

Abstract
Gut microbiota is considered as a human organ with its own specific functions and complexity. Development of novel techniques such as shut gun sequencing, metagenomics, and next-generation sequencing (NGS) has enabled bypassing the traditional culturedependent bias and has significantly expanded our understanding of the composition, diversity, and roles of the gut microbiota in human health and diseases. Although amplicon sequencing characterizes the taxonomic composition of the gut microbiome, it is impossible to cover the direct evidence of the microbial biological functions related to the gut microbial community. Hence, the critical next step for gut microbiome studies is shifting from gene/ genome-centric analysis to mechanism-centric techniques by integrating omics data with experimental results. Realizing gut microbial diversity and their bioactive metabolites function will provide insight into the clinical application of gut microbiota in diagnoses and treatments of several diseases. In this review, we focused on explaining the conventional and advanced microbiome analysis techniques regarding gut microbiota investigation with considering the advantages and disadvantages of the platforms.
Keywords: Gut microbiota, Microbiome analysis techniques, Advantages, Disadvantages
INTRODUCTION
1. Context
Gut microbiota
The new organ, the last human organ, a forgotten organ, or a missing organ, all of these names remark the important role of gut microbiota.1,2 They can be explained as a community of bacteria, archea, viruses, fungi, and protozoa. Predominating human gastrointestinal tract microbiota consists of firmicutes, proteobacteria, bacteroidetes , actinobacteria, fusobacteria, verromicrobia, and cyanobacteria.3 An altered gut microbiome composition not only has been shown linked to the gastrointestinal disorders such as colorectal polyps,4 colorectal cancer (CRC),5,6 and celiac disease,7 but also is associated with non-intestinal disorders such as allergy,8 asthma,9,10 obesity,11,12 non-alcoholic fatty liver,13 cardiovascular diseases,13,14 and neuropsychiatric diseases.15-17 These disorders can be attributed to the imbalance of the gut microbiota, which would be referred to as dysbiosis (dysbacteriosis) or dysregulation of gut microbiota.16
Conventional gut microbiome analysis techniques: advantages and disadvantages
A link to the gut microbiome and diseases was first postulated in the 20th century. However, until the 1990s, understanding of the gut microbiome was confined because the microbiological culture was the only way to investigate its composition. Actually, a small fraction of the gut microbiota has been cultured up to now and developments in culture-independent techniques have led to our belief of the complexity of this microenvironment. These techniques have clarified the gut microbiota diversity and composition and have shown that dysbacteriosis and dysregulation of gut microbiota are associated with several diseases. These latest techniques are rapid, accurate, and comfort high throughput to identify and enumerate uncultivable microorganisms.18 These techniques are based on sequence divergences of the small subunit ribosomal RNA (16S rRNA) that are well conserved between various species of bacteria and provide bacterial species identification and demonstrate gut microbiota diversity. Also, they provide qualitative and quantitative information on bacterial species and gut microbiota alteration in relation to diseases. Examples of these conventional analysis techniques are quantitative real-time polymerase chain reaction (q PCR), denaturing gradient gel electrophoresis (DGGE), terminal restriction fragment length polymorphism (T-RFLP), fluorescence in situ hybridization (FISH), 16S rRNA gene sequencing-based on cloning, direct sequencing of 16S rRNA amplicons, shotgun sequencing (table 1).19,20
Table 1. Conventional gut microbiome analysis techniques considering their strengths and weaknesses .
