

Abstract
Although second-generation HIV integrase strand-transfer inhibitors (INSTIs) are prescribed throughout the world, the mechanistic basis for the superiority of these drugs is poorly understood. We used single-particle cryo–electron microscopy to visualize the mode of action of the advanced INSTIs dolutegravir and bictegravir at near-atomic resolution. Glutamine-148→histidine (Q148H) and glycine-140→serine (G140S) amino acid substitutions in integrase that result in clinical INSTI failure perturb optimal magnesium ion coordination in the enzyme active site. The expanded chemical scaffolds of second-generation compounds mediate interactions with the protein backbone that are critical for antagonizing viruses containing the Q148H and G140S mutations. Our results reveal that binding to magnesium ions underpins a fundamental weakness of the INSTI pharmacophore that is exploited by the virus to engender resistance and provide a structural framework for the development of this class of anti-HIV/AIDS therapeutics.
One Sentence Summary:
Metal ion coordination underlies both strengths and weaknesses of HIV strand transfer inhibitors.
Despite immediate clinical impact, the first in-class INSTI raltegravir (RAL) suffered setbacks from the emergence of viral resistance (1). Although second-generation INSTIs dolutegravir (DTG) and bictegravir (BIC) display improved activity against RAL-resistant strains (2, 3), the advanced compounds are not immune to resistance (3–7). In particular, Q148H/G140S changes in HIV-1 IN are associated with complete or partial loss of efficacy across the entire drug class. The mode of INSTI binding to the IN active site was first visualized in the context of the prototype foamy virus (PFV) intasome (8). However, the limited ~15% amino acid sequence identity between PFV and HIV-1 INs greatly restricts the utility of PFV for studies of INSTI resistance and precludes its use as a template for structure-based lead optimization. Conversely, unfavorable biochemical properties of the HIV-1 intasome have impeded structural refinements to atomic resolution (9).
In order to establish a robust experimental system suitable for informing INSTI development, we evaluated IN proteins from primate lentiviruses that are highly related to circulating strains of HIV-1. The simian immunodeficiency virus from red-capped mangabeys (SIVrcm) is a direct ancestor of chimpanzee SIV (10, 11). Because the HIV-1 pol gene is originally derived from SIVrcm, the viruses share as much as 75% IN amino acid sequence identity (fig. S1). SIVrcm IN displayed robust strand transfer activity in vitro, which was stimulated by the lentiviral IN host factor LEDGF/p75 (12, 13). Reaction conditions were conducive for the formation of stable nucleoprotein complexes, which were competent for strand transfer activity and sensitive to INSTI inhibition (figs S2, S3A). Examination of the material by negative stain electron microscopy (EM) revealed a heterogenous population with the prominent presence of long linear polymers (hereafter referred to as stacks, Fig. 1A). Reference-free classification revealed 2D averages that were strikingly similar to those observed in maedi-visna virus (MVV) intasome preparations (Fig. 1A, fig. S4) (14). However, while the latter behaved as a near-monodispersed population with a predominance of hexadecamers (tetramer-of-tetramers) of IN, the flanking IN tetramers of SIVrcm intasomes were notably disordered, often nucleating stack formation. Although HIV-1 IN assembly was much less efficient, it yielded particles visually indistinguishable from SIVrcm intasomes (figs S3B, S5–6). These observations are consistent with polydispersity previously reported in HIV-1 intasomes assembled with a hyperactive IN mutant (9). 2D class averages apparently corresponding to the dodecameric assembly from that study were readily identified in our wild type HIV-1 and SIVrcm intasome images (Fig. 1A, fig. S5).
Fig. 1: Reconstruction of the SIVrcm intasome core.
