (Coulpier et al., 2002, Kawamoto et al., 2004). These results indicated that RET is a direct dephosphorylation target of PTPRA phosphatase and implied a potential enzyme-substrate relationship.
Figure 5.

PTPRA Dephosphorylates RET and Regulates RET Kinase Activity
(A) The in vitro dephosphorylation of recombinant RET by recombinant PTPRA (active) or GFP (control) is checked with immunoblotting using an anti-pTyr antibody, where overall tyrosine phosphorylation of RET was significantly lowered in the presence of PTPRA. Also, the site-specific tyrosine phosphorylation changes of recombinant RET are detected with LC-MS/MS analysis, where several known phosphotyrosine (Y) sites (vertical red bars) on RET are effectively dephosphorylated by the PTPRA. The bar graph illustrates the percentage phosphorylation level of detected pTyr-sites in RET + PTPRA sample compared with control (RET + GFP), where the phosphorylation in control sample (i.e., maximum) is set to 100% (green dotted line). pTyr sites directly involved in RET-Ras-MAPK activation are marked with an asterisk (*). CLD1-4, cadherin-like domains 1–4; CRD, cysteine-rich domain; TM, transmembrane region; JM, juxtamembrane region; and TK, tyrosine kinase domain.
(B) The MS-based in vitro RET dephosphorylation assay shows the clear global inhibition of RET-mediated phosphorylation of pTyr sites on the HEK cell lysate proteins by PTPRA phosphatase. Phosphosites with localization probability score ≥0.75 are shown and log2 of the phosphopeptide intensities are depicted in the heat-map (scale bar, 0–30). Few known RET substrates also show a clear decrease in their indicated pTyr sites (Y) (% phosphorylation level; RET + PTPRA versus RET + GFP) in the presence of active PTPRA, shown as a bar graph. The phosphorylation in RET + GFP control sample (i.e., maximum) is set to 100% (green dotted line). See also Figure S4 and Table S2.
(C) Both Y1062 and Y981 are required for the RET activity on the Ras-MAPK pathway as displayed by the reporter assay activity. The Y1062 seems to be critical for RET activity, but both sites are regulated by the PTPRA as shown by using tyrosine (Y) to non-phosphorylatable phenylalanine (F) mutants:Y1062F and Y981F. The effect of PTPRA is measured in the absence (blue bars) and presence (orange bars) of GDNF-GFRα1 co-complex (100–500 ng/mL; 24 h). The data represent average ratios (n = 5 replicates) of the relative luciferase unit (RLU), and the error bars indicate one standard deviation. The statistical significance is obtained by a two-tailed Student's t test (***p < 0.0001).
Then, we utilized a modified in vitro dephosphorylation assay coupled to mass spectrometry for obtaining the effects of PTPRA on cellular substrates of RET (Figures 5B and S4). Briefly, endogenous kinase deactivated (FSBA) cell lysate was incubated with recombinant RET and heavy ATP (ATP-18O4) in the presence or absence of recombinant PTPRA (Figure S4). The Ti4+-IMAC enriched phosphopeptides were then analyzed using LC-MS/MS, revealing that the global tyrosine phosphorylation-profile of various candidate RET substrates was diminished by the PTPRA phosphatase activity (Figure 5B and Table S2). To our interest, several pTyr-sites on previously known RET substrates, also found in our PTPRA-RET interactome as part of the Ras-MAPK module, were clearly afflicted. For example, the phosphorylation status of Y209, Y279, Y548, Y1253, and Y904 sites in GRB2, PTN1, PLCG1, and CTND1, respectively, was greatly reduced (Figure 5B and Table S2). Moreover, phosphotyrosine sites in EGFR (Y1197), CTND1 (Y257), PTN1 (Y546), KAP2 (Y282), as well as GRB2 (Y37) proteins were completely lost in the presence of PTPRA (Table S2).
Among the above detected PTPRA-induced broad and site-specific RET phosphoproteomic changes, the autophosphorylation of Y1062 and Y981 has been shown in discrete oncogenic RET forms and is critical for GDNF-GFRα1-mediated neuronal signaling where Y1062 is vital for commencing Ras-MAPK as well as PI3K-AKT pathways and Y981 for Src activation (Asai et al., 1996, Coulpier et al., 2002, Encinas et al., 2004, Kawamoto et al., 2004). Therefore, we then mutated these sites to generate tyrosine (Y)-to-phenylalanine (F) mutants (Y1062F and Y981F) of RET and used them with PTPRA to analyze RET-Ras-MAPK signaling in a ligand-dependent manner. In the luciferase-reporter assay, the Y1062F mutant displayed much lower RET activity than the Y981F mutant in response to GDNF-GFRα1, suggesting that Y1062 is imperative for Ras-MAPK activation and directly contributes to RET regulation (Figure 5C). Surprisingly, with both RET-YF mutants PTPRA still prevented (up to 2-fold) the ligand-activated pathway response, which pointed toward other subsidiary RET pTyr sites that are likely to be affected by PTPRA and supported the phosphosite results obtained via MS-analysis.
