

Abstract
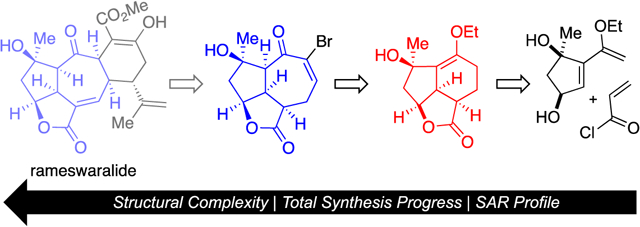
A Pharmacophore-Directed Retrosynthesis (PDR) strategy applied to rameswaralide provided simplified precursors bearing the common 5,5,6 (red) and 5,5,7 (blue) skeleton present in several cembranoid and norcembranoids from Sinularia soft corals. Key steps include a Diels-Alder lactonization organocascade delivering the common 5,5,6 core and a subsequent ring expansion affording a 5,5,7 core serviceable for the synthesis of rameswaralide. Initial structure-activity relationships of intermediates en route to the natural product has revealed interesting differential and selective cytotoxicity.
Graphical Abstract
Soft corals belonging to the genus Sinularia produce a number of cembranoid and norcembranoid diterpenes bearing a complex, caged, highly oxygenated tri- or tetracyclic skeleton as seen in several representative members 1–6 (Figure 1).1–6 However, the limited availability of these natural products from corals has hindered a broader understanding of their bioactivity. To date, moderate cytotoxicity of ineleganolide (5) toward the P388 leukemia cell line (11.6 μM ED50)5 was reported. The potential anti-inflammatory activity7 and cytotoxicity toward A549 human lung carcinoma epithelial cells (IC50 67 ± 3.7 μM)8 of rameswaralide (6) was also reported. Furthermore, with the exception of rameswaralide,8 the absolute stereochemistry of these diterpenoids has not yet been confirmed; however it was indirectly assigned based on the established absolute stereochemistry of a probable biosynthetic precursor.3,9 While several groups have pursued the synthesis of rameswaralide (6)10–14 and ineleganolide (5)15–21 which bear the 5,5,7 core (blue, Figure 1), to our knowledge, there are no reported total syntheses. Less synthetic work has been directed to members possessing the 5,5,6 core (red) such as that found in yonarolide, scabrolides, dissectolides, and sinulochmodin C (1-4).22–25
Figure 1.
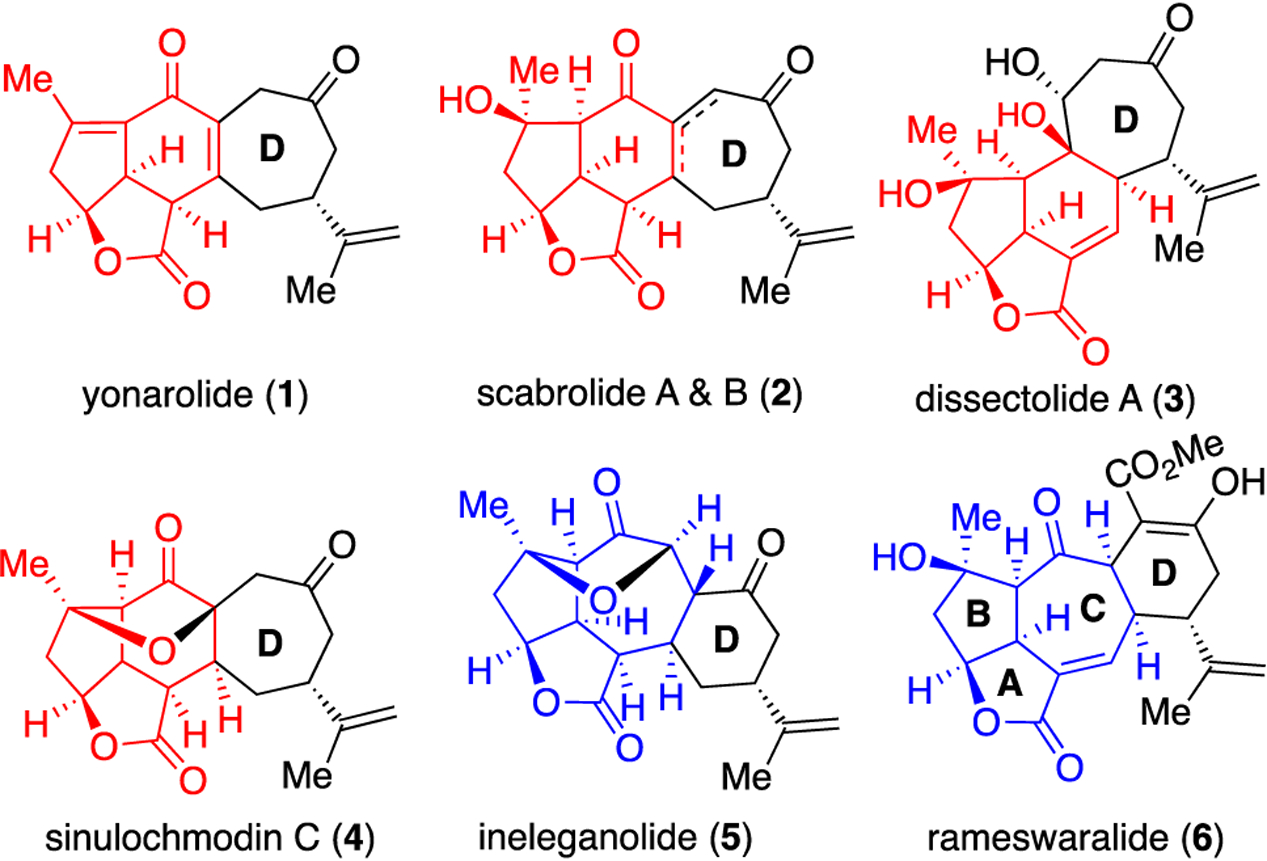
Structurally related, polycyclic, Sinularia diterpenoid natural products.
