

Latency of HSV-1 in host neurons enables long-term persistence from which reactivation may occur to cause recurrent diseases, such as blinding herpetic stromal keratitis. Latency is not antigenically silent, and viral proteins are sporadically expressed at low levels without full virion production. This protein expression is recognized by ganglion-resident HSV-1-specific CD8+ T cells that maintain a protective resident population. Since these T cells can influence lytic/latent decisions in reactivating neurons, we argue that improving their ganglionic retention and function may offer a strategy in vaccine design to reduce reactivation and recurrent disease. To understand factors driving the infiltration and retention of ganglionic CD8s, we examined several HSV recombinants that have different viral promoters driving expression of the immunodominant gB epitope. We show that the selection of epitope promoter influences CD8+ T cell population hierarchies and their function.
KEYWORDS: CD8 T cell, HSV-1, immunodominance, latency
ABSTRACT
Reactivation of herpes simplex virus 1 (HSV-1) from neurons in sensory ganglia such as the trigeminal ganglia (TG) is influenced by virus-specific CD8+ T cells that infiltrate the ganglia at the onset of latency and contract to a stable activated tissue-resident memory population. In C57BL/6 mice, half of HSV-specific CD8+ T cells (gB-CD8s) recognize one dominant epitope (residues 498 to 505) on glycoprotein B (gB498–505), while the remainder (non-gB-CD8s) recognize 19 subdominant epitopes from 12 viral proteins. To address how expression by HSV-1 influences the formation and ganglionic retention of CD8+ T cell populations, we developed recombinant HSV-1 with the native immunodominant gB epitope disrupted but then expressed ectopically from different viral promoters. In mice, the epitope expressed from the gB promoter restored full gB-CD8 immunodominance to 50%. Intriguingly, earlier expression from constitutive, immediate-early, and early promoters did not significantly increase immunodominance, indicating that these promoters cannot elicit more than half of the CD8 compartment. Epitope expressed from candidate viral promoters of “true late” HSV-1 genes either delayed or reduced the priming efficiency of gB-CD8s and their levels in the TG at early times. HSV expressing the epitope from the full latency-associated transcript promoter did not efficiently prime gB-CD8s; however, gB-CD8s primed by a concurrent wild-type flank infection infiltrated the TG and were retained long term, suggesting that latent epitope expression is sufficient to retain gB-CD8s. Taken together, the data indicate that viral promoters shape latent HSV-1-specific CD8+ T cell populations and should be an important consideration in future vaccine design.
IMPORTANCE Latency of HSV-1 in host neurons enables long-term persistence from which reactivation may occur to cause recurrent diseases, such as blinding herpetic stromal keratitis. Latency is not antigenically silent, and viral proteins are sporadically expressed at low levels without full virion production. This protein expression is recognized by ganglion-resident HSV-1-specific CD8+ T cells that maintain a protective resident population. Since these T cells can influence lytic/latent decisions in reactivating neurons, we argue that improving their ganglionic retention and function may offer a strategy in vaccine design to reduce reactivation and recurrent disease. To understand factors driving the infiltration and retention of ganglionic CD8s, we examined several HSV recombinants that have different viral promoters driving expression of the immunodominant gB epitope. We show that the selection of epitope promoter influences CD8+ T cell population hierarchies and their function.
INTRODUCTION
Herpesviruses are generally characterized by three types of infection, a primary lytic productive infection, a latent or quiescent infection where no virus is produced, and a renewed lytic infection originating from the latent state. In productive lytic infections, viral genes are expressed in a highly regulated cascade in which immediate-early (IE, α) genes are expressed first, early (E, β) genes are expressed after α proteins have been made, and late genes are expressed after viral DNA replication has initiated. The late genes can be divided into leaky late (L1, γ1) and true late (L2, γ2). γ1 genes show some expression prior to DNA replication, while γ2 genes are expressed only after viral DNA replication has begun.
Lytic infections can occur in multiple cell types and tissues. In contrast, herpes simplex virus 1 (HSV-1) latent infections occur only in sensory and autonomic neurons. In latency, the viral expression program becomes epigenetically silenced, concurrent with genome circularization and genome association with host chromatin (1, 2). Only one genomic region is abundantly transcribed during latency, encoding a family of transcripts termed the latency-associated transcripts (LATs). Extensive studies of LAT transcripts have not completely resolved their function; while they do not encode proteins, they do promote viral latency, neuronal survival, and efficiency of reactivation (3–6). It is now recognized that the latent viral state is not antigenically silent, as multiple lines of evidence have indicated that latency is a dynamic process in which there are low levels of sporadic viral protein expression that mostly do not result in virus production (7–12). Multiple and varied stress and reactivation stimuli result in neuronal signaling that influences chromatin states and epigenetic control of the viral genome. This may result in derepression of the genome with protein expression outside the normal lytic temporal cascade, which may then either (i) reverse to a silent state, or (ii) progress to full reactivation and virus production (8, 11). One example of a reactivation stimulus is the withdrawal of nerve growth factor (NGF), where the loss of NGF signaling in explanted HSV latently infected mouse ganglia promotes derepression and expression of viral genes and proteins (11). This scenario is consistent with several reports detecting rare viral lytic mRNA and proteins found in in unstimulated latently infected ganglia (12–16). The factors influencing lytic/latent decisions after such a derepression may be driven in part by nuclear entry of the transactivator of immediate-early genes, VP16 (11).
Evidence supporting some antigen expression (“prereactivation” events) in latently infected ganglia has also come from studies showing that there is immune recognition in ganglia of both mice and humans (14–17). At the primary HSV-1 infection stage, the adaptive immune responses primed include an HSV-specific CD8+ T cell response (14). In HSV-1 ocular infection models in mice and rabbits, the onset of latency triggers an immune cell infiltration of the ganglia that peaks and subsequently contracts as latency is established. However, a persistent HSV-specific CD8 population remains that is then maintained for the life of the host. Ganglia of latently infected humans have been shown to contain similar immune cell infiltrates (17).
The nature of the HSV-1-specific CD8+ T cell response has been particularly well studied in the C57BL/6 (B6) mouse model of ocular HSV-1 infection, where the entire antigen hierarchy to which CD8+ T cells are directed is now defined (18). CD8+ T cells to HSV show a remarkably skewed immunodominance hierarchy, in which approximately half of HSV-specific CD8+ T cells are directed to a single epitope (SSIEFARL, residues 498 to 505 of the γ1-regulated glycoprotein gB, here termed gB-CD8s), while the remainder (collectively termed non-gB-CD8s) recognize 19 subdominant epitopes on 12 viral proteins, of which most are expression regulated as β or γ1 (18). Few CD8+ T cells recognize γ2 proteins, and none recognize epitopes from α genes, though the recognition of gB by gB-CD8s occurs as rapidly as 2 h after the initiation of a lytic infection (19). This immunodominance hierarchy is maintained systemically in the lymphoid tissues and in the resident populations in the latently infected ganglia (20); gB-CD8s appear to be continuously activated and functional during latency, expressing proinflammatory cytokines, including gamma interferon (IFN-γ), in response to antigen stimulation (21–23), while non-gB-CD8s develop signs of functional exhaustion (20). Ganglionic HSV-1-specific gB-CD8s proximal to neurons develop immunological synapses with polarized T cell receptors, strongly suggesting that antigen recognition during latency occurs in the murine model (24).
Evidence now suggests that ganglionic CD8+ T cells can influence reactivation and lytic/latent decisions. Maintaining CD8+ T cell population viability in explant cultures of latently infected ganglia will greatly reduce the reactivation frequency of HSV, and this has been shown to occur through IFN-γ and perforin-dependent mechanisms (25). We have proposed that the tissue-resident ganglionic CD8+ T cell infiltrate surveys the ganglia for reactivation events; recognition is able to tip the lytic/latent decision process toward the latent state (26). As such, optimizing the number of ganglionic CD8+ T cells or their function during latency may be an ideal and attractive strategy to reduce reactivation events and the diseases that result. We particularly aim to prevent the inflammatory cascade in the cornea triggered by HSV recurrent infections, which result in a blinding stromal keratitis that is the leading infectious cause of blindness in westernized societies and also the leading cause of corneal transplantation (27).
Multiple factors influence the antigens seen by CD8+ T cell responses to a pathogen, including precursor frequency of T cells to a specific antigen, the level and chronicity of protein expression, and the presentation of antigens to the developing T cells in the lymphoid organs in the priming process (28–31). For HSV-1 in B6 mice, the powerfully immunodominant gB498–505 epitope is remarkable, and the factors defining this are not clear. To investigate such factors, we recently detailed a study of the CD8+ T cell response to a recombinant virus in which expression kinetics of the gB glycoprotein were altered to γ2 kinetics (32). This virus primed a diminished but effective immunodominant gB-CD8 response, but considerably fewer gB498–505-specific CD8+ T cells were retained in the ganglia throughout latency than in the response directed to a wild-type (WT) HSV strain (32). We concluded that delaying the expression of gB results in reduced antigenic exposure during latency and less ganglionic retention of gB-CD8s. However, interpretation of these results was complicated by a growth impairment of the virus, most likely as a result of the elimination of roles of gB early in the infectious process.
As such, a more thorough analysis of the contribution of viral promoters and different kinetics of expression on CD8+ T cell populations was warranted. Here, we evaluate the contributions of different viral promoters by further exploiting a recently described recombinant HSV-1 strain in which the native gB498–505 epitope is not present but gB is expressed normally and appears to maintain its functions (23). We derived recombinant viruses in which the missing epitope was expressed at an ectopic site under the control of different promoters. This approach permits us to alter the immunodominant gB498–505 epitope expression profile, independently of the essential gB protein. It allowed us to address the following two questions: (i) can the immunodominance of the CD8+ T cell population to gB498–505 be increased by increasing expression of this epitope or by expressing it from more active or earlier expressed viral promoters? And (ii) does epitope expression from candidate late promoters alter the development of the CD8+ T cell response or ganglionic retention during latency? Establishing the requirements for efficiently eliciting and maintaining an effective CD8+ T cell population throughout latency is vital for maintaining HSV-1 in its latent state. We report that the immunodominant fraction of CD8+ T cells beyond the WT levels of ∼50% is not increased by earlier viral promoter expression. However, expression from different late promoters can influence overall CD8+ T cell priming, ganglionic infiltration, and retention of CD8+ T cell populations associated with HSV-1 latency.