| Technique | Explanation | Strength | Weakness |
| Culture | Cultivation of selected bacteria on selective media | Inexpensive, semi-quantitative method | A small fraction of gut microbiota has been cultured up to now, labor-intensive |
| Quantitative real-time polymerase chain reaction (q PCR) | Quantification of 16S rRNA. Reaction mixture includes a compound that fluoresces when it binds to double-stranded DNA | Rapid, phylogenetic characterization, quantitative method | PCR bias, incapable of characterizing unknown species |
| Denaturing gradient gel electrophoresis (DGGE) | Gel separation of 16S rRNA amplicons using denaturant/ temperature | Semi-quantitative, bands could be applied for further analysis, rapid | PCR bias, no phylogenetic characterization |
| Terminal restriction fragment length polymorphism (T-RFLP) | Fluorescently labeled primers are quantified and then restriction enzymes are applied to digest the 16S rRNA amplicon. Digested fragments further separated by gel electrophoresis | Inexpensive, fast, semi-quantitative method | PCR bias, no phylogenetic characterization, low resolution |
|
Fluorescence in situ hybridization (FISH) |
Fluorescently labeled oligonucleotide probes hybridize complementary target 16S rRNA sequences. When hybridization occurs, fluorescence can be enumerated using flow cytometry | No PCR bias, Phylogenetic characterization, semi-quantitative method | Related to probe sequences— incapable of characterizing unknown species |
| 16S rRNA gene sequencing based on cloning | Sanger sequencing, capillary electrophoresis, cloning of full-length 16S rRNA amplicon | Phylogenetic characterization, quantitative method | Expensive, PCR bias, laborious, cloning bias |
| Direct sequencing of 16S rRNA amplicons | Large parallel sequencing of partial 16S rRNA amplicons | Rapid, phylogenetic characterization, quantitative method, capable of identifying unknown bacteria | PCR bias, expensive, laborious |
| Shotgun sequencing | Massive parallel sequencing of the whole genome | Phylogenetic characterization, quantitative method | Expensive, analysis of data is computationally intense |
Advance microbiome analysis techniques: advantages and disadvantages
Microbiome shotgun sequencing and metagenomics
Despite applying 16S rRNA technology has significantly improved our knowledge of gut microbiota composition and diversity; it has not illuminated the significant association between microbial patterns and disease initiation or progression. In fact, metagenomics demonstrates the newest growth in gut microbiota composition analysis. It has been widely used in studies including the human microbiome project (HMP).21-23 This technique is capable of sequencing all the DNA fragments in the sample rather than the sequencing of particular DNA fragments.24,25 This method is used to analyze the composition and diversity of gut microbiota by data sequencing from the merged genomes of the microbiota. The strength of the technique is that it is strong enough to identify new functional genes. On the other hand, the weakness of the technique is that it cannot provide gene expression profiles and predict how different conditions can regulate it also cannot distinguish DNA of live cells from DNA of dead cells.26,27
Whole-genome shotgun metagenomic sequencing is a widely used tool for characterizing the metagenomic content of gut microbiome samples. While whole-genome shotgun metagenomic sequencing data contains gene-level information, it can be an incentive to analyze the millions of microbial genes, which are typically found in microbiome experiments. It is performed by major parallel sequencing of the mixed DNA sample. It implicates random fragmentation of DNA, sequencing of DNA fragments and reconstruction of overlapping sequences to assemble them into a continuous sequence. The advantage of the technique is that information is assembled on the genetic diversity of the gut microbiota. Information on the genetic diversity and composition of the gut microbiota permits correlations to be made between gut microbiota and disease position. Disadvantages of shotgun metagenomics sequencing are that it is costly and also the analysis of a large amount of data is computationally intense and not easily performed in general laboratories.28,29
Next-generation sequencing (NGS) in gut microbiota study
NGS technologies in particular targeted amplicon sequencing of the 16S-seq and enabled the identification and quantification of human-resident microorganisms at unprecedented resolution, providing novel insights into the role of the microbiota in health and disease. The ability to rapidly sequence human genomes and to generate genetic, transcriptomic, and epigenetic data and other genome-wide data for a relatively small cost opens up numerous opportunities for translation into the clinic over the next few years.30,31 NGS as a high throughput procedure has revolutionized the human gut microbiota studies and enabled the exploration of uncultured gut microbial diversity communities as a sufficient and cost-effective technique.32,33 It consists of the following techniques: 454 GS FLX + (Roche), HiSeq 2000/2500 (lumina), 5500 xl SOLiD (Life Technologies), PacBio RS (Pacific Bioscience), Ion torrent (Life Technologies).34 Figure 1 depicts NGS technology upon PCR or non-PCR based.
