

Abstract
Gnathic fibro-osseous lesions are a diverse group of disease processes which share overlapping microscopic features characterized by fibroblastic stroma with variable cellularity and a range of bone forming pathological processes leading to woven, sclerotic and cementum-like structures. Some of the lesions are unique to craniofacial location and a combination of clinical, radiological and pathological correlation is often necessary for diagnostic accuracy. Gnathic osteosarcomas are rare tumors with differences in age distribution and behavior as compared to osteosarcoma of long bones. This review will discuss the clinicopathological and radiological features of gnathic fibro-osseous lesions and osteosarcoma with updates on current genetics and molecular pathogenesis.
Keywords: Fibro-osseous lesions, Ossifying fibroma, Cemento-ossifying fibroma, Juvenile trabecular ossifying fibroma, Juvenile psammomatoid ossifying fibroma, Cemento-osseous dysplasia, Diffuse sclerosing osteomyelitis, Fibrous dysplasia, Osteosarcoma
Fibro-Osseous Lesions
The term fibro-osseous lesions (FOL) describes a group of diseases with the shared microscopic finding of a deposited collagenous matrix containing variably sized collections of immature bone [1]. In the gnathic bones, FOL includes a range of mesenchymal proliferations from reactive to neoplastic. Some are unique to the craniofacial complex while others can be also be seen in the extragnathic skeleton [2]. The gnathic bones are unique because they arise from neural crest, in contrast to the mesodermal origin of most bones of the rest of the skeleton [3], a developmental feature that may in part, explain the unique pathologic features of some FOLs of the jaws. FOLs typically include cemento-ossifying fibroma (formerly ossifying fibroma) and its variants, cemento-osseous dysplasias and fibrous dysplasia. In a traditional sense, chronic sclerosing osteomyelitis and low-grade osteosarcoma are technically not FOL but are included in this review because the radiographic and pathologic features often overlap with FOL. The term “atypical fibro-osseous lesion” has been used descriptively to imply microscopic or radiographic features, while not overtly malignant, that do not clearly fit into a specific diagnostic category [4].
Ossifying Fibroma
Ossifying fibroma (OF) is a benign osteogenic tumor with a prominent fibrous matrix. In the gnathic bones, the category is divided into three specific clinicopathologic entities: cemento-ossifying fibroma (COF), juvenile trabecular ossifying fibroma and juvenile psammomatoid ossifying fibroma.
Cemento-Ossifying Fibroma (COF)
Clinical Features
Cemento-ossifying fibroma (COF) is the preferred term now adopted in the 4th edition of the World Health Organization‘s Classification of Head and Neck Tumours [5] COF is a neoplasm likely of odontogenic origin, predominantly affecting adults (peak third to fourth decade) and more common in women [6]. COF typically presents as an asymptomatic, solitary, slowly growing, symmetrically expansile mass in a tooth-bearing area especially in the mandibular premolar-molar area. Unusual presentations include multifocal lesions and transcortical spread resulting in mucosal ulceration.
Radiographic Features
The typical radiographic appearance of COF is a marginated, symmetrically expansile mass with a predominantly radiolucent quality. The density of the calcifications within the tumor determines the presence, and extent, of radio-opacity that can range from radiolucent (Fig. 1a) to radio-opaque with patchy radiolucencies. The roots of the adjacent teeth may be displaced or, rarely, resorbed [7].
Fig. 1.
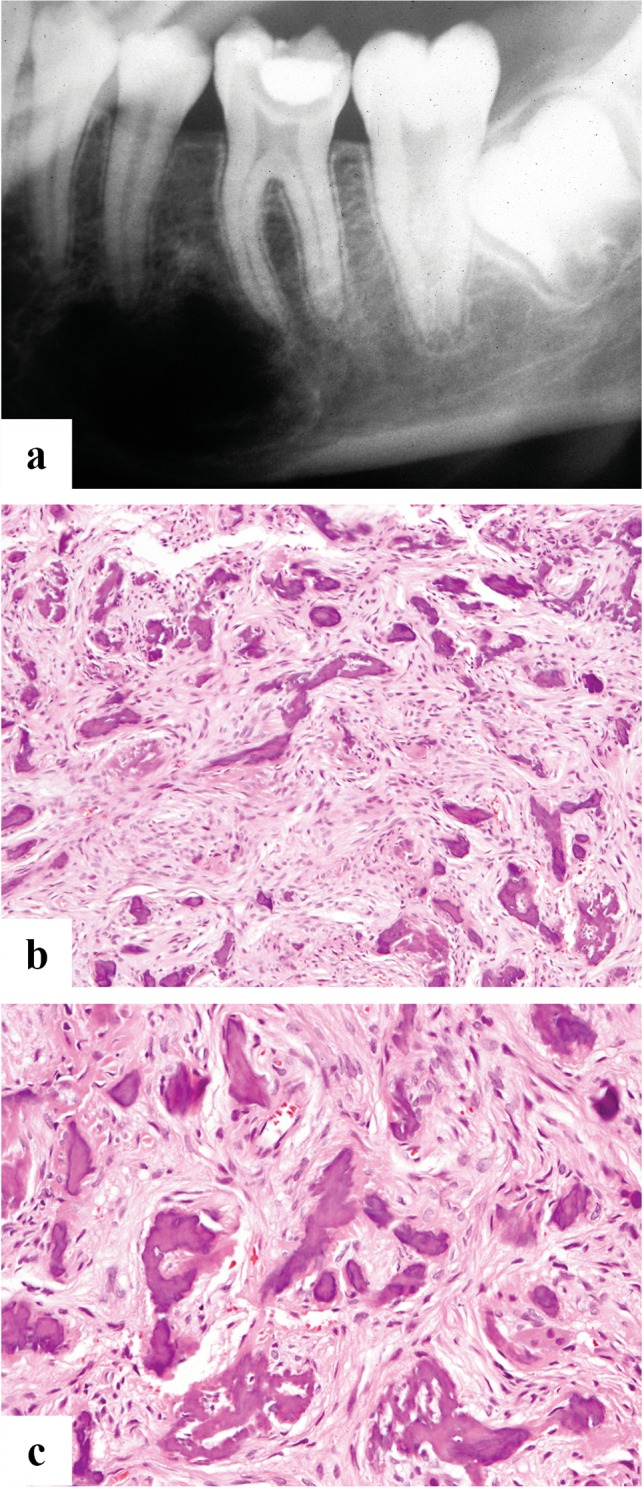
a A cropped panoramic radiograph showing a symmetrically expansile radiolucency below the roots of lower left first molar to the lower left first premolar (teeth numbers 19–21). b and c There are islands of osteoid and woven bone evenly distributed in a moderately to highly cellular component of bland bipolar to stellate cells. The bone is variably sized and shaped woven type
Pathological Features
The peripheral circumscription often permits surgical enucleation of the lesion producing a single yellow-tan fleshy mass with a gritty cut surface. The latter corresponds to the calcifications distributed through-out the lesion. Microscopically, the lesion is formed by a fibrous stroma with a moderately to highly cellular component of bland bipolar to stellate cells (Fig. 1b, c). The cells may show slight nuclear hyperchromasia with small nucleoli, but pleomorphism is absent. Mitotic activity is generally absent to low. The collagen is usually haphazardly arranged without a pattern. The bone component shows osteoblastic rimming with chiefly immature woven that can contain a small amount of mature lamellar bone in long standing lesions. The bone deposits form droplet-like spheroids, trabeculae and irregular islands. Some have suggested that the droplet deposits resemble cementum hence the name “cemento-ossifying fibroma” although similar appearing hard tissue can be seen in lesions in the long bones. The amount of bone varies from lesion to lesion but is uniformly distributed in a given lesion. Osteoclast-like giant cells and secondary cyst formation are uncommon and diagnostic findings that can help differentiate this lesion from juvenile trabecular ossifying fibroma.
