

Spliced X-box binding protein 1 (XBP-1s), part of the unfolded protein response and expressed in developing germinal center B cells, can induce Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic replication and directly activate viral interleukin-6 (vIL-6). We show here that XBP-1s can also directly activate KSHV ORF21, a lytic gene. ORF21 encodes KSHV thymidine kinase (TK), which increases the pool of dTTP for viral replication and enhances lytic replication. Direct activation of ORF21 by XBP-1s can enhance viral replication in germinal center B cells and contribute to the pathogenesis of KSHV multicentric Castleman disease (MCD). KSHV-MCD is characterized by systemic inflammation caused, in part, by lytic replication and overproduction of KSHV vIL-6 in XBP-1s-expressing lymph node plasmablasts. KSHV thymidine kinase can phosphorylate zidovudine and ganciclovir to toxic moieties, and direct activation of ORF21 by XBP-1s may also help explain the effectiveness of zidovudine and valganciclovir in the treatment of KSHV-MCD.
KEYWORDS: HHV-8, KSHV, Kaposi, ORF21, thymidine kinase, XBP-1, herpesvirus
ABSTRACT
Kaposi’s sarcoma-associated herpesvirus (KSHV) is the causative agent for Kaposi sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman disease (MCD). Like other herpesviruses, it has latent and lytic repertoires. However, there is evidence that some lytic genes can be directly activated by certain cellular factors. Cells undergoing endoplasmic reticulum stress express spliced X-box binding protein 1 (XBP-1s). XBP-1s is also present in large amounts in germinal center B cells. XBP-1s can activate the KSHV replication and transcription activator (RTA) and lytic replication. It can also directly activate KSHV-encoded viral interleukin-6 (vIL-6) and, thus, contribute to the pathogenesis of KSHV MCD. KSHV thymidine kinase (TK), the ORF21 gene product, can enhance the production of dTTP and is important for lytic replication. It can also phosphorylate zidovudine and ganciclovir to toxic moieties, enabling treatment of KSHV-MCD with these drugs. We show here that XBP-1s can directly activate ORF21 and that this activation is mediated primarily through two XBP-response elements (XRE) on the ORF21 promoter region. Deletion or mutation of these elements eliminated XBP-1s-induced upregulation of the promoter, and chromatin immunoprecipitation studies provide evidence that XBP-1s can bind to both XREs. Exposure of PEL cells to a chemical inducer of XBP-1s can induce ORF21 within 4 hours, and ORF21 expression in the lymph nodes of patients with KSHV-MCD is predominantly found in cells with XBP-1. Thus, XBP-1s may directly upregulate KSHV ORF21 and, thus, contribute to the pathogenesis of KSHV-MCD and the activity of zidovudine and valganciclovir in this disease.
IMPORTANCE Spliced X-box binding protein 1 (XBP-1s), part of the unfolded protein response and expressed in developing germinal center B cells, can induce Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic replication and directly activate viral interleukin-6 (vIL-6). We show here that XBP-1s can also directly activate KSHV ORF21, a lytic gene. ORF21 encodes KSHV thymidine kinase (TK), which increases the pool of dTTP for viral replication and enhances lytic replication. Direct activation of ORF21 by XBP-1s can enhance viral replication in germinal center B cells and contribute to the pathogenesis of KSHV multicentric Castleman disease (MCD). KSHV-MCD is characterized by systemic inflammation caused, in part, by lytic replication and overproduction of KSHV vIL-6 in XBP-1s-expressing lymph node plasmablasts. KSHV thymidine kinase can phosphorylate zidovudine and ganciclovir to toxic moieties, and direct activation of ORF21 by XBP-1s may also help explain the effectiveness of zidovudine and valganciclovir in the treatment of KSHV-MCD.
INTRODUCTION
Kaposi’s sarcoma-associated herpesvirus (KSHV), also called human herpesvirus-8 (HHV-8), is a gammaherpesvirus that is the causative agent of several tumors or hyperproliferative diseases, including Kaposi’s sarcoma (KS), a form of multicentric Castleman disease (MCD), and primary effusion lymphoma (PEL) (1–4). These diseases most often develop in patients with immunodeficiency from human immunodeficiency virus (HIV) infection, transplantation, or other causes but can also develop in patients without obvious immunodeficiency, especially in sub-Saharan Africa (5). Like other herpesviruses, the life cycle of KSHV is composed of latent and lytic phases (6–8). In latent infection, only a restricted number of genes are expressed. When induced into the lytic phase, the full genome is expressed and viral replication and cell lysis ensues. This switch is mediated by the activation of the replication and transcription activator (RTA), which turns on the lytic program by activating other KSHV genes in an orderly cascade as well as inducing more RTA in a positive feedback loop (7–9). RTA can be activated in infected cells by a variety of stimuli, including inflammatory cytokines, a variety of cell signaling pathways, hypoxia, and endompasmic reticulum (ER) stress (10–14). Recently, it has been shown that some of the lytic genes of KSHV can be directly activated by cellular factors without the involvement of RTA (13, 15, 16). In particular, it has been shown that hypoxia-inducible factors (HIFs), which accumulate in cells exposed to hypoxia and certain other stresses, can directly induce KSHV open reading frames (ORFs) 34 to 37 and ORF74 by binding to hypoxia response elements (HREs) on the promoter region of these genes (13, 16, 17).
Spliced X-box binding protein 1 (XBP-1s) is one of the key proteins in the unfolded protein response (UPR) to endoplasmic reticulum (ER) stress caused by factors such as an excess of protein production (18, 19). In cells undergoing ER stress, a 26-nucleotide intron is excised from the mRNA of unspliced XBP-1 (XBP-1u) by inositol-requiring enzyme α (IRE1α) and then translated to create XBP-1s (20) which then enters the nucleus where it can activate specific cellular genes of the UPR by binding to XBP-1 response elements (XREs) in their promoter regions. High levels of XBP-1s are expressed in B cells undergoing differentiation to plasma cells in lymph node germinal centers, where it protects against ER stress induced by immunoglobulin production (21). XBP-1s has been reported to activate KSHV RTA both in normoxic cells and in cells exposed to hypoxia (12, 22, 23). In addition, XBP-1 has been shown to induce gene activation in other gammaherpesviruses; in particular, it can induce lytic replication and the expression of LMP1, a latent oncoprotein, in Epstein-Barr virus and can also indirectly induce lytic activation of murine gammaherpesvirus 68 through activation of the latent protein M2 (24–28).
KSHV-MCD is a severe systemic illness characterized by inflammatory flares caused by the overproduction of a number of cytokines, including KSHV-encoded human interleukin-6 (hIL-6); viral IL-6 (vIL-6), an analog of hIL-6; and interleukin-10 (IL-10) (29–32). KSHV-MCD is characterized by intermittent flares and is usually fatal within 2 years if not treated. The key pathological finding is the presence of KSHV-infected plasmablasts in lymph nodes, especially in the mantle zones of the germinal centers (33). These plasmablasts express KSHV latency-associated nuclear antigen (LANA), and a subset also express KSHV vIL-6, a gene inducible in the lytic cycle that is sometimes expressed at low levels during latency (15, 29, 34–36). In some of these plasmablasts that express vIL-6, other KSHV lytic genes, such as ORF45, are also expressed; however, more than 60% of those expressing vIL-6 do not express ORF45 (15). The production of vIL-6 by plasmablasts without the full lytic cascade of KSHV is believed to be important in KSHV-MCD pathogenesis; otherwise, the KSHV-infected plasmablasts would lyse and the disease would be self-limiting. Our group recently showed that XBP-1s can directly upregulate vIL-6 production by binding to XRE on the promoter of vIL-6 (15), and this likely contributes to the pathogenesis of KSHV-MCD.
Two genes of KSHV, namely, ORF21 and ORF36, can phosphorylate certain antiviral drugs, leading to the production of their active triphosphate moieties. ORF21, a thymidine kinase, can phosphorylate both zidovudine (AZT) and ganciclovir (GCV) (37–39), and ORF36, a phosphotransferase, can phosphorylate GCV (37, 39–41). In addition to manifesting antiviral activity, the phosphorylated forms of these drugs are toxic to the KSHV-infected cells (39). Based on this observation and the expression of KSHV lytic genes by at least some KSHV-MCD plasmablasts, we explored the use of these drugs as a treatment for flares of KSHV-MCD and observed that 86% of patients had major clinical responses when treated with a combination of high-dose AZT and valganciclovir, an oral prodrug of GCV (42). This is now a recommended treatment for KSHV-MCD (43). To try to better understand why this regimen might be effective given the activation of vIL-6 in many MCD plasmablasts without other lytic genes, we explored the possibility that ORF21 and ORF36 might also be directly activated in KSHV-MCD lymph nodes. Given the high levels of XBP-1s in germinal center developing B cells, we hypothesized that XBP-1s might also play a direct role in activating KSHV ORF21 and/or ORF36.
