

Abstract
Fluorine labelling represents one promising approach to study proteins in their native environment due to efficient suppressing of background signals. Here, we systematically probe inherent thermodynamic and structural characteristics of the Cold shock protein B from Bacillus subtilis (BsCspB) upon fluorine labelling. A sophisticated combination of fluorescence and NMR experiments has been applied to elucidate potential perturbations due to insertion of fluorine into the protein. We show that single fluorine labelling of phenylalanine or tryptophan residues has neither significant impact on thermodynamic stability nor on folding kinetics compared to wild type BsCspB. Structure determination of fluorinated phenylalanine and tryptophan labelled BsCspB using X-ray crystallography reveals no displacements even for the orientation of fluorinated aromatic side chains in comparison to wild type BsCspB. Hence we propose that single fluorinated phenylalanine and tryptophan residues used for protein labelling may serve as ideal probes to reliably characterize inherent features of proteins that are present in a highly biological context like the cell.
Subject terms: Biophysical chemistry, X-ray crystallography, NMR spectroscopy
Introduction
Proteins are key operating elements of complex biological systems, such as cells. These macromolecules control a multiplicity of chemical processes and are a central characteristic of living systems1. The determination of the three-dimensional structure of a protein and the identification of its potential interacting partners is crucial to understand the function of these macromolecules in biological systems. However, most studies aiming to unravel the intracellular functionality of proteins are performed in vitro, using dilute experimental conditions that are distant from the crowded cellular environments that proteins experience in living organisms. The total intracellular macromolecular concentration can be estimated with up to about 400 g/l2,3 whereas individual proteins are often present in tiny amounts. Thus, performing protein studies using in vitro conditions neglects the intracellular environment and does not account for a variety of effects like e.g. crowding4, confinement5 or quinary interactions6. In the recent years, significant progress has been made on the study of proteins in native cellular environments. In this regard, high-resolution NMR spectroscopy is a promising technique that offers indispensable insights into the structure, dynamics, and stability of the protein under study in an intracellular context7–9. The specific labelling of proteins using fluorine10,11 may represent a successful strategy to perform in cellula NMR experiments. Once the protein has been successfully labelled using fluorine, the determination of kinetic and thermodynamic parameters (such as the change in enthalpy, ΔH, the change in heat capacity, ΔCP, upon protein unfolding, and the folding and unfolding rate constants) is possible even using cell-like conditions12–14. Thus, the labelling of proteins using fluorine allows conducting a broad variety of NMR spectroscopic experiments focusing on 19F resonances. This approach offers several advantages and simplifications compared to experiments commonly used in biomolecular NMR spectroscopy that focus on 1H, 13C, and 15N nuclei14,15. In this respect, fluorine has a natural abundance of 100%, a high sensitivity (83% compared to 1H), a large range of chemical shifts and no natural occurrence in proteins enabling to efficiently suppress undesired background signals. Thereby, structural, dynamical, and functional protein information can be obtained at atomic resolution in complex biological environments, even if the protein of interest is only present in a relatively low concentration12,15,16. We do not conceal that fluorine possesses large chemical shift anisotropy17 which may potentially impede assignment and interpretation of fluorine NMR spectra. However, as leading examples of the applicability of 19F-derivatized proteins, fluorotryptophan has been incorporated into the carbohydrate binding protein lectin from Ralstonia solanacearum enabling monitoring its interaction with ligands at atomic detail by X-ray crystallography18, whereas 19F NMR spectroscopy has been successfully applied to reveal the exchange dynamics present in antibody-antigen binding, an information that is not accessible using X-ray crystallography19. Furthermore, selective fluorine-labelling has been successfully used to derive distance restraints for a protein based on 19F paramagnetic relaxation enhancement experiments20 whereas the interaction sites present in protein-ligand complexes have been elucidated by applying 19F pseudocontact shift analysis21. An elegant combination of X-ray crystallography, NMR spectroscopy, and MD simulations has been recently applied to unravel the distribution of conformational states along the reaction pathway of a homodimeric enzyme22. Pomerantz and co-workers have successfully utilized fluoroaromatic amino acids for the implementation into proteins to report on the structure-activity relationship23 focusing on screening of small molecule-protein interactions based on protein-observed fluorine NMR spectroscopy24. Moreover, incorporation of difluorotyrosine has been successfully employed enabling to report on tyrosine phosphorylation25 or to probe distinct conformational states of a protein which are related to signalling by applying 19F NMR spectroscopy26. In a recent study, an NMR based comparison of the incorporation of 2- and 3-fluorotyrosine into a KIX domain has been presented27.
The outstanding question in the application of 19F-NMR based methodologies is how much does the 19F-modification impact the inherent properties of the protein under study, particularly its atomic three-dimensional structure, conformational dynamics, and its overall thermodynamic stability. Addressing this question is of high importance as the fluorine-labelled protein variant, and not the wild type, is used in in cellula NMR spectroscopy to report on the native structural and dynamical features of proteins in vivo. In this context, it has been shown that extensively fluorinated amino acids can be particularly effective in increasing protein stability28 due to the increase in buried hydrophobic surface area as identified in structures solved by X-ray crystallography29. Kitevski-LeBlanc and co-workers have shown that 19F enrichment in fluoro-phenyalanine labelled calmodulin results in an increasing protein disorder which can be diminished by a decreased level of fluorination30. Other work has shown that the structural integrity of a small single domain protein is conserved when one fluoro-phenylalanine is incorporated31 and that fluoro-tryptophan labelling of various sites in fluoroacetate dehalogenase does not modify its three-dimensional structural characteristics compared to the wild type22.
In previous work, we have used auxotrophic E. coli strains to incorporate fluorinated phenylalanine (Phe) or tryptophan (Trp) amino acid residues into the Cold shock protein B from Bacillus subtilis (BsCspB). Within these residues, we used different sites for the incorporation of the fluorine atoms, generating a total of three Phe and three Trp modified amino acids (Fig. 1)32. Using this tool kit, we were then able to successfully prepare six different 19F-labelled BsCspB variants (2-19F-Phe-, 3-19F-Phe-, 4-19F-Phe-, 4-19F-Trp-, 5-19F-Trp- and 6-19F-Trp-BsCpB) at a milligram scale and in high purity enabling high resolution one-dimensional 1H, 19F and two-dimensional 1H-15N correlation NMR spectroscopy of the samples mentioned above32. BsCspB is a relatively small protein of 67 amino acids length that belongs to the cold shock protein family33–35. It folds into five beta-strands that form a beta-barrel structure36,37, which binds single-stranded DNA and RNA38,39. Notably, BsCspB shows an overall thermodynamic stability of about ΔG° = 10 kJ mol−1 at T = 298 K and possesses fast unfolding, ku, and refolding, kf, rate constants of about ku = 12 s−1 and kf = 1070 s−1, respectively33. Here, we focus on the precise determination of thermodynamic and kinetic parameters of all six fluorine-labelled protein variants in comparison to wild type BsCspB. A combination of fluorescence, kinetic stopped flow, NMR spectroscopy, and X-ray crystallography has been applied to obtain an integrated understanding of potential effects of fluorine-labelling in proteins. Our findings show that fluorine-labelling of proteins utilizing singly modified amino acids like Phe or Trp does not cause a detectable alteration of thermodynamic and structural properties of BsCspB. The present study closes an important gap in the basic characterization of fluorine-labelled proteins and underlines that this methodology may serve as an optimal tool to study proteins in their native complex biological environment applying high-resolution NMR spectroscopy.
Figure 1.
Numbering of sites used for single fluorine labelling in 2-19F-, 3-19F-, and 4-19F-phenylalanine (left) and 4-19F-, 5-19F-, and 6-19F-tryptophan (right). The structures have been created by using ChemDraw18 (www.perkinelmer.com).
