

Abstract
N-arachidonoyl glycine (NAGly) is an endogenous lipid deriving from the endocannabinoid anandamide (AEA). Identified as a ligand of several G-protein coupled receptors (GPCRs), it can however exert biological responses independently of GPCRs. NAGly was recently shown to depress store-operated Ca2+ entry (SOCE) but its mechanism of action remains elusive. The major aim of this study was to gain a better knowledge on the NAGly-dependent impairment of SOCE in neurons of the central nervous system (CNS) from mice. First, we examined the expression of genes encoding for putative lipid sensing GPCRs using transcriptomic data publicly available. This analysis showed that the most abundant GPCRs transcripts present in the cerebral cortices of embryonic brains were coding for lysophosphatidic acid (LPA) and sphingosine-1 phosphate (S1P) receptors. Next, the presence of functional receptors was assessed with live-cell calcium imaging experiments. In primary cortical cells S1P and LPA mobilize Ca2+ from internal stores via a mechanism sensitive to the S1P and LPA receptor antagonists Ex26, H2L5186303, or Ki16425. However, none of these compounds prevented or attenuated the NAGly-dependent impairment of SOCE. We found no evidence for the requirement of lipid sensing GPCRs in this inhibitory process, indicating that NAGly is an endogenous modulator interfering with the core machinery of SOCE. Moreover, these data also raise the intriguing possibility that the depression of SOCE could play a role in the central effects of NAGly.
Subject terms: Lipid signalling, Cellular neuroscience, Ion channels in the nervous system
Introduction
N-arachidonoyl glycine (NAGly) is a lipid deriving from the endocannabinoid anandamide (AEA). It is naturally present in various tissues and organs like the brain1,2 but the physiological functions exerted by NAGly in the neural system are not yet fully characterized. This endogenous bioactive molecule influences pain perception and displays analgesic properties1,3–5. This led to the hypothesis that NAGly could be a natural modulator of pain6. The analgesic actions of NAGly have been studied principally on dorsal root ganglia (DRG) neurons and dorsal horn neurons from spinal cord slices7. For instance, NAGly inhibits low threshold voltage-gated Ca2+ channels (Cav3) in DRG neurons8 and the glycine uptake transporter GLYT2 in dorsal horn neurons, which contributes to enhance inhibitory glycinergic synaptic transmission in these cells7. NAGly has also been shown to depress excitatory NMDA-dependent synaptic transmission7. The effects of NAGly on neurons of the brain have however been much less characterized. In primary cortical neurons NAGly releases Ca2+ from the endoplasmic reticulum (ER), potentiates the passive leakage of Ca2+ out of the ER, and impairs the store-operated Ca2+ entry (SOCE)9.
One key issue when addressing the question of the cellular effects of NAGly is to determine whether it interferes directly with the activity of its targets or recruits dedicated G-protein coupled receptors (GPCRs) linked to downstream intracellular signaling cascades. NAGly has been proposed to act as an agonist of some orphan GPCRs like GPR1810, GPR5511, and GPR9212. It is however worthy of note that NAGly can influence the activity of some effectors without the requirement of GPCRs. This is for instance the case for the NAGly-dependent regulation of voltage-gated Ca2+ channels and Na+/Ca2+ exchanger activity8,13,14.
SOCE is an important physiological Ca2+ route of the plasma membrane. It is activated in response to the depletion of the ER Ca2+ stores15 and involves distinct actors: stromal interacting molecules (STIM1–2) and Orai1–3 channels16–18. STIM are ER resident proteins that seem to function as Ca2+ sensors19. The depletion of the ER Ca2+ stores governs the molecular interaction between STIM and plasma membrane Ca2+ channels of Orai type that are responsible for the SOCE response17,18. Recent studies identified NAGly as a potent inhibitor of SOCE in various cell lines (NIH-3T3 fibroblasts, human endothelial cell line EA.hy926, rat pancreatic β-cell line INS-1 832/13, rat basophilic leukemia cell line RBL-2H3) and in primary cultured neural cells9,20,21. Two hypotheses were put forward to explain the NAGly-dependent depression of SOCE20: (1) direct disruption of the coupling between STIM and Orai, or (2) recruitment of an intracellular signalling cascade activated downstream to NAGly-sensitive receptors and regulating negatively SOCE activity. The aim of the present work was to verify whether a lipid sensing GPCR is contributing to the NAGly-induced impairment of SOCE in cortical neurons. First, we analyzed a recent publicly available transcriptomic dataset obtained by RNAseq22 to characterize the expression of genes encoding for putative lipid sensing GPCRs in the cerebral cortices of embryonic mice. The most abundant transcripts were coding for lysophosphatidic acid (LPA) and sphingosine-1 phosphate (S1P) receptors. After having checked the presence of functional receptors, the contribution of LPA and S1P receptors to the NAGly-dependent inhibition of SOCE was evaluated using a pharmacological approach.
Material and Methods
Animal and ethical statement
C57Bl6/J (stock #000664) mice were obtained from the Jackson Laboratory (USA). They were housed in a temperature-controlled room under a 12 h light–12 h dark cycle with ad libitum access to food and water. An environmental enrichment consisting in the addition of tunnels was provided for all animals in accordance with the Animal Welfare Committee of the CEA Grenoble. Experimental procedures were approved by the animal care committee of the CEA’s Life Sciences Division (CETEA, A14-006). Experiments were conducted in compliance with the French legislation and the European Community Council Directive of 24 November 1986 (86/609/EEC).
Primary cultures of cortical neurons
Cells were dissociated from cerebral cortices collected from embryonic (E13) mice (with the vaginal plug as E0) according to9,23,24. Briefly, tissues were placed in a 1.5 mL Eppendorf tube containing 1 mL of an ice-cold Ca2+- and Mg2+-free Hank’s solution supplemented with 33 mM glucose, 4.2 mM NaHCO3, 10 mM HEPES, and 1% penicillin/streptomycin. Cells were isolated by a mechanical trituration of the medium containing the cerebral cortices. The cell suspension was filtered through a 40-µm cell strainer before plating the cells on 16 mm ∅ glass coverslips. They were kept in a Neurobasal medium supplemented with B27 (2%) and glutamine (500 µM) and maintained in a 5% CO2 atmosphere at 37 °C. All the experiments were conducted on cells kept 2 or 3 days in vitro.