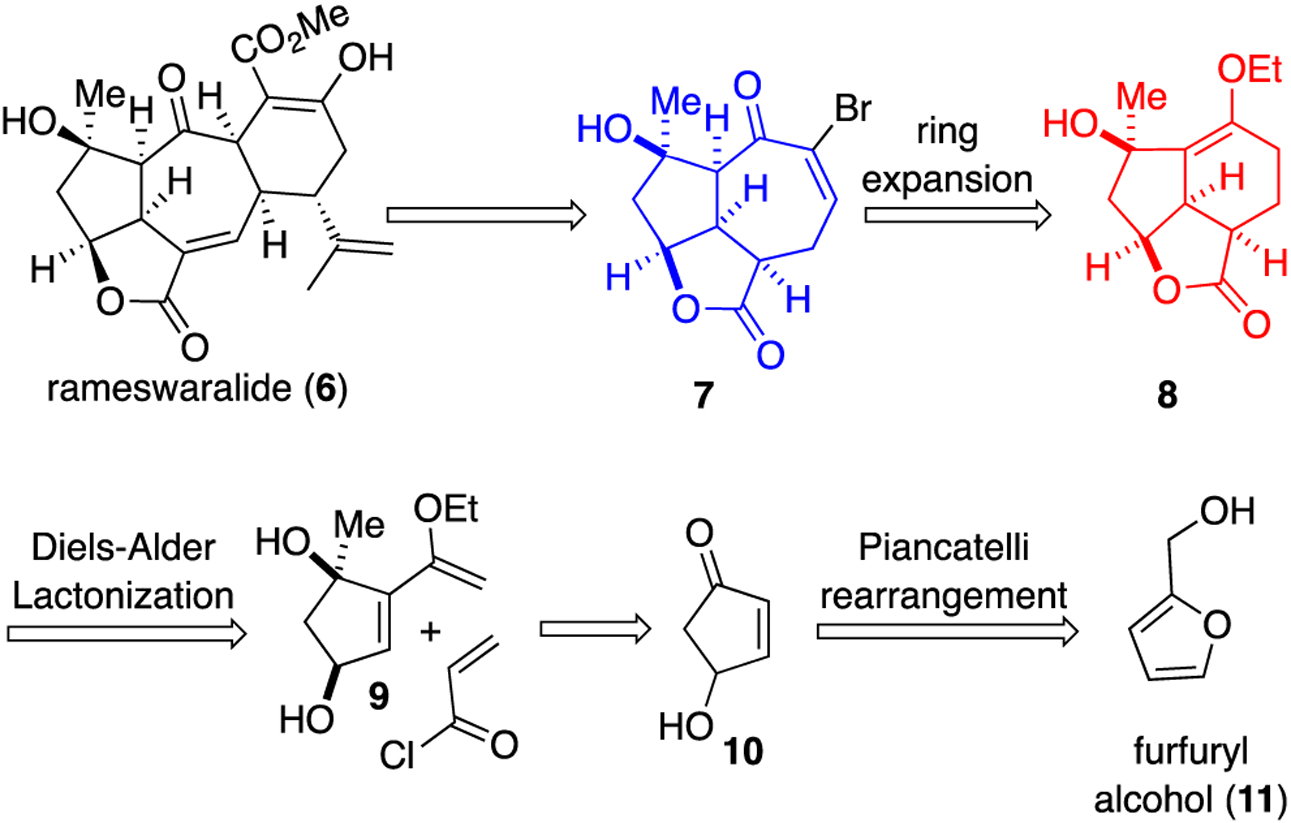
Our interest in ineleganolide (5) and rameswaralide (6) stems from their structural complexity and the lack of information regarding structure and activity relationships (SAR) for these diterpenoids. Toward the latter goal, herein we apply our recently disclosed Pharmacophore-Directed Retrosynthesis (PDR)26 strategy, to enable simultaneous collection of SAR for rameswaralide and congeners en route to a total synthesis. An initial hypothesis to guide our PDR approach is premised on the noted similarities in the common 5,5,7-core (blue, Figure 1) which we propose may encompass the pharmacophore of ineleganolide (5) and rameswaralide (6). Variations in this core structures derive from points of attachment of the fused 6-membered D ring to the C rings and a bridging ether in ineleganolide derived from a presumed oxa-Michael addition.9 Furthermore, sinulochmodin C (4) and scabrolides A&B (2) are constitutional isomers of ineleganolide (5), yet in cell lines tested, have shown moderate to no bioactivity.27,28 We propose that the 5,5,7 core structure imparts the greatest bioactivity to this class of diterpenoids. We therefore devised a retrosynthesis that would allow access to the 5,5,7 core through a ring expansion proceeding via a 5,5,6 core intermediate. This strategy would enable a systematic study of the impact of core ring size and location of attachment of the D-ring to the 5,5,7-core on bioactivity. Thus, annulation of various D-rings to these core structures and attendant functionality would provide valuable SAR information en route to members of this family. It is certainly possible that in these highly functionalized caged natural products, the entire molecule is required for bioactivity, nevertheless application of PDR facilitates and opens the possibility of identifying equipotent, simplified derivatives and also potential new bioactivities not previously observed with this class of diterpenoids.
We decided to set rameswaralide (6) as our initial target since it displays some of the most compelling bioactivity in this class of tetracyclic diterpenoids.6 Based on previous work by both Mehta10 and Trost,11 who independently synthesized a similar 5,5,7 core of rameswaralide, we anticipated that annulation of the D-ring would be challenging given the lack of further efforts toward this end. With this in mind, we proposed that α-halo enones like enone 7, could serve as highly versatile intermediates for several D-ring annulation strategies toward rameswaralide (6). The α-bromo enone 7 could be accessed via a cyclopropanation ring expansion strategy from 5,5,6 tricycle 8. Early access to both tricyclic intermediates 7 and 8 provides a divergence point towards several members of this natural product family and would enable a systematic study of potential varying bioactivity between 5,5,6 and 5,5,7 cores. We envisioned access to tricycle 8 through our recently described Diels-Alder Lactonization (DAL) organocascade process of diene 9 and acryloyl chloride.29,30 Diene 9 is available through a Stille cross-coupling and 1,2 addition to cyclopentenone 10, in turn available through a known Piancatelli rearrangement31 of furfuryl alcohol (11).
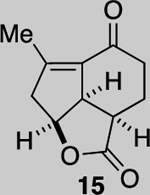
Our synthetic studies began with the preparation of diol 9 employing known iodide 12.32–34 The latter was subjected to a Stille coupling with ethoxy vinyl stannane 13 to afford racemic diene 14. Subsequent 1,2 addition with MeLi proceeded with high diastereoselectivity (>19:1 dr) and deprotection provided diol 9 in 45% yield over 2 steps. With the requisite diene in hand, we attempted the key DAL reaction initially employing racemic benzotetramisole, however, typical DAL conditions provided only enone 15 corresponding to the core of yonarolide (1). This product presumably results from elimination of the tertiary alcohol catalyzed by trace HCl; this same enone was recently obtained by Deng and co-workers through a different synthetic strategy.23 Extensive optimization to identify acid scavenging conditions that did not interfere with the DAL process ultimately led to replacement of the previously reported biphasic proton shuttle base system29,30 with only Hunig’s base (i-Pr2NEt). These conditions reliably provided the desired tertiary alcohol 8 in 77% yield with high diastereoselectivity (>19:1, 1H NMR). An optimized route to obtain enone 15 was also identified involving direct workup with 1 M HCl. The relative stereochemistry of this product was confirmed by X-ray crystallography (Scheme 2, inset). While the current route provides racemic material, our previous studies suggest the potential for an asymmetric variant of the key DAL process.29,30
Scheme 2.
