

Abstract
Mutations in PINK1 and Parkin/PRKN cause the degeneration of dopaminergic neurons in familial forms of Parkinson’s disease but the precise pathogenic mechanisms are unknown. The PINK1/Parkin pathway has been described to play a central role in mitochondrial homeostasis by signalling the targeted destruction of damaged mitochondria, however, how disrupting this process leads to neuronal death was unclear until recently. An elegant study in mice revealed that the loss of Pink1 or Prkn coupled with an additional mitochondrial stress resulted in the aberrant activation of the innate immune signalling, mediated via the cGAS/STING pathway, causing degeneration of dopaminergic neurons and motor impairment. Genetic knockout of Sting was sufficient to completely prevent neurodegeneration and accompanying motor deficits. To determine whether Sting plays a conserved role in Pink1/parkin related pathology, we tested for genetic interactions between Sting and Pink1/parkin in Drosophila. Surprisingly, we found that loss of Sting, or its downstream effector Relish, was insufficient to suppress the behavioural deficits or mitochondria disruption in the Pink1/parkin mutants. Thus, we conclude that phenotypes associated with loss of Pink1/parkin are not universally due to aberrant activation of the STING pathway.
Subject terms: Mechanisms of disease, Genetics of the nervous system
Introduction
Loss of function mutations in PINK1 and PRKN cause familial parkinsonism, an incurable neurodegenerative disorder predominantly associated with the progressive loss of dopaminergic neurons in substantia nigra leading to loss of motor control. PRKN encodes a cytosolic ubiquitin E3 ligase, Parkin, and PINK1 encodes a mitochondrially targeted kinase. Extensive evidence shows that they cooperate in signalling the targeted autophagic destruction of damaged mitochondria (mitophagy) as part of a homeostatic mitochondrial quality control process1,2.
Mitochondria are essential organelles that perform many critical metabolic functions but are also a major source of damaging reactive oxygen species and harbour pro-apoptotic factors. Hence, multiple homeostatic processes, such as mitophagy, operate to maintain mitochondrial integrity and prevent potentially catastrophic consequences. Such homeostatic mechanisms are particularly important for post-mitotic, energetically demanding tissues such as nerves and muscles.
The molecular details of PINK1/Parkin-induced mitophagy are well characterized in cultured cells, however, relatively little is known about mitophagy under physiological conditions in vivo3–5. Nevertheless, several studies provide evidence consistent with PINK1 and Parkin acting to remove mitochondrial damage in vivo. One study used a mass spectrometry-based analysis of mitochondrial protein turnover in Drosophila6, which revealed that fly PINK1 and Parkin selectively affect the degradation of certain mitochondrial proteins under physiological conditions. Another found that loss of Prkn in mice, which alone has very little phenotype7,8, exacerbated the phenotypic effects of a mitochondrial DNA mutator strain, provoking loss of dopaminergic neurons and motor deficits9.
Importantly, a subsequent study shed light on the mechanism by which loss of Pink1/Prkn leads to neurodegeneration in the presence of mtDNA mutations, or upon exposure to exhaustive exercise, as chronic or acute mitochondrial stresses, respectively10. This demonstrated that in the absence of Pink1/Prkn these mitochondrial stresses cause an aberrant inflammatory response mediated by the STING pathway, presumably via the release of mtDNA into the cytosol. Consequently, loss of STING completely prevented the inflammatory response and the resulting neurodegeneration and locomotor phenotypes10. These results strongly implicate the induction of STING-mediated inflammation in the pathogenic cause of Parkinson’s disease.
The recently identified Drosophila Sting ortholog has been shown to bind to cyclic-dinucleotides, in particular 2′3′-cGAMP, and trigger an immune response to bacterial and viral infection11–14, mediated by the IMD pathway and the transcription factor Relish (homologous to NF-κB). Consequently, Drosophila mutant for Sting showed a reduced survival upon infection. Interestingly, while aberrant activation of the IMD-Relish pathway has been shown to cause neurodegeneration and shortened lifespan in Drosophila15, transcriptional profiling has shown that innate immune signalling pathways are ectopically active in Drosophila parkin and Pink1 mutants16,17.
