

Kaposi’s sarcoma-associated herpesvirus (KSHV) is the etiological agent of three human malignancies: multicentric Castleman’s disease, primary effusion lymphoma, and Kaposi’s sarcoma. Kaposi’s sarcoma is a highly angiogenic tumor that arises from endothelial cells. It has been previously reported that KSHV infection of endothelial cells leads to an increase of integrin αVβ3, a molecule observed to be involved in the angiogenic process of several malignancies. Our data demonstrate that the KSHV protein viral interleukin-6 (vIL-6) can induce integrin β3 in an intracellular and paracrine manner. Furthermore, we showed that this induction is necessary for vIL-6-mediated cell adhesion and angiogenesis, suggesting a potential role of integrin β3 in KSHV pathogenesis and development of Kaposi’s sarcoma.
KEYWORDS: Kaposi’s sarcoma-associated herpesvirus, integrin β3, viral IL-6
ABSTRACT
Kaposi’s sarcoma-associated herpesvirus (KSHV) is the causative agent of two B-cell lymphoproliferative diseases and Kaposi’s sarcoma, an endothelial-cell-driven cancer. KSHV viral interleukin-6 (vIL-6) is a viral homolog of human IL-6 (hIL-6) that is expressed in KSHV-associated malignancies. Previous studies have shown that the expression of the integrin β3 (ITGB3) subunit is induced upon KSHV infection. Here we report that KSHV vIL-6 is able to induce the expression of ITGB3 and increase surface expression of the αVβ3 integrin heterodimer. We demonstrated using small interfering RNA (siRNA) depletion and inhibitor studies that KSHV vIL-6 can increase ITGB3 by inducing STAT3 signaling. Furthermore, we found that secreted vIL-6 is capable of inducing ITGB3 in endothelial cells in a paracrine manner. Importantly, the ability to induce ITGB3 in endothelial cells seems to be specific to vIL-6, as overexpression of hIL-6 alone did not affect levels of this integrin. Our lab and others have previously shown that vIL-6 can induce angiogenesis, and we investigated whether ITGB3 was involved in this process. We found that siRNA depletion of ITGB3 in vIL-6-expressing endothelial cells resulted in a decrease in adhesion to extracellular matrix proteins. Moreover, depletion of ITGB3 hindered the ability of vIL-6 to promote angiogenesis. In conclusion, we found that vIL-6 can singularly induce ITGB3 and that this induction is dependent on vIL-6 activation of the STAT3 signaling pathway.
IMPORTANCE Kaposi’s sarcoma-associated herpesvirus (KSHV) is the etiological agent of three human malignancies: multicentric Castleman’s disease, primary effusion lymphoma, and Kaposi’s sarcoma. Kaposi’s sarcoma is a highly angiogenic tumor that arises from endothelial cells. It has been previously reported that KSHV infection of endothelial cells leads to an increase of integrin αVβ3, a molecule observed to be involved in the angiogenic process of several malignancies. Our data demonstrate that the KSHV protein viral interleukin-6 (vIL-6) can induce integrin β3 in an intracellular and paracrine manner. Furthermore, we showed that this induction is necessary for vIL-6-mediated cell adhesion and angiogenesis, suggesting a potential role of integrin β3 in KSHV pathogenesis and development of Kaposi’s sarcoma.
INTRODUCTION
Integrins are heterodimeric membrane glycoproteins that consist of an alpha and a beta subunit. These cell surface proteins are receptors for extracellular matrix proteins, growth factors, cytokines, immunoglobulins, and matrix-degrading proteases (1). One of the beta subunits, β3, can dimerize with αIIb in platelets and αV in other cell types, including endothelial cells. Integrin αVβ3 binds to adhesive proteins such as von Willebrand factor, fibrinogen, and fibronectin. The binding of these ligands can induce “outside-inside” signaling through the integrin, resulting in endothelial cell migration (2), angiogenesis (2, 3), and transforming growth factor β1 (TGF-β1) signaling (4, 5). Integrin αVβ3 expression and function are of significant interest, as expression of integrin αVβ3 is upregulated in several forms of cancer and correlates with progression of malignancies (1, 6).
A receptor for cellular proteins, integrin αVβ3 has also been reported to be a receptor for viruses, including Kaposi’s sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV-8) (7–9). KSHV, a double-stranded DNA virus from the gammaherpesvirus family (10), was first isolated from a Kaposi’s sarcoma (KS) patient and found to be the causative agent of this cancer (11). Subsequently, it was demonstrated that KSHV also causes the B-cell malignancy primary effusion lymphoma (PEL) (12). KSHV is also associated with the proliferative disorder multicentric Castleman’s disease (MCD) (13). DiMaio and colleagues reported that latent infection of KSHV increases integrin β3 (ITGB3) expression in endothelial cells (14). This increase causes the infected cells to bind more strongly to particular extracellular matrix (ECM) components. Additionally, knockdown of ITGB3 results in a decreased ability of infected cells to form tubules in an in vitro model of angiogenesis. These data suggest that KSHV upregulates ITGB3, which can play a role in the angiogenic processes of KSHV-infected endothelial cells (14). However, the mechanism by which the virus mediates the induction of ITGB3 has not been characterized.
KSHV encodes over 80 open reading frames (ORFs), which consist of latent and lytic genes. Most infected cells contain a latent virus, expressing only a small subset of genes that encode proteins and noncoding RNAs. Upon reactivation, the virus starts expressing lytic genes in an ordered fashion and produces new viral progeny (15). ORF K2 encodes viral interleukin-6 (vIL-6), a homolog of human IL-6 (hIL-6) (16–18). During latency, vIL-6 expression is detected at low levels but significantly increases during lytic replication (19). Similar to hIL-6, secreted vIL-6 binds to the IL-6 receptor (IL-6R, composed of gp80 and gp130 subunits) and induces the JAK/STAT signaling cascade (18, 20). However, while the cell readily secretes hIL-6, a majority of vIL-6 is localized to the endoplasmic reticulum (ER). Here, vIL-6 interacts with gp130, a component of the IL-6R, and induces JAK/STAT signaling in an intracellular manner (21). Through activation of transcription factor STAT3, vIL-6 induces cell proliferation (18, 22, 23) as well as migration (23–25). vIL-6 also induces angiogenesis and hematopoiesis, aiding in the growth of tumors (26, 27).
We report that vIL-6 expressed from different cell types is capable of inducing ITGB3 at the mRNA and protein levels. Phosphorylation of STAT3 is required for this induction, as indicated by assays using drug inhibitors and small interfering RNA (siRNA) knockdowns. Interestingly, in endothelial cells, vIL-6 is a stronger inducer of ITGB3 compared to the human homolog, even though both are capable of inducing STAT3 signaling. Finally, we show how this phenotype may promote KSHV pathogenesis by contributing to angiogenesis. These studies highlight vIL-6 as a player in KSHV-induced ITGB3.
RESULTS
Viral IL-6-expressing cells have increased levels of ITGB3.