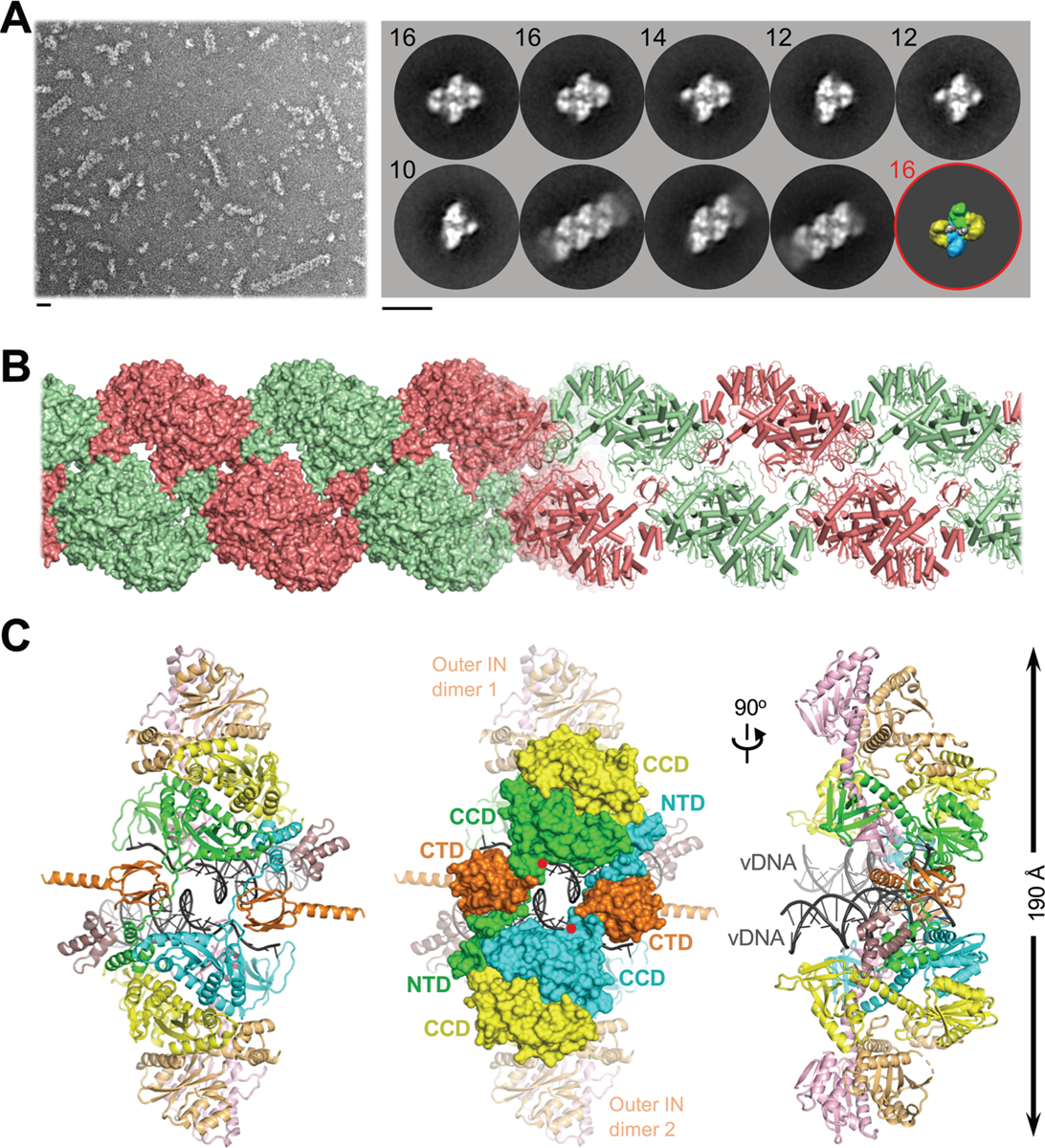
(A) Raw image (left) and 2D class averages (right) of negatively stained SIVrcm intasome particles; apparent numbers of IN subunits are indicated for non-stacked classes. Particle distributions are given in fig. S4. The envelope of the hexadecameric maedi-visna virus intasome (red circle; central and flanking IN tetramers in blue/green and yellow, respectively) is shown for comparison; scale bars are 0.2 nm. (B) Atomistic reconstruction of the SIVrcm intasome stack shown as space fill (left) and cartoons (right); separate repeat units are shown in alternating red and green colors. (C) Detailed view of a single intasomal repeat representing a pair of viral DNA ends (vDNA, grey cartoons) synapsed between a pair of IN tetramers (composed of yellow, orange, pink, and either green or cyan IN protomers; the active sites of the green and cyan molecules (red dots) catalyze DNA recombination). The repeat unit is completed by pairs of C-terminal (orange) and N-terminal (dark magenta) domains donated by IN chains belonging to neighboring repeats. These CTDs are critical to form the conserved intasome core (CIC), which is shown in space fill mode in the middle panel. CCD, catalytic core domain.
