

Abstract
Intraductal carcinomas (IDCs) of salivary gland are rare neoplasms. Here, we report a case of IDCs harboring TRIM27-RET fusion with mixed low grade and apocrine types. A 79-year-old male presented with slowly-growing left parotid mass for 2.5 years. Histologically, the tumor demonstrated two distinct morphologies; a classic intercalated duct and low grade apocrine component. The intercalated duct component was positive for S100, SOX10 and vimentin, and negative for DOG-1 and HER2. The apocrine component was positive for androgen receptor (AR) and focally positive for HER2. The tumor harbored a TRIM27-RET fusion by FISH, and was negative for ETV6 and PLAG1 rearrangements. This case is unusual in that it displays two true phenotypically distinct components, which has only rarely been reported. This is the first report of intraductal carcinoma with two true phenotypically distinct components composed of low-grade and apocrine types with RET rearrangement and TRIM27-RET fusion.
Keywords: Low-grade intraductal carcinomas, Low grade cribriform cystadenocarcinoma, RET rearrangement, TRIM27 rearrangement
Introduction
Intraductal carcinoma (IDC) is a recently re-classified salivary gland neoplasm in the World Health Organization (WHO) 2017 system [1], previously referred to as “low-grade salivary duct carcinoma” or “low-grade cribriform cystadenocarcinoma”. The understanding of IDCs’ morphology, immunophenotype and molecular genetics are continuing to evolve, as this tumor is extremely rare. IDCs typically show an “intercalated duct” phenotype, demonstrating S100 and SOX10 positivity, though varying levels of apocrine morphology have been noted, supported immunphenotypically by androgen receptor (AR) immunopositivity. Pure apocrine IDC are considered to be distinct biologically and are more closely related to conventional salivary duct carcinoma (SDC). Conversely, though rare, invasive adenocarcinomas of intercalated duct phenotype can arise from intercalated duct IDC. Classically, IDCs are entirely intraductal with nests and cysts of varying size formed by a cellular proliferation resembling atypical ductal hyperplasia or ductal carcinoma in situ of the breast [2]. IDCs are usually slow-growing and with an indolent clinical course and an excellent prognosis [3–8].
Studies investigating the molecular genetics of IDC showed recurrent rearrangements of the RET gene in 23–47% of IDCs [9, 10]. When queried, the NCOA4-RET appears to be the more common fusion but a novel TRIM27-RET has been described as well [10]. RET gene rearrangement was most common in IDCs with an intercalated duct type (6 of 14 cases tested) and also found in 1 IDC with an invasive adenocarcinoma of intercalated duct type. However, RET alterations are exceptionally rare in the pure apocrine subtype. In one study, two RET rearranged IDC cases had hybrid intercalated duct and apocrine features; the hybrid features in these cases were largely due to overlap of features in the same cell population (dual staining for S100 and AR) [9]. Herein we report a third mixed IDC with TRIM27-RET fusion, but which uniquely shows two morphologically and immunophenotypically distinct intercalated duct and apocrine cellular components. This report will further expand the current understanding of the pathologic and genetic spectrum of IDC.
Case Report
Clinical Presentation
A 79-year-old man with a past history of seminoma of the right testis, treated by surgery and radiation therapy 2 years previously, underwent follow-up imaging by positron emission tomography-computed tomography (PET CT) which demonstrated a 1.2 × 1.0 cm hypermetabolic lesion in the left parotid. He was asymptomatic and the lesion was non-palpable on exam at that time, so the clinical team decided to follow him with serial imaging. Two and a half years later, at routine follow-up the left parotid mass was now palpable on exam, and PET-CT showed mild interval enlargement of the mass with a 1.2 cm heterogeneously enhancing well-defined nodule in the inferior aspect of the superficial left parotid lobe (Fig. 1). Ultrasound guided fine-needle aspiration (FNA) of the tumor was performed which showed atypical cells. In view of the abnormal FNA result, the increasing size, and heterogenous enhancement, a superficial parotidectomy was performed. All margins were negative for neoplasm. At 1 year follow-up, the patient is doing well with no evidence of recurrence.
Fig. 1.