Fig.1.
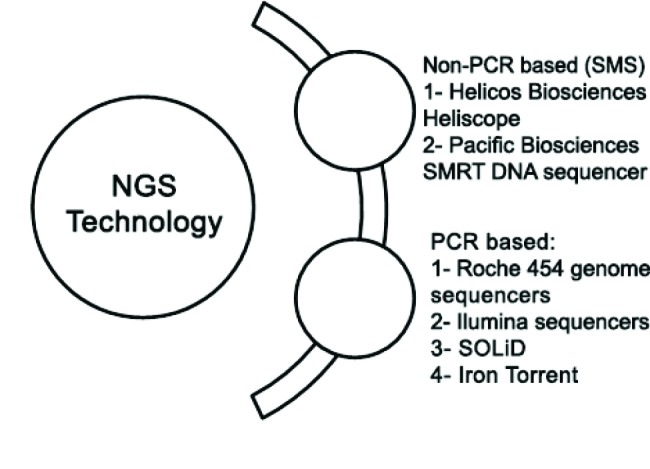
Diagram for different NGS platforms technologies
Actually the procedure is amplicon-based profiling and is applied as one of the most widely used techniques for defining gut microbiome diversity. Usually, 16S rRNA for bacteria and archaea as a taxonomically informative gene marker is prevalent for organisms to be studied, targeted, and amplified from the total DNA. The resulting amplicons are sequenced, and downstream bioinformatics analyses are performed to characterize the relative taxonomical abundances in the samples.34-36 There are many advantages and disadvantages related to applying NGS platforms (table 2).30
Table 2. NGS platforms considering the strengths and weaknesses34 .
| Machinery | Chemistry | Strengths | Weakness |
| 454 Gs Flx + (Roche) | Pyrosequencing | Read length is long, sensitive, analysis of multiple samples at the same time, no cloning bias | In hands-genes, expensive, the high error rate in homopolymers, short sequencing reads, extensive, bioinformatics analysis require |
|
HighSeq (2000/25000) (Illumina) |
Quantification of 16S rRNA. Reaction mixture includes a compound that fluoresces when it binds to double-stranded DNA |
Rapid, phylogenetic characterization, quantitative method |
PCR bias, incapable of characterizing unknown species |
|
5500 SOLiD (Life technologies) |
Ligation | Low error rate, high throughput | Short length, long process |
| PacBio RS (Pacific Bioscience) | Real-time sequencing | Easy preparation, cheap, long read length | High error rate, expensive, difficult installation |
| - Ion torrent (Life Technologies) | Proton detection | Short process, flexible chip reagents | Instrument under development |
In a one-run, pyrosequencing sequences 500 million bases with more than 99% accuracy rate.37 NGS platform compares gene marker profiles across samples in order to identify microbial diversity. Although amplicon sequencing characterizes the taxonomic composition of the gut microbiome, it is impossible to cover the direct evidence of the microbial biological metabolites and functions, which are related to the gut microbial community. Hence, important next step for gut microbiome studies is shifting from genome-based analysis to mechanism-based techniques by integrating omics data with these results.35 Alongside, metagenomics, metabolomics, proteomics, transcriptomics, and phenomics data are important for investigating gut microbiome characterization, functions, metabolites, proteins, and RNA.38,39
Omics techniques: advantages and disadvantages
By gathering metagenomics, metabolomics, proteomics, transcriptomics, and phenomics data we will reach more realization on the functioning besides diversity of the gut microbiota. The integrative analysis of these omics data will be critical for understanding the host-microbiome interaction mechanisms (table 3).