PTPRA Restrains Oncogenic Potential of the RET Mutant MEN2A but Not MEN2B
We continued to broaden the participation of PTPRA phosphatase in controlling RET by studying its tumorigenic missense mutants, MEN2A and MEN2B, responsible for multiple endocrine neoplasms (MEN) of the thyroid and adrenal glands (Figure 6A). Reportedly, the MEN2A mutation (C634W) triggers RET by inducing ligand-independent homodimerization through abnormal intermolecular disulfide linkage, whereas the MEN2B mutation (M918T) stimulates the activity of RET monomer (Figure 6A) (Iwashita et al., 1996). This unusual intrinsic RET activation is liable for its high transforming ability and onset of atypical neurocristopathies (familial medullary thyroid carcinoma, papillary thyroid carcinoma, and pheochromocytomas). Therefore, we pursued to evaluate whether PTPRA can remodel the molecular and functional properties of these cancer-associated RET proteins. The co-immunoprecipitation of both MEN2A and MEN2B with wild-type PTPRA as well as its DD mutants established that, similar to wild-type RET, these RET mutants bind efficiently to PTPRA and might also serve as dephosphorylation targets of PTPRA in human cancers (Figures 4B and 6B). Therefore, when the general in vivo tyrosine phosphorylation level of these oncogenic RET forms was measured under varying levels of PTPRA (2.5 and 5 μg plasmid) in HEK293-MSR cells, we noted that phosphorylation of RET and MEN2A, but not of MEN2B, gradually decreased as the amount of PTPRA increased (Figure 6C).
Figure 6.

RET-MEN2A and -MEN2B Cancer Mutations and their Regulation by PTPRA
(A) RET-linked human multiple endocrine neoplasia (MEN) type 2 carcinomas, affecting the thyroid and adrenal glands. The corresponding MEN2 tumorigenic mutations (vertical red bars), MEN2A (C634W) and MEN2B (M918T), are in the RET CRD and TK domains, respectively. CLD1-4, cadherin-like domains 1–4; CRD, cysteine-rich domain; TM, transmembrane region; JM, juxtamembrane region; and TK, tyrosine kinase domain.
(B) PTPRA co-immunoprecipitates with both RET-MEN2A and -MEN2B mutants. V5-tagged RET-MEN2A or -MEN2B and either StrepIII-HA-tagged wild-type (WT) or phosphatase domain-deleted (DD: ΔD1 and ΔD2) mutants of PTPRA were expressed in HEK293-MSR cells. The immunoprecipitated (IP) proteins, as well as cell lysates, were immunoblotted with anti-V5 and anti-HA antibodies to detect RET-MEN2s and PTPRA proteins, respectively. Tubulin is used as a loading control (* unspecific band).
(C) In vivo tyrosine phosphorylation of RET and MEN2A, but not of MEN2B, successively decreases with the increasing amounts of PTPRA (0, 2.5, and 5 μg) construct. The RET and MEN2 (A and B) proteins were precipitated from cell lysates with anti-HA beads and immunoblotted with general anti-phosphotyrosine (pTyr) antibody. Equal gel loading is verified by re-probing with anti-HA antibody, and relative intensity of pTyr level is analyzed by ImageJ software. PTPRA expression in cell lysate is determined by the anti-V5 antibody. Tubulin is used as a loading control.
(D) PTPRA efficiently inhibits the wild-type RET and MEN2A, whereas the MEN2B is less sensitive to the inhibition in the Ras-MAPK reporter assay, shown in the absence (blue bars) and presence (orange bars) of GDNF-GFRα1 ligands (100–500 ng/mL; 24 h). The bar graph is representative of two independent experiments, where all bars indicate average ratios (n = 5 replicates) of Firefly to Renilla luciferase counts (relative luciferase unit, RLU) and the error bars designate one standard deviation. The statistical significance is obtained by a two-tailed Student's t test (***p < 0.0001 and **p < 0.001). See also Table S3.
(E) Similarly to the wild-type RET, the ΔD2 mutant inhibits MEN2A but not MEN2B in the absence (blue bars) and presence (orange bars) of GDNF-GFRα1 ligands. Interestingly, the ΔD1 mutant slightly increases the MEN2B activity in our reporter assay. The bar graph is representative of two independent experiments, where all bars indicate average ratios (n = 5 replicates) of Firefly to Renilla luciferase counts (relative luciferase unit, RLU) and the error bars designate one standard deviation. The statistical significance is obtained by a two-tailed Student's t test (***p < 0.0001 and **p < 0.001).
We then conducted reporter assays with these oncogenic RET variants (MEN2s) in the presence of PTPRA and its DD mutants to check their individual effects on Ras-MAPK signaling. As expected, the baseline MEN2s-MAPK reporter activity was higher than the RET-MAPK activity, owing to the continuous signaling flux by the homodimeric MEN2A and monomeric MEN2B proteins (Figure 6D). Compared with luciferase intensity of wild-type RET alone, the MEN2A showed more pronounced (∼5.5-fold; p = 5.92 × 10−7) basal activity than MEN2B (∼3.8-fold; p = 0.0001) (Figure 6D). Additionally, the expression of PTPRA significantly attenuated MEN2A basal activation (∼2.6-fold; p = 6.7 × 10−6), whereas that of MEN2B remained unchanged (Figure 6D). Surprisingly, GDNF-GFRα1 treatment still caused hyper-activation of both MEN2A and MEN2B reporter signals (∼2.3- and ∼4.5-fold, respectively) (Figures 6D and 6E). This could be due to the existence of hybrid receptor species between wild-type RET and MEN2 mutants, causing the additive response to ligand activation. However, this needs to be clarified in the future. Nevertheless, the data under these ligand-activated conditions also correlate well with the basal pathway activation results, where MEN2A-MAPK pathway activity (but not MEN2B) was again significantly inhibited (∼2.5-fold; p = 2.39 × 10−9) by PTPRA co-expression (Figure 6D). Furthermore, in corresponding assays of PTPRA DD mutants (ΔD1 and ΔD2) with MEN2A and MEN2B proteins, PTPRA ΔD2 mutant inhibited the MEN2A-MAPK activity significantly (∼3.2-fold) (Figure 6E). Unexpectedly, ΔD1 mutant enhanced (∼1.9-fold; p = 5.14 × 10−5) the basal MEN2B-MAPK activity, for which the exact reason is not known and requires further investigation.