Calcium imaging experiments with Fluo-4
The culture medium was removed and replaced by a saline containing (in mM) 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 5.5 glucose, 10 HEPES (pH 7.4, NaOH). LPA- and S1P-induced Ca2+ responses were analyzed with Fluo-4. Cells were incubated with 5 μM Fluo-4/AM for 20 min following procedures described previously23,24. Images were obtained by a CCD CoolSnap HQ2 camera (Princeton Instruments, Roper Scientific, France) mounted on an inverted Zeiss A1 microscope (Carl Zeiss, France). Cells were excited at 470 nm and emission was collected at 525 nm using a DG-4 wavelength switcher (Princeton Instruments, Roper Scientific, France). MetaFluor (Universal Imaging, Roper Scientific, France) was used for image acquisition and analysis. All experimental procedures were conducted at room temperature. Time-lapse changes in Fluo-4 fluorescence intensity were collected at a frequency of 0.2 Hz from 30–45 cell bodies per dish and analyzed off-line by defining regions of measurements. Results were expressed as F/F0, with F being the fluorescence at each time point and F0 being the mean baseline fluorescence that was monitored at the beginning of each experiment for 1 min before the addition of any substance. Culture dishes were discarded at the end of the recording and never re-used. A positive LPA (or S1P)-induced calcium response was determined as one F/F0 greater than 0.02 that develops within 50 s upon the application of the agonist. Fluo-4 responses were measured as area under curve (AUC).
Calcium imaging experiments with Fura-2
The fluorescent Ca2+ probe Fura-2 was used to study store-operated Ca2+ entry (SOCE). The experimental conditions and setup were as above except that cells were incubated with 2.5 µM Fura-2 for 20 min at room temperature. They were then washed twice and kept in a Fura-2-free saline solution for >12 min at room temperature. A dual excitation at 340 and 380 nm was used and emission was collected at 515 nm. Images were acquired at a frequency of 0.2 Hz and analyzed off-line. The classical “Ca2+ add-back” protocol was used to study SOCE. Cells were bathed in a nominally Ca2+-free saline containing (in mM) 150 NaCl, 5 KCl, 3 MgCl2, 5.5 glucose, 10 HEPES (pH 7.4, NaOH). SOCE activation was triggered by depletion of the ER Ca2+ pool with 200 nM thapsigargin, which induced a transient elevation in intracellular Ca2+ concentration before re-admission of 2 mM external Ca2+. SOCE responses were analyzed in cells generating a rapid Ca2+ rise upon the application of a depolarizing saline containing 90 mM KCl. In cultures of embryonic cortical cells, KCl responding cells are identified as neurons whereas KCl-unresponding cells are considered as non-neuronal cells25. The depolarizing (K+ rich) medium had the following composition (in mM): 65 NaCl, 90 KCl, 1 MgCl2, 2 CaCl2, 5.5 glucose, 10 HEPES (pH 7.4, NaOH). Ca2+ changes as a function of time were expressed as delta ratio F340/F380 whereas total Ca2+ responses were measured as area under curve (AUC).
Stock solutions of Ex26, Ki16425, and BTP2 were prepared in dimethyl sulfoxide (DMSO). Methanol and ethanol were used for preparing stock solutions of S1P and NAGly, respectively. These stock solutions were diluted at least 1000-fold into the recording saline immediately before use so that the final concentration of vehicle never exceeded 0.1%. Control experiments were performed with DMSO, ethanol and methanol alone. None of the solvent altered cytosolic Ca2+ signals, at least at the concentrations used.
Analysis of gene expression by RNAseq
The RNASeq gene expression data derive from22. Raw fastq files are publicly available and can be found on the GEO repository under accession number: GSEXXX.
Data and Statistical analysis
Each experimental condition as well as its appropriate control were tested on the same batch of primary neuronal cell cultures. For the Ca2+ imaging experiments, all experiments were done ≥3 times (e.g. with ≥3 distinct biological samples) using distinct dishes from different batches of cells (e.g. from distinct pregnant mice). Data are presented as means ± standard error of the mean (SEM) with n being the number of biological replicates. SigmaPlot (version 10.0, Systat Software) and SigmaStat (version 3.5, Systat Software) were used for plotting graphs and statistical analysis, respectively. Differences between several groups of cells were tested using one-way analysis of variance (ANOVA) followed by a Bonferroni’s post hoc test. A P value < 0.05 was considered statistically significant.
Materials
Fluo-4/AM, Fura-2 and tissue culture media were from Molecular Probes (Invitrogen, France). N-arachidonoyl glycine (NAGly) was from Tocris (Bio-Techne, France). All the other reagents were obtained from Sigma-Aldrich (France).
Results
mRNA expression of lipid sensing GPCRs in the cerebral cortex of embryonic mice
In order to determine whether NAGly is acting via a GPCR, we analyzed the expression of genes encoding for putative lipid sensing GPCRs in the embryonic cerebral cortex. Table 1 provides the list of the 60 mouse genes selected26–30. The transcriptomic data were extracted from a recent RNAseq study22. The expression pattern of putative lipid sensing GPCRs was analyzed at 3 embryonic ages: E11, E13 and E17. Only genes for which the number of transcripts per million (TPM) was >2 were considered as significantly expressed31, therefore when the number of transcripts was <2 TPM, the gene was eliminated from the analysis. This resulted in the selection of 14 genes encoding for putative lipid sensing GPCRs (Fig. 1). In this RNAseq analysis the genes encoding for GPR18, GPR55 and GPR92, 3 putative targets of NAGly, were not expressed. Overall, the most abundant transcripts were coding for cannabinoid receptors type 1 (CB1) (Cnr1 gene), the orphan receptor GPR12, lysophosphatidic acid (LPA) and sphingosine-1 phosphate (S1P) receptors (Fig. 1). Of note, the abundance of CB1 and GPR12 transcripts increased markedly during the embryonic development of the cerebral cortex whereas the expression of genes encoding for LPA and S1P receptors was repressed. Since all the live-cell Ca2+ imaging reported previously were conducted on cortical cells isolated from E13 brain cerebral cortices9 we focused our attention on the most expressed lipid sensing GPCR genes at that embryonic age: S1pr1, Lpar2 and Lpar6 (vertical arrows, Fig. 1). They encode for S1P1, LPA2 and LPA6 receptors, respectively. CB1 was excluded from our analysis because NAGly has no affinity for CB1 receptors32 and the CB1 antagonist AM251 did not prevent the NAGly-induced responses in cortical neurons9, arguing against a role for these receptors. On the other hand, GPR12 was also not considered as a likely target of NAGly because the GPR12 gene was weakly expressed at E13 (Fig. 1). Its expression was strongly upregulated but only at the end of corticogenesis (E17).