Table 3. Omics gut microbiome analysis techniques considering the strengths and weaknesses .
| Techniques: Metaomics | Explanation | Strength | Weakness |
| Metagenomics |
Investigating gut microbiota genomes profiling in high resolution, characterization of genes structures of uncultivated microbiota |
Comprehensive sequence data, data of functional contributions of the microbiota, needless to cloning of specific genes | The function of gut microbiota are not generated |
| Metatranscriptomics |
Messenger RNA/cDNA sequencing for high-resolution gene expression profiling, differential microbial gene in expression various physiological/environmental situations |
High throughput, high sensitivity, quantification method, characterization of known and unknown gut microbiota, evaluation of microbial interaction |
Lack of unique protocol, low stability of microbial mRNA, representatively unknown/ multiple purification steps needed |
| Metaproteomics |
Proteins/Peptides are identified for high resolution protein monitoring and profiling, differential microbial proteins production under various physiological/environmental conditions |
Locating and monitoring new functional genes |
Lack of unique protocol, unknown proteins in databases, heterogeneous stability |
| Metabolomics | Metabolites are analyzed for microbial host metabolic profiling | Rapid and easy to perform on every low amount of specimens including faces/serum/urine, time-efficient, the impact of gut microbiota in health and disease |
Lack of unique protocol, unknown metabolites in databases, strict characterization of compound labor-intensive, the combination of host and bacterial molecules |
Data analysis and bioinformatics approaches
The first step for the analysis of the microbiota is to determine the evolutionary association among the microbiota in the gut. Both alignments dependent based and alignment independent based techniques are applied to specify the evolutionary relatedness between them. In the alignment-based analysis, the homologous positions in the gene sequence are identified through a multiple sequence alignment against databases such as Ribosomal Database Project II (RDPII). The most general method for manufacturing alignments is the CLUSTAL online software and databases such as NCBI and multivariate statistical analysis are employed in this way.
One of the advantages of the alignment dependent based method is that it is the most precise approach to permit a detailed map of the phylogenetic relations, but it is not suitable for analysis of large data sets. However alternative methods applied for large datasets can be cost-effective.
Presently, the most used method for alignment independent based technique analysis is principal component analysis.26
CONCLUSION
With the newest technologies including microbiome shotgun sequencing, metagenomics, and NGS platforms, a deep understanding of the gut microbiome diversity has been gained, but it is impossible to cover direct evidence of the microbial biological metabolites and functions, which are related to the gut microbial community, along with their role in initiation and progression of diseases. Hence, the critical next step for gut microbiome studies is shifting from genome-based analysis to mechanism-based techniques by integrating omics data with experimental results. Therefore, there is a critical demand to go beyond solely characterizing the gut microbiome composition, systematic modeling, and analysis of the gut microbiome metabolites and function. Generally, understanding gut microbial diversity and their bioactive metabolites function in different types of disorders will provide insight into the clinical application of gut microbiota in diagnoses and treatments of diseases.
Please cite this paper as:
Rezasoltani S, Ahmadi Bashirzadeh D, Nazemalhosseini Mojarad E, Asadzadeh Aghdaei H, Norouzinia M, Shahrokh S. Signature of Gut Microbiome by Conventional and Advanced Analysis Techniques: Advantages and Disadvantages. Middle East J Dig Dis 2020;12:5-11. doi: 10.15171/mejdd.2020.157.
Footnotes
ETHICAL APPROVAL There is nothing to be declared.
CONFLICT OF INTEREST The authors declare no conflict of interest related to this work.
References
- 1.Cherng-Shyang Ch, Cheng-Yuan K. Current understanding of the gut microbiota shaping mechanisms. J Biomed Sci. 2019;26:59. doi: 10.1186/s12929-019-0554-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rezasoltani S, Dabiri H, Asadzadeh-Aghdaei H, Sepahi AA, Modarressi MH, Nazemalhosseini-Mojarad E. The gut microflora assay in patients with colorectal cancer: in feces or tissue samples? Iran J Microbiol. 2019;11:1–6. doi: 10.18502/ijm.v11i1.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maukonen J, Characterization of the human predominant fecal microbiota-With special focus on the Clostridial clusters IV and XIVa, Dissertation. Espoo 2012; VTT Technical Research Centre of Finland.