Results and Discussion
Thermodynamic stability probed by fluorescence in equilibrium
Intrinsic fluorescence spectroscopy has been applied to probe the overall thermodynamic stability of the six differently fluorine-labelled variants of BsCspB. For this purpose, folded-to-unfolding transitions of BsCspB samples were chemically induced with increasing amounts of urea and monitored using fluorescence spectroscopy in equilibrium. Specifically, fluorescence emission spectra of wild type and 2-19F-Phe-, 3-19F-Phe-, 4-19F-Phe-, 4-19F-Trp-, 5-19F-Trp-, and 6-19F-Trp-labelled BsCspB were measured in urea concentrations ranging from curea = 0 M to curea = 8 M. Note that all seven intrinsic Phe residues Phe9, Phe15, Phe17, Phe27, Phe30, Phe38, and Phe45 present in BsCspB have been equally fluorine-labelled in 2-19F-Phe-, 3-19F-Phe- or 4-19F-Phe-modified protein samples32 (Fig. S1A) whereas 4-19F-Trp-, 5-19F-Trp-, and 6-19F-Trp-BsCspB possess Trp8 as single site of the modification (Fig. S1B). Fluorescence spectra showed that, upon unfolding, the maximal fluorescence emission intensity decreased (Figs. S2A–S8A) and shifted to significantly larger wavelengths (Figs. S2B–S7B) for both wild type and fluorine-labelled variants. The signal shifting to larger wavelengths regarding fluorescence emission of tryptophans is consistent with the aromatic group becoming more exposed to the polar solvent upon unfolding of the protein. The properties observed here for all six differently fluorine-labelled variants of BsCspB regarding fluorescence emission upon chemical unfolding have been also reported for the wild type33. Solely 6-19F-Trp-BsCspB does not show a dependence of the maximum wavelength in fluorescence emission, 𝜆max, on the concentration of urea (Fig. S8B) but preserving a decrease in fluorescence emission intensity by an increasing concentration of urea (Fig. S8A). The lack in the increase of the maximum wavelength in fluorescence emission observed for the 6-19F-Trp-labelled variant can be anticipated as 𝜆max is about 360 nm already in the absence of urea (Fig. S8B). However, 𝜆max observed for 6-19F-Trp-BsCspB has been also approached for remaining variants of BsCspB at high concentrations of urea (Figs. S2B–S7B). The fluorescence emission intensity is lowest for 4-19F-Trp-BsCspB among all probed fluorine-labelled protein variants (Fig. S6A). This observation is based on the inherent low fluorescence quantum yield of 4-19F-Trp40 which is about 100 times lower compared to Trp, 5-19F-Trp, and 6-19F-Trp (Fig. S9). For fluorescence data analysis the intensity emission averaged wavelength, < 𝜆 >, has been determined (Fig. 2). This procedure is based on two rationales. Firstly, the maximum of fluorescence emission of BsCspB variants shifts by an increasing concentration of urea to larger wavelengths (see above). Secondly, the maximum of fluorescence emission is dependent on the site used for inserting fluorine into free tryptophan residues (Fig. S9). Thus the free form of native Trp shows a maximum in fluorescence emission at 𝜆max = 353 nm, free 5-19F-Trp at 𝜆max = 360 nm, free 6-19F-Trp at 𝜆max = 360 nm, and free 4-19F-Trp 𝜆max = 376 nm using 𝜆 = 280 nm for excitation going along with observations on free fluorotryptophans performed before using 𝜆 = 295 nm for excitation40.
Figure 2.
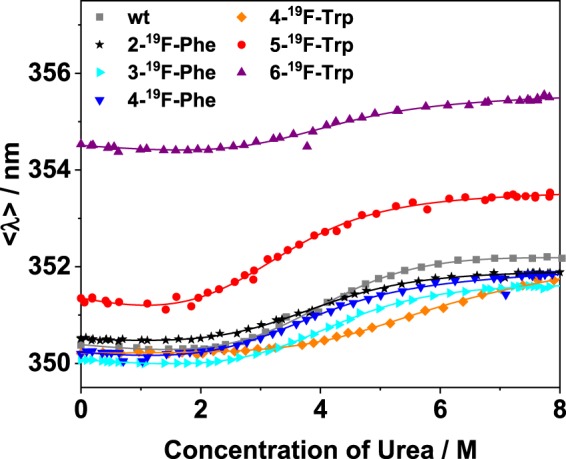
Dependence of average emission wavelength on the concentration of urea obtained from fluorescence spectra for all protein variants probed in this study (rectangles represent wild type protein, stars represent 2-19F-Phe-, triangles with the tip to right represent 3-19F-Phe-, triangles with the tip to bottom represent 4-19F-Phe-, diamonds represent 4-19F-Trp-, circles represent 5-19F-Trp-, and triangles with the tip to top represent 6-19F-Trp-variant). The straight lines represent best fits of Eq. (3) to the experimental data sharing the same value for the cooperativity of unfolding, m. Results of this fitting procedure are summarized in Table 1.
The intensity averaged emission wavelength < λ > is calculated using Eq. (1)
| 1 |
where Ii is the intensity of fluorescence emission at wavelength λi41.
This method of fluorescence data analysis accounts for both potential changes in the shape of acquired fluorescence emission spectra and a potential shift in wavelength of the maximum in fluorescence emission, respectively. Note that measuring the refractive index of the buffer accounts for the precise determination of the concentration of urea which has been individually used (Eq. 2).
Applying a two state folding-to-unfolding model42 (Eq. 3) to the experimentally obtained fluorescence data yields the free energy against unfolding, ΔG°, of wild type BsCspB and all six variants differing in the site used for fluorine labelling. The cooperativity of protein unfolding, m, is then used as a global fitting parameter enabling a direct comparison of the overall thermodynamic stability of all seven different protein variants probed in the present study. This approach results in m = −2.9 ± 0.1 kJ/(mol M), which agrees well with m values which have been reported for BsCspB before, m = −3.2 ± 0.1 kJ/(mol M)33,43. The free energy of protein unfolding ranges between ΔG° = 9.0 ± 0.5 kJ/mol obtained for 5-19F-Trp-BsCspB and ΔG° = 12.7 ± 1.1 kJ/mol for 4-19F-Trp-BsCspB (Table 1). Thus, the overall thermodynamic stability of fluorine-labelled BsCspB variants reproduces the stability of wild type BsCspB, determined here using the same type of data analysis (ΔG° = 11.1 ± 0.5 kJ/mol) (Table 1). Note that linear fitting of fluorescence emission intensities characterizing the transition region between folded and unfolded state has been also performed for all variants of BsCspB44 (Fig. S10, Table S1). This procedure ensures that the determination of the overall thermodynamic stability ΔG° is not primarily influenced by missing baselines representing the folded as well as the unfolded state as it has been shown by Alexander and Pace44. Notably, both approaches used for the analysis of fluorescence emission intensities yield consistent results for ΔG° (Table S1).
Table 1.
Analysis of equilibrium unfolding of wild type and fluorine-labelled variants of BsCspB using fluorescence spectroscopy (Fig. 2) to determine the overall thermodynamic stability, ΔG0. The cooperativity of protein unfolding has been used as global parameter in Eq. (3) taking all seven folded-to-unfolding transitions into account and has been determined to m = −2.9 ± 0.1 kJ/(mol M).
| Protein | ΔG0/kJ/mol | CM/M |
|---|---|---|
| wt BsCspB | 11.1 ± 0.5 | 3.8 ± 0.2 |
| 2-19F-Phe-BsCspB | 10.0 ± 0.7 | 3.5 ± 0.2 |
| 3-19F-Phe-BsCspB | 10.5 ± 0.6 | 3.6 ± 0.2 |
| 4-19F-Phe-BsCspB | 9.6 ± 0.6 | 3.3 ± 0.2 |
| 4-19F-Trp-BsCspB | 12.7 ± 1.1 | 4.4 ± 0.4 |
| 5-19F-Trp-BsCspB | 9.0 ± 0.5 | 3.1 ± 0.2 |
| 6-19F-Trp-BsCspB | 11.0 ± 0.7 | 3.8 ± 0.2 |
The midpoint of folded-to-unfolding transition, CM, has been calculated using CM = ΔG0/m.