Genetics
An association with inactivating mutations in the CDC73 gene (also known as HRPT2) has been proposed based on familial syndromes predisposed to multiple COF [8] and in sporadic tumors [9]. However, it is less clear whether such CDC73 inactivation plays a role in tumor initiation or tumor progression [10]. Mutations in CTNNB1 have been identified in rare cases [11]. Importantly, mutations in GNAS, the genetic basis of fibrous dysplasia, are absent [12, 13].
Treatment and Prognosis
COF is generally slowly growing with curettage or enucleation usually curative. Recurrence is rare. Malignant transformation has not been reported to date.
Juvenile Trabecular Ossifying Fibroma
Clinical Features
Juvenile trabecular ossifying fibroma (JTOF) is a rare tumor that chiefly affects children and adolescents (mean age 8.5–12 years) with equal gender predilection [14]. Patients older than 15 years may present with JTOF, but represent < 20% of cases. Previously reported using the terms “juvenile ossifying fibroma”, “juvenile active ossifying fibroma” and “juvenile aggressive ossifying fibroma”, JTOF is more common in the maxilla in contrast to other cemento-osseous dysplasias that are more common in mandible; maxillary JTOF can produce nasal obstructive symptoms or epistaxis. Surface and extra-osseous lesions are exceptional. Intraosseous JTOF presents as a rapidly expanding mass without pain.
Radiographic Features
The tumor shows a well-defined radiographic border but is more often radiolucent compared to COF (Fig. 2a) [14, 15]. Transcortical spread is rare. Tumors occurring primarily in soft tissue are very rare where they also show circumscription with a mixed lucent-opaque parenchyma.
Fig. 2.

a Coronal CT image of juvenile trabecular ossifying fibroma shows a symmetrically expansile, circumscribed mostly radiolucent lesion of the right maxilla pushing superiorly into the maxillary sinus with inferior displacement of a molar tooth. b Grossly, juvenile trabecular ossifying fibroma consists of yellow tan fibrous parenchyma within which are arrayed serpiginous hemorrhagic bands. The tumor has enveloped a tooth root. c and d Juvenile trabecular ossifying fibroma characteristically has a looser architecture than conventional ossifying fibroma composed of spindle cells with little collagen production. Osteoid appears to develop directly from the stroma forming trabeculae of osteoid and bone. e Juvenile trabecular ossifying fibroma can contain osteoclast type multinucleated giant cells in the stroma
Pathological Features
Grossly, the lesions are unencapsulated but circumscribed. The cut surface shows serpiginous hemorrhagic bands that impart a jigsaw puzzle like appearance (Fig. 2b) amid yellow-tan fibrous parenchyma and gritty calcified material.
Compared to COF, microscopically JTOF consists of a more cellular proliferation of plump bipolar to stellate cells that grow more loosely with a stroma containing scant collagen. Osteoid appears to form directly from the stroma as elongated, curved or branching trabeculae without osteoblastic rimming (Fig. 2c, d). The trabeculae may become ossified into immature woven bone. Osteoclast-type giant cells are relatively common and secondary cystic change has been reported particularly around the new bone [14, 16] (Fig. 2e).
Genetics
The genetic or molecular events that may drive JTOF tumorigenesis have not been characterized. CTNNB1 mutations have not been identified in JTOF [17].
Treatment and Prognosis
Following curettage or enucleation, recurrence is common but en bloc excision is usually curative [17]. Malignant transformation has not been described.
Juvenile Psammomatoid Ossifying Fibroma (JPOF)
Clinical Features
Despite the name, the rare juvenile psammomatoid ossifying fibroma (JPOF) shows a wider age range (3 months to 72 years) with a peak incidence slightly older than JTOF between (16 to 33 years) depending on the study [5, 18]. Males and females are equally affected. Unlike COF and JTOF, JPOF occurs chiefly in extragnathic sites, especially the paranasal sinuses, periorbital bones and skull base [1, 14]. Consequently, the chief complaint is often painless exophthalmos, sinonasal symptoms or epistaxis. Rarely, the tumor can affect the mandible or maxilla. Rapid growth of JPOF has been attributed to secondary aneurysmal bone cyst change [1, 14].
Radiographic Features
Radiographically JPOF is usually circumscribed and expansile, and is surrounded by a thin rim of residual cortex. The central parenchyma can vary from lucent to ground glass to more discrete calcifications [19] (Fig. 3a).
Fig. 3.

a Axial CT image of juvenile psammomatoid ossifying fibroma demonstrates an expansile mass centered within bilateral sphenoid sinuses with erosion of the clivus and pterygoids. It shows low radiodensity with a thin rim of retained native bone (arrow). b–d Juvenile psammomatoid ossifying fibroma is moderately to highly cellular with a haphazard growth pattern and innumerable deposits of bone with concentric calcification reminiscent of psammoma bodies
Pathological Features
Grossly, JPOF consists of an unencapsulated fleshy yellow-tan mass with conspicuous fine gritty calcifications. Microscopically, JPOF contains a moderately to highly cellular, compact proliferation of spindled of stellate cells with scant stroma and small rounded deposits of woven bone with central calcification resembling psammoma bodies or cementum. Osteoblastic rimming is not observed. Larger deposits of bone may coalesce to form angulated or linear trabeculae. Secondary cystic change is relatively common (Fig. 3b–d).