Because MEN2B-MAPK signaling was not modified by PTPRA (or DD mutants), irrespective of the ligand addition, we reasoned that PTPRA prefers dimeric configuration of MEN2A over the monomeric state of MEN2B to exert substrate inhibition. Alternatively, we evaluated the in vivo MS phosphorylation difference between RET and MEN2 mutants in the presence of GDNF-GFRα1 ligands as well as monitored whether their pTyr sites are impaired by PTPRA. The peptides and phosphosites (without phosphoenrichment) identified in these samples are listed in Table S3. Relatively more pTyr sites were detected for MEN2A (eight sites) than for RET and MEN2B (three sites each), which was in line with the RET and MEN2s phosphorylation levels in our reporter assays (Figures 6D and 6E and Table S3). Moreover, MEN2A and MEN2B harbored discrete phosphopeptide profiles. Under this setup, PTPRA reduced the phosphopeptide intensities of RET and MEN2A pTyr sites to a larger extent than MEN2B sites, demonstrating the basis of MEN2B resistance to PTPRA action (Table S3).
Also of interest is the likelihood that the MEN2B localization would also differ from the wild-type RET and MEN2A mutant. To assess these spatial prospects, the molecular context of PTPRA and RET variants was derived from the BioID data using mass spectrometry (MS)-microscopy approach, where they were predominantly enriched in the plasma membrane (Figure 7A and Table S1) (Liu et al., 2018). However, MEN2A exclusively displayed plasma membrane localization in a manner identical to the ligand-induced aggregation of receptors on the cell surface. Additionally, the co-localization was also inspected in HeLa cells with fluorescent microscopy, where PTPRA showed comparable colocalization signal with RET and MEN2A (Figure S5).
Figure 7.

PTPRA Inhibits the RET Mutant MEN2A-induced Invasion
(A) Mass spectrometry (MS)-microscopy analysis of RET, MEN2A-B, and PTPRA subcellular localization. The cellular localization context of RET, MEN2A-B, and PTPRA interactors identified through the BioID method. The circular polar plot is divided into 14 different cellular compartments: centrosome (CEN), chromatin (CHR), endosome (END), endoplasmic reticulum (ER), exosome (EXO), Golgi (GOL), lysosome (LYS), microtubule (MIC), mitochondria (MIT), nuclear envelope (NE), nucleolus (NUC), peroxisome (PER), plasma membrane (PM), and proteasome (PRO). The color gradient indicates the localization scores (scale bar, 0–1) calculated by the MS-microscopy tool. See also Figure S5.
(B) PTPRA inhibits the MEN2A-induced cell invasion. The light micrographs of the hematoxylin and eosin-stained MDCK cells co-expressing MEN2A or MEN2B along with control vector (mock) or PTPRA plasmids show reduced cell invasion when PTPRA is present (scale bar, 20 μm). The bar graph shows the quantitated relative invasion, where the invasion of mock cells is set to one. The error bars indicate mean ± SEM (standard error of the mean, n = 6) and **p < 0.001 defines the significance, calculated from a two-tailed Student's t test.
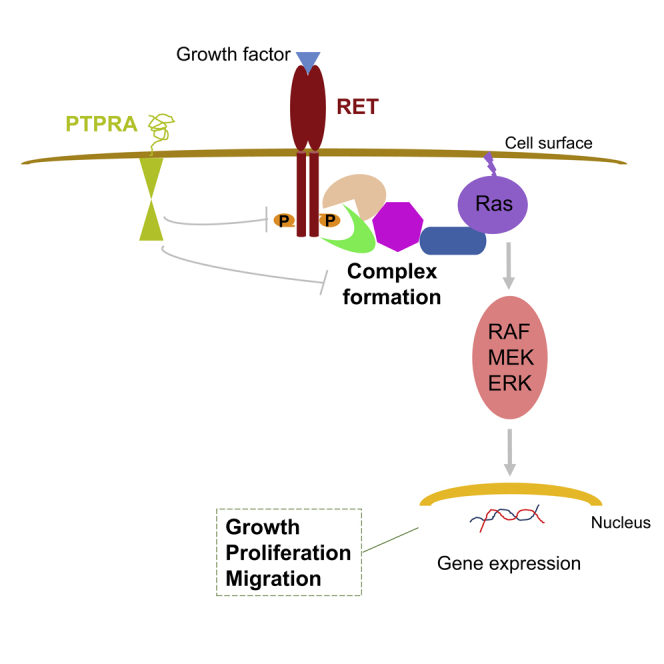
(C) Model for signaling regulation mediated by RET and PTPRA interaction. By binding to dimeric RET and oncogenic mutant MEN2A (C634W), PTPRA limits the RET and MEN2A phosphorylation along with downstream Ras-MAPK activation. However, monomeric MEN2B (M918T) RET mutant refrains from PTPRA inhibition. As a consequence, by hindering abnormal cancer traits, such as cell invasion, through RET regulation, this model demarks the plausible anti-cancer role of PTPRA in medullary thyroid cancer, pheochromocytomas, and parathyroid adenomas. CAD, cadherin-like domain; CYS, cysteine-rich domain; ECD, extracellular domain; JM, juxtamembrane region; P, phosphate group; PTPase, protein tyrosine phosphatase domain; TK, tyrosine kinase domain; and TM, transmembrane region.