Our lab has previously reported microarray data that indicated that human umbilical vein endothelial cells (HUVEC) stably expressing vIL-6 (vIL-6-HUVEC) had increased levels of ITGB3 mRNA compared to those of cells expressing the empty vector (EV-HUVEC) (25). High levels of ITGB3 expression in vIL-6-expressing HUVEC were confirmed with reverse transcription-quantitative PCR (RT-qPCR) (Fig. 1A). We next performed immunoblotting to probe for ITGB3 and found that the protein level was also increased in the vIL-6-HUVEC (Fig. 1B). Additionally, we wanted to know whether the higher levels of ITGB3 mRNA and protein were due to increased ITGB3 transcription. HEK293T cells were cotransfected with a vIL-6-expressing plasmid or the corresponding EV control and a luciferase reporter plasmid. Expression of vIL-6, as detected by immunoblotting, led to a significant increase in the expression of luciferase (Fig. 1C). The results suggest that vIL-6 promotes the activation of the ITGB3 promoter and consequently increases the ITGB3 mRNA and protein levels.
FIG 1.

HUVEC stably expressing vIL-6 have increased ITGB3 mRNA and protein levels. (A) Relative ITGB3 mRNA expression in stable HUVEC normalized to the expression levels in EV-HUVEC. (B) Integrin β3 protein expression in the total cell lysate of stable HUVEC. (C) (Top) Relative luciferase expression from a luciferase reporter under the control of an ITGB3-promoter transfected into HEK293T cells. (Bottom) Immunoblots for vIL-6 and actin from transfected HEK293T cells. (D) Integrin αV protein expression in the total cell lysate of stable HUVEC. (E) Surface expression of αVβ3 integrin in stable HUVEC was measured using flow cytometry. The gray histogram represents EV HUVEC, and the white histogram represents vIL-6 HUVEC. **, P < 0.01; ***, P < 0.001.
It has been suggested that the surface expression of the heterodimer αVβ3 is determined by the expression of ITGB3, as integrin αV (ITGAV) is usually excessively expressed in cells (28). To assess the levels of ITGAV, we performed immunoblotting with cell lysates from EV- and vIL-6-HUVEC. As expected, vIL-6-HUVEC showed a slight increase in the protein levels of ITGAV (Fig. 1D). Furthermore, to determine if the increase in total ITGB3 resulted in increased surface expression, we performed flow cytometry analysis using an αVβ3 antibody. Histograms comparing the geometric means of fluorescence intensity indicate that vIL-6-HUVEC had a 2-fold increase in levels of surface αVβ3 integrin (Fig. 1E). Together, these results suggest that expression of vIL-6 in HUVEC promotes an increase in the expression of ITGB3, leading to higher surface expression of integrin αVβ3.
Viral IL-6-expressing cells can induce ITGB3 expression in a paracrine manner.
We next wanted to determine whether vIL-6-expressing endothelial cells induce ITGB3 through paracrine signaling. To explore this possibility, we collected conditioned medium (CM) from both the EV- and vIL-6-HUVEC and added them to naive HUVEC at a 1:1 ratio with fully supplemented medium. After 24 h, we were able to detect an increase in ITGB3 mRNA (Fig. 2A) and protein (Fig. 2B) from the HUVEC that were treated with the vIL-6-containing conditioned medium.
FIG 2.
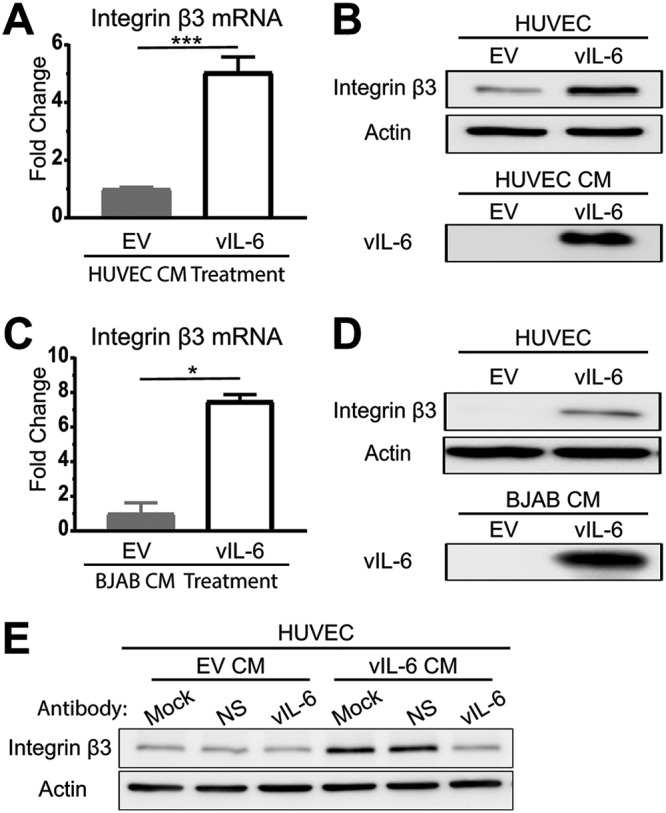
vIL-6 induces ITGB3 expression in a paracrine manner. (A and B) HUVEC were treated with conditioned medium from EV- or vIL-6-expressing HUVEC for 24 h, followed by the comparison of ITGB3 mRNA levels (A) and protein levels (B). (C and D) Similar experiments were conducted using conditioned medium from EV- and vIL-6-expressing BJABs. (E) Conditioned media were collected from EV- and vIL-6-HUVEC in the presence of nonspecific mouse IgG or mouse anti-vIL-6 IgG. This conditioned medium was then placed on HUVEC. After 24 h, lysates were collected, and immunoblotting was performed for actin and ITGB3. CM, conditioned medium; NS, nonspecific. *, P < 0.05; ***, P < 0.001.
In addition to endothelial cells, B cells are another cell type that is readily infected in vivo (29–32). In KS lesions, the cells that express the highest quantities of vIL-6 are from invading lymphocytes (33). For these reasons, we constitutively expressed EV or vIL-6 in BJAB cells, a B-cell line. Conditioned medium from these vIL-6-expressing BJAB cells induced ITGB3 mRNA and protein expression in HUVEC similarly to what we observed from the HUVEC-conditioned medium (Fig. 2C and D).
To determine whether secreted vIL-6 was necessary for the induction of ITGB3 or if it was another secreted factor from stable vIL-6 cells, we performed a neutralization assay (Fig. 2E). Conditioned media were created containing no antibody, mouse nonspecific IgG, or mouse anti-vIL-6 IgG. These conditioned media were then placed on naive HUVEC, further supplemented with antibody, and incubated for 24 h. As expected, cells treated with the EV-conditioned medium, regardless of the antibody supplement, did not induce ITGB3. On the other hand, cells that were treated with the mock or nonspecific-antibody-containing vIL-6-HUVEC-conditioned medium had increased levels of ITGB3. However, cells that were treated with the vIL-6-HUVEC-conditioned medium that contained the vIL-6-specific antibody had ITGB3 levels similar to those of the EV-conditioned-medium-treated cells. These results demonstrate that secreted vIL-6 is involved in the induction of ITGB3.