RESULTS
Ectopic expression of the gB494–509 peptide in HSV-1 primes a robust gB-CD8 response in B6 mice.
The goal of these studies was to determine how the activity of different constitutive, early, late and latency-active promoters influence the numerical and functional gB-CD8 response in the HSV latently infected ganglia of HSV-1-infected B6 mice. In particular, we sought to determine if the fraction of immunodominant gB-CD8s in the ganglionic CD8+ T cell population could be increased by using promoters that mediated earlier expression. We also postulated that the promoter of LAT may increase expression and influence the latent CD8+ T cell infiltrate since it is the only region abundantly transcribed during latency. We were also interested in further evaluating how true late γ2 promoters affect the hierarchy, since our previous data had suggested that while the majority of the HSV genes are from the late γ kinetic class, expression of the complete gB protein as a true late gene was poor at retaining a gB-CD8 population in the ganglia during latency (32). Given that the expression of HSV gB as a different kinetic gene affects pathogenicity and had a fitness cost to the virus (32), we sought to separate the immunodominant epitope from the gB protein.
We recently detailed recombinant HSV-1 that contains a single point mutation (S1L) in the gB498–505 epitope that fully eliminates priming and the development of gB498–505-specific CD8+ T cells in B6 mice, without altering virus fitness (23). HSV-1 S1L generates an equivalent-sized CD8+ T cell response in the ganglia of mice to that induced by HSV-1 WT, but gB-CD8s are not primed (Fig. 1A) (23), and in their absence, the non-gB-CD8s expand to fill the T cell compartment systemically and in the ganglia. A recombinant HSV-1 strain was developed in the S1L genome background that expressed, at the gC locus, a 4-mer of the gB494–509 peptide as a GFP fusion protein regulated under the control of the gB promoter (gBp, Fig. 1B). We have previously shown that the loss of gC does not pathogenically impair HSV-1 if the corneas are scarified before infection in the B6 ocular infection model (33). Mice infected by the ocular route with this recombinant virus (HSV gBp) efficiently primed an HSV-1-specific CD8+ T cell response, and the TG-infiltrating population contained gB498–505-specific CD8s (detected using gB498–505 H2Kb major histocompatibility complex class I [MHC-I] tetramers) at nearly half of the total ganglionic CD8+ T cells (Fig. 1C). This shows that the expression of the SSIEFARL immunodominant epitope can be separated from gB protein and allow manipulation of epitope expression without affecting the essential protein gB.
FIG 1.

Ectopic restoration of an immunodominant gB498–505-specific CD8+ T cell response. B6 mice were infected at 1 × 105 PFU/eye with either WT KOS strain, S1L, or the ectopically restored gBp virus. At 8 days postinfection, TG single-cell suspensions were stained with anti-CD45, anti-CD3, anti-CD8, and gB498–505 tetramer. Depiction is gated to show only CD8 staining cells and gB498–505 tetramer staining, and the fraction is the gB tetramer-stained cells as a fraction of the total ganglionic CD45+ CD3+ CD8+ T cells. These data are representative of one experiment that was repeated at least 3 times (n = 5 mice/group). (A) Representative flow cytometry demonstrating MHC-I gB498–505 tetramer staining of total ganglionic CD8+ T cells in mice infected with WT HSV or HSV with the S1L epitope point mutation in gB. (B) Depiction of ectopically restored gB peptide expressing virus. In a gB-S1L point mutation parental strain, the native gC promoter drives a monomeric red fluorescent protein (mRFP) transcriptionally terminated by a BGH signal, followed by an inserted gB promoter driving (gB494–509)4 linked in frame to EGFP. (C) Representative flow cytometry of gB498–505 tetramer staining of total ganglionic CD8+ T cells in mice infected with HSV S1L or HSV expressing gB498–505 epitope containing multimers linked to EGFP under the control of the gBp. TRL, terminal repeat long; UL, unique long region; IRL, inverted repeat long; IRS, inverted repeat short; TRS, terminal repeat short; prom, promoter.
Expression kinetics of the gB498–505 epitope from different promoters in recombinant HSV-1.
Since the gB-CD8 response could be restored by ectopic expression of the epitope from HSV S1L, the same strategy was employed to develop recombinant HSV-1 strains that expressed the gB498–505 epitope under different promoters (Table 1). One of the goals of this strategy was to determine if constitutive or more-active promoters might be capable of driving a more highly skewed CD8+ T cell response to the gB epitope over that made by the gB promoter in the WT virus. We were also intrigued by the observation that none of the HSV-1 α genes expressed an epitope that was recognized by CD8+ T cells in the B6 mouse background (18). We therefore evaluated the constitutive cytomegalovirus (CMV) immediate-early promoter (CMVp) and the immediate-early promoter driving the ICP0 protein (ICP0p); each is efficiently expressed during a viral lytic infection, and the CMV IE promoter may be active during latency. We also evaluated the promoter driving the VP16 (VP16p), which is expressed in a lytic infection with γ1 kinetics and has recently been indicated to be expressed in mice ganglia as a “phase 1” gene expressed before α genes in the context of murine HSV reactivation from latency (34). Three γ2 promoters were also evaluated to build on our previous studies suggesting that true late promoters driving gB drove a weaker ganglionic CD8+ T cell response (32). The strategy used here was without the complication of expressing the whole gB protein as a true late gene, as this resulted in a considerably less fit virus background (from the gC, UL38, and UL41 genes; HSV-gCp, UL38p, and UL41p, respectively). UL41 is reportedly expressed very late in the infectious cycle, based on transcription analyses (35). Finally, we developed virus in which gB epitope was expressed from the full latency-active promoter (LAP) which is responsible for expression of the LAT transcripts detected in latently infected neurons (36–38).
TABLE 1.
Promoters chosen for ectopic gB498–505 peptide expression
| Promoter | Description | Primer sequence (5′–3′) or description for direction: |
|
|---|---|---|---|
| Forward | Reverse | ||
| gBp | HSV-1 γ1 late promoter for glycoprotein B | GGATCCGGATCCGCACGACGGGCCCCCGTAG | AAGCTTAAGCTTTAATACAGCGCGGTGTTGG |
| CMVp | CMV α promoter, constitutive expression | GGATCCGGATCCCTGAGTCCGGTAGCGCTAGCGGATCTGACGGTTCACTAAA | AAGCTTAAGCTTTAGTTATTAATAGTAATCAATT |
| ICP0p | HSV-1 α promoter for ICP0 | GGATCCGGATCCGGGTCGTATGCGGCTGGAG | AAGCTTAAGCTTACTTGCAAGAGGCCTTGTTC |
| VP16p | HSV-1 γ1 late promoter for the VP16 transactivator | GGATCCGGATCCGGTTGATACGGGAAAGACGATATC | AAGCTTAAGCTTCACGGCGACTCGAGGGCGTTC |
| gCp | HSV-1 γ2 late promoter for glycoprotein C | Native promoter region | Detailed in Materials and Methods |
| UL38p | HSV-1 γ2 late promoter for UL38 capsid protein | GGATCCGGATCCTGGTCTTCATTGCGACCCCA | AAGCTTAAGCTTTGCCGTCTCGGCCGTGGG |
| UL41p | HSV-1 γ2 late promoter for UL41, virion host shutoff gene | GGATCCGGATCCGATGTCAGGTCAATTGTAA | AAGCTTAAGCTTCGGCGCCCTGCAGGACCAC |
| LAP | Full 2-kb HSV-1 LAP1/2 promoter for LAT | Fragment generated by restriction digestion | Detailed in Materials and Methods |
All recombinant HSV-1 were derived similarly as shown in Fig. 1, in which the epitope expression cassette was placed at the gC locus and selected on the basis of monomeric red fluorescent protein (mRFP) from the gC promoter after homologous recombination. Plaque-purified viruses homogeneous for mRFP were grown and DNA was evaluated for homogeneity by Southern blotting and sequencing. Virus was assessed by a single-step growth curve in Vero cells, which established that there were no differences in growth for each virus compared to the KOS WT parental strain and the HSV-1 S1L parental virus (Fig. 2).
FIG 2.

Characterization of growth and gB498–505-EGFP expression of recombinant viruses. (A) A total of 5 × 105 Vero cells were infected at an MOI of 1, and after 1 h of absorption, the cells were harvested at 0-, 2-, 4-, 8-, 12-, 18-, and 24-h time points. Cells were then freeze-thawed 3 times, and virus concentration was determined by plaque assay on Vero monolayers. Each data point represents a mean plaque count of n = 3. This experiment was repeated with similar results. (B and C) Vero cells were infected at an MOI of 10 in 24-well plates, with or without inhibitors. Cycloheximide (100 μg/ml) and phosphonoacetic acid (400 μg/ml) were both pretreated for 1 h before infection and maintained in inocula and media throughout infection. At the indicated time points, RNA was isolated, and qPCR was performed on cDNA using primers specific for EGFP. Data shown represent the fold change over inoculum at t = 0. Statistical significance was assessed by one-way analysis of variance (ANOVA) with Holm-Sidak’s multiple-comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
We next sought to establish the kinetics of expression of the gB epitope from these viruses via detection of the linked enhanced green fluorescent protein (EGFP) by quantitative reverse transcription-PCR (qRT-PCR) (Fig. 2B and C). In addition to detection of expression over time, verification of the kinetic class (α, γ1, or γ2 for the selected promoters) needed to be confirmed in vitro. Since α gene expression requires no new protein synthesis, the ribosomal inhibitor cycloheximide should not affect ICP0p. Conversely, since γ1 and especially γ2 genes not only need new protein synthesis but also ongoing viral DNA replication to be expressed fully, both cycloheximide and a viral DNA replication inhibitor, phosphonoacetic acid (PAA), should reduce late-gene expression. Indeed, Vero cells infected with the panel of recombinant promoter viruses in the presence of cycloheximide (4 and 8 hours postinfection [hpi]) or PAA (4, 8, and 16 hpi) showed no effect on ICP0p gB498–505-EGFP expression, but both drastically reduced the expression of γ1 (gBp and VP16p) and γ2 (gCp, UL38p, and UL41p) late genes (Fig. 2B and C). This would be consistent with previously published data indicating that LAP activity in lytic infections is also expressed with true late gene kinetics (36, 39, 40). It is also noteworthy that the CMVp is moderately affected by both inhibitors, suggesting a complex regulation. These data taken together demonstrate that each promoter behaves similarly to previous characterizations and are expressed over time with the expected kinetics in Vero cells.