PET-CT of the mass. CT scan in coronal view (a) and axial view (b) showed a mass with a 1.2 cm, well-defined, heterogeneously enhancing mass (arrows) in the inferior aspect of the superficial left parotid lobe
Materials and Methods
Cytology
Ultrasound guided fine-needle aspiration (UG-FNA) was performed by the patient’s otolaryngologist. One pass was performed with a #21 gauge needle and all material was rinsed into CytoLyt® solution. The needle rinse material was used for a ThinPrep® preparation; insufficient material was present for a cell block. The ThinPrep was stained with Papanicolaou stain.
Histology and Immunohistochemistry
The surgically resected tumor was fixed in buffered formalin and embedded for routine histological examination. Immunohistochemistry was performed on 4-micron formalin-fixed, paraffin-embedded sections at Cleveland Clinic as previously described [11, 12]. Antibodies used included CK7 (DAKO, OV-TL 12/30, 1:40), Mammaglobin (Cell Marque, 31A5, 1:40), CD117 (DAKO, polyclonal, 1:500), synaptophysin (Biogenex, snp88, predilute), chromogranin (DAKO, DAK-A3, 1:100), P63 (Ventana, 4A4, predulite), CK5/6 (Millipore, D5/16B4, 1:150), SMA (DAKO, 3E6, predilute), S100 (DAKO, polyclonal, 1:800), SOX10 (Biocare, polyclonal, predilute), Vimentin (Ventana, 3B4, predilute), DOG-1 (Leica, K9, 1:100), Androgen receptor (AR) (DAKO, AR441, 1:100), HER2 (Ventana, 4B5, predilute). Stains were performed on an automated immunostainer (Ventana Benchmark, Ventana Medical Systems, Tucson, AZ) and 3,3′-diaminobenzidine detection (DAB, Ventana) with appropriate negative and positive controls. A mucicarmine stain was also performed at the Cleveland Clinic.
Molecular Testing
Fluorescence in Situ Hybridization (FISH) studies for RET, ETV6 and PLAG1 were performed on 4 µm thick sections of formalin- fixed paraffin-embedded tissue at University of Pittsburgh as previously described [13]. Briefly, custom bacterial artificial chromosome (BAC) probes, flanking the RET gene, were obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA) (http://bacpac.chori.org) and were chosen according to the UCSC genome browser (http://genome.ucsc.edu). Briefly, DNA from individual BACs was isolated according to the manufacturer’s instructions and labeled with different fluorochromes in a nick translation reaction [13]. They were then denatured, hybridized to pretreated unstained coated slides, incubated, washed, and mounted with DAPI in an antifade solution. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes [13]. Two hundred nonoverlapping nuclei were scored using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems, Watertown, MA). The result was confirmed as positive for rearrangement when ≥ 10.6% of the nuclei examined showed a break-apart signal pattern (this cutoff represents 2 standards of deviation beyond the mean number of split signals seen in negative controls). FISH for ETV6 was performed using a commercially available probe (Abbot Molecular, Des Plains, IL) and analyzed similarly to those tested by BAC probes.
To investigate the RET rearrangement previously identified at University of Pittsburgh, further FISH studies were performed at the Mayo Clinic Cytogenetic Core Facility, examining the TRIM27 locus. Similar to the method mentioned above, 4 µm thick formalin- fixed paraffin-embedded tissue sections were used. For rearrangement of TRIM27, Agilent (Agilent Technologies, Inc. CA) custom designed 3′TRIM27 labeled with FITC and 5′TRIM27 labeled with Cy3 were combined as one break-apart probe set, while for TRIM27/RET fusion, Agilent (Agilent Technologies, Inc.) custom designed TRIM27 labeled with Cy3 and RET labeled with FITC were combined as one dual-fusion probe set. Slides were placed in a 90 °C oven for 15 min. Slides were then deparaffinized with xylene (2 times, 15 min each) at room temperature (RT), dehydrated in 100% ethanol for 5 min at RT, and placed in 10 mM Citric Acid (pH 6.0) and microwaved for 10 min. Following this, the slides were immersed in 2 × standard saline citrate (SSC) for 5 min at 37 °C followed by digestion in 0.2% pepsin working solution (1.2 g pepsin/600 mL 0.9% NaCl pH 1.5) at 37 °C for 48 min. Immediately after digestion, the slides were dehydrated using an ethanol series (70, 85, 100%) 2 min each at RT.