RESULTS
Identification of XREs in the ORF21 promoter.
To assess whether KSHV-encoded ORF21 and/or ORF36 could be upregulated by XBP-1s, we cotransfected HEK-293T cells with luciferase reporter plasmids for KSHV ORF21 (Fig. 1A), KSHV ORF36, KSHV vIL-6 (positive control), and pGL3basic control reporter plasmid, along with XBP-1s, XBP-1u, or a negative-control plasmid (pcDNA3.1). Consistent with our previous study (15), XBP-1s increased the promoter activity of the vIL-6 promoter by about 7-fold (P < 0.05) (Fig. 1B). In addition, XBP-1s, but not XBP-1u, increased the activity of the ORF21 promoter by about 5-fold (P < 0.01). In these experiments, some upregulation of the pGL3 basic promoter was also observed upon cotransfection with XBP-1s, but the upregulation of vIL-6 and ORF21 was substantially more. In contrast, no upregulation of the ORF36 promoter over that seen with the pGL3 basic promoter was seen upon cotransfection with XBP-1s (Fig. 1B). These results provide evidence that XBP-1s may activate the ORF21 promoter region but not the ORF36 promoter region.
FIG 1.

Schematic of ORF21 luciferase (LUC) promoter constructs and results of activation of various promoter constructs by XBP-1u and XBP-1s. (A) ORF21 promoter contains 4 XBP-1 response element (XRE) core sequences (5ʹ-ACGT-3ʹ) (44), including 2 consensus (5ʹ-NNGNTGACGTGKNNNWT-3ʹ) XRE sequences (3 and 4) within 1,239 bp upstream of the ORF21 start codon (nucleotide positions 34144 to 35382 of KSHV-BAC36; GenBank accession number HQ404500). Consensus XREs are indicated as black squares and the other two (core only) XREs as gray squares. XRE1, −8 to −5; XRE2, −262 to −259; XRE3, −321 to −318; XRE4, −629 to −626. Direction of each XRE is indicated with an arrow. Consensus core HREs are shown as black triangles. Constructs of promoters pORF21-624, pORF21-316, and pORF21-256 were made by sequential deletions as shown. (B) Comparison of the activation of the ORF21-1239, vIL-6, and ORF36 promoter luciferase reporter constructs by XBP-1 unspliced (XBP-1u) or spliced (XBP-1s). HEK-293T cells were cotransfected with 300 ng of different promoter luciferase plasmids and 50 ng of an internal β-Gal control plasmid (pGL3 basic empty vector) in the presence of 100 ng of an expression plasmid encoding XBP-1u, XBP-1s, or pcDNA3.1 expression plasmid control. Values are expressed as fold increase over the respective control reporter plasmid transfected with an empty expression vector (pcDNA3.1) and represent the mean of three independent experiments. Error bars denote the standard deviations, and asterisks show the P values (*P ≤ 0.05, **P ≤ 0.01) for the comparison shown with the pGL3B control. (C) Comparison of the activation of ORF21 and truncated forms of the ORF21 luciferase reporter by XBP-1s or pcDNA3.1 plasmid control. 293T cells were cotransfected with 300 ng of each ORF21 promoter and 50 ng of an internal β-Gal control plasmid in the presence of 100 ng of an expression plasmid encoding XBP-1s or pcDNA3.1 control. Values are expressed as fold increase over pGL3basic transfected with an empty expression vector (pcDNA3.1) and represent the mean of three independent experiments. Error bars denote the standard deviations, and asterisks show the P values as in (B).
We analyzed the promoter sequences of ORF21 and ORF36 for potential XRE and hypoxia response elements (HREs) (Fig. 1A) using MacVector software (version 14.5.3). Four XRE core sequences (ACGT) (44) were found within the 1,239-bp upstream region of the ORF21 start codon. Further analysis using MATCH (public version 1.0) showed that two of these potential XREs (XRE3 and XRE4) had full consensus XRE sequences. All four XREs are encoded in the 5′ to 3′ direction on the sense strand. In addition, the promoter region had 6 core HRE (RCGTG) sequences, where R is A or G. The ORF36 promoter region has 28 core XRE sequences but only 1 consensus XRE, and as previously described, it has 6 HREs (16). To further define the region of the ORF21 promoter that may mediate XBP-1-specific activation, we made truncated forms of the ORF21 promoter region, namely, pORF21-624 (lacking XRE4), pORF21-316 (lacking XRE3 and 4), and pORF21-256 (lacking XRE 2, 3, and 4). When cotransfected with XBP-1s, the upregulation of pORF21-624 was approximately half of that seen with pORF21-1239 (Fig. 1C). Also, pORF21-316 had a much lower level of XBP-1s-induced activity and the activity was completely lost in pORF21-256 (Fig. 1C). Taken together, these findings provide evidence that XRE3 and/or XRE4 are functional XREs that mediate XBP-1s upregulation of ORF21.
To further identify and characterize the functional XRE(s) that mediate the response to XBP-1, we performed site-directed mutagenesis of XRE3 and XRE4 on the full-length (1,239 bp) promoter. Two to four point mutations of the core XRE sequence of XRE3 were introduced into pORF21-1239WT to produce pORF21-1239XRE3M1, M2, and M3 and were introduced into the core sequence of XRE4 to produce pORF21-1239XRE4M1, M2, and M3 (Fig. 2A). All three mutations of XRE3 significantly decreased the XBP-1s-induced activation of pORF21-1239 in transfected HEK-293T cells, and mutation 3 virtually eliminated the XBP-1s-induced upregulation of the promoter (P ≤ 0.005) (Fig. 2B). In contrast, mutations of XRE4 had little or no effect (P > 0.05). These results indicate that XRE3 is the primary contributor to the response of the ORF21 promoter to XBP-1s, although XRE4 as well as other potential XREs may contribute to the full response.
FIG 2.
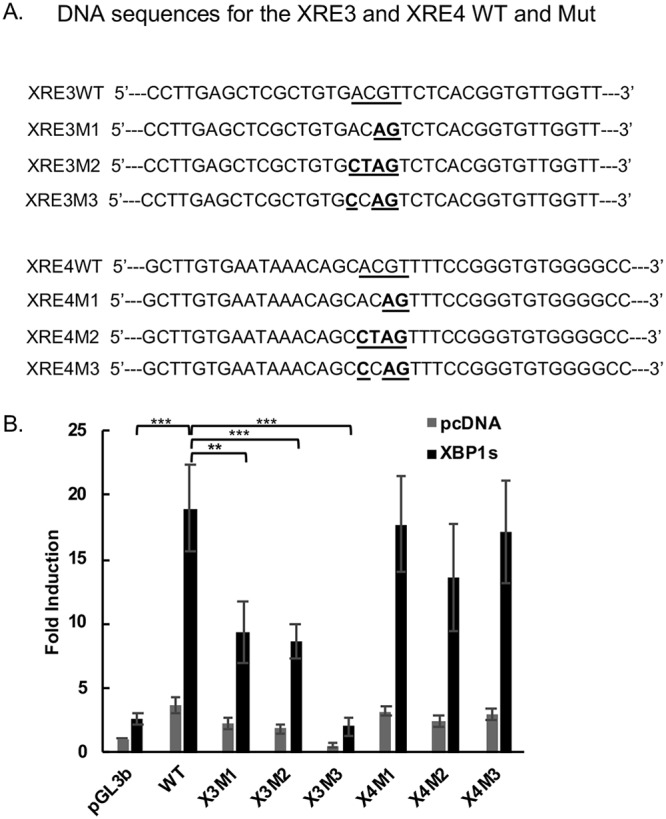
Effect of XRE3 and XRE4 mutations in the ORF21 promoter on the response to XBP1s. (A) Construct of the wild-type ORF21 and mutant reporter plasmids. Three different mutant reporters for each XRE were constructed in the pORF21-1239 full-length promoter, containing a 2- to 4-bp substitution within core XRE sequences. DNA sequences for the XRE3, XRE4 wild type, and mutant plasmids are shown. The underlined regions and bold letters indicate the mutations from wild type for each mutant construct. (B) Comparison of the activation of wild-type pORF21-1239 luciferase reporter with the XRE3 or XRE4 mutant luciferase reporters by XBP-1s. HEK-293T cells were cotransfected with 300 ng of pORF21-1239WT or X3 or X4 M1, 2, or 3 promoters and 50 ng of an internal β-Gal control plasmid in the presence of 100 ng of an expression plasmid encoding XBP-1s (black bars) or pcDNA3.1 expression plasmid control (gray bars). Values are expressed as fold increase over the pGL3basic reporter transfected with an empty expression vector (pcDNA3.1) and represent the mean of three independent experiments. Error bars denote the standard deviations. **P < 0.01 and ***P < 0.005 for the comparisons shown; none of the comparisons between the X4 mutations and wild type (WT) were significant (P > 0.05).