PTPRA Suppresses the MEN2A-Induced Invasion
Finally, to assess the possible role of PTPRA in limiting certain RET mutants’ cancer-causing traits, we performed a three-dimensional (3D) collagen invasion assay using the MDCK cell model. These cells display characteristic epithelial basolateral polarity and directional phenotypic transition in response to their growth environment (Zak et al., 2000). In this model, wild-type RET-expressing cells were minimally invasive, whereas both MEN2A- and MEN2B-expressing cells showed enhanced baseline invasiveness (up to 5-fold) towards GDNF-GFRα1 ligands (used as chemoattractants), which indicated the severity of these cancer-associated mutations in tumor progression (Figure 7B). This robust phenotype was antagonized by the co-incorporation of PTPRA, where it significantly decreased (3-fold) the migration potential of MEN2A mutant (Figure 7B). By contrast, the MEN2B-expressing cells remained insensitive to PTPRA-mediated suppression of chemotaxis and invasion. Because invasion across extracellular matrix constraint is prevalent to cancer metastasis, these facets of PTPRA effects on cell invasion in our 3D collagen model uncovers PTPRA as an immediate regulator with a probable anti-cancer role in steering RET-Ras-MAPK signaling.
Concluding Remark
PTPRA is expressed widely in human neuronal cells (brain, thyroid, testis, and kidney) and their associated cancers (astrocytoma, mixed glioma, thyroid carcinoma, renal cell carcinoma, and nephroblastoma). Previously, PTPRA has been shown to regulate a few growth factor-dependent signaling by dephosphorylating only a minority of cytoplasmic- and receptor-tyrosine kinases (Src, Fyn, and EGFR). Hence, the other critical functional primary cellular targets of PTPRA have remained as yet unknown. Thus, we investigated the role of PTPRA in GDNF-activated RET-Ras-MAPK signaling and obtained extensive physical and functional interactions between PTPRA and RET complexes. For example, neurotrophic factor GDNF and different adaptors (GRB2, GAB1, and FRS2) collectively interacted with both PTPRA and RET. These along with other adaptor proteins (Src, SOS1-2, SHC1, and SHB) have been reported as components of neuronal RET-Ras-MAPK signaling, and their association with PTPRA in our study proposed its relevance in coordinating RET intracellular signaling (Besset et al., 2000, Katz et al., 2007). Moreover, we demonstrated that PTPRA significantly inhibits the GDNF-GFRα1-induced RET-Ras-MAPK pathway activity, indicating the negative regulation imposed by PTPRA. We also showed the decreased phosphorylation levels of downstream MAPKs ERK1 and ERK2, suggesting that RET is a primary target of PTPRA. Of note, by using PTPRA Y789F mutant we established the role of proximate adaptor protein GRB2 in the context of RET-mediated Ras-MAPK signaling. Surprisingly, the response of PTPRA on overall Ras-MAPK signaling in our study contradicted with that of Yao et al. (2017), where PTPRA was shown to positively influence the EGFR-driven Ras-MAPK signaling. Nevertheless, the reason, to some extent, might lie in the composition of RET and EGFR signaling complexes, as evinced by their unique interactomes (∼10% overlap) obtained through proteomic analysis. Then, using phosphatase domain (PTPase) deletion (D1 and D2) and catalytically inactive CS point mutants, we not only established direct physical interaction between PTPRA and RET but also mapped the functional domain required for its molecular activity. We further revealed that PTPRA influences the overall (in vitro) tyrosine phosphorylation profile of RET and its candidate cellular substrates such as GRB2, PLCG1, EGFR, and KAP2, which displayed either complete loss or a significant decrease in their tyrosine phosphorylation. Additionally, many tyrosine sites on RET itself (Y826, Y981, Y1015, Y1062, and Y1096) were inflicted by PTPRA, suggesting that RET is directly dephosphorylated by this phosphatase. Besides that, the MS-based phosphoproteomic results of in vivo RET dephosphorylation by PTPRA after ligand-activation confirmed that the functional outcome of RET is indeed reshaped by PTPRA and explained the prevalent Ras-MAPK activation inhibition. We also inspected the plausible role of PTPRA in inhibiting the cancer-causing attributes of RET thyroid carcinoma mutants MEN2A and MEN2B. The results showed that PTPRA inhibited the MEN2A-Ras-MAPK activation and effectively dephosphorylated MEN2A, whereas MEN2B resisted this. Notably, MEN2B (M918T) mutation is located in the RET catalytic domain, which might disrupt its conformation and alter in vivo substrate specificity. This could explain the unresponsiveness of MEN2B toward PTPRA inhibition, despite its physical interaction with PTPRA. However, with MS we identified the distinct phosphorylation profiles (in vivo) of MEN2s post GDNF-GFRα1 activation, which revealed the grounds for their differential activity and response to PTPRA action. Finally, we disclosed the biological consequences of MEN2A and MEN2B dephosphorylation by PTPRA on the regulation of cell invasion, whereby it drastically reduced the MDCK cell invasion induced by MEN2A oncogene in 3D collagen, a matrix that typifies the primary and metastatic tumor tissue microenvironments. Because MEN2B is clinically more aggressive than other MEN2 subtypes, the inertness of this RET mutant receptor to overall PTPRA adjustment successfully justifies for its high oncogenic activity in thyroid carcinomas.
To conclude, our study provides compelling evidence that PTPRA regulates GDNF-dependent RET-Ras-MAPK growth signaling by inhibiting the molecular activities of both RET and MEN2A via dephosphorylation (Figure 7C), thus making PTPRA indispensable for balancing RET activity and function in both health and disease.