Results
Fine Needle Aspiration Biopsy
The ThinPrep was cellular and the background was clean with scattered macrophages and lymphocytes, and lacked necrotic debris or mucin. Two distinct cell populations were appreciated morphologically. The predominant cellular population had a relatively smaller size, uniform nuclei with dispersed chromatin and low nuclear/cytoplasmic ratio (Fig. 2a). The other cell population was larger with abundant cytoplasm granular and enlarged nuclei with central nucleoli suggestive of apocrine differentiation (Fig. 2b). Given the unusual appearance of a possible biphasic neoplasm, and the lack of sufficient material for further evaluation, a generic interpretation of “atypical cells present” was rendered. The differential diagnosis included benign neoplasms and low grade malignancy.
Fig. 2.

Papanicolaou stain on FNA sample. a Intercalated-type cells with relatively smaller size, uniform nuclei with dispersed chromatin and low nuclear/cytoplasmic ratio. b Apocrine type cells with abundant cytoplasm and enlarged nuclei and nucleoli features (Papanicolaou, 100 ×, bar = 25 µm)
Histology
In the surgical resection, a discRETe, well-circumscribed tan-white 1.7 cm mass was identified within the parotid gland. The mass was firm and solid with several small central cysts on cross section. Histologically, the mass contained two distinct cell types by H&E morphology (Fig. 3a). The predominant component was intercalated duct type, which had solid/nested growth with focal cribriform structures and scant cytoplasm with clear cell change (Fig. 3b). These cells were monomorphic with round to overall nuclei and dispersed chromatin. In some areas glandular and cribriform structures contained secretory material. The other component was apocrine, characterized by cribriform and micropapillary intraductal growth within larger cystic spaces (Fig. 3c). These cells had larger nuclei, hyperchromasia, variable nucleoli, and had abundant eosinophilic cytoplasm and occasional apocrine snouts. The apocrine component varied from bland to areas with considerable pleomorphism and scattered mitotic figures, but no necrosis or atypical mitotic figures were appreciated.
Fig. 3.

Surgical resection (H&E). a Low power view showed a discrete, well-circumscribed mass with solid component at the periphery with several small central microcysts. (H&E, 2 ×, bar = 3 mm); b high power view of the solid area showing it was composed of intercalated duct cell-type with solid/nested growth patten and scant cytoplasm with clear cell change. c High power view of the apocrine cells in the central area showing the cells had larger nuclei, hyperchromasia, and variable nucleoli, and had abundant eosinophilic cytoplasm and occasional apocrine snouts. (H&E, 40 ×, bar = 50 µm)
Immunohistochemistry
Immunohistochemical stains showed that the entire epithelial proliferation was intraductal, as demonstrated by an intact myoepithelial layer around the neoplastic cells, as highlighted by P63, CK5/6 and SMA; these stains were negative in both neoplastic cell components. Both components were strongly and diffusely positive for CK7 and mammaglobin (Fig. 4g, h), and negative for CD117, DOG-1, synaptophysin and chromogranin. The intercalated duct component was positive for S100 (Fig. 4c), SOX10 (Fig. 4e) and vimentin, and negative for AR (Fig. 4i), and HER2 (Fig. 4k). The apocrine component was positive for AR (Fig. 4j), focally positive for HER2 (Fig. 4l), and negative for S100 (Fig. 4d), SOX10 (Fig. 4f), and vimentin. Mucicarmine stain highlighted extracellular secretory material as well as rare cells with intracytoplasmic mucin, mainly in the intercalated duct component.
Fig. 4.