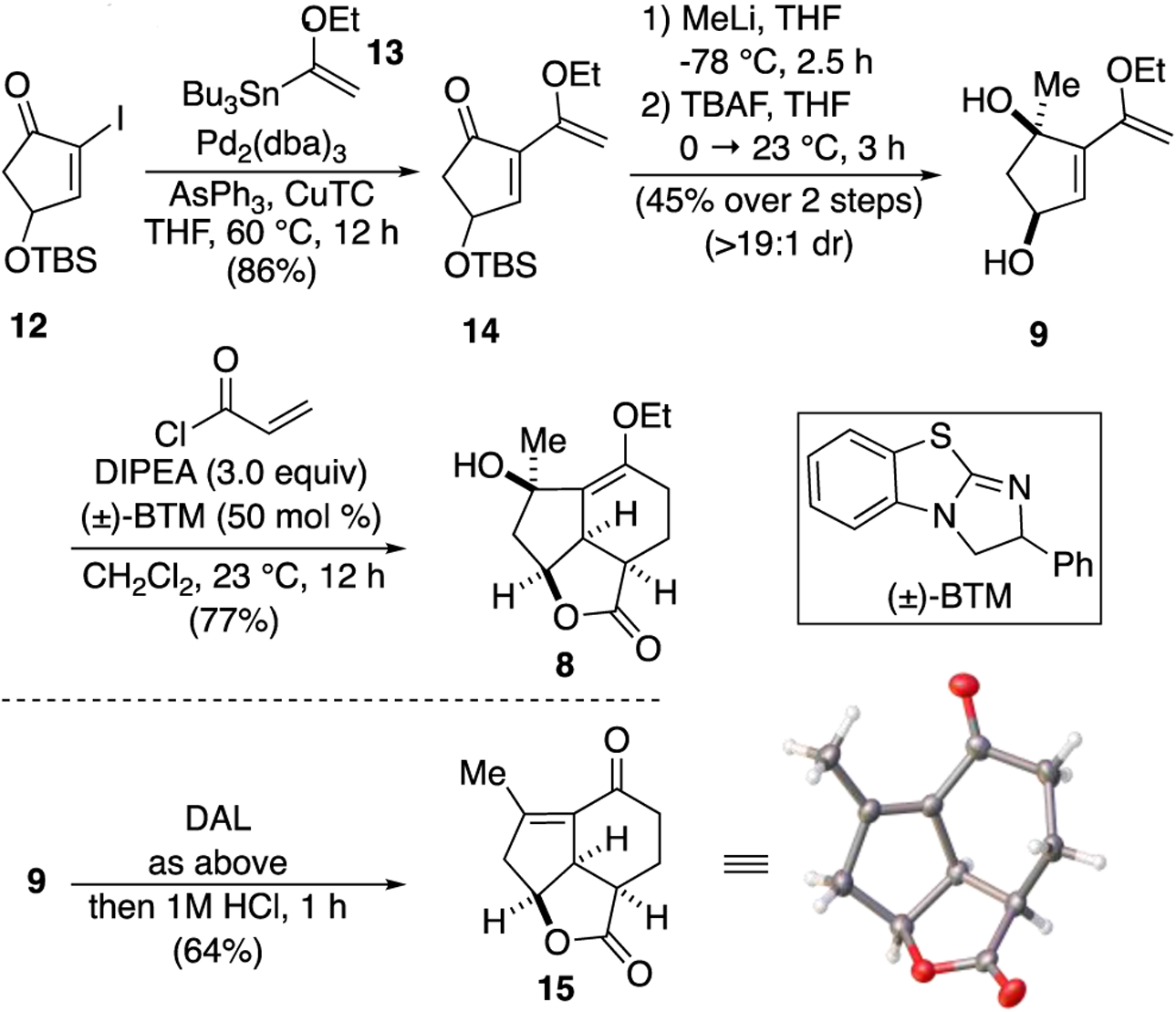
Synthesis of diene 9 and the 5,5,6 tricyclic cores 8 and 15 via the Diels-Alder Lactonization (DAL) organocascade.
Enol ether 8 is a regioisomer of an ideal alkene substrate for the planned ring expansion to the desired cycloheptanone ring. This realization coupled with the propensity for elimination of the tertiary alcohol leading to enone 15, under even mild acidic conditions, led us to consider strategies for direct transposition of the alkene through an electrophilic addition/alkene transposition pathway. Davies reported a similar alkene transposition utilizing N-bromosuccinimide (NBS) presumably driven by release of strain.35 Indeed, the desired transposition of the less stable ring-fused alkene was achieved under mild conditions with NBS providing the brominated, regioisomeric enol ether 16.
With an initial substrate for cyclopropanation in hand, we studied the Furukawa-modified, Simmons-Smith cyclopropanation (Scheme 3).36–38 However, not surprisingly, treatment of alcohol 16 to cyclopropanation conditions initially generates the tertiary alkoxide which cyclized to epoxide 21 as the sole product. Unfortunately, the instability of epoxide 21 precluded its use as a cyclopropanation substrate. Protection of the tertiary alcohol as a triethylsilyl (TES) ether 17 precluded epoxide formation, however the cyclopropanation still did not proceed and only starting material was recovered. We considered that the tertiary bromide may be preventing approach of the Zn-carbenoid to the more accessible convex face. Therefore, standard radical dehalogenation conditions provided enol ether 18. However, attempted cyclopropanation again led only to recovered starting material. We next considered removal of the TES protecting group which, based on models, may be forcing the ethyl group of the enol ether into the more accessible convex face through a gearing effect. The net result would be steric blocking of both faces of the enol ether. Furthermore, deprotection of the alcohol would enable a complex-induced proximity effect through formation of a pendant carbenoid proximal to the enol ether thereby increasing the rate of cyclopropanation through intramolecularity.39,40 Use of allylic alcohols in this manner through directed Furukawa-modified, Simmons-Smith cyclopropanation is precedented.41–45 Indeed, after removal of the TES protecting group, enol ether 19 participated in the desired cyclopropanation employing diiodomethane delivering cyclopropane 20 as a single diastereomer (>19:1 by 1H NMR, 600 MHz) in 38% yield, (unoptimized) sufficient for our initial studies of the ring expansion. The relative stereochemistry was confirmed by X-ray analysis and verifies that methylene transfer occurred exclusively on the sterically less accessible, concave face and that delivery of the zinc carbenoid is likely directed by the ideally situated, in situ generated tertiary alkoxide.
Scheme 3.