The ORF21 promoter contains potential HREs but does not respond to hypoxia.
It has been shown that germinal centers are hypoxic environments (45, 46). The ORF21 promoter (to bp 1,239) contains 6 hypoxia response element (HRE) consensus sequences ([A/G]CGTG), suggesting that it might also directly respond to hypoxia or hypoxia-inducible factors (HIFs) (47). To test this, cells were transfected with an ORF21 or ORF36 promoter, without or with an XBP-1s expression plasmid and then placed in a normoxia or hypoxia environment. While hypoxia induced a 2.0-fold upregulation of ORF36 in these experiments (P < 0.05), hypoxia did not significantly affect promoter activation of ORF21, suggesting that hypoxia does not activate the ORF21 promoter (Fig. 3A). Interestingly, there was a somewhat enhanced response of ORF21 to XBP-1s in the presence of hypoxia (29.6- versus 23.0-fold, P < 0.05) (Fig. 3A). To more specifically look at the effect of HIFs, we cotransfected the degradation-resistant HIF-1α (drHIF-1) with the pORF21-1239WT or pORF36 reporter plasmids. There was no increase in the ORF21-1239WT promoter response with dr-HIF-1 (Fig. 3B). In contrast, an ORF36 promoter construct (pORF36), used as a positive control, was upregulated 10-fold by dr-HIF-1 (Fig. 3B). We also determined if tunicamycin (TM), which increases levels of XBP-1s by inhibiting N-linked glycosylation, causing protein misfolding, and inducing ER stress (48), also upregulated HIF (and could potentially upregulate ORF36 or ORF21 by that mechanism). BCBL-1 cells were treated with TM (0.5 to 2.5 μg/ml); CoCl2 (75 μM), a hypoxia mimic, served as a positive control. While CoCl2 dramatically induced HIF-1α protein expression in BCBL-1 PEL cells, TM treatment increased XBP-1s protein (Fig. 3C and also seen in Fig. 5D of reference 15) but failed to increase HIF-1α protein expression in these cells (Fig. 3C). Therefore, TM does not appear to induce HIF-1α in BCBL-1 cells.
FIG 3.

The ORF21 promoter contains potential HREs but does not respond to hypoxia in the absence of XBP-1s. (A). Comparison of the activation of wild-type pORF21 and pORF36 luciferase reporter in response to hypoxia. HEK-293T cells were cotransfected with 300 ng of pORF21 or 36 luciferase promoter, 100 ng of an expression plasmid encoding XBP-1s (black bars) or pcDNA3.1 control (gray bars), and 50 ng of an internal β-Gal control plasmid, cultured in normoxia for 32 hours, and then cells were treated in normoxic or hypoxic (1% oxygen) conditions for 16 hours. Values are expressed as the fold increase over the value for the pGL3 basic reporter transfected with an empty expression vector (pcDNA3.1) in normoxia and represent the mean of three independent experiments. Error bars denote the standard deviations. (*P ≤ 0.05; NS, not significant). (B). Comparison of the activation of the wild-type pORF21-1239 luciferase reporter or the HIF-responsive pORF36 reporter in response to degradation-resistant HIF-1 (dr-HIF-1). HEK-293T cells were cotransfected with 300 ng of the pORF21-1239 luciferase reporter or pORF36 promoter and 50 ng of an internal-β-Gal control plasmid in the presence of 100 ng of an expression plasmid encoding dr-HIF-1 or the pcDNA3.1 expression plasmid control. Values are expressed as the fold increase over the value of the pGL3basic reporter transfected with an empty expression vector pcDNA3.1 for each reporter construct and represent the mean of three independent experiments. Error bars denote standard deviation; ****P ≤ 0.001; NS, not significant. (C) Western blot showing XBP-1s (55 kDa) and RTA (85 kDa) expression in BCBL-1 cells 48 hours after treatment with 0.5 μg/ml to 2.5 μg/ml of tunicamycin. As seen, TM induces XBP-1s and RTA but does not induce HIF-1α production in BCBL-1 cells. However, HIF-1α protein expression is seen in BCBCl-1 cells 48 hours after treatment with CoCl2 at 75 μM. Actin was used as the loading control, and TPA was a control for KSHV activation. (D) Comparison of the activation of the pORF21-1239 luciferase reporter by RTA, XBP-1s, or both. HEK-293T cells were cotransfected with 300 ng of pGL3b control reporter plasmid DNA or the pORF21-1239 promoter luciferase reporter and 50 ng of an internal β-Gal control plasmid in the presence of 10 ng of a DNA expression plasmid encoding RTA and 100 ng pcDNA3.1, 100 ng of an expression plasmid for XBP-1s and 10 ng pcDNA3.1, or both RTA and XBP-1s expression plasmids. Values are expressed as the fold increase over the value for the pGL3b basic reporter transfected with pcDNA3.1. Shown are the mean ± standard deviation of triplicate determinations from one representative experiment expressed as the fold change compared with the level of nontreated cells.
The ORF21 promoter responds to RTA, and RTA enhances its response to XBP-1s.
Since KSHV RTA is the principal lytic activator of KSHV that then induces many downstream viral promoters, we wanted to dissect the ability of RTA and XBP-1s to activate the ORF21 promoter. To this end, we cotransfected HEK-293T cells with an RTA-expression vector or a pcDNA3.1 control and pORF21-1239 full-length promoter in the presence or absence of the XBP-1s expression plasmid. RTA transfection induced an approximately 28-fold activation of the pORF21-1239 promoter compared with the pGL3b control. Moreover, activation of the pORF21 promoter was increased to 59-fold in RTA- and XBP-1s-cotransfected cells. (Fig. 3D) These results suggest that both RTA and XBP-1s can activate the pORF21 promoter and that XBP-1s can enhance the activation of the ORF21 promoter by RTA.
Functional binding of XBP-1s to the ORF21 promoter.
To further explore the activation of ORF21 by XBP-1s, we assessed the binding of XBP-1s to the promoter DNA by chromatin immunoprecipitation assay (ChIP). BCBL-1 cells were treated with 0.5 μg/ml TM for 48 hours to induce XBP-1s. Cells were treated with 37% formaldehyde to cross-link the DNA followed by DNA fragmentation. Immunoprecipitation was performed with rabbit anti-XBP-1 antibody or rabbit anti-histone H3 antibody, and reverse cross-linked immunoprecipitated DNA was amplified by real-time PCR using primers surrounding the XRE2, XRE3, and the XRE4 sites (Table 1; Fig. 4). DNA that was immunoprecipitated by the anti-XBP-1s antibody, but not control IgG antibody, was found to be enriched for the DNA sequences identified when amplified by primers surrounding XRE2, XRE3, and, to a somewhat lesser extent, XRE4 in TM-treated BCBL-1 cells (Fig. 4B) compared with dimethyl sulfoxide (DMSO) control (Fig. 4A). However, there was no amplification seen with primers directed against a region of ORF21 distant from the XRE (−56 to −210 bp from the start site) or a sequence in the ORF36 promoter (away from a putative XRE sequence). These results provide evidence that XBP-1s can bind to the ORF21 promoter in the region of XRE2, XRE3, and/or XRE4. It should be noted that this assay cannot distinguish which of these is the functional XRE, in part because XRE2, 3, and 4 are all within 370 bp of each other (Fig. 1) and fragments of DNA may extend for up to 900 bp or so and, thus, include more than one XRE after sonication. Also, the degree of binding to a given XRE may not directly correlate with XRE activity.
TABLE 1.
Primers used in CHIP-PCR
| Target | Primer direction | Sequence (5ʹ to 3ʹ) |
|---|---|---|
| ORF21NonXRE | Forward | 5′-CTGTGCCCTAGAGTCACCTCA-3′ |
| Reverse | 5′-AAAAGAGCAGGTAACCGGGCCCA-3′ | |
| ORF36NonXRE | Forward | 5′-TGTGACGGCTGAGCAGCATGT-3′ |
| Reverse | 5′-TTGTCCAGGTGTGTTCTCGC-3′ | |
| ORF21XRE2 | Forward | 5′-ATCGAGTCGGAGAGTTGGCAC-3′ |
| Reverse | 5′-TGAGGTGACTCTAGGGCACAG-3′ | |
| ORF21XRE3 | Forward | 5′-GTGATAGTCCACGCCTCGGTA-3′ |
| Reverse | 5′-GTAGAGCTCAAGACTTGTCTG-3′ | |
| ORF21XRE4 | Forward | 5′-TTCGCACGAGTTGGCTGCGCAGT-3′ |
| Reverse | 5′-TGCTGACATCAGAAAGGTCC-3′ |
FIG 4.