Lytic replication promotes the expression of ITGB3 in a paracrine manner.
Since vIL-6 expression in latently infected cells is relatively low (19), we wanted to determine whether induction of the lytic life cycle would promote the expression of ITGB3. To test this, we reactivated a KSHV-infected cell line, iSLK.219, for 24 h and harvested protein lysates. Although the induction of lytic replication significantly increased the expression of vIL-6, the levels of ITGB3 remained relatively similar to those in unreactivated cells (Fig. 3A). These results suggest that the contribution of vIL-6 to ITGB3 expression in infected cells may take place mostly during latency.
FIG 3.

vIL-6 from lytically replicating cells promotes the expression of ITGB3 in a paracrine manner. (A) iSLK.219 cells were reactivated with doxycycline for 24 h, and the levels of ITGB3, vIL-6, and actin were detected by immunoblotting. (B) Lysates from reactivated (24 h post-doxycycline treatment) iSLK.219 and vIL-6-expressing HUVEC were resolved by SDS-PAGE, and vIL-6 expression was detected by immunoblotting. (C) HUVEC were treated for 24 h with conditioned medium from latent or reactivated iSLK.219 (24 h post-doxycycline treatment). Immunoblotting was performed with HUVEC lysates and iSLK.219 conditioned medium. (D) Conditioned media were collected from TREx-Rta-BCBL-1 after 24 h of DMSO (latent) or doxycycline (reactivated [react.]) treatment. Conditioned medium was supplemented with nonspecific mouse IgG or mouse anti-vIL-6 IgG and placed on HUVEC. After 24 h, lysates were collected, and immunoblotting was performed. BCBL-1 cell conditioned medium was processed in the same manner.
Given that vIL-6 expression in iSLK.219 cells was significantly increased during the induction of the lytic life cycle (Fig. 3A), we asked whether these levels of vIL-6 were comparable to those in the vIL-6-expressing HUVEC. We found that expression of vIL-6 in the reactivated iSLK.219 cells was slightly higher than in the stable vIL-6-HUVEC (Fig. 3B). The minimal change in the expression of ITGB3 following lytic reactivation, despite vIL-6 expression, might be due to the difference in cell type, as iSLK.219 cells are considered epithelial cells.
The results from Fig. 2 showed that secreted vIL-6 induces the expression of ITGB3 in a paracrine manner. Since reactivated iSLK.219 cells express high levels of vIL-6, we investigated if secreted factors such as vIL-6 can induce the expression of ITGB3 in naive HUVEC. To address this question, we reactivated iSLK.219 and collected the conditioned medium 24 h post-doxycycline treatment. The conditioned medium from the unreactivated and reactivated iSLK.219 cells was mixed with HUVEC medium and added to naive HUVEC for 24 h. Importantly, vIL-6 was detected in the cell-free conditioned medium collected from reactivated cells but not from the dimethyl sulfoxide (DMSO)-treated cells. Similar to the results obtained with the conditioned medium of vIL-6-expressing-HUVEC (Fig. 2A and B), cells cultured in the conditioned medium from reactivated cells showed a higher expression of ITGB3 than cells cultured in the conditioned medium from unreactivated cells (Fig. 3C).
We also investigated whether vIL-6 secreted from primary effusion lymphoma cells was able to induce ITGB3 in a paracrine manner. We reactivated TREx-Rta-BCBL-1 (BCBL-1) cells for 24 h and collected conditioned medium. Filtered conditioned medium from reactivated cells contained vIL-6 and was able to promote the expression of ITGB3 in HUVEC (Fig. 3D). Importantly, vIL-6 antibodies blocked the induction of ITGB3 driven by the conditioned medium from reactivated BCBL-1 cells. Together, these results suggest that although lytically replicating cells do not induce the expression of ITGB3, secreted vIL-6 from iSLK.219 and BCBL-1 cells, can induce ITGB3 in neighboring uninfected endothelial cells.
STAT3 signaling is necessary for vIL-6-mediated ITGB3 induction.
A previous study examining the effect of high αVβ3 integrin levels in breast cancer cells found that STAT3 contributes to the invasiveness of these cells (34). This finding indicated to us a possible link between STAT3 and αVβ3 activity. Since it is well established that vIL-6 interacts with gp130 to induce JAK2/STAT3 signaling (20, 35), we wanted to examine the relationship between this signaling pathway and ITGB3 in vIL-6-expressing cells. To determine whether vIL-6 interacts with gp130 in HUVEC, we performed coimmunoprecipitations with FLAG-antibody-conjugated beads to immunoprecipitate FLAG-tagged vIL-6 (24). The proteins bound to vIL-6 were eluted with a FLAG peptide and resolved on an SDS-PAGE gel. The results showed that gp130 coimmunoprecipitated with vIL-6 in HUVEC (Fig. 4A). Furthermore, to assess the importance of the pathway in inducing ITGB3, we knocked down gp130 in EV- and vIL-6-HUVEC and performed immunoblotting. The depletion of gp130 reduced vIL-6-mediated induction of phosphorylated STAT3 and ITGB3, suggesting that activation of STAT3 may be essential in this process (Fig. 4B).
FIG 4.

JAK/STAT3 signaling is necessary for vIL-6-induced ITGB3. (A) Whole-cell lysates from EV- and vIL-6-HUVEC were immunoprecipitated with a FLAG (vIL-6) antibody. The eluates and inputs were resolved by SDS-PAGE, and immunoblotting was performed for the indicated proteins. (B) EV- and vIL-6-HUVEC were transfected with siRNAs for 72 h, and lysates were probed for the indicated proteins. (C) EV- and vIL-6-HUVEC were treated with the STAT3 inhibitor cryptotanshinone (0 or 20 μM) for 48 h. Lysates were then collected, and immunoblotting was performed for the indicated proteins. (D) EV- and vIL-6-HUVEC were transfected with siRNAs for 48 h, and lysates were probed for the same proteins as for panel C. (E) HUVEC were transfected with siRNAs against a nontargeting control or STAT3. Twenty-four hours posttransfection, cells were treated with conditioned medium from EV- or vIL-6-HUVEC and incubated for an additional 24 h before lysates were collected and used for immunoblotting. NTC, nontargeting control; H, HUVEC.
To further test whether STAT3 is necessary for this process, we treated EV- and vIL-6-HUVEC with the STAT3 inhibitor cryptotanshinone. After 48 h, we observed the expected decrease in STAT3 phosphorylation as well as a decrease in total ITGB3 expression (Fig. 4C). This result indicates that vIL-6-induced STAT3 signaling is essential for ITGB3 induction. To confirm these data, we transfected cells with a STAT3-specific siRNA and observed the same loss of ITGB3 levels in the vIL-6-HUVEC (Fig. 4D). We then performed a similar experiment in which naive HUVEC were first transfected with STAT3 siRNA, followed by treatment with EV- or vIL-6-conditioned medium. We observed that knockdown of STAT3 attenuated ITGB3 induction by the vIL-6-containing conditioned medium (Fig. 4E). Together, these results suggest that activation of STAT3 is necessary for the ability of vIL-6 to induce ITGB3 in endothelial cells.