We recorded micrograph movies of unstained SIVrcm intasome stacks in vitreous ice using a direct electron detector and refined the cryo-EM structure of an averaged intasome repeat unit. To prevent DNA binding to the target binding groove, which would occlude INSTI occupancy (15), the intasomes were prepared using A119D IN that precludes target DNA capture without affecting IN active site function (16–18). The overall resolution of the reconstruction throughout the conserved intasome core (CIC) was 3.3 Å, while the local resolution of the active site region approached 2.8 Å (figs S7–8, table S1). In agreement with the resolution metrics, the cryo-EM density map was sufficiently detailed to build and refine an atomic model (fig. S9). The resulting model encompassed two IN tetramers with associated viral DNA ends, as well as two pairs of C- and N-terminal domains (CTDs and NTDs) donated by flanking stack units (Fig. 1B, C). Exchange of NTDs and CTDs between neighboring intasomes forms the structural basis for stack formation (Fig. 1B, fig. S10).
Using available nucleotide sequence data (10), we engineered recombinant SIVrcm and evaluated its sensitivity to INSTIs (fig. S11A). First- (RAL) and second- (DTG, BIC) generation INSTIs inhibited HIV-1 and SIVrcm at similar half-maximal effective concentrations (EC50) (Fig. 2A). Q148H/G140S changes in IN rendered HIV-1 and SIVrcm significantly resistant to RAL, by a factor of >2000, whereas EC50 values of the second-generation INSTIs BIC and DTG increased similarly ~5- to-8-fold against HIV-1 and 40 to 73-fold against SIVrcm (fig. S11B). Importantly, the majority of residues that when altered confer INSTI resistance are conserved between HIV-1 and SIVrcm (fig. S1). An exception is Thr138: in HIV-1, E138T potentiates resistance of Q148H-containing viruses (19, 20). Concordantly, reverting Thr138 to Glu decreased DTG and BIC resistance of Q148H/G140S SIVrcm to the levels observed with HIV-1 Q148H/G140S. Moreover, T97A/L74M, which increase resistance of Q148H/G140S HIV-1 to second-generation INSTIs (7), exerted the same effect on SIVrcm (fig. S11B).
Fig. 2: Binding modes of second generation INSTIs in the IN active site.
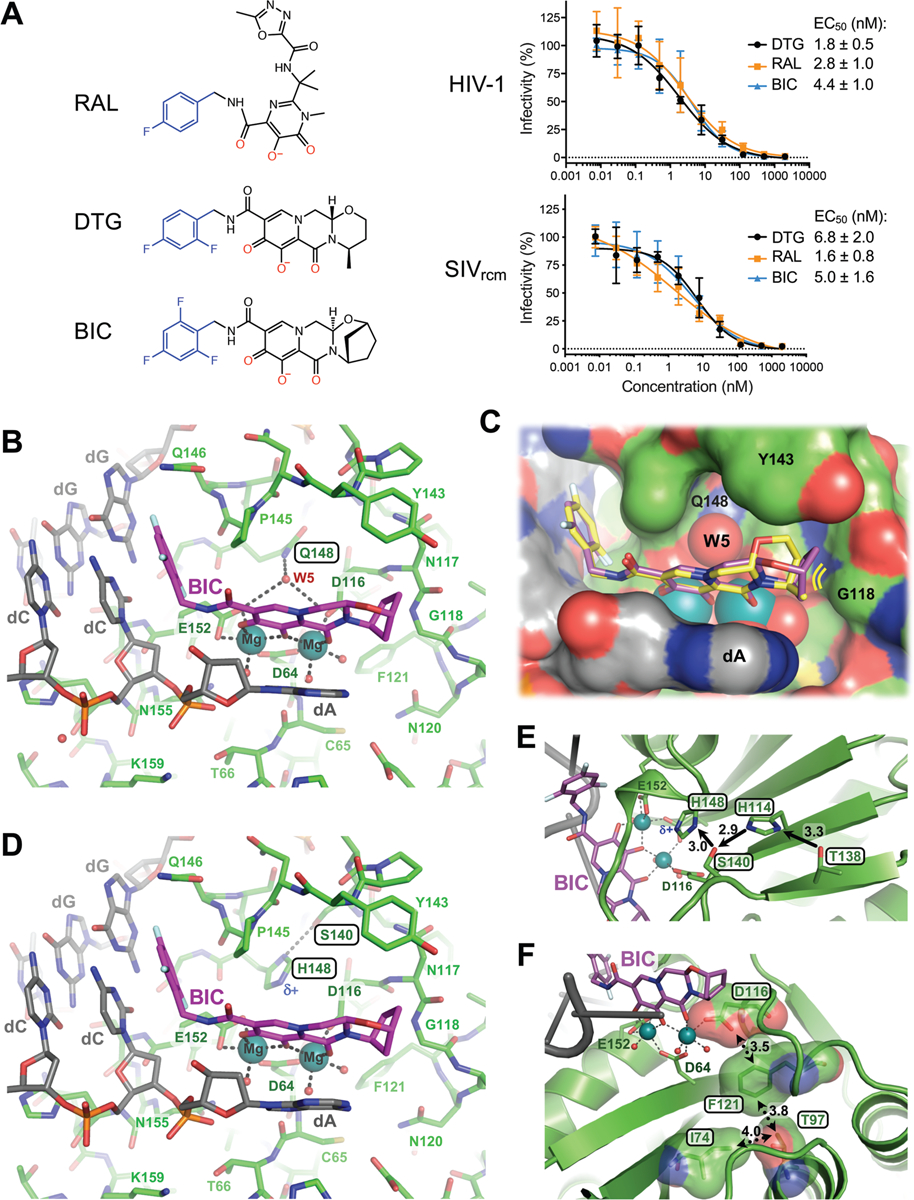