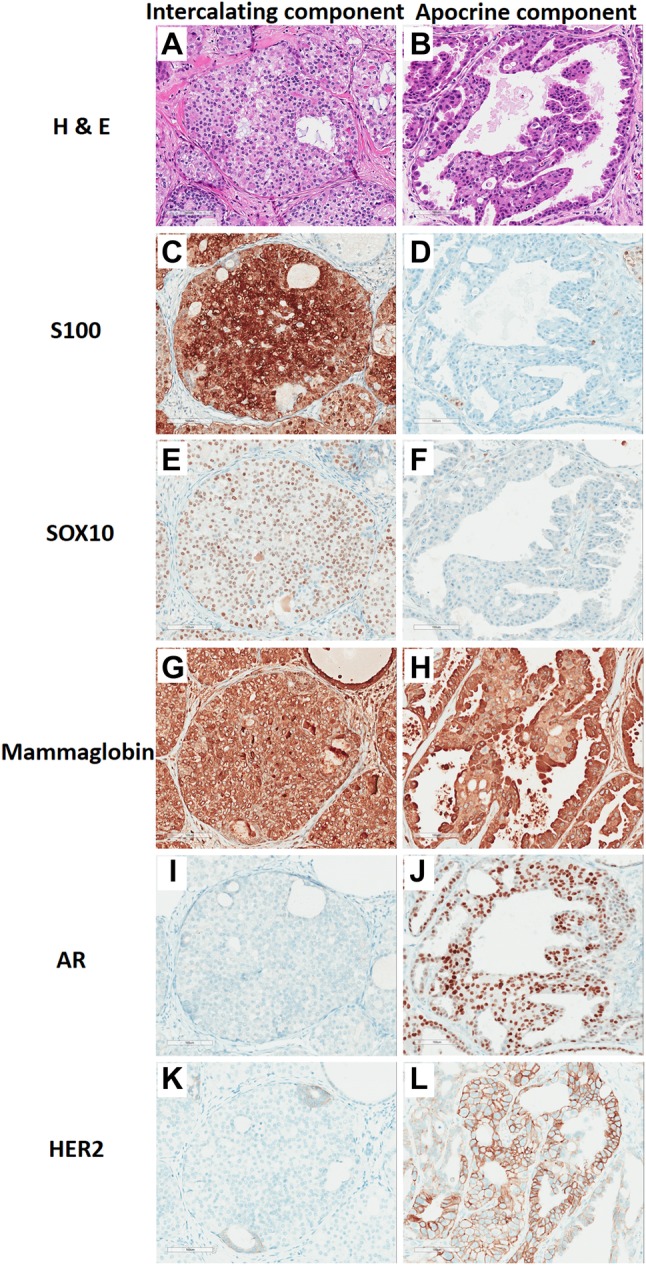
Surgical resection comparative immunohistochemistry. The intercalated duct component was monomorphic and bland with a nested clear cell pattern (a), and was strongly positive for S100 (c), SOX10 (e), and mammaglobin (g), and negative for AR (i) and HER2 (k). The apocrine component had larger nuclei, variable nucleoli, and had abundant eosinophilic cytoplasm (b), and not express S100 (d) and SOX10 (f), but strongly expressed mammaglobin (h) and AR (j) and partially expressed membranous HER2 (l). (20 ×, Bar = 50 µm)
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) detected RET rearrangement in 75.8% neoplastic cells of both cell types. Simultaneously, ETV6 and PLAG1 rearrangement were negative. Further FISH studies detected rearrangement of TRIM27 in 70% of cells (Fig. 5a) and TRIM27/RET fusion in 80% of cells (Fig. 5b).
Fig. 5.

Fusion interphase FISH assays for rearrangement of TRIM27 and for TRIM27/RET fusion. Separate green and red signals indicate break apart of the TRIM27 gene (a) and fusion yellow signals indicate TRIM27-RET fusion (b) (arrows: the cells with a rearrangement and fusion)
Discussion
Since the original description of salivary “intraductal carcinoma” in 1983 [4], multiple different names have been used to refer to this neoplasm in the literature including “low-grade salivary duct carcinoma,” “low-grade cribriform cystadenocarcinoma,” and “salivary duct carcinoma in situ.” [4, 14–16]. In 2005 the WHO classified these tumors as “low-grade cribriform cystadenocarcinoma” [14] because the relationship with salivary ductal carcinoma (SDC) was uncertain. Supporting this decision was the presence of diffuse strong S100 expression, good prognosis and predominant intraductal growth [15, 17], while conventional SDC is a high-grade adenocarcinoma with an aggressive clinical course that is S100 negative. In 2017 the WHO again reclassified these tumors as “intraductal carcinoma” to further separate IDC from SDC to prevent clinical confusion, as IDCs appear to be almost exclusively intraductal, and often lack the apocrine morphology seen in SDC [1].
With classification of these tumors based on intraductal location came recognition that different morphologic and immunophenotypic subtypes of IDC can occur. A recent series of 23 IDCs showed they mainly present as two histological and immunohistochemical phenotypes: intercalated duct type and apocrine type [9]. The intercalated duct type show identical features to the previously described “low-grade cribriform cystadenocarcinoma” according to WHO 2005, staining for S100 and mammaglobin and having a good prognosis. Histologically, intercalated duct type tumor cells are small, with scant to moderate eosinophilic to amphophilic cytoplasm, small nuclei, and lack nuclear atypia [9]. In contrast, IDCs with pure apocrine exhibit apocrine snouts and decapitation secretions, have more abundant cytoplasm, and often show higher-grade features including large nuclei with prominent nucleoli [9]. Pure apocrine IDCs lack S100 and SOX10 expression, and show AR expression. In addition, the majority of pure apocrine IDCs are identified in association with invasive carcinoma [9].