ChIP showing binding of XBP-1s to the ORF21 promoter in TM-treated cells. BCBL-1 cells were treated with DMSO (A) or TM treatment (0.5 μg/ml) (B) for 48 hours to induce XBP1s and then cross-linked. Chromatin IP of fragmented DNA was performed with anti-XBP-1 antibody, CHIP positive-control anti-histone H3 antibody, or control IgG. Precipitated DNA was assayed by qPCR with specific primers for amplification of XRE2, 3, or 4 of the ORF21 promoter and with primers for an ORF21, ORF36 non-XRE region as a negative control. The data were quantitated as described in the Materials and Methods. Results shown are the mean ± standard deviation of triplicate determinations from a typical experiment of three experiments performed. In controls performed at the same time, DNA immunoprecipitated with histone H3 antibody, but not XBP-1 antibody or control IgG, was enriched for RPL30 exon 3.
An ER stress inducer that promotes XBP-1s production upregulates ORF21 mRNA in BCBL-1 cells.
We next asked if induction of XBP-1s in PEL cells would lead to increased production of ORF21 mRNA, and if so, we assessed its time course. BCBL-1 cells were exposed to TM and harvested at 4, 8, or 24 hours. Real-time quantitative PCR showed that TM induces spliced XBP-1 (XBP-1s) and total XBP-1 mRNA within 4 hours in BCBL-1 cells, and they generally increased at later time points (Fig. 5A and B). There was a dose-dependent induction of total XBP-1 at TM levels ranging from 0.1 μg/ml to 2.5 μg/ml. However, at the highest dose of TM utilized (2.5 μg/ml), the levels of XBP-1s RNA decreased between 8 and 24 hours, and at 24 hours, the highest levels of XBP-1s RNA was seen at 1 μg/ml (Fig. 5A). This decrease may be because of the suppression of IRE1α-mediated splicing of XBP-1 mRNA by high levels of XBP-1s (49). Also, by 24 hours, TM was toxic at the highest dose tested; in a separate experiment, the number of viable cells at 24 hours was 96% of control with 0.25 μg/ml, 77% of control with 0.5 μg/ml, 53% with 1 μg/ml, and 20% of control with 2.0 μg/ml TM. In these same cells, TM exposure also induced production of ORF21 mRNA at 4 h, 8 h, and 24 h (Fig. 5C to E). In addition, consistent with previous results (15), TM induced the production of RTA and vIL-6 mRNA. XBP-1s alone or in combination with hypoxia has previously been reported to upregulate RTA (12, 22, 23), and we considered the possibility that the effects of TM on ORF21 were mediated solely through its upregulation of RTA. In this regard, the upregulation of ORF21 was seen as early as 4 hours (Fig. 5C), which is too early to see substantial secondary effects of RTA activation of ORF21 (9). Also, there was little or no induction of ORF21 by the lytic inducer 12-O-tetradecanoylphorbol-13-acetate (TPA) until 8 hours, and at 8 hours (Fig. 5D) and 24 hours (Fig. 5E), the upregulation of ORF21 by TM was substantially greater than the upregulation of RTA or the induction of ORF21 induced by TPA. Taken together, these results provide evidence that XBP-1 can directly induce ORF21 and that this may be particularly important soon after XBP-1 induction. At later timepoints, it is quite possible that XBP-1s and RTA work in tandem to upregulate ORF21.
FIG 5.

XBP-1, ORF21, RTA ,and vIL-6 mRNA upregulation mediated by TM, a chemical inducer of XBP-1s, in the BCBL-1 PEL line. BCBL-1 cells were treated with increasing doses of TM to induce ER stress; cells were also treated with TPA as an inducer of RTA or a DMSO control. Real-time quantitative PCR showing expression of spliced XBP-1 (A) and total XBP-1 (B) in BCBL-1 cells treated with the compounds shown for 4 h, 8 h, and 24 h. (C, D, and E) Real-time quantitative PCR showing expression of ORF21, vIL-6, and RTA mRNA in BCBL-1 cells cultured in the same way and harvested at 4 hours (C), 8 hours (D), and 24 hours (E). Shown are the mean ± the standard deviation of triplicate determinations from one representative experiment out of three expressed as the fold change compared with the DMSO control.
Tunicamycin enhances the cytotoxicity of AZT and GCV in BCBL-1 cells.
Since ORF21 can phosphorylate GCV and AZT, leading to toxic triphosphates, we next asked if these drugs might be more toxic in BCBL-1 cells exposed to TM. BCBL-1 cells were treated with different concentrations of the above drugs for 72 h, and cell viability was assessed by the ATP luminescence assay. Unlike previous studies on the effects of HIF on GCV and AZT in PEL (39), assessment of the effects of XBP-1s was complicated somewhat because TM (0.25 μg/ml) alone showed toxicity at this dose and resulted in a 23% decrease in viable cells. As shown in Fig. 6, AZT at 10 μM had little to no effect on cell viability (97% viable). However, in the presence of TM at 0.25 μg/ml, AZT decreased viability to 76% of the TM-only control. Also, TM enhanced the killing by GCV at 250 μM and 500 μM and in combination with AZT as well (Fig. 6). These data suggest that by increasing ORF21 gene expression, the phosphorylation of AZT and GCV is enhanced, leading to increased toxicity.
FIG 6.
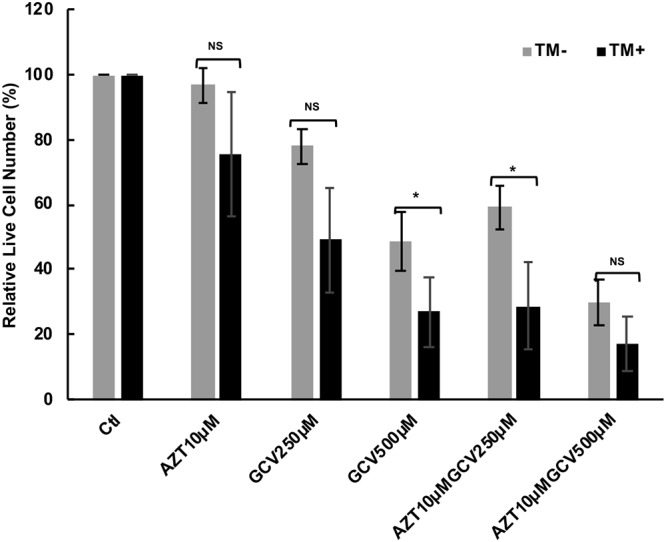
Toxicity of TM, AZT, and GCV in the BCBL-1 cell line after exposure to 0.25 μg/ml TM. BCBL-1 cells were put into culture in 200 μl wells at 100,000 cells ml−1 and incubated for 24 h. The cells were then treated with the indicated concentrations of AZT (10 μM) and GCV (250 μM or 500 μM) in the absence (gray bar) or presence (black bar) of TM at 0.25 μg/ml for 72 h. Cell viability was then determined using the ATP viability assay, and the relative live cell number was calculated against the PBS controls exposed or not exposed to TM. Bars show the average of treatments done in triplicate. * denotes a P value of <0.05 and ** denotes a P value of <0.01 comparing the relative decrease in control versus TM-treated cells. Note that in the TM control, the relative live cell number was decreased by 23% compared with the non-TM control.
ORF21 mRNA is detected predominantly in XBP-1-expressing cells in KSHV-MCD patients’ lymph nodes.
To determine if the induction of ORF21 expression is associated with the expression of XBP-1 in KSHV-MCD lymph nodes, we analyzed the expression of these 2 mRNA species by RNAscope. We examined lymph nodes from 2 patients with KSHV-MCD, and a considerable number of these cells expressed XBP-1. A smaller number of ORF21+ cells were found; these were seen predominantly in B cell follicles of these KSHV-MCD lymph nodes. Most of the cells expressing ORF21 also expressed XBP-1; in one node, 27 of 43 (62.8%) of counted cells expressing ORF21 also expressed XBP-1, and in the other node examined, 48 of 68 (70.1%) of counted cells expressing ORF21 also expressed XBP-1. Consistent with previous results, most of the cells expressing ORF21 failed to express CD20. Two representative pictures of one of these lymph nodes are shown (Fig. 7 A and B) along with a probe control stained for CD20 (Fig. 7C). It should be noted that only 75 of 1,110 (6.8%) counted cells expressing XBP-1 coexpressed ORF21; however, this was most likely because only a small percentage of the differentiating B cells in KSHV-MCD lymph nodes are infected with KSHV (15, 33).
FIG 7.