Table 1.
List of selected 60 murine genes encoding for lipid sensing G protein-coupled receptors (GPCRs).
| EnsemblID | Gene name | other names | Gene description |
|---|---|---|---|
| ENSMUSG00000044288 | Cnr1 | cannabinoid receptor 1 | |
| ENSMUSG00000062585 | Cnr2 | cannabinoid receptor 2 | |
| ENSMUSG00000046856 | Gpr1 | G protein-coupled receptor 1 | |
| ENSMUSG00000046856 | Gpr1 | G protein-coupled receptor 1 | |
| ENSMUSG00000044317 | Gpr4 | G protein-coupled receptor 4 | |
| ENSMUSG00000046922 | Gpr6 | G protein-coupled receptor 6 | |
| ENSMUSG00000041468 | Gpr12 | G-protein coupled receptor 12 | |
| ENSMUSG00000052229 | Gpr17 | G protein-coupled receptor | |
| ENSMUSG00000050350 | Gpr18 | G protein-coupled receptor 18 | |
| ENSMUSG00000053647 | Gpr30 | Gper1 | G protein-coupled estrogen receptor 1 |
| ENSMUSG00000071311 | Gpr31b | G protein-coupled receptor 31 | |
| ENSMUSG00000040229 | Gpr34 | P2Y12 | G protein-coupled receptor 34 |
| ENSMUSG00000026271 | Gpr35 | G protein-coupled receptor 35 | |
| ENSMUSG00000049608 | Gpr55 | G protein-coupled receptor 55 | |
| ENSMUSG00000040372 | Gpr63 | G protein-coupled receptor 63 | |
| ENSMUSG00000021886 | Gpr65 | TDAG8 | G-protein coupled receptor 65 |
| ENSMUSG00000047415 | Gpr68 | OGR1 | G protein-coupled receptor 68 |
| ENSMUSG00000049241 | gpr81 | Hcar1 | hydrocarboxylic acid receptor 1 |
| ENSMUSG00000063234 | Gpr84 | G protein-coupled receptor 84 | |
| ENSMUSG00000051431 | Gpr87 | G protein-coupled receptor 87 | |
| ENSMUSG00000045502 | Gpr109A | Hcar2 | hydroxycarboxylic acid receptor 2 |
| ENSMUSG00000051209 | Gpr119 | G-protein coupled receptor 119 | |
| ENSMUSG00000064272 | Gpr131 | Gpbar1 | G protein-coupled bile acid receptor 1 |
| ENSMUSG00000021298 | Gpr132 | G protein-coupled receptor 132 | |
| ENSMUSG00000073008 | Gpr174 | G protein-coupled receptor 174 | |
| ENSMUSG00000051212 | Gpr183 | G protein-coupled receptor 183 | |
| ENSMUSG00000034730 | Adgrb1 | Bai1 | adhesion G protein-coupled receptor B1 |
| ENSMUSG00000046908 | Ltb4r1 | leukotriene B4 receptor 1 | |
| ENSMUSG00000040432 | Ltb4r2 | leukotriene B4 receptor 2 | |
| ENSMUSG00000052821 | Cysltr1 | cysteinyl leukotriene receptor 1 | |
| ENSMUSG00000033470 | Cysltr2 | cysteinyl leukotriene receptor 2 | |
| ENSMUSG00000071489 | Ptgdr | prostaglandin D receptor | |
| ENSMUSG00000034117 | Ptgdr2 | prostaglandin D2 receptor 2 | |
| ENSMUSG00000019464 | Ptger1 | prostaglandin E receptor 1 | |
| ENSMUSG00000037759 | Ptger2 | prostaglandin E receptor 2 (subtype EP2) | |
| ENSMUSG00000040016 | Ptger3 | prostaglandin E receptor 3 (subtype EP3) | |
| ENSMUSG00000039942 | Ptger4 | prostaglandin E receptor 4 (subtype EP4) | |
| ENSMUSG00000044453 | Ffar1 | free fatty acid receptor 1 | |
| ENSMUSG00000051314 | Ffar2 | free fatty acid receptor 2 | |
| ENSMUSG00000051314 | Ffar2 | free fatty acid receptor 2 | |
| ENSMUSG00000054200 | Ffar4 | free fatty acid receptor 4 | |
| ENSMUSG00000028036 | Ptgfr | prostaglandin F receptor | |
| ENSMUSG00000052270 | Fpr2 | formyl peptide receptor 2 | |
| ENSMUSG00000043017 | Ptgir | prostaglandin I receptor | |
| ENSMUSG00000038668 | Lpar1 | lysophosphatidic acid receptor 1 | |
| ENSMUSG00000031861 | Lpar2 | lysophosphatidic acid receptor 2 | |
| ENSMUSG00000036832 | Lpar3 | lysophosphatidic acid receptor 3 | |
| ENSMUSG00000049929 | Lpar4 | lysophosphatidic acid receptor 4 | |
| ENSMUSG00000067714 | Lpar5 | lysophosphatidic acid receptor 5 | |
| ENSMUSG00000033446 | Lpar6 | lysophosphatidic acid receptor 6 | |
| ENSMUSG00000044819 | Gpr80 | Oxgr1, Gpr99, P2Y15 | oxoglutarate (alpha-ketoglutarate) receptor 1 |
| ENSMUSG00000056529 | Ptafr | platelet-activating factor receptor | |
| ENSMUSG00000050921 | P2ry10 | purinergic receptor P2Y, G-protein coupled 10 | |
| ENSMUSG00000045092 | S1pr1 | sphingosine-1-phosphate receptor 1 | |
| ENSMUSG00000043895 | S1pr2 | sphingosine-1-phosphate receptor 2 | |
| ENSMUSG00000067586 | S1pr3 | sphingosine-1-phosphate receptor 3 | |
| ENSMUSG00000044199 | S1pr4 | sphingosine-1-phosphate receptor 4 | |
| ENSMUSG00000045087 | S1pr5 | sphingosine-1-phosphate receptor 5 | |
| ENSMUSG00000027762 | Sucnr1 | succinate receptor 1 | |
| ENSMUSG00000034881 | Tbxa2r | thromboxane A2 receptor |
Figure 1.