The Drosophila models have been highly informative for interrogating the physiological role of PINK1/Parkin, primarily via genetic or pharmacological manipulations17–21 that can modify the robust neuromuscular phenotypes associated with loss of the Pink1/parkin orthologs22–25 (for review see26). Therefore, we sought to determine whether aberrant activation of the Sting-Relish immune signalling cascade may contribute to the neuromuscular degeneration phenotypes observed in Drosophila Pink1/parkin mutants. Surprisingly, we found that loss of Sting or Relish had no suppressing effect on the locomotor deficits or mitochondrial disruption in Pink1 or parkin mutants. Moreover, Sting knockout did not affect the behavioural phenotypes associated with a fly mtDNA mutator model, nor the combined effect of mtDNA mutations in a parkin background. Hence, the central role of Sting in the induction of Pink1/parkin mutant phenotypes proposed for mammals is not conserved in Drosophila.
Results
Drosophila Sting mutants have recently been generated and, consistent with Sting’s role in triggering an innate immune response, shown to be more susceptible to infection11–13. As other organismal phenotypes were not reported, we first assessed whether loss of Sting may induce additional phenotypes associated with the neuromuscular system that might confound further genetic interaction analysis. To this end, we examined the motor behaviour and muscle integrity in Sting loss of function conditions. We assessed the impact of RNAi-induced loss of function using previously validated RNAi lines expressed via the ubiquitous driver da-GAL4. A small impact on climbing ability in young flies was observed with one RNAi transgene, which was also seen in homozygous Sting null (StingΔRG5) mutants (Fig. 1A). Aged Sting-RNAi flies showed a consistent, modest impact on climbing ability, but this was not evident in Sting mutants (Fig. 1B). Microscopy analysis of muscle and mitochondrial integrity did not reveal any obvious disruption in Sting mutants (Fig. 1C). Since loss of Sting did not appear to grossly affect neuromuscular integrity, we next assessed whether the activity of Sting contributed to the neuromuscular phenotypes in Pink1/parkin mutants.
Figure 1.

Loss of Sting has limited impact on neuromuscular phenotypes. Locomotor assays analysing climbing ability (negative geotaxis) in (A) young and (B) older adult flies of control and Sting knockdown (RNAi) or null (StingΔRG5) mutants. Charts show mean ± 95% confidence interval (CI); number of animals analysed is shown in each bar. Significance was measured by Kruskal-Wallis test with Dunn’s post hoc correction for multiple comparisons; **p < 0.01, ****p < 0.0001; ns, non-significant. Control genotype is da-GAL4/+. (C) Representative confocal microscopy analysis of mitochondria in flight muscles, immunostained with anti-ATP5A, in control (w1118) and Sting heterozygous and homozygous mutants. Scale bar = 10 µm.
Combining all the manipulations of Sting (two RNAi transgenes, heterozygous and homozygous null mutations) with parkin null mutants (park25), we did not observe any modification (suppression or enhancement) of the parkin mutants climbing defect (Fig. 2A). Similarly, the thoracic indentations typically observed in park25 flies due to the degeneration of the underlying musculature, were still present in the absence of Sting (Fig. 2B). Consistent with this, we did not observe any improvement of the tissue or mitochondrial integrity in the flight muscles of parkin mutants by removal of Sting (Fig. 2C).
Figure 2.

Loss of Sting does not modify parkin mutant phenotypes. (A) Analysis of locomotor (climbing) ability, (B) thoracic indentations, and (C) mitochondrial morphology in young park25 mutants combined with Sting knockdown or null mutations. Charts show mean ± 95% confidence interval (CI); number of animals analysed is shown in each bar. Statistical significance was measured by Kruskal-Wallis test with Dunn’s post hoc correction for multiple comparisons; ****p < 0.0001; ns, non-significant. Confocal microscopy images show flight muscle mitochondria immunostained with anti-ATP5A. Scale bar = 10 µm. Control genotypes are da-GAL4/+ for climbing, and w1118 for thoracic indentation and microscopy. Ctrl RNAi is lacZ-RNAi in the mutant background.