Human IL-6 is not a strong inducer of ITGB3 as is vIL-6 in HUVEC.
Since vIL-6 and hIL-6 can both activate STAT3, we hypothesized that hIL-6 could induce ITGB3 as well. To test this hypothesis, we collected lysates from HUVEC expressing EV, vIL-6, or hIL-6 and performed immunoblotting (Fig. 5A). Surprisingly, hIL-6 did not affect levels of ITGB3. To confirm these results, stable HUVEC were supplemented for 48 h with recombinant hIL-6 (rhIL-6). The results indicate that even in the presence of rhIL-6, levels of ITGB3 only increase in vIL-6-expressing cells. We next prepared conditioned media from HUVEC and BJAB cells that contained EV, vIL-6, or hIL-6. These conditioned media were then used to treat naive HUVEC for 24 h, after which the lysates were collected (Fig. 5B). Again, we observed a substantial increase in ITGB3 protein levels in cells treated with the vIL-6-conditioned medium. Cells that were treated with the EV- or hIL-6-conditioned medium had no increase or a very modest increase in ITGB3 protein compared to EV control cells. These results suggest that hIL-6 is not a potent inducer of ITGB3 compared to vIL-6 in HUVEC.
FIG 5.

Human IL-6 is not a strong inducer of ITGB3 as is vIL-6 in HUVEC. (A) Immunoblots of total cell lysates from HUVEC expressing EV, vIL-6, or hIL-6 treated for 48 h in the presence of recombinant hIL-6 at the indicated concentrations. (B) Immunoblots of HUVEC treated with conditioned medium from EV-, vIL-6-, and hIL-6-expressing HUVEC and BJABs. (C) Similar to panel A, but EV- and vIL-6-HUVEC were grown for 24 h in the presence of recombinant hIL-6 (rhIL-6; 250 ng/ml), soluble IL-6Rα (sIL-6R; 250 ng/ml), or both.
The induction of signaling pathways by hIL-6 can occur through two different but similar mechanisms known as classical signaling and trans-signaling (36, 37). In classical signaling, hIL-6 binds to membrane-bound IL-6Rα (gp80), which interacts with gp130, leading to the intracellular activation of the JAK/STAT signaling pathway. This process is restricted to cells that express the membrane-bound receptor, which includes hepatocytes, some leukocytes, and some epithelial cells (36). In contrast, the trans-signaling pathway occurs when secreted hIL-6 binds to the soluble form of the IL-6Rα. This hIL-6/soluble IL-6R (sIL-6R) complex binds gp130 and induces activation of the JAK/STAT signaling pathway. Importantly, since gp130 is ubiquitously expressed, this process can take place even in cells that lack expression of the membrane-bound IL-6Rα.
Recently, it was shown that HUVEC could support both mechanisms, but activation of the trans-signaling pathway by treating cells with both recombinant hIL-6 and sIL-6R led to higher and longer activation of STAT3 (38). For this reason, we hypothesized that supplementing cells for 24 h with both hIL-6 and sIL-6Rα would result in high levels of activated STAT3 and, thus ITGB3. Immunoblots show that EV-HUVEC treated with both recombinant proteins displayed an increase in phosphorylated STAT3, confirming activation of the pathway (Fig. 5C). Importantly, this activation also increased ITGB3 to levels similar to those seen in the vIL-6-expressing cells (Fig. 5C). However, the addition of the recombinant proteins did not enhance vIL-6-mediated induction of ITGB3. Altogether, the data suggest that in endothelial cells, activation of IL-6R plays a crucial role in the induction of ITGB3, and in comparison to its cellular homolog, vIL-6 can accomplish this even in the absence of soluble IL-6Rα.
Integrin β3 expression increases vIL-6-HUVEC adhesion.
DiMaio et al. demonstrated that KSHV-infected endothelial cells adhere more readily to the ECM proteins fibronectin and vitronectin (14). This increase in adhesion could be inhibited by the addition of RGD peptides, which interfere with several integrin-ECM interactions. We wanted to examine if the increased amount of ITGB3 in vIL-6-HUVEC resulted in increased adherence to fibronectin and vitronectin. EV- and vIL-6-HUVEC were transfected with nontargeting control (NTC) or an ITGB3-targeting pool of siRNAs (Fig. 6A). Cells were stained with a fluorescent dye and then allowed to adhere to wells coated with fibronectin or vitronectin (Fig. 6B and C, respectively). We found that vIL-6-HUVEC transfected with NTC siRNAs had increased adherence to fibronectin-coated wells but had relatively the same adherence to vitronectin as EV-HUVEC. However, when ITGB3 was knocked down in vIL-6-HUVEC, the fluorescent signal, indicating the number of attached cells, significantly decreased for both ECM components, whereas the signal for EV-HUVEC did not show a significant decrease. These results indicate that vIL-6 expression makes cell attachment to the ECM components, fibronectin and vitronectin, more heavily dependent on ITGB3.
FIG 6.

ITGB3 aids in vIL-6-HUVEC adhesion to ECMs. (A) EV- and vIL-6-HUVEC were transfected for 48 h with nontargeting control or ITGB3 siRNAs, and cell lysates were collected for immunoblotting to confirm knockdown efficiency. (B) Cells treated as for panel A were stained with calcein-AM and plated for 30 min on wells precoated with fibronectin. Unattached cells were removed by gentle washes, and fluorescence was measured to quantify the relative amounts of cells adhered to the ECM component. (C) Similar to panel B, but with vitronectin-coated wells. Panels B and C represent the averages from three experiments, each with seven technical replicates. ***, P < 0.001.
Viral IL-6-induced tubule formation is mediated through ITGB3.
vIL-6 has been previously reported to aid in angiogenesis (26, 27, 39, 40). Since KS is highly vascularized, we sought to determine if this was the case for vIL-6-expressing cells as well. EV- and vIL-6-HUVEC were treated with NTC or ITGB3 siRNAs for 48 h. Cells were then placed on top of Matrigel and incubated for up to 4 h. The number of branching points was manually calculated from at least 32 images. The results showed that vIL-6-HUVEC transfected with NTC siRNA had significantly more branch points than EV-HUVEC (Fig. 7). The number of branching points, however, significantly decreased when ITGB3 was knocked down in vIL-6-HUVEC; this decrease was not observed in EV-HUVEC depleted of ITGB3. These results indicate that ITGB3 is involved in vIL-6-mediated endothelial tubule formation, suggesting a possible role for ITGB3 in KSHV-induced angiogenesis.
FIG 7.

ITGB3 contributes to vIL-6-induced tubule formation of endothelial cells. (A) Representative images from four experiments performed in duplicate with EV- and vIL-6-HUVEC treated with siRNAs against a nontargeting control or ITGB3. The center of the images was zoomed in for a better resolution of the tubules. (B) The average number of branching points per well (4 or 5 frames/well) in duplicates was calculated and represented in the scatterplot graph. **, P < 0.01.