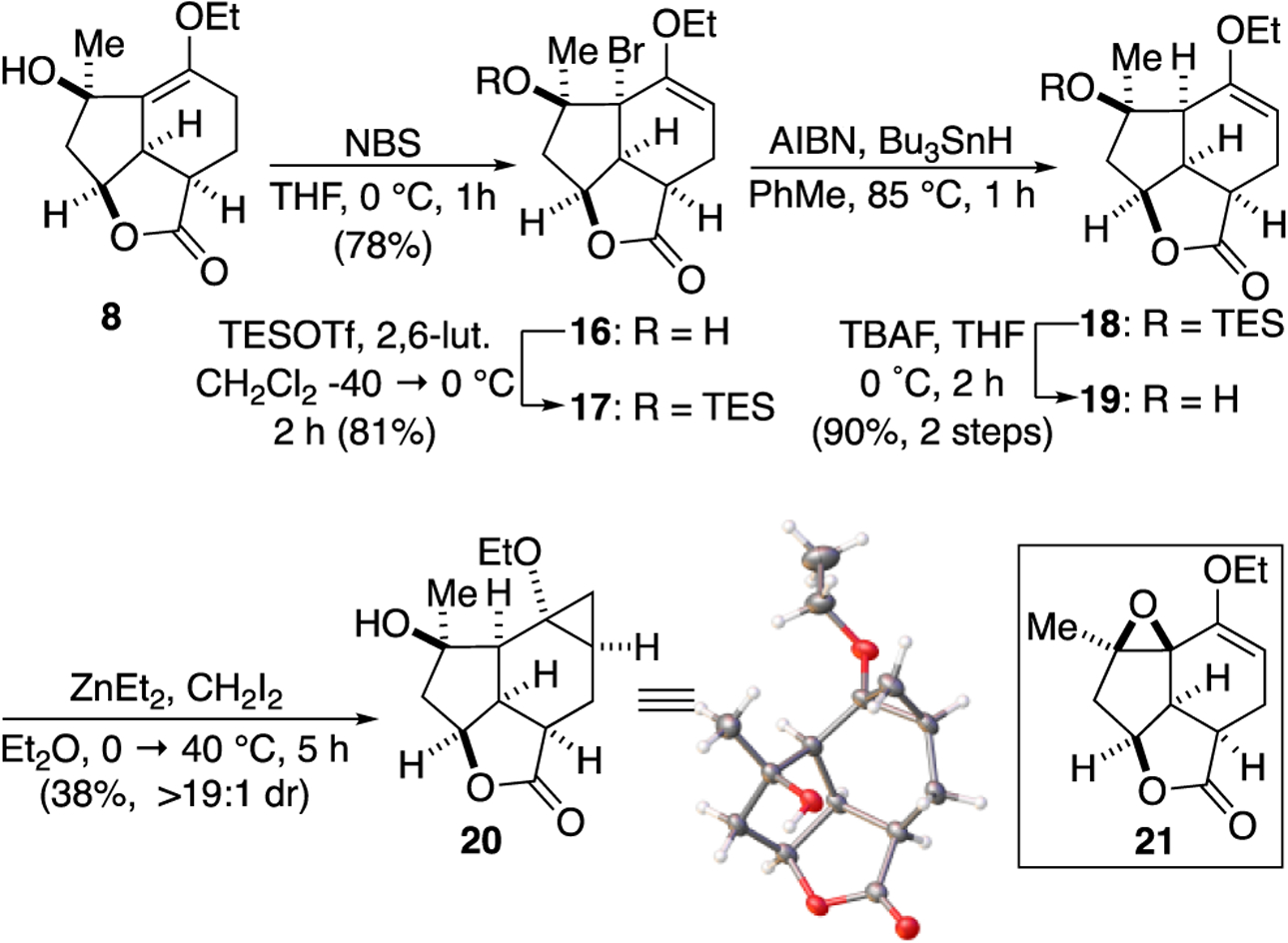
Alkene transposition of enol ether 8 and a complex-induced, proximity effect driven cyclopropanation.
With cyclopropane 20 in hand, we attempted radical mediated, oxidative ring expansions (Scheme 4). Under FeCl3 conditions reported by Seagusa for cyclopropyl TMS ethers,38 no reaction was observed. Use of ceric ammonium nitrate (CAN) with NaI,46,47 which presumably generates iodine radicals, led to cleavage of the cyclopropane providing primary iodide 22 which proved to be highly unstable. This suggest that the ethyl ether is not being oxidized directly but rather iodine radical is formed and attacks the least hindered carbon of the cyclopropane as proposed in Scheme 4.48
Scheme 4.
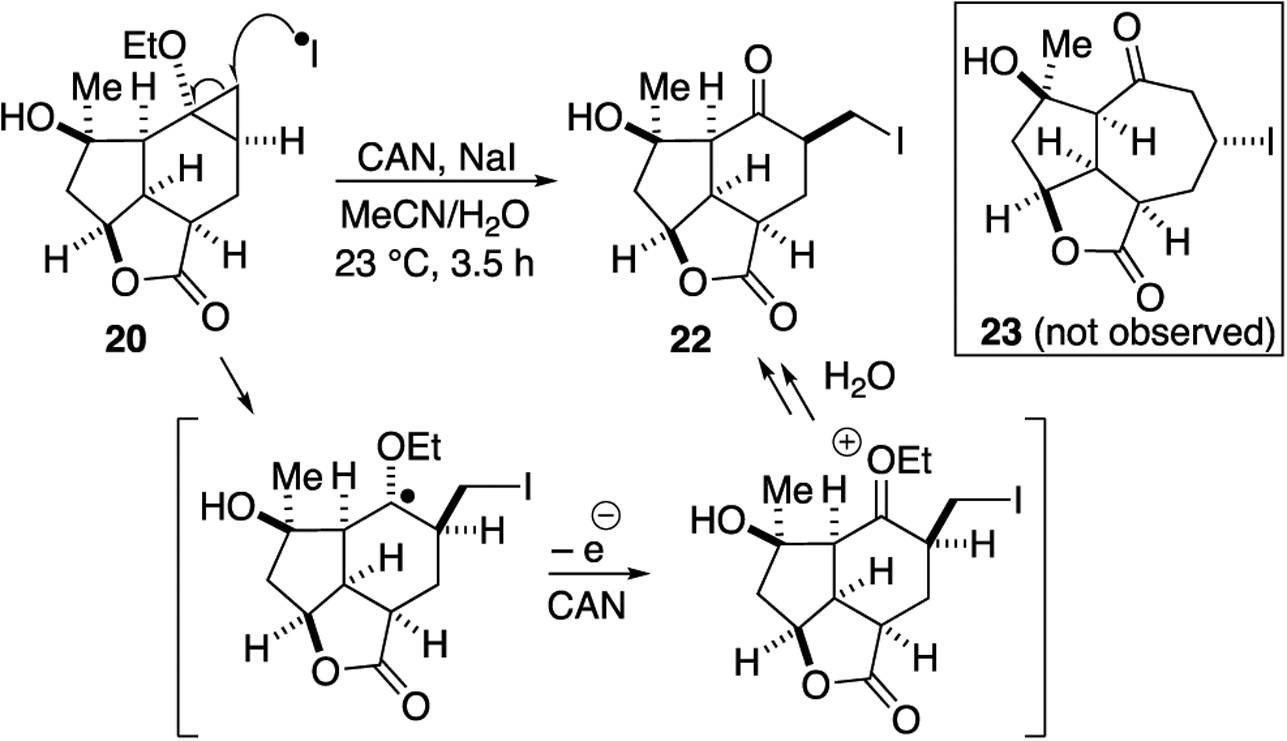
Attempted ring expansion of cyclopropane 20.