We next assessed the contribution of Sting function towards Pink1 mutant (Pink1B9) phenotypes. Similar to parkin mutants, loss of Sting failed to modify the climbing defect (Fig. 3A), thoracic indentations (Fig. 3B) or disruption of flight muscle and mitochondrial integrity (Fig. 3C) observed in Pink1B9 flies. Taken together, these results indicate that Sting does not contribute to the neuromuscular phenotypes observed in Pink1/parkin mutants.
Figure 3.

Loss of Sting does not modify Pink1 mutant phenotypes. (A) Analysis of locomotor (climbing) ability, (B) thoracic indentations, and (C) mitochondrial morphology in young Pink1B9 mutants combined with Sting knockdown or null mutations. Charts show mean ± 95% confidence interval (CI); number of animals analysed is shown in each bar. Statistical significance was measured by Kruskal-Wallis test with Dunn’s post hoc correction for multiple comparisons; ****p < 0.0001; ns, non-significant. Confocal microscopy images show flight muscle mitochondria immunostained with anti-ATP5A. Scale bar = 10 µm. Control genotypes are da-GAL4/+ for climbing, and w1118 for thoracic indentations and microscopy. Ctrl RNAi is lacZ-RNAi in the mutant background.
Considering that loss of STING in mouse completely abrogated the Pink1/Prkn-associated neurodegeneration and motor phenotypes provoked by additional mitochondrial stresses, we were surprised by the lack of suppression of Pink1/parkin phenotypes in flies. Therefore, to further interrogate the potential contribution of this pathway to Pink1/parkin pathology, we also analysed a downstream effector of the Sting-IMD pathway, the transcription factor Relish (Rel). While RNAi knockdown using two previously characterized transgenes11,13 elicited modest effect on climbing, Rel mutants (RelE20) displayed a strong locomotor defect (Fig. 4A, B). However, analysis of flight muscles in these mutants did not reveal any major disruption of mitochondrial integrity (Fig. 4C).
Figure 4.
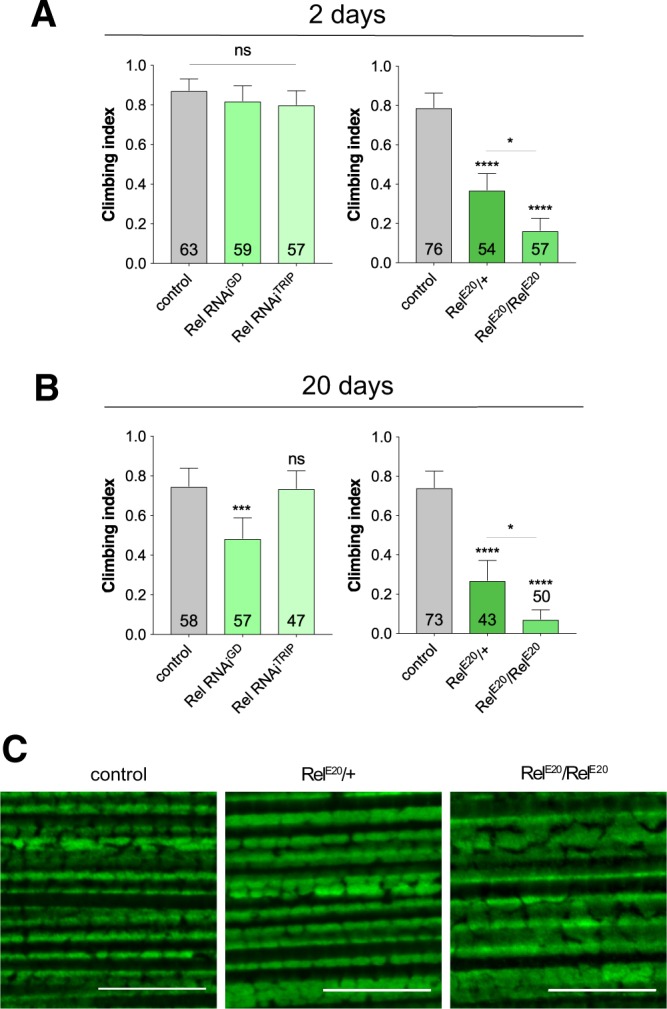
Loss of Relish causes mild locomotor deficits. Locomotor assays analysing climbing ability in (A) young and (B) older adult flies of control and RNAi knockdown or Relish mutant (RelE20). Charts show mean ± 95% confidence interval (CI); number of animals analysed is shown in each bar. Statistical significance was measured by Kruskal-Wallis test with Dunn’s post hoc correction for multiple comparisons; *p < 0.05, ***p < 0.001, ****p < 0.0001; ns, non-significant. Control genotype is da-GAL4/+. (C) Representative confocal microscopy analysis of mitochondria in flight muscles, immunostained with anti-ATP5A, in control (w1118) and Relish heterozygous and homozygous mutants. Scale bar = 10 µm.