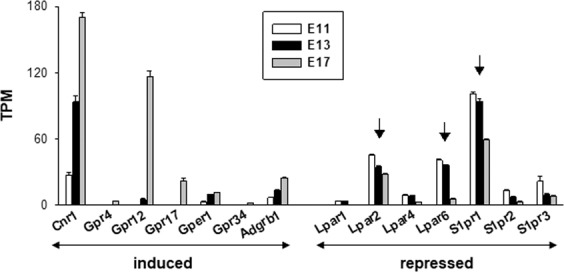
mRNA expression of putative lipid sensing GPCRs in the embryonic murine cortex. The data used to plot this graph were extracted from a previous whole-genome transcriptomic analysis22. A total of 60 genes encoding for putative lipid sensing GPCRs were selected (see Table 1). Transcripts of only 14 genes (out of 60) could be detected (e.g. having TPM values ≥ 2). The graph shows the temporal pattern of the mRNA abundance of these 14 genes at 3 embryonic ages: E11, E13 and E17. Genes that were induced (Cnr1, Gpr4, Gpr12, Gpr17, Gper1, Gpr34, Adgrb1) are shown on the left whereas genes that were repressed (Lpar1, Lpar2, Lpar4, Lpar6, S1pr1, S1pr2, S1pr3) appear on the right. Vertical arrows indicate the 3 most abundant transcripts at E13 (except CB1, see text for further details).
Presence of functional LPA and S1P receptors
The activation of S1P and LPA receptors mobilizes Ca2+ from internal stores33–36. We thus performed live-cell Ca2+ imaging fluorescent microscopy experiments with Fluo-4 to assess the presence of functional S1P and LPA receptors. Several cell populations are present in the primary cultures. For instance, 80–85% of the cells express βIII-tubulin (a marker of post-mitotic neurons) and possess voltage-gated Ca2+ channels37,38, indicating that most cells display a post-mitotic neuronal phenotype. First, the presence of functional LPA and S1P receptors was assessed in the entire cell population. LPA (10 µM, Fig. 2A) and S1P (10 µM, Fig. 2C) evoked prominent Ca2+ rises in ⁓15% (61/416 cells) and ⁓13% (39 out of 303) of the cells tested, respectively. The LPA-induced Ca2+ signals were partially blocked by 10 µM H2L5186303, a selective LPA2/3 receptor antagonist39, and nearly completely suppressed by 10 µM Ki16425, a LPA1/2/3 receptor antagonist39–42 (Fig. 2B). The percentage of cells responding to LPA was 12% (29/238 cells) and <1% (1/169 cells) with H2L5186303 and Ki16425, respectively. Therefore, H2L5186303 diminished the peak of the Ca2+ rise without affecting the number of LPA responsive cells whereas Ki16425 affected both parameters.
Figure 2.

Presence of functional LPA- and S1P-sensitive receptors. The presence of functional LPA and S1P receptors was verified with the fluorescent Ca2+ probe Fluo-4. In these experiments, cells were maintained in a nominally Ca2+ free saline. Panels A and C show somatic Fluo-4 signals (F/F0) as a function of time in response to 10 µM LPA (n = 10) (panel A) and 10 µM S1P (n = 9) (panel C). Panel B shows the LPA-induced Ca2+ rises measured as area under the curve (AUC) in the absence (white bar, n = 10) or presence of H2L5186303 (10 µM, n = 5), Ki16425 (10 µM, n = 7), or after the application of thapsigargin (Tg, 200 nM, n = 5). *p < 0.05 vs LPA, one-way ANOVA followed by a Bonferroni’s post hoc test. Panel D shows the Fluo-4 responses (measured as area under the curve, AUC) induced by S1P alone (10 µM, n = 9), S1P + Ex26 (1 µM, n = 7), and S1P applied after thapsigargin (Tg, 200 nM, n = 5), with *p < 0.05 vs S1P, one-way ANOVA followed by a Bonferroni’s post hoc test. Antagonists of LPA and S1P receptors were added 4–7 min before time 0 and remained present throughout the recordings. LPA and S1P can stimulate store-released Ca2+. Pre-depleting the ER Ca2+ with Tg prevents any response to LPA or S1P.
The S1P1 receptor antagonist Ex26 (1 µM)43 reduced the peak amplitude of the S1P-induced Ca2+ signals and diminished the number of responsive cells with only 12 cells out 220 tested (⁓5%) generating a Ca2+ signal in response to 10 µM S1P (Fig. 2D). In each instance, depleting the ER with thapsigargin prevented the development of a Ca2+ rise upon LPA or S1P application (Fig. 2B,D).
Previous reports showed that LPA and S1P receptors are mainly found in proliferative regions of the immature cerebral cortex, with few post-mitotic neurons responding to LPA and S1P35. This latter point was checked by using a depolarizing saline solution containing 90 mM KCl to evoke KCl-dependent Ca2+ rises. Acutely cultured cells were undifferentiated cells. When cultured for several days, some of these differentiate into neurons (post-mitotic) responding to high-K+ whereas non-differentiated cells are not high-K+ sensitive. In cultures of embryonic cortical cells, KCl responding cells are identified as neurons whereas KCl-unresponding cells are considered as non-neuronal cells25. Overall, only 10 of 67 LPA sensitive cells (⁓15%) generated an intracellular Ca2+ rise in response to KCl. These data are consistent with a previous report showing that in the embryonic cerebral cortex LPA receptors are predominantly expressed by neural precursor cells with only a small minority of neurons responding to LPA35. On the other hand, 5 of 25 S1P sensitive cells (20%) were KCl-responsive cells. This indicates that the S1P-sensitive cells are also mainly found in KCl-insensitive cells34. Taken together, LPA or S1P mobilizes Ca2+ from the ER in a subset of cells (<20%). These functional LPA- and S1P-sensitive receptors are essentially expressed by non-neuronal cells35,44.