- 4.Rezasoltani S, Asadzadeh Aghdaei H, Dabiri H, Akhavan Sepahi A, Modarressi MH, Nazemalhosseini Mojarad E. The association between fecal microbiota and different types of colorectal polyp as precursors of colorectal cancer. Microb Pathog. 2018;124:244–249. doi: 10.1016/j.micpath.2018.08.035. [DOI] [PubMed] [Google Scholar]
- 5.Rezasoltani S, Sharafkhah M, Asadzadeh Aghdaei H, Nazemalhosseini Mojarad E, Dabiri H, Akhavan Sepahi A. et al. Applying simple linear combination, multiple logistic and factor analysis methods for candidate fecal bacteria as novel biomarkers for early detection of adenomatous polyps and colon cancer, 2018. J Microbiol Methods. 2018;155:82–88. doi: 10.1016/j.mimet.2018.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Rezasoltani S, Asadzadeh-Aghdaei H, Nazemalhosseini-Mojarad E, Dabiri H, Ghanbari R, Zali MR. Gut microbiota, epigenetic modification and colorectal cancer. Iran J Microbiol. 2017;9:55–63. doi: 10.18502/ijm.v11i1.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rostami Nejad M, Ishaq S, Dulaimi DA, Zali MR, K Rostami K. The Role of infectious mediators and gut microbiome in the pathogenesis of celiac disease. Arch Iran Med. 2015;18:244–9. [PubMed] [Google Scholar]
- 8.van den Elsen LWJ, Garssen J, Burcelin R, Verhasselt V. Shaping the gut microbiota by breastfeeding: the gateway to allergy prevention? Front Pediatr. 2019;7:47. doi: 10.3389/fped.2019.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S. et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med. 2015;7:307ra152. doi: 10.1126/scitranslmed.aab2271. [DOI] [PubMed] [Google Scholar]
- 10.Sokolowska M, Frei R, Lunjani N, Akdis CA, O’Mahony L. Microbiome and asthma. Asthma Res Pract. 2018;4:1. doi: 10.1186/s40733-017-0037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, Hardt PD. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 2010;18:190–5. doi: 10.1038/oby.2009.167. [DOI] [PubMed] [Google Scholar]
- 12.Sanz Y, Santacruz A, Gauffin P. Gut microbiota in obesity and metabolic disorders. Proc Nutr Soc. 2010;69:434–41. doi: 10.1017/s0029665110001813. [DOI] [PubMed] [Google Scholar]
- 13.Sanduzzi Zamparelli M, Compare D, Coccoli P, Rocco A, Nardone OM, Marrone G. et al. The metabolic role of gut microbiota in the development of nonalcoholic fatty liver disease and cardiovascular disease. Int J Mol Sci. 2016;17:1225. doi: 10.3390/ijms17081225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jie Z, Xia H, Zhong SL, Feng Q, Li S, Liang S. et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. 2017;8:845. doi: 10.1038/s41467-017-00900-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yarandi SS, Peterson DA, Treisman GJ, Moran TH, Pasricha PJ. Modulatory effects of gut microbiota on the central nervous system: how gut could play a role in neuropsychiatric health and diseases. J Neurogastroenterol Motil. 2016;22:201–12. doi: 10.5056/jnm15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rezasoltani S, Nazemalhosseini Mojarrad E, Norouzinia M, Asadzadeh Aghdaei H. The necessity of gut microbiome characterization in diseases prevention and therapy. Gastroenterol Hepatol Bed Bench. 2017;10:150–1. [PMC free article] [PubMed] [Google Scholar]
- 17.Grochowska M, Wojnar M, Radkowski M. The gut microbiota in neuropsychiatric disorders. Acta Neurobiol Exp (Wars) 2018;78:69–81. doi: 10.21307/ane-2018-008. [DOI] [PubMed] [Google Scholar]
- 18.Fraher MH, O’Toole PW, Quigley EM. Techniques used to characterize the gut microbiota: a guide for the clinician. Nat Rev Gastroenterol Hepatol. 2012;9:312–22. doi: 10.1038/nrgastro.2012.44. [DOI] [PubMed] [Google Scholar]
- 19.Rajilic-Stojanovic M, Smidt H, de Vos WM. Diversity of the human gastrointestinal tract microbiota revisited, Environ. Microbiol. 2007;9:2125–36. doi: 10.1111/j.1462-2920.2007.01369.x. [DOI] [PubMed] [Google Scholar]