(A) Chemical structures of select first- (RAL) and second-generation (DTG, BIC) INSTIs (left; halo-benzyl groups in blue and metal-chelating oxygen atoms in red) and viral sensitivities (right). Results are averages and standard deviations of minimally n = 2 experiments, with each experiment conducted in triplicate; EC50 values are noted. (B) Active site of the SIVrcm intasome in complex with BIC; protein, DNA, and drug are shown as sticks. Blue spheres are Mg2+ ions, water molecules are shown as small red spheres. (C) Superposition of BIC (magenta) and DTG (yellow) bound structures with protein and DNA shown in space-fill mode. Yellow lines accentuate proximity to IN β4-α2 connector. (D) Q148H/G140S active site bound to BIC. δ+ indicates increased electropositivity of the His148 Nε2 proton. (E) The extended hydrogen bond network that couples Thr138 to His148 in the Q148H/G140S SIVrcm intasome. Black arrows indicate hydrogen bond donation; the corresponding interatomic distances are given in Ångstroms. Cryo-EM map of the same region is shown in fig. S20. (F) Long-range interactions of Ile74 and Thr97 with the metal chelating cluster via Phe121. Key amino acid residues are shown as sticks and semi-transparent van der Waals surfaces. Contacts between side chain atoms are indicated by double-headed dotted arrows with distances given in Ångstroms. Cryo-EM map showing definition of the side chain rotamers and the effects of I74M/T97A substitutions on the Phe121 side chain are shown in fig. S21.
Encouraged by these results, we acquired cryo-EM data on SIVrcm intasomes vitrified in the presence of INSTIs and Mg2+ ions. DTG- and BIC-bound structures were reconstructed to resolutions of 3.0 and 2.6 Å across the CIC, with local resolutions within active site regions of 2.8 and 2.4 Å, respectively (figs S7–8, table S1). The inhibitors were defined remarkably well in density maps, allowing their refinements with bound Mg2+ ions and associated water molecules (fig. S12–13, movie S1). The invariant IN active site carboxylates Asp64, Asp116, and Glu152 coordinate a pair of Mg2+ ions, which in turn interact with the metal chelating cores of the INSTIs (Fig. 2B, C). As previously observed in PFV intasome crystals (8), the drugs displace the 3′ viral DNA nucleotide, which stacks against the central body of the INSTI (fig. S14). In agreement with low-level amino acid sequence identity, there are considerable differences in the environment of the small molecules in the SIVrcm and PFV structures (fig. S15).