DISCUSSION
The heterodimer αVβ3 integrin is hypothesized to be important in several cancers and viral infections. A role for αVβ3 integrin has been suggested in KSHV pathogenesis, specifically in viral infection (7–9) and angiogenesis (14). Though KSHV infection has been demonstrated to induce ITGB3 (14), and the viral protein gB has been shown to interact with integrin αVβ3 (8), no specific KSHV protein has been identified as responsible for ITGB3 induction. In this report, we demonstrate that ITGB3 is highly upregulated in vIL-6-HUVEC and demonstrate that stable expression of this viral protein in endothelial cells results in an increase of total and cellular-surface-targeted ITGB3 protein (Fig. 8). Interestingly, the levels of ITGB3 gene expression in the vIL-6-HUVEC were similar to what DiMaio and colleagues detected following KSHV infection of endothelial cells (14) despite the generally low levels of vIL-6 expression seen upon infection. To our surprise, the induction of the lytic life cycle, which significantly increased vIL-6 expression, did not affect the levels of ITGB3 in KSHV-infected cells. We hypothesized that this is due to the persistent activation of STAT3 in latently infected cells (41). Thus, the activation of STAT3 by vIL-6 and other viral factors during latency may induce maximal levels of ITGB3 in latently infected cells.
FIG 8.

Model of vIL-6 induction of ITGB3. Expression of vIL-6 augments JAK/STAT3 activation increasing the levels of ITGB3, which results in higher surface expression of the heterodimer αVβ3 integrin. This process promotes vIL-6-induced endothelial cell adhesion to the ECM components fibronectin and vitronectin and promotes tubule formation. The illustration was designed using BioRender.
We have also demonstrated that vIL-6 is secreted from endothelial, epithelial, and B cells and intercellularly induces ITGB3 at the mRNA and protein levels in naive endothelial cells. Importantly, lytically replicating cells secreted high levels of vIL-6, which induced ITGB3 in a paracrine manner. These results are relevant because vIL-6 can be found circulating in the blood of KSHV- and HIV-coinfected individuals (42, 43), and its paracrine signaling is believed to play a significant role in viral pathogenesis (44).
Since vIL-6 is known to induce the activation of the JAK/STAT signaling pathway (20, 35), we sought to determine if this pathway is involved in ITGB3 induction. First, we found, as has been shown in other cell types (21, 24), that vIL-6 coimmunoprecipitates with gp130 in HUVEC and that siRNA-mediated knockdown of gp130 reduced vIL-6-induced activation of STAT3 and ITGB3 expression. Additionally, we found that phosphorylated STAT3 is required for vIL-6 specific induction of ITGB3. To our knowledge, this is the first report to demonstrate a mechanism of ITGB3 expression that requires STAT3 signaling.
Furthermore, neither overexpression of the cellular IL-6 homolog nor treatment with recombinant hIL-6 in endothelial cells induced ITGB3 expression. A possible explanation for this result is that HUVEC are more responsive to hIL-6 trans-signaling activation that relies on the binding of the cytokine to soluble, rather than to membrane-bound, IL-6Rα (37, 38). Despite the slight activation of STAT3, levels of ITGB3 were not increased in the presence of hIL-6. Importantly, this was circumvented when cells were supplemented with recombinant, soluble IL-6Rα that increased the levels of ITGB3 to levels seen in vIL-6-expressing cells, confirming that induction of the pathway plays a significant role in modulating the expression of ITGB3. In our studies, activation of STAT3 signaling by hIL-6 alone in endothelial cells is not sufficient for ITGB3 induction, suggesting that vIL-6 overcomes the need for IL-6Rα by consistently activating the pathway through gp130. Given that sIL-6R can be found circulating in human blood (37) and KSHV-infected individuals have high levels of circulating viral and cellular IL-6, we speculate that in patients, activation of the signaling pathway by both proteins may induce the expression of ITGB3.
Viral IL-6-HUVEC treated with a nontargeting siRNA pool have a statistically significant higher binding affinity to the ECM proteins, fibronectin and vitronectin, than do vIL-6-HUVEC that have ITGB3 knocked down. This significant decrease in binding with ITGB3 knockdown was unique to the vIL-6-HUVEC. Furthermore, utilizing these siRNAs, we performed a tubule formation assay, an in vitro approach to measure angiogenic phenotype. The vIL-6-HUVEC that still expressed ITGB3 had more branching points than either EV-HUVEC or vIL-6-HUVEC treated with the ITGB3 siRNA. The ability of vIL-6-HUVEC to have increased binding to fibronectin, which aids in the stability and growth of microvessels (45, 46), and increased endothelial cell branching only when ITGB3 is expressed hints at the importance of this induction for angiogenesis.
Altogether, our data identify the KSHV vIL-6 protein as a bona fide inducer of ITGB3. We hypothesize that ITGB3 plays an important role in the capacity of the virus to induce angiogenesis and endothelial cell migration. The ability of vIL-6 to be secreted from KSHV-infected cells to affect cells in a paracrine manner further confirms the essential role that infiltrating cells might play in KS lesions by enhancing cellular processes such as angiogenesis. In conclusion, this report further characterizes the role of vIL-6 in endothelial cells and its contribution to viral pathogenesis.
MATERIALS AND METHODS
Cell culture.
All endothelial-cell-based assays used human telomerase reverse transcriptase (hTERT)-immortalized human umbilical vein endothelial cells (HUVEC) cultured in EBM-2 (Lonza) or ECGM2 (PromoCell) and their respective supplement kits as described previously (47). BJAB and TREx-Rta-BCBL-1 cells were maintained in RPMI 1640 medium (Corning). The cell line iSLK.219 (harboring latent rKSHV.219) was maintained in Dulbecco modified Eagle medium (DMEM; Corning) in the presence of 1 μg/ml of puromycin (Corning), 250 μg/ml of G418 (Sigma), and 400 μg/ml of hygromycin (Corning). All media were supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin-streptomycin, and 1% l-glutamine. The iSLK.219 and TREx-Rta-BCBL-1 cells were reactivated by supplementing the media with 1 μg/ml of doxycycline for up to 24 h.
Production of lentivirus vectors and stable cell lines.
Production of the empty vector (EV) and vIL-6 lentivirus and the construction of stable cell lines were described previously (24). The hIL-6 lentiviral vector was cloned similarly. Briefly, hIL-6 was inserted into a lentivirus vector with puromycin resistance. Lentivirus was produced using the ViraPower lentiviral expression system (Invitrogen), and cells were transduced by spin inoculation in the presence of Polybrene (8 μg/ml). BJAB cells expressing EV, vIL-6, or hIL-6 were made using the same lentivirus and spin inoculation procedure.
RNA isolation and real-time qPCR.
RNA was isolated from cells using the RNeasy Plus minikit (Qiagen). cDNA was obtained from 1 μg of total RNA using the iScript cDNA synthesis kit (Bio-Rad). At least three biological replicates were performed for each condition used in experiments, with three technical replicates for each sample. Real-time qPCR was performed on a Quantstudio 6 Flex real-time PCR machine (Thermo Fisher) using SensiFAST SYBR Lo-Rox real-time PCR master mix (Bioline). PCR primer sequences used for ITGB3 were obtained from reference 48. To amplify actin cDNA, the forward primer 5′-TCATGAAGTGTGACGTGGACATC-3′ and reverse primer 5′-CAGGAGGAGCAATGATCTTGATCT-3′ were used.