Limitations of the Study
We have demonstrated many common interacting proteins, statistically obtained in a data-dependent manner, for both RET and PTPRA using AP-MS and BioID proteomic analysis. A subset of interacting proteins included several cell-surface receptors and docking proteins, as well as proteins involved in neuronal development, polarization, axon guidance, pathfinding, and pattern/axis formation. It may be important to validate these interactors for GDNF-associated RET-Ras-MAPK signaling. Besides, we have shown that PTPRA directly dephosphorylates RET and modulates the GDNF-activated RET-Ras-MAPK pathway; however, one may check the reaction (dephosphorylation) kinetics.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We acknowledge Sini Miettinen and Jenni Montonen for excellent technical assistance. This work was supported by grants from the Academy of Finland (Nos. 288475 and 294173), Sigrid Juselius Foundation (to M.V., K.L., and M.S.), Finnish Cancer Foundation (to M.V. and K.L.), University of Helsinki three-year research grant, Biocentrum Helsinki, Biocentrum Finland, and Instrumentarium Research Foundation.
Author Contributions
M.V. and L.Y. conceived and designed the study. L.Y. performed majority of the experiments, analyzed, and organized the results. A.K.M. and Y.S. participated in PTPRA intervention experiments. T.Ö., E.P., K.L., and X.L. performed and interpreted the in vitro dephosphorylation assay, cell invasion assay, and fluorescent microscopy, respectively. L.Y. and M.V. wrote the manuscript with the input from T.Ö., K.L., and M.S.
Declaration of Interests
The authors declare no competing interest.
Published: February 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100871.
Data and Code Availability
All reagents are available within Transparent Methods section of the accompanying Supplemental Information text file. The requests for resources and reagents may be directed to and will be fulfilled by the Lead Contact, Markku Varjosalo (markku.varjosalo@helsinki.fi). The plasmids are made available via Addgene.org.
Supplemental Information
List of AP-MS and BioID identified interactors of PTPRA, RET, GDNF, and GRB2 bait proteins. The data are filtered against respective GFP controls with preys showing frequency ≤3 and average PSM <1.5 were excluded. Preys appearing in either AP-MS or BioID purifications and showing average PSM fold change (avg. GFP/avg. prey) of at least >1.5 are retained for network construction. The novel (uk = 1), known (k = 2), and prey-to-prey (pp = 3) interactions were retrieved from the PINA interaction database.
List of phosphotyrosine sites that are obtained with a high localization probability score of ≥0.75, under two reaction conditions RET + GFP (control) and RET + PTPRA. The MS-raw intensities are converted to log2 values for plotting heatmap.
List of RET and MEN2s (A and B) peptides and phosphotyrosine sites that were identified through MaxQuant in the absence and presence of PTPRA phosphatase after GDNF-GFRα1 activation. (- = site not detected and 0 = site dephosphorylated)
References
- Airaksinen M.S., Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Alberti L., Borrello M.G., Ghizzoni S., Torriti F., Rizzetti M.G., Pierotti M.A. Grb2 binding to the different isoforms of Ret tyrosine kinase. Oncogene. 1998;17:1079–1087. doi: 10.1038/sj.onc.1202046. [DOI] [PubMed] [Google Scholar]
- Amoresano A., Incoronato M., Monti G., Pucci P., De Franciscis V., Cerchia L. Direct interactions among Ret, GDNF and GFRalpha1 molecules reveal new insights into the assembly of a functional three-protein complex. Cell Signal. 2005;17:717–727. doi: 10.1016/j.cellsig.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Arighi E., Borrello M.G., Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16:441–467. doi: 10.1016/j.cytogfr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Asai N., Murakami H., Iwashita T., Takahashi M. A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs their transforming activity and association with Shc adaptor proteins. J. Biol. Chem. 1996;271:17644–17649. doi: 10.1074/jbc.271.30.17644. [DOI] [PubMed] [Google Scholar]
- Besset V., Scott R.P., Ibanez C.F. Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J. Biol. Chem. 2000;275:39159–39166. doi: 10.1074/jbc.M006908200. [DOI] [PubMed] [Google Scholar]
- Bodrikov V., Leshchyns'ka I., Sytnyk V., Overvoorde J., Den Hertog J., Schachner M. RPTPalpha is essential for NCAM-mediated p59fyn activation and neurite elongation. J. Cell Biol. 2005;168:127–139. doi: 10.1083/jcb.200405073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.C., Khanna R.S., Bessette D.C., Samayawardhena L.A., Pallen C.J. Protein tyrosine phosphatase-alpha complexes with the IGF-I receptor and undergoes IGF-I-stimulated tyrosine phosphorylation that mediates cell migration. Am. J. Physiol. Cell Physiol. 2009;297:C133–C139. doi: 10.1152/ajpcell.00110.2009. [DOI] [PubMed] [Google Scholar]