While our design was to express the gB epitope as EGFP fusions, virus plaques did not fluoresce at EGFP wavelengths under fluorescence microscopy, and immunoblot analyses for EGFP in recombinant virus-infected cell extracts showed little full-length protein (data not shown). This suggested that gB498–505-EGFP was unstable. However, we reasoned that this would also not be detrimental to rapid surface MHC-I presentation of peptide for CD8+ T cell recognition. It has been shown that surface MHC-I peptide expression can be quantified using a modified gB-CD8 T cell stimulation assay, which was sensitive to detect the very early (<2 h) expression of gB in a lytic HSV infection (19). Accordingly, monolayers of the MHC-I-compatible B6 mouse-derived fibroblast cell line B6WT3 were infected with each virus, and infections were halted at 4, 8, and 24 hpi by a precalibrated exposure to 254-nm UV light under sterile conditions. Cells were then cocultured with 1 × 105 splenic gB-CD8+ T cells/culture derived from HSV-infected gB-T mice at 8 days postinfection (dpi) (Fig. 3A). The gB-T1 mice are transgenic for the gB498–505-specific T cell receptor, and the majority of activated CD8+ T cells are able to recognize the gB498–505 epitope (41). The T cell stimulation assay was done in the presence of brefeldin A, allowing intracellular cytokine accumulation of tumor necrosis factor alpha (TNF-α) and IFN-γ for immunodetection and flow cytometry quantification. In cells gated for CD45+, CD3+, and CD8+ (Fig. 3B), each recombinant HSV-1 strain stimulated gB498–505-specific CD8+ T cells in a manner consistent with the promoter chosen.
FIG 3.

Viral gB498–505 peptide from selected promoters is detected by gB495-505-specific CD8+ T cells. (A) Depiction of experimental setup. Confluent 12-wells of B6WT3 cells were infected with approximately 1 × 106 PFU at an MOI of 1 with WT HSV-1, S1L, or each promoter virus. At 4, 8, or 24 h, each plate was UV inactivated (with less than 10 PFU remaining per well) and combined into coculture with 1 × 105 splenic cells derived from an infected gB-T mouse at 8 dpi for 6 to 8 h in the presence of brefeldin A. Cocultures were then stained for CD45, CD8, IFN-γ, and TNF-α. (B) Flow cytometry controls, including isotype antibodies for IFN-γ and TNF-α, a phorbol myristate acetate (PMA)/ionomycin positive control, and an uninfected B6WT3 fibroblast negative control. (C) Representative flow cytometric analysis from each group of intracellular staining for IFN-γ and TNF-α. (D) Quantitation of data representative of five replicates from at least two separate experiments for each virus, with the mean number of IFN-γ+ CD8+ T cells per coculture stimulation shown. The dotted line is indicative of the average maximal response of WT HSV-1 after a 24-h costimulation.
At 4 h postinfection, a small number of gB-CD8s recognized WT HSV-1 KOS-infected B6WT3 fibroblasts, and the fraction responding increased at 8 and 24 h, consistent with leaky-late expression kinetics expected for gBp. In contrast, cells infected with HSV S1L failed to stimulate any detectable cytokine production in gB498–505-specific CD8+ T cells. This establishes the specificity of the assay for MHC-I-presented gB498–505 peptide. Similar timing and kinetics of gB-CD8 activation were stimulated by cells infected by HSV-gBp but were consistently higher at every time point, likely due to the peptide cassette expression of a 4-mer. Cells infected with HSV making peptides driven by the constitutive CMV IE promoter or the ICP0 promoter were found to strongly activate gB-CD8s at 4 h and stimulated a higher fraction of CD8s to become activated and express IFN-γ and TNF-α cytokines than that stimulated by cells infected with WT HSV, making the gB peptide from the gB promoter at 4 and 8 hpi. The gB-CD8s also responded at higher frequencies to cells infected with HSV expressing the epitope under the VP16p, reaching levels similar to those expressed ectopically from the gB promoter at each time point.
In contrast, expression from the true late promoters gCp, 38p, and 41p resulted in lower levels of gB-CD8 activation at 4 h, although by 24 h postinfection, the levels of activated CD8+ T cells were similar (38p) or higher (gCp) than those generated by WT HSV-1 KOS-infected cells at the same time point. These results were consistent with expected true late gene expression kinetics. Finally, the LAP showed a considerably weaker activation of gB-CD8s than WT infections by 24 hpi (Fig. 3C). These data were consistent with gB498–505-EGFP expression levels shown in Fig. 2, with minor differences likely due to cell type and translation/MHC-I presentation efficiency.
Equivalent ganglionic CD8+ T cell responses are generated by HSV expressing gB498–505 from different promoters but contain altered gB-CD8s immunodominance.
Since we are using the KOS strain of HSV-1, there was little corneal disease present in any of the groups. Therefore, we next examined the panel of recombinant HSV-1 promoter-peptide viruses for in vivo fitness by measuring their ability to (i) establish a ganglionic latent load in the murine TG after ocular infection and (ii) induce a strong ganglionic CD8+ T cell response at the onset of latency. Our previous studies with parental S1L virus established that it induced ganglionic viral load at day 8 that was not significantly different from mice infected with HSV-1 KOS. It also induced an equivalent-sized ganglionic CD8+ T cell infiltrate (23). Corneas of B6 mice were infected with 1 × 105 PFU of HSV-1, the S1L virus, or each promoter virus, and ganglionic loads were determined using quantitative PCR (qPCR) real-time methods at 8 dpi, using a well-characterized primer set recognizing sequences in gH (32). As expected, S1L and KOS viral DNA loads in the ganglia at day 8 were similar, and all new recombinant HSV strains generated from S1L yielded a ganglionic DNA load at least as robust as that of WT HSV S1L. This establishes that the viruses are robust and can establish latent genome loads similar to those of parent S1L and WT HSV (Fig. 4). Analyses of the T cell populations also indicated that the total numbers of CD8+ T cells infiltrating the ganglia at the peak infiltrate time of 8 dpi (Fig. 5A) were similar for each virus, and the contracted infiltrates at latency (day 30 to 35) were similar to those induced by WT HSV-1 (Fig. 5B). We take these data to indicate that the recombinant viruses used in this study were not significantly attenuated in establishing latency, inducing the peak ganglionic CD8+ T cell infiltrate at the onset of latency, or retaining a small population of gB-CD8s after contraction in the latently infected ganglia.
FIG 4.
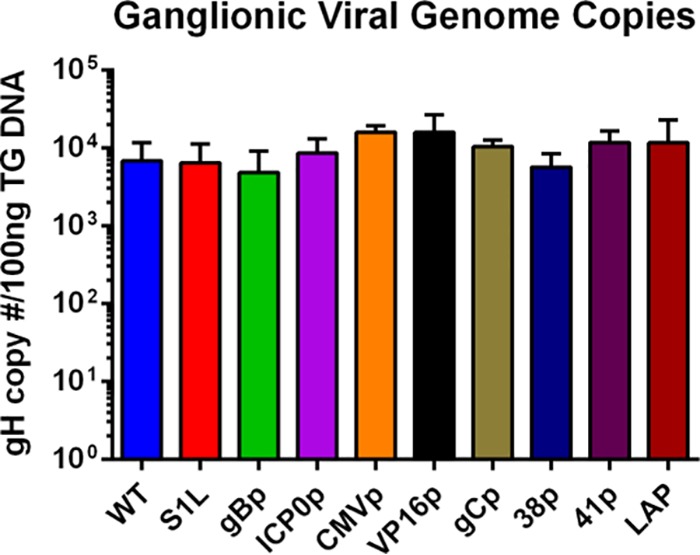
Ganglionic viral genome copy number determined by qPCR in the TG of mice ocularly infected with 1 × 105 PFU of HSV-1 WT, S1L, or gB498–505 promoter viruses at day 8 postinfection (n = 4 to 10 per group). Values are representative of the total copies per 100 ng of TG DNA recovered. No significant differences from the WT was found. HSV-1 KOS strain bacterial artificial chromosome (BAC) standards were used as a positive control for gH detection, as well as calculations for viral copy number. No significant differences between means were found, as analyzed by one-way ANOVA.
FIG 5.

gB498–505-specific CD8+ T cell responses to selected gB498–505 promoter viruses. Corneas of mice were infected with 1 × 105 PFU/eye of HSV-1 WT, S1L, or a recombinant HSV-1 expressing gB498–505 from the indicated promoter. At 8 dpi (peak CD8+ T cell infiltrate) or 30 dpi (latency), TG and spleen samples were dissociated into single-cell suspensions and surface stained with antibodies to CD45, CD3, CD8, and with MHC-I gB498–505 tetramer, as detailed in Materials and Methods. (A and B) Cells were subsequently analyzed by flow cytometry and show total CD8+ T cells per TG at 8 dpi (A) or 30 dpi (B). (C) The total number of gB498–505 tetramer-positive CD8+ T cells in each spleen at 8 dpi are also shown. (D) The fraction of gB498–505 tetramer-positive cells among the total CD8+ T cells in the TG at day 30 is shown. The data shown are pooled from 2 to 3 identical experiments or are representative of one of at least two repeats of n = 3 to 5 mice per group. Bars represent the mean and standard deviation for each group. Significant differences by one-way ANOVA from the WT group are indicated *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. tet, tetramer.
Intriguingly, the gB-CD8 compartments in the splenic and ganglionic population differed for several of the viruses (Fig. 5C and D). The splenic gB-CD8 levels for five of the viruses (CMVp, gBp, ICP0p, VP16p, and gCp) were strong, suggesting efficient priming of a systemic gB-CD8. Three of the viruses, namely, the 38p, 41P, and LAP viruses, induced only a weak splenic gB-CD8 response by day 8, suggesting poor priming. In the infected TG at 8 dpi when HSV begins to enter latency, those viruses priming a substantial gB splenic CD8+ T cell response all showed a remarkably similar fraction of the ganglionic infiltrate to be gB498–505 specific, and in all cases, the gB-CD8s represented more than 40% of the ganglionic CD8 population, as seen when induced by WT HSV-1 infections. An exception was that driven by HSV-1 gCp, where few ganglionic gB-CD8s were seen at day 8, despite an apparent systemic priming. Not surprisingly, few gB-CD8s were detected in ganglia latently infected with viruses that failed to prime a significant systemic gB-specific CD8 response.