Genetics
Only rare cases have been studied with no mutations in GNAS identified [20].
Treatment and Prognosis
Treatment consists of curettage or enucleation but recurrence is common (~ 30%) and sometimes multiple. Resection is generally curative [19]. Malignant transformation has not been reported.
Cemento-Osseous Dysplasia
Clinical Features
Cemento-osseous dysplasia is a group of fibro-osseous lesions that includes periapical cemento-osseous dysplasia (PCD), focal cemento-osseous dysplasia and florid cemento-osseous dysplasia. These represent a diverse group of reactive, dysplastic and neoplastic lesions characterized microscopically by the replacement of normal bone with a collagenous matrix containing trabeculae of immature bone and in some instances hard tissue resembling cementum. As a group, cemento-osseous dysplasia (COD) is believed to arise from cells of the periodontal ligament.
PCD characteristically involves the apices (roots) of teeth, typically one or several mandibular incisors. Focal COD is a solitary lesion usually not associated with a tooth and florid COD involves two or more quadrants of bone [20]. COD is the most common FOL of the jaws with a strong predilection to middle aged women of African descent [5, 21]. The lesions are usually asymptomatic and identified incidentally on radiographs. The teeth in the region of PCD will test vital in contrast to a periapical dental infection where the teeth are nonvital. Florid COD may be expansile and thus present with pain particularly if the lesion becomes infected [21].
Radiographic Features
COD is a non-expansile intramedullary lesion that evolves from a lucent to mixed to sclerotic radio-opacity as the lesion matures. PCD surrounds the root apices of vital teeth while focal COD shows similar signal characteristics localized to non-tooth bearing regions of the jaw. Late, diffusely sclerotic lesions may have a lucent rim. Florid COD is multifocal, involving both maxilla and mandible often in a symmetric fashion [22] (Fig. 4a).
Fig. 4.

a Coronal CT scan of a patient with florid cemento-osseous dysplasia showing bilateral maxillary and mandibular lesions. b–d All three variants of cemento-osseous dysplasia (periapical, focal and florid) microscopically show identical features. Specifically, fragments of variably cellular fibrous stroma containing small blood vessels and variably thickened, curvilinear and rounded trabeculae of woven bone and cementum-like material with minimal osteoblastic rimming
Pathologic Features
Grossly, COD consists of gritty tan-brown parenchyma. All three variants are histologically identical. Specifically, COD is composed of a highly vascular collagenous stroma with a heterogeneously distributed plump spindle cell component. As the lesion evolves, increasingly abundant spicules of woven bone and cementum (with numerous reversal lines) of varying sizes appear, usually lined by cementoblasts or osteoblasts (Fig. 4b–d). In late stage disease, dense sclerotic bone with only rare marrow spaces. Secondary cystic change is more common in florid COD [22].
Genetics
No underlying genetic event has been thus far identified for COD. Very rare familial forms with autosomal dominant inheritance have been described using the terms “familial florid osseous dysplasia” or “familial gigantiform cementoma [23, 24]. These are more common in the maxilla and in contrast to other cemento-osseous dysplasias that are more common in whites.
Treatment and Prognosis
No treatment is required for asymptomatic lesions. The teeth remain vital throughout the evolution of the lesion to the sclerotic stage and should not be extracted or treated endodontically [1]. Biopsy of cemento-osseous dysplasia, particularly large and sclerotic lesions can lead to infection and secondary osteomyelitis of the mandible that can be difficult to treat with antibiotics because the bone is sclerotic and relatively avascular.
Diffuse Sclerosing Osteomyelitis
Clinical Features
Diffuse sclerosing osteomyelitis (DSO) is an inflammatory reaction in the gnathic bones ostensibly to an infection by low virulence bacteria although cultures are often negative. However, recent evidence suggests that a proportion of these lesions may be auto-inflammatory and part of the spectrum of chronic recurrent multifocal osteomyelitis (CRMO) in children and “synovitis, acne, pustulosis, hyperostosis, osteitis” (SAPHO) syndrome in adults [25]. Severe, irregular jaw pain with swelling and trismus are common symptoms. Unilateral involvement of the mandibular angle or condyle is most common [26, 27]. The disease affects a wide age range and both genders equally.
Radiographic Features
DSO presents as an ill-defined heterogeneous but mainly sclerotic lesion affecting a large portion of the mandible, predominantly the angle or condyle. In contrast to the cemento-osseous dysplasias, there is no lamina lucida around the lesion. Eventually sclerosis predominates [28] (Fig. 5a).
Fig. 5.

a Diffuse sclerosing osteomyelitis (axial CT image) shows a smoothly contoured expansion of the right mandible by a heterogeneous density. b Diffuse sclerosing osteomyelitis demonstrates a loose fibrous stroma with bland spindle cells and abundant anastomosing trabeculae of woven bone. c Osteoclastic activity may be prominent in DSO. d Unlike other fibro-osseous lesions, DSO often features inflammatory cells, usually chronic in nature. Note the prominent osteoclastic activity and scalloping of trabeculae
Pathological Features
Grossly, the lesion consists of dense tan-yellow fibrous tissue mixed with granular calcified material that increases depending on the chronicity of the condition. Microscopically, DSO shows fibrous stroma replacing the marrow with a moderately cellular, loosely arranged proliferation of bipolar to stellate cells. Unlike most other FOLs, inflammation can be prominent, usually lymphoplasmacytic but occasionally neutrophilic. A rich vascular proliferation may be present. Irregularly anastomosing trabeculae of woven bone with conspicuous osteoblastic rimming are common. Osteoclastic activity is also usually present (Fig. 5b–d).
Genetics
The genetics of DSO have not been specifically studied. However, familial clustering of SAPHO syndrome has been documented. Mouse models of CRMO suggest that Fgr, a member of the Src family kinases may be a susceptibility gene [28–30]
Treatment and Prognosis
Treatment protocols have varied over the years and include antibiotics, debridement and corticosteroids. Recent reports suggest good response to anti-TNF therapy [31], and denosumab [32] further supporting the notion that DSO is an inflammatory rather than infectious process.