RNAscope analysis of ORF21 and XBP-1 in representative sections of a lymph node from a patient with KSHV-MCD. (A and B) A paraformaldehyde-fixed paraffin-embedded lymph node from a patient with KSHV-MCD was analyzed for ORF21 (green) and XBP-1 (red) mRNA as described in the Materials and Methods. The white arrows denote cells that express both ORF21 and XBP-1, while the pink arrow denotes a cell that expresses ORF21 only. In addition, CD20 protein expression is identified by immunohistofluorescence (blue), and nuclei identified by 4′,6-diamidino-2-phenylindole (DAPI) is shown in gray. (C) A probe-control section stained for CD20. It is worth noting that most KSHV plasmablasts do not express CD20. The scale bar is 100 μm.
DISCUSSION
In this study, we show that KSHV-encoded ORF21 can be directly activated by XBP-1s and that this activation is mediated primarily through two XRE sequences in the promoter region of ORF21, one (XRE4) with its core sequence at bp −629 to −626, and one (XRE3) with its core sequence at bp −321 to −318. ChIP studies provide evidence that XBP-1s can bind to both XREs; however, mutagenesis studies show that much of this activity is mediated by XRE3. We also show that exposure of cells to TM, an inducer of ER stress, can upregulate ORF21 within 4 hours and that the toxicity of the combination of AZT and GCV is enhanced in the presence of ER stress. Finally, we show that in the lymph nodes of patients with KSHV-MCD, ORF21 mRNA is primarily observed in cells in which XBP-1 is expressed.
There is substantial evidence that KSHV infection of B cells in germinal centers is important in the transmission and biology of the virus as well as in the pathogenesis of KSHV-induced diseases, especially KSHV-MCD (50–52). The early steps of KSHV infection often involve the tonsils, and infection of B cells in the tonsillar germinal centers and their release can then enable the systemic spread of the virus throughout the body (50, 51). Developing germinal center B cells express high levels of XBP-1s (19, 53). In addition, the germinal centers of lymph nodes are a hypoxic environment with elevated levels of HIF (45, 46). Interestingly, KSHV has evolved to respond to the cellular expression of HIF and XBP-1s in a variety of ways.
It was previously shown that HIF and XBP-1s can each activate KSHV RTA (12–14, 23, 54), and this, in turn, can induce the lytic repertoire. It was also shown that certain lytic genes can be directly activated by these stimuli without the requirement for RTA activation; hypoxia can directly induce ORF34-37 and ORF74 (13, 16, 17), and XBP-1s can directly induce vIL-6 (15). In this work, we expand this observation to show that ORF21 can be directly activated by XBP-1s. Genes that respond to cellular factors, such as HIF-1α or XBP-1s, often have multiple response elements for these factors in their promoter regions, with one of these elements sometimes mediating most of the effect. As seen here, ORF21 has 4 potential XRE elements in its promoter region, of which 2 (XRE3 and XRE4) are consensus XREs (44). While truncation experiments suggested that XRE3 or 4 might mediate the effect and ChIP binding suggested that XBP-1s might bind XRE2, 3, or 4, mutagenesis experiments provided evidence for a dominant role for XRE3. However, it is possible that XRE4 also plays a contributory role.
Given that XBP-1s can also activate RTA, a challenge is separating out the direct effect on ORF21 from an indirect effect through RTA activation. In this regard, we found that substantial ORF21 upregulation by TM, an XBP-1s inducer, was observed as early as 4 hours; in contrast, when BCBL-1 cells are induced to lytic activation, for example by TPA, substantial upregulation of ORF21 is not seen until 8 hours (Fig. 5) (9). The rapid and robust upregulation of ORF21 in response to TM suggests that this is, at least in part, a direct effect through XBP-1s. In addition, as seen in Fig. 3C, XBP-1s can work together with RTA to induce an even more robust upregulation of ORF21 than either alone.
This raises the question of what evolutionary pressure led to the activation of ORF21 by XBP-1s. Like other herpesvirus thymidine kinases, thymidine kinase phosphorylates thymidine, ultimately leading to the triphosphate form that is needed for DNA replication. Both XBP-1s and hypoxia can also activate RTA and lytic replication of KSHV (11–13, 15, 16, 22, 23). Direct activation of ORF21 by XBP-1s can enable the relatively earlier production of thymidine-triphosphate in these cells, which is especially important for viral replication in nondividing cells that have relatively little dTTP. The activation of pancreatic endoplasmic reticulum kinase (PERK) as part of the unfolded protein response can lead to cell cycle arrest, and induction of KSHV-induced thymidine phosphorylation in these situations can act to ensure an adequate supply of dTTP in such cells (which otherwise may have insufficient thymidine triphosphate). KSHV-encoded proteins may have a variety of functions. There is recent evidence that in addition to acting as a thymidine kinase, the gene carried by ORF21 can function as a tyrosine kinase, autophosphorylating itself; this leads to an interaction with Crk family proteins and the p85 regulatory unit of P-12 kinase (55, 56). Also, the ORF21 gene product leads to extensive cell contraction and membrane blebbing (55). Whether these effects are advantageous to the virus or a bystander effect is unclear at this point. RTA activation not infrequently leads to incomplete lytic gene activation of KSHV (57). Thus, having these otherwise lytic genes directly activated by XBP-1s (or coactivated by XBP-1s and RTA) may serve to enhance lytic replication in certain environments and may have other advantages for KSHV that are not yet fully understood. Enhancement of lytic replication by the ORF21 gene product may also play an important role in the pathogenesis of KSHV-MCD.
Based on the observations that KSHV ORF36 could phosphorylate GCV (37, 39–41) and that ORF21 could phosphorylate AZT and GCV to yield toxic moieties (37–39), our group showed that patients with KSHV-MCD flares could be treated with virus-activated cytotoxic therapy using the combination of high-dose AZT and valganciclovir (42). This is now a recommended therapy for KSHV-MCD (43). This said, it was somewhat puzzling in retrospect that this approach was successful, given the subsequent finding that many KSHV-MCD plasmablasts express vIL-6 but not the full KSHV lytic repertoire (15). The results of the current study provide a clue to explain this puzzle by showing that KSHV ORF21 in the KSHV-MCD plasmablasts can be directly activated by XBP-1s and also that XBP-1s can enhance RTA-induced activation of ORF21. We also show that treatment of PEL cells with TM to induce ER stress rapidly upregulates the production of XBP-1s, increases the amount of ORF21 in as little as 4 hours, and enhances the toxicity AZT, GCV, or their combination. However, interpretation of these latter experiments is complicated by the toxic effects of TM in these cells at doses that induce XBP-1s and the potential for XBP-1s-induced RTA activation, and it is unclear just how much direct ORF21 activation contributes to the activity of this regimen in patients. Even so, as we have seen here, RTA and XBP-1s can also work together to upregulate ORF21. Moreover, there is evidence that germinal centers can be a hypoxic environment, and this may contribute to the activity of this regimen through upregulation of ORF36 and phosphorylation of GCV (16, 45, 46).
In summary, we show in this work that XBP-1s, a mediator of the unfolded protein response, can directly activate ORF21 through binding to XRE in its promoter region and can also work with RTA to induce a robust activation of ORF21. This finding may help explain the activity of high-dose AZT plus valganciclovir in the treatment of KSHV-MCD. Moreover, it can provide insights into the pathogenesis of KSHV-MCD and the ways that KSHV has evolved to take advantage of the environment of lymph node germinal centers.
MATERIALS AND METHODS
Cell lines, drugs, and reagents.
HEK-293T cells (American Type Culture Collection [ATCC], Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (heat inactivated; HyClone, Logan, UT) and penicillin (Pen)/streptomycin (Strep)/glutamine (Invitrogen Corp., Carlsbad, CA). BCBL-1 (NIH AIDS Research and Reference Reagent Program) cells were maintained in RPMI 1640 medium (Gibco, Grand Island, NY) supplemented with 10% FBS and pen/strep/glutamine. All cells were utilized within 30 passages of procurement or less than 3 months of culture. Except where indicated, cultures were maintained in 95% air and 5% CO2 at 37°C. AZT, GCV, and cobalt chloride (CoCl2) (Sigma, St. Louis, MO) were prepared in phosphate-buffered saline (PBS). TM and TPA were prepared in dimethyl sulfoxide (DMSO) (all from Sigma). DMSO at an equivalent final concentration was used as a vehicle control where appropriate.
Plasmid DNA construction and site-directed mutagenesis.
Expression plasmids encoding unspliced (pcDNA-XBP1u) and spliced XBP-1 (pcDNA-XBP1s) were gifts from Kazatoshi Mori (Kyoto, Japan) (18). An expression plasmid encoding KSHV RTA (pcDNA-RTA) was a kind gift from Keiji Ueda (Osaka University Graduate School of Medicine, Osaka, Japan). The expression plasmid encoding a degradation-resistant form of hypoxia-inducible factor 1α (dr-HIF-1) has been described previously (16, 58). pcDNA3.1 empty vector was used as a control. All plasmids were purified with a maxiprep kit (Qiagen, Valencia, CA) and inserts verified by DNA sequencing.