Before testing the contribution of LPA and S1P receptors in the NAGly-dependent alteration of SOCE, it was important to check whether the receptor antagonists Ki16425 and Ex26 could alter SOCE on their own. In the following experiments, the ratiometric Ca2+ probe Fura-2 was used to analyze SOCE in cells that responded to the KCl challenge (i.e. post-mitotic neurons). Cells, bathed in a nominally Ca2+-free medium, were challenged with thapsigargin to deplete ER Ca2+ stores. A subsequent re-admission of external Ca2+ was followed by an intracellular elevation of Ca2+ (open circles, Fig. 3A)9,24. This entry of Ca2+ was sensitive to the CRAC channel blocker BTP245,46 (1 µM, gray up triangles, Fig. 3A). The thapsigargin-evoked Ca2+ release was unaffected by Ex26 (1 µM, filled down triangles) or Ki16425 (10 µM, gray squares) (Fig. 3A). The SOCE response was however upregulated by Ki16425 but not by Ex26. This is further illustrated in Fig. 3B showing the Ca2+ release and entry analyzed as area under the curve for each condition tested. Ki16426 enhanced the SOCE signal by nearly 30% (n = 5, p < 0.05) (Fig. 3B, gray bar). Altogether, these data show that the LPA and S1P receptor antagonists used did not alter the ER Ca2+ release. The SOCE response was also unaffected by Ex26 but augmented by Ki16426. This potentiating effect was not investigated further.
Figure 3.
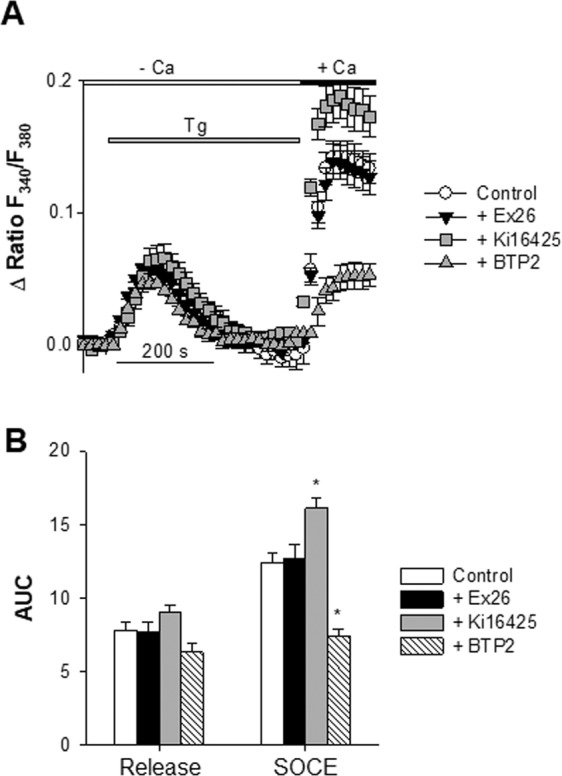
Effects of Ex26, Ki16425, and BTP2 on the thapsigargin-evoked Ca2+ release and SOCE. SOCE responses were analysed with Fura-2. Cells were kept in a nominally Ca2+-free medium. ER Ca2+ stores were depleted with thapsigargin (Tg, 200 nM) before re-introducing external Ca2+. The resulting increase in intracellular Ca2+ is due to Ca2+ entering via the plasma membrane. Panel A shows somatic Ca2+ responses (expressed as Δ ratio F340/F380) as a function of time, and generated by the sequential addition of Tg (200 nM, horizontal gray bar) followed by the readmission of 2 mM external Ca2+ (horizontal black bar). Four conditions are shown: without antagonists of LPA and S1P receptors (Control, open circles, n = 7), with 1 µM Ex26 (gray triangles, n = 5), with 10 µM Ki16425 (filled squares, n = 5), and with 1 µM BTP2 (symbols, n = 5). When tested, Ex26 (or Ki16425) and BTP2 were added 4–7 and 11–12 min, respectively, before time 0 and were also present during the recordings. One time point out of 3 is shown. Panel B shows the thapsigargin-evoked Ca2+ release and SOCE measured as area under the curve (AUC). Mean ± SEM.
NAGly depresses SOCE independently of LPA and S1P receptors
After having shown the presence of functional receptors sensitive to LPA and S1P, their involvement in the NAGly-induced impairment of SOCE was considered. In the following set of experiments, Fura-2-loaded cells were first stimulated with a K+-rich saline (90 mM KCl) before recording SOCE responses in neurons (i.e. in KCl-responsive cells). Figure 4A shows SOCE without NAGly (open circles) and in the presence of NAGly (10 µM, gray down triangles). As already illustrated9, NAGly exerts complex actions on neuronal Ca2+ signalling: (i) it induces a release of cations (Ca2+ and Zn2+) that develops prior to thapsigargin addition (phase ➀, Fig. 4A); (ii) it upregulates the thapsigargin-dependent Ca2+ release (phase ➁); and (iii) reduces the amplitude of SOCE (phase ③). Even in the presence of 1 µM Ex26 (gray up triangles) or 10 µM Ki16425 (filled squares, Fig. 4A), NAGly elevated the Fura-2 fluorescence on its own (phase ➀) and potentiated the thapsigargin-evoked Ca2+ release (phase ➁). The NAGly-induced inhibition of SOCE (phase ③) was also not affected by Ex26 or Ki16425 (Fig. 4A). NAGly had however no inhibitory action on the entry of Ca2+ when added together with BTP2 (open squares, Fig. 4A).
Figure 4.