Immunoblotting.
Cells were collected, and lysates were prepared from washed pellets using NP-40 lysis buffer (0.1% NP-40, 150 mM NaCl, 50 mM Tris HCl [pH 8.0], 30 mM β-glycerophosphate, 50 mM NaF, 1 mM Na3VO4, and 1 Roche protease inhibitor tablet per 50 ml). Samples were clarified by centrifugation at 16,000 × g for 10 to 15 min, and protein concentration was determined by Bradford assay (Bio-Rad). Lysates were resolved on acrylamide SDS-PAGE gels. ITGB3 (4702S), ITGAV (4711S), gp130 (3732S), pSTAT3 Y705 (9131S), total STAT3 (4904S), and secondary horseradish peroxidase (HRP)-conjugated antibodies (anti-rabbit [7074] and anti-mouse [7076]) were purchased from Cell Signaling Technology (CST). Human IL-6 antibodies were obtained from Origene (TA300413) and CST (12153S). The vIL-6 antibody was purified from the supernatant of v6m 12.1.1 hybridomas (ATCC) (49) using magnetic protein A/G beads (Thermo Fisher). Actin (sc-47778) antibody conjugated with HRP was purchased from Santa Cruz Biotechnology.
Coimmunoprecipitation.
Cell lysates containing equal amounts of proteins from EV- and vIL-6 (FLAG-tagged)-HUVEC were precleared with EZview red protein A affinity gel (P6486; Sigma) and mouse nonspecific IgG (sc-2025; Santa Cruz Biotechnology). Supernatants were collected and incubated with 40 μl of EZview red anti-FLAG M2 affinity gel (F2426; Sigma) overnight at 4°C. Beads were pelleted and washed twice with lysis buffer and twice with cold Tris-buffered saline. Bound proteins were eluted with 3× FLAG peptide (F4799; Sigma). All immunoprecipitation inputs and eluted proteins were resolved in SDS-PAGE gels, and immunoblottings were performed as described above.
Luciferase reporter assay.
Two hundred thousand HEK293T cells were plated per well into a 24-well plate. Twenty-four hours postseeding, cells were cotransfected with 500 ng/well of a vIL-6-expressing plasmid or the corresponding EV backbone (a gift from Britt Glausinger) (50) and 150 ng/well of ITGB3-luciferase reporter plasmid (HPRM23183-PG04) purchased from GeneCopoeia. Transfection was performed with Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s protocol. Supernatants were collected 48 h posttransfection. The Gaussia luciferase and the internal control, secreted embryonic alkaline phosphatase (SEAP), were measured using the Secrete-Pair dual-luminescence assay kit (GeneCopoeia) according to the manufacturer’s protocol. To confirm vIL-6 expression, protein lysates were prepared and immunoblotting was performed as described above.
Flow cytometry.
Endothelial cells were plated after trypsinization and incubated for 48 h. Cells were then collected using Versene and counted. Five hundred thousand cells per sample were washed and resuspended in 80 μl of FACS buffer (phosphate-buffered saline [PBS], 2% FBS, 2 mM EDTA) along with 20 μl of human Fc receptor (FcR) blocking reagent (MACS Miltenyi Biotec) and then allowed to incubate for 10 min at 4°C. Cells were then spun down and stained with 2 μg of αVβ3 antibodies (MAB1976; Millipore Sigma) in 100 μl of fluorescence-activated cell sorter (FACS) buffer and incubated on ice in the dark for 30 min. After primary staining, cells were washed three times and then stained for 30 min on ice in the dark with 300 ng of goat anti-mouse antibody conjugated with Alexa Fluor 488 (AF488) fluorophore (Thermo Fisher). After washing off the excess secondary stain, cells were fixed in FACS buffer containing 1% formaldehyde. Samples were run on a MACSQuant VYB flow cytometer (Miltenyi Biotec). The analysis was conducted using FlowJo software.
Conditioned-medium preparation and treatment.
HUVEC and BJAB stable cell lines were incubated for 24 h in serum- or supplement-free medium. TREx-Rta-BCBL-1 and iSLK.219 cells were reactivated for 24 h in complete medium. The conditioned medium was then collected, centrifuged, filtered, and then added to naive HUVEC cells supplemented with completed medium. Lysates were harvested 24 h posttreatment.
Neutralization antibody assay.
Conditioned medium from stable HUVEC and BCBL-1 cells were created as described above except for certain wells that received 10 μg of mouse nonspecific IgG (Santa Cruz Biotechnology) or 10 μg of purified vIL-6 antibody. Conditioned medium was then added to immortalized HUVEC and resupplemented with the antibody. After a 24-h incubation, lysates were collected, and immunoblotting was performed.
siRNA transfections and drug inhibitor treatment.
siRNAs were transfected into cells using Lipofectamine RNAiMax reagent (Thermo Fisher) and allowed to incubate for up to 72 h before lysate collection or cell use in other assays. ON-TARGETplus SMARTpool siRNAs for human ITGB3 (L004124), IL-6ST/gp130 (L005166), as well as the nontargeting control (D001810) siRNAs, were purchased from Dharmacon. Silencer Select siRNA targeting STAT3 (4390824), as well as the respective negative control, was obtained from Thermo Fisher. For STAT3 drug inhibition, cells were treated with 20 μM cryptotanshinone (MedChemExpress) or a vehicle (DMSO). Cells were incubated for 48 h posttreatment before lysates were collected.
Recombinant human IL-6 and sIL-6Rα treatment.
For hIL-6 treatment, HUVEC were plated on six-well plates with complete medium. Twenty-four hours after plating, the medium was replaced with fresh medium containing rhIL-6 (Peprotech) at the desired concentrations. After incubating cells for 48 h with rhIL-6, lysates were prepared and immunoblotting was performed as described above. For the experiment with sIL-6Rα, cells were treated for 24 h similarly to the above description but in the presence or absence of sIL-6Rα (GenScript).
Adhesion assay.
Cells were plated in 100-mm dishes and transfected the next day with siRNAs as described above. Cells were removed from the plate using Versene and washed in PBS twice. Cells were resuspended in 500 μl of serum-free medium at a concentration of 106/ml. Two and a half microliters of the fluorescent dye calcein-AM, obtained from Invitrogen Vybrant cell adhesion kit (Thermo Fisher), was added to the cell suspension and incubated for 30 min at 37°C. During the incubation, 8-well strips that contained either fibronectin or vitronectin (Millicoat Cell Adhesion strips; Millipore) were allowed to warm up to room temperature and washed in PBS. After the 30-min incubation, cells were washed in prewarmed medium three times and then diluted to 105/ml, and 100 μl was added to each well. The cells were allowed to adhere to the ECM components for 30 min at 37°C. Nonadherent cells were removed by gentle washing (repeated five times) in a warmed medium. After the final wash, 200 μl of PBS was added to each well, and the fluorescence was measured on a CLARIOstar plate reader (BMG Labtech).