Similar to the Sting manipulations, RNAi knockdown of Rel did not modify the climbing deficit of parkin or Pink1 mutants (Fig. 5A), nor did it noticeably affect the mitochondrial integrity in flight muscles (Fig. 5B). Indeed, in contrast to expectation, genetic loss of Rel enhanced the Pink1 locomotor defect (Fig. 5A), although the mitochondrial integrity was not noticeably worsened in Pink1B9; RelE20 flies (Fig. 5B).
Figure 5.

Loss of Relish does not rescue Pink1 or parkin mutant phenotypes. (A) Analysis of locomotor (climbing) ability and (B) mitochondrial morphology in young park25 or Pink1B9 mutants combined with Relish knockdown or null mutations. Charts show mean ± 95% confidence interval (CI); number of animals analysed is shown in each bar. Statistical significance was measured by Kruskal-Wallis test with Dunn’s post hoc correction for multiple comparisons; ****p < 0.0001; ns, non-significant. Confocal microscopy images show flight muscle mitochondria immunostained with anti-ATP5A. Scale bar = 10 µm. Control genotypes are da-GAL4/+ for climbing, and w1118 for microscopy. Ctrl RNAi is lacZ-RNAi in the respective mutant background.
In a final effort to assess whether the Drosophila Pink1/parkin-Sting axis acts in an analogous fashion to mice, we sought to recapitulate the conditions assessed by Sliter et al.10 and test the role of Sting when an additional mitochondrial stress is combined with parkin loss-of-function. To do this, we used our previously established mtDNA mutator model (mito-APOBEC1), which generates high levels of deleterious mtDNA mutations in somatic tissues, disrupting mitochondrial function and causing motor defects and shortened lifespan27. Notably, the loss of parkin or Sting did not exacerbate the impact of mito-APOBEC1 alone on locomotor function (Fig. 6A). Furthermore, the combination of the mtDNA mutator in a parkin; Sting double mutant background, in stark contrast to the results in mice10, enhanced the climbing deficit (Fig. 6A). Similarly, while loss of Sting alone did not affect normal lifespan, it significantly enhanced the shortened lifespan of the mito-APOBEC1 model or the combination of mito-APOBEC1 with parkin loss-of-function (Fig. 6B), consistent with the locomotor analysis.
Figure 6.