Summing up, fluorine labelling of either tryptophan or phenylalanine residues does not have a major effect on the overall thermodynamic stability of cold shock protein B when monitoring fluorescence emission dependent on an increasing concentration of urea. The overall thermodynamic stability determined for 4-19F-Trp-BsCspB is highest comparing all variants probed in the present study (Table 1) and deviates in ΔG° by 1.6 kJ/mol from wild type BsCspB. This property of 4-19F-Trp-BsCspB has been one rationale to elucidate the structural features of this fluorine-labelled variant of BsCspB by using X-ray crystallography in more detail. Focusing on phenylalanine modified variants, 4-19F-Phe-BsCspB deviates most in ΔG° from wild type BsCspB by 1.5 kJ/mol. This property of 4-19F-Phe-BsCspB has been one rationale for the crystallization of this variant enabling to obtain atomically resolved information (see below).
Thermodynamic stability probed by NMR spectroscopy
The characterization of the thermodynamic stability of differently fluorine-labelled variants of BsCspB has been expanded to folded-to-unfolding transitions induced by increasing the temperature. The thermal denaturation of the six 19F-labelled BsCspB variants and wild type BsCspB has been monitored by one-dimensional 1H (Fig. S11) and 19F NMR spectroscopy (holds for 4-19F-Phe-BsCspB) to independently verify the experimental results obtained for chemical denaturation monitored using intrinsic fluorescence emission (see above). Advantageously, NMR spectroscopy provides direct spectroscopic information about the folded-to-unfolding transition of the entire protein under investigation whereas fluorescence spectroscopy operates as a local reporter of aromatic residues only. Thus, one-dimensional 1H and 19F NMR spectra have been acquired for different variants of BsCspB including the wild type in the temperature range T = 291 K to T = 330 K (Figs. S11A,B–S17A,B). The fitting of Eq. (4) to the experimentally obtained data representing the fraction of unfolded protein gives access to the change in heat capacity, ΔCP, the change in enthalpy, ΔH, and the temperature midpoint characterizing the folded-to-unfolding transition, TM, for all variants of BsCspB which have been probed here. Note that the change in heat capacity has been fixed to ΔCP = 5.8 kJ/(mol K) here as it has been specifically determined for BsCspB using 1H NMR spectroscopy before relying on both heat and cold denaturing45. The change in enthalpy has been used as a global parameter for all variants of BsCspB when fitting Eq. (4) to the experimental data acquired by one-dimensional 1H spectroscopy whereas TM has been individually determined for all variants (Fig. 3A, Table 2). This procedure leads to ΔH = 197 ± 2 kJ/mol and values for TM ranging between TM = 315.6 K (2-19F-Phe-BsCspB) and TM = 320.8 K (4-19F-Trp-BsCspB). Note that wild type BsCspB shows TM = 316.8 K indicating that there exists no significant difference in thermodynamic stability to the variously fluorine-labelled variants of BsCspB. This result independently verifies the observation made for the chemical denaturation of BsCspB variants presented before. The fitting of Eq. (4) to the data obtained for 4-19F-Phe-BsCspB using one-dimensional 19F NMR spectroscopy yield TM = 318.4 K and ΔH = 176 ± 6 kJ/mol (Fig. 3B) confirming the thermodynamic analysis done by using 1H NMR spectroscopic data which has illuminated TM = 318.5 K (Table 2). Note that a precise thermodynamic analysis of 19F NMR detected folding-to-unfolding transitions of 2-19F-Phe-, 3-19F-Phe-, 4-19F-Trp-, 5-19F-Trp-, and 6-19F-Trp-BsCspB has been prohibited due to spectral indistinguishability of signals representing either the native state or the unfolded ensemble of the protein under study.
Figure 3.

Folded-to-unfolding transitions monitored by using one-dimensional 1H (A) and 19F (B) NMR spectroscopy. The thermal denaturation has been probed for wild type (rectangles), 2-19F-Phe- (stars), 3-19F-Phe- (triangles with the tip to right), 4-19F-Phe- (triangles with the tip to bottom), 4-19F-Trp- (diamonds), 5-19F-Trp- (circles), and 6-19F-Trp-variant (triangles with the tip to top). The straight lines represent best fits of Eq. (4) to the experimental data sharing the same value for the change in enthalpy, ΔH. Results of this fitting procedure are summarized in Table 2.
Table 2.
Analysis of equilibrium unfolding of wild type and fluorine-labelled variants of BsCspB using one-dimensional 1H and 19F NMR spectroscopy to determine the midpoint of folded-to-unfolding transition, TM.
| Protein | TM/K |
|---|---|
| wt BsCspB | 316.8 ± 0.2 |
| 2-19F-Phe-BsCspB | 315.6 ± 0.1 |
| 3-19F-Phe-BsCspB | 318.7 ± 0.1 |
| 4-19F-Phe-BsCspB | 318.5 ± 0.1 |
| 4-19F-Trp-BsCspB | 320.8 ± 0.1 |
| 5-19F-Trp-BsCspB | 317.1 ± 0.1 |
| 6-19F-Trp-BsCspB | 316.6 ± 0.1 |
| 4-19F-Phe-BsCspB (19F) | 318.4 ± 0.2 |
The change in enthalpy of protein unfolding at TM, ΔH(TM), has been used as global parameter in Eq. (4) taking all seven 1H-detected folded-to-unfolding transitions into account and has been determined to ΔH(TM) = 197 ± 2 kJ/mol. For 19F detection, the change in enthalpy has been determined to ΔH(TM) = 176 ± 6 kJ/mol. The change in heat capacity has been fixed to ΔCP = 5.8 kJ/(mol K) as it has been specifically determined for BsCspB using NMR spectroscopy before relying on both heat and cold denaturing45.
The thermodynamic parameters calculated using thermal denaturation probed with NMR spectroscopy (Table 2) show excellent agreement with those derived by chemical denaturation monitored using intrinsic fluorescence (Table 1). Thus, 4-19F-Trp-BsCspB shows a small increase in overall thermodynamic stability (approx. 4 Kelvin) compared to wild type BsCspB whereas 2-19F-Phe-BsCspB and 5-19F-Trp-BsCspB variants show a small decrease or uniformity in ΔG° (Table 1) and TM (Table 2), respectively.