Tubule formation assay.
EV- or vIL-6-HUVEC were plated and transfected with siRNAs as indicated above. Forty-eight hours posttransfection, cells were detached from the plates and counted, and 1.25 × 105 cells in a total of 1 ml of complete medium were seeded on top of 300 μl of Matrigel (Corning) in the wells of a 24-well plate. At least four images were taken per well between 3 and 4 h postseeding, and the number of branching points was manually calculated using ImageJ.
Statistics.
Statistical tests (Student’s t test for RT-qPCR, Mann-Whitney test for luciferase assay, and Kruskal-Wallis for adhesion and tubule formation assays) were calculated using GraphPad.
ACKNOWLEDGMENTS
We are thankful to the Damania lab members and Whitney Tevebaugh for revising and editing the manuscript and Zhigang Zhang for technical advice.
This work was supported by Public Health Service grants CA096500, CA019014, DE028211, and CA163217. B.D. is a Leukemia and Lymphoma Society Scholar and a Burroughs Wellcome Fund Investigator in Infectious Disease. R.R.-S. was supported by NIH grant GM007092 and holds a Graduate Diversity Enrichment Program Award from the Burroughs Wellcome Fund.
The funders had no role in the design of the study or in the collection or interpretation of the data.
REFERENCES
- 1.Danhier F, Le Breton A, Préat V. 2012. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm 9:2961–2973. doi: 10.1021/mp3002733. [DOI] [PubMed] [Google Scholar]
- 2.Brooks PC, Clark RA, Cheresh DA. 1994. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 3.Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, Klier G, Cheresh DA. 1994. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 4.Feng XX, Liu M, Yan W, Zhou ZZ, Xia YJ, Tu W, Li PY, Tian DA. 2013. β3 integrin promotes TGF-β1/H2O2/HOCl-mediated induction of metastatic phenotype of hepatocellular carcinoma cells by enhancing TGF-β1 signaling. PLoS One 8:e79857. doi: 10.1371/journal.pone.0079857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rapisarda V, Borghesan M, Miguela V, Encheva V, Snijders AP, Lujambio A, O’Loghlen A. 2017. Integrin beta 3 regulates cellular senescence by activating the TGF-beta pathway. Cell Rep 18:2480–2493. doi: 10.1016/j.celrep.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieberler M, Reuning U, Reichart F, Notni J, Wester H-J, Schwaiger M, Weinmüller M, Räder A, Steiger K, Kessler H, Nieberler M, Reuning U, Reichart F, Notni J, Wester H-J, Schwaiger M, Weinmüller M, Räder A, Steiger K, Kessler H. 2017. Exploring the role of RGD-recognizing integrins in cancer. Cancers (Basel) 9:116. doi: 10.3390/cancers9090116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veettil MV, Sadagopan S, Sharma-Walia N, Wang FZ, Raghu H, Varga L, Chandran B. 2008. Kaposi’s sarcoma-associated herpesvirus forms a multimolecular complex of integrins (alphaVbeta5, alphaVbeta3, and alpha3beta1) and CD98-xCT during infection of human dermal microvascular endothelial cells, and CD98-xCT is essential for the postentry stage of infection. J Virol 82:12126–12144. doi: 10.1128/JVI.01146-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garrigues HJ, Rubinchikova YE, Dipersio CM, Rose TM. 2008. Integrin alphaVbeta3 binds to the RGD motif of glycoprotein B of Kaposi’s sarcoma-associated herpesvirus and functions as an RGD-dependent entry receptor. J Virol 82:1570–1580. doi: 10.1128/JVI.01673-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garrigues HJ, DeMaster LK, Rubinchikova YE, Rose TM. 2014. KSHV attachment and entry are dependent on alphaVbeta3 integrin localized to specific cell surface microdomains and do not correlate with the presence of heparan sulfate. Virology 464–465:118–133. doi: 10.1016/j.virol.2014.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renne R, Lagunoff M, Zhong W, Ganem D. 1996. The size and conformation of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J Virol 70:8151–8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 12.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 13.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86:1276–1280. doi: 10.1182/blood.V86.4.1276.bloodjournal8641276. [DOI] [PubMed] [Google Scholar]
- 14.DiMaio TA, Gutierrez KD, Lagunoff M. 2011. Latent KSHV infection of endothelial cells induces integrin beta3 to activate angiogenic phenotypes. PLoS Pathog 7:e1002424. doi: 10.1371/journal.ppat.1002424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damania B, Cesarman E. 2013. Kaposi’s sarcoma-associated herpesvirus, p 2080–2128. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Willams & Wilkins, Philadelphia, PA. [Google Scholar]
- 16.Moore PS, Boshoff C, Weiss RA, Chang Y. 1996. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274:1739–1744. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- 17.Neipel F, Albrecht JC, Ensser A, Huang YQ, Li JJ, Friedman-Kien AE, Fleckenstein B. 1997. Human herpesvirus 8 encodes a homolog of interleukin-6. J Virol 71:839–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicholas J, Ruvolo VR, Burns WH, Sandford G, Wan X, Ciufo D, Hendrickson SB, Guo HG, Hayward GS, Reitz MS. 1997. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat Med 3:287–292. doi: 10.1038/nm0397-287. [DOI] [PubMed] [Google Scholar]
- 19.Sakakibara S, Tosato G. 2011. Viral interleukin-6: role in Kaposi’s sarcoma-associated herpesvirus: associated malignancies. J Interferon Cytokine Res 31:791–801. doi: 10.1089/jir.2011.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molden J, Chang Y, You Y, Moore PS, Goldsmith MA. 1997. A Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J Biol Chem 272:19625–19631. doi: 10.1074/jbc.272.31.19625. [DOI] [PubMed] [Google Scholar]
- 21.Chen D, Sandford G, Nicholas J. 2009. Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J Virol 83:722–733. doi: 10.1128/JVI.01517-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burger R, Neipel F, Fleckenstein B, Savino R, Ciliberto G, Kalden JR, Gramatzki M. 1998. Human herpesvirus type 8 interleukin-6 homologue is functionally active on human myeloma cells. Blood 91:1858–1863. doi: 10.1182/blood.V91.6.1858. [DOI] [PubMed] [Google Scholar]
- 23.Wu J, Xu Y, Mo D, Huang P, Sun R, Huang L, Pan S, Xu J. 2014. Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 promotes cell proliferation and migration by upregulating DNMT1 via STAT3 activation. PLoS One 9:e93478. doi: 10.1371/journal.pone.0093478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giffin L, Yan F, Ben Major M, Damania B. 2014. Modulation of Kaposi’s sarcoma-associated herpesvirus interleukin-6 function by hypoxia-upregulated protein 1. J Virol 88:9429–9441. doi: 10.1128/JVI.00511-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giffin L, West JA, Damania B. 2015. Kaposi’s sarcoma-associated herpesvirus interleukin-6 modulates endothelial cell movement by upregulating cellular genes involved in migration. mBio 6:e01499-15. doi: 10.1128/mBio.01499-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aoki Y, Jaffe ES, Chang Y, Jones K, Teruya-Feldstein J, Moore PS, Tosato G. 1999. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood 93:4034–4043. doi: 10.1182/blood.V93.12.4034.412k38_4034_4043. [DOI] [PubMed] [Google Scholar]