The map of the BIC complex revealed an interaction between the side chain amide of Gln148 and the carboxylates of metal-chelating residues Glu152 and Asp116 via a water molecule (W5, Fig. 2B). Molecular dynamics simulations confirmed stability of this hydrogen bonding network (fig. S16A). DTG and BIC intimately contact the backbone atoms of Asn117 and Gly118 from the IN β4-α2 connector, making respectively 8 and 12 contacts with interatomic distances ≤5 Å. Moreover, BIC makes three contacts with interatomic distances of 3.9–4.0 Å within this region of the active site. We obtained a truncated INSTI derivative lacking the heterocycle involved in these interactions to test their importance to drug potency (analogue 1, Fig. 3A). This modification was not expected to impact the metal chelating properties of the compound or its ability to stack with DNA bases, and indeed analogue 1 and DTG similarly inhibited HIV-1 infection. However, in contrast to DTG, analogue 1 was ~80-fold less effective against HIV-1 Q148H/G140S (Fig. 3B). In agreement with published work (21), the amino acid substitutions increased the dissociative rate of DTG from HIV-1 intasomes, while their impact on the truncated derivative was much greater (Fig. 3A). Collectively, these data implicate contacts with the β4-α2 connector as a crucial feature of the second-generation INSTIs.
Fig. 3: Effects of Q148H/G140S substitutions on DTG and analogue 1 activities.
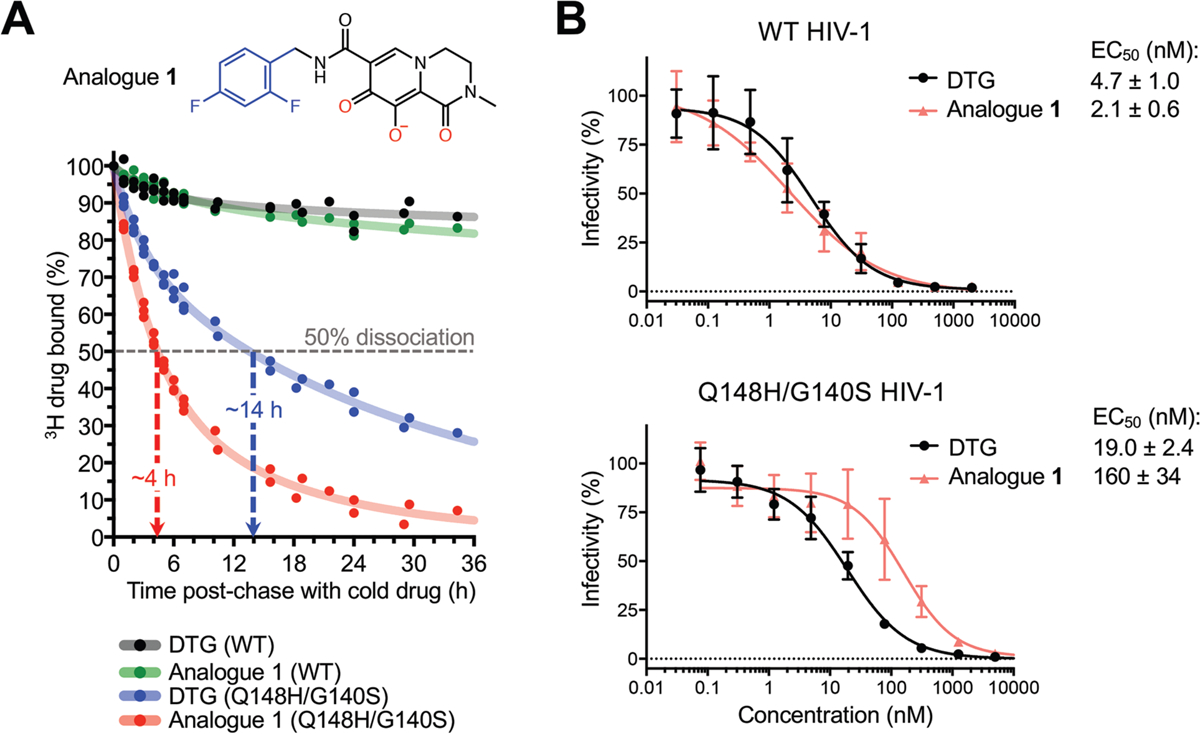
(A) Structure of analogue 1 (top; colors as in Fig. 2A) and a time course of 3H-DTG and analogue 1 dissociation from wild type and Q148H/G140S HIV-1 intasomes (bottom). Results from three independent experiments are plotted; each data point is an average of two measurements done in parallel; trendlines are for illustration purpose. Apparent INSTI dissociative half-times from the mutant intasome are indicated. (B) Activities of DTG and analogue 1 against wild type (top) and Q148H/G140S (bottom) HIV-1. Results are averages and standard deviations of two independent experiments, with each experiment conducted in triplicate.