Unfolding and refolding kinetics probed by kinetic stopped-flow fluorescence
The thermodynamic analysis of all six differently fluorine-labelled variants has been further extended to kinetic experiments using a stopped-flow fluorescence device. This setup enables to monitor the change in fluorescence on a millisecond time scale induced by rapid mixing of two solutions representing either folding or unfolding conditions46. A monoexponential function has been applied to obtain the apparent rate constant, kobs, to account for refolding (Fig. S18) and unfolding (Fig. S19) kinetics of fluorine-labelled variants and wild type BsCspB. Having values for kobs representing final concentrations of urea in the mixing cell ranging between curea = 2.3 M and curea = 7.3 M in hands, Eq. (5) has been applied to obtain rate constants for refolding, kf, and unfolding, ku, in absence of any denaturant, respectively. Moreover, Eq. (5) enables the determination of the slope of the unfolding, mu, and the refolding limb, mf, respectively. The fitting of Eq. (5) to the experimental data has been performed assuming a global value for the cooperativity of folding, m = mf + mu, for all differently fluorine-labelled variants of BsCspB including wild type protein. The Chevron plot analysis reveals a two-state folding process for all variants of BsCspB which have been kinetically probed (Fig. 4). The cooperativity of folding has been determined to m = −2.8 ± 0.8 kJ/(mol M) matching the value of m = −2.9 ± 0.1 kJ/(mol M) which has been independently determined in the present study by monitoring the folded-to-unfolding transition of all variants of BsCspB using equilibrium fluorescence (Fig. 2, Table 1). Comparing the kinetic rate constants for unfolding of fluorine-labelled variants of BsCspB (31 s−1 ≤ ku ≤ 57 s−1) does not reveal a significant difference to wild type BsCspB (ku = 40 ± 1 s−1, Table 3). We note that refolding kinetics for differently fluorine-labelled variants of BsCspB using curea < 2.3 M could not be reliably acquired. However, obtaining refolding rate constants for all differently fluorine-labelled variants of BsCspB by applying Eq. (5) enables a comparison among individual kf values in a qualitative manner as all kinetic data have been analyzed in the same way. Thus, the refolding rate constants which have been determined for all fluorine-labelled variants of BsCspB are slightly higher (except for 5-19F-Trp-BsCspB) compared to kf = 1050 ± 20 s−1 obtained for wild type BsCspB (Table 3). The determination of the rate constants accounting for refolding und unfolding enables to apply Eq. (6) to compute the difference in free energy, ΔG°, of all variants of BsCspB. As a result, these values are highly similar (Table 3) ranging between ΔG° = 6.4 ± 0.1 kJ/mol (5-19F-Trp-BsCspB) and ΔG° = 9.5 ± 0.1 kJ/mol (4-19F-Trp-BsCspB) covering the thermodynamic stability determined for wild type (ΔG° = 8.1 ± 0.1 kJ/mol). Comparing the values of ΔG° determined for all differently fluorine-labelled variants of BsCspB by applying a kinetic setup with the overall thermodynamic stability obtained in equilibrium using chemical (Fig. 2, Table 1) or thermal denaturation (Fig. 3, Table 2) reveals two main features. Firstly, the overall thermodynamic stability of BsCspB does not change significantly if fluorine has been attached either to all seven phenylalanine residues or to its single tryptophan residue. Secondly, it has been consistently shown by three independent experimental methods that 4-19F-Trp-BsCspB exhibits a small increase in ΔG° by about 1 kJ/mol and in TM by about 4 Kelvin whereas 5-19F-Trp-BsCspB shows a moderate decrease in ΔG° by about 1.5 kJ/mol and a conserved value of TM compared to wild type BsCspB. Focusing on fluorine labelled phenylalanine variants, 4-19F-Phe-BsCspB has a thermodynamic stability which is about 1 kJ/mol lower as the value for ΔG° which has been determined for 2-19F-Phe-BsCspB and 3-19F-Phe-BsCspB, respectively (Tables 1 and 3).
Figure 4.
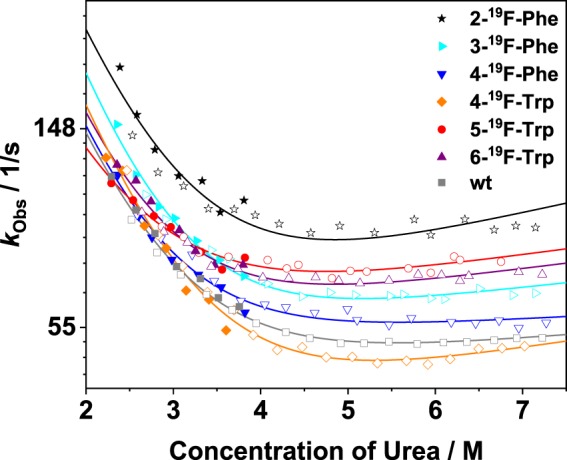
Dependence of the apparent rate constant on the concentration of urea characterizing unfolding (open symbols) and refolding (closed symbols) of fluorine-labelled variants of BsCspB (stars represent 2-19F-Phe-, triangles with the tip to right represent 3-19F-Phe-, triangles with the tip to bottom represent 4-19F-Phe-, diamonds represent 4-19F-Trp-, circles represent 5-19F-Trp-, and triangles with the tip to top represent 6-19F-Trp-variant), and wild type protein (represented using rectangles). The straight lines represent best fits of Eq. (5) to the experimental data sharing the same value for the cooperativity of folding, m = mf + mu. Results of this fitting procedure are summarized in Table 3.
Table 3.
Analysis of unfolding and refolding kinetics of wild type and fluorine-labelled variants of BsCspB using kinetic stopped-flow fluorescence.
| Protein | ku/s−1 | kf/s−1 | mu/kJ/(molM) | mf/kJ/(molM) | ΔG0/kJ/mol |
|---|---|---|---|---|---|
| wt BsCspB | 40 ± 1 | 1050 ± 20 | 0.1 | −2.9 | 8.1 ± 0.1 |
| 2-19F-Phe-BsCspB | 56 ± 2 | 2000 ± 100 | 0.2 | −3.0 | 8.9 ± 0.2 |
| 3-19F-Phe-BsCspB | 42 ± 1 | 1600 ± 40 | 0.2 | −3.0 | 9.0 ± 0.1 |
| 4-19F-Phe-BsCspB | 44 ± 1 | 1050 ± 30 | 0.1 | −2.9 | 7.9 ± 0.1 |
| 4-19F-Trp-BsCspB | 31 ± 1 | 1450 ± 30 | 0.2 | −3.0 | 9.5 ± 0.1 |
| 5-19F-Trp-BsCspB | 57 ± 1 | 770 ± 20 | 0.1 | −2.9 | 6.4 ± 0.1 |
| 6-19F-Trp-BsCspB | 48 ± 1 | 1160 ± 30 | 0.2 | −3.0 | 7.9 ± 0.1 |
We have extended the analysis of the kinetic data set to the information included in the amplitude of the monoexponential function following the refolding and unfolding of differently fluorine-labelled variants of BsCspB. Thus, we have observed that the final values of the kinetics reporting on refolding and unfolding of fluorine-labelled variants do not converge as illustrated by 4-19F-Phe-BsCspB (Fig. S20A,B). The refolding kinetics for all fluorine variants probed here show consistently lower amplitudes compared to unfolding kinetics (Fig. S20C–G). Note that the endpoint analysis performed for wild type BsCspB based on kinetic stopped-flow data shows an unfolding transition which is anticipated (Fig. S20H). Consequently, we have examined the feature of non-converging endpoints seen for refolding and unfolding of fluorine-labelled BsCspB variants by performing a set of control experiments. Firstly, 4-19F-Phe-BsCspB has been mixed with denaturing buffer leading to curea = 2.6 M. Furthermore, unfolded 4-19F-Phe-BsCspB present in curea = 7 M has been mixed with native buffer leading to curea = 2.6 M, too. The fluorescence emission of both unfolded and refolded 4-19F-Phe-BsCspB has been immediately measured. As a result, the fluorescence intensity representing refolded 4-19F-Phe-BsCspB is lower compared to the fluorescence intensity observed for unfolded 4-19F-Phe-BsCspB (Fig. S21A). Quantitatively, the fluorescence intensity monitoring unfolding of 4-19F-Phe-BsCspB is about 30% higher compared to the fluorescence intensity monitored for protein refolding (Fig. S21A). The higher fluorescence intensity observed for unfolding of 4-19F-Phe-BsCspB compared to refolding matches the difference seen in fluorescence amplitude observing folding kinetics of 4-19F-Phe-BsCspB independently acquired at the stopped-flow instrument (Fig. S20A). Secondly, potential slow unfolding or refolding of 4-19F-Phe-BsCspB taking place on a second-to-minute time scale cannot be observed as the fluorescence emission spectra monitored directly after mixing and after 30 minutes look identical (Fig. S21A). Thirdly, monitoring refolding and unfolding of wild type BsCspB by using identical experimental conditions reveals a different result. Both refolding and unfolding of wild type BsCspB observed in curea = 2.6 M lead to almost identical profiles in fluorescence emission intensity (Fig. S22). Fourthly, we have applied high-resolution NMR spectroscopy to further unravel the origin of the gap present in amplitude analysis of the kinetic data observed for fluorine-labelled BsCspB variants (Fig. S21B). Thus, the reversibility of folded-to-unfolding reaction of 4-19F-Phe-BsCspB has been quantitatively probed by acquiring one-dimensional 1H NMR data on both unfolded and refolded samples, respectively (Fig. S21B). The fraction of native 4-19F-Phe-BsCspB, fN, can be determined by calculating the ratio fN = IN/(IN + IN+U) using integrals representing native, IN, as native and unfolded, IN+U, signals of BsCspB. The analysis of both the unfolding and the refolding reaction of 4-19F-Phe-BsCspB leads to almost identical values for fN, namely fN = 0.191 and fN = 0.189, respectively. This result shows that 4-19F-Phe-BsCspB folds fully reversibly and that there is no apparent long-term folding rate constant additionally present which may explain the gap observed in the amplitude analysis of the kinetic data. Taken these results together, we conclude that adding singly fluorinated tryptophan or phenylalanines to BsCspB inherently changes the properties in tryptophan fluorescence emission of this protein. This change in fluorescence emission can be observed when comparing fluorine-labelled BsCspB which has been refolded with the unfolded counterpart. Contrary, applying high-resolution NMR spectroscopy does not reveal structural differences in fluorine-labelled BsCspB when comparing refolded with unfolded protein which has been modified by fluorotryptophan or fluorophenylalanines.