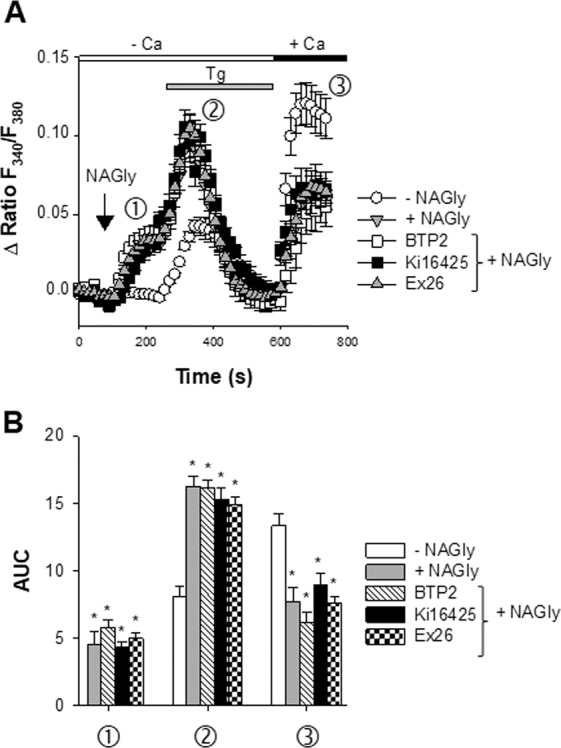
Ex26 and Ki16425 do not reverse the NAGly-induced depression of SOCE. Panel A shows Fura-2 responses (Δ ratio F340/F380) as a function of time before, during the transient application of 200 nM thapsigargin (Tg, horizontal gray bar) to cells kept in a nominally Ca2+-free medium (depletion of ER Ca2+ stores, phase ➁), and after the readmission of 2 mM external Ca2+ (horizontal black bar) (SOCE, phase ➂). Open circles: control conditions (without NAGly) (n = 7). When indicated, 10 µM NAGly was added (vertical arrow) prior to thapsigargin. This elevated the Fura-2 fluorescence (phase ➀) (black triangles, n = 6). Similar experiments were conducted in the presence of NAGly + 10 µM Ki16425 (gray squares, n = 5), NAGly + 10 µM Ex26 (open triangles, n = 4), and NAGly + 1 µM BTP2 (symbol, n = 3). As in Fig. 3, Ki16425 (or Ex26) and BTP2 were added 4–7 and 11–12 min before time 0 and remained present throughout the recordings. One time point out of 3 is shown. Mean ± SEM. Panel B: Area under curve (AUC) measurements of Fura-2 signals under the different conditions tested. Three phases were considered: Ca2+ signals prior to the addition of thapsigargin (phase ➀), the thapsigargin-induced Ca2+ release (phase ➁) and SOCE (phase ➂). *p < 0.05 vs NAGly-untreated cells, one-way ANOVA followed by a Bonferroni’s post hoc test.
The Fura-2 Ca2+ signals (phases ➀, ➁, ③) observed without NAGly (white bars), with NAGly (gray bars), NAGly + Ki16425 (black bars), NAGly + Ex26 (black/white bars), and NAGly + BTP2 (hatched bars) were analyzed as area under curve (AUC) (Fig. 4B). In conclusion, Ki16425 and Ex26 failed to affect the NAGly-evoked Ca2+ rise (phase ➀). These blockers also did not influence the potentiation of the thapsigargin-induced Ca2+ signal induced by NAGly (phase ➁) and the NAGly-dependent depression of SOCE (phase ③). It is proposed that NAGly inhibits a BTP2-sensitive Ca2+ entry pathway without recruiting LPA or S1P-sensitive receptors.
Discussion
NAGly inhibits SOCE20. This impairment has been observed in every cell type and cell line tested so far like fibroblasts, neurons, EA.hy926 (human endothelial cell line), INS-1 832/13 (rat pancreatic β-cell line), and RBL-2H3 cells (rat basophilic leukemia cell line)9,20,21. However, the mechanism by which NAGly alters SOCE is unclear. In the present study we addressed the question of the contribution of lipid sensing GPCRs as targets of NAGly with the aim to gain a better knowledge on neuronal SOCE functioning and regulation. To reach that goal, we took advantage of a recent transcriptomic analysis of the whole murine genome by RNA-seq.22. This allowed us to consider the mRNA expression of 60 putative lipid sensing GPCRs26–30. Overall, transcripts of 14 genes (⁓25%) were detected. Their abundance varied during embryonic development with 7 genes being induced (Cnr1, Gpr4, Gpr12, Gpr17, Gper1, Gpr34, Adgrb1) and 7 genes being repressed (Lpar1, Lpar2, Lpar4, Lpar6, S1pr1, S1pr2, S1pr3). At E13, age at which cerebral cortices were collected to perform the Ca2+ imaging experiments9, the most abundant mRNAs were those coding for CB1 and S1P1 receptors, followed by LPA2 and LPA6 receptors. Since the cannabinoid receptor CB1 does not seem to mediate the NAGly-dependent impairment of SOCE9, only the contribution of S1P and LPA receptors in the NAGly-mediated modulation of SOCE was investigated.
Five subtypes of S1P receptors are known (S1P1–5). They belong to the group of GPCRs and mediate most of the biological actions of the bioactive sphingolipid S1P30. Embryonic cerebral cortices displayed a high mRNA level of S1P1 receptors that declined during embryonic brain development. In addition, cultured cortical cells expressed functional receptors coupled to the release of Ca2+ from the ER and sensitive to the S1P1 antagonist Ex26. These findings are in line with previous reports showing that S1P1 is the major S1P receptor of the murine embryonic brain, followed by S1P2 and S1P3 receptors. It is detected as early as E14, highly expressed in proliferative regions (neurogenic ventricular zone) but its expression decreases at E16 and E1847. The activation of S1P1 receptors is coupled to the mobilization of Ca2+ 33.
LPA receptors constitute another important family of GPCRs sensitive to bioactive lipids30,39. LPA signalling is of particular physiological relevance for the embryonic brain cortex48. At E12.5 the most abundant transcripts in the telencephalon are LPA1, LPA2 and LPA435. In the present work, the main genes expressed at E13 were encoding for LPA2 and LPA6. Moreover, the application of LPA caused the release of Ca2+ from the ER. These responses were highly sensitive to the LPA1/3 antagonist Ki16425 but moderately affected by the LPA2/3 antagonist H2L518630330,39. The pharmacological dissection of the LPA-induced Ca2+ signalling pointing to LPA1/3 as the likely receptors responding to LPA is difficult to reconcile with the gene analysis showing that LPA1 and LPA3 are, respectively, very weakly expressed and undetected. The pharmacological properties of native LPA receptors of cortical neurons may differ from those of LPA receptors heterogeneously expressed.
After having shown the presence of functional LPA and S1P receptors, their contribution to the NAGly-dependent depression of SOCE was evaluated. The pharmacological blockade of S1P and LPA receptors with Ex26 or Ki16425 did not abolish or attenuate the NAGly-dependent impairment of SOCE. Some cellular responses of NAGly have been shown to be mediated by the orphan receptor GPR5511. However, we found no evidence for the presence of significant levels of GPR55 mRNA. Furthermore, the GPR55 agonist AM25149, which induces a GPR55-dependent mobilization of Ca2+ with an EC50 of ~0.6 µM50, fails to evoke any Ca2+ release when applied to cortical cells at 10 µM. This further suggests that GPR55 does not participate in the NAGly-induced alteration of neuronal Ca2+ signalling.