The KSHV ORF21 luciferase promoter reporter constructs from pORF21-1239 were created spanning nucleotides 1,239 bp upstream of the ATG start site of ORF21. DNA fragments were amplified by PCR from KSHV-infected BCBL-1 cells induced with sodium butyrate using gene-specific primers containing NheI and BglII sites to the 5′ and 3′ end, respectively (Fig. 1A). Three deletions of ORF21 promoter luciferase reporter constructs (pORF21-624, pORF21-316, and pORF21-256) were amplified by PCR using pORF21-1239 plasmid DNA as a template with 5′ primers (5′-AGTTATGCTAGCTTCCGGGTGTGGGGC-3′), (5′-AGTCATGCTAGCCTCACGGTGTTGGTT-3′), (5′-AGTCATGCTAGCCATACGGGCTGATGC-3′), which contained NheI sites and the common 3′ primer 5′-AGCTGCAAGCTTTGGTACGTCCAACCG-3′ which contained a HindIII site. PCR fragments were purified and cloned into the corresponding sites of the reporter vector pGL3basic (Promega Corporation, Madison, WI). (Recognition sequences are underlined.) The ORF21 promoter luciferase reporters pORF21-1239X3-M1, M2, M3 and pORF21-1239X4-M1, M2, and M3 containing mutagenized XREs were constructed from pORF21-1239 wild type using the GeneArt site-directed mutagenesis system (Invitrogen Corp., Carlsbad, CA) (15, 58, 59). These mutant plasmids contained 2 to 4 nucleotide substitutions (from ACGT to ACAG or CTAG or CCAG) in XREs. Primer sequences used to mutate XRE3 mutation 1 were 5′-CCTTGAGCTCGCTGTGACAGTCTCACGGTGTTGGTT 3′ and complementary 3′ primer 5′-AACCAACACCGTGAGACTGTCACAGCGAGCTCAAGG-3′; sequences for mutating XRE3 mutation 2 were 5′-CCTTGAGCTCGCTGTGCTAGTCTCACGGTGTTGGTT-3′ and complementary 3′ primer 5′-AACCAACACCGTGAGACTAGCACAGCGAGCTCAAGG-3′. Primers for mutation 3 were 5′-CCTTGAGCTCGCTGTGCCAGTCTCACGGTGTTGGTT-3′ and 5′-AACCAACACCGTGAGACTGGCACAGCGAGCTCAAGG-3′. Primer sequences used to mutate XRE4 mutation 1 were 5′-GCTTGTGAATAAACAGCACAGTTTCCGGGTGTGGGGCC-3′ and complementary 3′ primer 5′-GGCCCCACACCCGGAAACTGTGCTGTTTATTCACAAGC-3′; for mutating XRE4 mutation 2, primers were 5′-GCTTGTGAATAAACAGCCTAGTTTCCGGGTGTGGGGCC-3′ and complementary 3′ primer 5′-GGCCCCACACCCGGAAACTAGGCTGTTTATTCACAAGC-3′. Primers for mutation 3 were 5′-GCTTGTGAATAAACAGCCCAGTTTCCGGGTGTGGGGCC-3′ and 5′-GGCCCCACACCCGGAAACTGGGCTGTTTATTCACAAGC-3′. The KSHV ORF36 luciferase promoter reporter construct was described previously (16); it was called in that paper the ORF35-37 (35-37P1/1891 or 35-37P1) reporter plasmid. Reactions and transformation were performed according to the manufacturer’s protocol. All constructs were confirmed by DNA sequencing.
Transfection and luciferase reporter assays.
HEK-293T cells were transfected using Fugene6 transfection reagent (Promega Corporation, Madison, WI) following the manufacturer’s protocol. For luciferase reporter experiments, 1 × 105 HEK-293T cells/well were plated in a 12-well plate and the following day were cotransfected with 300 ng/well of reporter plasmid DNA, and either expression plasmid DNA or a control (pcDNA3.1) of 100 ng/well in the presence of 50 ng DNA of an internal control plasmid, pSV-β-Gal (Promega Corporation, Madison, WI), was used to normalize for transfection efficiency. Expression plasmids encoding unspliced (pcDNA-XBP-1u) and spliced XBP-q (pcDNA-XBP-1s) have been described previously (15, 59). The pcDNA3.1 empty expression vector was used as a control. All plasmid DNA was purified with a maxiprep kit (Qiagen, Valencia, CA). The amount of each expression plasmid DNA used was 100 ng/well for pcDNA-XBP1u, 100 ng/well for pcDNA-XBP1s, and 5 ng/well for pcDNA-RTA. The amount of control plasmid pcDNA3.1 and expression plasmid used was equal. Cells were incubated for 48 hours, lysed with 250 μl per well of 1× reporter lysis buffer (Promega Corporation), and freeze-thawed once. After centrifugation at 13,000 × g for 8 minutes, 20 μl and 50 μl of cell lysates were used to determine luciferase and β-galactosidase (β-Gal) in the extracts. Means and standard deviations were calculated, and conditions were compared using the 2-tailed Student’s t test for paired values.
Chromatin immunoprecipitation assay (ChIP).
Chromatin immunoprecipitation assays were performed using the SimpleChIP enzymatic chromatin IP kit (no. 9003; Cell Signaling Technologies). BCBL-1 cells were treated with 0.5 μg/ml TM or DMSO control for 48 h at 37°C and then cross-linked with 37% formaldehyde at a final concentration of 1% at room temperature for 10 min. Fragmented chromatin was prepared by nuclease and sonication. Chromatin immunoprecipitation was performed with rabbit anti-XBP-1 polyclonal antibody (5 μg; TA328002; OriGene, Rockville MD), rabbit anti-histone H3 (a technical positive control; 1:50; no. 4620; Cell Signaling Technologies) monoclonal antibody, and normal rabbit IgG (a negative control; 5 μg; no. 2729; Cell Signaling Technologies). After reverse cross-linking and DNA purification, immunoprecipitated DNA was quantified by real-time PCR using power SYBR green mix (no. 4367659; Applied Biosystems) with primers for XBP-1 binding sites in the ORF21 promoter, XRE2, XRE3, and XRE4. In addition, primers for regions of ORF21 and ORF36 lacking the XREs were used as negative controls, while RPL30 exon 3 (no. 7014; Cell Signaling Technologies) was used for a technical positive control. The CHIP-PCR primers used are listed in Table 1. Fold enrichment was calculated based on the threshold cycle (CT) value of the IgG control using the comparative CT method (15).
RNA isolation, reverse transcriptase PCR (RT-PCR), real-time quantitative PCR.
Total cellular RNA was isolated from cells using the RNeasy minikit (Qiagen, Hilden, Germany). For real-time quantitative PCR, cDNA was synthesized using the reverse transcriptase enzyme SuperScript II (Invitrogen Corp., Carlsbad, CA) (15, 59). Real-time PCR was performed with SYBR green (no. 4367659; Applied Biosystems); the gene-specific PCR primers used are listed in Table 2. Samples were run in triplicate, and PCR was performed using an ABI StepOnePlus thermocycler (Applied Biosystems). Actin was used as a housekeeping gene, and quantitative PCR (qPCR data) (CT) values were analyzed using the ΔΔCT method. The qPCR data are presented as the fold change in target gene expression compared with the control ± standard error of mean.
TABLE 2.
Primers used in real-time PCR
| Target | Primer direction | Sequence (5ʹ to 3ʹ) |
|---|---|---|
| ORF21 | Forward | 5′-CCCTGAGGAGAGGAAACCACTAAC-3′ |
| Reverse | 5′-CCGACTGGCAAAAATGCTGC-3′ | |
| RTA | Forward | 5′-TTGCCAAGTTTGTACAACTGCT-3′ |
| Reverse | 5′-ACCTTGCAAAGACCATTCAGAT-3′ | |
| vIL-6 | Forward | 5′-CTGTTACCGTACCGGCATCT-3′ |
| Reverse | 5′-GGGTGGACTGTAGTGCGTCT-3′ | |
| XBP-1s | Forward | 5′-GTCTGCTGAGTCCGCAGCAGG-3′ |
| Reverse | 5′-TCCTTCTGGGTAGACCTCTGGGAG-3′ | |
| XBP-1t | Forward | 5′-GCAGGTGCAGCCCAGTTGTCAC-3′ |
| Reverse | 5′-CCCCACTGACAGAGAAAGGGAGG-3′ | |
| VEGF | Forward | 5′-CCTTGCTGCTCTACCTCCAC-3′ |
| Reverse | 5′-AGCTGCGCTGATAGACATCC-3′ | |
| Actin | Forward | 5′-CCTTCCTGGGCATGGAGT-3′ |
| Reverse | 5′-CAGGGCAGTGATCTCCTTCT-3′ |
Immunoblotting.