Structure determination of 4-19F-Phe-BsCspB and 4-19F-Trp-BsCspB by X-ray crystallography
Probing fluorine-labelled variants of BsCspB using fluorescence and NMR spectroscopy has independently shown that 4-19F-Trp-BsCspB has a slightly increased thermodynamic stability, whereas ΔG° of 4-19F-Phe-BsCspB is slightly decreased compared to wild type protein. To investigate potential structural changes in 4-19F-Trp-BsCspB and 4-19F-Phe-BsCspB, we have elucidated their atomic structures using X-ray crystallography to 2.05 Å and 2.1 Å resolution, respectively (Fig. 5, Table S2). The samples crystallized in a closely related crystallographic lattice, containing one single molecular copy of modified BsCspB in the asymmetric unit and sharing a same space group symmetry. The global RMSD for all Cα atoms for the 4-19F-Trp-BsCspB and 4-19F-Phe-BsCspB structures calculated in this work is 0.23 Å, indicating that the fold trace of the two variants is virtually identical. A comparison of the variants here studied with a previously reported crystal structure of wild type BsCspB elucidated in a different crystallographic symmetry (PDB ID: 1CSP) revealed RMSDCα values of 0.52 Å and 0.37 Å for 4-19F-Trp-BsCspB and 4-19F-Phe-BsCspB, respectively (Fig. 5). The excellent agreement across all structures confirmed that 19F derivatization did not induce any detectable alterations of the fold. Furthermore, wild type and 19F-derivative structures displayed identical conformational rotamers for the modified Trp and Phe residues, indicating that local structural distortions had also not taken place (Fig. 5). When comparing wild type and derivatives, the RMSD values for all atoms in the modified residues were: 0.48 Å for residue Trp8, 0.35 Å for Phe9, 0.30 Å for Phe15, 0.15 Å for Phe17, 0.43 Å for Phe27, 0.2 Å for Phe30, 0.65 Å for Phe38, and 0.3 Å for Phe49. Based on this excellent agreement, we conclude that the introduction of the 19F-labels in BsCspB did not induce structural changes of significance even when seven residues were modified.
Figure 5.

Three-dimensional structures of 19F-derivatized BsCspB. (A) The crystal structures of 4-19F-Trp-BsCspB (colored in orange) and 4-19F-Phe-BsCspB (colored in blue) are shown superimposed onto wild type BsCspB (pdb code 1NMG, colored in grey). The side chains of 19F-derivatized residues in this study are displayed and labelled according to their sequence numbering. (B,C) Electron density maps of residues 19F-Trp8 (B) and 19F-Phe49 (as representative) are shown. (2mFobs − DFcalc)αcalc maps contoured at 1σ are colored in grey. (mFobs − DFcalc) maps calculated using phases from refined models with omitted 19F-derivatized residues are shown in green color, contoured at 3σ. The structures have been created by using Chimera59, version 1.14 (www.cgl.ucsf.edu/chimera).
Conclusion
Synergistic combination of equilibrium and kinetic fluorescence with NMR spectroscopy and X-ray crystallography
We have presented an integrated approach to structurally and dynamically characterize fluorine-labelled proteins using a repertoire of orthogonal biophysical methodologies. Fluorescence experiments conducted in equilibrium following the folded-to-unfolding reaction of fluorine-labelled BsCspB induced by chemical denaturation as well as monitoring the refolding and unfolding of these samples by fluorescence emission in a time-resolved manner elucidated only small differences in overall thermodynamic stability. This conservation in thermodynamic stability regarding wild type behaviour could be confirmed by monitoring the thermal denaturation of fluorine-labelled BsCspB using one-dimensional 1H and 19F NMR spectroscopy. The crystallization and structure determination of fluorine modified BsCspB corroborated the findings presented above by showing an almost perfect overlay between wild type and 19F-Phe or 19F-Trp modified protein variants taking both backbone and side chain atoms into account. To our knowledge, this represents the first three-dimensional structure determination of a fluorophenylalanine-labelled protein possessing more than a single fluorination site which has been reported so far. Note that the solution structure of a fluorinated side chain labelled villin headpiece subdomain comprising 35 residues has been presented before31. Thus, the coherent characterization of fluorine-labelled BsCspB done here consistently show only mild effects protein labelling causes on structural and dynamic properties by using singly fluorinated amino acids, even when multiple sites in the protein have been used for modification. The integrated approach for the determination of the impact fluorine-labelling has on proteins converges to the main finding. In fact, incorporation of singly fluorinated Trp or Phe into BsCspB induces, if at all, only slight changes in structural and dynamic parameters. Interestingly, these slight changes are independent of the number and sites of fluorine atoms which have been inserted into BsCspB: one fluorine atom used for 19F-Trp labelling (sequence position 8) or seven fluorine atoms used for 19F-Phe labelling (sequence positions 9, 15, 17, 27, 30, 38, and 49). This modest impact of protein labelling using fluorine has been partially presented for other proteins but only by using either a limited number of experimental techniques or a reduced number of different fluorinated amino acids which have been used for protein labelling. In this regard, a combination of CD and NMR spectroscopy showed that the three-dimensional structure and thermodynamic stability of GB1 protein is not significantly affected when 5-19F-Trp has been used for labelling47. Moreover, the structure of 5-19F-Trp- and 6-19F-Trp-labelled annexin V has been determined by X-ray crystallography48 elucidating only minimal changes in the local protein geometry compared to non-labelled protein and slight changes in thermal melting observed by CD spectroscopy which are on the same order as elucidated here for fluorine-labelled BsCspB. Similarly, the crystal structure of 5-19F-Trp-labelled triosephosphate isomerase showed no discrepancies in local and global structural properties comparing labelled with non-labelled protein49, the same property has been reported for anthrax protective antigen indicating that 5-19F-Trp-labelling minimally perturb structural properties seen for wild type protein50 or for fluoroacetate dehalogenase utilizing also 5-19F-Trp-labelling22. Specialized studies have focused e.g. on relaxation properties of fluorinated amino acids in free and protein-bound form51, general NMR parameters of isolated fluorine-labelled amino acids52 or the application of homonuclear 19F-19F EXSY NMR on a fluorine-labelled receptor protein53. We are aware that we have solely probed one protein in this study. However, the experimental design has been done following a highly systematic strategy by incorporating six different fluorinated amino acids into BsCspB. Such fluorine-labelled variants of BsCspB have been subsequently probed applying various orthogonal biophysical techniques which led to a consistent result. For this reason, we believe that the present study closes an important gap in the characterization of fluorine-labelled proteins by obtaining a convergent view on the impact that the insertion of fluorine into proteins has. We propose that single fluorine-labelled tryptophan and phenylalanine residues may serve as ideal candidates for the incorporation into proteins enabling experiments to understand protein performance in highly biological contexts like cell lysates or even in cellula.