To direct the cyclopropane ring expansion, we targeted the monobromocyclopropane 24 (Scheme 5). We anticipated that the bromide would enable ring expansion via an ionic mechanism or, alternatively, it could block addition of a radical to the undesired cyclopropyl carbon. The same cyclopropanation conditions employed previously were performed with tribromomethane and led to a very sluggish conversion to the desired cyclopropane. The presence of oxygen has been reported to increase the rate of cyclopropanation by aiding in the formation of the bromozinc carbenoid.49,50 Thus, addition of a balloon of O2 after alkoxide formation and addition of tribromomethane greatly enhanced the reaction rate and led to reproducible yields of cyclopropane 24 with high diastereoselectivity (>19:1, 1H NMR). The relative stereochemistry of the cyclopropane ring was assigned by analogy to cyclopropane 20 and coupling constant analysis of the cyclopropane protons.
Scheme 5.
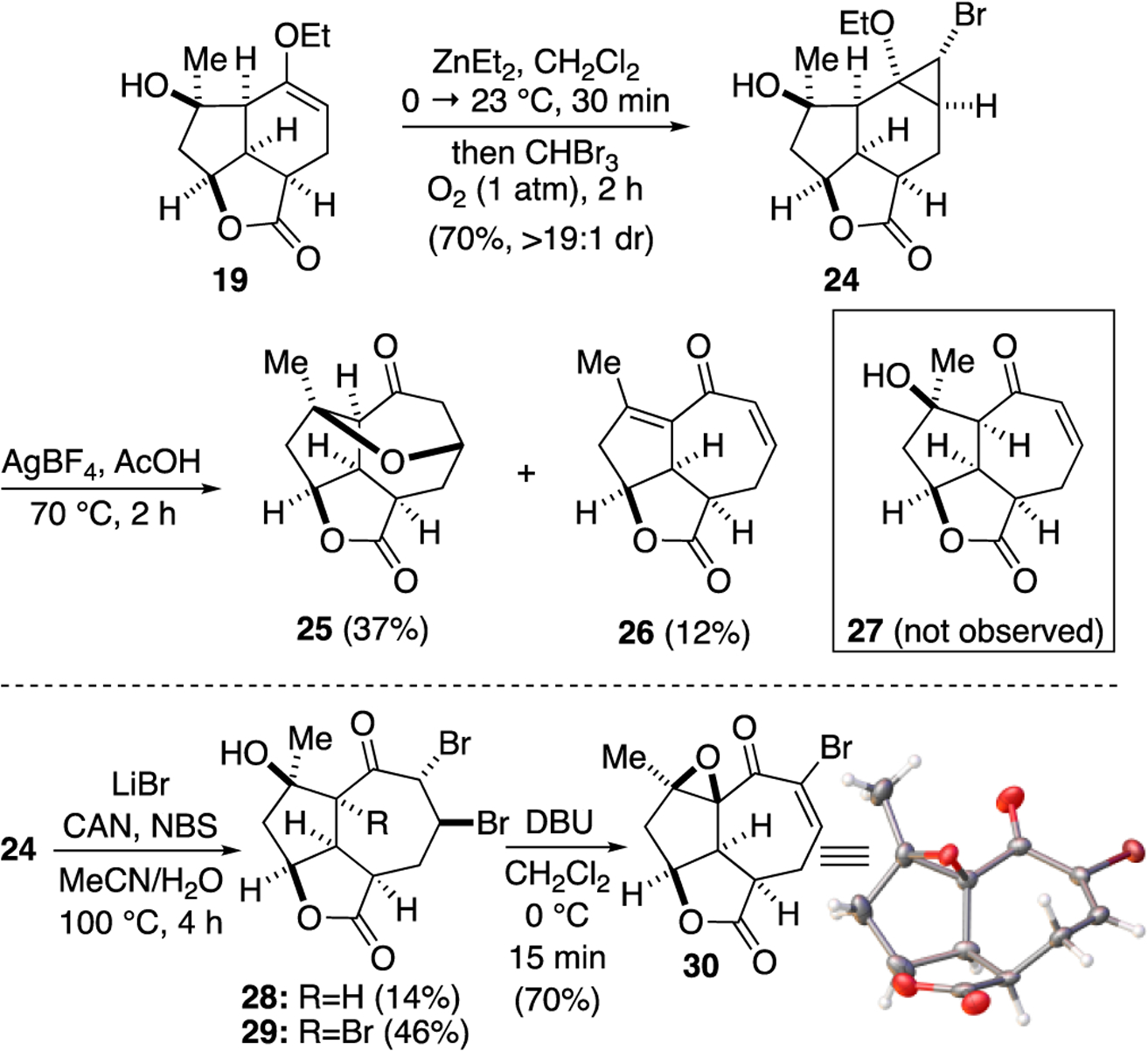
Synthesis of bromocyclopropane 24 and ring-expansions to functionalized 5,5,7 tricyclic ring systems.
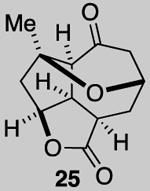
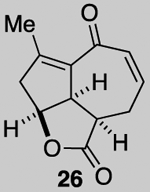
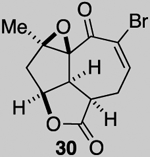
We next attempted the ring expansion utilizing various silver salts but were unsuccessful in obtaining the desired enone 27 (Scheme 5) but instead obtained pyranone 25, derived from ring expansion followed by oxa-Michael, and enone 26 derived from elimination. Since an ionic mechanism failed, we again turned to addition of a halo radical and in particular, a bromide radical through oxidation of Br- with CAN. This reaction afforded the desired ring expanded product 28 along with significant quantities of tribromide 29 (relative stereochemistry assigned by X-Ray, see SI). The latter product presumably results from the desired ring expansion followed by α-bromination. While this α-bromination was not expected, it proved extremely useful. Treatment of dibromo ketone 28 under several basic conditions to give the α-bromo enone 7 led to complex mixtures. However, treatment of tribromo ketone 29 with DBU provides the epoxy, α-bromo enone 30 in which the tertiary alkoxide again cyclizes to form an epoxide and serves to prevent elimination of the tertiary alcohol.