In WT virus infections, latently infected ganglia contain a contracted CD8+ T cell population that remains stable for the life of the host. At latency, our viruses could be divided into one of two groups (Fig. 5D). Viruses that effectively primed a systemic gB498–505-specific CD8+ T cell response, including HSV-gCp, developed a ganglionic infiltrate that was typically dominated by gB-CD8s to approximately half of the total CD8 in the ganglia, as seen in mice infected with WT HSV-1. Given that these ectopic peptide-promoter viruses expressed the gB498–505 epitope on the surface of lytic infected cells at levels greater than with the WT virus (as measured by the in vitro levels of T cell activation; Fig. 3), we conclude that earlier expression in the context of the lytic HSV replication cycle cannot result in a higher proportional gB-CD8 response in the ganglia. Even under the CMV and HSV ICP0 α promoters, which expressed peptide efficiently, the ganglionic population was not significantly different from that of the WT. The second group of viruses (38p, 41p, and LAP) were those in which few gB-CD8s were primed and showed comparatively little gB-CD8 infiltrate in the latently infected ganglia. However, we pursued these further because we wanted to ask the question as to whether gB-CD8s, if they were primed and present, could enter the ganglia and be retained.
In a WT HSV infection, the retained ganglionic gB-CD8 population shows a higher degree of functionality than do the non-gB-CD8 populations, which develop signs of functional impairment in that they express less granzyme B, produce less IFN-γ and TNF-α upon antigen stimulation, and express more of the exhaustion marker PD-1 on their cell surface. In contrast, the ganglionic gB-specific CD8s show maintained functionality from onset through HSV-1 latency, even up to 90 days postinfection (14, 22). One factor influencing CD8+ T cell functionality is the chronicity of antigen expression during viral persistence (42). As such, we next addressed the functionality of the gB-CD8 and non-gB-CD8s in the TG at latency for the five viruses in which a gB-CD8 response was primed to evaluate if the different promoters altered the functional make-up of the gB-CD8 and non-gB-CD8 populations. Ganglionic CD8+ T cells at long-term latent infections (90 dpi) of each virus were assessed for surface expression of PD-1 and intracellular staining for the functional marker granzyme B. Indeed, all viruses maintained an immunodominant gB-CD8 infiltrate accounting for about half of the ganglionic CD8+ T cells (Fig. 6A). The WT HSV-1 ganglionic gB-CD8s showed very low levels of surface PD-1, while the non-gB-CD8 population had considerably more cells with surface PD-1 expression, reaching average levels of more than 21% (Fig. 6B). A reverse correlation was seen for granzyme B in that most gB-CD8s stained positive for granzyme B, while less than 20% of the non-gB-CD8s were granzyme B positive (Fig. 6C). The different promoter-epitope viruses showed an overall similar phenotype, with gB-CD8s showing high functionality and low PD-1, while non-gB-CD8 levels were consistently higher in PD-1 than that with gB-CD8s. There were some statistically significant differences in that the fractions of granzyme B-positive gB-CD8s were higher for the WT and HSV-1 gBp viruses but were marginally lower in ganglionic gB-CD8s of all other promoter viruses stimulating a strong ganglionic gB-CD8 response. The CMVp virus gB-CD8s were also significantly higher in PD-1 expression, which may be consistent with higher epitope exposure at late times. We take these results to indicate that expression from different promoters does not greatly affect hierarchical trends of ganglion-resident memory gB-CD8 versus non-gB-CD8+ T cells that are functional, and in all cases, T cells that recognize the gB epitope retain a more functional phenotype than do the non-gB-CD8s.
FIG 6.

gB-specific CD8+ T cells remain more activated during long-term latency than do subdominant ones. Mice infected by the corneal route with 1 × 105 PFU/eye of HSV-1 WT KOS, gBp, CMVp, ICP0p, VP16p, or gCp were assessed at long-term latency at 90 dpi. TG were dissociated into single-cell suspensions and surface stained with antibodies to CD45, CD3, CD8, and PD-1 and MHC-I gB498–505 tetramer, stained intracellularly for granzyme B (GzB), as detailed in Materials and Methods, and analyzed by flow cytometry. (A to C) Data are presented as the mean ± standard error of the mean (SEM) (n = 5 to 10 mice per group [10 to 20 TGs per group]) of the percentage of gB tetramer-positive of total CD8+ T cells per TG (A) and of gB498–505 tetramer-positive (gBs) or -negative (gB-nons) CD8+ T cells in each TG that are PD-1 (B) or GzB positive (C). The data shown are representative of two separate experiments. Bars represent the mean and standard deviation for each group. *, P < 0.05; **, P < 0.01, by one-way ANOVA with multiple comparisons to WT within that group.
Epitope expression from late promoters results in poor gB-CD8 priming.
We first addressed the ganglionic CD8+ T cell populations developing in mice infected with HSV expressing the gB epitope from gCp, because this virus clearly primed a systemic a gB-CD8 response, but few gB-CD8s infiltrated the ganglia at day 8, while a typical half fraction of total CD8s at latency were gB498–505 specific (Fig. 7A). In animals infected with this virus, the ganglionic CD8+ T cell infiltrate began to contract from day 10 onwards, as seen for WT virus, but intriguingly, the fraction of CD8s specific for gB increased so that by 12 dpi, the gB-CD8 fraction was at the level of immunodominance seen for WT HSV-1 (Fig. 7B). These data suggest that expression driven by the gC promoter leads to preferential retention of the gB-CD8 population infiltrating the ganglia to reach the half fraction of the total CD8s seen at latency. As our previous data have shown that antigen expression in part drives a preferential retention of CD8+ T cell populations (23), the gC promoter may express at a low level throughout the establishment and long-term latent state.
FIG 7.

gB498–505 expressed from gCp results in a delayed gB-CD8+ T cell immunodominance in ganglia. Corneas of mice were infected as described previously, with 1 × 105 PFU/eye of HSV-1 WT or recombinant HSV-1 expressing gB498–505 from the gC promoter. At 8, 10, 12, 16, 21, and 30 dpi, TG were dissociated and stained with antibodies to CD45, CD3, and CD8 and with MHC-I gB498–505 tetramer, as detailed in Materials and Methods. (A) The total number of TG-infiltrating gB-specific CD8s is shown over time in a gCp infection. (B) The fraction of gB498–505 tetramer-positive cells among the total CD8+ T cells in the TG is shown over time in each infection. Bars represent the mean and standard deviation for each group. *, P < 0.05; **, P < 0.01; ****, P < 0.0001, by unpaired t test between groups at each time point.
The HSV strains expressing the gB epitope from the UL38, UL41, and LAT promoters showed a defect in priming of the gB-CD8 response, despite the fact that gB peptide is presented at levels similar to those in WT HSV-1 in vitro after 24 hpi (Fig. 3). Reduced or absent priming from protein antigens expressed late in the growth cycle has been reported in other viruses (43, 44). We note that while true late genes form the majority of the HSV proteome, they are vastly underrepresented in the antigen hierarchy recognized by HSV-1-specific T cell responses in B6 mice (18, 23).
Regarding HSV expressing the gB epitope from the LAP, while it did not induce a robust gB-specific immune population, a small but significant fraction of gB-CD8s were detected in the systemic response (Fig. 5C) and also in the latently infected ganglia at later times postinfection over that seen for S1L (Fig. 5D). While the LAP is weakly expressed in a lytic infection with γ2 kinetics, it should more active during latency. Quantification of the total gB-CD8s by tetramer staining over time revealed that while no gB-CD8s were seen at the peak of the infiltrate at day 8 postinfection, a small but significant fraction of ganglionic gB-CD8s developed by 20 dpi, which represented approximately 11% of the total contracted CD8 populations in the ganglia during latency (Fig. 8A and B) that was maintained out to 60 dpi. This low late-developing population could have been due to the expansion of a very small population infiltrating the ganglia at the onset of latency, or it could be due to the reentry of gB-CD8s at later stages. We performed BrdU pulse labeling for 2 days to identify dividing CD8+ T cells in the ganglia at 20 and 60 dpi. In mice infected with WT HSV-1, few cells were labeled at day 20 and day 60, indicating that the majority of the CD8+ T cells were not proliferating in the period of latency in which labeling was performed. However, the ganglia of mice infected with the LAP virus contained significantly more replicating CD8+ T cells at 20 dpi, and analyses with tetramer revealed the majority of this was proliferating gB-CD8 populations at 20 dpi (Fig. 8C and D). These cells were still undergoing marginal proliferation at day 60, significantly more than those for WT virus at both short- and long-term latency (Fig. 8D). These results suggest that the LAP was active, and the antigen stimulated the division of the few gB-CD8s that were present.
FIG 8.

Latent expression of gB498–505 results in reduced and delayed gB-specific CD8+ T cell infiltrate. Corneas of mice were infected as described previously, with 1 × 105 PFU/eye of HSV-1 WT or recombinant HSV-1 expressing gB498–505 from the LAT promoter. Two days prior to harvest at 20 and 60 dpi, mice were pulsed with 1 mg/mouse bromodeoxyuridine (BrdU). At 9, 20, 30, and 60 dpi, TG were dissociated and stained with antibodies to CD45, CD3, CD8, and BrdU and MHC-I gB498–505 tetramer, as detailed in Materials and Methods. (A) The total number of TG-infiltrating gB-specific CD8s is shown over time. (B) The fraction of gB498–505 tetramer-positive cells among the total CD8+ T cells in the TG is shown over time in each infection. (C and D) BrdU staining of total CD8s (C) and gB498–505-specific CD8s (D) is shown at 20 and 60 dpi. Bars represent the mean and standard deviation for each group. *, P < 0.05; **, P < 0.01; ****, P < 0.0001, by unpaired t test between groups at each time point.
True late promoters and the LAP can retain gB-CD8+ T cell infiltrates in the latently infected ganglia.