Methods
Protein expression and purification
The six variants of the cold shock proteins B from Bacillus subtilis differing in the position of the fluorine (2-19F-Phe-, 3-19F-Phe-, 4-19F-Phe-, 4-19F-Trp-, 5-19F-Trp-, and 6-19F-Trp-BsCpB, Figs. 1 and S1) were expressed in E. coli cells by using pET24a CspB and pAR1219 vectors as described previously32 (DSMZ 12779 strain was used for the three Phe variants whereas the strain CAG 18455 7371 was used for the three Trp variants). Subsequently, an established protocol for protein purification was applied32. The concentration, c, of the purified proteins was determined by measuring the absorbance at λ = 280 nm, A280, of the protein solution in a d = 1 cm long cuvette in an UV/Vis spectrometer (Agilent 8453 UV-visible Spectroscopy System, Agilent Technologies) employing extinction coefficients of ε°280 = 2705 M−1 cm−1 (4-19F-Trp BsCspB), ε280 = 2887 M−1 cm−1 (5-19F-Trp BsCspB), ε280 = 2575 M−1 cm−1 (6-19F-Trp BsCspB), and ε280 = 5800 M−1 cm−1 (2-19F-Phe BsCspB, 3-19F-Phe BsCspB, 4-19F-Phe BsCspB, and wild type BsCspB)32. The Lambert Beer law, A280 = c*d*ε280, was then applied to the determination of c. Fluorine labelling efficiency can be specified with >95%32.
Fluorescence spectroscopy in steady state
Fluorescence experiments performed in steady state were conducted on a FP-8500 Spectrofluorometer (Jasco). The final concentration of the protein was set to c = 1 µM in all experiments. Samples were thermally equilibrated for at least 30 minutes and measured under stirring condition at a temperature T = 298 K, using 20 mM sodiumcacodylate, pH 7.0. The individual folded-to-unfolding transitions of BsCspB variants were monitored between c = 0 M and c = 8 M urea applying 34 data points. Fluorescence spectra were acquired as triplicates in the wavelength (λ) range 290 nm to 400 nm in steps of 0.5 nm by using a wavelength for excitation of λ = 280 nm. The concentration of urea, curea, was determined by measuring the refractive index (Eq. (2))54.
| 2 |
where Δn reflects the difference in the refractive index of the buffer solution taking absence and presence of urea into account.
All equilibrium protein folded-to-unfolding transitions were background subtracted and measured in duplicate.
The Eq. (3) was applied to determine the difference in free energy between the folded and the unfolded state of BsCspB, ΔG0, and the cooperativity of protein unfolding, m
| 3 |
where represents the intensity averaged emission wavelength41, gN/U and mN/U account for the baselines of the folded and the unfolded state, R is the universal gas constant, curea is the concentration of urea, and T is the absolute temperature42.
NMR spectroscopy
All one-dimensional 1H NMR spectra were acquired at protein concentrations ranging between c = 200 µM and c = 650 µM and measured in 20 mM sodiumcacodylate containing 90% H2O and 10% D2O (v/v) at pH = 7.0. NMR data were collected on an 800 MHz Bruker Avance NEO NMR spectrometer equipped with a TCI cryogenically cooled probe possessing a proton channel which allows tuning of the 19F resonance at ωL19F = 753 MHz. The proton resonance frequency of trimethylsilylproanoic acid (TMSP) was used for direct referencing of all 1H spectra. One-dimensional 19F NMR spectra were indirectly referenced by using the information obtained for referencing of protons. The processing of NMR data used TOPSPIN 4.0.3 software (Bruker Biospin, Germany).
The determination of the thermodynamic stability of the different variants of BsCspB was monitored by one-dimensional 1H- and 19F- NMR spectroscopy (holds for 4-19F-Phe-BsCspB) at different temperatures ranging between T = 291 K and T = 330 K. Two ranges differing in chemical shifts were used for the determination of the fraction of unfolded protein, fU, being present at different temperatures. The first range, IN, covers chemical shifts between 0.59 ppm and 0.14 ppm representing signals seen for the folded state. The second range, IN+U, covers chemical shifts between 0.697 ppm and 1.064 ppm representing signals indicating both the folded and the unfolded state, respectively. Using the ratio IN/(IN + IN+U) enables the precise determination of the total population of the folded state, fN, at any temperature45. The population of the unfolded state, fU, can now be determined assuming a two-state folding scenario as described for BsCspB, fU = 1 − fN33. The fraction of unfolded protein, fU, has been subsequently used to determine the temperature midpoint, TM, of the folded-to-unfolding transition of different variants of BsCspB by using Eq. (4)
| 4 |
where ΔHU represents the van’t Hoff enthalpy of unfolding at TM, ΔCP the change in heat capacity between folded und unfolded state, R is the universal gas constant, and T is the absolute temperature45.
Kinetic measurements using fluorescence stopped-flow methodology
Kinetic fluorescence spectroscopic data were collected by using a SX20 Stopped Flow Spectrometer (Applied Photophysics, UK). After excitation at a wavelength of 280 nm the folding kinetics was recorded by the change of fluorescence above a wavelength of 320 nm using a cut off filter. All single mixing experiments were performed in 20 mM sodiumcacodylate at pH = 7.0 and T = 298 K. The protein solution of c = 15 µM present in the native buffer was diluted 11-fold with urea solutions (in 20 mM sodiumcacodylate, pH = 7.0) of different concentrations leading to final concentrations of urea in the measuring cell ranging between curea = 2.6 M and 7.3 M to detect kinetics of protein unfolding. Refolding kinetics of unfolded protein of c = 15 µM present in curea = 7 M and 20 mM sodiumcacodylate, pH = 7.0 were followed by an 11-fold dilution with urea solutions (in 20 mM sodiumcacodylate, pH = 7.0) at different concentrations, leading to final concentrations of urea in the measuring cell ranging between curea = 0.6 M and 3.9 M. All folding and refolding kinetics were measured 12 times under identical conditions and averaged. Data processing used the Pro Data Viewer software (Applied Photophysics, UK).
Kinetic data were analysed using a monoexponential function. The apparent rate constant, kobs, has been plotted logarithmically as a function of curea. Data analysis applying Eq. (5) enabled the determination of the rate constants of protein refolding kf and unfolding ku, respectively, assuming a two state folding process
| 5 |
Here, mf and mu represent the limbs for protein refolding and unfolding leading to the cooperativity of folding m = mf + mu, which was independently determined before using Eq. (3), R is the universal gas constant, and T is the absolute temperature33.
Finally, the rate constants for refolding, kf, and unfolding ku, can be used to determine the overall thermodynamic stability, ΔG0, of the protein under investigation. Thus the difference in free energy of a protein sensing folding or unfolding conditions can be obtained by using Eq. (6)
| 6 |
where K is the equilibrium constant, R is the universal gas constant, and T is the absolute temperature33.