- 27.Rivera-Soto R, Damania B. 2019. Modulation of angiogenic processes by the human gammaherpesviruses, Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus. Front Microbiol 10:1544. doi: 10.3389/fmicb.2019.01544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. 2005. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J Immunol 175:7708–7718. doi: 10.4049/jimmunol.175.11.7708. [DOI] [PubMed] [Google Scholar]
- 29.Whitby D, Howard MR, Tenant-Flowers M, Brink NS, Copas A, Boshoff C, Hatzioannou T, Suggett FE, Aldam DM, Denton AS, Miller RF, Weller IVD. 1995. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi’s sarcoma. Lancet 346:799–802. doi: 10.1016/s0140-6736(95)91619-9. [DOI] [PubMed] [Google Scholar]
- 30.Huang YQ, Li JJ, Poiesz BJ, Kaplan MH, Friedman-Kien AE. 1997. Detection of the herpesvirus-like DNA sequences in matched specimens of semen and blood from patients with AIDS-related Kaposi’s sarcoma by polymerase chain reaction in situ hybridization. Am J Pathol 150:147–153. [PMC free article] [PubMed] [Google Scholar]
- 31.Ambroziak JA, Blackbourn DJ, Herndier BG, Glogau RG, Gullett JH, McDonald AR, Lennette ET, Levy JA. 1995. Herpes-like sequences in HIV-infected and uninfected Kaposi’s sarcoma patients. Science 268:582–583. doi: 10.1126/science.7725108. [DOI] [PubMed] [Google Scholar]
- 32.Staskus KA, Sun R, Miller G, Racz P, Jaslowski A, Metroka C, Brett-Smith H, Haase AT. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. J Virol 73:4181–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cannon JS, Nicholas J, Orenstein JM, Mann RB, Murray PG, Browning PJ, DiGiuseppe JA, Cesarman E, Hayward GS, Ambinder RF. 1999. Heterogeneity of viral IL-6 expression in HHV-8-associated diseases. J Infect Dis 180:824–828. doi: 10.1086/314956. [DOI] [PubMed] [Google Scholar]
- 34.Mierke CT. 2013. The integrin alphav beta3 increases cellular stiffness and cytoskeletal remodeling dynamics to facilitate cancer cell invasion. New J Phys 15:015003. doi: 10.1088/1367-2630/15/1/015003. [DOI] [Google Scholar]
- 35.Wan X, Wang H, Nicholas J. 1999. Human herpesvirus 8 interleukin-6 (vIL-6) signals through gp130 but has structural and receptor-binding properties distinct from those of human IL-6. J Virol 73:8268–8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. 2011. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 1813:878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 37.Scheller J, Garbers C, Rose-John S. 2014. Interleukin-6: from basic biology to selective blockade of pro-inflammatory activities. Semin Immunol 26:2–12. doi: 10.1016/j.smim.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 38.Zegeye MM, Lindkvist M, Falker K, Kumawat AK, Paramel G, Grenegard M, Sirsjo A, Ljungberg LU. 2018. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6 trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Commun Signal 16:55. doi: 10.1186/s12964-018-0268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou F, Xue M, Qin D, Zhu X, Wang C, Zhu J, Hao T, Cheng L, Chen X, Bai Z, Feng N, Gao SJ, Lu C. 2013. HIV-1 Tat promotes Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6-induced angiogenesis and tumorigenesis by regulating PI3K/PTEN/AKT/GSK-3β signaling pathway. PLoS One 8:e53145. doi: 10.1371/journal.pone.0053145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu X, Guo Y, Yao S, Yan Q, Xue M, Hao T, Zhou F, Zhu J, Qin D, Lu C. 2014. Synergy between Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 and HIV-1 Nef protein in promotion of angiogenesis and oncogenesis: role of the AKT signaling pathway. Oncogene 33:1986–1996. doi: 10.1038/onc.2013.136. [DOI] [PubMed] [Google Scholar]
- 41.Punjabi AS, Carroll PA, Chen L, Lagunoff M. 2007. Persistent activation of STAT3 by latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells. J Virol 81:2449–2458. doi: 10.1128/JVI.01769-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aoki Y, Yarchoan R, Wyvill K, Okamoto S, Little RF, Tosato G. 2001. Detection of viral interleukin-6 in Kaposi sarcoma-associated herpesvirus-linked disorders. Blood 97:2173–2176. doi: 10.1182/blood.v97.7.2173. [DOI] [PubMed] [Google Scholar]
- 43.Brousset P, Cesarman E, Meggetto F, Lamant L, Delsol G. 2001. Colocalization of the viral interleukin-6 with latent nuclear antigen-1 of human herpesvirus-8 in endothelial spindle cells of Kaposi’s sarcoma and lymphoid cells of multicentric Castleman’s disease. Hum Pathol 32:95–100. doi: 10.1053/hupa.2001.21131. [DOI] [PubMed] [Google Scholar]
- 44.Giffin L, Damania B. 2014. KSHV: pathways to tumorigenesis and persistent infection. Adv Virus Res 88:111–159. doi: 10.1016/B978-0-12-800098-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mongiat M, Andreuzzi E, Tarticchio G, Paulitti A. 2016. Extracellular matrix, a hard player in angiogenesis. Int J Mol Sci 17:E1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neve A, Cantatore FP, Maruotti N, Corrado A, Ribatti D. 2014. Extracellular matrix modulates angiogenesis in physiological and pathological conditions. Biomed Res Int 2014:756078. doi: 10.1155/2014/756078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang L, Wakisaka N, Tomlinson CC, DeWire SM, Krall S, Pagano JS, Damania B. 2004. The Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) K1 protein induces expression of angiogenic and invasion factors. Cancer Res 64:2774–2781. doi: 10.1158/0008-5472.can-03-3653. [DOI] [PubMed] [Google Scholar]
- 48.Principe M, Borgoni S, Cascione M, Chattaragada MS, Ferri-Borgogno S, Capello M, Bulfamante S, Chapelle J, Di Modugno F, Defilippi P, Nistico P, Cappello P, Riganti C, Leporatti S, Novelli F. 2017. Alpha-enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. J Hematol Oncol 10:16. doi: 10.1186/s13045-016-0385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aoki Y, Narazaki M, Kishimoto T, Tosato G. 2001. Receptor engagement by viral interleukin-6 encoded by Kaposi sarcoma-associated herpesvirus. Blood 98:3042–3049. doi: 10.1182/blood.v98.10.3042. [DOI] [PubMed] [Google Scholar]
- 50.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jager S, Shales M, Horner J, Hernandez RD, Krogan NJ, Glaunsinger BA. 2015. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57:349–360. doi: 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]