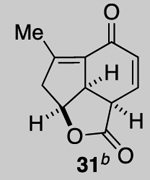
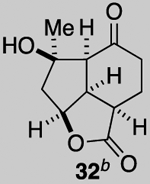
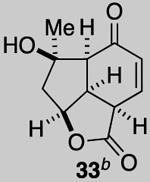
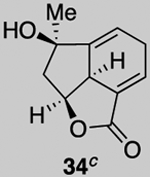
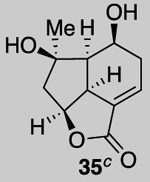
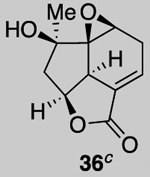
Our described PDR strategy toward rameswaralide (6) provided access to both 5,5,6- and 5,5,7-tricyclic intermediates, bearing resemblance to Sinularia natural products, that were assayed for bioactivity. Furthermore, intermediates 15 and 19 were readily transformed into three additional analogs 31-33 providing additional SAR data (see SI for synthetic details).51 In collaboration with Prof. Brian Stoltz’s group (Caltech), three additional tricyclic analogs 34-36 were assayed to widen the SAR profile.52
Cell viability assays (alamar blue viability assay, 72 h incubation) employing three cancer cell lines were performed with the described simplified Sinularia natural product derivatives (Table 1). Rameswaralide previously showed cytotoxicity against the A549 (human lung epithelial carcinoma) cell line with an IC50 of 67 ± 3.7 μM.8 We therefore assayed this cell line along with triple-negative breast cancer (MDA-MB-231) and colorectal carcinoma (HCT116) cell lines along with primary Human Umbilical Vein Endothelial cells (HUVEC) to obtain information regarding selective cytotoxicity. The presence of α,β-enones, as a result of our synthetic strategy and also present in some Sinularia family members opens the possibility of covalent modifying compounds via thio-Michael additions. However, when selectivity can be achieved with these Michael acceptors, they have found utility for drug design and identification of novel cellular targets.53
Table 1.
Cell viability data (alamar blue assay, 72 h incubation) of select intermediates toward Sinularia natural products.a
 |
 |
 |
 |
 |
|
| MDA MB 231 (EC50, μM) | >100 | >100 | >100 | >100 | >100 |
| HCT 116 (EC50, μM) | >100 | 28.90 ± 6.36 | >100 | >100 | >100 |
| A549 (EC50, μM) | >100 | >100 | >100 | >100 | >100 |
| HUVEC (EC50, μM) | >100 | >100 | >100 | >100 | >100 |
 |
 |
 |
 |
 |
|
| MDA MB 231 (EC50, μM) | 77.70 ± 21.63 | 45.40 ± 7.97 | >100 | 45.20 ± 10.19 | 10.52 ± 2.21 |
| HCT 116 (EC50, μM) | >100 | >100 | >100 | 2.97 ± 0.50 | 0.39 ± 0.03 |
| A549 (EC50, μM) | >100 | >100 | >100 | 5.69 ± 0.75 | 3.26 ± 0.35 |
| HUVEC (EC50, μM) | 62.91 ± 6.32 | 48.66 ± 4.07 | >100 | 6.88 ± 0.73 | 3.89 ± 0.33 |
While a complete SAR profile is not yet available, several interesting observations can be made from initial assays performed on the described tricyclic intermediates. Not surprisingly, the presence of α,β-enones on both the 5,5,6 and 5,5,7 core structures (i.e. 26, 30, 31) led to the greatest cytotoxicity toward all cancer cell lines however with some interesting and unexpected selectivities observed. A notable example involves the most potent analog tested, our key synthetic intermediate α-bromo enone 30, which uniquely displays nanomolar activity (0.39 ± 0.03 μM) against the HCT116 cell line with 8–27X greater potency over other cancer cell lines and also primary HUVEC. In addition, the cross-conjugated dienone 31, which displays micromolar activity against HCT116 cells (28.90 ± 6.36 μM) but is inactive up to 100 μM toward other cancer cell lines tested including primary HUVEC. Furthermore, it appears that α,β-enones present in the cyclopentane ring (C ring) do not inherently result in cytotoxicity given that enone 15, possessing a β-disubstituted enone, is inactive (up to 100 μM) toward all cell lines tested while dienone 31, possessing a β-monosubstituted cyclohexanone, displays cytotoxicity toward HCT116 cells. However, the tertiary alcohol in enone 33 appears to reduce toxicity compared to dienone 31. Overall, it is interesting to note that the 5,5,6-tricyclic cores are generally less cytotoxic to all cell lines tested compared to the 5,5,7-variants (cf. dienones 31 and 26). However, the α,β-unsaturated lactones 35 and 36 display selective cytotoxicity toward MDA-MB-231 cells but comparable cytotoxicity to HUVEC cells. The SAR gathered to date provides preliminary validation for our initial hypothesis that the 5,5,7-tricyclic core present in some of the most cytotoxic members of this family may be important for activity. However, further support for this hypothesis and the relevance of the observed bioactivities to the natural products themselves must await further studies including eventual proteomics studies to identify cellular targets.
In summary, application of a PDR strategy that employed the use of a Diels-Alder lactonization organocascade has enabled rapid access to both 5,5,6 and 5,5,7 tricyclic cores present in several Sinularia soft coral-derived natural products including rameswaralide. Preliminary cytotoxicity studies revealed some interesting cytotoxicities and selectivities that will be further verified and elaborated upon throughout our total synthesis efforts and subsequent in-depth biological studies. We anticipate that the versatile intermediate, α-bromo enone 30, will serve as a useful springboard toward several members of the Sinularia class of natural products bearing the 5,5,7-tricyclic core.
Supplementary Material
Scheme 1.
Pharmacophore Directed Retrosynthetic (PDR) approach toward rameswaralide
ACKNOWLEDGMENT
Support from NIH NIGMS (R37 GM052964 and R37 GM052964S1 to D.R. and J.O.L. (sub-contract), FARI and NCI (P30CA006973) to J.O. L., and a Ruth L. Kirschstein Predoctoral Fellowship, F31 GRANT12692796 to N.J.T is gratefully acknowledged. thank Dr. Kevin Klausmeyer for all X-ray analysis. We thank Prof. Brian Stoltz, Nicholas Hafeman, Steven Loskot, Chris Reimann, and Beau Pritchett (Caltech) for the warm collaboration leading to the more complete preliminary SAR profile presented herein.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Procedures for all synthetic transformations and characterization data for all new compounds including 1H and 13C NMR, IR, HRMS, and crystallographic data for compounds 15, 20, 29, 30 (PDF). Materials and methods for biological assays.
Crystallographic data contained in this article have been deposited at the Cambridge Crystallograpic Data Centre under deposition number CCDC 1937890 (15), 1937891 (20), 1937892 (29), 1937893 (30). The data can be accessed free of charge at https://www.ccdc.cam.ac.uk/structures/.
The authors declare no competing financial interest.