- Chen Y.M., Wang Q.J., Hu H.S., Yu P.C., Zhu J., Drewes G., Piwnica-Worms H., Luo Z.G. Microtubule affinity-regulating kinase 2 functions downstream of the PAR-3/PAR-6/atypical PKC complex in regulating hippocampal neuronal polarity. Proc. Natl. Acad. Sci. U S A. 2006;103:8534–8539. doi: 10.1073/pnas.0509955103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini F., Shakya R. GDNF/Ret signaling and the development of the kidney. Bioessays. 2006;28:117–127. doi: 10.1002/bies.20357. [DOI] [PubMed] [Google Scholar]
- Coulpier M., Anders J., Ibanez C.F. Coordinated activation of autophosphorylation sites in the RET receptor tyrosine kinase: importance of tyrosine 1062 for GDNF mediated neuronal differentiation and survival. J. Biol. Chem. 2002;277:1991–1999. doi: 10.1074/jbc.M107992200. [DOI] [PubMed] [Google Scholar]
- Daum G., Regenass S., Sap J., Schlessinger J., Fischer E.H. Multiple forms of the human tyrosine phosphatase RPTP alpha. Isozymes and differences in glycosylation. J. Biol. Chem. 1994;269:10524–10528. [PubMed] [Google Scholar]
- Den Hertog J., Tracy S., Hunter T. Phosphorylation of receptor protein-tyrosine phosphatase alpha on Tyr789, a binding site for the SH3-SH2-SH3 adaptor protein GRB-2 in vivo. EMBO J. 1994;13:3020–3032. doi: 10.1002/j.1460-2075.1994.tb06601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denardo B.D., Holloway M.P., Ji Q., Nguyen K.T., Cheng Y., Valentine M.B., Salomon A., Altura R.A. Quantitative phosphoproteomic analysis identifies activation of the RET and IGF-1R/IR signaling pathways in neuroblastoma. PLoS One. 2013;8:e82513. doi: 10.1371/journal.pone.0082513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denhertog J., Hunter T. Tight association of GRB2 with receptor protein-tyrosine phosphatase alpha is mediated by the SH2 and c-terminal SH3 domains. EMBO J. 1996;15:3016–3027. [PMC free article] [PubMed] [Google Scholar]
- Drinkut A., Tillack K., Meka D.P., Schulz J.B., Kugler S., Kramer E.R. Ret is essential to mediate GDNF's neuroprotective and neuroregenerative effect in a Parkinson disease mouse model. Cell Death Dis. 2016;7:e2359. doi: 10.1038/cddis.2016.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eketjall S., Fainzilber M., Murray-Rust J., Ibanez C.F. Distinct structural elements in GDNF mediate binding to GFRalpha1 and activation of the GFRalpha1-c-Ret receptor complex. EMBO J. 1999;18:5901–5910. doi: 10.1093/emboj/18.21.5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M., Crowder R.J., Milbrandt J., Johnson E.M., Jr. Tyrosine 981, a novel ret autophosphorylation site, binds c-Src to mediate neuronal survival. J. Biol. Chem. 2004;279:18262–18269. doi: 10.1074/jbc.M400505200. [DOI] [PubMed] [Google Scholar]
- Eng C., Mulligan L.M. Mutations of the RET proto-oncogene in the multiple endocrine neoplasia type 2 syndromes, related sporadic tumours, and Hirschsprung disease. Hum. Mutat. 1997;9:97–109. doi: 10.1002/(SICI)1098-1004(1997)9:2<97::AID-HUMU1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Geiger T., Wehner A., Schaab C., Cox J., Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014050. M111 014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman K.M., Kjaer S., Beuron F., Knowles P.P., Nawrotek A., Burns E.M., Purkiss A.G., George R., Santoro M., Morris E.P. RET recognition of GDNF-GFRalpha1 ligand by a composite binding site promotes membrane-proximal self-association. Cell Rep. 2014;8:1894–1904. doi: 10.1016/j.celrep.2014.08.040. [DOI] [PubMed] [Google Scholar]
- Hennige A.M., Lammers R., Hoppner W., Arlt D., Strack V., Teichmann R., Machicao F., Ullrich A., Haring H.U., Kellerer M. Inhibition of Ret oncogene activity by the protein tyrosine phosphatase SHP1. Endocrinology. 2001;142:4441–4447. doi: 10.1210/endo.142.10.8453. [DOI] [PubMed] [Google Scholar]
- Huang J., Yao L., Xu R., Wu H., Wang M., White B.S., Shalloway D., Zheng X. Activation of Src and transformation by an RPTPalpha splice mutant found in human tumours. EMBO J. 2011;30:3200–3211. doi: 10.1038/emboj.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbrici P., Tucker S.J., D'adamo M.C., Pessia M. Role of receptor protein tyrosine phosphatase alpha (RPTPalpha) and tyrosine phosphorylation in the serotonergic inhibition of voltage-dependent potassium channels. Pflugers Arch. 2000;441:257–262. doi: 10.1007/s004240000406. [DOI] [PubMed] [Google Scholar]
- Imgrund S., Hartmann D., Farwanah H., Eckhardt M., Sandhoff R., Degen J., Gieselmann V., Sandhoff K., Willecke K. Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J. Biol. Chem. 2009;284:33549–33560. doi: 10.1074/jbc.M109.031971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T., Taniguchi M., Asai N., Ohkusu K., Nakashima I., Takahashi M. cDNA cloning of mouse ret proto-oncogene and its sequence similarity to the cadherin superfamily. Oncogene. 1993;8:1087–1091. [PubMed] [Google Scholar]
- Iwashita T., Asai N., Murakami H., Matsuyama M., Takahashi M. Identification of tyrosine residues that are essential for transforming activity of the ret proto-oncogene with MEN2A or MEN2B mutation. Oncogene. 1996;12:481–487. [PubMed] [Google Scholar]