Since the true late promoters were not able to induce a robust gB-specific splenic response, we then asked if these viral promoters in the latent HSV genome were able to attract and retain a gB-CD8 response if such a response was efficiently primed at the onset of infection. We exploited the published observation that, at the time of the peak infiltrate into the ganglia at day 8, any activated systemic CD8+ T cells in the systemic population will enter the acutely infected ganglia, independent of the antigen to which the cells are directed (45). To evaluate this property, we employed a dual-infection strategy in which the ocular route was used for infection by the late promoter viruses, but a simultaneous flank infection with WT HSV-1 was used to systemically prime a gB-CD8 response (Fig. 9A). At day 8, all ganglia infected with the nonpriming HSV-1 recombinants contained gB-CD8s, consistent with the notion that recently activated CD8+ T cells will traffic to sites of infection (Fig. 9B). As the CD8 population contracts as viral latency is established, only ganglia that harbor the cognate gB498–505 antigen should retain a gB-CD8 infiltrate. Greatly reduced levels of the gB-CD8s were seen in ganglia latently infected with S1L, which does not express a recognizable gB epitope at any time. The mice infected with viruses expressing the gB epitope under the control of promoters that fail to prime a large initial response (UL38p, UL41p, and LAP) retained flank-primed gB-CD8s in the ganglia at a level significantly higher than the gB-CD8s retained in the ganglia for S1L (Fig. 9C). The levels were much lower for the viruses expressing the immunogenic peptide from UL38 and UL41 promoters than for the viruses expressing peptide under the control of the latency-active promoter (LAP). We conclude from these data that expression of the peptide from the two true late promoters is not sufficient to prime a significant gB-CD8 response, but when a gB-CD8 population is effectively primed from the flank, there is a low level of γ2 late-gene expression within the ganglia that is sufficient to retain a small number of CD8+ T cells within the latently infected ganglia. In contrast, the LAP can retain a much larger gB-CD8 response, consistent with the idea that this promoter expresses during CD8+ T cell contraction and latency. Taken together with the data from Fig. 8, this indicates that expression from the LAP in a lytic infection is not sufficient to induce antigen levels sufficient for priming a large CD8+ T response. However, during latency, LAP-driven antigens will mediate efficient ganglionic retention driven by ongoing antigen expression. Our data show that poor priming and generation of tissue-resident memory gB-CD8s in the ganglia can be bypassed by priming the responses at another location on the animal at the onset of latency.
FIG 9.

True late or latent promoters deficient in priming gB-CD8s are capable of maintaining ganglionic gB-CD8s at latency. (A) Representation of an infection model in which mice received bilateral corneal infections with S1L, UL38p, or UL41p HSV-1 and flank infection with WT HSV-1. All corneal infections were with 1 × 105 PFU/scarified cornea, and flank infections were with 1 × 106 PFU on a scarified flank. (B and C) At 8 (acute) or 30 (latent) dpi, TG suspensions were analyzed by flow cytometry for CD45, CD3, and CD8 and the gB498–505 tetramer. Shown is the frequency of CD3+ CD8+ T cells in the TG that are gB tetramer positive. Five to 15 mice were analyzed per group. Bars represent the mean and standard deviation for each group. Statistical significance (P values) by one-way ANOVA is displayed above each group at latency.
DISCUSSION
Pathogens have many potential antigens to which adaptive immune responses could develop but only a fraction prime the CD8+ T cell responses or act as their recognition target. The identity of these antigens and the hierarchical order of the CD8+ T cell populations to them are of interest in developing vaccines, but they are governed by complex and multifactorial influences (46). The HSV-1-infected B6 mouse model is a powerful model to examine such factors because viral antigens and their dominance hierarchy are known, and the hierarchy is dominated by a single gB498–505 epitope (18). Here, we assessed aspects of how the dominant CD8+ T cell population is influenced by promoters driving the immunodominant epitope in HSV-infected B6 mice. We were particularly interested to determine if the proportion of gB-CD8s in the latent ganglia could be increased by the use of more active promoters, because gB-CD8s are functionally active and could control reactivation events in HSV infections of B6 mice (23).
Our strategy used HSV-1 with a recently detailed mutation of the gB498–505 epitope that ablates gB-CD8+ T cell priming and recognition (23). Our strategy to use the same site of ectopic expression also ensured that viruses that were comparatively fit in the murine model. In a previous study, we attempted to modify the CD8+ T cell responses by modifying the whole gB protein, but this resulted in a considerable cost to viral fitness (32). While all viruses were eventually inserted into and disrupted the expression of gC, we have previously established that this does not affect infection of B6 mice when infections are accompanied by corneal scarification (33, 47). All recombinant viruses used here induced ganglionic latent loads at least as efficiently as the WT virus and a total peak of the immune infiltrate in the ganglia similar to WT-equivalent total CD8+ T cells. While this strategy does have a caveat that it may not completely reflect the influence of local genomic environment on the expression of the native promoter, it greatly simplified our assessment.
One conclusion is that the gB promoter is not necessarily a “Goldilocks” promoter, which is expressed at just the right levels and strength to induce an immunodominant CD8+ T cell response. Several lines of evidence have indicated that retention of the CD8+ T cell population in the HSV-1 latent state requires antigen expression (20, 23), most likely through sporadic gene derepression during HSV-1 latency (12, 13, 16). We considered it possible that the immunodominance of gB498–505 in the latent infected ganglia was due to the gB promoter activity being at an optimal level not only for CD8+ T cell priming but also sporadic gene expression during the latent state. However, we show here that other promoters can easily generate a strongly primed gB-immunodominant CD8+ T cell population that differs from that of the WT systemically yet results in a near-identical fraction of gB-CD8s in the ganglia at latency, even in the case of the gC promoter, where there was apparently delayed priming and ganglionic infiltration (Fig. 7). Our data also suggest that increased lytic expression of the epitope does not necessarily lead to increased levels of CD8+ T cell immunodominance. WT HSV-1 normally expresses gB as a γ1 leaky-late gene that can be recognized in MHC-I-matched cells by gB-CD8s within hours postinfection (19). HSV with the epitope expressed from the CMV IEp and the α-regulated ICP0p showed only marginal differences in the priming of the splenic gB-CD8 response, despite being able to activate over five times as many CD8+ T cells by 4 h postinfection using in vitro activation assays over that of WT HSV-1 (Fig. 2). While we do not know if these promoters are more active than the gB promoter in the latently infected murine ganglia, the CMV IE promoter is reportedly active from the HSV genome for up to 30 days in the murine TG (48). HSV with the epitope driven by the VP16 promoter was also evaluated, since VP16 has been implicated as a possible pre-α gene that drives α genes in sporadic reactivation events (11, 34). It was as active as a gB promoter in driving the gB epitope in a lytic infection, and the 4-mer multimeric peptide made in a lytic infection resulted in high target recognition. However, it did not result in a greater immunodominant response than that with the WT. We conclude that this subset of viruses achieves a minimal level of ganglionic expression that is required to retain the gB-CD8 infiltrate during latency and that expression above this minimal level neither primes a more immunodominant CD8+ T cell response nor maintains a higher level of immunodominance during latency. The factors affecting this limit are not yet clear.
An additional conclusion regarded the observation that the CD8+ T cell repertoire to HSV in B6 mice contained no substantial representative CD8 populations targeting epitopes on α proteins. ICP0 was evaluated to address the possibility that this kinetic protein group has unique properties that act to prevent their ability to prime the CD8+ T cell response. However, this does not seem to be the case, since α promoter expression of the epitope stimulated a robust and strong CD8+ T cell immune response. Taken together, while one of the goals of this project was to create a virus that rapidly expressed a large amount of gB498–505 epitope and elicit a CD8+ T cell response that remains particularly active and capable of effector functions throughout latency in B6 mice, it seems that this is not achievable by increasing epitope expression alone. The mechanism governing this peak may involve other factors, such as cytokine competition within the TG, competition for antigen, viral immune evasion strategies, and other factors (43, 46). One possibility is that, as viral gB498–505 antigen is detected and shut down by gB-CD8s, there is less antigen available for other gB-CD8s to detect, leading to an inability to rise above a certain threshold of gB-CD8s. From a host evolutionary perspective, it also makes sense not to focus all attention on one epitope, which would provide opportunities for viral escape mutants, and therefore, there may be interactions among specific CD8 populations preventing full immunodominance within the tissue.
This work does indicate that the choice of promoter used for expressing the epitope in the context of HSV infection can influence CD8+ T cell priming and long-term retention of CD8 populations in the latently infected murine ganglia. In contrast to the early and strong lytic promoters, the use of γ2 late-promoter-driven epitopes from UL38p or UL41p demonstrated a reduced ability in priming gB-CD8 responses. These promoters were also reduced in the ability to retain gB-CD8s in the ganglia when the gB498–505 response is exogenously primed by a WT infection. These two promoters were clearly active during lytic infections, since in vitro by 24 hpi, both viruses could elicit gB-CD8+ T cell activation levels that equaled or exceeded the WT virus response (Fig. 3). Neither virus was able to activate a significant fraction of the gB-CD8s at 4 h postinfection, consistent with the delayed expression expected for a true late promoter. The non-gB-CD8s that make up the rest of the response have been thoroughly characterized for S1L (18), and these expand when gB-CD8s fail to prime (23). We suspect that a similar compensation occurs for the viruses. Why these promoters did not efficiently prime a response is not clear. However, it has come to light that generating effector memory CD8+ T cells requires several naive T cell interactions with dendritic cells (DCs) in the draining lymph nodes (49, 50), with epitope being presented on specific DC subsets and cross-presented by other DC subsets being vital steps in generating memory CD8+ T cell responses. Our altering both the expression kinetics of the peptide as well as the location within the protein (as a terminal peptide multimer instead of a residue within the gB gene) may very well change the amount of epitope that these DC populations have access to via either direct or cross-presentation. Finally, we note that both γ2 promoters could retain an exogenously primed gB-CD8 T cell response but at levels nowhere near that seen for the active viruses detailed above. This suggests that these promoters might be less active during latency than others and indicates that there is a low level of γ2 gene expression within the ganglia. This would fit with our previous observation that the subdominant population hierarchies retained in the S1L latently infected TG are different from the systemic response, suggesting that some promoters are more active during latency than others, suggesting that their cognate CD8+ T cell populations are selectively retained. Different rates of sporadic gene expression for different promoters were also seen by Russell and Tscharke (12), a finding which conflicts with the general derepression model in sporadic reactivation events put forward by Wilson and colleagues (11). This implies that there are genomic regions that are preferentially active in sporadic reactivation events, and that this in turn affects local adaptive immunity.