In conclusion, our data show that NAGly inhibits a BTP2-sensitive Ca2+ entry, which is most likely a SOCE. This occurs independently of GPR55, LPA and S1P receptors (present report), and via a mechanism insensitive to the pertussis toxin9. It is worth recalling that NAGly regulates voltage-gated Ca2+ channel activity without acting on GPCRs8,13. Although we cannot exclude the possibility that NAGly acts on an orphan lipid sensing GPCR that was not considered in the present study, our report suggests that NAGly disturbs the coupling of the core components of the SOCE machinery (STIM-Orai)20. This inhibitory process does not seem to develop in response to an intracellular signalling cascade. These past9 and present data show that the phytocannabinoid cannabidiol, the endocannabinoid AEA and its derivative NAGly are potent inhibitors of neuronal SOCE. This indicates that NAGly and endocannabinoids are endogenous SOCE modulators, and raises the possibility that the depression of SOCE could play a role in the neuro-behavioural effects of cannabinoids and signalling lipids.
Acknowledgements
The study was supported by a grant from l’Agence Nationale de la Recherche (ANR-16-CE29-0024 to AB). We also acknowledge support from the Centre National de la Recherche Scientifique (CNRS), the Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), the Université de Grenoble Alpes (UGA), and the animal facility platform supported by GRAL, financed within the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). We wish to thank Drs C. Blugeon and S. Lemoine from the Institut de Biologie de l’Ecole Normale Supérieure (IBENS, Paris) for their help with the gene analysis. We also appreciate comments on this manuscript prior to submission from Dr J. Gibon (University of British Columbia, Kelowna). We also thank Dr A. Journet, E. Taillebourg and G. Courtois for reading the manuscript.
Author contributions
A.D., J.H. and A.B. performed experiments and analyzed data. A.B. designed the study and wrote the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Huang SM, et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J. Biol. Chem. 2001;276:42639–42644. doi: 10.1074/jbc.M107351200. [DOI] [PubMed] [Google Scholar]
- 2.Bradshaw HB, et al. The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem. 2009;10:14. doi: 10.1186/1471-2091-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbara G, et al. T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J. Neurosci. 2009;29:13106–13114. doi: 10.1523/JNEUROSCI.2919-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Staton PC, et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain. 2008;139:225–236. doi: 10.1016/j.pain.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Deliu E, et al. The Lysophosphatidylinositol Receptor GPR55 Modulates Pain Perception in the Periaqueductal Gray. Mol. pharmacology. 2015;88:265–272. doi: 10.1124/mol.115.099333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burstein SH. The cannabinoid acids, analogs and endogenous counterparts. Bioorganic medicinal Chem. 2014;22:2830–2843. doi: 10.1016/j.bmc.2014.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeong HJ, Vandenberg RJ, Vaughan CW. N-arachidonyl-glycine modulates synaptic transmission in superficial dorsal horn. Br. J. pharmacology. 2010;161:925–935. doi: 10.1111/j.1476-5381.2010.00935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chemin J, Cazade M, Lory P. Modulation of T-type calcium channels by bioactive lipids. Pflug. Arch. 2014;466:689–700. doi: 10.1007/s00424-014-1467-5. [DOI] [PubMed] [Google Scholar]
- 9.Bouron A. Phyto and endocannabinoids exert complex actions on calcium and zinc signaling in mouse cortical neurons. Biochem. Pharmacol. 2018;152:244–251. doi: 10.1016/j.bcp.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 10.McHugh D, et al. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010;11:44. doi: 10.1186/1471-2202-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Console-Bram L, et al. N-arachidonoyl glycine, another endogenous agonist of GPR55. Biochemical biophysical Res. Commun. 2017;490:1389–1393. doi: 10.1016/j.bbrc.2017.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oh DY, et al. Identification of farnesyl pyrophosphate and N-arachidonylglycine as endogenous ligands for GPR92. J. Biol. Chem. 2008;283:21054–21064. doi: 10.1074/jbc.M708908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu VB, Puhl HL, III., Ikeda SR. N-Arachidonyl glycine does not activate G protein-coupled receptor 18 signaling via canonical pathways. Mol. Pharmacol. 2013;83:267–282. doi: 10.1124/mol.112.081182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bondarenko AI, et al. N-Arachidonoyl glycine suppresses Na(+)/Ca(2)(+) exchanger-mediated Ca(2)(+) entry into endothelial cells and activates BK(Ca) channels independently of GPCRs. Br. J. pharmacology. 2013;169:933–948. doi: 10.1111/bph.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Putney JW, et al. The functions of store-operated calcium channels. Biochimica et biophysica acta. Mol. Cell Res. 2017;1864:900–906. doi: 10.1016/j.bbamcr.2016.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parekh AB. Store-operated CRAC channels: function in health and disease. Nat. Rev. Drug. Discov. 2010;9:399–410. doi: 10.1038/nrd3136. [DOI] [PubMed] [Google Scholar]
- 17.Yeung PS, Yamashita M, Prakriya M. Pore opening mechanism of CRAC channels. Cell Calcium. 2017;63:14–19. doi: 10.1016/j.ceca.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hogan PG, Rao A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015;460:40–49. doi: 10.1016/j.bbrc.2015.02.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stathopulos PB, Ikura M. Structure and function of endoplasmic reticulum STIM calcium sensors. Curr. Top. Membr. 2013;71:59–93. doi: 10.1016/B978-0-12-407870-3.00003-2. [DOI] [PubMed] [Google Scholar]
- 20.Deak AT, et al. The endocannabinoid N-arachidonoyl glycine (NAGly) inhibits store-operated Ca2+ entry by preventing STIM1-Orai1 interaction. J. Cell Sci. 2013;126:879–888. doi: 10.1242/jcs.118075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson Heather, Leech Colin, Kopp Richard, Roe Michael. Interplay between ER Ca2+ Binding Proteins, STIM1 and STIM2, Is Required for Store-Operated Ca2+ Entry. International Journal of Molecular Sciences. 2018;19(5):1522. doi: 10.3390/ijms19051522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasna J, Bohic S, Lemoine S, Blugeon C, Bouron A. Zinc Uptake and Storage During the Formation of the Cerebral Cortex in Mice. Mol. Neurobiol. 2019;56:6928–6940. doi: 10.1007/s12035-019-1581-7. [DOI] [PubMed] [Google Scholar]