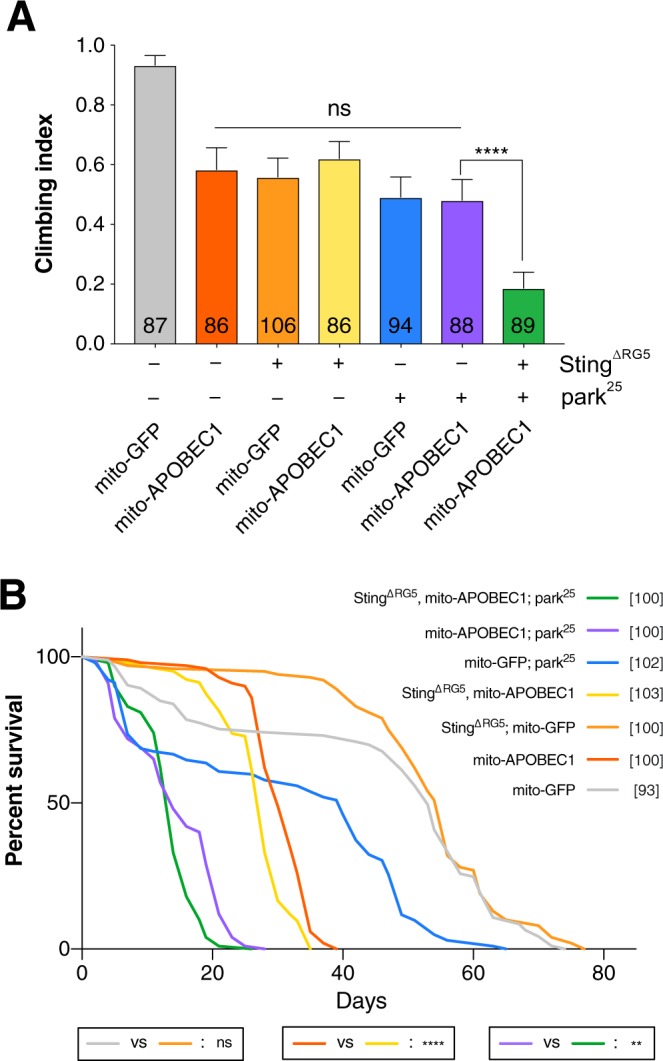
Loss of Sting does not ameliorate mtDNA mutator; parkin mutant combinations. Analysis of (A) locomotor (climbing) ability in young adults and (B) lifespan in flies combining mito-APOBEC1 mtDNA mutator expression with or without parkin and/or Sting. Transgene expression was driven by da-GAL4. Mito-GFP expression was used as a control. Charts show mean ± 95% confidence interval (CI); number of animals analysed is shown in each bar for climbing, and beside the genotype key for lifespan. For climbing, statistical significance was determined by Kruskal-Wallis test with Dunn’s post hoc correction for multiple comparisons; and for lifespan, by Log-rank (Mantel-Cox) test; **p < 0.01, ****p < 0.0001; ns, non-significant.
Thus, together the above data suggest that the Sting pathway, although proposed to be mediating motor and neurodegenerative defects in Prkn−/− mice, do not similarly contribute to the neuromuscular defects observed in Pink1/parkin mutant flies.
Discussion
Understanding the pathogenic mechanisms by which loss of function mutations in PINK1 and Parkin lead to neurodegeneration in Parkinson’s disease is central to defining better disease-modifying therapies. While tremendous advances have been made in uncovering the molecular mechanisms of PINK1/Parkin function in vitro and in cell culture models, understanding the consequences of this dysfunction on neuronal demise must be studied in vivo, in the complex milieu of organismal biology. This has been severely hampered by the lack of robust phenotypes in Pink1/Prkn knockout mice. In contrast, Drosophila models have provided substantial insights in this realm as fly Pink1/parkin mutants exhibit extensive disruption of the neuromuscular system presenting, amongst other phenotypes, profound deficits in locomotor behaviours, apoptotic degeneration of flight muscles, progressive degeneration of dopaminergic neurons, all accompanied by morphological and functional breakdown of mitochondria. Consequently, genetic studies using the fly models, primarily using suppression or enhancement of the mutant phenotypes as a sensitive readout, have elucidated several important and conserved features of PINK1/Parkin biology17–21,26.