X-ray crystallography
Protein samples 4-19F-Phe-BsCspB and 4-19F-Trp-BsCspB were concentrated to 20 mg/ml in 20 mM sodium cacodylate pH 7.0 and 100 mM NaCl and crystallized using a Gryphon liquid dispenser (Art Robbins instruments) on 96-well Intelli-plates (Art Robbins instruments) employing the sitting drop method at 18 °C. Crystals of 4-19F-Phe-BsCspB grew from 100 mM CHES pH 9.5, 1 M sodium citrate tribasic. Crystals of 4-19F-Trp-BsCspB grew from 0.1 M BIS-TRIS propane pH 7.0, 1.2 M sodium citrate tribasic dehydrate. Drop consisted of a 150 nl:150 nl protein:precipitate ratio. For X-ray data cryo-collection, crystals were vitrified in LN2 in mother liquor supplemented with 25% [v/v] glycerol. X-ray diffraction data were collected at the Swiss Light Source synchrotron (Villigen) and processed using XDS/XSCALE55. Phasing was by molecular replacement in PHASER56 using the crystal structure of wild type CspB (PDB ID: 1CSP) as search model. Manual model building was in COOT57 and refinement used PHENIX.refine58. Chemical libraries for the modified residues 4-19F-Phe (PFF) and 4-19F-Trp (4FW) were obtained from the Protein Data Bank. Visual structure comparisons and calculations of root-mean-square deviations (RMSD) were performed in Chimera59.
Supplementary information
Acknowledgements
We thank Birgit Köhn for providing samples of wild type BsCspB and Professor Magnus Wolf-Watz for fruitful discussion. Financial support from the Juniorprofessurenprogramm handled by the Baden-Württemberg Stiftung (grant ID: 647/16) is gratefully acknowledged.
Author contributions
M.K. designed the study. H.W., T.Z., and X.M. performed experiments and collected data. H.W., T.Z., O.M., and M.K. analyzed data. H.W., and M.K. wrote the manuscript. All authors have read and critically revised drafts for intellectual contents and provided approval for publication.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-59446-w.
References
- 1.Vendruscolo M, Zurdo J, MacPhee CE, Dobson CM. Protein folding and misfolding: a paradigm of self-assembly and regulation in complex biological systems. Philos. T R. Soc. A. 2003;361:1205–1222. doi: 10.1098/rsta.2003.1194. [DOI] [PubMed] [Google Scholar]
- 2.Zimmerman SB, Trach SO. Estimation of Macromolecule Concentrations and Excluded Volume Effects for the Cytoplasm of Escherichia-Coli. J. Mol. Biol. 1991;222:599–620. doi: 10.1016/0022-2836(91)90499-V. [DOI] [PubMed] [Google Scholar]
- 3.Mika JT, Poolman B. Macromolecule diffusion and confinement in prokaryotic cells. Curr. Opin. Biotech. 2011;22:117–126. doi: 10.1016/j.copbio.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 2001;26:597–604. doi: 10.1016/S0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 5.Zhou HX, Rivas GN, Minton AP. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophysics. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gnutt D, et al. Stability effect of quinary interactions reversed by single point mutations. J. Am. Chem. Soc. 2019;141:4660–4669. doi: 10.1021/jacs.8b13025. [DOI] [PubMed] [Google Scholar]
- 7.Inomata K, et al. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nat. 2009;458:106. doi: 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- 8.Theillet F-X, et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nat. 2016;530:45. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka, T. et al. High‐Resolution Protein 3D Structure Determination in Living Eukaryotic Cells. Angewandte Chemie International Edition (2019). [DOI] [PubMed]
- 10.Frieden C, Hoeltzli SD, Bann JG. The preparation of 19F-labeled proteins for NMR studies. Methods enzymology. 2004;380:400. doi: 10.1016/S0076-6879(04)80018-1. [DOI] [PubMed] [Google Scholar]
- 11.Gerig J. Fluorine NMR of proteins. Prog. Nucl. Magnetic Reson. Spectrosc. 1994;26:293–370. doi: 10.1016/0079-6565(94)80009-X. [DOI] [Google Scholar]
- 12.Li CG, et al. Protein F-19 NMR in Escherichia coli. J. Am. Chem. Soc. 2010;132:321–327. doi: 10.1021/ja907966n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun X, Dyson HJ, Wright PE. Fluorotryptophan Incorporation Modulates the Structure and Stability of Transthyretin in a Site-Specific Manner. Biochemistry-Us. 2017;56:5570–5581. doi: 10.1021/acs.biochem.7b00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuler B, Kremer W, Kalbitzer HR, Jaenicke R. Role of entropy in protein thermostability: Folding kinetics of a hyperthermophilic cold shock protein at high temperatures using F-19 NMR. Biochemistry-Us. 2002;41:11670–11680. doi: 10.1021/bi0262931. [DOI] [PubMed] [Google Scholar]
- 15.Crowley PB, Kyne C, Monteith WB. Simple and inexpensive incorporation of F-19-Tryptophan for protein NMR spectroscopy. Chem. Commun. 2012;48:10681–10683. doi: 10.1039/c2cc35347d. [DOI] [PubMed] [Google Scholar]
- 16.Li HL, Frieden C. Observation of sequential steps in the folding of intestinal fatty acid binding protein using a slow folding mutant and F-19 NMR. P Natl Acad. Sci. USA. 2007;104:11993–11998. doi: 10.1073/pnas.0705253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hull WE, Sykes BD. Fluorotyrosine Alkaline Phosphatase: Internal Mobility of Individual Tyrosines and the Role of Chemical Shift Anisotropy as a ~gFNuclear Spin Relaxation Mechanism in Proteins. J. Mol. Biol. 1975;98:121–153. doi: 10.1016/S0022-2836(75)80105-7. [DOI] [PubMed] [Google Scholar]
- 18.Tobola F, et al. Effect of Noncanonical Amino Acids on Protein-Carbohydrate Interactions: Structure, Dynamics, and Carbohydrate Affinity of a Lectin Engineered with Fluorinated Tryptophan Analogs. Acs Chem. Biol. 2018;13:2211–2219. doi: 10.1021/acschembio.8b00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acchione M, et al. Specific Fluorine Labeling of the HyHEL10 Antibody Affects Antigen Binding and Dynamics. Biochemistry-Us. 2012;51:6017–6027. doi: 10.1021/bi300455t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matei E, Gronenborn AM. 19F paramagnetic relaxation enhancement: a valuable tool for distance measurements in proteins. Angew. Chem. Int. Ed. 2016;55:150–154. doi: 10.1002/anie.201508464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimmermann K, et al. Localization of ligands within human carbonic anhydrase II using 19 F pseudocontact shift analysis. Chem. Sci. 2019;10:5064–5072. doi: 10.1039/C8SC05683H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim TH, et al. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Sci. 2017;355:eaag2355. doi: 10.1126/science.aag2355. [DOI] [PubMed] [Google Scholar]
- 23.Divakaran A, Kirberger SE, Pomerantz WCK. SAR by (Protein-Observed) 19F NMR. Acc. Chem. Res. 2019;52:3407–3418. doi: 10.1021/acs.accounts.9b00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gee CT, Arntson KE, Koleski EJ, Staebell RL, Pomerantz WCK. Dual Labeling of the CBP/p300 KIX domain for 19F NMR leads to identification of a new small molecule binding site. ChemBioChem. 2018;19:963–969. doi: 10.1002/cbic.201700686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li F, et al. A Genetically Encoded 19F NMR Probe for Tyrosine Phosphorylation. Angew. Chem. Int. Ed. 2013;52:3958–3962. doi: 10.1002/anie.201300463. [DOI] [PubMed] [Google Scholar]