To visualize the impact of the Q148H/G140S substitutions on drug binding, we imaged mutant SIVrcm intasomes in complex with BIC to a local resolution of 2.8 Å (figs S7–8, S17A, table S1). Ser140 and His148 side chains directly interact, and the latter positioned within 3.3 Å of the metal-chelating Glu152 carboxylate (Fig. 2D, fig. S17B). In the refined model, steric clashes between the side chains are avoided by a 0.5-Å shift at the His148 Cα atom. Importantly, local crowding due to insertion of the mutant His148 side chain expelled water molecule W5 (fig. S16B), thus disturbing the secondary coordination shell of the Mg2+ ions. We also note that the amino acid changes caused a ~0.5-Å shift in the position of the bound drug; while arguably minor given the resolution of the cryo-EM map, the observed displacement agrees precisely with predictions by computational chemistry, illustrating the effect of the substitutions on drug binding. The Nε2 atom of His148 intimately contacts the carboxylate of Glu152 (3.3 Å, fig. S17), which is involved in bidentate coordination with one of the Mg2+ atoms. Importantly, the acidity of His148 Nε2 is increased due to hydrogen bonding of Nδ1 with Ser140 (Fig. 2D). The Ser140-His148-Glu152 coupling is strikingly reminiscent of the non-catalytic Ser-His-Glu triad proposed as a stability determinant in α-amylases, representing a reversal of the charge relay system in hydrolase active sites (22, 23). However, hydrogen bonding would require reorientation of IN Glu152 and His148 side chains, which would be incompatible with Mg2+ ion coordination and drug binding, suggesting an empirical interpretation of the INSTI resistance mechanism.
Our simulations show that analogue 1 is considerably more dynamic in the active site compared to the full-sized molecule, the mobility of which is restricted through interactions with the β4-α2 connector (fig. S18). The additional degree of freedom is expected to allow more extensive re-orientation of the truncated inhibitor, which may permit His148 to withdraw more electron density from the Mg2+-ligand cluster. Our natural bond orbital analysis illustrates the changes of atomic charge distribution within the cluster in response to polarization by protonated His148 Nε2 and subtle conformational adaptations (fig. S19). It is easy to extend these observations to the other substitutions at position 148, Lys and Arg, both of which introduce electropositive functionalities to yield high-level INSTI resistance (4).
Further work will be required to unravel long-range interactions involved in boosting INSTI resistance by secondary changes such as E138T and L74M/T97A (19, 20). As a start, we analyzed respective side chains in our SIVrcm Q148H/G140S intasome structure. Thr138 is ideally positioned to hydrogen bond with Nδ1 of conserved residue His114, prompting it to donate its Nε2 proton to Ser140 (Fig. 2E, fig. S20). This extended network, which may form a proton wire, is expected to reinforce Ser140 as a hydrogen bond donor for its interaction with His148 Nδ1, explaining why the E138T substitution can enhance the resistance of Q148H/G140S HIV-1 (19, 20). SIVrcm IN residues Ile74 (the position occupied by Leu or Ile in HIV-1 strains) and Thr97 are in close proximity to the side chain of conserved Phe121, which is involved in van der Waals interactions with the metal-chelating carboxylate of Asp116 (Fig. 2F, fig. 21A). Readjustment of the Phe121 side chain in response to changes in its local packing environment serves as a likely conduit to perturb the structural integrity of the metal-chelating cluster (fig. S21B).