REFERENCES
- (1).Iguchi K; Kajiyama K; Yamada Y Yonarolide: A New Marine Norditerpenoid Possessing a Novel Tricyclic Skeleton, from the Okinawan Soft Coral of the Genus, Sinularia. Tetrahedron Lett. 1995, 36, 8807–8808. [Google Scholar]
- (2).Sheu J-H; Ahmed AF; Shiue R-T; Dai C-F; Kuo Y-H Scabrolides A–D, Four New Norditerpenoids Isolated from the Soft Coral Sinularia Scabra. J. Nat. Prod 2002, 65, 1904–1908. [DOI] [PubMed] [Google Scholar]
- (3).Tseng Y-J; Ahmed AF; Dai C-F; Chiang MY; Sheu J-H Sinulochmodins A–C, Three Novel Terpenoids from the Soft Coral Sinularia Lochmodes. Org. Lett 2005, 7, 3813–3816. [DOI] [PubMed] [Google Scholar]
- (4).Kobayashi M; Appa Rao KMC; Krishna MM; Anjaneyulu V Marine Terpenes and Terpenoids. Part 19. Structure of a Tetracyclic Norcembranolide Derivative Isolated from the Soft Coral Sinularia Dissecta. J. Chem. Research (.S) 1995, 188–189. [Google Scholar]
- (5).Duh C-Y; Wang S-K; Chia M-C; Chiang MY A Novel Cytotoxic Norditerpenoid from the Formosan Soft Coral Sinularia Inelegans. Tetrahedron Lett. 1999, 40, 6033–6035. [Google Scholar]
- (6).Ramesh P; Reddy NS; Venkateswarlu Y; Reddy MVR; Faulkner DJ Rameswaralide, a Novel Diterpenoid from the Soft Coral Sinularia Dissecta. Tetrahedron Lett. 1998, 39, 8217–8220. [Google Scholar]
- (7).Faulkner DJ; Venkateswarlu Y; Raghavan KV; Yadav JS Rameswaralide and Rameswaralide Derivatives. US6300371B1, October 9, 2001. [Google Scholar]
- (8).Chitturi BR; Tatipamula VB; Dokuburra CB; Mangamuri UK; Tuniki VR; Kalivendi SV; Bunce RA; Yenamandra V Pambanolides A–C from the South Indian Soft Coral Sinularia Inelegans. Tetrahedron 2016, 72, 1933–1940. [Google Scholar]
- (9).Li Y; Pattenden G Perspectives on the Structural and Biosynthetic Interrelationships between Oxygenated Furanocembranoids and Their Polycyclic Congeners Found in Corals. Nat. Prod. Rep 2011, 28, 1269–1310. [DOI] [PubMed] [Google Scholar]
- (10).Mehta G; Lakshminath S Synthetic Studies towards the Novel Diterpenoid Rameswaralide: RCM Mediated Acquisition of the Tricyclic Core. Tetrahedron Lett. 2006, 47, 327–330. [Google Scholar]
- (11).Trost BM; Nguyen HM; Koradin C Synthesis of a Tricyclic Core of Rameswaralide. Tetrahedron Lett. 2010, 51, 6232–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Srikrishna A; Dethe DH Synthetic Approaches to Guanacastepenes. Enantiospecific Syntheses of BC and AB Ring Systems of Guanacastepenes and Rameswaralide. Org. Lett 2004, 6, 165–168. [DOI] [PubMed] [Google Scholar]
- (13).Pattenden G; Winne JM An Intramolecular [4+3]-Cycloaddition Approach to Rameswaralide Inspired by Biosynthesis Speculation. Tetrahedron Lett. 2009, 50, 7310–7313. [Google Scholar]
- (14).Palframan MJ; Pattenden G Elaboration of the Carbocyclic Ring Systems in Plumarellide and Rameswaralide Using a Coordinated Intramolecular Cycloaddition Approach, Based on a Common Biosynthesis Model. Tetrahedron Lett. 2013, 54, 324–328. [Google Scholar]
- (15).Moeller KD Intramolecular Anodic Olefin Coupling Reactions: Using Radical Cation Intermediates to Trigger New Umpolung Reactions. Synlett 2009, 1208–1218. [Google Scholar]
- (16).Tang F; Moeller KD Intramolecular Anodic Olefin Coupling Reactions: The Effect of Polarization on Carbon–Carbon Bond Formation. J. Am. Chem. Soc 2007, 129, 12414–12415. [DOI] [PubMed] [Google Scholar]
- (17).Tang F; Moeller KD Anodic Oxidations and Polarity: Exploring the Chemistry of Olefinic Radical Cations. Tetrahedron 2009, 65, 10863–10875. [Google Scholar]
- (18).Horn EJ; Silverston JS; Vanderwal CD A Failed Late-Stage Epimerization Thwarts an Approach to Ineleganolide. J. Org. Chem 2016, 81, 1819–1838. [DOI] [PubMed] [Google Scholar]
- (19).Roizen JL; Jones AC; Smith RC; Virgil SC; Stoltz BM Model Studies To Access the [6,7,5,5]-Core of Ineleganolide Using Tandem Translactonization–Cope or Cyclopropanation–Cope Rearrangements as Key Steps. J. Org. Chem 2017, 82, 13051–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Craig Robert A. I. I.; Roizen, J. L; Smith, R. C; Jones, A. C; Virgil, S. C; Stoltz, B. M Enantioselective, Convergent Synthesis of the Ineleganolide Core by a Tandem Annulation Cascade. Chem. Sci 2017, 8, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Craig RA; Smith RC; Roizen JL; Jones AC; Virgil SC; Stoltz BM Development of a Unified Enantioselective, Convergent Synthetic Approach Toward the Furanobutenolide-Derived Polycyclic Norcembranoid Diterpenes: Asymmetric Formation of the Polycyclic Norditerpenoid Carbocyclic Core by Tandem Annulation Cascade. J. Org. Chem 2018, 83, 3467–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ueda Y; Abe H; Iguchi K; Ito H Synthetic Study of Yonarolide: Stereoselective Construction of the Tricyclic Core. Tetrahedron Lett. 2011, 52, 3379–3381. [Google Scholar]
- (23).Deng M; Zhang X; Li Z; Chen H; Zang S; Liang G Rapid Construction of the Common [5–5–6] Tricyclic Ring Skeleton in Polycyclic Cembranoids and Norcembranoids via Intramolecular 1,3-Dipolar Cycloaddition. Org. Lett 2019, 21, 1493–1496. [DOI] [PubMed] [Google Scholar]
- (24).Liu G Beta-Lactones as Synthetic Vehicles in Natural Product Synthesis: Total Syntheses of Schulzeines B & C and Omphadiol, and Studies toward the Total Syntheses of Scabrolides A & B and Sinulochmodin C. Thesis, 2012. [Google Scholar]