- 20.Zoetendal EG, Vaughan E E, de Vos W M. A microbial world within us. Mol Microbiol. 2006;59:1639–50. doi: 10.1111/j.1365-2958.2006.05056.x. [DOI] [PubMed] [Google Scholar]
- 21.NIH HMP Working Group, Peterson J, Garges S, Giovanni M, McInnes P, Wang L. et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–23. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA. et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 23.Breitbart M, Hewson I, Felts B, Mahaffy JM, Nulton J, Salamon P. et al. Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol. 2003;185:6220–3. doi: 10.1128/jb.185.20.6220-6223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sekirov I, Russell S L, Antunes LC, Finlay BB. Gut microbiota in health and disease, Physiol Rev 2010;90: 859–904. Gut microbiota in health and disease, Physiol Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 25.Wang WL, Xu SY, Ren ZG, Tao L, Jiang JW, Zheng SS. Application of metagenomics in the human gut microbiome. World J Gastroenterol. 2015;21:803–14. doi: 10.3748/wjg.v21.i3.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong J, Yang Ch. Advances in the methods for studying gut microbiota and their relevance to the research of dietary fiber functions. Food Res Int. 2012;48 doi: 10.1016/j.foodres.2011.12.027. [DOI] [Google Scholar]
- 27.Teeling H, Glöckner FO. Current opportunities and challenges in microbial metagenome analysis—a bioinformatic perspective. Brief Bioinform. 2012;13:728–42. doi: 10.1093/bib/bbs039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Vos WM. Mining the microbes the human microbiome as model Microb. Biotechnol. 2009;؟؟:153–4. doi: 10.1111/j.1751-7915.2009.00090_20.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun. 2016;469:967–77. doi: 10.1016/j.bbrc.2015.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finotello F, Mastrorilli E, Di Camillo B. Measuring the diversity of the human microbiota with targeted next-generation sequencing. Brief Bioinform. 2018;19:679–92. doi: 10.1093/bib/bbw119. [DOI] [PubMed] [Google Scholar]
- 31.Panek M, Čipčić Paljetak H, Barešić A, Perić M, Matijašić M, Lojkić I. et al. Methodology challenges in studying human gut microbiota–effects of collection, storage, DNA extraction and next generation sequencing technologies. Sci Rep. 2018;8:5143. doi: 10.1038/s41598-018-23296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeller G, Tap J, Voigt AY, Sunagaw a Sh, Roat Kultima J, Costea P. et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol. 2014;10:766. doi: 10.15252/msb.20145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malla MA, Dubey A, Kumar A, Yadav S, Hashem A, Abd Allah EF. Exploring the Human Microbiome: The Potential Future Role of Next-Generation Sequencing in Disease Diagnosis and Treatment. Front Immunol. 2018;9:2868. doi: 10.3389/fimmu.2018.02868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- 35.Boyang Ji, Jens N. From next-generation sequencing to systematic modeling of the gut microbiome. Front Genet. 2015;6:219. doi: 10.3389/fgene.2015.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F. et al. A metagenome-wide association study of gut microbiota in type2 diabetes. Nature. 201;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 37.Zhou X, Ren L, Li Y, Zhang M, Yu Y, Yu J. The next-generation sequencing technology: a technology review and future perspective. Sci China Life Sci. 2010;53:44–57. doi: 10.1007/s11427-010-0023-6. [DOI] [PubMed] [Google Scholar]
- 38.Vanessa Aguiar P, Wenrui H, Victoria Suarez U, Trevor C, Kalai M, Giri N. Metagenomics, Metatranscriptomics, and Metabolomics Approaches for Microbiome Analysis. Evol Bioinform Online. 2016;12:5–16. doi: 10.4137/EBO.S36436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hassa J, Maus I, Off S, Pühler A, Scherer P, Klocke M, Schlüter A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl Microbiol Biotechnol. 2018;102:5045–63. doi: 10.1007/s00253-018-8976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]