Fibrous Dysplasia
Clinical Features
Fibrous dysplasia (FD) is a non-hereditary benign fibro-osseous lesion that involves the medullary cavity; the condition can be monostotic (single bone) or polyostotic (multiple bones). In the polyostotic form the association with endocrine abnormalities and cutaneous pigmentation (café-au-lait spot) is the hallmark of McCune Albright Syndrome that is also non-hereditary with a female preponderance [33]. In the jaws, FD can affect either the maxilla or mandible with maxillary cases predominating [34]. FD affects individuals in young adulthood (20–30 years) and tends to show a slight preponderance towards female gender. The bony expansion is usually painless resulting in facial asymmetry. Involvement of orbit, paranasal sinuses and base of skull can present with symptoms such as headache, hearing loss, proptosis etc. [35, 36]. In the gnathic location the lesion can extend beyond one bone across suture lines to involve adjacent bones and the term craniofacial fibrous dysplasia has been used [2].
Radiographic Features
Radiographic findings are variable with early lesions being more radiolucent and later lesions more opaque. A mixed lucent/sclerotic and characteristic “ground glass” or “peau d’orange” appearance is common (Fig. 6a). Margins are not easily defined since the lesions blend with the adjacent normal bone [37]. MRI appearance is also variable depending on the degree of lucent and sclerotic areas [37].
Fig. 6.
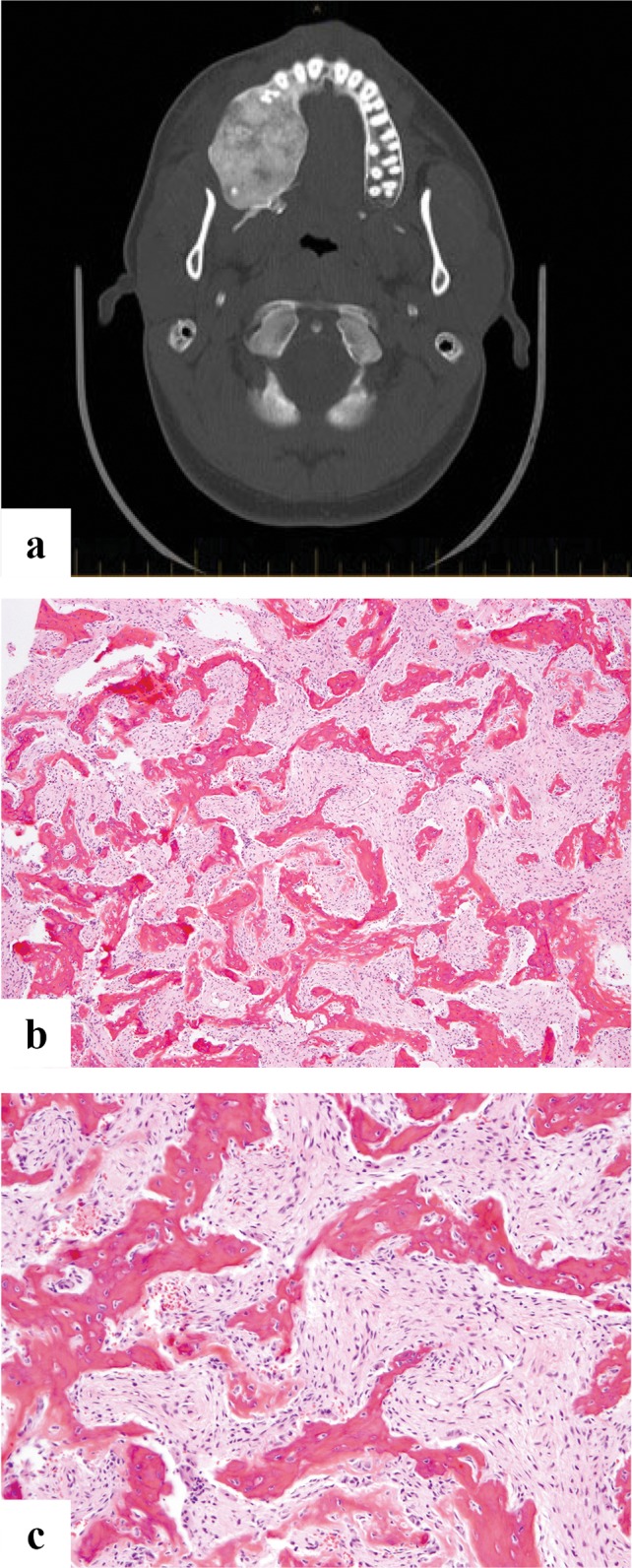
a Axial CT scan showing a well-defined, expansile intramedullary lesion of the right mandible, containing extensive high-attenuation “ground-glass” matrix. b Microscopic image showing curvilinear woven bone in a fibrous stroma. The bone trabeculae are disorderly and irregularly intersect with each other. c High power view shows the woven bone is not rimmed by osteoblasts
Pathologic Features
Grossly FD has a tan white and gritty appearance. Microscopically the marrow is replaced by irregularly intersecting bony trabeculae that are embedded in fibrous stroma. The stroma can be cellular or loosely textured containing randomly arranged delicate bony trabeculae that are made up of immature or woven bone without osteoblastic rimming (Fig. 6b, c). The disordered arrangement of woven bone in the fibrous stroma has been variously described as an “alphabet soup” or “Chinese script letters”. The fabric of the woven bone is seen in continuity with the stroma which is more readily visible under polarized light. Maturation leading to lamellar bone formation is very rarely seen at this site [35, 36]. Presence of cartilage islands (fibrocartilaginous dysplasia) is even rarer than in long bones [37, 38]. In one of the reported series [37] it was seen in association with Ollier disease. Correlation with imaging and clinical findings are of utmost importance.
Genetics
The genetic hallmark of FD is the presence of activating missense mutations of the GNAS gene (ch20q13) encoding the Gs alpha subunit of the heterotrimeric G protein complex. These mutations occur post zygotically leading to a focal somatic mosaic state within the lesional tissue [39, 40]. The mutations commonly involve codon 201 of exon 8 of the GNAS gene and lead to substitution of arginine by histidine (R201H) or a cysteine (R201C) [41, 42]. All forms of FD including McCune Albright’s syndrome contain this mutation [42] that leads to constitutive GS alpha adenyl cyclase activity and overproduction of cyclic adenosine monophosphate (cAMP) in dysplastic cells [43–45]. Due to the somatic “mosaic” state of the mutation in lesional tissue the sensitivity of the various molecular methods to detect mutation in FD cases is variable and caution should be exercised in interpretation of negative results [46].
Treatment and Prognosis
The management of the gnathic FD is based, in part, on the patient’s age and skeletal maturity. Many lesions become quiescent after puberty and in such cases, if they are asymptomatic, observation and monitoring may be an acceptable treatment modality. Surgical contouring and/or resection may be performed in cases with deformity. Regular follow up is warranted to monitor recurrence of lesion and deformity [47]. Rare malignant transformation (< 1%) in the form of osteosarcoma, fibrosarcoma, chondrosarcoma and undifferentiated pleomorphic sarcoma (so-called malignant fibrous histiocytoma) [49] has been described. Radiation therapy is contraindicated in fibrous dysplasia due to the risk of transformation to high grade sarcoma [48, 49].