Recent studies have shed new light on the in vivo role of PINK1/Parkin in vertebrates, and the context in which loss of Pink1/Prkn can reveal pathogenic phenotypes. First, combining Prkn knockout mutants with a mtDNA mutator strain selectively led to degeneration of nigral dopaminergic neurons, decline in motor ability and increased mitochondrial dysfunction9. Extending these observations, Sliter et al.10 revealed that this Prkn−/−; mutator combination (or Pink1−/−; mutator) provoked an aberrant innate immune response mediated by the STING pathway, suggesting that the systemic inflammatory response ultimately caused the dopaminergic neurodegeneration and motor deficits. Indeed, genetic loss of STING was sufficient to completely prevent the inflammation, motor defect and neurodegeneration in the Prkn−/−; mutator mice. These findings established the STING pathway and, more broadly, aberrant innate immune signalling, as a pathogenic cause and a highly attractive therapeutic target. Moreover, additional work has also implicated Pink1/Prkn mutations in inducing aberrant inflammation, albeit via adaptive immunity28. However, while the PINK1/Parkin pathway is clearly an ancient mechanism regulating mitochondrial quality control, our data indicate that Sting does not appear to be a fundamental, conserved feature of PINK1/Parkin biology.
The question arises why loss of Sting does not suppress Pink1/parkin phenotypes in flies when it is capable of completely preventing pathology in mice? At this stage, the answer is unknown and rather puzzling given that innate immune signalling is dysregulated in Pink1/parkin mutants16,17, and Sting performs an analogous function in flies as it does in vertebrates11. One possibility is that the aberrant innate immune activation observed in parkin and Pink1 mutant flies is not mediated by the presence of cytosolic DNA or activation of the Sting pathway. Moreover, investigating whether induction of mtDNA mutations is required to trigger the innate immune response, as indicated by Sliter et al., our data show that even in the presence of a mtDNA mutator, the Sting immune cascade did not contribute to the neuromuscular phenotypes caused by loss of Pink1/parkin in flies. An alternative interpretation is that the Pink1/parkin phenotypes are not due to aberrant immune signalling and this may be an epiphenomenon. Supporting this view, many studies have established that loss of Pink1/parkin in flies causes catastrophic mitochondrial disruptions, triggering cell-autonomous apoptosis22–24.
Considering this, it isn’t clear from current data why either exhaustive exercise or increased mtDNA mutations should trigger an innate immune response that is mitigated by PINK1/Parkin in mice. In the mouse model, the involvement of STING implicates the presence of cytosolic DNA as a trigger. The evidence from Sliter et al. suggests that exhaustive exercise or mtDNA mutations is sufficient to induce mitophagy, which if not properly executed by Pink1/Parkin leads to the release of mtDNA and activation of STING signalling. However, it remains unclear how these mitochondrial stresses in the absence of Pink1/Prkn lead to release of mtDNA – presumably by loss of integrity and rupture of the mitochondrial boundary membranes. The observed increase in mitophagy in mouse cardiac muscle upon exhaustive exercise is again intriguing as this tissue shares striking structural and functional homology with Drosophila flight muscles, further increasing the puzzle as to why the role of Sting does not appear to be a conserved feature of Pink1/parkin biology in flies. Clearly, further work is necessary in order to fully understand the mechanisms linking mitochondrial disruption and immune activation across species.