- 26.Yang F, et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and 19F-NMR. Nature. Commun. 2015;6(8202):1–15. doi: 10.1038/ncomms9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ycas, P. D., Wagner, N., Olsen, N. M., Fu, R. & Pomerantz, W. C. K. 2‐Fluorotyrosine is a valuable but understudied amino acid for protein‐observed 19F NMR. Journal of Biomolecular NMR, 10.1007/s10858-019-00290-0 (2019). [DOI] [PubMed]
- 28.Buer BC, Meagher JL, Stuckey JA, Marsh ENG. Comparison of the structures and stabilities of coiled-coil proteins containing hexafluoroleucine and t-butylalanine provides insight into the stabilizing effects of highly fluorinated amino acid side-chains. Protein Sci. 2012;21:1705–1715. doi: 10.1002/pro.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buer BC, Meagher JL, Stuckey JA, Marsh ENG. Structural basis for the enhanced stability of highly fluorinated proteins. P Natl Acad. Sci. USA. 2012;109:4810–4815. doi: 10.1073/pnas.1120112109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitevski-LeBlanc JL, Evanics F, Prosser RS. Optimizing 19F NMR protein spectroscopy by fractional biosynthetic labeling. J. Biomolecular NMR. 2010;48:113–121. doi: 10.1007/s10858-010-9443-7. [DOI] [PubMed] [Google Scholar]
- 31.Cornilescu G, et al. Solution structure of a small protein containing a fluorinated side chain in the core. Protein Sci. 2007;16:14–19. doi: 10.1110/ps.062557707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welte H, Kovermann M. Targeted expression and purification of fluorine labelled cold shock protein B by using an auxotrophic strategy. Protein Expr. Purif. 2019;157:86–91. doi: 10.1016/j.pep.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 33.Schindler T, Herrler M, Marahiel MA, Schmid FX. Extremely Rapid Protein-Folding in the Absence of Intermediates. Nat. Struct. Biol. 1995;2:663–673. doi: 10.1038/nsb0895-663. [DOI] [PubMed] [Google Scholar]
- 34.Schindelin H, Herrler M, Willimsky G, Marahiel MA, Heinemann U. Overproduction, Crystallization, and Preliminary-X-Ray Diffraction Studies of the Major Cold Shock Protein from Bacillus-Subtilis, Cspb. Proteins. 1992;14:120–124. doi: 10.1002/prot.340140113. [DOI] [PubMed] [Google Scholar]
- 35.Schnuchel A, et al. Structure in Solution of the Major Cold-Shock Protein from Bacillus-Subtilis. Nat. 1993;364:169–171. doi: 10.1038/364169a0. [DOI] [PubMed] [Google Scholar]
- 36.Schindelin H, Marahiel MA, Heinemann U. Universal Nucleic Acid-Binding Domain Revealed by Crystal-Structure of the Bacillus-Subtilis Major Cold-Shock Protein. Nat. 1993;364:164–168. doi: 10.1038/364164a0. [DOI] [PubMed] [Google Scholar]
- 37.Schindler, T. et al. Surface-exposed phenylalanines in the RNP1/RNP2 motif stabilize the cold-shock protein CspB from Bacillus subtilis. Proteins30, 401–406, doi: 10.1002/(Sici)1097-0134(19980301)30:4<401::Aid-Prot7>3.0.Co;2-L (1998). [DOI] [PubMed]
- 38.Zeeb M, et al. Recognition of T-rich single-stranded DNA by the cold shock protein Bs-CspB in solution. Nucleic Acids Res. 2006;34:4561–4571. doi: 10.1093/nar/gkl376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sachs R, Max KE, Heinemann U, Balbach J. RNA single strands bind to a conserved surface of the major cold shock protein in crystals and solution. Rna. 2012;18:65–76. doi: 10.1261/rna.02809212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong C-Y, Eftink MR. Incorporation of tryptophan analogues into staphylococcal nuclease, its V66W mutant, and Δ137− 149 fragment: Spectroscopic studies. Biochemistry-Us. 1998;37:8938–8946. doi: 10.1021/bi971862o. [DOI] [PubMed] [Google Scholar]
- 41.Royer CA, Mann CJ, Matthews CR. Resolution of the Fluorescence Equilibrium Unfolding Profile of Trp Aporepressor Using Single Tryptophan Mutants. Protein Sci. 1993;2:1844–1852. doi: 10.1002/pro.5560021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santoro MM, Bolen DW. Unfolding Free-Energy Changes Determined by the Linear Extrapolation MethodMethod .1. Unfolding of Phenylmethanesulfonyl Alpha-Chymotrypsin Using Different Denaturants. Biochemistry-Us. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 43.Köhn Birgit, Kovermann Michael. Macromolecular Crowding Tunes Protein Stability by Manipulating Solvent Accessibility. ChemBioChem. 2019;20(6):759–763. doi: 10.1002/cbic.201800679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alexander SS, Pace CN. Comparison of Denaturation of Bovine Beta-Lactoglobulins-a and B and Goat Beta-Lactoglobulin. Biochemistry-Us. 1971;10:2738-+. doi: 10.1021/bi00790a013. [DOI] [PubMed] [Google Scholar]
- 45.Szyperski T, Mills JL, Perl D, Balbach J. Combined NMR-observation of cold denaturation in supercooled water and heat denaturation enables accurate measurement of Delta C-p of protein unfolding. Eur. Biophys. J. Biophy. 2006;35:363–366. doi: 10.1007/s00249-005-0028-4. [DOI] [PubMed] [Google Scholar]
- 46.Khan F, Chuang JI, Gianni S, Fersht AR. The kinetic pathway of folding of barnase. J. Mol. Biol. 2003;333:169–186. doi: 10.1016/j.jmb.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 47.Campos-Olivas R, Aziz R, Helms GL, Evans JN, Gronenborn AM. Placement of 19F into the center of GB1: effects on structure and stability. Febs Lett. 2002;517:55–60. doi: 10.1016/S0014-5793(02)02577-2. [DOI] [PubMed] [Google Scholar]
- 48.Minks C, Huber R, Moroder L, Budisa N. Atomic mutations at the single tryptophan residue of human recombinant annexin V: effects on structure, stability, and activity. Biochemistry-Us. 1999;38:10649–10659. doi: 10.1021/bi990580g. [DOI] [PubMed] [Google Scholar]
- 49.Rozovsky S, Jogl G, Tong L, McDermott AE, Solution-state NMR. investigations of triosephosphate isomerase active site loop motion: ligand release in relation to active site loop dynamics. J. Mol. Biol. 2001;310:271–280. doi: 10.1006/jmbi.2001.4673. [DOI] [PubMed] [Google Scholar]
- 50.Chadegani F, et al. 19F nuclear magnetic resonance and crystallographic studies of 5-fluorotryptophan-labeled anthrax protective antigen and effects of the receptor on stability. Biochemistry-Us. 2014;53:690–701. doi: 10.1021/bi401405s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu, M., Ishima, R., Polenova, T. & Gronenborn, A. M. 19 F NMR relaxation studies of fluorosubstituted tryptophans. J Biomol Nmr, 1–9 (2019). [DOI] [PMC free article] [PubMed]
- 52.Dürr UHN, Grage SL, Witter R, Ulrich AS. Solid state 19F NMR parameters of fluorine-labeled amino acids. Part I: Aromatic substituents. J. Magnetic Reson. 2008;191:7–15. doi: 10.1016/j.jmr.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 53.Sušac L, Eddy MT, Didenko T, Stevens RC, Wüthrich K. A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl Acad. Sci. 2018;115:12733–12738. doi: 10.1073/pnas.1813649115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warren JR, Gordon JA. On Refractive Indices of Aqueous Solutions of Urea. J. Phys. Chem-Us. 1966;70:297-&. doi: 10.1021/j100873a507. [DOI] [Google Scholar]
- 55.Kabsch W. Xds. Acta Crystallogr. Sect. D: Biol. Crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCoy, A., Grosse-Kunstleve, R., Adams, P., Winn, M. & Storoni, L. PHASER crystallographic software. J. Appl. Cryst (2007). [DOI] [PMC free article] [PubMed]
- 57.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. Sect. D: Biol. Crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adams PD, et al. The Phenix software for automated determination of macromolecular structures. Methods. 2011;55:94–106. doi: 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.