Gnathic Osteosarcoma
Clinical Features
Craniofacial osteosarcomas (CFOS) comprise about 6–10% of all osteosarcomas and 1% of all head and neck cancers [50, 51]. Males and females are equally affected. In contrast to osteosarcomas of the long bones that affect adolescent patients, the disease most often presents in the fourth to fifth decades. The mandible and maxilla are the most common sites with the mandibular body more affected than ramus; in the maxilla the alveolar ridge is the common site [52, 53]. Subtypes of CFOS include low grade intramedullary and parosteal, and high grade conventional osteosarcomas. The presenting symptoms include paresthesia, pain, loosening of teeth and swelling. Previous radiation therapy is a predisposing factor for developing osteosarcoma in craniofacial bones [54]. Paget’s disease is also a known predisposing condition [55].
Radiographic Features
Radiological presentation can be lytic or sclerotic or mixed lytic and sclerotic. Tumor matrix mineralization is frequently seen. Periosteal reaction is more commonly seen in mandibular tumors where a “sunburst” pattern is characteristic. Aggressive bone destruction and soft tissue extension is common in conventional high grade osteosarcomas [56] (Fig. 7a). Intraosseous low grade osteosarcomas usually expand the bone with ill-defined lytic or mixed lytic and sclerotic lesions, but can extend into soft tissue and produce periosteal reaction [57].
Fig. 7.

a Axial CT scan showing an expansile lytic lesion of left hemimandible with cortical destruction, containing foci of osseous calcification. b Microscopic image of high-grade osteosarcoma, osteoblastic type showing neoplastic bone rimmed by malignant osteoblasts. c Microscopic image of a high grade chondroblastic osteosarcoma showing cartilage differentiation with chondrocytes in lacunar spaces in a background of hyaline matrix. d Microscopic image of a high-grade fibroblastic osteosarcoma with atypical and pleomorphic spindle cells accompanied by neoplastic osteoid
Pathological Features
Pathological sub-types of conventional high-grade osteosarcomas are similar to OS of long bones and include osteoblastic, chondroblastic, fibroblastic, telangiectatic, small cell, giant cell rich and epithelioid subtypes. The most common type of CFOS is the chondroblastic form. Histologically the tumor shows pleomorphic cells producing bone with mitotic activity. The osteoid can be lace-like or sclerotic (Fig. 7b). Myxoid areas can be present. The chondroblastic type (Fig. 7c) can be so predominantly cartilaginous with very little tumor bone that in small biopsy specimens the diagnosis is challenging. In an atypical chondroid tumor, the pathologist should maintain a high index of suspicion for osteosarcoma and ensure careful scrutiny for osteoid produced by the tumor cells. The extreme rarity of chondrosarcomas of craniofacial bones is to be remembered in such instances [58]. A high grade spindle cell component predominates in fibroblastic osteosarcoma (Fig. 7d). Small cell variant shows primitive round cells and in these cases appropriate ancillary studies are needed to exclude Ewing Sarcoma and other round cell tumors. Low grade intramedullary and parosteal osteosarcomas resemble their counterparts in long bones, however, can be challenging due to their resemblance to other FOLs especially, fibrous dysplasia that shows ill-defined margins radiographically at this site. Histologically, low grade osteosarcomas contain woven bone without osteoblastic rimming, (not curvilinear), relatively bland spindle cells with minimal atypia and a streaming growth pattern that permeates into preexisting cancellous or cortical bone (Fig. 8a). The tumor can be accompanied low grade hyaline cartilage [57]. Radiation associated osteosarcomas are always high grade [54].
Fig. 8.
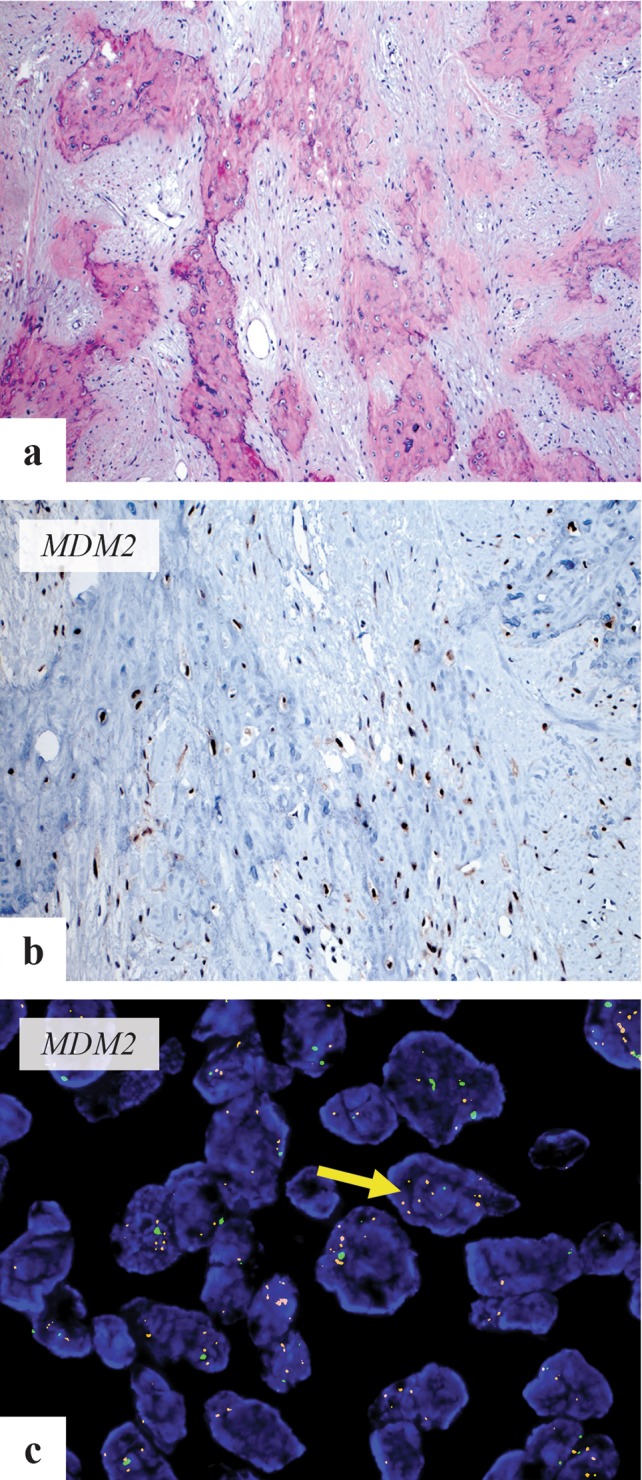
a Microscopic image of low-grade osteosarcoma showing minimally atypical spindle cells infiltrating broad seams of woven bone trabeculae. b MDM2 immunohistochemical stain shows positive nuclear staining in tumor cells. c Fluorescent in situ hybridization using MDM2 probe (Vysis) shows amplification of MDM2 (arrow-red signals). Image courtesy Dr. W. Patrick Devine (UCSF)
Genetics
Only limited data is available on the genetics of CFOS. In a series of mandibular osteosarcomas, MDM2 and RASAL1 amplification was seen by qPCR. All 5 cases were high grade fibroblastic type and 3 had co amplification of MDM2 and RASAL1. Two cases showed low grade with dedifferentiation to high grade osteosarcoma [59]. CDK4 and MDM2 amplification has been reported in 79% of parosteal and 29% of low grade intraosseous osteosarcomas and is detectable by immunohistochemistry or Fluorescent in situ hybridization (Fig. 8b, c) [58].