The interactions with Mg2+ ions, which are nearly covalent in nature, are partly responsible for the extraordinary tight binding of INSTIs. Our results reveal that the chink in the armor of this drug class, exploited by the virus, is the extreme sensitivity of metal ions for the precise geometry and electronic properties of the ligand cluster (24, 25). Each DNA-bound IN active site within the intasome catalyzes just one strand transfer event, allowing the virus to balance INSTI resistance by detuning its active site while retaining sufficient replication capacity. However, extending the small molecules towards the IN backbone helps to stabilize optimal binding geometry and improve the resilience of the drug in the face of INSTI resistance mutations. We note that although DTG and BIC maximally extend to the β4-α2 connector, they leave substantial free space in the IN active site, which is occupied by solute molecules in our structures (movie S1). Extension of the INSTI scaffolds to fill this space should be explored for the development of improved compounds.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to R. Carzaniga for the maintenance of Vitrobot and Tecnai G2 microscope and user training; P. Walker, A. Purkiss and M. Oliveira for computer and software support; A. Costa, P. Rosenthal, and J. Locke for generous advice and help with cryo-EM screening; A. Costa for critical reading of the manuscript.
Funding: This research was funded by US National Institutes of Health grant P50 AI150481 (P.C. and A.N.E.), R01 AI070042 (A.N.E.), and the Francis Crick Institute (P.C.), which receives its core funding from Cancer Research UK (FC001061), the UK Medical Research Council (FC001061), and the Wellcome Trust (FC001061). E.R. acknowledges funding from EPSRC (EP/R013012/1) and ERC (project 757850 BioNet). This project made use of time on ARCHER granted via the UK High-End Computing Consortium for Biomolecular Simulation, supported by EPSRC (EP/R029407/1).
Footnotes
Competing interests: A.N.E. reports fees from ViiV Helathcare Co.; no other authors declare competing financial interests.
Data and materials availability: All manuscript data are available. The cryo-EM maps were deposited with the Electron Microscopy Data Bank (accession codes 10041, 10042, 10043, and 10044) and the refined models with the Protein Data Bank (6RWL, 6RWM, 6RWN, and 6RWO).
Supplementary Materials:
Materials and Methods
Figs S1–S27
Table S1
Movie S1
References (26–64)
REFERENCES AND NOTES
- 1.Anstett K, Brenner B, Mesplede T, Wainberg MA, Retrovirology 14, 36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johns BA et al. , J Med Chem 56, 5901 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Oliveira M et al. , Retrovirology 15, 56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith SJ, Zhao XZ, Burke TR Jr., Hughes SH, Retrovirology 15, 37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pham HT et al. , J Infect Dis 218, 698 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wijting IEA et al. , J Infect Dis 218, 688 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Zhang WW et al. , J Infect Dis 218, 1773 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P, Nature 464, 232 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Passos DO et al. , Science 355, 89 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahuka-Mundeke S et al. , J Gen Virol 91, 2959 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharp PM, Shaw GM, Hahn BH, J Virol 79, 3891 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cherepanov P, Nucleic Acids Res 35, 113 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hare S et al. , PLoS Pathog 5, e1000259 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballandras-Colas A et al. , Science 355, 93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Espeseth AS et al. , Proc Natl Acad Sci U S A 97, 11244 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konsavage WM Jr., Burkholder S, Sudol M, Harper AL, Katzman M, J Virol 79, 4691 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nowak MG, Sudol M, Lee NE, Konsavage WM Jr., Katzman M, Virology 389, 141 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Maertens GN, Hare S, Cherepanov P, Nature 468, 326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shafer RW, J Infect Dis 194 Suppl 1, S51 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George JM et al. , Open Forum Infect Dis 5, ofy221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hightower KE et al. , Antimicrob Agents Chemother 55, 4552 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blow D, Nature 343, 694 (1990). [DOI] [PubMed] [Google Scholar]
- 23.Marx JC, Poncin J, Simorre JP, Ramteke PW, Feller G, Proteins 70, 320 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Maguire ME, Cowan JA, Biometals 15, 203 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Harding MM, Acta Crystallogr D Biol Crystallogr 57, 401 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.