Recent papers examining the molecular genetics of IDCs have shown recurring alterations in the RET gene, most commonly NCOA4-RET fusion, but rarely TRIM27-RET [10]. A limited number of IDCs showing a hybrid (some features of both in the same cell population) or mixed (two distinct cell populations) intercalated duct and apocrine phenotypes have been reported. Weinreib and colleagues reported a series of IDC with apocrine phenotype in 2006 [2], of which two cases had a hybrid phenotype positive for both S100 and AR and both were low-grade by histologic criteria; one of these was shown to have a RET rearrangement with an unknown partner in their 2018 study (Case 2) [9]. Weinreib and colleagues also reported an additional case of hybrid IDC in 2018, which showed hybrid intercalated duct and apocrine features, with staining for both S100 and AR in the same cells, although it did show focal loss of S100 in the areas with greatest apocrine differentiation. This case was also positive for a RET alteration suggestive of a break-apart signal with an unknown partner (Case 15) [9], and was included in the Skalova paper published in AJSP in 2018 [10], which showed TRIM27-RET fusion by next generation sequencing. One other IDC has been reported to show TRIM27-RET fusion, and was reported to show hybrid apocrine features, was positive for S100 and mammogloblin, but had a high nuclear grade with minimal invasion (Case 3) [10]. Our current case is the third reported IDC with a TRIM27-RET fusion, and the first case of a mixed IDC (showing two distinct morphologic intercalated duct and apocrine components with distinct immunophenotypes) rather than hybrid morphology and immunohistochemistry in the same population of cells.
The cytologic features of intraductal carcinoma (previously reported as low grade cribriform cystadenocarcinoma) have been varied, although the most common pattern is that of cells with a low nuclear:cytoplasmic ratio cuboidal cells with bland round nuclei and coarse chromatin [18–20]. The solid/nested intercalated component in the current case showed similar cytologic features to these prior reports [18, 19]. However, other reports have shown differing cytologic patterns, including apocrine-like or mucin-producing epithelial cells arranged in cystic, pseudopapillary or cribriform structures [20, 21]. The apocrine component in the current case was similar to these other reports, with occasional apical snouts. The reported background of IDCs on FNA was also variable from clean background without necrosis and mucin [20] to widespread cellular necrosis [21]. In the current case, the background was clean without necrotic debris or mucin. Scattered inflammatory cells composed of macrophages and rare lymphocytes suggested a possible cystic component.
Histologically, IDC can mimic other entities in salivary glands including sclerosing polycystic adenosis (SPA), (high-grade) salivary duct carcinoma (SDC) and secretory carcinoma (SC) (formerly mammary analogue secretory carcinoma, or MASC). SPA is a reactive, inflammatory lesion of the salivary glands resulting in degenerativa fibrocystic changes and adenosis [22]. It frequently harbors intraductal epithelial proliferations with low grade atypia [22] similar to that seen in IDC. However, the predominant cells are acinar and apocrine without atypia [21]. Moreover, the stroma harbors a variably intense chronic inflammatory infiltrate which may contain lymphoid follicles with germinal centers [22]. Immunophenotypically, SPA expresses AE1/AE3, Cam 5.2, cytokeratin 7 in both ductal and acinar cells [23, 24]. A myoepithelial cell layer was demonstrated by immunopositivity for anti-smooth muscle, p63 and S100 protein [25]. Dilated ducts lined by apocrine cells expressed strong nuclear immunoreactivity for AR, whereas ducts lined by proliferative epithelial cells do not exhibit AR positivity. Estrogen receptor, progesterone receptor and HER2 were completely negative [23]. Salivary duct carcinomas are typically widely invasive with high grade cytological features including prominent nucleoli, coarse chromatin, necrosis, and significant mitotic activity, including atypical mitotic figures [26]. Importantly, they are essentially defined by an apocrine phenotype and are almost invariably AR positive. They can also express mammaglobin, but S100 is typically negative and HER2 protein is commonly overexpressed [26]. SDCs have not been shown to have RET rearrangement or ETV6 rearrangement while SDC are characterized by a myriad of molecular alterations including ERB2 amplification, mutations involving the MAP/PI3K pathways and TP53 mutations [27]. Secretory carcinomas can mimic IDC cytologically and architecturally. They have secretory features similar to apocrine component in IDC, and architecturally, they can have varying proportions of solid, microcystic, tubular, papillary, and cribriform growth patterns akin IDC [26]. Immunohistochemically, SCs typically express S100, mammaglobin and vimentin, and are negative for AR and HER2. However, SC are defined by recurrent rearrangements involving the ETV6 gene, and are infiltrative despite focal intraductal growth, which can facilitate distinguishing them from IDC [26].