Treatment and Prognosis
Unlike conventional osteosarcoma of long bones, the incidence of distant metastases is lower, at about 20–30%, but local disease is difficult to control often leading to treatment failure [60]. In a study of 214 well characterized osteosarcomas of the jaws with long term follow up, metastases was seen in 17.6% of the cases and complete surgical resection resulted in excellent long term survival (83.2% after 10 years). Also in contradistinction to osteosarcoma of the long bones, neoadjuvant therapy, applied to a smaller set of patients, failed to show any additional benefit [61]. In a meta-analysis, positive resection margin was poor prognostic sign and chemotherapy improved survival in CFOS patients with adverse factors such as positive margin, high grade tumors and local recurrence [62].
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Regezi J, Sciubba J, Jordan R. Odontogenic tumors. Oral pathology: clinical pathology correlates. 6. Amsterdam: Elsevier; 2012. pp. 285–287. [Google Scholar]
- 2.Eversole R, Su L, ElMofty S. Benign fibro-osseous lesions of the craniofacial complex. A review. Head Neck Pathol. 2008;2:177–202. doi: 10.1007/s12105-008-0057-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuratani S. Evolution of the vertebrate jaw: comparative embryology and molecular developmental biology reveal the factors behind evolutionary novelty. J Anat. 2004;205:335–347. doi: 10.1111/j.0021-8782.2004.00345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koury ME, Regezi JA, Perrott DH, Kaban LB. “Atypical” fibro-osseous lesions: diagnostic challenges and treatment concepts. Int J Oral Maxillofac Surg. 1995;24:162–169. doi: 10.1016/S0901-5027(06)80094-9. [DOI] [PubMed] [Google Scholar]
- 5.El Mofty SK, Nelson B, Toyosawa S. Fibro-osseous and osteochondromatous lesions. In: WHO Classification of Head and Neck Tumors. Lyon: IARC Press; 2017. pp. 251–255.
- 6.Eversole LR, Leider AS, Nelson K. Ossifying fibroma: a clinicopathologic study of sixty-four cases. Oral Surg Oral Med Oral Pathol. 1985;60:505–511. doi: 10.1016/0030-4220(85)90239-7. [DOI] [PubMed] [Google Scholar]
- 7.Regezi J, Sciubba J, Jordan R. Benign non-odontogenic tumors. In: Regezi J, Sciubba J, Jordan R, editors. Oral pathology: clinical pathology correlates. 6. Amsterdam: Elsevier; 2012. pp. 294–298. [Google Scholar]
- 8.Carpten JD, Robbins CM, Villablanca A, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32:676–680. doi: 10.1038/ng1048. [DOI] [PubMed] [Google Scholar]
- 9.Pimenta FJ, Gontijo Silveira LF, Tavares GC, et al. HRPT2 gene alterations in ossifying fibroma of the jaws. Oral Oncol. 2006;42:735–739. doi: 10.1016/j.oraloncology.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 10.de Mesquita Netto AC, Gomez RS, Diniz MG, Fonseca-Silva T, Campos K, De Marco L, Carlos R, Gomes CC. Assessing the contribution of HRPT2 to the pathogenesis of jaw fibrous dysplasia, ossifying fibroma, and osteosarcoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:359–367. doi: 10.1016/j.oooo.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Shi RR, Li XF, Zhang R, Chen Y, Li TJ. GNAS mutational analysis in differentiating fibrous dysplasia and ossifying fibroma of the jaw. Mod Pathol. 2013;26:1023–1031. doi: 10.1038/modpathol.2013.31. [DOI] [PubMed] [Google Scholar]
- 12.Eh A, Cj R. Fibro-osseous lesions of the craniofacial bones: beta-catenin immunohistochemical analysis and CTNNB1 and APC mutation analysis. Head Neck Pathol. 2014;8:291–297. doi: 10.1007/s12105-014-0535-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pereira T, Diniz MG, Franca JA, et al. The Wnt/beta-catenin pathway is deregulated in cemento-ossifying fibromas. Oral Surg Oral Med Oral Pathol Oral Radiol. 2018;125:172–178. doi: 10.1016/j.oooo.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 14.El-Mofty S. Psammomatoid and trabecular juvenile ossifying fibroma of the craniofacial skeleton: two distinct clinicopathologic entities. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;93:296–304. doi: 10.1067/moe.2002.121545. [DOI] [PubMed] [Google Scholar]
- 15.Slootweg PJ, Muller H. Juvenile ossifying fibroma. Report of four cases. J Craniomaxillofac Surg. 1990;18:125–129. doi: 10.1016/S1010-5182(05)80329-4. [DOI] [PubMed] [Google Scholar]
- 16.Slootweg PJ, Panders AK, Koopmans R, Nikkels PG. Juvenile ossifying fibroma. An analysis of 33 cases with emphasis on histopathological aspects. J Oral Pathol Med. 1994;23:385–388. doi: 10.1111/j.1600-0714.1994.tb00081.x. [DOI] [PubMed] [Google Scholar]
- 17.Chrcanovic BR, Gomez RS. Juvenile ossifying fibroma of the jaws and paranasal sinuses: a systematic review of the cases reported in the literature. Int J Oral Maxillofac Surg. 2019 doi: 10.1016/j.ijom.2019.06.029. [DOI] [PubMed] [Google Scholar]
- 18.Wang K, Ma XJ, Hao SY, Du J, Zhang LW, Zhang JT, Wu Z. Skull base juvenile psammomatoid ossifying fibroma: clinical characteristics, treatment, and prognosis. World Neurosurg. 2019;125:e843–e848. doi: 10.1016/j.wneu.2019.01.197. [DOI] [PubMed] [Google Scholar]
- 19.Hasselblatt M, Jundt G, Greiner C, Rama B, Schmal F, Iglesias-Rozas JR, van de Nes JA, Paulus W. Juvenile psammomatoid ossifying fibroma of the neurocranium. Report of four cases. Journal of neurosurgery. 2005;102:1151–1154. doi: 10.3171/jns.2005.102.6.1151. [DOI] [PubMed] [Google Scholar]