Nuclear extracts were prepared from BCBL-1 using the NE-PER nuclear and cytoplasmic protein extraction reagent (Pierce, Rockford, IL). Protein concentrations were determined using the Pierce bicinchoninic acid (BCA) protein assay kit. A total of 40 μg of nuclear protein was electrophoresed on 4% to 12% NuPAGE gels (Invitrogen Corp., Carlsbad, CA). Proteins were transferred to a nitrocellulose membrane using the iBlot dry blotting system, and the membrane was blocked with 50% Li-Cor blocking buffer diluted with 1× Tris-buffered saline with Tween 20 (TBST) (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 0.05% Tween 20) for 1 hour at room temperature. Blots were then incubated with 1:1,000 anti-HIF-1α mouse monoclonal antibody (BD Biosciences, San Jose, CA), 1:1,000 XBP-1s rabbit polyclonal antibody (Biolegend, San Diego, CA), 1:10,000 RTA rabbit polyclonal antibody (a gift from Muzmmel Haque, Louisiana State University Agricultural Center), and mouse anti-β-actin at a 1:50,000 dilution (Sigma, St. Louis, MO) overnight at 4°C. After being washed with washing buffer, the blots were incubated with anti-mouse or anti-rabbit IRDye 800CW secondary antibody (Li-Cor Biosciences). Western blots were visualized using an Odyssey scanner and analyzed using ImageStudio (Li-Cor Biosciences) (59).
Determination of GCV and AZT cytotoxicity with and without TM activation of XBP-1.
BCBL-1 cells were seeded at 100,000 cells/ml in 96-well plates (200 μl/well) and incubated for 24 hours. Cells were treated with PBS, tunicamycin, GCV, or AZT or with different combinations of the drugs. Cells were incubated for an additional 72 hours in a normoxic incubator. Cell viability assays were assessed using the Cell-Glo luminescent cell viability assay (Promega). Luminesce was read by the VictorX3 instrument (Perkin-Elmer) (39).
RNAscope analysis.
Formalin-fixed paraffin-embedded lymph node biopsy specimens were obtained from patients with KSHV-MCD on protocols of the HIV and AIDS Malignancy Branch; these protocols were approved by the National Cancer Institute Institute Institutinal Review Board, and all patients gave written informed consent. ORF21 (probe number ACD 559011; ACD Bio, Newark, CA) and XBP-1 (probe number ACD 436251) RNA was detected using next-generation, ultrasensitive in situ hybridization technology as previously described (60). We combined RNAscope with immunofluorescence staining, targeting a subset of B cells using anti-CD20 antibody (Dako; clone L26; Agilent, Santa Clara, CA). To quantify the number of ORF21-positive cells harboring XBP-1, high-magnification confocal images were collected across lymph nodes and manually counted using an Olympus FV10i confocal microscope using a 60× phase contrast oil-immersion objective and imaging using sequential mode to separately capture the fluorescence from the different fluorochromes at an image resolution of 1,024 × 1,024 pixels. To maximize the size of the tissue to be assessed, we stained and quantified a total of 2 sections (5 μm) per sample.
Statistics.
Where indicated, the mean and standard deviation were calculated for experiments repeated 3 or more times. Statistical comparisons reported were performed using the Student’s two-tailed paired t test.
ACKNOWLEDGMENTS
This research was supported by the International Research Program of the NIH National Cancer Institute and, in part, with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin M, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86:1276–1280. doi: 10.1182/blood.V86.4.1276.bloodjournal8641276. [DOI] [PubMed] [Google Scholar]
- 4.Yarchoan R, Uldrick TS. 2018. HIV-associated cancers and related diseases. N Engl J Med 378:1029–1041. doi: 10.1056/NEJMra1615896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cesarman E, Damania B, Krown SE, Martin J, Bower M, Whitby D. 2019. Kaposi sarcoma. Nat Rev Dis Primers 5:9. doi: 10.1038/s41572-019-0060-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarid R, Wiezorek JS, Moore PS, Chang Y. 1999. Characterization and cell cycle regulation of the major Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J Virol 73:1438–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- 8.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 95:10866–10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paulose-Murphy M, Ha N-K, Xiang C, Chen Y, Gillim L, Yarchoan R, Meltzer P, Bittner M, Trent J, Zeichner S. 2001. Transcription program of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus). J Virol 75:4843–4853. doi: 10.1128/JVI.75.10.4843-4853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye F, Lei X, Gao S-J. 2011. Mechanisms of Kaposi’s sarcoma-associated herpesvirus latency and reactivation. Adv Virol 2011:193860. doi: 10.1155/2011/193860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R. 2001. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 97:3244–3250. doi: 10.1182/blood.v97.10.3244. [DOI] [PubMed] [Google Scholar]
- 12.Dalton-Griffin L, Wilson SJ, Kellam P. 2009. X-box binding protein 1 contributes to induction of the Kaposi’s sarcoma-associated herpesvirus lytic cycle under hypoxic conditions. J Virol 83:7202–7209. doi: 10.1128/JVI.00076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haque M, Davis DA, Wang V, Widmer I, Yarchoan R. 2003. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) contains hypoxia response elements: relevance to lytic induction by hypoxia. J Virol 77:6761–6768. doi: 10.1128/jvi.77.12.6761-6768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai IY-C, Farrell PJ, Kellam P. 2011. X-box binding protein 1 induces the expression of the lytic cycle transactivator of Kaposi’s sarcoma-associated herpesvirus but not Epstein-Barr virus in co-infected primary effusion lymphoma. J Gen Virol 92:421–431. doi: 10.1099/vir.0.025494-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu D, Wang V, Yang M, Abdullah S, Davis DA, Uldrick TS, Polizzotto MN, Veeranna RP, Pittaluga S, Tosato G, Yarchoan R. 2015. Induction of Kaposi’s sarcoma-associated herpesvirus-encoded viral interleukin-6 by X-box binding protein 1. J Virol 90:368–378. doi: 10.1128/JVI.01192-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haque M, Wang V, Davis DA, Zheng ZM, Yarchoan R. 2006. Genetic organization and hypoxic activation of the Kaposi’s sarcoma-associated herpesvirus ORF34-37 gene cluster. J Virol 80:7037–7051. doi: 10.1128/JVI.00553-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh RK, Lang F, Pei Y, Jha HC, Robertson ES. 2018. Metabolic reprogramming of Kaposi’s sarcoma associated herpes virus infected B-cells in hypoxia. PLoS Pathog 14:e1007062. doi: 10.1371/journal.ppat.1007062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 19.Iwakoshi NN, Lee A-H, Glimcher LH. 2003. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev 194:29–38. doi: 10.1034/j.1600-065x.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- 20.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 21.Gass JN, Jiang H-Y, Wek RC, Brewer JW. 2008. The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol Immunol 45:1035–1043. doi: 10.1016/j.molimm.2007.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu F, Feng J, Harada JN, Chanda SK, Kenney SC, Sun R. 2007. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi’s sarcoma-associated herpesvirus. FEBS Lett 581:3485–3488. doi: 10.1016/j.febslet.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 23.Wilson SJ, Tsao EH, Webb BLJ, Ye H, Dalton-Griffin L, Tsantoulas C, Gale CV, Du M-Q, Whitehouse A, Kellam P. 2007. X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J Virol 81:13578–13586. doi: 10.1128/JVI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee DY, Sugden B. 2008. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 111:2280–2289. doi: 10.1182/blood-2007-07-100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhende PM, Dickerson SJ, Sun X, Feng W-H, Kenney SC. 2007. X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. J Virol 81:7363–7370. doi: 10.1128/JVI.00154-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsiao J-R, Chang K-C, Chen C-W, Wu SY, Su I-J, Hsu M-C, Jin Y-T, Tsai S-T, Takada K, Chang Y. 2009. Endoplasmic reticulum stress triggers XBP-1-mediated up-regulation of an EBV oncoprotein in nasopharyngeal carcinoma. Cancer Res 69:4461–4467. doi: 10.1158/0008-5472.CAN-09-0277. [DOI] [PubMed] [Google Scholar]