In summary, the current report illustrates a unique mixed type IDC case with morphologically and immunophenotypically distinct intercalated duct and apocrine type components, which harbored a TRIM27-RET gene fusion. Additional cases of IDC, particularly those showing hybrid or mixed features, will be needed to better understand the clinical, pathologic and genetic spectrum of this rare disease.
Funding
No funding was received for this work from any organizations.
Compliance with Ethical Standards
Conflict of interest
All the authors have no conflict of interest to disclose.
References
- 1.Loening T, Leivo I, Simpson RHW, et al. Intraductal carcinoma. In: El-Naggar AK, Chan JKC, Grandis JR, Takata T, Slootweg PJ, et al., editors. WHO classification of head and neck tumours. Lyon: IARC Press; 2017. pp. 170–171. [Google Scholar]
- 2.Weinreb I, Tabanda-Lichauco R, Van der Kwast T, Perez-Ordoñez B. Low-grade intraductal carcinoma of salivary gland: report of 3 cases with marked apocrine differentiation. Am J Surg Pathol. 2006;30:1014–1021. doi: 10.1097/00000478-200608000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Anderson C, Muller R, Piorkowski R, et al. Intraductal carcinoma of major salivary gland. Cancer. 1992;69:609–614. doi: 10.1002/1097-0142(19920201)69:3<609::AID-CNCR2820690302>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 4.Chen KT. Intraductal carcinoma of the minor salivary gland. J Laryngol Otol. 1983;97:189–191. doi: 10.1017/S002221510009397X. [DOI] [PubMed] [Google Scholar]
- 5.Chen KT. Cytology of salivary duct carcinoma. Diagn Cytopathol. 2000;22:132–134. doi: 10.1002/(SICI)1097-0339(200002)22:2<132::AID-DC17>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 6.Khurana KK, Pitman MB, Powers CN, et al. Diagnostic pitfalls of aspiration cytology of salivary duct carcinoma. Cancer. 1997;81:373–378. doi: 10.1002/(SICI)1097-0142(19971225)81:6<373::AID-CNCR12>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 7.Tatemoto Y, Ohno A, Osaki T. Low malignant intraductal carcinoma on the hard palate: a variant of salivary duct carcinoma? Eur J Cancer B. 1996;32:275–277. doi: 10.1016/0964-1955(95)00092-5. [DOI] [PubMed] [Google Scholar]
- 8.Watatani K, Shirasuna K, Aikawa T, et al. Intraductal carcinoma of the tongue: report of a case. Int J Oral Maxillofac Surg. 1991;20:175–176. doi: 10.1016/S0901-5027(05)80011-6. [DOI] [PubMed] [Google Scholar]
- 9.Weinreb I, Bishop JA, Chiosea SI, et al. Recurrent RET gene rearrangements in intraductal carcinomas of salivary gland. Am J Surg Pathol. 2018;42(4):442–452. doi: 10.1097/PAS.0000000000000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skálová A, Vanecek T, Uro-Coste E, Bishop JA, Weinreb I, Thompson LDR, de Sanctis S, Schiavo-Lena M, Laco J, Badoual C, Santana Conceiçao T, Ptáková N, Baněčkova M, Miesbauerová M, Michal M. Molecular profiling of salivary gland intraductal carcinoma revealed a subset of tumors harboring NCOA4-RET and novel TRIM27-RET fusions: a report of 17 cases. Am J Surg Pathol. 2018;42(11):1445–1455. doi: 10.1097/PAS.0000000000001133. [DOI] [PubMed] [Google Scholar]
- 11.Tubbs RR, Sheibani K. Immunohistology of lymphoproliferative disorders. Semin Diagn Pathol. 1984;1:272–284. [PubMed] [Google Scholar]
- 12.Goldblum JR, Shannon R, Kaljiam EP, et al. Immunohistochemical assessment of proliferative activity in adrenocortical neoplasms. Mod Pathol. 1993;6:663–668. [PubMed] [Google Scholar]