The gC promoter induces a different pattern of priming and retention. gC is a γ2 late gene that strictly requires new viral DNA synthesis for expression, but it appears to be expressed at levels higher than the UL38 and UL41 promoters by 24 hpi in gB-CD8 T cell activation assays (Fig. 3). In mice, the primed splenic responses were considerably reduced in comparison to the WT, gBp, CMVp, and ICP0p (Fig. 4C). More strikingly, TG infiltration of gB-CD8s was delayed compared to the WT, with only about 10% of the ganglionic CD8s being gB specific at day 8 (Fig. 7) compared to 50% for a WT infection. Our data show that gB-CD8+ T cells are retained very efficiently for this virus and become a typical immunodominant population seen during latency. This suggests that the gC promoter is sufficiently active to drive priming of the gB-CD8 population, but the activity of the promoter during latency is above the minimal level required for the 50% immunodominance just discussed. This fits well with data from studies in which both gC RNA and protein were both found to be expressed during latency (12, 51, 52).
Finally, the LAP also shows a unique pattern in priming and ganglionic retention. The full 2-kb LAT promoter used here to drive the expression of gB498–505 from an ectopic locus induced expression by 24 hpi in the gB-CD8 activation assay, although a small number of in vitro gB-CD8s were activated compared to that by the WT virus (Fig. 3). HSV LAT is active not only during latency but also during lytic infection, with kinetics of a late-regulated gene (40). The LAT promoter virus induced little priming during the initial infection of B6 mice, and the splenic response was not significantly different from that with the epitope knockout (S1L, Fig. 3). However, mice infected with this virus over time develop a small population of gB-CD8s into the murine TG (Fig. 8A). This population likely reflected an expansion, as suggested by the increased BrdU uptake by these gB-CD8s, and demonstrates that they are likely seeing expressed antigen (Fig. 8C and D). This gB-CD8 expansion results in gB-CD8s becoming a significant percentage (∼10%) of the CD8 response throughout latency, despite virtually undetectable priming (Fig. 8B). This suggests that the levels of epitope during initial priming are insufficient to compete with all other viral antigens and prime a gB-CD8 response. However, the continuous expression of gB peptide under LAP may result in CD8+ T cells being primed during a time when there are few other antigens to compete. When exogenous priming is done by flank infection, the TG retains the gB-CD8 in the ganglia quite efficiently. This suggests that the LAP could be used to maintain a CD8+ T cell population in the ganglia but only when the T cells are efficiently primed first (23).
The function and exhaustion of CD8+ T cells are influenced by both chronicity of expression and the levels of antigen expression in the lymphocytic choriomeningitis virus (LCMV) models (53). Previous studies with WT HSV show that gB-CD8s are more functional and demonstrate better antiviral effector properties than do their non-gB-CD8 counterparts, and that while many classic exhaustion markers are unreliable in this model, PD-1 expression is highly correlated with reduced functionality (18, 21, 23, 54). We found that there was a global trend for increased granzyme B and reduced PD-1 on gB-CD8s generated by all gB promoter viruses over the non-gB-CD8s (Fig. 6). This suggests that even with an apparent difference in peptide expression levels, the immunodominant gB-CD8s generally remain more activated and functional than their subdominant counterparts in the TG. The gB-CD8s retained by the CMVp virus show a higher level of PD-1 and lower level of granzyme B, which suggests they may have started to become functionally impaired due to more epitope expression within the TG.
Taken together, these data begin to paint a picture in which viral antigen expression kinetics play a role in shaping ganglionic CD8+ T cell responses associated with HSV-1 latency. Strong lytic promoters can dictate the efficiency of priming of the effector CD8+ T cell responses, but it also becomes apparent that promoter activity driving expression within the TG itself is important for selective retention of these responses. Furthermore, the TG-resident/tissue-specific CD8+ T cell responses associated with HSV latency do not necessarily reflect the early splenic and systemic response. This apparent mismatch between systemic and ganglionic CD8+ T cell memory responses, and how peptide expression affects this, will be of particular importance in designing efficacious ganglionic vaccine responses. As latency progresses, sporadic viral gene expression becomes an important factor in shaping epitope recognition hierarchies in the protective ganglion-resident CD8+ T cell populations.
MATERIALS AND METHODS
Virus and cells.
Vero cells (ATCC, Manassas, VA) and an SV40-transformed B6 embryo fibroblast cell line, B6WT3 (MHC-I compatible with C57BL/6 mice) (55), were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin G (100 units/ml), and streptomycin (100 mg/ml). All HSV-1 strains are based on the KOS strain originally obtained from a master stock originally obtained from P. Schaffer (designated HSV WT or WT). The KOS strain used has been sequenced (GenBank accession no. JQ780693.1) (56). HSV-1 lacking the immunodominant epitope through the mutation of residue 498 of gB (SSIEFARL to LSIEFARL) is termed HSV S1L or S1L, and the characterization of the T cell infiltrates to this virus was recently described (23).
Virus purification.
All HSV-1 cells used in these studies were purified from infected Vero cell monolayers showing cytopathic effect in >90% of cells after low multiplicity of infection (MOI; 0.01). NaCl (5 M) was added to the media and cells to reach a final concentration of 0.45 M, and cells were rocked for 1 h, dislodged by shaking, and spun at 2,000 × g for 10 min. Supernatants filtered through a 0.8-μm filter were layered onto a 10-ml 50% sucrose cushion made in phosphate-buffered saline (PBS) and pelleted at 142,000 × g for 1 h at 4°C. The supernatant was removed, and the virus pellet was carefully resuspended in a small volume of the remaining sucrose cushion, aliquoted, stored at –80°C, and titrated. Virus titers of purified stocks between 1 × 109 and 3 × 1010 were used for animal studies to minimize the storage buffer added for in vivo infection.
Construction of HSV-1 with gB498–505 expressed from various promoters.
DNAs were all generated by amplified PCR using a proofreading polymerase (PrimeSTAR [TaKaRa]) with reaction mixtures containing 4% dimethyl sulfoxide (DMSO), as detailed previously (32). The constructs used to derive recombinant viruses were developed in a manner similar to that detailed previously (32). Gene cassettes were developed to recombine into the HSV gene designated UL44 that encodes the nonessential gC glycoprotein, which we have previously shown does not influence the pathogenesis of HSV-1 KOS in the mouse corneal scarification infection model (32). The plasmid vector p.gCp-EGFP previously detailed contains the gC promoter driving expression of EGFP inserted in frame with the ATG of UL44, followed by a translational stop, a poly(A) signal, and part of the gC open reading frame (ORF) in the vector pUC9 (57). To develop a gB498–505 epitope fusion, two complementary oligonucleotides, the sense oligonucleotide 5′-GATCCCACCATGGCGCATAAGACCACCTCCTCCATCGAGTTCGCCAGGCTGCAGTTTACGTACAACCACAA-3′ and the antisense oligonucleotide 5′-GATCTTGTGGTTGTACGTAAACTGCAGCCTGGCGAACTCGATGGAGGAGGTGGTCTTATGCGCCATGGTGG-3′ (the underlined sequences are overhangs in the annealed primers), were purchased from IDT, Inc.; these were hybridized together at 70°C for 5 min, slow cooled, phosphorylated using T4 polynucleotide kinase, and ligated together overnight with T4 DNA ligase at room temperature to form multimers. Reaction mixtures were heat inactivated and digested with BamHI and BglII to remove head-to-head and tail-to-tail concatemers. A gel-purified multimer of four copies was ligated into a unique BamHI site immediately upstream of EGFP in the plasmid p.gCp-EGFP. A clone (gCp-pep4-EGFP) containing four copies was selected and sequenced to confirm in-frame expression of the gB494–509 peptides with EGFP (Fig. 1) (-TMAHIKTTSSIEFARLQFTYNHKDP-)4-GFP (the boldface sequence represents the initiating methionine for translation; underlining denotes the immunodominant peptide). All promoters used in this work, with the exception of that driving LAT, were generated by PCR and contained a minimum of 500 bp of sequence immediately upstream of the respective ORF ATG, using primers shown in Table 1. The primers contained 5′ additional sequences to add a flanking unique HindIII site distally and a unique BamHI site at the proximal end of the promoter. These were cloned into the vector gCp-pep4-EGFP cut with BamHI and HindIII. Finally, DNA containing monomeric red fluorescent protein (mRFP) with its bovine growth hormone (BGH) polyadenylation A signal was generated from the plasmid pgC-RFP/pgB-EGFP detailed previously (Ramachandran et al. [33]) and cloned into unique NheI and HindIII sites, resulting in placing mRFP-poly(A) downstream of the gC promoter and upstream of the ectopic promoter-peptide EGFP gene. The HSV-1 LAT promoter was derived using a SacI-BamHI DNA fragment of the vector pBS-KS:LAP1 (a gift of David Bloom, University of Florida) representing nucleotides (nt) 117396 to 119289 of HSV strain 17+, which includes the full LAT promoter and LAT 5′ exon region. It was cloned into the gCp-pep4-EGFP cut with BamHI and SacI prior to cloning of the mRFP as just detailed above. In summary, each construct contained the promoter of gC driving the expression of the mRFP, a BGH poly(A) signal, and then the selected promoter sequence driving a 4-mer repeat of the gB494–509 peptide in frame with the EGFP. The flanking gC promoter and open reading frame provide sequences for homologous recombination into the gC locus.
Recombinant viruses were derived by cotransfection of linearized plasmids (using SspI or EcoRI) with HSV-1 KOS S1L infectious DNA prepared as detailed previously (23, 32). Virus plaques were selected based on the gain of red fluorescence and plaque purified to homogeneity. EGFP could not be used, as the peptides fused to EGFP reduced fluorescence to minimal levels. Each virus was grown and purified from Vero cells as described above (23). Final recombinant sequences were verified by sequencing insertional junctions, and recombinant promoters and EGFP location were verified by Southern blotting.
Relative expression analysis by quantitative reverse transcription-PCR.