- 20.Das BK, Das SN, Gupta A, Nayak S. Florid cemento-osseous dysplasia. J Oral Maxillofac Pathol. 2013;17:150. doi: 10.4103/0973-029X.110735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenerty S, Shaw W, Verma R, Syed AB, Kuklani R, Yang J, Ali S. Florid cemento-osseous dysplasia: review of an uncommon fibro-osseous lesion of the jaw with important clinical implications. Skeletal Radiol. 2017;46:581–590. doi: 10.1007/s00256-017-2590-0. [DOI] [PubMed] [Google Scholar]
- 22.Mahomed F, Altini M, Meer S, Coleman H. Cemento-osseous dysplasia with associated simple bone cysts. J Oral Maxillofac Surg. 2005;63:1549–1554. doi: 10.1016/j.joms.2005.05.322. [DOI] [PubMed] [Google Scholar]
- 23.Sedano HO, Kuba R, Gorlin RJ. Autosomal dominant cemental dysplasia. Oral Surg Oral Med Oral Pathol. 1982;54:642–646. doi: 10.1016/0030-4220(82)90078-0. [DOI] [PubMed] [Google Scholar]
- 24.Coleman H, Altini M, Kieser J, Nissenbaum M. Familial florid cemento-osseous dysplasia–a case report and review of the literature. J Dent Assoc S Afr. 1996;51:766–770. [PubMed] [Google Scholar]
- 25.Kahn MF, Hayem F, Hayem G, Grossin M. Is diffuse sclerosing osteomyelitis of the mandible part of the synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO) syndrome? Analysis of seven cases. Oral Surg Oral Med Oral Pathol. 1994;78:594–598. doi: 10.1016/0030-4220(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 26.Kodama Y, Tanaka R, Kurokawa A, Ohnuki H, Sultana S, Hayashi T, Iizuka T, Takagi R. Severe destruction of the temporomandibular joint with complete resorption of the condyle associated with synovitis, acne, pustulosis, hyperostosis, and osteitis syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:e128–e133. doi: 10.1016/j.oooo.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Regezi J, Scriubba J, Jordan R. Inflammatory jaw lesions. In: Regezi J, Scriubba J, Jordan R, editors. Oral pathology clinical pathology correlates. 6. Amsterdam: Elsevier; 2012. pp. 324–325. [Google Scholar]
- 28.Muller-Richter UD, Roldan JC, Mortl M, Behr M, Reichert TE, Driemel O. SAPHO syndrome with ankylosis of the temporomandibular joint. Int J Oral Maxillofac Surg. 2009;38:1335–1341. doi: 10.1016/j.ijom.2009.03.724. [DOI] [PubMed] [Google Scholar]
- 29.Byrd L, Grossmann M, Potter M, Shen-Ong GL. Chronic multifocal osteomyelitis, a new recessive mutation on chromosome 18 of the mouse. Genomics. 1991;11:794–798. doi: 10.1016/0888-7543(91)90002-V. [DOI] [PubMed] [Google Scholar]
- 30.Abe K, Cox A, Takamatsu N, et al. Gain-of-function mutations in a member of the Src family kinases cause autoinflammatory bone disease in mice and humans. Proc Natl Acad Sci USA. 2019;116:11872–11877. doi: 10.1073/pnas.1819825116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mari A, Morla A, Melero M, Schiavone R, Rodriguez J. Diffuse sclerosing osteomyelitis (DSO) of the mandible in SAPHO syndrome: a novel approach with anti-TNF therapy. Systematic review. J Craniomaxillofac Surg. 2014;42:1990–1996. doi: 10.1016/j.jcms.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Hallmer F, Korduner M, Moystad A, Bjornland T. Treatment of diffuse sclerosing osteomyelitis of the jaw with denosumab shows remarkable results—a report of two cases. Clin Case Rep. 2018;6:2434–2437. doi: 10.1002/ccr3.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapurlat RD, Orcel P. Fibrous dysplasia of bone and McCune-Albright syndrome. Best Pract Res Clin Rheumatol. 2008;22:55–69. doi: 10.1016/j.berh.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 34.Lucas R. Pathology of tumors of oral tissues. 5. London: Churchill Livingstone; 1998. [Google Scholar]
- 35.Waldron CA. Fibro-osseous lesions of the jaws. J Oral Maxillofac Surg. 1993;51:828–835. doi: 10.1016/S0278-2391(10)80097-7. [DOI] [PubMed] [Google Scholar]
- 36.El-Mofty S. Bone lesions. In: Gnepp D, editor. Diagnostic surgical pathology of the head and neck. Philadelphia: Saunders, Elsevier; 2009. [Google Scholar]
- 37.Fitzpatrick KA, Taljanovic MS, Speer DP, Graham AR, Jacobson JA, Barnes GR, Hunter TB. Imaging findings of fibrous dysplasia with histopathologic and intraoperative correlation. AJR Am J Roentgenol. 2004;182:1389–1398. doi: 10.2214/ajr.182.6.1821389. [DOI] [PubMed] [Google Scholar]
- 38.Clauser L, Marchetti C, Piccione M, Bertoni F. Craniofacial fibrous dysplasia and Ollier’s disease: combined transfrontal and transfacial resection using the nasal-cheek flap. J Craniofac Surg. 1996;7:140–144. doi: 10.1097/00001665-199603000-00012. [DOI] [PubMed] [Google Scholar]
- 39.MacMahon H. Albright’s syndrome—thirty years later (polyostotic fibrous dysplasia) Pathol Annu. 1976;6:81–146. [PubMed] [Google Scholar]
- 40.Shenker A, Weinstein LS, Sweet DE, Spiegel AM. An activating Gs alpha mutation is present in fibrous dysplasia of bone in the McCune-Albright syndrome. J Clin Endocrinol Metab. 1994;79:750–755. doi: 10.1210/jcem.79.3.8077356. [DOI] [PubMed] [Google Scholar]
- 41.Happle R. The McCune-Albright syndrome: a lethal gene surviving by mosaicism. Clin Genet. 1986;29:321–324. doi: 10.1111/j.1399-0004.1986.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 42.Tabareau-Delalande F, Collin C, Gomez-Brouchet A, et al. Diagnostic value of investigating GNAS mutations in fibro-osseous lesions: a retrospective study of 91 cases of fibrous dysplasia and 40 other fibro-osseous lesions. Mod Pathol. 2013;26:911–921. doi: 10.1038/modpathol.2012.223. [DOI] [PubMed] [Google Scholar]