- 13.Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-sixcases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1gene. Genes Chromosomes Cancer. 2010;49:1114–1124. doi: 10.1002/gcc.20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandwein-Gensler MS, Gnepp DR. WHO classification of tumours. In: Barnes L, Eveson JW, Reichart P, Sidransky D, editors. Pathology and genetics of head and neck tumours. Lyon: IARC Press; 2005. p. 233. [Google Scholar]
- 15.Delgado R, Klimstra D, Albores-Saavedra J. Low grade salivary duct carcinoma. A distinctive variant with a low grade histology and a predominant intraductal growth pattern. Cancer. 1996;78:958–967. doi: 10.1002/(SICI)1097-0142(19960901)78:5<958::AID-CNCR4>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 16.Simpson RH, Desai S, Di Palma S. Salivary duct carcinoma in situ of the parotid gland. Histopathology. 2008;53:416–425. doi: 10.1111/j.1365-2559.2008.03135.x. [DOI] [PubMed] [Google Scholar]
- 17.Brandwein-Gensler M, Hille J, Wang BY, et al. Low-grade salivary duct carcinoma: description of 16 cases. Am J Surg Pathol. 2004;28:1040–1044. doi: 10.1097/01.pas.0000128662.66321.be. [DOI] [PubMed] [Google Scholar]
- 18.Nakazawa T, Kondo T, Yuminomochi T, et al. Fine-needle aspiration biopsy of low-grade cribriform cystadenocarcinoma of the salivary gland. Diagn Cytopathol. 2011;39:218–222. doi: 10.1002/dc.21405. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Liu Y, Lin X, et al. Low-grade cribriform cystadenocarcinoma of salivary glands: report of two cases and review of the literature. Diagn Pathol. 2013;8:28–33. doi: 10.1186/1746-1596-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohta Y, Hirota Y, Kohno Y, et al. Cytology of low-grade cribriform cystadenocarcinoma in salivary glands: cytological and immunohistochemical distinctions from other salivary gland neoplasms. Diagn Cytopathol. 2016;44:241–245. doi: 10.1002/dc.23411. [DOI] [PubMed] [Google Scholar]
- 21.Barizzi J, Merlo E, Schönegg R, et al. Pure intraductal carcinoma of the parotid gland: cytologic findings on FNA sample. Report of one case. Diagn Cytopathol. 2017;45(11):1046–1049. doi: 10.1002/dc.23770. [DOI] [PubMed] [Google Scholar]
- 22.Petersson F. Sclerosing polycystic adenosis of salivary glands: a review with some emphasis on intraductal epithelial proliferations. Head Neck Pathol. 2013;7(Suppl 1):97–106. doi: 10.1007/s12105-013-0465-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manojlović S, Virag M, Milenović A, et al. Sclerosing polycystic adenosis of parotid gland: a unique report of two cases occurring in two sisters. Pathol Res Pract. 2014;210(6):342–345. doi: 10.1016/j.prp.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Shilpi A, Ansari F, Bahadur S, et al. Sclerosing polycystic adenosis: a rare tumor misdiagnosed as retention cyst on fine needle aspiration cytology. Diagn Cytopathol. 2017;45(7):640–644. doi: 10.1002/dc.23701. [DOI] [PubMed] [Google Scholar]
- 25.Espinosa CA, Rua L, Torres HE, Del FernándezValle Á, Fernandes RP, Devicente JC. Sclerosing polycystic adenosis of the parotid gland: a systematic review and report of 2 new cases. J Oral Maxillofac Surg. 2017;75(5):984–993. doi: 10.1016/j.joms.2016.10.031. [DOI] [PubMed] [Google Scholar]
- 26.Stevens TM, Kovalovsky AO, Velosa C, et al. Mammary analog secRETory carcinoma, low-grade salivary duct carcinoma, and mimickers: a comparative study. Mod Pathol. 2015;28(8):1084–1100. doi: 10.1038/modpathol.2015.64. [DOI] [PubMed] [Google Scholar]
- 27.Williams L, Thompson LD, Seethala RR, et al. Salivary duct carcinoma: the predominance of apocrine morphology, prevalence of histologic variants, and androgen receptor expression. Am J Surg Pathol. 2015;39(5):705–713. doi: 10.1097/PAS.0000000000000413. [DOI] [PubMed] [Google Scholar]