- 23.Gibon J, et al. The antidepressant hyperforin increases the phosphorylation of CREB and the expression of TrkB in a tissue-specific manner. Int. J. Neuropsychopharmacol. 2013;16:189–198. doi: 10.1017/S146114571100188X. [DOI] [PubMed] [Google Scholar]
- 24.Chauvet, S. et al. Pharmacological Characterization of the Native Store-Operated Calcium Channels of Cortical Neurons from Embryonic Mouse Brain. Frontiers in Pharmacology7, 10.3389/fphar.2016.00486 (2016). [DOI] [PMC free article] [PubMed]
- 25.Gruszczynska-Biegala J, Sladowska M, Kuznicki J. AMPA Receptors Are Involved in Store-Operated Calcium Entry and Interact with STIM Proteins in Rat Primary Cortical Neurons. Front. Cell. Neurosci. 2016;10:251. doi: 10.3389/fncel.2016.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Im DS. Discovery of new G protein-coupled receptors for lipid mediators. J. lipid Res. 2004;45:410–418. doi: 10.1194/jlr.R300006-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Im DS. Intercellular Lipid Mediators and GPCR Drug Discovery. Biomolecules therapeutics. 2013;21:411–422. doi: 10.4062/biomolther.2013.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irving A, et al. Cannabinoid Receptor-Related Orphan G Protein-Coupled Receptors. Adv. pharmacology. 2017;80:223–247. doi: 10.1016/bs.apha.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Pertwee R. G., Howlett A. C., Abood M. E., Alexander S. P. H., Di Marzo V., Elphick M. R., Greasley P. J., Hansen H. S., Kunos G., Mackie K., Mechoulam R., Ross R. A. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharmacological Reviews. 2010;62(4):588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 2014;171:3575–3594. doi: 10.1111/bph.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner GP, Kin K, Lynch VJ. A model based criterion for gene expression calls using RNA-seq data. Theory Biosci. = Theorie den. Biowissenschaften. 2013;132:159–164. doi: 10.1007/s12064-013-0178-3. [DOI] [PubMed] [Google Scholar]
- 32.Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J. medicinal Chem. 1997;40:659–667. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- 33.Mattie M, Brooker G, Spiegel S. Sphingosine-1-phosphate, a putative second messenger, mobilizes calcium from internal stores via an inositol trisphosphate-independent pathway. J. Biol. Chem. 1994;269:3181–3188. [PubMed] [Google Scholar]
- 34.Shirakawa H, et al. Sphingosine-1-phosphate induces Ca(2+) signaling and CXCL1 release via TRPC6 channel in astrocytes. Glia. 2017;65:1005–1016. doi: 10.1002/glia.23141. [DOI] [PubMed] [Google Scholar]
- 35.Dubin AE, Herr DR, Chun J. Diversity of lysophosphatidic acid receptor-mediated intracellular calcium signaling in early cortical neurogenesis. J. Neurosci. 2010;30:7300–7309. doi: 10.1523/JNEUROSCI.6151-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hildebrandt JP, Hildebrandt P. Lysophosphatidic acid depletes intracellular calcium stores different from those mediating capacitative calcium entry in C6 rat glioma cells. Glia. 1997;19:67–73. doi: 10.1002/(SICI)1098-1136(199701)19:1<67::AID-GLIA7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 37.Bouron A, Altafaj X, Boisseau S, De Waard M. A store-operated Ca2+ influx activated in response to the depletion of thapsigargin-sensitive Ca2+ stores is developmentally regulated in embryonic cortical neurons from mice. Brain Res. Dev. Brain Res. 2005;159:64–71. doi: 10.1016/j.devbrainres.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Bouron A, Boisseau S, De Waard M, Peris L. Differential down-regulation of voltage-gated calcium channel currents by glutamate and BDNF in embryonic cortical neurons. Eur. J. Neurosci. 2006;24:699–708. doi: 10.1111/j.1460-9568.2006.04946.x. [DOI] [PubMed] [Google Scholar]
- 39.Yung YC, Stoddard NC, Chun J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014;55:1192–1214. doi: 10.1194/jlr.R046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Im DS. Pharmacological tools for lysophospholipid GPCRs: development of agonists and antagonists for LPA and S1P receptors. Acta pharmacologica Sin. 2010;31:1213–1222. doi: 10.1038/aps.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoddard NC, Chun J. Promising pharmacological directions in the world of lysophosphatidic Acid signaling. Biomolecules therapeutics. 2015;23:1–11. doi: 10.4062/biomolther.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 2010;62:579–587. doi: 10.1124/pr.110.003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cahalan SM, et al. Sphingosine 1-phosphate receptor 1 (S1P(1)) upregulation and amelioration of experimental autoimmune encephalomyelitis by an S1P(1) antagonist. Mol. Pharmacol. 2013;83:316–321. doi: 10.1124/mol.112.082958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dubin AE, Bahnson T, Weiner JA, Fukushima N, Chun J. Lysophosphatidic Acid Stimulates Neurotransmitter-Like Conductance Changes that Precede GABA and L-Glutamate in Early, Presumptive Cortical Neuroblasts. J. Neurosci. 1999;19:1371–1381. doi: 10.1523/JNEUROSCI.19-04-01371.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishikawa J, et al. A Pyrazole Derivative, YM-58483, Potently Inhibits Store-Operated Sustained Ca2+ Influx and IL-2 Production in T Lymphocytes. J. Immunol. 2003;170:4441–4449. doi: 10.4049/jimmunol.170.9.4441. [DOI] [PubMed] [Google Scholar]
- 46.Zitt C, et al. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J. Biol. Chem. 2004;279:12427–12437. doi: 10.1074/jbc.M309297200. [DOI] [PubMed] [Google Scholar]
- 47.McGiffert C, Contos JJ, Friedman B, Chun J. Embryonic brain expression analysis of lysophospholipid receptor genes suggests roles for s1p(1) in neurogenesis and s1p(1-3) in angiogenesis. FEBS Lett. 2002;531:103–108. doi: 10.1016/S0014-5793(02)03404-X. [DOI] [PubMed] [Google Scholar]
- 48.Yung YC, Stoddard NC, Mirendil H, Chun J. Lysophosphatidic Acid signaling in the nervous system. Neuron. 2015;85:669–682. doi: 10.1016/j.neuron.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryberg E, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. pharmacology. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henstridge CM, et al. The GPR55 ligand L-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB journal: Off. Publ. Federation Am. Societies Exp. Biol. 2009;23:183–193. doi: 10.1096/fj.08-108670. [DOI] [PubMed] [Google Scholar]