- Kaplan R., Morse B., Huebner K., Croce C., Howk R., Ravera M., Ricca G., Jaye M., Schlessinger J. Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proc. Natl. Acad. Sci. U S A. 1990;87:7000–7004. doi: 10.1073/pnas.87.18.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp K., Metzinger E., Kellerer M., Haring H.U., Lammers R. The protein tyrosine phosphatase alpha modifies insulin secretion in INS-1E cells. Biochem. Biophys. Res. Commun. 2003;311:361–364. doi: 10.1016/j.bbrc.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Katz M., Amit I., Yarden Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta. 2007;1773:1161–1176. doi: 10.1016/j.bbamcr.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto Y., Takeda K., Okuno Y., Yamakawa Y., Ito Y., Taguchi R., Kato M., Suzuki H., Takahashi M., Nakashima I. Identification of RET autophosphorylation sites by mass spectrometry. J. Biol. Chem. 2004;279:14213–14224. doi: 10.1074/jbc.M312600200. [DOI] [PubMed] [Google Scholar]
- Larrivee B., Freitas C., Trombe M., Lv X., Delafarge B., Yuan L., Bouvree K., Breant C., Del Toro R., Brechot N. Activation of the UNC5B receptor by Netrin-1 inhibits sprouting angiogenesis. Genes Dev. 2007;21:2433–2447. doi: 10.1101/gad.437807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Su Y., Zhao C., Zhao H., Liu G., Wang J., Xu Q. The role of Ret receptor tyrosine kinase in dopaminergic neuron development. Neuroscience. 2006;142:391–400. doi: 10.1016/j.neuroscience.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Liu X., Salokas K., Tamene F., Jiu Y., Weldatsadik R.G., Ohman T., Varjosalo M. An AP-MS- and BioID-compatible MAC-tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat. Commun. 2018;9:1188. doi: 10.1038/s41467-018-03523-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo R.M., Santoro M., Ong S.H., Billaud M., Fusco A., Hadari Y.R., Schlessinger J., Lax I. Docking protein FRS2 links the protein tyrosine kinase RET and its oncogenic forms with the mitogen-activated protein kinase signaling cascade. Mol. Cell Biol. 2001;21:4177–4187. doi: 10.1128/MCB.21.13.4177-4187.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosevitsky M.I. Nerve ending "signal" proteins GAP-43, MARCKS, and BASP1. Int. Rev. Cytol. 2005;245:245–325. doi: 10.1016/S0074-7696(05)45007-X. [DOI] [PubMed] [Google Scholar]
- Mulligan L.M. RET revisited: expanding the oncogenic portfolio. Nat. Rev. Cancer. 2014;14:173–186. doi: 10.1038/nrc3680. [DOI] [PubMed] [Google Scholar]
- Norman R.X., Ko H.W., Huang V., Eun C.M., Abler L.L., Zhang Z., Sun X., Eggenschwiler J.T. Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through inhibition of Hedgehog signaling. Hum. Mol. Genet. 2009;18:1740–1754. doi: 10.1093/hmg/ddp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris K., Norris F., Kono D.H., Vestergaard H., Pedersen O., Theofilopoulos A.N., Moller N.P. Expression of protein-tyrosine phosphatases in the major insulin target tissues. FEBS Lett. 1997;415:243–248. doi: 10.1016/s0014-5793(97)01133-2. [DOI] [PubMed] [Google Scholar]
- Ohiwa M., Murakami H., Iwashita T., Asai N., Iwata Y., Imai T., Funahashi H., Takagi H., Takahashi M. Characterization of Ret-Shc-Grb2 complex induced by GDNF, MEN 2A, and MEN 2B mutations. Biochem. Biophys. Res. Commun. 1997;237:747–751. doi: 10.1006/bbrc.1997.7225. [DOI] [PubMed] [Google Scholar]
- Pachnis V., Mankoo B., Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119:1005–1017. doi: 10.1242/dev.119.4.1005. [DOI] [PubMed] [Google Scholar]
- Perrinjaquet M., Vilar M., Ibanez C.F. Protein-tyrosine phosphatase SHP2 contributes to GDNF neurotrophic activity through direct binding to phospho-Tyr687 in the RET receptor tyrosine kinase. J. Biol. Chem. 2010;285:31867–31875. doi: 10.1074/jbc.M110.144923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrone A., Battaglia F., Wang C., Dusa A., Su J., Zagzag D., Bianchi R., Casaccia-Bonnefil P., Arancio O., Sap J. Receptor protein tyrosine phosphatase alpha is essential for hippocampal neuronal migration and long-term potentiation. EMBO J. 2003;22:4121–4131. doi: 10.1093/emboj/cdg399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S., Morales D., Croteau L.P., Krawchuk D., Palmesino E., Morton S., Cloutier J.F., Charron F., Dalva M.B., Ackerman S.L. Synergistic integration of Netrin and ephrin axon guidance signals by spinal motor neurons. Elife. 2015;4 doi: 10.7554/eLife.10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao S., Iwashita T., Furukawa T., Yamamoto M., Sobue G., Takahashi M. Differential effects of leukocyte common antigen-related protein on biochemical and biological activities of RET-MEN2A and RET-MEN2B mutant proteins. J. Biol. Chem. 2001;276:9460–9467. doi: 10.1074/jbc.M008744200. [DOI] [PubMed] [Google Scholar]
- Richardson D.S., Rodrigues D.M., Hyndman B.D., Crupi M.J., Nicolescu A.C., Mulligan L.M. Alternative splicing results in RET isoforms with distinct trafficking properties. Mol. Biol. Cell. 2012;23:3838–3850. doi: 10.1091/mbc.E12-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M., Melillo R.M., Carlomagno F., Vecchio G., Fusco A. Minireview: ret: normal and abnormal functions. Endocrinology. 2004;145:5448–5451. doi: 10.1210/en.2004-0922. [DOI] [PubMed] [Google Scholar]