- 27.Matar CG, Rangaswamy US, Wakeman BS, Iwakoshi N, Speck SH. 2014. Murine gammaherpesvirus 68 reactivation from B cells requires IRF4 but not XBP-1. J Virol 88:11600–11610. doi: 10.1128/JVI.01876-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. 2009. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog 5:e1000677. doi: 10.1371/journal.ppat.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oksenhendler E, Carcelain G, Aoki Y, Boulanger E, Maillard A, Clauvel J-P, Agbalika F. 2000. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric Castleman disease in HIV-infected patients. Blood 96:2069–2073. doi: 10.1182/blood.V96.6.2069. [DOI] [PubMed] [Google Scholar]
- 30.Aoki Y, Yarchoan R, Wyvill K, Okamoto S, Little RF, Tosato G. 2001. Detection of viral interleukin-6 in Kaposi sarcoma-associated herpesvirus-linked disorders. Blood 97:2173–2176. doi: 10.1182/blood.v97.7.2173. [DOI] [PubMed] [Google Scholar]
- 31.Polizzotto MN, Uldrick TS, Wang V, Aleman K, Wyvill KM, Marshall V, Pittaluga S, O'Mahony D, Whitby D, Tosato G, Steinberg SM, Little RF, Yarchoan R. 2013. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood 122:4189–4198. doi: 10.1182/blood-2013-08-519959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suthaus J, Stuhlmann-Laeisz C, Tompkins VS, Rosean TR, Klapper W, Tosato G, Janz S, Scheller J, Rose-John S. 2012. HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric Castleman disease in mice. Blood 119:5173–5181. doi: 10.1182/blood-2011-09-377705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cronin DM, Warnke RA. 2009. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol 16:236–246. doi: 10.1097/PAP.0b013e3181a9d4d3. [DOI] [PubMed] [Google Scholar]
- 34.Parravicini C, Chandran B, Corbellino M, Berti E, Paulli M, Moore PS, Chang Y. 2000. Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am J Pathol 156:743–749. doi: 10.1016/S0002-9440(10)64940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aoki Y, Tosato G, Fonville TW, Pittaluga S. 2001. Serum viral interleukin-6 in AIDS-related multicentric Castleman disease. Blood 97:2526–2527. doi: 10.1182/blood.v97.8.2526. [DOI] [PubMed] [Google Scholar]
- 36.Staskus KA, Sun R, Miller G, Racz P, Jaslowski A, Metroka C, Brett-Smith H, Haase AT. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. J Virol 73:4181–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cannon JS, Hamzeh F, Moore S, Nicholas J, Ambinder RF. 1999. Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J Virol 73:4786–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gustafson EA, Schinazi RF, Fingeroth JD. 2000. Human herpesvirus 8 open reading frame 21 is a thymidine and thymidylate kinase of narrow substrate specificity that efficiently phosphorylates zidovudine but not ganciclovir. J Virol 74:684–692. doi: 10.1128/jvi.74.2.684-692.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis DA, Singer KE, Reynolds IP, Haque M, Yarchoan R. 2007. Hypoxia enhances the phosphorylation and cytotoxicity of ganciclovir and zidovudine in Kaposi’s sarcoma-associated herpesvirus infected cells. Cancer Res 67:7003–7010. doi: 10.1158/0008-5472.CAN-07-0939. [DOI] [PubMed] [Google Scholar]
- 40.Park J, Lee D, Seo T, Chung J, Choe J. 2000. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) open reading frame 36 protein is a serine protein kinase. J Gen Virol 81:1067–1071. doi: 10.1099/0022-1317-81-4-1067. [DOI] [PubMed] [Google Scholar]
- 41.Hamza MS, Reyes RA, Izumiya Y, Wisdom R, Kung HJ, Luciw PA. 2004. ORF36 protein kinase of Kaposi’s sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J Biol Chem 279:38325–38330. doi: 10.1074/jbc.M400964200. [DOI] [PubMed] [Google Scholar]
- 42.Uldrick TS, Polizzotto MN, Aleman K, O'Mahony D, Wyvill KM, Wang V, Marshall V, Pittaluga S, Steinberg SM, Tosato G, Whitby D, Little RF, Yarchoan R. 2011. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood 117:6977–6986. doi: 10.1182/blood-2010-11-317610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zelenetz AD, Gordon LI, Abramson JS, Advani RH, Bartlett NL, Caimi PF, Chang JE, Chavez JC, Christian B, Fayad LE, Glenn MJ, Habermann TM, Lee Harris N, Hernandez-Ilizaliturri F, Kaminski MS, Kelsey CR, Khan N, Krivacic S, LaCasce AS, Mehta A, Nademanee A, Rabinovitch R, Reddy N, Reid E, Roberts KB, Smith SD, Snyder ED, Swinnen LJ, Vose JM, Dwyer MA, Sundar H. 2019. NCCN guidelines insights: B-cell lymphomas, version 4.2019. J Natl Compr Canc Netw 17:650–661. doi: 10.6004/jnccn.2019.0029. [DOI] [PubMed] [Google Scholar]
- 44.Clauss IM, Chu M, Zhao J-L, Glimcher LH. 1996. The basic domain/leucine zipper protein hXBP-1 preferentially binds to and transactivates CRE-like sequences containing an ACGT core. Nucleic Acids Res 24:1855–1864. doi: 10.1093/nar/24.10.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abbott RK, Thayer M, Labuda J, Silva M, Philbrook P, Cain DW, Kojima H, Hatfield S, Sethumadhavan S, Ohta A, Reinherz EL, Kelsoe G, Sitkovsky M. 2016. Germinal center hypoxia potentiates immunoglobulin class switch recombination. J Immunol 197:4014–4020. doi: 10.4049/jimmunol.1601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, Thomas JW, Hiebert S, Haase VH, Boothby MR. 2016. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 537:234–238. doi: 10.1038/nature19334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wenger RH, Stiehl DP, Camenisch G. 2005. Integration of oxygen signaling at the consensus HRE. Sci STKE 2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 48.Duksin D, Mahoney WC. 1982. Relationship of the structure and biological activity of the natural homologues of tunicamycin. J Biol Chem 257:3105–3109. [PubMed] [Google Scholar]
- 49.Chalmers F, van Lith M, Sweeney B, Cain K, Bulleid NJ. 2017. Inhibition of IRE1alpha-mediated XBP1 mRNA cleavage by XBP1 reveals a novel regulatory process during the unfolded protein response. Wellcome Open Res 2:36. doi: 10.12688/wellcomeopenres.11764.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schulz TF, Cesarman E. 2015. Kaposi sarcoma-associated herpesvirus: mechanisms of oncogenesis. Curr Opin Virol 14:116–128. doi: 10.1016/j.coviro.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 51.Dittmer DP, Damania B. 2016. Kaposi sarcoma-associated herpesvirus: immunobiology, oncogenesis, and therapy. J Clin Invest 126:3165–3175. doi: 10.1172/JCI84418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goncalves PH, Ziegelbauer J, Uldrick TS, Yarchoan R. 2017. Kaposi sarcoma herpesvirus-associated cancers and related diseases. Curr Opin HIV AIDS 12:47–56. doi: 10.1097/COH.0000000000000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwakoshi NN, Lee A-H, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. 2003. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol 4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 54.Cai Q, Lan K, Verma SC, Si H, Lin D, Robertson ES. 2006. Kaposi’s sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 alpha to upregulate RTA expression during hypoxia: latency control under low oxygen conditions. J Virol 80:7965–7975. doi: 10.1128/JVI.00689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gill MB, Turner R, Stevenson PG, Way M. 2015. KSHV-TK is a tyrosine kinase that disrupts focal adhesions and induces Rho-mediated cell contraction. EMBO J 34:448–465. doi: 10.15252/embj.201490358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mariggiò G, Koch S, Schulz TF. 2017. Kaposi sarcoma herpesvirus pathogenesis. Philos Trans R Soc Lond B Biol Sci 372:20160275. doi: 10.1098/rstb.2016.0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scholz BA, Harth-Hertle ML, Malterer G, Haas J, Ellwart J, Schulz TF, Kempkes B. 2013. Abortive lytic reactivation of KSHV in CBF1/CSL deficient human B cell lines. PLoS Pathog 9:e1003336. doi: 10.1371/journal.ppat.1003336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang V, Davis DA, Veeranna RP, Haque M, Yarchoan R. 2010. Characterization of the activation of protein tyrosine phosphatase, receptor-type, Z polypeptide 1 (PTPRZ1) by hypoxia inducible factor-2 alpha. PLoS One 5:e9641. doi: 10.1371/journal.pone.0009641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang V, Davis DA, Yarchoan R. 2017. Identification of functional hypoxia inducible factor response elements in the human lysyl oxidase gene promoter. Biochem Biophys Res Commun 490:480–485. doi: 10.1016/j.bbrc.2017.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deleage C, Wietgrefe SW, Del Prete G, Morcock DR, Hao XP, Piatak M Jr., Bess J, Anderson JL, Perkey KE, Reilly C, McCune JM, Haase AT, Lifson JD, Schacker TW, Estes JD. 2016. Defining HIV and SIV Reservoirs in lymphoid tissues. Pathog Immun 1:68–106. doi: 10.20411/pai.v1i1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]