- 43.Lee SE, Lee EH, Park H, et al. The diagnostic utility of the GNAS mutation in patients with fibrous dysplasia: meta-analysis of 168 sporadic cases. Hum Pathol. 2012;43:1234–1242. doi: 10.1016/j.humpath.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 44.Lietman SA, Ding C, Levine MA. A highly sensitive polymerase chain reaction method detects activating mutations of the GNAS gene in peripheral blood cells in McCune-Albright syndrome or isolated fibrous dysplasia. J Bone Joint Surg Am Vol. 2005;87:2489–2494. doi: 10.2106/JBJS.E.00160. [DOI] [PubMed] [Google Scholar]
- 45.Mariot V, Wu JY, Aydin C, Mantovani G, Mahon MJ, Linglart A, Bastepe M. Potent constitutive cyclic AMP-generating activity of XLalphas implicates this imprinted GNAS product in the pathogenesis of McCune-Albright syndrome and fibrous dysplasia of bone. Bone. 2011;48:312–320. doi: 10.1016/j.bone.2010.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jour G, Oultache A, Sadowska J, Mitchell T, Healey J, Nafa K, Hameed M. GNAS Mutations in Fibrous Dysplasia: a Comparative Study of Standard Sequencing and Locked Nucleic Acid PCR Sequencing on Decalcified and Nondecalcified Formalin-fixed Paraffin-embedded Tissues. Appl Immunohistochem Mol Morphol. 2016;24:660–667. doi: 10.1097/PAI.0000000000000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JS, FitzGibbon EJ, Chen YR, Kim HJ, Lustig LR, Akintoye SO, Collins MT, Kaban LB. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012;7(Suppl 1):S2. doi: 10.1186/1750-1172-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruggieri P, Sim FH, Bond JR, Unni KK. Malignancies in fibrous dysplasia. Cancer. 1994;73:1411–1424. doi: 10.1002/1097-0142(19940301)73:5<1411::AID-CNCR2820730516>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 49.Tanner HC, Jr, Dahlin DC, Childs DS., Jr Sarcoma complicating fibrous dysplasia. Probable role of radiation therapy. Oral Surg Oral Med Oral Pathol. 1961;14:837–846. doi: 10.1016/S0030-4220(61)80014-5. [DOI] [PubMed] [Google Scholar]
- 50.Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer. 2009;125:229–234. doi: 10.1002/ijc.24320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tran LM, Mark R, Meier R, Calcaterra TC, Parker RG. Sarcomas of the head and neck. Prognostic factors and treatment strategies. Cancer. 1992;70:169–177. doi: 10.1002/1097-0142(19920701)70:1<169::AID-CNCR2820700127>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 52.Fernandes R, Nikitakis NG, Pazoki A, Ord RA. Osteogenic sarcoma of the jaw: a 10-year experience. J Oral Maxillofac Surg. 2007;65:1286–1291. doi: 10.1016/j.joms.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 53.Sato M, Tanaka N, Sato T, Amagasa T. Oral and maxillofacial tumours in children: a review. Br J Oral Maxillofac Surg. 1997;35:92–95. doi: 10.1016/S0266-4356(97)90682-3. [DOI] [PubMed] [Google Scholar]
- 54.McHugh JB, Thomas DG, Herman JM, Ray ME, Baker LH, Adsay NV, Rabah R, Lucas DR. Primary versus radiation-associated craniofacial osteosarcoma: biologic and clinicopathologic comparisons. Cancer. 2006;107:554–562. doi: 10.1002/cncr.22019. [DOI] [PubMed] [Google Scholar]
- 55.Cheng YS, Wright JM, Walstad WR, Finn MD. Osteosarcoma arising in Paget’s disease of the mandible. Oral Oncol. 2002;38:785–792. doi: 10.1016/S1368-8375(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 56.Lee YY, Van Tassel P, Nauert C, Raymond AK, Edeiken J. Craniofacial osteosarcomas: plain film, CT, and MR findings in 46 cases. AJR Am J Roentgenol. 1988;150:1397–1402. doi: 10.2214/ajr.150.6.1397. [DOI] [PubMed] [Google Scholar]
- 57.Demicco EG, Deshpande V, Nielsen GP, Kattapuram SV, Rosenberg AE. Well-differentiated osteosarcoma of the jaw bones: a clinicopathologic study of 15 cases. Am J Surg Pathol. 2010;34:1647–1655. doi: 10.1097/PAS.0b013e3181ea50b2. [DOI] [PubMed] [Google Scholar]
- 58.Baumhoer D. Bone-related lesions of the jaws. Surg Pathol Clin. 2017;10:693–704. doi: 10.1016/j.path.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 59.Guerin M, Thariat J, Ouali M, et al. A new subtype of high-grade mandibular osteosarcoma with RASAL1/MDM2 amplification. Hum Pathol. 2016;50:70–78. doi: 10.1016/j.humpath.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 60.Ferrari D, Codeca C, Battisti N, et al. Multimodality treatment of osteosarcoma of the jaw: a single institution experience. Med Oncol. 2014;31:171. doi: 10.1007/s12032-014-0171-9. [DOI] [PubMed] [Google Scholar]
- 61.Baumhoer D, Brunner P, Eppenberger-Castori S, Smida J, Nathrath M, Jundt G. Osteosarcomas of the jaws differ from their peripheral counterparts and require a distinct treatment approach. Experiences from the DOESAK Registry. Oral Oncol. 2014;50:147–153. doi: 10.1016/j.oraloncology.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 62.Liang L, Zhang T, You Y, He Q, Fan Y, Liao G. An individual patient data meta-analysis on the effect of chemotherapy on survival in patients with craniofacial osteosarcoma. Head Neck. 2019;41:2016–2023. doi: 10.1002/hed.25668. [DOI] [PubMed] [Google Scholar]