Methods
Drosophila stocks and husbandry
Flies were raised under standard conditions in a humidified, temperature-controlled incubator with a 12 h:12 h light:dark cycle at 25 °C, on food consisting of agar, cornmeal, molasses, propionic acid and yeast. Transgene expression was induced using the ubiquitous da-GAL4 driver. The following strains were obtained from the Bloomington Drosophila Stock Center (RRID:SCR_006457): w1118 (RRID:BDSC_6326), da-GAL4 (RRID:BDSC_55850), StingTRiP (RRID:BDSC_31565), RelishTRiP (RRID:BDSC_33661), RelishE20 (RRID:BDSC_9457), UAS-mito-HA-GFP (RRID:BDSC_8443); and the Vienna Drosophila Resource Center (RRID:SCR_013805): StingGD (P{GD1905}v4031), RelishGD (P{GD1199}v49413), and lacZ-RNAi (P{GD936}v51446) used as a control RNAi. Other lines were kindly provided as follows: StingΔRG5 from A. Goodman11, Pink1B9 mutants from J. Chung24, and the park25 mutants and UAS-mito-APOBEC1 have been described previously23,27. All experiments were conducted using male flies.
Locomotor and lifespan assays
The startle induced negative geotaxis (climbing) assay was performed using a counter-current apparatus. Briefly, 20–23 males were placed into the first chamber, tapped to the bottom, and given 10 s to climb a 10 cm distance. This procedure was repeated five times (five chambers), and the number of flies that has remained into each chamber counted. The weighted performance of several group of flies for each genotype was normalized to the maximum possible score and expressed as Climbing index23.
For lifespan experiments, flies were grown under identical conditions at low-density. Progeny were collected under very light anaesthesia and kept in tubes of approximately 20 males each, around 100 in total. Flies were transferred every 2–3 days to fresh medium and the number of dead flies recorded. Percent survival was calculated at the end of the experiment after correcting for any accidental loss.
Immunohistochemistry and sample preparation
For immunostaining, adult flight muscles were dissected in PBS and fixed in 4% formaldehyde for 30 min, permeabilized in 0.3% Triton X-100 for 30 min, and blocked with 0.3% Triton X-100 plus 1% bovine serum albumin in PBS for 1 h at RT. Tissues were incubated with ATP5A antibody (Abcam Cat# ab14748, RRID:AB_301447; 1:500), diluted in 0.3% Triton X-100 plus 1% bovine serum albumin in PBS overnight at 4 °C, then rinsed 3 times 10 min with 0.3% Triton X-100 in PBS, and incubated with the appropriate fluorescent secondary antibodies overnight at 4 °C. The tissues were washed 2 times in PBS and mounted on slides using Prolong Diamond Antifade mounting medium (Thermo Fischer Scientific).
Microscopy
Fluorescence imaging was conducted using a Zeiss LSM 880 confocal microscope (Carl Zeiss MicroImaging) equipped with Nikon Plan-Apochromat 100×/1.4 NA oil immersion objectives. Images were prepared using Fiji software (Fiji, RRID:SCR_002285). For thoracic indentations, images were acquired using a Leica DFC490 camera mounted on a Leica MZ6 stereomicroscope.
Statistical analysis
For behavioural analyses, Kruskal-Wallis non-parametric test with Dunn’s post-hoc correction for multiple comparisons was used. For statistical analyses of lifespan experiment a Log-rank (Mantel-Cox) test was used. Analyses were performed using GraphPad Prism 8 software (RRID:SCR_002798).
Acknowledgements
This work is supported by MRC core funding (MC_UU_00015/4, MC-A070-5PSB0 and MC_UU_00015/6) and ERC Starting grant (DYNAMITO; 309742). J.J.L. was supported by an MRC PhD Studentship awarded via the MRC MBU, and S.A. was supported by an MRC Career Development Fellowship. Stocks were obtained from the Bloomington Drosophila Stock Center which is supported by grant NIH P40OD018537. We also thank Alan Goodman (Washington State University) for kindly providing the Sting mutant line. In particular, we thank V. Hewitt and L. Garcia for developing an algorithm to aid the lifespan analysis, and all members of the Whitworth lab for discussions.
Author contributions
J.J.L. designed and performed experiments, and analysed data, with assistance from S.A. A.J.W. conceived the study, designed experiments, analysed the data and supervised the work. A.J.W. wrote the manuscript with input from all authors.