- (25).Craig RA; Stoltz BM Polycyclic Furanobutenolide-Derived Cembranoid and Norcembranoid Natural Products: Biosynthetic Connections and Synthetic Efforts. Chem. Rev 2017, 117, 7878–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Abbasov ME; Alvariño R; Chaheine CM; Alonso E; Sánchez JA; Conner ML; Alfonso A; Jaspars M; Botana LM; Romo D Simplified Immunosuppressive and Neuroprotective Agents Based on Gracilin A. Nat. Chem 2019, 11, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cheng S-Y; Chuang C-T; Wen Z-H; Wang S-K; Chiou S-F; Hsu C-H; Dai C-F; Duh C-Y Bioactive Norditerpenoids from the Soft Coral Sinularia Gyrosa. Bioorg. Med. Chem 2010, 18, 3379–3386. [DOI] [PubMed] [Google Scholar]
- (28).Thao NP; Nam NH; Cuong NX; Quang TH; Tung PT; Dat LD; Chae D; Kim S; Koh Y-S; Kiem PV; et al. Anti-Inflammatory Norditerpenoids from the Soft Coral Sinularia Maxima. Bioorg. Med. Chem. Lett 2013, 23, 228–231. [DOI] [PubMed] [Google Scholar]
- (29).Abbasov ME; Hudson BM; Tantillo DJ; Romo D Acylammonium Salts as Dienophiles in Diels–Alder/Lactonization Organocascades. J. Am. Chem. Soc 2014, 136, 4492–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Abbasov ME; Hudson BM; Tantillo DJ; Romo D Stereodivergent, Diels–Alder-Initiated Organocascades Employing α,β-Unsaturated Acylammonium Salts: Scope, Mechanism, and Application. Chem. Sci 2017, 8, 1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Piancatelli G; Scettri A; Barbadoro S A Useful Preparation of 4-Substituted 5-Hydroxy-3-Oxocyclopentene. Tetrahedron Lett. 1976, 17, 3555–3558. [Google Scholar]
- (32).Song L; Yao H; Zhu L; Tong R Asymmetric Total Syntheses of (−)-Penicipyrone and (−)-Tenuipyrone via Biomimetic Cascade Intermolecular Michael Addition/Cycloketalization. Org. Lett 2013, 15, 6–9. [DOI] [PubMed] [Google Scholar]
- (33).Saitman A; Theodorakis EA Synthesis of a Highly Functionalized Core of Verrillin. Org. Lett 2013, 15, 2410–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yang P; Yao M; Li J; Li Y; Li A Total Synthesis of Rubriflordilactone B. Angew. Chem. Int. Ed 2016, 55, 6964–6968. [DOI] [PubMed] [Google Scholar]
- (35).Davies HML; Calvo RL; Townsend RJ; Ren P; Churchill RM An Exploratory Study of Type II [3 + 4] Cycloadditions between Vinylcarbenoids and Dienes. J. Org. Chem 2000, 65, 4261–4268. [DOI] [PubMed] [Google Scholar]
- (36).Simmons HE; Smith RD A New Synthesis of Cyclopropanes from Olefins. J. Am. Chem. Soc 1958, 80, 5323–5324. [Google Scholar]
- (37).Furukawa J; Kawabata N; Nishimura J Synthesis of Cyclopropanes by the Reaction of Olefins with Dialkylzinc and Methylene Iodide. Tetrahedron 1968, 24, 53–58. [Google Scholar]
- (38).Ito Y; Fujii S; Saegusa T Reaction of 1-Silyloxybicyclo[n.1.0]Alkanes with Iron(III) Chlorides. A Facile Synthesis of 2-Cycloalkenones via Ring Enlargement of Cyclic Ketones. J. Org. Chem 1976, 41, 2073–2074. [Google Scholar]
- (39).Beak P; Meyers AI Stereo- and Regiocontrol by Complex Induced Proximity Effects: Reactions of Organolithium Compounds. Acc. Chem. Res 1986, 19, 356–363. [Google Scholar]
- (40).Denmark SE; Edwards JP; Wilson SR Solution- and Solid-State Structural Studies of (Halomethyl)Zinc Reagents. J. Am. Chem. Soc 1992, 114, 2592–2602. [Google Scholar]
- (41).Kim HY; Salvi L; Carroll PJ; Walsh PJ Highly Enantio- and Diastereoselective One-Pot Methods for the Synthesis of Halocyclopropyl Alcohols. J. Am. Chem. Soc 2009, 131, 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chan JHH; Rickborn B Relative Rates and Stereochemistry of the Iodomethylzinc Iodide Methylenation of Some Hydroxy- and Methoxy-Substituted Cyclic Olefins. J. Am. Chem. Soc 1968, 90, 6406–6411. [Google Scholar]
- (43).Takahashi H; Yoshioka M; Ohno M; Kobayashi S A Catalytic Enantioselective Reaction Using a C2-Symmetric Disulfonamide as a Chiral Ligand: Cyclopropanation of Allylic Alcohols by the Et2Zn-CH2I2-Disulfonamide System. Tetrahedron Lett. 1992, 33, 2575–2578. [Google Scholar]
- (44).Denmark SE; O’Connor SP Enantioselective Cyclopropanation of Allylic Alcohols. The Effect of Zinc Iodide. J. Org. Chem 1997, 62, 3390–3401. [DOI] [PubMed] [Google Scholar]
- (45).Lebel H; Marcoux J-F; Molinaro C; Charette AB Stereoselective Cyclopropanation Reactions. Chem. Rev 2003, 103, 977–1050. [DOI] [PubMed] [Google Scholar]
- (46).Nair V; Panicker SB; Mathai S Bromination of Cyclopropanes Using Potassium Bromide and Cerium(IV) Ammonium Nitrate (CAN): Synthesis of 1,3-Dibromides. Res. Chem. Intermed 2003, 29, 227–231. [Google Scholar]
- (47).Jiao J; Nguyen LX; Patterson DR; Flowers RA An Efficient and General Approach to β-Functionalized Ketones. Org. Lett 2007, 9, 1323–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tsuchida H; Tamura M; Hasegawa E Cyclization and Ring-Expansion Processes Involving Samarium Diiodide Promoted Reductive Formation and Subsequent Oxidative Ring Opening of Cyclopropanol Derivatives. J. Org. Chem 2009, 74, 2467–2475. [DOI] [PubMed] [Google Scholar]
- (49).Miyano S; Hashimoto H Cyclopropanes from Olefins by the Oxygen-Accelerated Zinc-Carbenoid Reaction. Bull. Chem. Soc. Jpn 1973, 46, 892–897. [Google Scholar]
- (50).Miyano S; Hashimoto H Oxygen-Accelerated Generation of the Bromocarbenoid Reagent of Zinc from Diethylzinc and Bromoform. Bromocyclopropanes from Olefins. Bull. Chem. Soc. Jpn 1975, 48, 3665–3668. [Google Scholar]
- (51).Morrill LA; Susick RB; Chari JV; Garg NK Total Synthesis as a Vehicle for Collaboration. J. Am. Chem. Soc 2019, 141, 12423–12443. [DOI] [PubMed] [Google Scholar]
- (52).Unpublished results of: Hafeman NJ; Loskot SA; Reimann CE; Pritchett BP; Stoltz BM.
- (53).Jackson PA; Widen JC; Harki DA; Brummond KM Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem 2017, 60, 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.