Twenty-four-well plates of Vero cell monolayers were infected with recombinant HSV-1 at an MOI of 10 PFU/cell for 1 h at room temperature (25°C) in normal medium with or without 100 μg/ml cycloheximide or 400 μg/ml PAA (Lancaster Synthesis, Pelham, NH). Following medium replacement under the same conditions, the monolayers were incubated at 37°C. Cells were harvested at 4, 8, and 16 h postinfection, and whole-cell RNA was extracted using the RNeasy midi kit (Qiagen, Valencia, CA). RNA concentrations were measured using a NanoDrop spectrophotometer and concentrations normalized to make cDNA (high-capacity cDNA reverse transcription kit; Applied Biosystems). qRT-PCR was performed using SYBR green (SYBR Select; Applied Biosystems) for the detection of EGFP (EGFP-F, 5′-GAACCGCATCGAGCTGAA; EGFP-R, 5′-TGCTTGTCGGCCATGATATAG), on equivalent amounts of total cDNA from each condition. qRT-PCRs and analysis/melt curves were performed using an Applied Biosystems StepOnePlus system.
Quantitative real-time PCR for genome copy number.
The HSV-1 genome copy number in infected TG was determined by quantitative real-time PCR, as previously described, using primers that recognize the sequences of the gH gene (58).
T cell stimulation assay for quantification of peptide expression.
Confluent 12-wells of MHC-I homologous B6WT3 cells were infected with each virus at an MOI of 1 with HSV-1 WT, S1L, or each promoter virus. At 4, 8, or 24 h postinfection, a plate was removed from incubation, UV inactivated using a 254-nm hood UV light (at a dose time precalibrated to result in >1 × 106-fold reduction in infectivity under the same conditions), and combined into a coculture with 1 × 105 splenic CD8+ gB-T cells derived from a gB-T mouse that had been infected with HSV-1 for 8 days. gB-T mice are transgenic for the T cell receptor to gB498–505 and were a gift of Francis Carbone (University of Melbourne, Australia). Cells were incubated at 37°C for 6 to 8 h in the presence of brefeldin A to prevent secretion. T cells in cocultures were then antibody stained for CD45, CD8, and intracellular IFN-γ and TNF-α after permeabilization as detailed below. Experiments were repeated five times and collated.
Animal infection, tissue preparation, and ex vivo reactivation studies.
Six- to 8-week-old female C57BL/6 mice (purchased from Jackson Laboratory) or gB-T1 mice (41) were used in these studies. All animal experiments were conducted in accordance with protocols approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Corneas of anesthetized mice were scarified using a 30-gauge needle prior to HSV-1 corneal infections, as detailed previously (32). HSV-1 (1 × 105 PFU of purified HSV-1 diluted in 2 to 3 μl RPMI medium) was applied to each eye and the lids held closed for 5 min. Infections of the mouse flank were in a small (5 mm by 5 mm) region of skin previously denuded of hair and abraded using the same amount of virus. Infections were allowed to proceed until the select time without any treatment. Tissues extracted for flow cytometry analyses were obtained from anesthetized mice, injected intraperitoneally (i.p.) with 0.3 ml of 1,000 U/ml heparin (Sigma-Aldrich, St. Louis, MO), and then euthanized by exsanguination. Lymph nodes, TG, and/or spleen tissue were removed and subsequently digested in RPMI containing 10% fetal bovine serum and 400 U/ml collagenase type I (Sigma-Aldrich) for 45 to 60 min at 37°C. Tissues were then mechanically dissociated and triturated into single-cell suspensions, filtered through a 40-μm nylon cell strainer (BD Biosciences, Bedford, MA), and treated with red blood cell lysis buffer (BD Pharm Lyse) for 3 min prior to analyses and staining.
Antibody reagents and flow cytometry.
T cell phenotypic characterization by flow cytometry was performed essentially as detailed previously (18). Single-cell suspensions were stained with antibodies to CD45, CD3, and CD8α and with gB498–505 tetramers for 1 h at room temperature prior to fixing for 20 min with the Cytofix/Cytoperm kit (BD Biosciences, Bedford, MA). Washed cells were then analyzed by flow cytometry. Where indicated, cells were also stained with phycoerythrin (PE)-conjugated H-2Kb tetramer, complexed with the gB498–505 peptide (provided by the National Institute of Allergy and Infectious Diseases Tetramer Core Facility, Emory University Vaccine Center, Atlanta, GA). Efluor-450-conjugated anti-CD3 (clone 17A2) was purchased from eBioscience; the Pacific-Blue-conjugated anti-CD8α (clone 53-6.7), antigen-presenting cell (APC)-conjugated anti-IFN-γ (clone XMG1.2), peridinin chlorophyll protein (PerCP)-conjugated anti-CD45 (clone 30-F11), PE-Cy7-conjugated anti-TNFα (clone MP6-XT22), APC-conjugated anti-granzyme B (clone GB11), and BD Cytofix/Cytoperm fixation/permeabilization solution kit were all purchased from BD Pharmingen (San Diego, CA). The appropriate isotype control antibodies were purchased from the same company used for the reactive antibody and used where appropriate to establish gates and cutoffs. All flow cytometry samples were collected on BD FACSAria cytometer and analyzed by the FlowJo software.
BrdU staining.
Latently infected mice (28 to 34 dpi) were pulsed for 2 days prior to harvest with 1 mg/mouse BrdU in 0.1 ml PBS, delivered via intraperitoneal injection. The TGs were harvested at 48 h later, and cells were prepared for flow cytometric analysis using a fluorescein isothiocyanate (FITC) BrdU Flow kit (BD Pharmingen, San Diego, CA), as per the manufacturer’s instructions and as follows. TG cells were first washed twice with fluorescence-activated cell sorting (FACS) buffer (1% FBS, 0.1% sodium azide in PBS) prior to surface antibody staining in the presence of Fc block. Following surface staining, cells were permeabilized with Cytofix/Cytoperm solution for 20 min on ice and then Cytofix/Cytoperm Plus for 10 min on ice. After being washed, cells were treated with 30 μg of DNase for 1 h at 37°C and washed with 1× BD Perm/Wash solution, and cells were then incubated for 30 min in BrdU antibody diluted in 1× BD Perm/Wash. After labeling, suspensions were washed with 1× Perm/Wash and resuspended in FACS buffer for analysis by flow cytometry.
Statistical analysis.
All statistical analyses were performed using the GraphPad Prism software package. The specific statistical applications are shown in the legends to each figure.
Ethics statement.
All animal experiments were conducted in accordance with protocol number 15076444, approved by the University of Pittsburgh Institutional Animal Care and Use Committee. This protocol meets the standards for humane animal care and use as set by the Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants R01 EY015291 (to P.R.K.), R01 EY05945 (to R.L.H.), and R00 EY025761 (to A.J.S.), predoctoral fellowships under T32 EY017272 (to B.R.T. and S.M.B.) and T32 AI049820 (to B.R.T.), core grant P30 EY08098, and unrestricted awards from the Research to Prevent Blindness, Inc., and the Eye & Ear Foundation of Pittsburgh.
The funders had no role in study design, data collection, analysis, or the decision to prepare and publish the manuscript.
REFERENCES
- 1.Lee S, Ives AM, Bertke AS. 2015. Herpes simplex virus 1 reactivates from autonomic ciliary ganglia independently from sensory trigeminal ganglia to cause recurrent ocular disease. J Virol 89:8383–8391. doi: 10.1128/JVI.00468-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preston CM, Efstathiou S. 2007. HSV-1 and 2: molecular basis of HSV latency and reactivation, p 602–615. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 3.Bloom DC. 2004. HSV LAT and neuronal survival. Int Rev Immunol 23:187–198. doi: 10.1080/08830180490265592. [DOI] [PubMed] [Google Scholar]
- 4.Thompson RL, Sawtell NM. 2001. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J Virol 75:6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 6.Nicoll MP, Hann W, Shivkumar M, Harman LE, Connor V, Coleman HM, Proenca JT, Efstathiou S. 2016. The HSV-1 latency-associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PLoS Pathog 12:e1005539. doi: 10.1371/journal.ppat.1005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du T, Zhou G, Roizman B. 2013. Modulation of reactivation of latent herpes simplex virus 1 in ganglionic organ cultures by p300/CBP and STAT3. Proc Natl Acad Sci U S A 110:E2621–E2628. doi: 10.1073/pnas.1309906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du T, Zhou G, Roizman B. 2011. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc Natl Acad Sci U S A 108:18820–18824. doi: 10.1073/pnas.1117203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roizman B, Zhou G, Du T. 2011. Checkpoints in productive and latent infections with herpes simplex virus 1: conceptualization of the issues. J Neurovirol 17:512–517. doi: 10.1007/s13365-011-0058-x. [DOI] [PubMed] [Google Scholar]
- 10.Wilson AC, Mohr I. 2012. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol 20:604–611. doi: 10.1016/j.tim.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog 8:e1002540. doi: 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell TA, Tscharke DC. 2016. Lytic promoters express protein during herpes simplex virus latency. PLoS Pathog 12:e1005729. doi: 10.1371/journal.ppat.1005729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC. 2014. Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell-intrinsic transcriptional response. PLoS Pathog 10:e1004237. doi: 10.1371/journal.ppat.1004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. 2003. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18:593–603. doi: 10.1016/s1074-7613(03)00112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Lint AL, Kleinert L, Clarke SR, Stock A, Heath WR, Carbone FR. 2005. Latent infection with herpes simplex virus is associated with ongoing CD8+ T-cell stimulation by parenchymal cells within sensory ganglia. J Virol 79:14843–14851. doi: 10.1128/JVI.79.23.14843-14851.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Velzen M, Jing L, Osterhaus AD, Sette A, Koelle DM, Verjans GM. 2013. Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently infected human trigeminal ganglia. PLoS Pathog 9:e1003547. doi: 10.1371/journal.ppat.1003547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verjans GM, Hintzen RQ, van Dun JM, Poot A, Milikan JC, Laman JD, Langerak AW, Kinchington PR, Osterhaus AD. 2007. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci U S A 104:3496–3501. doi: 10.1073/pnas.0610847104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.St. Leger AJ, Peters B, Sidney J, Sette A, Hendricks RL. 2011. Defining the herpes simplex virus-specific CD8+ T cell repertoire in C57BL/6 mice. J Immunol 186:3927–3933. doi: 10.4049/jimmunol.1003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller SN, Jones CM, Chen W, Kawaoka Y, Castrucci MR, Heath WR, Carbone FR. 2003. The early expression of glycoprotein B from herpes simplex virus can be detected by antigen-specific CD8+ T cells. J Virol 77:2445–2451. doi: 10.1128/jvi.77.4.2445-2451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheridan BS, Khanna KM, Frank GM, Hendricks RL. 2006. Latent virus influences the generation and maintenance of CD8+ T cell memory. J Immunol 177:8356–8364. doi: 10.4049/jimmunol.177.12.8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeon S, St. Leger AJ, Cherpes TL, Sheridan BS, Hendricks RL. 2013. PD-L1/B7-H1 regulates the survival but not the function of CD8+ T cells in herpes simplex virus type 1 latently infected trigeminal ganglia. J Immunol 190:6277–6286. doi: 10.4049/jimmunol.1300582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.St. Leger AJ, Jeon S, Hendricks RL. 2013. Broadening the repertoire of functional herpes simplex virus type 1-specific CD8+ T cells reduces viral reactivation from latency in sensory ganglia. J Immunol 191:2258–2265. doi: 10.4049/jimmunol.1300585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Treat BR, Bidula SM, Ramachandran S, St. Leger AJ, Hendricks RL, Kinchington PR. 2017. Influence of an immunodominant herpes simplex virus type 1 CD8+ T cell epitope on the target hierarchy and function of subdominant CD8+ T cells. PLoS Pathog 13:e1006732. doi: 10.1371/journal.ppat.1006732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. 2008. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322:268–271. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. 2000. CD8+ T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med 191:1459–1466. doi: 10.1084/jem.191.9.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linderman JA, Kobayashi M, Rayannavar V, Fak JJ, Darnell RB, Chao MV, Wilson AC, Mohr I. 2017. Immune escape via a transient gene expression program enables productive replication of a latent pathogen. Cell Rep 18:1312–1323. doi: 10.1016/j.celrep.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farooq AV, Shukla D. 2012. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol 57:448–462. doi: 10.1016/j.survophthal.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kotturi MF, Scott I, Wolfe T, Peters B, Sidney J, Cheroutre H, von Herrath MG, Buchmeier MJ, Grey H, Sette A. 2008. Naive precursor frequencies and MHC binding rather than the degree of epitope diversity shape CD8+ T cell immunodominance. J Immunol 181:2124–2133. doi: 10.4049/jimmunol.181.3.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolpert EZ, Grufman P, Sandberg JK, Tegnesjo A, Karre K. 1998. Immunodominance in the CTL response against minor histocompatibility antigens: interference between responding T cells, rather than with presentation of epitopes. J Immunol 161:4499–4505. [PubMed] [Google Scholar]
- 30.Kedl RM, Rees WA, Hildeman DA, Schaefer B, Mitchell T, Kappler J, Marrack P. 2000. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med 192:1105–1113. doi: 10.1084/jem.192.8.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrington LA, Smith TA, Grey F, Hill AB, Snyder CM. 2013. Competition for antigen at the level of the APC is a major determinant of immunodominance during memory inflation in murine cytomegalovirus infection. J Immunol 190:3410–3416. doi: 10.4049/jimmunol.1203151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramachandran S, Davoli KA, Yee MB, Hendricks RL, Kinchington PR. 2010. Delaying the expression of herpes simplex virus type 1 glycoprotein B (gB) to a true late gene alters neurovirulence and inhibits the gB-CD8+ T-cell response in the trigeminal ganglion. J Virol 84:8811–8820. doi: 10.1128/JVI.00496-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramachandran S, Knickelbein JE, Ferko C, Hendricks RL, Kinchington PR. 2008. Development and pathogenic evaluation of recombinant herpes simplex virus type 1 expressing two fluorescent reporter genes from different lytic promoters. Virology 378:254–264. doi: 10.1016/j.virol.2008.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sawtell NM, Thompson RL. 2016. De novo herpes simplex virus VP16 expression gates a dynamic programmatic transition and sets the latent/lytic balance during acute infection in trigeminal ganglia. PLoS Pathog 12:e1005877. doi: 10.1371/journal.ppat.1005877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aguilar JS, Devi-Rao GV, Rice MK, Sunabe J, Ghazal P, Wagner EK. 2006. Quantitative comparison of the HSV-1 and HSV-2 transcriptomes using DNA microarray analysis. Virology 348:233–241. doi: 10.1016/j.virol.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 36.Chen X, Schmidt MC, Goins WF, Glorioso JC. 1995. Two herpes simplex virus type 1 latency-active promoters differ in their contributions to latency-associated transcript expression during lytic and latent infections. J Virol 69:7899–7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Batchelor AH, O'Hare P. 1990. Regulation and cell-type-specific activity of a promoter located upstream of the latency-associated transcript of herpes simplex virus type 1. J Virol 64:3269–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobson AT, Sederati F, Devi-Rao G, Flanagan WM, Farrell MJ, Stevens JG, Wagner EK, Feldman LT. 1989. Identification of the latency-associated transcript promoter by expression of rabbit beta-globin mRNA in mouse sensory nerve ganglia latently infected with a recombinant herpes simplex virus. J Virol 63:3844–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu J, Kang W, Marquart ME, Hill JM, Zheng X, Block TM, Fraser NW. 1999. Identification of a novel 0.7-kb polyadenylated transcript in the LAT promoter region of HSV-1 that is strain specific and may contribute to virulence. Virology 265:296–307. doi: 10.1006/viro.1999.0057. [DOI] [PubMed] [Google Scholar]
- 40.Spivack JG, Fraser NW. 1988. Expression of herpes simplex virus type 1 latency-associated transcripts in the trigeminal ganglia of mice during acute infection and reactivation of latent infection. J Virol 62:1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mueller SN, Heath W, McLain JD, Carbone FR, Jones CM. 2002. Characterization of two TCR transgenic mouse lines specific for herpes simplex virus. Immunol Cell Biol 80:156–163. doi: 10.1046/j.1440-1711.2002.01071.x. [DOI] [PubMed] [Google Scholar]
- 42.Kahan SM, Wherry EJ, Zajac AJ. 2015. T cell exhaustion during persistent viral infections. Virology 479–480:180–193. doi: 10.1016/j.virol.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kastenmuller W, Gasteiger G, Gronau JH, Baier R, Ljapoci R, Busch DH, Drexler I. 2007. Cross-competition of CD8+ T cells shapes the immunodominance hierarchy during boost vaccination. J Exp Med 204:2187–2198. doi: 10.1084/jem.20070489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tewalt EF, Grant JM, Granger EL, Palmer DC, Heuss ND, Gregerson DS, Restifo NP, Norbury CC. 2009. Viral sequestration of antigen subverts cross presentation to CD8+ T cells. PLoS Pathog 5:e1000457. doi: 10.1371/journal.ppat.1000457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheridan BS, Cherpes TL, Urban J, Kalinski P, Hendricks RL. 2009. Reevaluating the CD8 T-cell response to herpes simplex virus type 1: involvement of CD8 T cells reactive to subdominant epitopes. J Virol 83:2237–2245. doi: 10.1128/JVI.01699-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yewdell JW. 2006. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity 25:533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 47.Sears AE, McGwire BS, Roizman B. 1991. Infection of polarized MDCK cells with herpes simplex virus 1: two asymmetrically distributed cell receptors interact with different viral proteins. Proc Natl Acad Sci U S A 88:5087–5091. doi: 10.1073/pnas.88.12.5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halford WP, Schaffer PA. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J Virol 75:3240–3249. doi: 10.1128/JVI.75.7.3240-3249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, Garbi N, Kaisho T, Germain RN, Kastenmuller W. 2015. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell 162:1322–1337. doi: 10.1016/j.cell.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, Kurts C, Garbi N, Barchet W, Iannacone M, Klauschen F, Kolanus W, Kaisho T, Colonna M, Germain RN, Kastenmuller W. 2017. CD8+ T cells orchestrate pDC-XCR1+ dendritic cell spatial and functional cooperativity to optimize priming. Immunity 46:205–219. doi: 10.1016/j.immuni.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. 2002. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc Natl Acad Sci U S A 99:978–983. doi: 10.1073/pnas.022301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kramer MF, Chen SH, Knipe DM, Coen DM. 1998. Accumulation of viral transcripts and DNA during establishment of latency by herpes simplex virus. J Virol 72:1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Probst HC, Tschannen K, Gallimore A, Martinic M, Basler M, Dumrese T, Jones E, van den Broek MF. 2003. Immunodominance of an antiviral cytotoxic T cell response is shaped by the kinetics of viral protein expression. J Immunol 171:5415–5422. doi: 10.4049/jimmunol.171.10.5415. [DOI] [PubMed] [Google Scholar]
- 54.Carroll KL, Avery L, Treat BR, Kane LP, Kinchington PR, Hendricks RL, St. Leger AJ. 2019. Differential expression of immune checkpoint molecules on CD8+ T cells specific for immunodominant and subdominant herpes simplex virus 1 epitopes. J Virol 94:e01132-19. doi: 10.1128/JVI.01132-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pretell J, Greenfield RS, Tevethia SS. 1979. Biology of simian virus 40 (SV40) transplantation antigen (TrAg). V In vitro demonstration of SV40 TrAg in SV40 infected nonpermissive mouse cells by the lymphocyte mediated cytotoxicity assay. Virology 97:32–41. doi: 10.1016/0042-6822(79)90370-2. [DOI] [PubMed] [Google Scholar]
- 56.Bowen CD, Renner DW, Shreve JT, Tafuri Y, Payne KM, Dix RD, Kinchington PR, Gatherer D, Szpara ML. 2016. Viral forensic genomics reveals the relatedness of classic herpes simplex virus strains KOS, KOS63, and KOS79. Virology 492:179–186. doi: 10.1016/j.virol.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Decman V, Kinchington PR, Harvey SA, Hendricks RL. 2005. Gamma interferon can block herpes simplex virus type 1 reactivation from latency, even in the presence of late gene expression. J Virol 79:10339–10347. doi: 10.1128/JVI.79.16.10339-10347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freeman ML, Sheridan BS, Bonneau RH, Hendricks RL. 2007. Psychological stress compromises CD8+ T cell control of latent herpes simplex virus type 1 infections. J Immunol 179:322–328. doi: 10.4049/jimmunol.179.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]