Data availability
All data that support the findings of this study are available on reasonable request to the corresponding author. The contributing authors declare that all relevant data are included in the paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamano K, Matsuda N, Tanaka K. The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. Embo Rep. 2016;17:300–316. doi: 10.15252/embr.201541486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cummins N, Gotz J. Shedding light on mitophagy in neurons: what is the evidence for PINK1/Parkin mitophagy in vivo? Cell Mol Life Sci. 2017;75:1151–1162. doi: 10.1007/s00018-017-2692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodger CE, McWilliams TG, Ganley IG. Mammalian mitophagy - from in vitro molecules to in vivo models. FEBS J. 2017;285:1185–1202. doi: 10.1111/febs.14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitworth AJ, Pallanck LJ. PINK1/Parkin mitophagy and neurodegeneration-what do we really know in vivo? Curr Opin Genet Dev. 2017;44:47–53. doi: 10.1016/j.gde.2017.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Vincow ES, et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y., Dawson V. L., Dawson T. M. Animal Models of Parkinson's Disease: Vertebrate Genetics. Cold Spring Harbor Perspectives in Medicine. 2012;2(10):a009324–a009324. doi: 10.1101/cshperspect.a009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pickrell AM, et al. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron. 2015;87:371–381. doi: 10.1016/j.neuron.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sliter DA, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Martin M, Hiroyasu A, Guzman RM, Roberts SA, Goodman AG. Analysis of Drosophila STING Reveals an Evolutionarily Conserved Antimicrobial Function. Cell Rep. 2018;23:3537–3550 e3536. doi: 10.1016/j.celrep.2018.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goto A, et al. The Kinase IKKbeta Regulates a STING- and NF-kappaB-Dependent Antiviral Response Pathway in Drosophila. Immunity. 2018;49:225–234 e224. doi: 10.1016/j.immuni.2018.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, et al. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe. 2018;24:57–68 e53. doi: 10.1016/j.chom.2018.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai, H. et al. 2′3′-cGAMP triggers a STING and NF-κB dependent broad antiviral response in Drosophila. bioRxiv, 852319, 10.1101/852319 (2019). [DOI] [PubMed]
- 15.Kounatidis I, et al. NF-kappaB Immunity in the Brain Determines Fly Lifespan in Healthy Aging and Age-Related Neurodegeneration. Cell Rep. 2017;19:836–848. doi: 10.1016/j.celrep.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ. Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum Mol Genet. 2005;14:799–811. doi: 10.1093/hmg/ddi074. [DOI] [PubMed] [Google Scholar]
- 17.Tufi R, et al. Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson’s disease. Nature cell biology. 2014;16:157–166. doi: 10.1038/ncb2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poole AC, et al. The PINK1/Parkin pathway regulates mitochondrial morphology. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vos M, et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science (New York, N.Y. 2012;336:1306–1310. doi: 10.1126/science.1218632. [DOI] [PubMed] [Google Scholar]
- 20.Biosa A, et al. Superoxide dismutating molecules rescue the toxic effects of PINK1 and parkin loss. Hum Mol Genet. 2018;27:1618–1629. doi: 10.1093/hmg/ddy069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bingol B, et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 22.Clark IE, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 23.Greene JC, et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park J, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 25.Whitworth AJ, et al. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc Natl Acad Sci USA. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hewitt VL, Whitworth AJ. Mechanisms of Parkinson’s Disease: Lessons from Drosophila. Curr Top Dev Biol. 2017;121:173–200. doi: 10.1016/bs.ctdb.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Andreazza S, et al. Mitochondrially-targeted APOBEC1 is a potent mtDNA mutator affecting mitochondrial function and organismal fitness in Drosophila. Nat Commun. 2019;10:3280. doi: 10.1038/s41467-019-10857-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matheoud D, et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(−/−) mice. Nature. 2019;571:565–569. doi: 10.1038/s41586-019-1405-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data that support the findings of this study are available on reasonable request to the corresponding author. The contributing authors declare that all relevant data are included in the paper.