- Schedl A. Renal abnormalities and their developmental origin. Nat. Rev. Genet. 2007;8:791–802. doi: 10.1038/nrg2205. [DOI] [PubMed] [Google Scholar]
- Soba P., Han C., Zheng Y., Perea D., Miguel-Aliaga I., Jan L.Y., Jan Y.N. The Ret receptor regulates sensory neuron dendrite growth and integrin mediated adhesion. Elife. 2015;4 doi: 10.7554/eLife.05491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M., Buma Y., Taniguchi M. Identification of the ret proto-oncogene products in neuroblastoma and leukemia cells. Oncogene. 1991;6:297–301. [PubMed] [Google Scholar]
- Telley L., Cadilhac C., Cioni J.M., Saywell V., Jahannault-Talignani C., Huettl R.E., Sarrailh-Faivre C., Dayer A., Huber A.B., Ango F. Dual function of NRP1 in axon guidance and subcellular target recognition in cerebellum. Neuron. 2016;91:1276–1291. doi: 10.1016/j.neuron.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Torban E., Wang H.J., Groulx N., Gros P. Independent mutations in mouse Vangl2 that cause neural tube defects in looptail mice impair interaction with members of the Dishevelled family. J. Biol. Chem. 2004;279:52703–52713. doi: 10.1074/jbc.M408675200. [DOI] [PubMed] [Google Scholar]
- Tremper-Wells B., Resnick R.J., Zheng X., Holsinger L.J., Shalloway D. Extracellular domain dependence of PTPalpha transforming activity. Genes Cells. 2010;15:711–724. doi: 10.1111/j.1365-2443.2010.01410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truffi M., Dubreuil V., Liang X., Vacaresse N., Nigon F., Han S.P., Yap A.S., Gomez G.A., Sap J. RPTPalpha controls epithelial adherens junctions, linking E-cadherin engagement to c-Src-mediated phosphorylation of cortactin. J. Cell Sci. 2014;127:2420–2432. doi: 10.1242/jcs.134379. [DOI] [PubMed] [Google Scholar]
- Wang Y., Pallen C.J. The receptor-like protein tyrosine phosphatase HPTP alpha has two active catalytic domains with distinct substrate specificities. EMBO J. 1991;10:3231–3237. doi: 10.1002/j.1460-2075.1991.tb04886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Buist A., Den Hertog J., Zhang Z.Y. Comparative kinetic analysis and substrate specificity of the tandem catalytic domains of the receptor-like protein-tyrosine phosphatase alpha. J. Biol. Chem. 1997;272:6994–7002. doi: 10.1074/jbc.272.11.6994. [DOI] [PubMed] [Google Scholar]
- Xiao Q., Rongfei W., Lingqiang Z., Fuchu H. The roles of signaling pathways in regulating kidney development. Yi Chuan. 2015;37:1–7. doi: 10.16288/j.yczz.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Yadav L., Tamene F., Goos H., Van Drogen A., Katainen R., Aebersold R., Gstaiger M., Varjosalo M. Systematic analysis of human protein phosphatase interactions and dynamics. Cell Syst. 2017;4:430–444.e5. doi: 10.1016/j.cels.2017.02.011. [DOI] [PubMed] [Google Scholar]
- Yao Z., Darowski K., St-Denis N., Wong V., Offensperger F., Villedieu A., Amin S., Malty R., Aoki H., Guo H. A global analysis of the receptor tyrosine kinase-protein phosphatase interactome. Mol. Cell. 2017;65:347–360. doi: 10.1016/j.molcel.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak J., Schneider S.W., Eue I., Ludwig T., Oberleithner H. High-resistance MDCK-C7 monolayers used for measuring invasive potency of tumour cells. Pflugers Arch. 2000;440:179–183. doi: 10.1007/s004240000282. [DOI] [PubMed] [Google Scholar]
- Zheng X.M., Resnick R.J., Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. EMBO J. 2000;19:964–978. doi: 10.1093/emboj/19.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.M., Resnick R.J., Shalloway D. Mitotic activation of protein-tyrosine phosphatase alpha and regulation of its Src-mediated transforming activity by its sites of protein kinase C phosphorylation. J. Biol. Chem. 2002;277:21922–21929. doi: 10.1074/jbc.M201394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of AP-MS and BioID identified interactors of PTPRA, RET, GDNF, and GRB2 bait proteins. The data are filtered against respective GFP controls with preys showing frequency ≤3 and average PSM <1.5 were excluded. Preys appearing in either AP-MS or BioID purifications and showing average PSM fold change (avg. GFP/avg. prey) of at least >1.5 are retained for network construction. The novel (uk = 1), known (k = 2), and prey-to-prey (pp = 3) interactions were retrieved from the PINA interaction database.
List of phosphotyrosine sites that are obtained with a high localization probability score of ≥0.75, under two reaction conditions RET + GFP (control) and RET + PTPRA. The MS-raw intensities are converted to log2 values for plotting heatmap.
List of RET and MEN2s (A and B) peptides and phosphotyrosine sites that were identified through MaxQuant in the absence and presence of PTPRA phosphatase after GDNF-GFRα1 activation. (- = site not detected and 0 = site dephosphorylated)
Data Availability Statement
All reagents are available within Transparent Methods section of the accompanying Supplemental Information text file. The requests for resources and reagents may be directed to and will be fulfilled by the Lead Contact, Markku Varjosalo (markku.varjosalo@helsinki.fi). The plasmids are made available via Addgene.org.